

Abstract
Background
A 31-gene expression profile (GEP) test that provides risk classification of cutaneous melanoma (CM) patients has been validated in several retrospective studies. The objective of the reported study was a prospective evaluation of the GEP performance in patients enrolled in two clinical registries.
Methods
Three-hundred twenty two CM patients enrolled in the EXPAND (NCT02355587) and INTEGRATE (NCT02355574) registries met the criteria of age ≥ 16 years, successful GEP result and ≥1 follow-up visit for inclusion in this interim analysis. Primary endpoints were recurrence-free (RFS), distant metastasis-free (DMFS), and overall survival (OS).
Results
Median follow-up was 1.5 years for event-free patients. Median age for subjects was 58 years (range 18–87) and median Breslow thickness was 1.2 mm (range 0.2–12.0). Eighty-eight percent (282/322) of cases had stage I/II disease and 74% (237/322) had a SLN biopsy. Seventy-seven percent (248/322) had class 1 molecular profiles. 1.5-year RFS, DMFS, and OS rates were 97 vs. 77%, 99 vs. 89%, and 99 vs. 92% for class 1 vs. class 2, respectively (p < 0.0001 for each). Multivariate Cox regression showed Breslow thickness, mitotic rate, and GEP class to significantly predict recurrence (p < 0.01), while tumor thickness was the only significant predictor of distant metastasis and overall survival in this interim analysis.
Conclusions
Interim analysis of patient outcomes from a combined prospective cohort supports the 31-gene GEP’s ability to stratify early-stage CM patients into two groups with significantly different metastatic risk. RFS outcomes in this real-world cohort are consistent with previously published analyses with retrospective specimens. GEP testing complements current clinicopathologic features and increases identification of high-risk patients.
Trial registration
ClinicalTrials.gov, NCT02355574 and NCT02355587
Electronic supplementary material
The online version of this article (10.1186/s13045-017-0520-1) contains supplementary material, which is available to authorized users.
Keywords: Gene expression profiling, DecisionDx-Melanoma, Cutaneous melanoma, Metastasis, Prognosis, Staging
Background
The majority of cutaneous melanoma (CM) patients are diagnosed with early-stage (AJCC stage I or II) disease and are considered to have a favorable prognosis [1, 2]. However, two-thirds of all melanoma deaths occur in patients initially included in this “low-risk” group, which indicates that metastatic risk is underestimated for a substantial number of early-stage patients [1–4]. A recent study reporting data from the Surveillance, Epidemiology, and End Results (SEER) program reported that the mortality rate for cutaneous melanoma is increasing faster than the incidence rate, highlighting the need for additional prognostic tools to supplement standard clinicopathologic factors and improve identification of high-risk disease [5]. More vigilant follow-up for high-risk melanoma patients, including the addition of imaging and increasing clinical assessment frequency, leads to earlier detection of asymptomatic distant metastatic disease, when tumor burden is lower, and when surgical approaches as well as contemporary therapies have greater benefit [5–11]. Distinguishing those CM patients with high-risk tumor biology from those who are categorized as low risk by TNM staging alone is therefore a clinically important goal.
A prognostic gene expression profile (GEP) test (DecisionDx-Melanoma, Castle Biosciences, Inc.) that evaluates 31 gene targets expressed in the primary melanoma tumor to provide a binary classification of low (class 1) or high (class 2) risk of metastasis within 5 years of the initial diagnosis has been previously reported [12]. The test assesses the expression of three control genes, four genes with proven prognostic utility for uveal melanoma tumors [13], and 24 gene targets previously reported to be differentially expressed in metastatic compared to primary tumors [14–20]. Performance of the test has been evaluated in several retrospective validation studies, showing that it accurately prognosticates survival independent of clinical or pathologic assessment alone [12, 21, 22], and though it was originally developed to identify the distant metastasis risk associated with stage I and II melanoma tumors, the prognostic capability of the test has recently been reported in patients with stage III disease. In order to prospectively evaluate outcomes for a real-world representative cohort of CM patients, two multi-center registry studies (EXPAND and INTEGRATE) were initiated. Here, we present an interim analysis of the survival outcomes for 322 patients enrolled in these registries.
Methods
Patients
Patients were enrolled in one of two prospective studies, EXPAND and INTEGRATE (Clinicaltrials.gov identifiers: NCT02355587 and NCT02355574). The protocols have identical enrollment criteria and aims, including (1) the tracking of outcomes of patients for whom GEP testing was completed, and (2) documentation of the clinical application of test results. The studies differ only in clinical data collection requirements based upon the administrative capabilities of the contributing centers. While a combined analysis of the two studies was originally planned based upon the similarity of protocols and aims, an interim analysis of this cohort was not included in the initial study design. Based upon the 1.1-year median time to recurrence observed for class 2 patients in previous validation studies, an unplanned interim analysis of this combined cohort of patients is warranted and expected to answer the hypothesis that class 1 and class 2 risk groups will have significantly different recurrence-free (RFS), distant metastasis-free (DMFS), and overall survival (OS) rates [12, 21, 22].
Eleven US dermatologic and surgical centers participated after receiving protocol approval from their Institutional Review Boards. The number and diversity of centers were selected to reduce bias arising from single centers. Physicians enrolled patients with a diagnosis of cutaneous melanoma who were ≥16 years of age and had a successful GEP test result. The GEP test was performed in a central CAP/CLIA laboratory using standard, published protocols as previously described [12].
Sample size calculations were originally developed from survival outcomes observed in a limited, retrospective analysis of the GEP test, with assumptions of 0.05 for alpha and a stringent power of 0.95. Based upon the rapid, significant separation of risk profiles for class 1 and class 2 cohorts reported in expanded analyses of the test since the inception of the studies (January, 2014), recruitment was discontinued prior to accrual of the patient numbers specified within the protocols. Clinical follow-up will continue through 5-year post-diagnosis for each patient (n = 322).
At the time of the initial patient evaluation, prior to GEP testing, the treating physician assessed each patient’s baseline data, including Breslow thickness (BT), ulceration status, T stage and mitotic rate. The majority of centers reported mitotic rate in mm2; for those that did not, high-powered fields (HPF) were converted to mm2 according to a conversion rate of 4 HPF per mm2 specified by the AJCC v7 guidelines [1, 23]. BT and mitotic rate were used as continuous variables while ulceration, SLN status and GEP class result were treated as categorical variables. All data were entered into a secure case report form and were abstracted and assembled independently of statistical analysis. Last censor date for clinical data was December 30, 2016.
Statistical analyses
Accuracy of the test was measured by sensitivity or specificity. The former was measured as the number of cases with an event [recurrence (including in transit and regional nodal recurrences), distant metastasis or death] that were identified as class 2, while specificity was the number of cases that did not have a documented event that were classified as low-risk class 1 that did not have a documented event. Primary survival endpoints of RFS (time to regional or distant metastasis), DMFS (time to any metastatic event beyond the regional nodal basin), and OS (time from diagnosis to documented death of any cause) were assessed using Kaplan-Meier and Cox regression analyses. For multivariate analyses, only complete cases were considered. In 26 cases, ulceration, BT and/or mitotic rate were not known or specified and these cases were excluded from multivariate analyses. Cases without a documented SLN biopsy result were categorized as clinically node-negative.
As an interim analysis at year 3 of an expected 5-year study, the critical alpha level (p value) was established at 0.01. Statistical associations were evaluated using the F test, chi-squared test, T test, and ANOVA, where appropriate. Statistical analyses were performed in R version 3.3.2 (University of Auckland, NZ).
Results
Clinical and demographical comparison between GEP classes
A total of 335 patients were enrolled in the studies at the last censor date. Of these, 322 patients had completed at least one follow-up visit and were available for inclusion in this interim analysis. A GEP class 1 profile was observed in 77% (248 of 322) of samples. Table 1 provides demographic variables for the entire cohort, stratified by GEP class. The median age of subjects was 58 years (range 18–87 years) and the median Breslow thickness was 1.2 mm (range 0.2–12.0 mm). Within the group, 88% (282 of 322) of the cases had stage I or II disease, and SLN biopsy was performed for 74% (237 of 322) of the subjects. A class 2 profile was observed in 23% (74 of 322) of cases, and was associated with older age, male gender, higher Breslow thickness, ulceration, advanced clinical stage, and positive SLN status (all p < 0.01). However, only half of class 2 patients had ulcerated tumors and a quarter of them had a positive SLN biopsy. Mitotic rate, primary tumor location, and location of first recurrence were not associated with a class 2 profile.
Table 1.
Demographic characteristics of 322 patients enrolled in two prospective registries and correlation with GEP Class
| All cases (n* = 322) | Class 1 (n = 248) | Class 2 (n = 74) | p value*** | |
|---|---|---|---|---|
| Age, median (range) | 58 (18–87) | 57 (18–87) | 65 (23–85) | 0.003 |
| Gender | ||||
| Female | 146 (45%)** | 123 (50%) | 23 (31%) | 0.005 |
| Male | 176 (55%) | 125 (50%) | 51 (69%) | |
| Breslow thickness, median (range) | 1.2 (0.2–12.0) | 1.0 (0.2–7.0) | 2.5 (0.4–12.0) | <0.001 |
| Ulceration | ||||
| Absent | 238 (74%) | 204 (82%) | 34 (46%) | <0.001 |
| Present | 58 (18%) | 23 (9%) | 35 (47%) | |
| Unknown | 26 (8%) | 21 (9%) | 5 (7%) | |
| Mitotic rate | ||||
| ≤1/mm2 | 222 (69%) | 176 (71%) | 46 (62%) | 0.151 |
| >1/mm2 | 100 (31%) | 72 (29%) | 28 (38%) | |
| Node status | ||||
| Negative | 286 (89%) | 228 (92%) | 58 (78%) | 0.007 |
| Positive | 36 (11%) | 20 (8%) | 16 (22%) | |
| Primary tumor location | ||||
| Extremity | 178 (55%) | 133 (54%) | 45 (61%) | 0.547 |
| Head and neck | 58 (18%) | 46 (19%) | 12 (16%) | |
| Trunk | 86 (27%) | 69 (28%) | 17 (23%) | |
| AJCC stage | ||||
| None | 3 (1%) | 3 (1%) | 0 (0%) | <0.001 |
| I | 1 (0%) | 1 (0%) | 0 (0%) | |
| IA | 63 (20%) | 62 (25%) | 1 (1%) | |
| IB | 145 (45%) | 129 (52%) | 16 (22%) | |
| IIA | 39 (12%) | 23 (9%) | 16 (22%) | |
| IIB | 26 (8%) | 9 (4%) | 17 (23%) | |
| IIC | 8 (2%) | 0 (0%) | 8 (11%) | |
| III | 30 (9%) | 16 (6%) | 14 (19%) | |
| IIIA | 3 (1%) | 3 (1%) | 0 (0%) | |
| IIIB | 3 (1%) | 1 (0%) | 2 (3%) | |
| IV | 1 (0%) | 1 (0%) | 0 (0%) | |
| Location of first recurrence | ||||
| Nodal/regional | 13 | 3 | 10 | 0.69 |
| Distant | 12 | 2 | 10 | |
*n = number of samples
**All percentages are reflective of the indicated group out of the total number of samples in that column
***p values reflect differences between class 1 and 2 patients and were calculated using chi-squared or F tests as appropriate
Interim analysis of outcomes
At 1.5 years of follow-up, regional metastasis was the first site of recurrence in 52% (13 of 25) of cases, and 77% (10 of 13) of those were identified as high-risk class 2. Similarly, for the 12 patients for whom the site of first recurrence was a distant metastasis, 83% (10 of 12) were identified as class 2.
The GEP test was the most sensitive prognostic factor for all endpoints assessed, including SLN biopsy and ulceration, with a high-risk class 2 result showing 80% sensitivity for recurrences, 83% for distant metastases, and 73% for death (Table 2). Overall, recurrence rates were 2% (5 of 248) for class 1 versus 27% (20 of 74) for class 2 patients, distant metastasis rates were 1% (2 of 248) versus 14% (10 of 74), and overall survival rates were 1% (3 of 248) versus 11% (8 of 74), respectively.
Table 2.
Outcomes in the 322-patient prospective cohort and correlation with GEP class, SLN status, and ulceration
| Recurrence (n* = 25) | Distant metastasis (n = 12) | Death (n = 11) | |
|---|---|---|---|
| Class 1 (n = 248) | 5 (2%) | 2 (1%) | 3 (1%) |
| Class 2 (n = 74) | 20 (27%) | 10 (14%) | 8 (11%) |
| p value** | <0.0001 | <0.0001 | 0.0001 |
| SLN− (n = 286) | 15 (5%) | 6 (2%) | 10 (3%) |
| SLN+ (n = 36) | 10 (28%) | 6 (17%) | 1 (3%) |
| p value | <0.0001 | <0.001 | 1 |
| Ulceration absent (n = 264) | 10 (4%) | 3 (1%) | 6 (2%) |
| Ulceration present (n = 58) | 15 (26%) | 9 (16%) | 5 (9%) |
| p value | <0.0001 | <0.0001 | 0.04 |
*n = number of samples
**p values reflect differences between high and low-risk groups for each prognostic factor (χ 2 test)
Median recurrence event times for class 2 patients in prior retrospective studies is 1.1 years after diagnosis [12, 21, 22]. With a median follow-up time for non-event samples that exceeds 1.5 years for all endpoints, interim analysis of outcomes in this population is possible. The GEP test was significantly associated with recurrence (p < 0.001), distant metastasis (p < 0.001), and death (p < 0.001). The 1.5-year RFS, DMFS and OS rates for class 1 versus class 2 were 97% (95–100%) versus 77% (67–87%), 99% (97–100%) versus 87% (81–96%), and 99% (97%–100%) versus 92% (86–99%), respectively (Fig. 1). The GEP test was also significantly associated with each endpoint analyzed in the group of subjects with stage I or II melanoma (n = 282, Additional file 1: Figure S1).
Fig. 1.
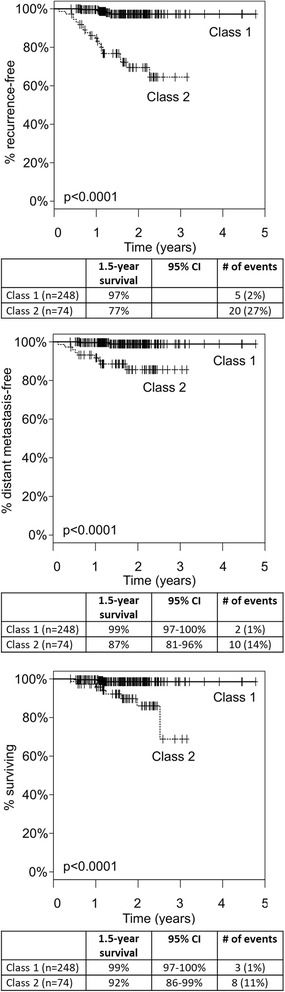
Kaplan-Meier analysis of RFS (top), DMFS (middle), and OS (bottom) by GEP class. Significant separation of class 1 and class 2 risk (p < 0.0001) is observed for each endpoint
Multivariate Cox regression modeling (n = 296 complete cases) showed that BT, mitotic rate, and GEP class were independent predictors of RFS outcome (Table 3; p < 0.01, BT – HR: 1.43 [95% confidence interval (CI) 1.18–1.73], mitotic rate – HR: 1.05 [95% CI 1.01–1.08], GEP – HR: 7.15 [95% CI 1.99–25.8]). For RFS, node status was not significant in these interim analyses at an alpha of 0.01 [p = 0.035, HR: 2.46 (95% CI 1.07–5.68)]. For DMFS and OS, only Breslow thickness was significant [p < 0.001, Additional file 2: Table S1].
Table 3.
Multivariate Cox regression analysis of RFS
| HR (95% CI) | p value | |
|---|---|---|
| Breslow thickness | 1.43 (1.18–1.73) | 0.001 |
| GEP class 2 | 7.15 (1.99–25.8) | 0.003 |
| Mitotic rate | 1.05 (1.01–1.08) | 0.005 |
| SLN positivity | 2.46 (1.07–5.68) | 0.035 |
| Ulceration present | 1.89 (0.75–4.72) | 0.17 |
Comparison to retrospective cohorts
Table 4 summarizes the outcomes data for this prospective trial and previously reported retrospective analyses. The original report of the GEP test performance included 104 samples, with median follow-up time of 8.2 years for non-event samples and 1.7 years for samples with an event (Table 4) [12]. The 1.5-year RFS rates were 98% (95% CI 95–100%) for class 1 and 70% (95% CI 58–85%) for class 2. The most recent validation dataset included 523 stage I-III long-term follow-up cases [22]. In this dataset, median follow-up time for cases without an event was 7.2 years and median time to a recurrence event was 1.2 years. For all cumulative cases that have been reported in retrospective validations (total for all cases: n = 782), the median time to a recurrence event was 1.3 years (1.1 years for the class 2 cohort alone). The 1.5-year RFS rates were 95% (95% CI 93–97%) for class 1 cases and 67% (95% CI 62–73%) for class 2 cases. While the median follow-up time is shorter for this prospective analysis, with 1.5 years of follow-up for cases without an event and 1.0 year for cases with an event, the observed RFS rates of 97% for class 1 and 77% for class 2 are consistent with 1.5-year event rates in prior retrospective datasets (Table 4, Fig. 2).
Table 4.
Summary table of RFS rates observed in the prospective registry cohort and retrospective archival studies
| Study | n | Non-event follow-up time | Event time | Class 1 1.5-year RFS | Class 2 1.5-year RFS |
|---|---|---|---|---|---|
| Median, years | Median, years | Percent (95% CI*) | Percent (95% CI) | ||
| Initial validation (Gerami, 2015) | 104 | 8.2 | 1.7 | 98% (95–100%) | 70% (58–85%) |
| Independent validation | 523 | 7.2 | 1.2 | 95% (92–97%) | 67% (61–74%) |
| All retrospective studies to date | 782 | 6.9 | 1.3 | 95% (93–97%) | 67% (62–73%) |
| Current study | 322 | 1.5 | 1.0 | 97% (95–100%) | 77% (67–87%) |
*CI confidence interval
Fig. 2.
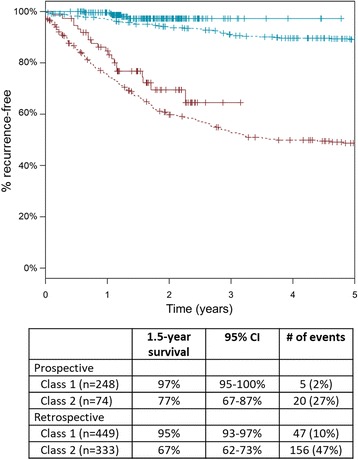
Kaplan-Meier recurrence-free survival curves for class 1 (blue) and class 2 (red) subjects in the prospective registry cohort (solid lines; n = 322) and collective retrospective studies (dashed lines; n = 782)
Discussion
An important unmet clinical need in melanoma is the accurate identification of early-stage patients who harbor a higher risk of developing advanced disease. While Breslow thickness and ulceration are strongly associated with metastasis and outcome, and sentinel lymph node status is a proven prognostic tool that identifies a subset of the patients with high-risk disease, two of three patients who die from melanoma are originally diagnosed with stage I or II tumors [24]. As these clinicopathologic staging features are used to drive management decisions, molecular tools that supplement and improve current methods of prognostication should have significant clinical impact.
To improve upon the identification of high-risk stage I and II patients, a gene expression profile test was developed to provide prognostic information based on the expression of 31 genes in the primary melanoma tumor [12]. A limitation of this interim analysis could be the impact of the short follow-up time. However, consistent with previous reports, the GEP test was a significant predictor of RFS, DMFS, and OS, in both the larger cohort of 322 patients (Fig. 1), and in 282 stage I and II CM cases (Additional file 1: Figure S1). The risk of metastasis and death was significantly higher for patients with a class 2 result compared to class 1 patients. The GEP accurately identified 80% of the recurrences detected during the study, and only 2% of the patients who were predicted to be low risk (class 1) by the test developed recurrent disease. Although multivariate analysis showed BT to be the only independent prognostic factor for DMFS and OS in this cohort, this is likely to be due to the limited number of events for these two outcomes. In all prior studies the GEP has shown independent prognostic value along with Breslow thickness and SLN status.
In this study, 83% (10 of 12) of patients who developed distant metastases were identified as high risk by the GEP test, compared to only 50% (6 of 12) who had a SLN-positive result. Additionally, we found that the median time to recurrence for those who developed advanced disease was only 1 year. National melanoma guidelines specify that patient management should be tailored to an individual’s probability of recurrence. Frequent follow-up and intensified surveillance with imaging is recommended for patients with high-risk clinicopathologic features, while management of low-risk patients is generally restricted to clinical follow-up at 3–12 month intervals [25]. A growing body of evidence supports cross-sectional radiographic imaging as the most effective method for detecting asymptomatic distant metastatic disease in patients with stage II and III melanoma [7, 10, 11]. In this study, 6 of 12 patients with distant metastasis were originally diagnosed with stage II disease. Of those, five were identified as class 2, including two patients with stage IIA tumors. While direct evidence of a benefit from surveillance has not been published, considering the rapid time to event observed in this interim analysis, and the accuracy of risk prediction by the GEP test, increased surveillance with imaging for class 2 patients might be useful, especially in those patients who would not be offered surveillance options based on stage.
The advantages of molecular testing for enhanced prognosis are well documented for other diseases, including breast cancer and ocular melanoma [26, 27]. Consistent with previous studies, the results from this prospective analysis indicate that the 31-gene GEP test for melanoma, in combination with standard clinicopathologic factors, can strengthen risk determination and improve patient management. Better risk prediction is particularly critical when considering the recent advances in therapeutics for melanoma and the evidence supporting better efficacy of contemporary therapies if treatment is administered when tumor burden is low [28, 29].
Conclusions
This constitutes the first report of performance for the prognostic 31-gene GEP test in a prospective population of patients with cutaneous melanoma. In concordance with prior retrospective studies, the test showed robust ability to predict recurrence, distant metastasis and death. While this report encompasses only an interim analysis and, therefore, it is expected that additional events will accrue in this population, the strong statistical association with outcomes even at this early time point provides assurance of the test’s prognostic value.
Additional files
Survival outcomes for stage I/II patients with molecular classification by the 31-gene expression profile test. A) Recurrence-free survival, B) distant metastasis-free survival, and C) overall survival for Class 1 and Class 2 subjects with stage I or stage II disease (n = 282). (TIFF 192 kb)
Cox regression analysis for distant metastasis-free (DMFS) and overall survival (OS) in the 322-subject cohort. (DOCX 12 kb)
Acknowledgements
The authors wish to thank the physicians and clinical staff at each of the contributing institutions, including Martin Fleming, MD, Adam Berger, MD, Elizabeth Liotta, MD, Abdallah Khourdaji, MD, and Charles St. Hill, MD for their contributions to the study. We would also like to recognize the significant contributions to clinical data review and tissue processing by Jeff Wilkinson, PhD, Natalie Lassen, PhD, John Stone, PhD, and Kristen Oelschlager, RN.
Funding
This study was partially sponsored by Castle Biosciences, Inc., which provided financial compensation to those centers contributing melanoma tissue to the study.
Availability of data and materials
The dataset analyzed during the current study is not publicly available due to ongoing accrual of patient data.
Abbreviations
- AJCC
American Joint Committee on Cancer
- ANOVA
Analysis of variance
- CAP/CLIA
College of American Pathologists/Clinical Laboratory Improvement Amendments
- CI
Confidence interval
- CM
Cutaneous melanoma
- DMFS
Distant metastasis-free survival
- GEP
Gene expression profile
- OS
Overall survival
- RFS
Recurrence-free survival
- SEER
Surveillance, Epidemiology, and End Results
- SLN
Sentinel lymph node
Authors’ contributions
RWC, CJ, and ECS conceptualized the study. RWC validated the study. KRC, RWC, and BM formally analyzed the study. KRC, RWC, and BM wrote the original draft. ECH, JRD, JL, JJS, KRC, BM, CJ, RWC, CLS, and KMM wrote, reviewed, and edited the manuscript. RWC, BM, and KRC contributed to the visuals of the study. RWC supervised the study. All authors have read and approved this manuscript.
Ethics approval and consent to participate
The current study was approved by the following Institutional Review Boards:
Thomas Jefferson Office of Human Research Institutional Review Board
University of Virginia Institutional Review Board for Health Sciences Research
University of Louisville Human Subjects Protection Program
The University of Tennessee Health Science Center IRB
Western Institutional Review Board (Northside Hospital, St. Louis University, University of Cincinnati, Fairmont Dermatology, Elizabeth Liotta, MD, LLC, South Carolina Skin Cancer Center, University of Nevada School of Medicine)
Written informed patient consent was obtained from study participants before enrollment in accordance with the Declaration of Helsinki.
Consent for publication
Not applicable
Competing interests
ECH and JJS are on Castle Biosciences’ Speaker Bureau. ECH, JJS, JL, and CLS have received honoraria for advisory board participation. KRC, BM, CJ, and RWC are employees of Castle Biosciences, Inc. and hold stock in the company. All remaining authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s13045-017-0520-1) contains supplementary material, which is available to authorized users.
Contributor Information
Eddy C. Hsueh, Email: hsuehec@slu.edu
James R. DeBloom, Email: debloomj@yahoo.com
Jonathan Lee, Email: jonathan.lee@northside.com.
Jeffrey J. Sussman, Email: sussmaj@ucmail.uc.edu
Kyle R. Covington, Email: kcovington@castlebiosciences.com
Brooke Middlebrook, Email: bmiddlebrook@castlebiosciences.com.
Clare Johnson, Email: cjohnson@castlebiosciences.com.
Robert W. Cook, Phone: 1-832-558-1791, Email: rcook@castlebiosciences.com
Craig L. Slingluff, Jr, Email: CLS8H@virginia.edu.
Kelly M. McMasters, Email: kmmcma01@exchange.louisville.edu
References
- 1.Balch CM, Gershenwald JE, Soong SJ, Thompson JF, Atkins MB, Byrd DR, et al. Final version of 2009 AJCC melanoma staging and classification. J Clin Oncol. 2009;27:6199–6206. doi: 10.1200/JCO.2009.23.4799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Edge SB, Compton CC. The American joint committee on cancer: the 7th edition of the AJCC cancer staging manual and the future of TNM. Ann Surg Oncol. 2010;17:1471–1474. doi: 10.1245/s10434-010-0985-4. [DOI] [PubMed] [Google Scholar]
- 3.Shaikh WR, Dusza SW, Weinstock MA, Oliveria SA, Geller AC, Halpern AC. Melanoma thickness and survival trends in the United States, 1989 to 2009. J Natl Cancer Inst. 2015; 10.1093/jnci/djv294. [DOI] [PMC free article] [PubMed]
- 4.Whiteman DC, Baade PD, Olsen CM. More people die from thin melanomas (1 mm) than from thick melanomas (>4 mm) in Queensland. Aust J Invest Dermatol. 2015;135:1190–1193. doi: 10.1038/jid.2014.452. [DOI] [PubMed] [Google Scholar]
- 5.Glazer AM, Winkelmann RR, Farberg AS, Rigel DS. Analysis of trends in US melanoma incidence and mortality. JAMA Dermatol. 2017;153:225–226. doi: 10.1001/jamadermatol.2016.4512. [DOI] [PubMed] [Google Scholar]
- 6.Andtbacka RH, Kaufman HL, Collichio F, Amatruda T, Senzer N, Chesney J, et al. Talimogene Laherparepvec improves durable response rate in patients with advanced melanoma. J Clin Oncol. 2015;33:2780–2788. doi: 10.1200/JCO.2014.58.3377. [DOI] [PubMed] [Google Scholar]
- 7.Bhutiani N, Egger ME, McMasters KM. Optimizing follow-up assessment of patients with Cutaneous melanoma. Ann Surg Oncol. 2017;24:861–863. doi: 10.1245/s10434-017-5771-0. [DOI] [PubMed] [Google Scholar]
- 8.Leon-Ferre RA, Kottschade LA, Block MS, McWilliams RR, Dronca RS, Creagan ET, et al. Association between the use of surveillance PET/CT and the detection of potentially salvageable occult recurrences among patients with resected high-risk melanoma. Melanoma Res. 2017; doi:10.1097/CMR.0000000000000344. [DOI] [PubMed]
- 9.Nishino M, Giobbie-Hurder A, Ramaiya NH, Hodi FS. Response assessment in metastatic melanoma treated with ipilimumab and bevacizumab: CT tumor size and density as markers for response and outcome. J Immunother Cancer. 2014; doi:10.1186/s40425-014-0040-2. [DOI] [PMC free article] [PubMed]
- 10.Park TS, Phan GQ, Yang JC, Kammula U, Hughes MS, Trebska-McGowan K, et al. Routine computer tomography imaging for the detection of recurrences in high-risk melanoma patients. Ann Surg Oncol. 2017;24:947–951. doi: 10.1245/s10434-017-5768-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Podlipnik S, Carrera C, Sanchez M, Arguis P, Olondo ML, Vilana R, et al. Performance of diagnostic tests in an intensive follow-up protocol for patients with American joint committee on cancer (AJCC) stage IIB, IIC, and III localized primary melanoma: a prospective cohort study. J Am Acad Dermatol. 2016;75:516–524. doi: 10.1016/j.jaad.2016.02.1229. [DOI] [PubMed] [Google Scholar]
- 12.Gerami P, Cook RW, Wilkinson J, Russell MC, Dhillon N, Amaria RN, et al. Development of a prognostic genetic signature to predict the metastatic risk associated with cutaneous melanoma. Clin Cancer Res. 2015;21:175–183. doi: 10.1158/1078-0432.CCR-13-3316. [DOI] [PubMed] [Google Scholar]
- 13.Onken MD, Worley LA, Tuscan MD, Harbour JW. An accurate, clinically feasible multi-gene expression assay for predicting metastasis in uveal melanoma. J Mol Diagn. 2010;12:461–468. doi: 10.2353/jmoldx.2010.090220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Onken MD, Worley LA, Ehlers JP, Harbour JW. Gene expression profiling in uveal melanoma reveals two molecular classes and predicts metastatic death. Cancer Res. 2004;64:7205–7209. doi: 10.1158/0008-5472.CAN-04-1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jaeger J, Koczan D, Thiesen HJ, Ibrahim SM, Gross G, Spang R, et al. Gene expression signatures for tumor progression, tumor subtype, and tumor thickness in laser-microdissected melanoma tissues. Clin Cancer Res. 2007;13:806–815. doi: 10.1158/1078-0432.CCR-06-1820. [DOI] [PubMed] [Google Scholar]
- 16.Bittner M, Meltzer P, Chen Y, Jiang Y, Seftor E, Hendrix M, et al. Molecular classification of cutaneous malignant melanoma by gene expression profiling. Nature. 2000;406:536–540. doi: 10.1038/35020115. [DOI] [PubMed] [Google Scholar]
- 17.Haqq C, Nosrati M, Sudilovsky D, Crothers J, Khodabakhsh D, Pulliam BL, et al. The gene expression signatures of melanoma progression. Proc Natl Acad Sci U S A. 2005;102:6092–6097. doi: 10.1073/pnas.0501564102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mauerer A, Roesch A, Hafner C, Stempfl T, Wild P, Meyer S, et al. Identification of new genes associated with melanoma. Exp Dermatol. 2011;20:502–507. doi: 10.1111/j.1600-0625.2011.01254.x. [DOI] [PubMed] [Google Scholar]
- 19.Scatolini M, Grand MM, Grosso E, Venesio T, Pisacane A, Balsamo A, et al. Altered molecular pathways in melanocytic lesions. Int J Cancer. 2010;126:1869–1881. doi: 10.1002/ijc.24899. [DOI] [PubMed] [Google Scholar]
- 20.Smith AP, Hoek K, Becker D. Whole-genome expression profiling of the melanoma progression pathway reveals marked molecular differences between nevi/melanoma in situ and advanced-stage melanomas. Cancer Biol Ther. 2005;4:1018–1029. doi: 10.4161/cbt.4.9.2165. [DOI] [PubMed] [Google Scholar]
- 21.Gerami P, Cook RW, Russell MC, Wilkinson J, Amaria RN, Gonzalez R, et al. Gene expression profiling for molecular staging of cutaneous melanoma in patients undergoing sentinel lymph node biopsy. J Am Acad Dermatol. 2015;72:780–785. doi: 10.1016/j.jaad.2015.01.009. [DOI] [PubMed] [Google Scholar]
- 22.Zager JS, Gastman BR, Messina J, Sondak VK, Ferris L, Cook RW, et al. Performance of a 31-gene expression profile in a previously unreported cohort of 334 cutaneous melanoma patients. J Clin Oncol. 2016;34(Suppl 15):9581. [Google Scholar]
- 23.Burton AL, Egger ME, Gilbert JE, Stromberg AJ, Hagendoorn L, Martin RC, et al. Assessment of mitotic rate reporting in melanoma. Am J Surg. 2012;204:969–975. doi: 10.1016/j.amjsurg.2012.05.021. [DOI] [PubMed] [Google Scholar]
- 24.Morton DL, Thompson JF, Cochran AJ, Mozzillo N, Nieweg OE, Roses DF, et al. Final trial report of sentinel-node biopsy versus nodal observation in melanoma. N Engl J Med. 2014;370:599–609. doi: 10.1056/NEJMoa1310460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Coit DG, Thompson JA, Algazi A, Andtbacka R, Bichakjian CK, Carson WE, 3rd, et al. Melanoma, version 2.2016, NCCN clinical practice guidelines in oncology. J Natl Compr Cancer Netw. 2016;14:450–473. doi: 10.6004/jnccn.2016.0051. [DOI] [PubMed] [Google Scholar]
- 26.Onken MD, Worley LA, Char DH, Augsburger JJ, Correa ZM, Nudleman E, et al. Collaborative ocular oncology group report number 1: prospective validation of a multi-gene prognostic assay in uveal melanoma. Ophthalmology. 2012;119:1596–1603. doi: 10.1016/j.ophtha.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van de Vijver MJ, He YD, van't Veer LJ, Dai H, Hart AA, Voskuil DW, et al. A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med. 2002;347:1999–2009. doi: 10.1056/NEJMoa021967. [DOI] [PubMed] [Google Scholar]
- 28.Kaufman H, Amatruda T, Nemunaitis JJ, Chesne JA, Delman KA, Spitler LE, et al. Tumor size and clinical outcomes in melanoma patients (MEL pts) treated with talimogene laherparepvec (T-VEC) J Clin Oncol. 2015;33(Suppl 15):9074. [Google Scholar]
- 29.Ribas A, Hamid O, Daud A, Hodi FS, Wolchok JD, Kefford R, et al. Association of pembrolizumab with tumor response and survival among patients with advanced melanoma. JAMA. 2016;315:1600–1609. doi: 10.1001/jama.2016.4059. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Survival outcomes for stage I/II patients with molecular classification by the 31-gene expression profile test. A) Recurrence-free survival, B) distant metastasis-free survival, and C) overall survival for Class 1 and Class 2 subjects with stage I or stage II disease (n = 282). (TIFF 192 kb)
Cox regression analysis for distant metastasis-free (DMFS) and overall survival (OS) in the 322-subject cohort. (DOCX 12 kb)
Data Availability Statement
The dataset analyzed during the current study is not publicly available due to ongoing accrual of patient data.