


Abstract
Myotonic dystrophy (DM1) is the most common form of adult muscular dystrophy and is inherited as an autosomal dominant trait. The genetic basis of DM1 is the expansion of a CTG repeat in the 3′ untranslated region of a protein kinase gene (DMPK). The molecular mechanism by which this expanded repeat produces the pathophysiology of DM1 remains unknown. Transcripts from the expanded allele accumulate as foci in the nucleus of DM1 cells and it has been suggested that these transcript foci sequester cellular proteins that are required for normal nuclear function. We have investigated the role of three RNA-binding proteins, CUG-BP, hnRNP C and MBNL, as possible sequestered factors. Using a combination of indirect immunofluorescence to detect endogenous proteins and overexpression of proteins with green fluorescent protein (GFP) tags we have shown that CUG-BP and hnRNP C do not co-localise with expanded repeat foci in DM1 cell lines. However, GFP-tagged MBNL does itself form foci in DM1 cell lines and co-localises with the foci of expanded repeat transcripts. GFP-tagged MBNL does not appear as foci in non-DM1 cell lines. This work provides further support for the involvement of MBNL in DM1.
INTRODUCTION
Myotonic dystrophy (DM1) is the most common form of adult muscular dystrophy, with an incidence of 1 in 8000. It is inherited as an autosomal dominant trait and is associated with defects in many tissues, including skeletal muscle myotonia, progressive myopathy, cataracts and abnormalities in the heart, brain and endocrine systems. Clinical expression of the disorder is variable, with some patients showing only development of cataracts in middle-age, while others show severe neonatal hypotonia. The classical presentation is a progressive muscular dystrophy affecting distal muscles more than proximal, often associated with the inability to relax muscles appropriately (myotonia) (1). DM1 is caused by the expansion of a CTG repeat in the 3′ untranslated region (3′-UTR) of protein kinase (DMPK) which maps to 19q13.3. The normal DMPK gene contains 5–37 copies of the CTG repeat whereas in DM1 patients the repeat is expanded in the range of 50 to several thousand CTGs. Repeat expansion shows a positive correlation with the severity of the disease and an inverse correlation with age at onset (2). Moreover, there is somatic mosaicism, for example CTG repeat lengths in muscle cells are longer than those in circulating lymphocytes (3).
There are three proposed mechanisms to explain the molecular basis of DM1. First, the expansion affects the level of DMPK expression in cis by altering its transcription, or by the retention of CUG expanded transcripts, which may lead to haploinsufficiency (4). Second, the expansion mutation may alter chromatin structure and affect the expression of both DMPK and other neighbouring genes (5–8). It has been shown that deficiency of Six5 (encoded by the Six5 gene which is immediately downstream of Dmpk) in mice contributes to the cataract phenotype that is common in DM1 patients (9). The third model involves a dominant RNA mutation in which RNAs from the expanded allele create a gain-of-function mutation. Recent experimental data from transgenic mice, expressing an untranslated expanded CUG repeat under the control of the human skeletal actin promoter, showed that expanded CUG repeats are sufficient to generate a DM1 phenotype (10). It has been shown that the transcripts containing CUG expanded repeats accumulate in the nucleus of both cultured cells (11,12) and biopsy tissues from patients with DM1 (13). Thus, the repeat expansion could also cause a toxic effect on nuclear metabolism by removing specific cellular proteins that are required for normal functions of the cell, such as pre-mRNA processing or export.
Initial attempts to identify proteins that may be sequestered by CTG repeats produced a CUG-binding protein (CUG-BP). This was initially isolated using a band-shift assay with a (CUG)8 repeat probe (14). In subsequent studies the possible role of CUG-BP in DM1 was strengthened by the finding that the cellular distribution of two different phosphorylated forms of CUG-BP was different in cells derived from DM1 patients and in mice lacking Dmpk (15). In addition, the observation that CUG-BP played a role in RNA processing and that alternative splicing of one of its targets, cTNT, was affected by DM1 status, added further weight to its central role in this disorder (16). However, there is also evidence against the direct interaction of CUG-BP and expanded repeat units. The demonstration that CUG-BP only bound to the base of a stem–loop structure in EM studies (17) raised the possibility that this protein was not sequestered as a direct consequence of repeat expansion, but perhaps that the effects noted were a secondary consequence of the binding of an alternative protein.
Other proteins have now been proposed as candidates for sequestration by retained DMPK transcripts. hnRNPs are an abundant family of proteins that associate with pre-mRNAs during transcription and/or remain associated with nuclear mRNAs after splicing is completed. It has been suggested that mRNA export is regulated by interplay between different hnRNPs, with hnRNP C promoting nuclear retention and hnRNP A mediating nuclear export (18). As hnRNP C has also been shown to bind to the DMPK 3′-UTR (19) it could play a central role in the retention of expansion derived transcripts. A third candidate protein has now been identified as a triplet repeat expansion-associated protein; MBNL/EXP was found to be sequestered in the nucleus of DM1 cells (20). These proteins are homologous to Drosophila muscleblind protein which is required for terminal differentiation of muscle and photoreceptor cells (21,22).
In order to clarify the role of CUG-BP, hnRNP C and MBNL/EXP in DM1, we have examined the localisation of these proteins with respect to the foci of triplet repeat transcripts that are formed in cells derived from patients with DM1. While our data indicate that CUG-BP and hnRNP C do not co-localise with triplet repeat foci, we show for the first time that MBNL/EXP co-localises with these transcript foci in the nuclei of DM1 cells.
MATERIALS AND METHODS
Green fluorescent protein (GFP)/CUG-BP construct
To obtain a GFP/CUG-BP fusion construct, plasmid tg-cugbp containing the coding fragment of the CUG-BP (hNab50) cDNA (gift from T. Cooper) was digested to completion with BamHI and XbaI. The coding fragment was gel-purified and subcloned into pEGFP-C1 (Clontech). The sequence of the resulting in-frame GFP/CUG-BP fusion construct was verified by restriction digestion and sequence analysis.
GFP/hnRNP C construct
To produce a GFP/hnRNP C fusion construct, the hnRNP C coding region was amplified by PCR from I.M.A.G.E clone: 3358683 (HGMP Resource Centre) using primers 5′-CCATCGAATTCGATGGCCAGCAACGTTACCAAC-3′ and 5′-TAATGGGATCCGATTTCTAAACCCCACTATGTGC-3′. The resulting 926 bp PCR product was cloned into pGEM-T (Promega) and sequenced. The open reading frame of hnRNP C was removed from this vector on an EcoRI and BamHI fragment and subcloned into pEGFP-C1.
GFP/MBNL construct
Total mRNA was extracted from HeLa cells and subjected to reverse transcription using random decamers according to the manufacturer’s instructions (Reverse-iT first strand synthesis kit, Abgene). Two MBNL fragments were subsequently generated by PCR using Platinum Pfx DNA polymerase (Gibco Life Technologies). Primer pairs 5′-ATGGCTGGTACCGTCACACCAATTCGGGACAC-3′ and 5′-CAAGAGCAGGCCTCTTTGGTAATG-3′ were used to generate the 5′ part of MBNL and 5′-GCTGCCATGACTCAGTCGGCTGTC-3′, 5′-CATCTGGGATCCATACTTGTGGCTAGTCAGATGTTC-3′ to generate the 3′ part of MBNL. The PCR products obtained were cloned into pGEM-T and to obtain the full MBNL coding sequence, the two fragments were digested and combined using an internal EcoRI site. The complete MBNL coding sequence was then subcloned into KpnI/BamHI digested pEGFP-C1 plasmid. This GFP/MBNL construct was verified by sequencing.
Cell culture and transfection
DM1 and control fibroblast cell lines were grown on coverslips for 24 h in Dulbecco’s minimum essential media (DMEM) (Gibco Life Technologies) containing 20% (v/v) fetal bovine serum (Gibco Life Technologies) and antibiotics penicillin and streptomycin (0.2 U and 0.2 µg/ml, respectively) (Gibco Life Technologies). After 24 h the cells were transiently transfected with 0.6 µg DNA construct and 15 µl Effectene reagent according to the manufacturer’s instructions (Qiagen). To avoid cytotoxicity the Effectene–DNA complexes were replaced by complete medium after 18 h and the cells were incubated for a further 30–42 h.
In-situ hybridisation
This modified method is based on that described in Taneja et al. (13). Briefly, cells on coverslips were washed with Hanks’ Balanced Salt Solution (HBSS) (Gibco Life Technologies) and fixed for 10 min at room temperature in 4% (w/v) paraformaldehyde/PBS, 5 mM MgCl2. After fixation, cells were treated with 40% (v/v) formamide in 2× SSC for 10 min at room temperature. Cells were then hybridised for 3 h at 37°C with 15 ng Cy3-labelled (CAG)10-oligonucleotide (Operon) in 150 µl total volume containing 40% (v/v) formamide, 2× SSC, 0.2% (w/v) BSA, 10% (w/v) dextran sulfate, 2 mM vanadyl adenosine complex, 1 mg/ml Escherichia coli transfer RNA and 1 mg/ml salmon sperm DNA. After hybridisation cells were washed twice with PBS for 5 min and mounted on slides. To check that hybridisation with Cy3-labelled (CAG)10-oligonucleotide was specific, control cells without expanded tracts were analysed in parallel with DM1 cells. Foci were never observed in control cells. In addition, Taneja et al. (13) reported an extensive investigation into the subcellular distribution of DMPK transcripts using several DMPK-specific probes from different regions of the transcript. From their findings, foci of DMPK transcripts are not found in control cells. Thus, the foci that are observed in DM1 cells, using a Cy3-labelled (CAG)10-oligonucleotide, are a feature of these cells and are not due simply to the presence of more target sequence as a consequence of repeat expansion.
Cell immunofluorescence and in-situ hybridisation
Based on a method originally developed by M. Mahadevan (personal communication), control and DM1 fibroblast cells were grown on coverslips for 24 h. Cells were fixed with 4% (w/v) paraformaldehyde and treated with 70% ethanol for 10 min and rehydrated in PBS for 10 min. For binding of primary antibody, cells were incubated for 1 h with 100 µl of the appropriate monoclonal antibody (1/300 dilution for anti CUG-BP, mAb 3B1, and 1/1000 dilution for anti-hnRNP C, mAb 4F4) and washed three times with PBS, each for 5 min. Indirect detection of primary antibody was achieved by incubation with 1/200 diluted secondary antibody (Alexa 488-labelled goat anti-mouse IgG; Molecular Probes) for 1 h. Cells were subjected to three 5 min washes with PBS. In-situ hybridisation was then performed as described above.
Fluorescence microscopy and quantification of colour intensity
Fluorescent staining was examined using an Olympus BX60 microscope equipped with a cooled digital CCD camera (Princeton Instruments). The Alexa 488-labelled goat anti-mouse IgG and GFP fluorescence were analysed using a 480/40 nm excitation and a 535/50 nm emission filter combination. The Cy3 fluorescence was analysed using a 545/30 nm excitation and a 610/75 nm emission filter combination. The images were processed using IP Lab software (Scanalytics). To quantify intensity of colour in different areas of the cells, an area of background or transcript foci was selected on a merged image and the mean intensity of colour per pixel automatically calculated for each channel according to software manuals.
RESULTS
Intracellular distribution of endogenous CUG-BP
In view of the suggested role for CUG-BP in DM1, we set out to examine whether this protein might be sequestered by the expanded-repeat-containing DMPK transcripts in vivo. CUG-BP was initially identified as a protein that bound to a CUG repeat (14); the hypothesis behind its involvement in DM1 is that binding of this protein to large numbers of target sites, produced by expansion of the repeat unit in DMPK, would lead to its sequestration in the nucleus. Although there is indirect evidence against a simple sequestration model for CUG-BP (17,19), there has been no direct evidence that formally disproves this hypothesis in vivo. In order to test the CUG-BP-sequestration hypothesis, we set out to determine whether there was an increase in the concentration of CUG-BP at sites in the nucleus that corresponded to the foci of transcripts containing the repeat expansion alleles of DMPK that can be detected by in-situ hybridisation. To determine whether there was an increase in the amount of CUG-BP co-localising with the foci of expanded transcripts, the transcript foci and the amount of protein at these transcript foci were visualised simultaneously by in-situ hybridisation and immunofluorescence in DM1 patient derived fibroblast cell lines. To quantify the amounts of immunoreactive protein at the transcript foci, mean intensities of fluorescence were measured for all transcript foci in 42 cells from four different DM1 cell lines. In order to establish the overall distribution of CUG-BP in the nucleus the mean intensities for regions adjacent to each focus were also determined as controls. Analysis of the variation in intensity of CUG-BP fluorescence indicated that distribution of CUG-BP was similar in DM1 cells to control cells, there were no distinct foci of CUG-BP protein, and there was no significant increase in the amount of immunoreactive protein at the foci of expanded repeats (Figs 1 and 2).
Figure 1.
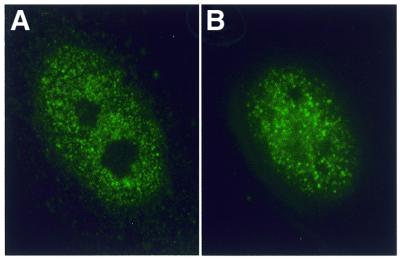
Distribution of the CUG-BP in control and DM1 fibroblast cells. Indirect immunofluorescence with mAb 3B1 (anti CUG-BP) of (A) control and (B) DM1 fibroblast cells.
Figure 2.

Quantification of endogenous and GFP-tagged proteins in DM1 cells. Indirect immunofluorescence or fluorescence from GFP-tagged proteins was quantified using digital images in which mean intensities were estimated per unit pixel. There was no significant difference between the levels of protein at the transcript foci and in the background for CUG-BP or hnRNP C. The level of GFP/MBNL in the transcript foci was significantly higher than background in DM1 cells. (number of transcript foci: 92 P < 0.01, t-test).
Intracellular distribution of endogenous hnRNP C
Analysis of the distribution patterns for hnRNP C in DM1 cells can answer two questions. First, whether hnRNP C is sequestered by the expanded repeat, thereby revealing a direct role for loss of hnRNP C in the formation of DM1. Second, whether the nature of the transcripts (in terms of their state of RNA processing) in the foci can be determined by analysing associated hnRNPs in vivo. hnRNP C is a member of a subset of hnRNPs whose roles do not appear to involve shuttling RNA between the nucleus and the cytoplasm, but are more likely to be involved with a nuclear-specific processing event (18). hnRNP C has also been shown to have a high affinity for pyrimidine-rich sequences and has been found to be associated with the 3′-UTR of DMPK (19). Thus, we set out to determine whether the foci of expanded repeat transcripts that are present in the nucleus of DM1 cells, sequester or are associated with hnRNP C. We examined the intracellular distribution of DMPK expanded transcripts using in-situ hybridisation for the detection of transcript foci simultaneously with immunofluorescence to detect hnRNP C using mAb 4F4. Endogenous hnRNP C was found to be diffusely distributed through the cell with a higher concentration in the nucleus in DM1 and control cells; there were no visible protein foci in the nucleus of DM1 cells (Fig. 3). The nuclei of 23 cells from two DM1 patients were analysed, and the mean intensity of fluorescence measured in transcript foci and the background as described above. No significant increase in the amount of immunoreactive hnRNP C was observed at the transcript foci (Fig. 2).
Figure 3.
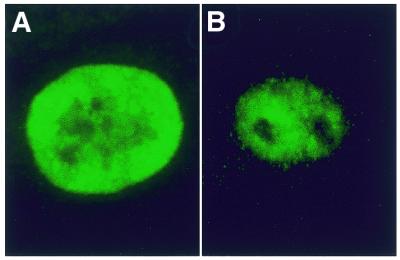
Distribution of hnRNP C in control and DM1 fibroblast cells.Endogenous hnRNP C was detected using mAb 4F4 (anti-hnRNP C) in (A) control and (B) DM1 cells.
Intracellular distribution of a GFP/CUG-BP fusion protein
As levels of CUG-BP are relatively low in fibroblast cells, lack of enrichment could be due to lack of protein. To test the in vivo capacity for CUG-BP to interact with foci of transcripts, we produced an expression construct of GFP/CUG-BP. This produces a fusion protein comprising full-length CUG-BP-tagged with enhanced GFP at the N-terminus. Control and DM1 fibroblast cells were transiently transfected with the fusion construct and analysed for the distribution of fusion protein by direct visualisation of GFP, and expanded repeat transcripts by in-situ hybridisation with a Cy3 probe (Fig. 4A–D). Measurements of fluorescence of transcript foci in 44 DM1 cells from three different DM1 patients indicated that there was no significant increase in the amount of fusion protein present at the transcript foci compared to background (Fig. 2). Despite the large size of the GFP tag, the fusion protein has a similar nuclear distribution pattern to endogenous CUG-BP (Fig. 1). Due to overexpression of the fusion protein, some control cells (Fig. 4A) and DM1 cells (Fig. 4B) showed bright spots of GFP that were also visible by phase contrast microscopy. These bright spots might represent pre-nuclear bodies that have previously been reported by others (23). However, they also provided a mechanism to test whether expanded transcripts could be drawn towards high concentrations of CUG-BP protein. Analysis (Fig. 2) of the amount of fluorescence from the (CAG)10 Cy3 in-situ probe indicated that there was no significant increase in the amount of CUG-rich transcript present in these concentrated spots of fusion protein (Fig. 4B–D).
Figure 4.

Transient expression of GFP-tagged proteins in control and DM1 fibroblast cells. (A–D) Cells transfected with GFP/CUG-BP, (E–H) cells transfected with GFP/hnRNP C and (I–L) cells transfected with GFP/MBNL. (A, E and I) Control fibroblasts, (B–D, F–H and J–L) DM1 cell lines. (B, F and J) GFP-tagged protein distribution in DM1 fibroblast cells. (C, G and K) Location of DMPK expanded transcripts using in-situ hybridization with (CAG)10-Cy3 probe. (D, H and L) Merged images which show that GFP/CUG-BP (D) and GFP/hnRNP C (H) do not co-localise with foci of expanded repeat transcripts. The merge image of GFP/MBNL and the (CAG)10-Cy3 probe (L) shows that GFP/MBNL co-localises with the foci of expanded DMPK transcripts.
Intracellular distribution of a GFP/hnRNP C fusion protein
To determine whether overexpression of hnRNP C gave a similar distribution pattern and to circumvent any problems associated with non-specificity of antibody, we generated a construct to produce a GFP/hnRNP C fusion protein. GFP/hnRNP C was transiently expressed in control and DM1 cells, which were then analysed for co-localisation of the fusion protein, by direct visualisation of GFP, and foci of expanded repeats using in-situ hybridisation with a (CAG)10 Cy3 probe (Fig. 4E–H). Analysis of 28 cells from two DM1 patients indicated that there were no visually detectable foci of GFP/hnRNP C protein neither was there a significant increase in the amount of fusion protein at the site of the transcript foci when analysed quantitatively (Fig. 2).
Intracellular distribution of GFP/MBNL fusion protein
The identification of proteins with affinity for expanded CUG repeats has provided indirect evidence for an interaction between retained DMPK transcripts and the human homologue of the Drosophila muscleblind in vivo (20). However, for technical reasons, Miller et al. (20) were unable to co-localise the foci of transcripts that are a hallmark of DM1 cells with the increased concentrations of MBNL that were noted in DM1 cells. In order to formally establish whether there is a direct interaction between this RNA binding protein and the expanded repeats in DM1 cells in vivo, we generated a construct to produce a GFP/MBNL fusion protein. Following transient transfection of this construct into control and DM1 cells, we examined the distribution of the fusion protein in relation to the foci of repeat expansion transcripts as determined by in-situ hybridisation with a (CAG)10 Cy3 probe. In control cells, GFP/MBNL is distributed through the nucleus and cytoplasm with greater concentration in the nucleus (Fig. 4I). In DM1 cells GFP/MBNL is distributed similarly, but crucially there are multiple nuclear protein foci that represent high concentration of GFP/MBNL which are absent from the nucleus of control cells (Fig. 4I and J). To determine whether these concentrated spots of GFP/MBNL co-localise with the foci of expanded transcripts we measured the intensity of GFP at the transcript foci (Fig. 4K and I). The intensities of GFP were measured at all 92 transcript foci from 31 cells taken from two DM1 patients. A significant increase in intensity of GFP, above the background, was noted for regions of the nucleus that are shown to contain foci of expanded repeats by in-situ hybridisation (Fig. 2).
DISCUSSION
According to the dominant RNA mutation model, the molecular pathology of DM1 arises as a consequence of nuclear proteins (or nucleic acids) interacting with DMPK expanded repeat transcripts retained within the nucleus of DM1 cells. We set out to examine the relationship between three proteins, CUG-BP, hnRNP C and MBNL, and DMPK transcripts containing expanded repeats in cell lines from DM1 patients using assays that allow the simultaneous detection of each protein and the expanded repeat transcript. Two types of assay were used, the first involved the detection of protein using indirect immunofluorescence, the second used a more direct tracking method with exogenous GFP-tagged proteins.
Initial studies into the sequestration of protein by expanded repeats suggested a role for CUG-BP (14,16), thus we set out to test whether CUG-BP co-localised to expanded triplet repeats in vivo. Our results indicated that neither endogenous CUG-BP, visualised by indirect immunofluorescence, nor GFP-tagged CUG-BP co-localised to expanded repeat transcripts. This result suggests that the sequestration of CUG-BP by nuclear retained transcripts is not a primary feature of DM1 and therefore questions the direct role of this protein in DM1 pathophysiology. It also indicates that the alterations in CUG-BP phosphorylation state reported by others (15) may be a secondary effect of nuclear retention. Studies using electron microscopy and thermal melting have shown that long CUG repeats form double-stranded RNA hairpins (24,25). Similar studies have also shown that CUG-BP does not bind along the stem of the duplex but is localised to the base of the RNA hairpin (17). Furthermore, evidence from yeast three hybrid analysis (26) indicates that CUG-BP is strongly and specifically associated with UG dinucleotide repeats. In addition, if CUG-BP is directly associated with DMPK transcripts, a slight increase in the level of this protein might be expected at transcript foci, due to a concentration effect, as estimates indicate that foci contain 15–230 DMPK transcripts (27). As no increase was noted, this may question whether CUG-BP has any interaction with DMPK in vivo. Thus, the absence of CUG-BP binding to double-stranded RNA is consistent with the results presented here showing no sequestration of CUG-BP in foci of expanded DMPK repeat transcripts.
The finding that CUG-BP did not co-localise with the expanded repeat transcripts led us to investigate two other proteins. As hnRNP C has been shown to play a role in RNA processing/export (18) we analysed the distribution of this protein in DM1 cell lines. The rationale for this analysis was that if repeat expansion transcripts were not fully processed they should still be associated with hnRNP C which may in turn be preventing their export from the nucleus. However, in both antibody staining and GFP tag experiments hnRNP C was not enriched at the site of expanded repeat transcripts. This result indicates either that transcripts are processed, but are unable to be exported, perhaps because of steric hindrance by binding of another protein, or the binding of another protein may prevent the normal association of hnRNP C with these transcripts. In either case, binding by hnRNP C does not appear to be the cause of the nuclear retention.
The third protein analysed in this study was MBNL. Using indirect immunofluorescence, Miller et al. (20) demonstrated the presence of concentrated regions of MBNL in the nuclei of DM1 cells. However, due to technical difficulties possibly due to the exclusion of the Cy3-labelled probe by the presence of antibody bound protein, they were not able to demonstrate co-localisation of protein with foci of DMPK transcripts. In order to overcome these technical problems we generated a GFP-tagged form of MBNL. Our results showed that the foci of expanded DMPK transcripts always co-localised to protein foci containing high amounts of GFP-tagged MBNL. The intensely green spots of GFP/MBNL that were a feature of the DM1 cells were never seen in control cells that had been transfected with the fusion construct. These results support a direct/primary role for MBNL in DM1.
From these results we conclude that one of the primary events in DM1 is the sequestration of MBNL protein by the repeat expansion. Whether this is a cause or an effect of the nuclear retention remains to be elucidated.
ACKNOWLEDGEMENTS
We thank Garry Morgan for his comments and suggestions. We also thank Tom Cooper, University of Texas, for his gift of plasmid containing CUG-BP sequence; Gideon Dreyfuss, University of Pennsylvania, for his gift of anti-hnRNP C (mAb 4F4) antisera; Maurice Swanson, University of Florida, for anti-CUG-BP (mAb 3B1) antisera, and Mani Mahadevan, University of Wisconsin–Madison, for the in-situ hybridisation and immunofluorescence protocol. This work was supported by the Muscular Dystrophy Campaign, the Muscular Dystrophy Association USA and the Myotonic Dystrophy Patients Support Group.
References
- 1.Harper P.S. (1989) Myotonic dystrophy, 2nd edn. Saunders, London; Philadelphia.
- 2.Brook J.D., McCurrach,M.E., Harley,H.G., Buckler,A.J., Church,D., Aburatani,H., Hunter,K., Stanton,V.P., Thirion,J.P., Hudson,T. et al. (1992) Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell, 68, 799–808. [DOI] [PubMed] [Google Scholar]
- 3.Krahe R., Ashizawa,T., Abbruzzese,C., Roeder,E., Carango,P., Giacanelli,M., Funanage,V.L. and Siciliano,M.J. (1995) Effect of myotonic dystrophy trinucleotide repeat expansion on DMPK transcription and processing. Genomics, 28, 1–14. [DOI] [PubMed] [Google Scholar]
- 4.Fu Y.H., Friedman,D.L., Richards,S., Pearlman,J.A., Gibbs,R.A., Pizzuti,A., Ashizawa,T., Perryman,M.B., Scarlato,G., Fenwick,R.G. et al. (1993) Decreased expression of myotonin-protein kinase messenger RNA and protein in adult form of myotonic dystrophy. Science, 260, 235–238. [DOI] [PubMed] [Google Scholar]
- 5.Jansen G., Groenen,P.J., Bachner,D., Jap,P.H., Coerwinkel,M., Oerlemans,F., van den Broek,W., Gohlsch,B., Pette,D., Plomp,J.J. et al. (1996) Abnormal myotonic dystrophy protein kinase levels produce only mild myopathy in mice. Nat. Genet., 13, 316–324. [DOI] [PubMed] [Google Scholar]
- 6.Wang Y.H. and Griffith,J. (1995) Expanded CTG triplet blocks from the myotonic dystrophy gene create the strongest known natural nucleosome positioning elements. Genomics, 25, 570–573. [DOI] [PubMed] [Google Scholar]
- 7.Thornton C.A., Wymer,J.P., Simmons,Z., McClain,C. and Moxley,R.T.,III (1997) Expansion of the myotonic dystrophy CTG repeat reduces expression of the flanking DMAHP gene. Nat. Genet., 16, 407–409. [DOI] [PubMed] [Google Scholar]
- 8.Klesert T.R., Otten,A.D., Bird,T.D. and Tapscott,S.J. (1997) Trinucleotide repeat expansion at the myotonic dystrophy locus reduces expression of DMAHP. Nat. Genet., 16, 402–406. [DOI] [PubMed] [Google Scholar]
- 9.Klesert T.R., Cho,D.H., Clark,J.I., Maylie,J., Adelman,J., Snider,L., Yuen,E.C., Soriano,P. and Tapscott,S.J. (2000) Mice deficient in Six5 develop cataracts: implications for myotonic dystrophy. Nat. Genet., 25, 105–109. [DOI] [PubMed] [Google Scholar]
- 10.Mankodi A., Logigian,E., Callahan,L., McClain,C., White,R., Henderson,D., Krym, M. and Thornton,C.A. (2000) Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat. Science, 289, 1769–1773. [DOI] [PubMed] [Google Scholar]
- 11.Hamshere M.G., Newman,E.E., Alwazzan,M., Athwal,B.S. and Brook,J.D. (1997) Transcriptional abnormality in myotonic dystrophy affects DMPK but not neighboring genes. Proc. Natl Acad. Sci. USA, 94, 7394–7399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davis B.M., McCurrach,M.E., Taneja,K.L., Singer,R.H. and Housman,D.E. (1997) Expansion of a CUG trinucleotide repeat in the 3′ untranslated region of myotonic dystrophy protein kinase transcripts results in nuclear retention of transcripts. Proc. Natl Acad. Sci. USA, 94, 7388–7393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Taneja K.L., McCurrach,M., Schalling,M., Housman,D. and Singer,R.H. (1995) Foci of trinucleotide repeat transcripts in nuclei of myotonic dystrophy cells and tissues. J. Cell Biol., 128, 995–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Timchenko L.T., Timchenko,N.A., Caskey,C.T. and Roberts,R. (1996) Novel proteins with binding specificity for DNA CTG repeats and RNA CUG repeats: implications for myotonic dystrophy. Hum. Mol. Genet., 5, 115–121. [DOI] [PubMed] [Google Scholar]
- 15.Roberts R., Timchenko,N.A., Miller,J.W., Reddy,S., Caskey,C.T., Swanson,M.S. and Timchenko,L.T. (1997) Altered phosphorylation and intracellular distribution of a (CUG)n triplet repeat RNA-binding protein in patients with myotonic dystrophy and in myotonin protein kinase knockout mice. Proc. Natl Acad. Sci. USA, 94, 13221–13226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Philips A.V., Timchenko,L.T. and Cooper,T.A. (1998) Disruption of splicing regulated by a CUG-binding protein in myotonic dystrophy. Science, 280, 737–741. [DOI] [PubMed] [Google Scholar]
- 17.Michalowski S., Miller,J.W., Urbinati,C.R., Paliouras,M., Swanson,M.S. and Griffith,J. (1999) Visualization of double-stranded RNAs from the myotonic dystrophy protein kinase gene and interactions with CUG-binding protein. Nucleic Acids Res., 27, 3534–3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nakielny S. and Dreyfuss,G. (1999) Transport of proteins and RNAs in and out of the nucleus. Cell, 99, 677–690. [DOI] [PubMed] [Google Scholar]
- 19.Tiscornia G. and Mahadevan,M.S. (2000) Myotonic dystrophy: the role of the CUG triplet repeats in splicing of a novel DMPK exon and altered cytoplasmic DMPK mRNA isoform ratios. Mol. Cell, 5, 959–967. [DOI] [PubMed] [Google Scholar]
- 20.Miller J.W., Urbinati,C.R., Teng-Umnuay,P., Stenberg,M.G., Byrne,B.J., Thornton,C.A. and Swanson,M.S. (2000) Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. EMBO J., 19, 4439–4448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Begemann G., Paricio,N., Artero,R., Kiss,I., Perez-Alonso,M. and Mlodzik,M. (1997) Muscleblind, a gene required for photoreceptor differentiation in Drosophila, encodes novel nuclear Cys3His-type zinc-finger-containing proteins. Development, 124, 4321–4331. [DOI] [PubMed] [Google Scholar]
- 22.Artero R., Prokop,A., Paricio,N., Begemann,G., Pueyo,I., Mlodzik,M., Perez-Alonso,M. and Baylies,M.K. (1998) The muscleblind gene participates in the organization of Z-bands and epidermal attachments of Drosophila muscles and is regulated by Dmef2. Dev. Biol., 195, 131–143. [DOI] [PubMed] [Google Scholar]
- 23.Timchenko L.T., Miller,J.W., Timchenko,N.A., DeVore,D.R., Datar,K.V., Lin,L., Roberts,R., Caskey,C.T. and Swanson,M.S. (1996) Identification of a (CUG)n triplet repeat RNA-binding protein and its expression in myotonic dystrophy. Nucleic Acids Res., 24, 4407–4414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tian B., White,R.J., Xia,T., Welle,S., Turner,D.H., Mathews,M.B. and Thornton,C.A. (2000) Expanded CUG repeat RNAs form hairpins that activate the double-stranded RNA-dependent protein kinase PKR. RNA, 6, 79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Napieraa M. and Krzyosiak,W.J. (1997) CUG repeats present in myotonin kinase RNA form metastable ‘slippery’ hairpins. J. Biol. Chem., 272, 31079–31085. [DOI] [PubMed] [Google Scholar]
- 26.Takahashi N., Sasagawa,N., Suzuki,K. and Ishiura,S. (2000) The CUG-binding protein binds specifically to UG dinucleotide repeats in a yeast three-hybrid system. Biochem. Biophys. Res. Commun., 277, 518–523. [DOI] [PubMed] [Google Scholar]
- 27.Taneja K.L. (1998) Localization of trinucleotide repeat sequences in myotonic dystrophy cells using a single fluorochrome-labeled PNA probe. Biotechniques, 24, 472–476. [DOI] [PubMed] [Google Scholar]