

Abstract
In the last few decades, perturbation in methyl-group and homocysteine (Hcy) balance have emerged as independent risk factors in a number of pathological conditions including neurodegenerative disease, cardiovascular dysfunction, cancer development, autoimmune disease, and kidney disease. Recent studies report Hcy to be a newly recognized risk factor for osteoporosis. Elevated Hcy levels are known to modulate osteoclastgenesis by causing detrimental effects on bone via oxidative stress induced metalloproteinase-mediated extracellular matrix degradation and decrease in bone blood flow. Evidence from previous studies also suggests that the decreased chondrocytes mediated bone mineralization in chick limb-bud mesenchymal cells and during the gestational period of ossification in rat model. However, Hcy imbalance and its role in bone loss, regression in vascular invasion, and osteoporosis, are not clearly understood. More investigations are required to explore the complex interplay between Hcy imbalance and onset of bone disease progression. This article reviews the current body of knowledge on regulation of Hcy mediated oxidative stress and its role in bone remodeling, vascular blood flow and progression of bone disease.
Methyl group and homocysteine (Hcy) metabolism have risen as important metabolic processes that cause profound effects on health and causes several diseases when disrupted (Newberne and Rogers, 1986; Scott et al., 1990; Kang et al., 1992; Mattson and Shea, 2003; Williams and Schalinske, 2010; Schalinske and Smazal, 2012). Hcy is a naturally occurring non-proteogenic, sulfur-containing amino acid derived from methionine metabolism via methyl group metabolism. Blood plasma Hcy concentrations highly depend on appropriate intracellular Hcy metabolism in the liver and kidney (Schalinske and Smazal, 2012). It has been shown that Hcy could be associated as an independent risk factor for heart, kidney and brain disease (Lominadze et al., 2006; Sen et al., 2009). Recently, high Hcy levels have also been associated with increased risk of fractures. However, the exact contributing factors that cause this fracture risk have not been fully studied (Meurs et al., 2004). Homocystinuria is a rare autosomal recessive disease characterized by increased circulating Hcy levels with several clinical manifestations. However, it is also reported that adults without homocystinuria are also subjected to risk of fractures with high levels of plasma Hcy (Meurs et al., 2004; Raisz, 2004). Nutritional factors such as vitamin B12, B6, and folate are cofactors in Hcy metabolism. Dietary supplementation with these vitamins may inversely alter plasma Hcy levels. Several studies found that cobalamin and folate are related to bone mineral density and fracture risk. The work of Sato et al. (1994) observed that fracture incidence was shown to reduce in patient who received high doses of vitamin B12 and folate. In addition, genetic polymorphism in relation to Hcy metabolism may also result in high Hcy level (Selhub et al., 1993; Jacques et al., 1999; Lee et al., 2008).
Osteoporosis is a major social health problem, causing considerable morbidity and mortality worldwide. The annual expenditures for the direct medical care of osteoporotic fractures in the United States in sum total at $13.8 billion in 1995, $17.5 billion in 2002, and $22 billion in 2008 (Herrmann et al., 2005a; Blume and Curtis, 2011). Little is known about the role of this innocent bystander “Hcy” in bone disease. More research is needed to illuminate the mechanism linking Hcy and bone disease. The first ever study about this was demonstrated by McKusick (1966) and then this particular area were further explored by others too (McKusick, 1966; Lubec et al., 1996; Masse et al., 2003). In this review, we emphasize the role of Hcy that elicits bone disease. More specifically, how elevated levels of plasma Hcy causes bone remodeling by activating osteoclasts, decreasing bone mineralization, and leading to onset osteoporotic fractures.
Homocysteine Mediates Oxidative Stress and Enhances Osteoclastgenesis
Hcy has been shown to be a potent stimulator for oxidant signaling in both in vivo and in vitro studies (Lentz, 1997; Voutilainen et al., 1999; Eberhardt et al., 2000; Tyagi et al., 2005; Koh et al., 2006). Osteoclasts lineage and its formation are very sensitive to increased generation of reactive oxygen species (ROS) (Garrett et al., 1990; Bax et al., 1992; Lean et al., 2003). Earlier work reports that Hcy increases intracellular ROS, which enhances osteoclast differentiation (OCs) and its activity in both primary mouse bone marrow cells (BMCs) in the femora and tibias of ICR mice and also in peripheral blood mononuclear cells (Markus et al., 2005; Koh et al., 2006; Blume and Curtis, 2011). The differentiated OCs activity was measured by tartrate-resistant acid phosphatase (TRAP), creatine phosphokinase (CP-K), and dentine resorbing activity. In addition, Hcy also suppress the apoptosis of osteoclasts (Selander et al., 1996; Koh et al., 2006). The increased Hcy impairs the cellular and molecular mechanism of bone marrow-derived osteoclasts by causing imbalance between phosphorylation and de-phosphorylation state of various protein kinases that modulates bone cell remodeling. Hcy has been reported to increased phosphorylation of P38 mitogen-activated protein kinases (MAPK) in receptor activator of nuclear factor kappa-B ligand (RANKL) dependent manner, but it does not have any effects on phosphorylation of extracellular signal-regulated kinases(ERK), c-Jun N-terminal kinases (JNK), and nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor (NFKBi), alpha (IκBα) (Koh et al., 2006). As osteoblasts are important for bone formation, studies showed that its activity is decreased with increased Hcy concentration (Herrmann et al., 2008).
It is reported that activation of peroxisome proliferator-activated receptor gamma (PPARγ) is associated with bone loss and increased fracture risk by positively regulating osteoclastgenesis (Wan et al., 2007). Moreover, Hcy acts as ligand to PPAR-γ receptors in bone cells and might affect bone resorption by activating detrimental oxidizing environments (Itzstein et al., 2001; Stunes et al., 2011). In the mouse model of hyperhomocysteinemia (HHcy), Akita mice, it is suggested that PPAR-γ, tissue inhibitor of metalloproteinase-4 (TIMP-4) and anti-oxidant thioredoxin are attenuated, whereas matrix metalloproteinase (MMP)-9, TIMP-3, and NADPH oxidase 4 (NOX4) are induced (Itzstein et al., 2001; Mishra et al., 2010). Moreover, the activation of PPAR receptor further increases cellular antioxidant enzymes like superoxide dismutase (SOD) and catalase, while decreasing NOX4 but under total homocysteine (tHcy) conditions, this antioxidant mechanism is disturbed by creating detrimental oxidative stress (Inoue et al., 1998).
Chenu et al. (1998) investigated the possibility that whether bone cells express N-methyl-D-aspartate (NMDA) receptors (NMDARI), ionotropic N-methyl-D-aspartate (NMDA) receptor-I subunit (INMDARI) or not. They further characterized if they are important for osteoclast activity via immunocytochemistry analysis of rat bone sections (Chenu et al., 1998). It is shown that Hcy increases oxidative stress-mediated ROS generation by activation of NMDA-R followed by intracellular calcium (Ca+2) overload. This increase in ROS will further activate the MMPs. The activation of MMPs may play an important role in matrix degradation (Vacek et al., 2013) (Fig. 1).
Fig. 1.
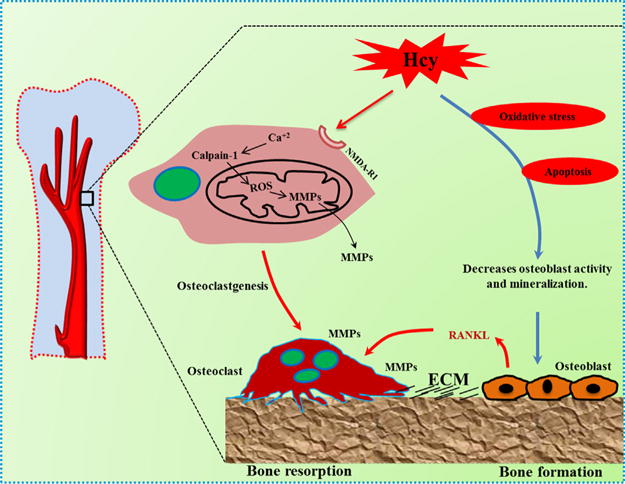
Schematic mechanism of homocysteine-mediated bone remodeling via oxidative stress. The increased homocysteine level elevates intracellular calcium by agonizing NMDA-RI. This in turn increases activation of calcium dependent calpain-I, which disrupts the mitochondrial membrane potential. Thus increasing reactive oxygen species (ROS) which further activates MMPs, resulting in matrix degradation. Increased homocysteine levels also induces apoptosis via ROS-mediated oxidative pathways which further switches off osteoblast activity and mineralization, by up regulating RANKL expression, leading to osteoporosis by reducing bone formation.
In another study, it was found that Hcy induces apoptosis via ROS-mediated mitochondrial pathway in NF-kappaB activated primary human bone marrow stromal cells and HS-5 cell line. This leads to release of cytochrome-c and subsequent activation of caspase-3 and 9. This suggests it would have a similar apoptotic effect on osteoblasts, leading to osteoporosis by reducing bone formation. This physiological perturbation may provide the basis for evidence that Hcy is involved in osteoporotic bone loss (Kim et al., 2006).
Homocysteine-Mediated Mitochondrial MMP Activation and Collagen Matrix Degradation
Mitochondria are important cellular organelles involved in aerobic respiration; producing ATP through oxidative phosphorylation. They serve an essential role in minimizing cellular ROS from the electron transport chain (ETC). MMPs are a series of enzymes involved in matrix degradation that are activated in mitochondria in response to cellular generation of ROS (Fu et al., 2001). One important mechanism for MMP-9 activation occurs via calpain-I in mitochondria. The mechanism of Hcy-induced extracellular matrix remodeling by MMP-9 activation, via mitochondrial pathway, is largely unknown. In this scenario, the work of Moshal et al. (2006) is very relevant which showed that Hcy was shown to activate MMP-9 via calpain-I mitochondrial translocation in heart microvascular endothelial cells in rats, resulting in intra-mitochondrial oxidative stress response. Prior to this downstream mechanism, Hcy binding to NMDA-R leads to increased intracellular Ca+2 and MAPK activation (Moshal et al., 2006). As NMDA-R was discovered in bone forming osteocytes (Itzstein et al., 2001), a similar mechanism may be harnessed for sequent activation of MMP via ROS-Calpain-I axis; therein activated MMPs are exited from mito-surface and cause matrix degradation process by entering the extracellular matrix (ECM). In this context, it concludes that mitochondria play an essential role in MMP activation and through this mechanism, is involved in bone turnover and repair (Fajardo et al., 2010).
Specific MMPs have their specific functions during physiological needs. Some MMPs have been ascertained to serve regulatory roles. For example, MMP-9/gelatinase B is a key regulator of growth plate angiogenesis and apoptosis of hypertrophic chondrocytes (Vu et al., 1998); MMP-9 and MMP-13 expressed by osteoclasts, endothelial cells, and bone marrow stromal cells plays crucial role in endochondral ossification (Pasternak and Aspenberg, 2009); MMP-14 regulates MMP-2, which promotes osteogenesis from osteoblasts (Sato et al., 1994).
Homocysteine Negatively Regulates Chondrocyte-Mediated Bone Matrix Mineralization and Gestational Ossification
Several reported studies have assessed the role of collagen cross-links in the process of matrix mineralization. β-aminopropionitrile (BAPN), an inhibitor of collagen crosslinks, causes a decrease in bone mineralization both in vitro and in vivo by inhibiting the enzyme responsible for the generation of Lysl oxidase; the reactive aldehyde involved in collagen cross-linking (Reddi and Sullivan, 1979; Carrington et al., 1984). Hcy is also known to cause decreased matrix maturation and bone mineralization. The work of Khan et al. (2001) showed differentiation chick limb-bud mesenchymal cells under Hcy treatment, plated in micro-mass cultures and allowed for chondrocyte-mediated matrix maturation and mineralization, demonstrated that exogenous Hcy supplementation was known to be teratogenic in the developing chick (Rosenquist et al., 1996); it reduced mineralization was also shown in the differentiating chick limb-bud mesenchymal cells. Hcy administration causes multifactorial effects on the cellular system. It reduces formation of pyridinoline and deoxypyridinoline cross-links, decreases proteoglycan content thereby directly altering matrix organization, and negatively affects mineralization by reducing Ca+2 uptake. Electron microscope results indicate that Hcy treatment results in irregular deposition of collagen in the extracellular microenvironment, and fibrils were found to be smaller in cross-section than those in control cultures. The abnormal collagen deposition leads to abnormal cell-matrix interaction. As a result, chondrocytes may not be receiving the appropriate extracellular signals from surrounding environment for their differentiation and mineralization (Khan et al., 2001).
Proper longitudinal bone growth depends on the production of a mineralized cartilage template by the epiphyseal plate. Alternations in the biochemical composition of growth plate lead to abnormalities in organization of cartilage septae thus contributing to reduced bone mass. Most experimental studies evaluating the effects of pronounced hyperhomocysteine have shown it to produce temporal shortening of the long bones (Robert et al., 2005; Herrmann et al., 2007). The work of Herrmann et al. (2007) reported moderate to severe reduction in trabecular bone mass in rats treated with moderate hyperhomocysteine- or methionine-rich diet. However, the possible effects of Hcy on growth plates in bone microarchitecture have not been fully reported. The work of Azizi et al. (2010) demonstrated in rat model of moderate HHcy condition. In this study, experimental groups were received Hcy in their drinking water before mating and for the total period of pregnancy. The histologic and histomorphometric analysis of tibia, radius, and vertebra were significantly decreased and that growth plate structure was abnormal in the offspring of homocysteine treated mothers. Trabecular bone volume was observed to possess abnormal cartilaginous structure and abnormal surface area in developing rat embryo and enlargement of the hypertrophic zone due to delayed resorption of epiphyseal cartilage during endochondral ossification and subsequent hypertrophy in hypertrophic zone (Azizi et al., 2010). They found that the increased number of chondrocytes rather than an increase in cell or extracellular matrix size may be responsible for enlargement of hypertrophic zone. Overall, the moderate HHcy condition induces micro-architectural deterioration in growth plate and bone development during endochondral ossification.
Effect of Homocysteine on Bone Loss and Osteoporosis
Bone homeostasis is maintained by a balance between bone resorption by osteoclasts and bone formation by osteoblasts. Osteoblasts only play a central role in bone formation, by producing multiple bone matrix, but also regulating osteoclast activity by soluble mediators resulting in bone resorption. To determine the detrimental effects of tHcy on bone, several biochemical bone turnover markers are used in studies, such as, hydroxyproline, N-terminal collagen-I telopeptides, and non-collagenous matrix protein osteocalcin (Ozdem et al., 2007). The work of Herrmann et al. (2005b) showed direct interaction of tHcy with collagen bone matrix. The following markers were observed to define the relationship between tHcy and organic/inorganic bone resorption. These are (i) bone mineral density at total hip (BMD-HIP); (ii) bone mineral density at lumbar spine (BMD-LS); (iii) serum Ca+2; (iv) urinary deoxypyridinoline (DPD) cross-links; (v) fasting venous blood and urine sampling; (vi) osteocalcin measurement; (vii) osteoprotegerin (OPG); and (viii) soluble receptor activator of NF-κB ligand (sRANKL) (McLean et al., 2004). There was a significant relationship found between Hcy and calcium Ca+2 measurements (r = 0.170, P = 0.045) as well as Hcy and deoxypyridinoline (DPD) measurements (r = 0.193, P = 0.022); however, the others did not found a significant relationship with Hcy (Herrmann et al., 2005a).
It is evidenced that tHcy has more effect on femoral neck fractures by observing parameters like collagen cross-link ratio and bone mineralization density distribution (BMD) which suggests significant correlation between HHcy and collagen crosslink ratio in primary mineralized bone area (P < 0.0001). However, there was no correlation obtained between HHcy and BMD (P < 0.05). Hence it could be possible that HHcy alters the bone quality by interfering with collagen cross-linking (Herrmann et al., 2005b; Blouin et al., 2009). In addition, another key bone protein marker, β-catenin, which mediates Wnt signalling pathways in bone metabolism. It controls both differentiation of osteoblasts and osteoclasts. A study by Kramer et al. (2010) observed that low bone mass phenotype had increased numbers of osteoclast, and osteoclast activity, in β-catenin-knockout mouse model. Hcy is also shown to have a direct effect on bone biomechanical properties by accumulating in bone surface and reducing trabecular or spongy bone density. The study observed that 65% of tHcy binds to collagen matrix and decreases bone strength (Herrmann et al., 2009). Hcy also suppresses the expression of the collagen crosslinkers lysyl oxidase involving IL-6, Fli 1, and epigenetic DNA methylation; all of which contribute to decrease bone quality (Thaler et al., 2011).
Osteoporosis is a complex multifactorial disease with characteristics of low bone mass. Strong genetic components and lifestyle factors are also involved in maintenance of bone mass (Vaes et al., 2009). Nutrition is one of the factors for maintaining proper bone health. During the early decades of life, adequate intake of nutrients (Ca+2 and vitamin D) is essential to establish peak bone mass. Nutritional deprivation of such nutrients can cause impaired skeletal development during childhood. In adults, these deficiencies have been associated with loss of bone quality, making bones more prone to fracture risk (Palacios, 2006; Prentice et al., 2006; Cashman, 2007). A challenge in nutrition research is identifying the key food components that maintain bone health. In the last few decades, it has been found that key vitamins, such as vitamin B12, B6, and folate, regulate methionine metabolism. Deficiencies in these key vitamins negatively regulates the methionine cycle leading to plasma accumulation of Hcy. A number of epidemiological studies have reported that plasma Hcy level associated with decreased intake of key vitamins is correlated with reduced bone quality. Elevated Hcy could be a possible independent factor for fracture risk (Dhonukshe-Rutten et al., 2005; Morris et al., 2005; Baines et al., 2007). The work of Perier et al. (2007) showed that fracture risk was higher in women with increased plasma tHcy in the highest quartile, independent of age. The tHcy in serum could be a frailty marker, modulated by factors such as nutritional state and physical activity (Perier et al., 2007). In another study, it was shown that Hcy impairs fracture repair in a bone fracture mouse model after feeding high Hcy diet for 3 weeks. The parameters like decreased bending stiffness of femora, smaller diameter with no difference in tissue formation (Claes et al., 2009). It is well known that estrogen receptors are important for female reproductive function and bone formation. An in vivo and in vitro study suggests that tHcy can promote hyper-methylation of the promoter region of estrogen receptor a, leading to decreased mRNA transcription. Hence, estrogen receptor α transcription is down regulated by increased levels of Hcy, causing deterioration of biomechanical properties of bone (Aaron et al., 2009). In a study of women between the ages of 70 and 58 years, result showed that higher tHcy was associated with a greater hip BMD loss over 4 years (−2.8%) compared to the lowest percentiles of tHcy (−1.2%). This concludes that increased tHcy in elderly patients is associated more with significant bone loss rather than incidence of bone fracture (Perier et al., 2007). In a HHcy mouse model, mice fed with high methionine for 3 months, results showed increased bone fragility of the femoral neck by 18% in methionine fed rats. In conclusion, Hcy causes a decrease in biomechanical properties through deteriorating bone loss and the relation between bone resorption and bone formation indicates a shift toward bone resorption, which might be a plausible explanation for osteoporosis (Ozdem et al., 2007) (Fig. 2).
Fig. 2.
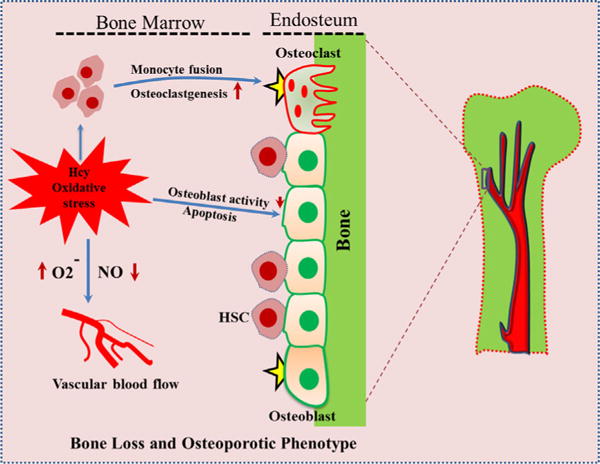
Mechanism of homocysteine that contribute to the development of osteoporosis. Homocysteine level is associated with increased oxidative stress in bone microenvironment. Increased reactive oxygen species (ROS) induces osteoblast apoptosis thereby decreasing osteoblastgenesis. This increase in oxidative stress further reduces NO bioavailability, by producing superoxide anions ( ), which could also decrease bone blood flow and possibly angiogenesis. The ROS generated by this process activates osteoclastgenesis, by monocyte fusion, further contributing to loss in bone density; leading to osteoporosis.
Effects of Homocysteine on Vasculature
It is known that bone formation is dependent on a well-organized vascular network. Vascular blood flow is recognised as a vital basis of normal bone growth and repair, as bones are living tissue entities responsible for the maintenance and support of life processes (Fleming et al., 2001; Vacek et al., 2013). The blood supply to bone, by nutrient arteries, is delivered to the endosteal cavity and then flows through marrow sinusoids before exiting via numerous small vessels that ramify through the bone cortex (Marenzana and Arnett, 2013). The quality of vascular blood supply to bone tends to decline with age and may be compromised in common pathological settings including; diabetes, bone metastasis, anemias, and chronic airway diseases. Reductions in vascular supply are associated with bone loss. This may be due to regional hypoxia, which blocks osteoblast-mediated matrix mineralization and bone formation and reciprocally activates bone resorptive osteoclast formation; subsequently causing bone loss. Common regulatory factors like nitrate, produced during endothelial nitric oxide synthase (eNOS) group enzyme signaling, are potent vasodilators which exerts their osteogenic effects on bone via vasculature (Marenzana and Arnett, 2013). It was also reported that mice deficient in endothelial nitric oxide synthase (NOS) showed diminished bone development, parallel with mice treated with the nitric oxide (NO) inhibitor, amino-guanidine (L-NAME) (Sunyer et al., 1997).
Recent studies have shown that HHcy can alter the ability of vasculature to dilate by decreasing the bioavailability of NO, which ultimately increases vascular resistance. Considering the evidence behind HHcy-mediated increase in vascular resistance; Tyagi et al. (2011) demonstrated that bone blood flow is decreased in HHcy conditions in experimental mice. The blood flow index in tibia of control groups was significantly higher compared with the Hcy-treated group, which potentially compromised mechanical bone properties. However, tibia bone density was unchanged among the both groups (Tyagi et al., 2011). It is suggested that Hcy increases oxidative stress mechanism and decreases the bioavailability of NO (Banfi et al., 2008). This increase in oxidative stress that further reduces NO bioavailability, could also decrease bone blood flow and lead to osteoporosis (Sanchez-Rodriguez et al., 2007). In a follow-up experiment on mice deficient in cystathionine β-synthase (CBS), a gene involved in tHcy clearance, tibia blood flow was observed to be decreased relative to control mice. In summary, the above studies provide strong evidence that HHcy is involved in decreased blood flow, thereby causing weakening of the bone (Fig. 2). Future studies are needed to understand the HHcy-mediated molecular mechanisms that disrupt bone vessel architecture and integrity.
Conclusion
It is clearly evidenced that Hcy directly activates osteoclast formation and activity, in vitro, via increased oxidative mechanism and mitochondrial MMP activation causing bone matrix degradation and alterations in the biomechanical properties of bone. Furthermore, it is evidenced that Hcy decreases bone blood flow, as a consequence of decreased bioavailability of NO, affecting bone structure. These findings suggest that increased bone resorptive action by osteoclasts may contribute to osteoporosis in individuals with mild to moderate HHcy. Our lab is interested in the mechanisms of osteoclast activation, osteoblastic dysfunction and vascular network-mediated decrease in bone blood flow, under mild to moderate HHcy conditions. We welcome further research in these proposed areas.
Acknowledgments
This work was supported by National Institutes of Health grants: HL-107640 to N.T. and AR-067667 to S.C.T.
Literature Cited
- Aaron WJ, Alexander AT, Brugmann SA, Xu Y, Carre AL, Leucht P, Hamilton K, Korach KS, Longaker MT. Estrogen/estrogen receptor a signaling in mouse postero-frontal cranial suture fusion. PLoS ONE. 2009;4:e7120. doi: 10.1371/journal.pone.0007120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azizi ZA, Zamani A, Omrani LR, Omrani L, Dabaghmanesh MH, Mohammadi A, Namavar MR, Omrani GR. Effects of hyperhomocysteinemia during the gestational period on ossification in rat embryo. Bone. 2010;46:1344–1348. doi: 10.1016/j.bone.2009.11.027. [DOI] [PubMed] [Google Scholar]
- Baines M, Kredan MB, Usher J, Davison A, Higgins G, Taylor W, West C, Fraser WD, Ranganath LR. The association of homocysteine and its determinants MTHFR genotype, folate, vitamin B12and vitamin B6 with bone mineral density in postmenopausal British women. Bone. 2007;40:730–736. doi: 10.1016/j.bone.2006.10.008. [DOI] [PubMed] [Google Scholar]
- Banfi G, Iorio EL, Corsi MM. Oxidative stress, free radicals and bone remodeling. Clin Chem Lab Med. 2008;46:1550–1555. doi: 10.1515/CCLM.2008.302. [DOI] [PubMed] [Google Scholar]
- Bax BE, Alam AS, Banerji B, Bax CM, Bevis PJ, Stevens CR, Moonga BS, Blake DR, Zaidi M. Stimulation of osteoclastic bone resorption by hydrogen peroxide. Biochem Biophys Res Commun. 1992;183:1153–1158. doi: 10.1016/s0006-291x(05)80311-0. [DOI] [PubMed] [Google Scholar]
- Blouin S, Thaler HW, Korninger C, Schmid R, Hofstaetter JG, Zoehrer R. Bone matrixquality and plasma homocysteine levels. Bone. 2009;44:959–964. doi: 10.1016/j.bone.2008.12.023. [DOI] [PubMed] [Google Scholar]
- Blume SW, Curtis JR. Medical costs of osteoporosis in the elderly medicare population. Osteoporos Int. 2011;22:1835–1844. doi: 10.1007/s00198-010-1419-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrington M, Bird T, Levene C. The inhibition of lysyl oxidase in vivo by isoniazid and its reversal by pyridoxal. Effect on collagen cross-linking in the chick embryo. Biochem J. 1984;221:837–843. doi: 10.1042/bj2210837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cashman KD. Diet, nutrition, and bone health. J Nutr. 2007;137:2507S–2512S. doi: 10.1093/jn/137.11.2507S. [DOI] [PubMed] [Google Scholar]
- Chenu C, Serre CM, Raynal C, Burt-Pichat B, Delmas PD. Glutamate receptors are expressed by bone cells and are involved in bone resorption. Bone. 1998;22:295–299. doi: 10.1016/s8756-3282(97)00295-0. [DOI] [PubMed] [Google Scholar]
- Claes L, Schmalenbach J, Herrmann M, Olku I, Garcia P, Histing T, Obeid R, Schorr H, Herrmann W, Pohlemann T, Menger MD, Holstein JH. Hyperhomocysteinemia is associated with impaired fracture healing in mice. Calcif Tissue Int. 2009;85:17–21. doi: 10.1007/s00223-009-9262-6. [DOI] [PubMed] [Google Scholar]
- Dhonukshe-Rutten RA, Pluijm SM, de Groot LC, Lips P, Smit JH, Van Staveren WA. Homocysteine and vitamin B12 status relate to bone turnover markers, broadband ultrasound attenuation, and fractures in healthy elderly people. J Bone Miner Res. 2005;20:921–929. doi: 10.1359/JBMR.050202. [DOI] [PubMed] [Google Scholar]
- Eberhardt RT, Forgione MA, Cap A, Leopold JA, Rudd MA, Trolliet M, Heydrick S, Stark R, Klings ES, Moldovan NI, Yaghoubi M, Goldschmidt-Clermont PJ, Farber HW, Cohen R, Loscalzo J. Endothelial dysfunction in a murine model of mild hyperhomocyst(e) inemia. J Clin Invest. 2000;106:483–491. doi: 10.1172/JCI8342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fajardo M, Liu CJ, Ilalov K, Egol KA. Matrix metalloproteinases that associate with and cleave bone morphogenetic protein-2 in vitro are elevated in hypertrophic fracture nonunion tissue. J Orthop Trauma. 2010;24:557–563. doi: 10.1097/BOT.0b013e3181ed294c. [DOI] [PubMed] [Google Scholar]
- Fleming JT, Barati MT, Beck DJ, Dodds JC, Malkani AL, Parameswaran D, Soukhova GK, Voor MJ, Feitelson JB. Bone blood flow and vascular reactivity. Cells Tissues Organs. 2001;169:279–284. doi: 10.1159/000047892. [DOI] [PubMed] [Google Scholar]
- Fu X, Kassim SY, Parks WC, Heinecke JW. Hypochlorous acid oxygenates the cysteine switch domain of pro-matrilysin (MMP-7): A mechanism for matrix metalloproteinase activation and atherosclerotic plaque rupture by myeloperoxidase. J Biol Chem. 2001;276:41279–41287. doi: 10.1074/jbc.M106958200. [DOI] [PubMed] [Google Scholar]
- Garrett IR, Boyce BF, Oreffo RO, Bonewald L, Poser J, Mundy GR. Oxygen-derived free radicals stimulate osteoclastic bone resorption in rodent bone in vitro and in vivo. J Clin Invest. 1990;85:632–639. doi: 10.1172/JCI114485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann M, Kraenzlin M, Pape G, Sand-Hill M, Herrmann W. Relation between homocysteine and biochemical bone turnover markers and bone mineral density in peri-and postmenopausal women. Clin Chem Lab Med. 2005a;43:1118–1123. doi: 10.1515/CCLM.2005.195. [DOI] [PubMed] [Google Scholar]
- Herrmann M, Widmann T, Colaianni G, Colucci S, Zallone A, Herrmann W. Increased osteoclast activity in the presence ofincreased homocysteine concentrations. Clin Chem. 2005b;52:2348–2353. doi: 10.1373/clinchem.2005.053363. [DOI] [PubMed] [Google Scholar]
- Herrmann M, Wildemann B, Claes L, Klohs S, Ohnmacht M, Taban-Shomal O. Experimental hyperhomocysteinemia reduces bone quality in rats. Clin Chem. 2007;53:1455–1461. doi: 10.1373/clinchem.2007.086272. [DOI] [PubMed] [Google Scholar]
- Herrmann M, Umanskaya N, Wildemann B, Colaianni G, Widmann T, Zallone A. Stimulation of osteoblast activity by homocysteine. J Cell Mol Med. 2008;12:1205–1210. doi: 10.1111/j.1582-4934.2008.00104.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann M, Tami A, Wildemann B, Wolny M, Wagner A, Schorr H. Hyperhomocysteinemia induces a tissue specific accumulation of homocysteine in bone by collagen binding and adversely affects bone. Bone. 2009;44:467–475. doi: 10.1016/j.bone.2008.10.051. [DOI] [PubMed] [Google Scholar]
- Homocysteine Studies Collaboration. Homocysteine and risk of ischemic heart disease and stroke—A meta-analysis. J Am Med Assoc. 2002;288:2015–2022. doi: 10.1001/jama.288.16.2015. [DOI] [PubMed] [Google Scholar]
- Inoue I, Noji S, Awata T, Takahashi K, Nakajima T, Sonoda M. Bezafibrate has an antioxidant effect: Peroxisome proliferator-activated receptor α is associated with Cu 2+, Zn2+ superoxide dismutase in the liver. Life Sci. 1998;63:135–144. doi: 10.1016/s0024-3205(98)00249-5. [DOI] [PubMed] [Google Scholar]
- Itzstein C, Cheynel H, Burt-Pichat B, Merle B, Espinosa L, Delmas PD, Chenu C. Molecular identification of NMDA glutamate receptors expressed in bone cells. J Cell Biochem. 2001;82:134–144. doi: 10.1002/jcb.1114. [DOI] [PubMed] [Google Scholar]
- Jacques PF, Selhub J, Bostom AG, Wilson PW, Rosenberg IH. The effect of folic acid fortification on plasma folate and total homocysteine concentrations. N Engl J Med. 1999;340:1449–1454. doi: 10.1056/NEJM199905133401901. [DOI] [PubMed] [Google Scholar]
- Kang SS, Wong PW, Malinow MR. Hyperhomocyst(e)inemia as a risk factor for occlusive vascular disease. Annu Rev Nutr. 1992;12:279–298. doi: 10.1146/annurev.nu.12.070192.001431. [DOI] [PubMed] [Google Scholar]
- Khan M, Yamauchi M, Srisawasdi S, Stiner D, Doty S, Paschalis EP, Boskey AL. Homocysteine decreases chondrocyte-mediated matrix mineralization in differentiating chick limb-bud mesenchymal cell micro-mass cultures. Bone. 2001;28:387–398. doi: 10.1016/s8756-3282(01)00409-4. [DOI] [PubMed] [Google Scholar]
- Kim DJ, Koh JM, Lee O, Kim NJ, Lee YS, Kim YS. Homocysteine enhances apoptosis in human bone marrow stromal cells. Bone. 2006;39:582–590. doi: 10.1016/j.bone.2006.03.004. [DOI] [PubMed] [Google Scholar]
- Koh JM, Lee YS, Kim YS, Kim DJ, Kim HH, Park JY. Homocysteine enhances bone resorption by stimulation of osteoclast formation and activity through increased intracellular ROS generation. J Bone Miner Res. 2006;21:1003–1011. doi: 10.1359/jbmr.060406. [DOI] [PubMed] [Google Scholar]
- Kramer I, Halleux C, Keller H, Pegurri M, Gooi JH, Weber PB. Osteocyte Wnt/β-cateninsignalingis requiredfornormal bone homeostasis. Mol Cell Biol. 2010;30:3071–3075. doi: 10.1128/MCB.01428-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lean JM, Davies JT, Fuller K, Jagger CJ, Kirstein B, Partington GA, Urry ZL, Chambers TJ. A crucial role for thiol antioxidants in estrogen-deficiency bone loss. J Clin Invest. 2003;112:915–923. doi: 10.1172/JCI18859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JS, Lacroix AZ, Wu L, Cauley JA, Jackson RD, Kooperberg C, Leboff MS, Robbins J, Lewis CE, Bauer DC, Cummings SR. Associations of serum sex hormone-binding globulin and sex hormone concentrations with hipfracture risk in postmenopausal women. J Clin Endocrinol Metab. 2008;93:1796–1803. doi: 10.1210/jc.2007-2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lentz SR. Homocysteine and vascular dysfunction. Life Sci. 1997;61:1205–1215. doi: 10.1016/s0024-3205(97)00392-5. [DOI] [PubMed] [Google Scholar]
- Lominadze D, Roberts AM, Tyagi N, Moshal KS, Tyagi SC. Homocysteine causes cerebrovascular leakage in mice. Am J Physiol Heart Cir Physiol. 2006;290:1206–1213. doi: 10.1152/ajpheart.00376.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubec B, Fang-Kircher S, Lubec T, Blom HJ, Boers GH. Evidence for McKusick’s hypothesis of deficient collagen cross-linking in patients with homocystinuria. Biochim Biophys Acta. 1996;1315:159–162. doi: 10.1016/0925-4439(95)00119-0. [DOI] [PubMed] [Google Scholar]
- Marenzana M, Arnett TR. The key role of the blood supply to bone. Bone Res. 2013;3:203–215. doi: 10.4248/BR201303001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masse PG, Boskey AL, Ziv I, Hauschka P, Donovan SM, Howell DS. Chemical and biomechanical characterization of hyperhomocysteinemic bone disease in an animal model. BMC Musculoskelet Disord. 2003;4:2. doi: 10.1186/1471-2474-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Shea TB. Folate and homocysteine metabolism in neural plasticity and neurodegenerative disorders. Trends Neurosci. 2003;26:137–146. doi: 10.1016/S0166-2236(03)00032-8. [DOI] [PubMed] [Google Scholar]
- McKusick VA. Heritable disorders of connective tissue. 4th. St. Louis: CV Mosby Company; 1966. p. 878. [Google Scholar]
- McLean RR, Jacques PF, Selhub J, Tucker KL, Samelson EJ, Broe KE, Hannan MT, Cupples LA, Kiel DP. Homocysteine as a predictive factor for hip fracture in older persons. N Engl J Med. 2004;350:2042–2049. doi: 10.1056/NEJMoa032739. [DOI] [PubMed] [Google Scholar]
- Meurs van JB, Dhonukshe-Rutten RA, Pluijm SM, van der Klift M, de Jonge R, Lindemans J, de Groot LC, Hofman A, Witteman JC, van Leeuwen JP, Breteler MM, Lips P, Pols HA, Uitterlinden AG. Homocysteine levels and the riskofosteoporotic fracture. N Engl J Med. 2004;350:2033–2041. doi: 10.1056/NEJMoa032546. [DOI] [PubMed] [Google Scholar]
- Mishra PK, Tyagi N, Sen U, Joshua IG, Tyagi SC. Synergism in hyperhomocysteinemia and diabetes: Role of PPAR gamma and tempol. Cardiovasc Diabetol. 2010;9:49. doi: 10.1186/1475-2840-9-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris MS, Jacques PF, Selhub J. Relation between homocysteine and B-vitamin status indicators and bone mineral density in older Americans. Bone. 2005;37:234–242. doi: 10.1016/j.bone.2005.04.017. [DOI] [PubMed] [Google Scholar]
- Moshal KS, Singh M, Sen U, Rosenberger DS, Henderson B, Tyagi N. Homocysteine mediated activation and mitochondrial translocation of calpain regulates MMP-9 in MVEC. Am J Physiol Heart Circ Physiol. 2006;291:2825–2835. doi: 10.1152/ajpheart.00377.2006. [DOI] [PubMed] [Google Scholar]
- Newberne PM, Rogers AE. Labile methyl groups and the promotion of cancer. Annu Rev Nutr. 1986;6:407–432. doi: 10.1146/annurev.nu.06.070186.002203. [DOI] [PubMed] [Google Scholar]
- Ozdem S, Samanci S, Tasatargil A, Yildiz A, Sadan G, Donmez L. Experimental hyperhomocysteinemia disturbs bone metabolism in rats. Scand J Clin Lab Invest. 2007;67:748–756. doi: 10.1080/00365510701342088. [DOI] [PubMed] [Google Scholar]
- Palacios C. The role of nutrients in bone health, from A to Z. Crit Rev Food Sci Nutr. 2006;46:621–628. doi: 10.1080/10408390500466174. [DOI] [PubMed] [Google Scholar]
- Pasternak B, Aspenberg P. Metalloproteinases and their inhibitors-diagnostic and therapeutic opportunities in orthopedics. Acta Orthop. 2009;80:693–703. doi: 10.3109/17453670903448257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perier MA, Gineyts E, Munoz F, Sornay-Rendu E, Delmas PD. Homocysteine and fracture risk in postmenopausal women: The OFELY study. Osteoporos Int. 2007;18:1329–1336. doi: 10.1007/s00198-007-0393-1. [DOI] [PubMed] [Google Scholar]
- Prentice A, Schoenmakers I, Laskey MA, de Bono S, Ginty F, Goldberg GR. Nutrition and bone growth and development. Proc Nutr Soc. 2006;65:348–360. doi: 10.1017/s0029665106005192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raisz LG. Homocysteine and osteoporotic fractures—Culprit or bystander? N Engl J Med. 2004;350:2089–2090. doi: 10.1056/NEJMe048030. [DOI] [PubMed] [Google Scholar]
- Reddi AH, Sullivan NE. Inhibition of mineralization by experimental lathyrism during matrix-induced endochondral bone differentiation. Proc Soc Exp Biol Med. 1979;162:445–448. doi: 10.3181/00379727-162-40701. [DOI] [PubMed] [Google Scholar]
- Robert K, Maurin N, Vayssettes C, Siauve N, Janel N. Cystathionine beta synthase deficiency affects mouse endochondral ossification. Anat Rec A Discov Mol cell Evol Biol. 2005;282:1–7. doi: 10.1002/ar.a.20145. [DOI] [PubMed] [Google Scholar]
- Rosenquist TH, Tatashak SA, Selhub J. Homocysteine induces congenital defects of the heart and neural tube: Effect of folic acid. Proc Natl Acad Sci USA. 1996;93:15227–15232. doi: 10.1073/pnas.93.26.15227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Rodriguez MA, Ruiz-Ramos M, Correa-Munoz E, Mendoza-Nunez VM. Oxidative stress as a risk factor for osteoporosis in elderly Mexicans as characterized by antioxidant enzymes. BMC Musculoskel Disord. 2007;19:124. doi: 10.1186/1471-2474-8-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato H, Takino T, Okada Y, Cao J, Shinagawa A, Yamamoto E. A matrix metalloproteinase expressed on the surface of invasive tumour cells. Nature. 1994;370:61. doi: 10.1038/370061a0. [DOI] [PubMed] [Google Scholar]
- Schalinske KL, Smazal AL. Homocysteine imbalance: A pathological metabolic marker. Adv Nutr. 2012;3:755–762. doi: 10.3945/an.112.002758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott JM, Kirke PN, Weir DG. The role of nutrition in neural tube defects. Annu Rev Nutr. 1990;10:277–295. doi: 10.1146/annurev.nu.10.070190.001425. [DOI] [PubMed] [Google Scholar]
- Selander KS, Monkkonen J, Karhukorpi EK, Harkonen P, Hannuniemi R, Vaananen HK. Characteristics of clodronate-induced apoptosis in osteoclasts and macrophages. Mol Pharmacol. 1996;50:1127–1138. [PubMed] [Google Scholar]
- Selhub J, Jacques PF, Wilson PW, Rush D, Rosenberg IH. Vitamin status and intake as primary determinants of homocysteinemia in an elderly population. JAMA. 1993;270:2693–2698. doi: 10.1001/jama.1993.03510220049033. [DOI] [PubMed] [Google Scholar]
- Sen U, Basu P, Abe OA, Giwimani S, Tyagi N, Metreveli N. Hydrogen sulfide ameliorates hyperhomocysteinemia-associated chronic renal failure. Am J Physiol Renal Physiol. 2009;297:410–419. doi: 10.1152/ajprenal.00145.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stunes AK, Westbroek I, Gustafsson BI, Fossmark R, Waarsing JH, Eriksen EF. The peroxisome proliferator-activated receptor (PPAR) a agonist fenofibrate maintains bone mass, while the PPARγ agonist pioglitazone exaggerates bone loss, in ovariectomized rats. BMC Endocr Disord. 2011;11:11. doi: 10.1186/1472-6823-11-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunyer T, Rothe L, Kirsch D, Jiang X, Anderson F, Osdoby P, Collin-Osdoby P. Ca2+ or phorbol ester but not inflammatory stimuli elevate inducible nitric oxide synthase messenger ribonucleic acid and nitric oxide (NO) release in avian osteoclasts: Autocrine NOmediates Ca2+-inhibited bone resorption. Endocrinology. 1997;138:2148–2162. doi: 10.1210/endo.138.5.5144. [DOI] [PubMed] [Google Scholar]
- Thaler R, Agsten M, Spitzer S, Paschalis EP, Karlic H, Klaushofer K, Varga F. Homocysteine suppresses the expression of the collagen cross-linker lysyl oxidase involving IL-6, Fli I, and epigenetic DNA methylation. J Biol Chem. 2011;286:5578–5588. doi: 10.1074/jbc.M110.166181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyagi N, Sedoris KC, Steed M, Ovechkin AV, Moshal KS, Tyagi SC. Mechanisms of homocysteine-induced oxidative stress. Am J Physiol Heart Circ Physiol. 2005;289:2649–2656. doi: 10.1152/ajpheart.00548.2005. [DOI] [PubMed] [Google Scholar]
- Tyagi N, Vacek TP, Fleming JT, Vacek JC, Tyagi SC. Hyperhomocysteinemia decreases bone blood flow. Vasc Health Risk Manag. 2011;7:31–35. doi: 10.2147/VHRM.S15844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vacek TP, Kalani A, Voor MJ, Tyagi SC, Tyagi N. The role of homocysteine in bone remodeling. Clin Chem Lab Med. 2013;51:579–590. doi: 10.1515/cclm-2012-0605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaes BL, Lute C, Blom HJ, Bravenboer N, de Vries TJ, Everts V, Dhonukshe-Rutten RA, Müller M, de Groot LC, Steegenga WT. Vitamin B(12) deficiency stimulates osteoclastogenesis via increased homocysteine and methylmalonic acid. Calcif Tissue Int. 2009;84:413–422. doi: 10.1007/s00223-009-9244-8. [DOI] [PubMed] [Google Scholar]
- Voutilainen S, Morrow JD, Roberts LJ, II, Alfthan G, Alho H, Nyyssonen K, Salonen JT. Enhanced in vivo lipid peroxidation at elevated plasma total homocysteine levels. Arterioscler Thromb Vasc Biol. 1999;19:1263–1266. doi: 10.1161/01.atv.19.5.1263. [DOI] [PubMed] [Google Scholar]
- Vu TH, Shipley JM, Bergers G, Berger JE, Helms JA, Hanahan D. MMP-9/gelatinase B is a key regulator of growth plate angiogenesis and apoptosis of hypertrophic chondrocytes. Cell. 1998;93:411–422. doi: 10.1016/s0092-8674(00)81169-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Y, Chong LW, Evans RM. PPAR-gamma regulates osteoclastogenesis in mice. Nat Med. 2007;13:1496–1503. doi: 10.1038/nm1672. [DOI] [PubMed] [Google Scholar]
- Williams KT, Schalinske KL. Homocysteine metabolism and its relation to health and disease. Biofactors. 2010;36:19–24. doi: 10.1002/biof.71. [DOI] [PubMed] [Google Scholar]