

Abstract
There is no effective treatment for amyotrophic lateral sclerosis (ALS), a devastating motor neuron disease. However, discovery of a G4C2 repeat expansion in the C9ORF72 gene as the most common genetic cause of ALS has opened up new avenues for therapeutic intervention for this form of ALS. G4C2 repeat expansion RNAs and proteins of repeating dipeptides synthesized from these transcripts are believed to play a key role in C9ORF72-associated ALS (c9ALS). Therapeutics that target G4C2 RNA, such as antisense oligonucleotides (ASOs) and small molecules, are thus being actively investigated. A limitation in moving such treatments from bench to bedside is a lack of pharmacodynamic markers for use in clinical trials. We explored whether poly(GP) proteins translated from G4C2 RNA could serve such a purpose. Poly(GP) proteins were detected in cerebrospinal fluid (CSF) and in peripheral blood mononuclear cells from c9ALS patients and, notably, from asymptomatic C9ORF72 mutation carriers. Moreover, CSF poly(GP) proteins remained relatively constant over time, boding well for their use in gauging biochemical responses to potential treatments. Treating c9ALS patient cells or a mouse model of c9ALS with ASOs that target G4C2 RNA resulted in decreased intracellular and extracellular poly(GP) proteins. This decrease paralleled reductions in G4C2 RNA and downstream G4C2 RNA–mediated events. These findings indicate that tracking poly(GP) proteins in CSF could provide a means to assess target engagement of G4C2 RNA–based therapies in symptomatic C9ORF72 repeat expansion carriers and presymptomatic individuals who are expected to benefit from early therapeutic intervention.
INTRODUCTION
Amyotrophic lateral sclerosis (ALS) is a rapidly progressive motor neuron disease that typically results in muscle atrophy, paralysis, and eventually death within 5 years of onset. Up to 50% of ALS patients develop cognitive and behavioral impairments, and ~15% fulfill the criteria for frontotemporal dementia (FTD), which is characterized by changes in personality, behavior, and language (1).
Only one minimally effective drug, riluzole, is approved for ALS despite more than 30 clinical trials conducted since 1995. The dearth of ALS therapeutics stems partly from an incomplete understanding of the causative pathomechanisms. However, discovery of a G4C2 repeat expansion in the C9ORF72 gene as the most common genetic cause of ALS and FTD (2, 3) has resulted in impressive efforts toward elucidating how this mutation causes c9ALS (C9ORF72-associated ALS) or c9FTD, collectively referred to as c9ALS/FTD. Although the normal number of C9ORF72 G4C2 repeats is lower than 30, c9ALS/FTD patients have several hundred to several thousand (4). Putative pathomechanisms associated with G4C2 repeat expansions include loss of C9ORF72 function as well as toxicity stemming from the accumulation of sense and antisense transcripts of the expanded repeats. These RNA transcripts assemble into structures called foci, aberrantly interact with RNA binding proteins, and cause defects in nucleocytoplasmic transport (5, 6). They additionally serve as templates for the synthesis of proteins of repeating dipeptides through repeat associated non-ATG (RAN) translation (7–11). Poly(GP), poly(GA), and poly(GR) proteins are produced from sense G4C2-containing transcripts, whereas poly(GP), poly(PA), and poly(PR) proteins are produced from antisense G2C4-containing transcripts. Neuronal inclusions of these so-called c9RAN proteins are pathognomonic to c9ALS/FTD, and studies show that certain c9RAN proteins, such as poly(GA), poly(GR), and poly(PR), are toxic in in vitro and in vivo overexpression models (5, 6). Potential mechanisms of toxicity associated with c9RAN proteins include nucleolar stress, impaired proteasomal function, and, as with G4C2 repeat RNA, impaired nucleocytoplasmic transport (5, 6).
On the basis of the expanding body of evidence supporting the role of G4C2 repeat RNA and c9RAN proteins in c9ALS/FTD pathogenesis, therapeutic approaches that target G4C2 RNA are being actively pursued. For example, antisense oligonucleotides (ASOs) complementary to G4C2 RNA or C9ORF72 transcripts (c9ASOs) decrease G4C2-containing RNA and consequently decrease the number of cells with RNA foci, as well as mitigate abnormalities in gene expression and nucleocytoplasmic transport in neurons differentiated from c9ALS patient-derived induced pluripotent stem cells (iPSCs) (12–14). In primary neurons and brain tissues from c9BAC mice expressing expanded G4C2 repeats, c9ASOs decrease G4C2 repeat–containing RNA, foci formation, and production of poly(GP) proteins (15, 16). Moreover, small-molecule binders of G4C2 RNA inhibit foci formation and RAN translation in patient-derived cell models (17).
Because possible therapeutics for c9ALS/FTD are being developed for clinical trials, it is paramount to address barriers in moving a treatment from bench to bedside. Chief among these is the lack of markers capable of predicting disease progression, monitoring the response to therapy, and confirming target engagement. Given that c9RAN proteins are synthesized from G4C2 repeat RNA, the target of therapeutic interventions under investigation, we anticipate that c9RAN proteins in cerebrospinal fluid (CSF) will reflect target engagement and biochemical responses to treatment. Although three c9RAN proteins are produced from G4C2 RNA, namely, poly(GP), poly(GA), and poly(GR), we believe that poly(GP) may be an especially suitable marker candidate. Both poly(GP) and poly(GA) are more highly expressed in the central nervous system (CNS) of c9ALS/FTD patients than poly(GR) (18). However, poly(GP) is more likely to be accurately measured in biospecimens because it is more soluble than poly(GA) (19). Indeed, in a small cohort of c9ALS patients, we established that poly(GP) can be detected in CSF (17). Thus, to prepare for upcoming clinical trials for c9ALS, the present study used patient CSF and several preclinical models to investigate the hypothesis that poly(GP) proteins could serve as an urgently needed pharmacodynamic marker for developing and testing therapies for treating c9ALS.
RESULTS
Poly(GP) is detected in the CSF of asymptomatic and symptomatic C9ORF72 repeat expansion carriers
To test our hypothesis, we used an international sampling of subjects (table S1) to (i) replicate our finding that poly(GP) is present in CSF from C9ORF72 mutation carriers (17), (ii) compare poly(GP) proteins in CSF between asymptomatic and symptomatic carriers, and (iii) examine the longitudinal profile of CSF poly(GP). Given these three primary analyses, P ≤ 0.017 was considered significant after Bonferroni adjustment.
Our CSF series comprised samples from 83 c9ALS patients [71 with c9ALS alone and 12 with comorbid FTD (c9ALS-FTD)] and 27 asymptomatic C9ORF72 repeat expansion carriers. CSF collected longitudinally was available for 33 of these subjects. Also included were samples from 24 C9ORF72 repeat expansion carriers clinically diagnosed with diseases other than c9ALS or c9ALS-FTD [c9FTD (n = 20), Alzheimer’s disease (n = 2), bipolar disease (n = 1), and dementia with Lewy bodies (n = 1)] and from 120 individuals without the C9ORF72 mutation. The latter encompassed patients with ALS (n = 57) or other neurological diseases [FTD (n = 4), Alzheimer’s disease (n = 10), and primary lateral sclerosis (n = 1)], as well as healthy controls (n = 48). Subject characteristics are provided in Table 1.
Table 1. Subject characteristics according to C9ORF72 repeat expansion status and disease group.
The sample median (minimum, 25th percentile, 75th percentile, and maximum) is given for continuous variables. The 15 subjects without a C9ORF72 repeat expansion in the “Other diseases” subgroup comprised 4 patients with FTD, 10 with Alzheimer’s disease, and 1 with primary lateral sclerosis. The 24 subjects with a C9ORF72 repeat expansion in the “Other diseases” subgroup comprised 20 patients with FTD, 2 with Alzheimer’s disease, 1 with dementia with Lewy bodies, and 1 diagnosed with bipolar disease. Information was unavailable regarding age of disease onset [n = 12; 11 non-C9ORF72 repeat expansion carriers (n = 1 with ALS and n = 10 with a disease other than ALS or ALS-FTD) and 1 C9ORF72 repeat expansion carrier with FTD], disease onset to CSF collection (n = 11; 10 non-C9ORF72 repeat expansion carriers with a disease other than ALS or ALS-FTD and 1 C9ORF72 repeat expansion carrier with FTD), and onset site (n = 3; one c9ALS and two c9ALS-FTD). For ALSFRS-R scores, data were missing from 10 ALS or ALS-FTD patients without the C9ORF72 repeat expansion, 19 asymptomatic subjects with the expansion, and 23 patients with c9ALS or c9ALS-FTD. N/A, not applicable.
| Characteristic | Non-C9ORF72 repeat expansion carriers | C9ORF72 repeat expansion carriers | ||||
|---|---|---|---|---|---|---|
| Healthy controls (n = 48) | ALS (n = 57) | Other diseases (n = 15) | Asymptomatic (n = 27) | ALS or ALS-FTD (n = 83) | Other diseases (n = 24) | |
| Age at disease onset (years) | N/A | 54 (24, 43, 60, 78) | 62 (39, 51, 74, 76) | N/A | 56 (33, 52, 62, 74) | 61 (20, 54, 64, 75) |
| Age at CSF collection (years) | 57 (23, 41, 64, 85) | 57 (25, 47, 64, 79) | 60 (51, 53, 64, 77) | 46 (28, 32, 56, 63) | 59 (35, 54, 63, 76) | 63 (33, 57, 67, 77) |
| Gender (male) | 17 (35.4%) | 39 (68.4%) | 9 (60.0%) | 8 (29.6%) | 51 (61.4%) | 12 (50.0%) |
| Disease onset to CSF collection (months) | N/A | 26 (0, 12, 51, 204) | 12 (0, 12, 12, 240) | N/A | 24 (0, 12, 31, 132) | 47 (0, 25, 63, 160) |
| Onset site | ||||||
| —Bulbar | N/A | 11 (19.3%) | N/A | N/A | 25 (31.3%) | N/A |
| —Limb | N/A | 45 (78.9%) | N/A | N/A | 51 (63.8%) | N/A |
| —Other | N/A | 1 (1.8%) | N/A | N/A | 4 (5.0%) | N/A |
| ALSFRS-R score | N/A | 36 (14, 28, 40, 47) | N/A | 48 (40, 45, 48, 48) | 36 (7, 28, 42, 46) | N/A |
| Poly(GP) (ng/ml) | 0.0 (0.0, 0.0, 0.0, 0.0) | 0.0 (0.0, 0.0, 0.0, 0.1) | 0.0 (0.0, 0.0, 0.0, 0.0) | 0.5 (0.0, 0.2, 1.0, 4.0) | 0.8 (0.0, 0.5, 1.6, 5.2) | 1.1 (0.0, 0.4, 1.9, 5.3) |
Poly(GP) in CSF was measured in a blinded manner using our previously described Meso Scale Discovery–based immunoassay (17, 19). We have validated that measures of poly(GP) in CSF determined using this assay significantly correlate with measures of poly(GP) assessed using a different antibody pair (Spearman’s r = 0.99, P < 0.0001, n = 14; fig. S1A) or using a different immunoassay platform, the Simoa HD-1 Analyzer (Spearman’s r = 0.98, P < 0.0001, n = 14; fig. S1B).
As anticipated on the basis of our previous study of 14 c9ALS patients (17), poly(GP) was detected in CSF from C9ORF72 mutation carriers in the large sample series used in the present study (Fig. 1A, Table 1, and tables S2 to S4). Poly(GP) proteins were significantly higher in individuals with the expansion (n = 134) than in those without (n = 120) in unadjusted analysis (P < 0.0001) and analyses adjusted for age at CSF collection, gender, and disease group (P < 0.0001). Notably, poly(GP) was detected in CSF from both asymptomatic and symptomatic C9ORF72 mutation carriers (Fig. 1B, Table 1, and table S2). In comparing asymptomatic individuals (n = 27) and patients with c9ALS or c9ALS-FTD (n = 83), there was nominal evidence of higher poly(GP) in the symptomatic subgroup (median, 0.8 ng/ml versus 0.5 ng/ml; P = 0.047), but this did not remain significant after correction for multiple comparisons or when adjusting for age at CSF collection and gender (P = 0.42) (Fig. 1B and tables S3 and S5).
Fig. 1. Poly(GP) is detected in CSF from asymptomatic and symptomatic C9ORF72 repeat expansion carriers.

(A) Poly(GP) in CSF from C9ORF72 repeat expansion carriers (C9+; n = 134) and noncarriers (C9−; n = 120). ****P < 0.0001, as assessed by van Elteren stratified Wilcoxon rank sum test. (B) CSF poly(GP) concentrations in asymptomatic C9ORF72 mutation carriers (ASX; n = 27) and symptomatic c9ALS patients with or without comorbid FTD (SX; n = 83). No significant difference in poly(GP) between ASX and SX subjects was observed using a linear regression model adjusted for gender and age at CSF collection. Red lines denote the median.
CSF poly(GP) is stable over time
We next evaluated whether CSF poly(GP) changes over time in asymptomatic (n = 9) C9ORF72 mutation carriers and patients with c9ALS or c9ALS-FTD (n = 24) (Fig. 2 and table S6). For these 33 subjects for whom longitudinally collected CSF was available, the median length of time between the first and last poly(GP) measurement was 12.9 months (range, 4.4 to 22.6 months); 24 subjects had two measurements, 6 had three measurements, 2 had four measurements, and 1 had five measurements. As depicted in Fig. 2, poly(GP) concentrations for a given individual remained largely constant. Although variations were seen in some subjects, there was no evidence of a significant change in poly(GP) over the time frame examined (P = 0.84).
Fig. 2. Longitudinal trajectory of poly(GP) in CSF.

Poly(GP) in CSF collected longitudinally from 33 C9ORF72 repeat expansion carriers who either were asymptomatic or had c9ALS or c9ALS-FTD. Twenty-four subjects had two measurements, 6 had three measurements, 2 had four measurements, and 1 had five measurements. One patient (denoted by red circles) converted from a clinical diagnosis of ALS to ALS-FTD between the first and second CSF collection.
Poly(GP) is not a prognostic marker
To gain insight into potential relationships between CSF poly(GP) and patient characteristics, we made 10 secondary comparisons. We first examined associations between poly(GP) in patients with c9ALS or c9ALS-FTD and either age at CSF collection, time from disease onset to CSF collection, or gender. Only gender showed a significant association with poly(GP), with levels being lower in females compared to males (P = 0.004; table S7).
In evaluating associations between CSF poly(GP) and age at disease onset, onset site, disease group (c9ALS versus c9ALS-FTD), or ALSFRS-R (Amyotrophic Lateral Sclerosis Functional Rating Scale – Revised) score (an indicator of the functional status of ALS patients; fig. S2A), no associations were found (table S7). We also investigated whether poly(GP) associates with survival after disease onset. Of the 83 patients with c9ALS or c9ALS-FTD, 79 had survival data. The median length of follow-up after disease onset was 3.3 years (range, 1.0 to 11.8 years), and 47 patients (60%) died. No association between poly(GP) and survival after disease onset was observed in unadjusted analysis [hazard ratio (HR) per each doubling, 0.94; 95% confidence interval (CI), 0.77 to 1.15; P = 0.55] or when adjusting for age at disease onset, gender, and onset site (HR, 0.95; 95% CI, 0.77 to 1.17; P = 0.60).
The findings above are in line with our observation that poly(GP) proteins in the frontal cortex or cerebellum do not associate with age at disease onset or survival after onset (19). However, we did find that cerebellar poly(GP) is associated with cognitive impairment in c9ALS patients (19). We thus examined whether CSF poly(GP) similarly correlated with cognitive or behavioral impairment. Patients with c9ALS or c9ALS-FTD seen at the Mayo Clinic or the National Institutes of Health (NIH) were included in these studies. These patients were systematically screened for cognitive and behavioral function using a battery of validated neuropsychological tests (table S8). Each subject was scored as normal or impaired with respect to behavior (n = 29) or cognition (n = 30) based on test scores by evaluators blinded to poly(GP) status. There was no significant difference in CSF poly(GP) between subjects with or without behavioral impairment (fig. S2B and tables S7 and S9). CSF poly(GP) trended higher in patients with cognitive impairment compared to those without (median, 1.5 versus 0.5; P = 0.018), but this finding lost significance when adjusting for age at CSF collection, gender, and years of education (P = 0.12; fig. S2C and tables S7 and S10).
Poly(GP) in immortalized peripheral blood mononuclear cells reflects a c9ASO-induced decrease in G4C2 repeat RNA
Our discovery that poly(GP) is detected in CSF from C9ORF72 expansion carriers, and that it is stable over 6 to 18 months, supports the potential use of poly(GP) as a pharmacodynamic marker. To investigate this further, we used patient-derived cell models to probe the utility of poly(GP) in gauging biochemical responses to therapeutics that target G4C2 RNA. To this end, we first generated lymphoblastoid cell lines by immortalizing peripheral blood mononuclear cells (PBMCs) from C9ORF72 mutation carriers, given our finding that they express poly(GP) (Fig. 3A and tables S11 and S12). Notably, poly(GP) was detected in lysates from lymphoblastoid cell lines from C9ORF72 mutation carriers and in media bathing these cells (Fig. 3B), suggesting that poly(GP) is secreted from cells. As shown in Fig. 3C, a significant correlation was observed between extracellular and intracellular poly(GP) (Spearman’s r = 0.77, P = 0.004, n = 12).
Fig. 3. Poly(GP) is detected in PBMCs from C9ORF72 mutation carriers, and c9ASO-1 treatment decreases poly(GP) in lymphoblastoid cell lines.

(A to C) Poly(GP) in lysates from PBMCs from C9ORF72 mutation carriers (C9+; n = 36) or noncarriers (C9−; n = 34) (A) or in lysates and media from lymphoblastoid cell lines (LCLs) from C9+ (n = 12) or C9− (n = 7) subjects. For (A) and (B), the red line indicates the median. **P < 0.01, ****P < 0.0001, Wilcoxon rank sum test. For (C), a significant correlation was observed between extracellular and intracellular poly(GP) (Spearman’s r = 0.77, P = 0.004, n = 12). (D to F) Two C9+ LCLs were treated with 5 μM nontargeting control ASO (CTL ASO) or one that targets G4C2 repeat–containing RNA (c9ASO-1) for 10 days. After treatment, RNA was extracted from cells to measure mRNA of C9ORF72 variants (D), sister cells were subjected to RNA fluorescence in situ hybridization for the detection of G4C2 RNA–positive foci (E), or cell lysates were prepared for analysis of poly(GP) (F). For (D) to (F), *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, unpaired, two-tailed t test. Error bars represent SEM for the three replicates for each cell line. a.u., arbitrary units. Scale bar, 10 μm.
Next, we treated one lymphoblastoid cell line from a c9ALS patient and one line from an asymptomatic C9ORF72 mutation carrier with a nontargeting control ASO or a ribonuclease H (RNase H)–active ASO that targets G4C2 repeat RNA (c9ASO-1). Exposure of the two lymphoblastoid cell lines to 5 μM c9ASO-1 for 10 days almost completely eliminated mRNA expression of C9ORF72 variants 1 and 3, the pre-mRNA of which contain the repeat, but had no effect on C9ORF72 variant 2 for which the repeat is not transcribed (Fig. 3D). Also observed was a significant reduction in the number of cells bearing foci formed of G4C2 repeat RNA (Fig. 3E) and of poly(GP) in cell lysates (Fig. 3F). The data indicate that poly(GP) production mirrors expression of repeat-containing C9ORF72 transcripts in lymphoblastoid cell lines.
c9ASO treatment decreases intracellular and extracellular poly(GP) in c9ALS iPSC-derived neurons
To validate our findings in a second patient-derived cell model, we used neurons differentiated from c9ALS iPSCs (iPSNs), which recapitulate the genetic, transcriptional, and biochemical signatures of c9ALS patient brain tissue (table S13) (12, 14). As a proxy for CSF, we examined whether poly(GP) could be detected in conditioned media bathing c9ALS iPSNs and whether levels of extracellular poly(GP) reflected those of intracellular poly(GP), as was seen for lymphoblastoid cell lines. As anticipated, poly(GP) was detected in cell lysates and media from c9ALS iPSNs but not iPSNs lacking the repeat expansion (Fig. 4A). A significant correlation was observed between extracellular and intracellular poly(GP) (Spearman’s r = 0.86, P = 0.02, n = 7; Fig. 4B).
Fig. 4. c9ASO-2 treatment decreases intracellular and extracellular poly(GP) in c9ALS iPSC neurons.

(A) Poly(GP) in lysates and media from cultured iPSNs derived from C9ORF72 repeat expansion carriers (C9+; n = 7) and noncarriers (C9−; n = 3). Red horizontal lines indicate the median. *P < 0.05, Wilcoxon rank sum test. (B) A significant correlation was observed between extracellular and intracellular poly(GP) in the c9ALS iPSN lines (Spearman’s r = 0.86, P = 0.02, n = 7). (C to E) At day 45 of differentiation, three c9ALS iPSN lines were treated with a control ASO or an ASO targeting intron 1 of C9ORF72 (c9ASO-2) at the indicated concentrations. Media were collected before treatment (day 0) and every 5 days thereafter. On day 20, RNA or protein was prepared from cells. (C) Total C9ORF72 or repeat-containing C9ORF72 mRNA transcripts. *P < 0.05, ***P < 0.001, one-way analysis of variance (ANOVA) followed by Tukey’s multiple comparisons test. (D) Poly(GP) in c9ALS iPSN media at different time points after treatment initiation (**P < 0.01, ***P < 0.001, ****P < 0.0001, two-way ANOVA) or in cell lysates after the 20-day exposure to c9ASO-2 (**P < 0.01, ***P < 0.001, ****P < 0.0001, one-way ANOVA followed by Tukey’s multiple comparisons test). (E) Intracellular and extracellular poly(GP) in c9ALS iPSNs treated with ASO was significantly correlated (Spearman’s r = 0.99, P < 0.0001, n = 12).
Next, we treated three lines of c9ALS iPSNs for 20 days with control ASO or with an ASO that targets intron 1 of C9ORF72 (c9ASO-2) and that dose-dependently decreases G4C2 repeat–containing transcripts (C9ORF72 variants 1 and 3) while having limited effect on total C9ORF72 mRNA (Fig. 4C). Consistent with these results, intracellular poly(GP) was dose-dependently decreased in c9ALS iPSN lysates after the 20-day c9ASO-2 treatment (Fig. 4D). Similarly, poly(GP) was reduced in media in a time- and dose-dependent manner (Fig. 4D). By 10 days of treatment, a significant decrease in extracellular poly(GP) was observed in cells exposed to 3 μM c9ASO-2 (P < 0.01), and by 15 days of treatment, a significant decrease in poly(GP) was observed in media from cells treated with 0.3 μM c9ASO-2 (P < 0.01). Again, a significant correlation was seen between intracellular and extracellular poly(GP) (Spearman’s r = 0.99, P < 0.0001, n = 12; Fig. 4E). These data support the notion that extracellular poly(GP) may serve as a surrogate marker for intracellular G4C2 RNA accumulation and suggest that extracellular poly(GP) in CSF can be used as a pharmacodynamic marker for therapies that target G4C2 RNA.
CSF poly(GP) associates with c9ASO-induced decreases in G4C2 repeat–containing RNA, foci burden, and c9RAN proteins in mice expressing an expanded G4C2 repeat
To explore the use of extracellular poly(GP) as a pharmacodynamic marker in vivo, we assessed the effects of c9ASO-1 on poly(GP) in mice that expressed an expanded (G4C2)66 repeat through adeno-associated virus transgenesis (20). We previously reported that by 6 months of age, RNA foci and c9RAN protein pathology were prevalent in (G4C2)66 mice (20). Therefore, at 4 to 4.5 months of age, a single bolus of 500 μg of c9ASO-1 in phosphate-buffered saline (PBS) or PBS alone was delivered into the right ventricle of the CNS of (G4C2)66 mice or control (G4C2)2 mice. Eight weeks later, CSF was collected from mice, and the brains were harvested. As shown in Fig. 5A, c9ASO-1 treatment significantly decreased repeat-containing mRNA in (G4C2)66 mice (P = 0.0004), whereas it had no effect on endogenous mouse C9orf72 mRNA (P = 0.85; Fig. 5B). As anticipated, a concomitant decrease in cells bearing RNA foci was seen throughout the brain (Fig. 5C and fig. S3A), with quantitative analysis of foci-bearing cells in the motor cortex supporting this observation (P < 0.0001; Fig. 5D). Likewise, we noted a marked decrease in the number of inclusions immunopositive for poly(GR), poly(GA), or poly(GP) (Fig. 5, E and F, and fig. S3B). Significant reductions in poly(GA) (P = 0.0025) or poly(GP) (P < 0.0001) in brain homogenates, as assessed by immunoassay, were also observed (Fig. 5G and fig. S4A. Of particular importance, poly(GP) was detected in CSF of (G4C2)66 mice, and CSF poly(GP) was significantly decreased after c9ASO-1 treatment (P = 0.0035; Fig. 5H and fig. S4B). CSF poly(GP) not only correlated with brain poly(GP) in (G4C2)66 mice treated or not treated with c9ASO-1, but both CSF and brain poly(GP) were associated with levels of repeat-containing transcripts, the percentage of motor cortex cells bearing RNA foci, and the amount of poly(GA) in brain homogenates (Table 2).
Fig. 5. Decreased CSF and brain poly(GP) in (G4C2)66 mice treated with c9ASO-1.
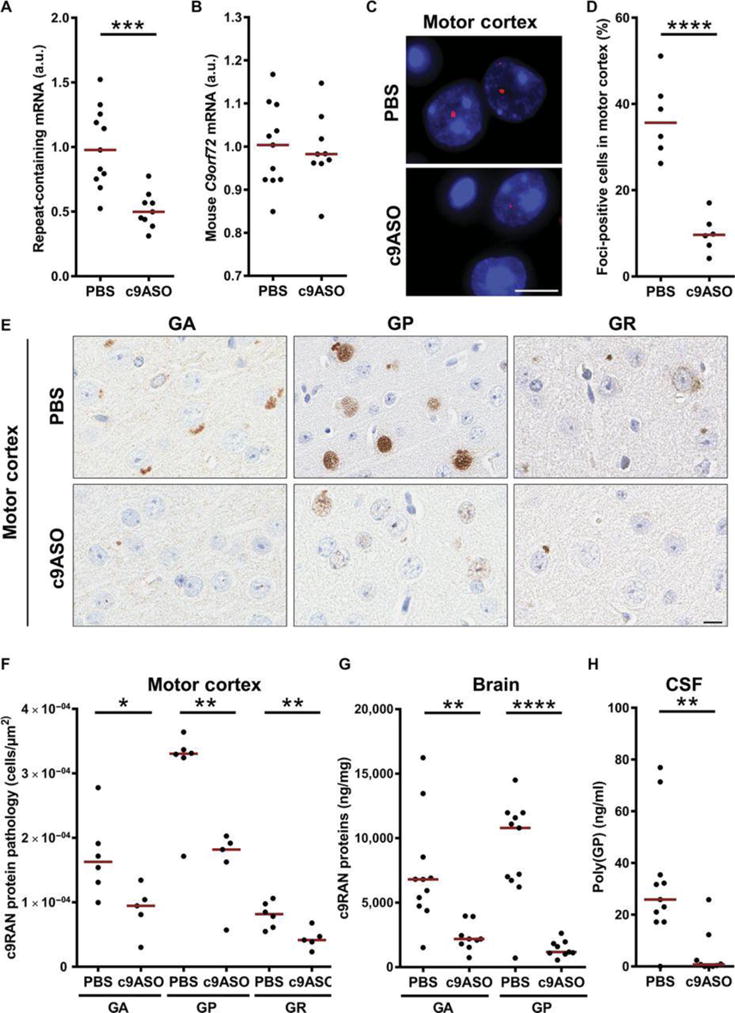
At 4 to 4.5 months of age, (G4C2)2 mice and (G4C2)66 mice were treated with a single intracerebroventricular bolus injection of PBS or c9ASO-1. Eight weeks later, CSF and tissues were harvested from mice for biochemical and immunohistochemical analyses. (A and B) Amount of repeat-containing mRNA or endogenous mouse C9orf72 mRNA in brain tissue from (G4C2)66 mice treated with PBS (n = 11) or c9ASO-1 (n = 9). (C and D) Representative images of RNA foci in the motor cortex of (G4C2)66 mice and quantitative analysis of the percentage of foci-positive cells (n = 6 per group). (E and F) Immunohistochemical analysis and quantification of poly(GA), poly(GP), or poly(GR) pathology in the motor cortex of (G4C2)66 mice treated with PBS (n = 6) or c9ASO-1 (n = 5). Scale bar, 10 μm. (G and H) Poly(GA) or poly(GP) in brain homogenates or CSF of (G4C2)66 mice treated with PBS (n = 11) or c9ASO-1 (n = 9). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, unpaired, two-tailed t test. Red horizontal lines indicate the median. See also related figs. S3 and S4.
Table 2. Associations of brain or CSF poly(GP) with c9ALS-like pathological features in (G4C2)66 mice.
Spearman’s r correlation coefficients, 95% CIs, and P values are presented.
| Characteristic | Brain poly(GP) | CSF poly(GP) | ||
|---|---|---|---|---|
| Spearman’s r (95% CI) | P | Spearman’s r (95% CI) | P | |
| Brain poly(GP) (n = 20) | — | — | 0.74 (0.42–0.89) | 0.0002 |
| Brain poly(GA) (n = 20) | 0.92 (0.80–0.97) | <0.0001 | 0.75 (0.44–0.90) | 0.0002 |
| G4C2 repeat–containing mRNA (n = 20) | 0.87 (0.69–0.95) | <0.0001 | 0.64 (0.26–0.85) | 0.0025 |
| Cells with foci in motor cortex (%) (n = 12) | 0.78 (0.35–0.94) | 0.004 | 0.82 (0.45–0.95) | 0.0017 |
DISCUSSION
Despite intense efforts, and more than 50 clinical trials conducted in the past half-century, there remains only one minimally effective therapy for ALS, one of the most rapidly progressive neurodegenerative diseases. Drug development for ALS, like for many neurological disorders, is hampered by numerous factors, including an incomplete understanding of the causative pathological mechanisms and the existence of different forms of the disease (for example, sporadic ALS versus ALS caused by different gene mutations). Perhaps most important, trials have been hindered by the lack of pharmacodynamic markers to monitor target engagement and therapeutic efficacy. However, with the discovery of C9ORF72 G4C2 repeat expansions as a major cause of ALS (2, 3), c9ALS may well become among the first forms of ALS with a treatment, provided that a concerted effort is made to tackle multiple aspects of the drug development process.
Strategies that target G4C2 repeat–containing RNA are actively being explored as therapeutics for c9ALS/FTD based on the premise that eliminating or neutralizing these transcripts will block detrimental downstream events initiated by the transcripts themselves or by the c9RAN proteins that they produce. As these approaches become closer to clinical trials, the need for a means to measure target engagement and biochemical responses to treatment becomes paramount. Spurred by our findings that poly(GP) is associated with G4C2 repeat–containing transcripts in the cerebellum of C9ORF72 mutation carriers (19, 21) and that small molecules, ASOs, and genetic modifiers that target G4C2 repeat RNA attenuate poly(GP) production in yeast (Saccharomyces cerevisiae), worms (Caenorhabditis elegans), c9ALS patient cell lines, and c9BAC mice (12, 14–17, 22, 23), the present study examined the utility of poly(GP) as a pharmacodynamic marker. Through the combined use of three model systems, we cross-validated our observation that extracellular poly(GP) mirrors intracellular levels and confirmed that c9ASOs are potent inhibitors of c9ALS-associated defects. The lymphoblastoid cell lines and iPSNs allowed us to study poly(GP) as a marker in human models and under the genetic context of the C9ORF72 mutation. A greater than 90% loss of poly(GP) was observed in lymphoblastoid cell lines treated with 5 μM c9ASO-1 for 10 days and in iPSNs treated with 3 μM c9ASO-2 for 20 days. Although both sense and antisense transcripts encode poly(GP), these results suggest that sense transcripts are the dominant producer of poly(GP) in these cells. Our (G4C2)66 mice provided an in vivo model that enabled us to examine relationships between CSF poly(GP) and c9ALS-associated features in brain tissue. We found that a single intracerebroventricular injection of c9ASO-1 in (G4C2)66 mice resulted in robust decreases in c9RAN proteins and RNA foci in the brain, despite their abundance in this model. Of particular importance, we detected significant associations between CSF poly(GP) in (G4C2)66 mice and repeat RNA, frequency of foci, and c9RAN protein expression in mouse brain. Evaluating similar associations between poly(GP) in antemortem CSF and neuropathological features in postmortem brain from C9ORF72 mutation carriers will be an important future study.
The data from our preclinical models suggest that monitoring poly(GP) before and during treatment of patients participating in clinical trials presents a feasible approach to gauge target engagement. It is thus noteworthy that CSF poly(GP) remained relatively stable over intervals of 6 to 18 months, which is expected to facilitate the detection of c9ASO-induced changes in poly(GP), even in patients with relatively low poly(GP). We do note that a few patients had very low poly(GP). We are currently exploring alternative strategies that we expect will markedly increase assay sensitivity and dynamic range. However, should this not be the case, this issue will need to be taken into consideration in the design of any clinical trial.
Our observation that CSF poly(GP) is detected in asymptomatic C9ORF72 mutation carriers is consistent with the emergence of brain c9RAN protein pathology before symptom onset (24). It also suggests that the primary source of poly(GP) in CSF is not the release of this protein from dying cells but rather its secretion from living cells. It is also important to bear in mind that increases in CSF poly(GP) resulting from neurodegeneration may be difficult to discern from steady-state levels, given that c9RAN proteins are widely expressed throughout the CNS of C9ORF72 mutation carriers (7), but a relatively small proportion of cells degenerate in ALS relative to all cells of the brain. Consequently, although our longitudinal studies suggest less utility for poly(GP) as a marker of disease progression, they in no way discount the potential contribution of G4C2 RNA, c9RAN proteins, or downstream events to disease progression, nor do they affect the utility of poly(GP) as a pharmacodynamic marker.
Many clinical trials for neurodegenerative disorders are believed to have failed, at least in part, because treatment was not initiated sufficiently early. Asymptomatic C9ORF72 carriers from a large family with a history of ALS show changes in brain morphology outside the primary motor cortex (25), and structural imaging studies of asymptomatic adults at risk for c9FTD suggest that the thalamus and posterior cortical areas are affected early, with the cerebellum also showing presymptomatic involvement (26). These studies suggest that the disease process precedes symptom onset; as such, asymptomatic C9ORF72 mutation carriers represent a population likely to benefit from early therapeutic intervention. That poly(GP) is detected in CSF from asymptomatic C9ORF72 mutation carriers makes the inclusion of these individuals in clinical trials for G4C2 RNA–targeting therapies more practical by providing a pharmacodynamic marker.
When investigating potential relationships between CSF poly(GP) and patient characteristics, we found that poly(GP) was higher in male C9ORF72 mutation carriers compared to females. Although the biological significance of this modest difference in poly(GP) remains unknown, it is intriguing that disease penetrance among C9ORF72 mutation carriers is reportedly higher in males (27, 28). We also detected a trend of higher CSF poly(GP) in patients with c9ALS or c9ALS-FTD compared to asymptomatic C9ORF72 mutation carriers, but this finding was not statistically significant. We did observe one asymptomatic individual with much higher CSF poly(GP) compared to the others, and it will be of interest to follow this subject for signs of onset of clinical disease in comparison to the other carriers. Because our longitudinal studies included only nine asymptomatic individuals, additional studies on repeated collections of CSF from asymptomatic subjects are advisable to compare the trajectory of poly(GP) before and after carriers convert to the symptomatic stage.
We additionally observed a trend of elevated CSF poly(GP) in c9ALS patients with cognitive impairment, but this finding did not meet statistical significance, perhaps because information on cognitive impairment was available for only a small number of patients (n = 30). Although the sample size accrued for these analyses is large for a study of C9ORF72 repeat expansion carriers, it is relatively small for conducting statistical tests of association. As a result, power to detect differences in poly(GP) between groups may be low, and there is a possibility of a type II error (that is, a false-negative association). It should also be noted that our study focused only on poly(GP) because of its abundance in the CNS and its solubility (19). Although cerebellar poly(GP) was associated with poly(GA) (19), and the same is true for poly(GP) and poly(GA) in (G4C2)66 mouse brain (Table 2), measurements of CSF poly(GA) and poly(GR), which show evidence of toxicity (6), could uncover associations with clinical features not observed for poly(GP). Given our discovery that poly(GP) is detected in PBMCs from C9ORF72 repeat expansion carriers, cross-sectional and longitudinal studies examining associations between PBMC c9RAN proteins and clinical features may also uncover relationships of potential prognostic value. Also of importance will be comparing poly(GP) in CSF to poly(GP) in PBMCs from the same patient. Finally, the fact that G4C2 repeat length varies among C9ORF72 mutation carriers, which presumably affects the length of c9RAN proteins, must be considered. Because longer poly(GP) proteins have more anti-GP antibody binding sites, this could influence poly(GP) quantification and comparison among patients. However, we found no association between poly(GP) and repeat length in the frontal cortex or cerebellum of 55 C9ORF72 repeat expansion carriers (19). Furthermore, when using CSF poly(GP) as a pharmacodynamic marker in clinical trials, each patient would serve as their own control, with poly(GP) being measured before and during treatment; thus, differences in repeat length among patients would not affect the use of poly(GP) in evaluating target engagement.
The findings from the present study have several important implications and also highlight areas in need of additional investigation to prepare for forthcoming clinical trials. Our data suggest that levels of extracellular and intracellular poly(GP) can be used in preclinical models to screen G4C2 RNA–targeting drugs and inform the rational selection of dose and schedule. In addition, this study and others provide ample evidence that c9ASOs efficiently block pathological features associated with c9ALS/FTD in preclinical models (12, 14–16, 22). Moreover, our data offer persuasive support that CSF poly(GP) represents a promising pharmacodynamic marker for c9ASOs and other therapeutic approaches, such as small molecules (17), that target G4C2 RNA. Boding well for the use of ASOs to treat c9ALS/FTD, intrathecal administration of ASOs against superoxide dismutase 1 was well tolerated in a phase 1 study (29), and an ASO for spinal muscular atrophy has been approved for use by the U.S. Food and Drug Administration. Overall, our identification of poly(GP) as a potential pharmacodynamic marker will aid in the development and clinical testing of therapeutics for patients with c9ALS, as well as patients with c9FTD and other diseases associated with C9ORF72 repeat expansions.
MATERIALS AND METHODS
Study design
The goals of this study were as follows: (i) to investigate poly(GP) as a pharmacodynamic marker using CSF from C9ORF72 mutation carriers and noncarriers and (ii) to determine whether poly(GP) proteins are predictive of target engagement in preclinical models of c9ALS. The three primary analyses in our clinical studies were to replicate the detection of poly(GP) in CSF from C9ORF72 mutation carriers, compare CSF poly(GP) between asymptomatic and symptomatic carriers, and examine the longitudinal profile of poly(GP) proteins in CSF. Ten secondary comparisons were made between CSF poly(GP) and various clinical features, as detailed in the “Statistical analysis” section and the Supplementary Materials. Our analyses focused on C9ORF72 mutation carriers that were asymptomatic or clinically diagnosed with ALS with or without comorbid FTD. However, to ensure that poly(GP) was measured in a blinded manner to genotype and disease subtype, CSF from C9ORF72 mutation carriers with diseases other than c9ALS or c9ALS-FTD was included, as were samples from individuals lacking the mutation (Table 1). Sample size was based on availability of CSF from multiple institutes (table S1). Written informed consent was obtained from all participants or their legal next of kin if they were unable to give written consent, and biological samples were obtained with ethics committee approval. For our preclinical studies, we examined whether c9ASO-induced decreases in G4C2 repeat–associated features were accompanied by decreases in extracellular and intracellular poly(GP) in lymphoblastoid cell lines, iPSNs, and (G4C2)66-expressing mice.
Immunoassay analysis of poly(GP)
Poly(GP) in all samples was measured using a previously characterized Meso Scale Discovery–based immunoassay (17, 19). See the Supplementary Materials for details on the preparation of all sample types and assay conditions.
Testing the effect of c9ASOs in preclinical models
Patient-derived lymphoblastoid cell lines and (G4C2)66-expressing mice were treated with c9ASO-1, which targets the G4C2 repeat sequence (Integrated DNA Technologies) (12) and has the following properties: (i) sequence, CCGGCCCCGGCCCCGGCCCC; (ii) modification, 5-10-5, 2′-O-methyl RNA, phosphorothioate backbone; and (iii) function, RNase H activation. Patient-derived iPSNs were treated with c9ASO-2, a modified 2′-O-methoxyethyl/DNA ASO generated by Ionis Pharmaceuticals (12, 22). The control ASO (CCTTCCCTGAAGGTTCCTCC) was also generated by Ionis Pharmaceuticals. Details on preclinical models, treatment conditions, and end-point evaluations are provided in the Supplementary Materials.
Statistical analysis
For our clinical studies using CSF, continuous variables were summarized with the sample median, minimum, 25th percentile, 75th percentile, and maximum. All analyses except the examination of poly(GP) over time used only the first measurement of poly(GP) for each patient to satisfy the statistical assumption of independent measurements. For the comparison of CSF poly(GP) between carriers and noncarriers of the C9ORF72 mutation, a nonparametric approach was needed because values equal to zero in the noncarriers resulted in a skewed distribution. Therefore, a Wilcoxon rank sum test was used in unadjusted analysis, whereas a van Elteren stratified Wilcoxon rank sum test was used in adjusted analysis (30), where a 12-level variable was created on the basis of age at CSF collection (less than the median or greater than or equal to the median), gender, and disease group (asymptomatic, ALS, ALS-FTD, or other), and the test was stratified by this variable.
For all other comparisons of poly(GP) between various groups, these involved C9ORF72 mutation carriers and were made using unadjusted and adjusted linear regression models, where poly(GP) was considered on the logarithm scale (after adding a constant of 0.1 to all values to avoid values of zero) because of its skewed distribution. Comparisons of poly(GP) between asymptomatic and ALS or ALS-FTD C9ORF72 mutation carriers were adjusted for age at CSF collection and gender. Comparisons of poly(GP) according to cognitive or behavioral impairment were adjusted for age at CSF collection, gender, and years of education. Comparisons of poly(GP) according to other patient characteristics [age at CSF collection, age of onset, gender, time from onset to CSF collection, onset site, disease group, or ALSFRS-R (a validated rating instrument for monitoring the progression of disability in ALS patients)] in c9ALS or c9ALS-FTD patients were adjusted for any variable associated with poly(GP) with a P value of 0.05 or lower in unadjusted analysis.
To evaluate whether poly(GP) changes over time, the slope from a linear regression model [where poly(GP) was the response and time after baseline poly(GP) measure was the predictor variable] was calculated separately for each patient. These slopes were tested for difference from a value of zero [indicating no change in poly(GP) over time] using a one-sample t test.
The association between poly(GP) and survival after disease onset in c9ALS or c9ALS-FTD patients was evaluated using Cox proportional hazards regression models, where an unadjusted model was examined as well as a model adjusted for age of disease onset, gender, and onset site (bulbar, limb, or other). Poly(GP) was again considered on the logarithmic scale, and HRs and 95% CIs were estimated.
The primary analyses of our clinical studies involved three different statistical tests; to adjust for multiple testing for these primary analyses, we used a Bonferroni correction, after which P values of 0.017 or lower are considered as statistically significant. P values of 0.05 or lower were considered as statistically significant for all statistical tests that involved secondary aims. All statistical tests were two-sided. Statistical analyses for these studies were performed using SAS (version 9.2, SAS Institute Inc.) and R statistical software (version 2.14.0, R Foundation for Statistical Computing).
For our preclinical studies, statistical analyses were performed with GraphPad Prism software. For the comparison of poly(GP) in PBMCs and lymphoblastoid cell line lysates between C9ORF72 mutation carriers and noncarriers, a Wilcoxon rank sum test was used. The effect of control ASO versus c9ASO-1 on mRNA of C9ORF72 variants, percentage of cells with RNA foci, and poly(GP) were assessed by unpaired, two-tailed t tests. For comparison of poly(GP) in media or lysates from iPSNs with or without the C9ORF72 expansion, a Wilcoxon rank sum test was used. Spearman’s test of correlation was used to compare poly(GP) in media and lysates. A one-way ANOVA followed by Tukey’s multiple comparison test was used to compare total C9ORF72 mRNA, repeat-containing mRNA, or poly(GP) in lysates from three iPSN lines treated with control ASO or various doses of c9ASO. A two-way ANOVA was used to compare poly(GP) in media in three lines of iPSNs treated with control ASO or various doses of c9ASO-2 over different durations of treatment. Unpaired, two-tailed t tests were used to compare the effect of control ASO versus c9ASO-1 in (G4C2)66 mice on repeat-containing mRNA or C9orf72 mRNA, the percentage of cells with RNA foci, density of c9RAN protein pathology, poly(GA) or poly(GP) in brain lysates, or poly(GA) or poly(GP) in CSF (Fig. 5). A one-way ANOVA was used to conduct the same analyses but also including (G4C2)2 mice treated with control ASO or c9ASO-1 (fig. S4). Spearman’s test of correlation was used to compare poly(GP) in the brain or CSF with brain poly(GP), brain poly(GA), repeat-containing transcripts, and percentage of foci-bearing cells in the motor cortex.
Supplementary Material
Fig. S1. Validation of poly(GP) detection in CSF.
Fig. S2. Relationships between CSF poly(GP) and ALSFRS-R score, behavioral impairment, and cognitive impairment.
Fig. S3. Decreases in RNA foci and c9RAN protein pathology are observed in (G4C2)66 mice treated with c9ASO-1.
Fig. S4. Decreases in brain and CSF poly(GP) are observed in (G4C2)66 mice treated with c9ASO-1.
Fig. S5. Validation of poly(GA) Meso Scale Discovery sandwich immunoassay.
Table S1. Distribution of the 254 subjects among the sites that provided CSF.
Table S2. Poly(GP) in CSF from C9ORF72 repeat expansion carriers and noncarriers.
Table S3. Associations of poly(GP) with C9ORF72 repeat expansion and disease status.
Table S4. Characteristics according to C9ORF72 repeat expansion carrier status.
Table S5. Characteristics for asymptomatic C9ORF72 repeat expansion carriers and patients with c9ALS or c9ALS-FTD.
Table S6. Poly(GP) in CSF collected longitudinally from C9ORF72 repeat expansion carriers.
Table S7. Secondary comparisons of CSF poly(GP) in patients with c9ALS or c9ALS-FTD.
Table S8. Neuropsychological test battery for Mayo Clinic and NIH.
Table S9. Characteristics according to behavioral impairment in patients with c9ALS or c9ALS-FTD.
Table S10. Characteristics according to cognitive impairment in patients with c9ALS or c9ALS-FTD.
Table S11. Subject characteristics for PBMCs.
Table S12. Subject characteristics for lymphoblastoid cell lines.
Table S13. Subject characteristics for iPSC lines.
Homing in on poly(GP) proteins.
A mutation in the C9ORF72 gene causes amyotrophic lateral sclerosis (ALS) through the accumulation of G4C2 RNA. Therapeutics that target G4C2 RNA are thus being developed. Testing these therapeutics in patients with “c9ALS” will depend on finding a marker to monitor the effect of treatments on G4C2 RNA. Gendron et al. demonstrate that poly(GP) proteins produced from G4C2 RNA are present in cerebrospinal fluid from c9ALS patients. Furthermore, using patient cell models and a mouse model of c9ALS, they report that poly(GP) proteins correlate with G4C2 RNA, suggesting that poly(GP) could be used to test potential treatments for c9ALS in upcoming clinical trials.
Acknowledgments
We thank all patients who donated samples and the NIH centers and programs that made this possible. We thank T. Hyman for sample processing at the Washington University and C. Henderson for insight on our study.
Funding: This work was supported by the NIH/National Institute on Aging (P01AG017586 to M.G. and J.Q.T., K23AG042856 to W.T.H., and AG10124 to J.Q.T.), the NIH/National Institute of Neurological Disorders and Stroke (R21NS089979 to K.B.B. and T.F.G.; R25NS065729 to L.R.H.; R01NS078398 to T.M.M.; R35NS097273 to L. Petrucelli; R21NS084528 to L. Petrucelli; P01NS084974 to L. Petrucelli, D.W.D., K.B.B., and R.R.; R01NS088689 to R.H.B. and L. Petrucelli; and R01NS085207 and U54NS091046 to J.D.R.), the intramural research program of the NIH/National Institute of Neurological Disorders and Stroke (Z01NS003146 to M.K.F.), the U.S. Department of Defense (Amyotrophic Lateral Sclerosis Research Program AL130125 to L. Petrucelli), Mayo Clinic Foundation (to L. Petrucelli), Mayo Clinic Center for Individualized Medicine (to K.B.B., T.F.G., and L. Petrucelli), Amyotrophic Lateral Sclerosis Association (to K.B.B., M.B., J.D.G., T.F.G., L.R.H., L. Petrucelli, M.P., J.W., and Y.-J.Z.), the Robert Packard Center for ALS Research at Johns Hopkins (to J.D.R. and L. Petrucelli), Target ALS (to J.D.R. and L. Petrucelli), Association for Frontotemporal Degeneration (to L. Petrucelli), Biogen (to L. Petrucelli), the ALS Therapy Alliance (to J.D.B. and J.D.G), ALS Finding A Cure Foundation (to J.D.B.), the Brain Science Institute (to J.D.R.), the Muscular Dystrophy Association (#416137 to T.F.G.; #4365 and #172123 to M.B. and J.W.), the Italian Ministry of Health (RF-2013-02355764 to C.T., C.M., B.P., F.S., A. Ratti, and V.S.) and STRENGTH project funded by EU Joint Programme–Neurodegenerative Disease Research (to C.T., C.M., B.P., A. Ratti, and V.S.), and Clinical Research in ALS and Related Disorders for Therapeutic Development (CReATe) (U54-NS-092091 to M.B. and J.W.) and Advancing Research and Treatment for Frontotemporal Lobar Degeneration (ARTFL) (U54-NS-092089 to A.L.B.) consortia, which are part of the Rare Diseases Clinical Research Network, an initiative of the Office of Rare Diseases Research, National Center for Advancing Translational Sciences (NCATS). CReATe and ARTFL are funded through a collaboration between NCATS and National Institute of Neurological Disorders and Stroke.
Footnotes
Competing interests: Anti-GP antibodies generated by T.F.G. and L. Petrucelli that were used in this study have been licensed to commercial entities. T.F.G. and L. Petrucelli have a U.S. patent #9,448,232 entitled “Methods and materials for detecting C9ORF72 hexanucleotide repeat expansion positive frontotemporal lobar degeneration or C9ORF72 hexanucleotide repeat expansion positive amyotrophic lateral sclerosis.” A.L.B. is an advisory board member for Biogen and Ionis Pharmaceuticals, is a paid consultant for Ionis Pharmaceuticals, and collaborated with and received research funding from Biogen. A.M. has restricted stock units in Biogen, is a full-time employee of the company, and shared antibodies for this study. B.L.M. is the director of an NIH-sponsored Alzheimer’s Disease Research Center; receives support from University of California, San Francisco/Quest Diagnostics; is the scientific director of the Tau Consortium, the Consortium for Frontotemporal Dementia Research, and the John Douglas French Foundation; is a medical adviser for the Larry L. Hillblom Foundation; and is a scientific advisory board member for the Cambridge Biomedical Research Centre and its subunit, the Biomedical Research Unit in Dementia. B.J.T. has a paid consulting relationship with the Brain Science Institute, Department of Neurology, Johns Hopkins; receives intramural funding from the NIH (Z01-AG000949); and has a patent EP #2751284 B1 entitled “Method for diagnosing a neurodegenerative disease.” C.A. holds shares in Biogen stock. F.R. is a paid employee of Ionis Pharmaceuticals and provided ASOs for the study. J.D.B. is an unpaid adviser to ALS One (a Massachusetts ALS nonprofit organization), an unpaid member of the executive committee for the Northeast ALS Consortium, and an unpaid expert panel member of the U.S. National ALS Registry; has been a paid consultant to Neuraltus Pharmaceuticals and Biogen; and holds the Massachusetts General Hospital–Voyager Therapeutics academic-industry fellow position (a research collaboration paid through appointment at Massachusetts General Hospital). M.G. is a governing board member of the International Society for Frontotemporal Dementia and is on the medical advisory board of the Association for Frontotemporal Dementias. M.K.F. has a paid consulting relationship with Department of Neurology, Uniformed Services University of Health Sciences as an adjunct associate professor. R.B. is the president of and holds stock options in Iron Horse Diagnostics Inc. R.H.B. is affiliated with the Angel Fund for ALS Research. S.L. has a paid consulting relationship with Barrow Neurological Institute and is an associate professor of Neurology in the University of Arizona College of Medicine, the Creighton University Medical School, and Sanofi Genzyme, for unrelated disorders. T.M.M. is a paid consultant for Cytokinetics and receives research support from Ionis Pharmaceuticals and Biogen Idec. V.S. serves on the board of Cytokinetics for the VITALITY trial in ALS and has received consultation fees. All other authors declare that they have no competing interests.
SUPPLEMENTARY MATERIALS
Materials and Methods
Author contributions: L. Petrucelli, K.B.B., J.D.R., T.F.G., J.C., J.N.S., L.R.H., and Y.-J.Z. contributed to the conception and design of the study. T.F.G., M.P., L.M.D., L. Pregent, V.B., M.v.B., Y.S., R.R., B.K.R., O.P., A.M.C., P.D., C.P., A. Robertson, E.W., M.T., L.B., J.F., D.L., S.L., C.N.F., L.F.M., L.B.E., J.B.T., J.D.M., C.T., C.M., B.P., F.S., A.P., J.W., J.J.-B., J.C.V.S., B.L.M., A.L.B., R.H.B., R.B., T.M.M., J.Q.T., M.G., J.D.B., W.T.H., A. Ratti, B.J.T., M.B., V.S., J.D.G., M.K.F., J.D.R., K.B.B., and L. Petrucelli contributed to the clinical studies through the collection and processing of patient samples, provided clinical data, and provided insight. All statistical analyses for clinical studies were carried out by M.G.H. and N.N.D. Studies on lymphoblastoid cell lines were conducted by Y.-J.Z., M.P., T.F.G., A.O., L.M.D., L. Pregent, and V.B. L.R.H., T.F.G., L.M.D., M.v.B., R.R., and F.R. contributed to the iPSN studies. J.C., J.N.S., T.F.G., K.J.-W., E.A.P., L.M.D., C.W.L., L.J.T., M.Y., J.T., M.C.-C., L.R., V.P., D.W.D., and J.D.F. contributed to the in vivo studies. Statistical analyses related to the preclinical models were carried out by T.F.G., J.C., J.N.S., L.R.H., and Y.-J.Z. Assay validation was made possible through the efforts of T.F.G., C.C., Y.C., C.A., A.D., W.Y., D.R., G.A., A.M., and M.D.D. The manuscript was written by T.F.G., and all authors read it and provided comments and revisions.
Data and materials availability: All requests for reagents (that is, antibodies and adeno-associated virus) should be made to L. Petrucelli. They will be made available through appropriate administrative channels (material transfer agreements).
References
- 1.Strong MJ, Grace GM, Freedman M, Lomen-Hoerth C, Woolley S, Goldstein LH, Murphy J, Shoesmith C, Rosenfeld J, Leigh PN, Bruijn L, Ince P, Figlewicz D. Consensus criteria for the diagnosis of frontotemporal cognitive and behavioural syndromes in amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2009;10:131–146. doi: 10.1080/17482960802654364. [DOI] [PubMed] [Google Scholar]
- 2.DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, Kouri N, Wojtas A, Sengdy P, Hsiung GYR, Karydas A, Seeley WW, Josephs KA, Coppola G, Geschwind DH, Wszolek ZK, Feldman H, Knopman DS, Petersen RC, Miller BL, Dickson DW, Boylan KB, Graff-Radford NR, Rademakers R. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72:245–256. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Renton E, Majounie E, Waite A, Simón-Sánchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, Swieten JC van, Myllykangas L, Kalimo H, Paetau A, Abramzon Y, Remes AM, Kaganovich A, Scholz SW, Duckworth J, Ding J, Harmer DW, Hernandez DG, Johnson JO, Mok K, Ryten M, Trabzuni D, Guerreiro RJ, Orrell RW, Neal J, Murray A, Pearson J, Jansen IE, Sondervan D, Seelaar H, Blake D, Young K, Halliwell N, Callister JB, Toulson G, Richardson A, Gerhard A, Snowden J, Mann D, Neary D, Nalls MA, Peuralinna T, Jansson L, Isoviita V-M, Kaivorinne A-L, Hölttä-Vuori M, Ikonen E, Sulkava R, Benatar M, Wuu J, Chio A, Restagno G, Borghero G, M SabatelliITALSGEN Consortium. Heckerman D, Rogaeva E, Zinman L, Rothstein JD, Sendtner M, Drepper C, Eichler EE, Alkan C, Abdullaev Z, Pack SD, Dutra A, Pak E, Hardy J, Singleton A, Williams NM, Heutink P, Pickering-Brown S, Morris HR, Tienari PJ, Traynor BJ. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72:257–268. doi: 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van Blitterswijk M, Dejesus-Hernandez M, Niemantsverdriet E, Murray ME, Heckman MG, Diehl NN, Brown PH, Baker MC, Finch NA, Bauer PO, Serrano G, Beach TG, Josephs KA, Knopman DS, Petersen RC, Boeve BF, Graff-Radford NR, Boylan KB, Petrucelli L, Dickson DW, Rademakers R. Association between repeat sizes and clinical and pathological characteristics in carriers of C9ORF72 repeat expansions (Xpansize-72): A cross-sectional cohort study. Lancet Neurol. 2013;12:978–988. doi: 10.1016/S1474-4422(13)70210-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gendron TF, Belzil VV, Zhang YJ, Petrucelli L. Mechanisms of toxicity in C9FTLD/ALS. Acta Neuropathol. 2014;127:359–376. doi: 10.1007/s00401-013-1237-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gitler D, Tsuiji H. There has been an awakening: Emerging mechanisms of C9orf72 mutations in FTD/ALS. Brain Res. 2016;1647:19–29. doi: 10.1016/j.brainres.2016.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ash PEA, Bieniek KF, Gendron TF, Caulfield TF, Lin W-L, Dejesus-Hernandez M, Blitterswijk MM van, Jansen-West K, Paul JW, III, Rademakers R, Boylan KB, Dickson DW, Petrucelli L. Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron. 2013;77:639–646. doi: 10.1016/j.neuron.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gendron TF, Bieniek KF, Zhang YJ, Jansen-West K, Ash PEA, Caulfield T, Daughrity L, Dunmore JH, Castanedes-Casey M, Chew J, Cosio DM, van Blitterswijk M, Lee WC, Rademakers R, Boylan KB, Dickson DW, Petrucelli L. Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS. Acta Neuropathol. 2013;126:829–844. doi: 10.1007/s00401-013-1192-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mori K, Arzberger T, Grässer FA, Gijselinck I, May S, Rentzsch K, Weng S-M, Schludi MH, van der Zee J, Cruts M, Van Broeckhoven C, Kremmer E, Kretzschmar HA, Haass C, Edbauer D. Bidirectional transcripts of the expanded C9orf72 hexanucleotide repeat are translated into aggregating dipeptide repeat proteins. Acta Neuropathol. 2013;126:881–893. doi: 10.1007/s00401-013-1189-3. [DOI] [PubMed] [Google Scholar]
- 10.Mori K, Weng SM, Arzberger T, May S, Rentzsch K, Kremmer E, Schmid B, Kretzschmar HA, Cruts M, Van Broeckhoven C, Haass C, Edbauer D. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science. 2013;339:1335–1338. doi: 10.1126/science.1232927. [DOI] [PubMed] [Google Scholar]
- 11.Zu T, Liu Y, Bañez-Coronel M, Reid T, Pletnikova O, Lewis J, Miller TM, Harms MB, Falchook AE, Subramony SH, Ostrow LW, Rothstein JD, Troncoso JC, Ranum LPW. RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc Natl Acad Sci USA. 2013;110:E4968–E4977. doi: 10.1073/pnas.1315438110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Donnelly CJ, Zhang PW, Pham JT, Haeusler AR, Mistry NA, Vidensky S, Daley EL, Poth EM, Hoover B, Fines DM, Maragakis N, Tienari PJ, Petrucelli L, Traynor BJ, Wang J, Rigo F, Bennett CF, Blackshaw S, Sattler R, Rothstein JD. RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron. 2013;80:415–428. doi: 10.1016/j.neuron.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sareen D, O’Rourke JG, Meera P, Muhammad AKMG, Grant S, Simpkinson M, Bell S, Carmona S, Ornelas L, Sahabian A, Gendron T, Petrucelli L, Baughn M, Ravits J, Harms MB, Rigo F, Bennett CF, Otis TS, Svendsen CN, Baloh RH. Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci Transl Med. 2013;5:208ra149. doi: 10.1126/scitranslmed.3007529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang K, Donnelly CJ, Haeusler AR, Grima JC, Machamer JB, Steinwald P, Daley EL, Miller SJ, Cunningham KM, Vidensky S, Gupta S, Thomas MA, Hong I, Chiu SL, Huganir RL, Ostrow LW, Matunis MJ, Wang J, Sattler R, Lloyd TE, Rothstein JD. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature. 2015;525:56–61. doi: 10.1038/nature14973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.O’Rourke JG, Bogdanik L, Muhammad AKMG, Gendron TF, Kim KJ, Austin A, Cady J, Liu EY, Zarrow J, Grant S, Ho R, Bell S, Carmona S, Simpkinson M, Lall D, Wu K, Daughrity L, Dickson DW, Harms MB, Petrucelli L, Lee EB, Lutz CM, Baloh RH. C9orf72 BAC transgenic mice display typical pathologic features of ALS/FTD. Neuron. 2015;88:892–901. doi: 10.1016/j.neuron.2015.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jiang J, Zhu Q, Gendron TF, Saberi S, McAlonis-Downes M, Seelman A, Stauffer JE, Jafar-Nejad P, Drenner K, Schulte D, Chun S, Sun S, Ling SC, Myers B, Engelhardt J, Katz M, Baughn M, Platoshyn O, Marsala M, Watt A, Heyser CJ, Ard MC, De Muynck L, Daughrity LM, Swing DA, Tessarollo L, Jung CJ, Delpoux A, Utzschneider DT, Hedrick SM, de Jong PJ, Edbauer D, Van Damme P, Petrucelli L, Shaw CE, Bennett CF, Da Cruz S, Ravits J, Rigo F, Cleveland DW, Lagier-Tourenne C. Gain of toxicity from ALS/FTD-linked repeat expansions in C9ORF72 is alleviated by antisense oligonucleotides targeting GGGGCC-containing RNAs. Neuron. 2016;90:535–550. doi: 10.1016/j.neuron.2016.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Su Z, Zhang Y, Gendron TF, Bauer PO, Chew J, Yang WY, Fostvedt E, Jansen-West K, Belzil VV, Desaro P, Johnston A, Overstreet K, Oh SY, Todd PK, Berry JD, Cudkowicz ME, Boeve BF, Dickson D, Floeter MK, Traynor BJ, Morelli C, Ratti A, Silani V, Rademakers R, Brown RH, Rothstein JD, Boylan KB, Petrucelli L, Disney MD. Discovery of a biomarker and lead small molecules to target r(GGGGCC)-associated defects in c9FTD/ALS. Neuron. 2014;83:1043–1050. doi: 10.1016/j.neuron.2014.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schludi MH, May S, Grässer FA, Rentzsch K, Kremmer E, Küpper C, Klopstock T, German Consortium for Frontotemporal Lobar Degeneration, Bavarian Brain Banking Alliance. Arzberger T, Edbauer D. Distribution of dipeptide repeat proteins in cellular models and C9orf72 mutation cases suggests link to transcriptional silencing. Acta Neuropathol. 2015;130:537–555. doi: 10.1007/s00401-015-1450-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gendron TF, van Blitterswijk M, Bieniek KF, Daughrity LM, Jiang J, Rush BK, Pedraza O, Lucas JA, Murray ME, Desaro P, Robertson A, Overstreet K, Thomas CS, Crook JE, Castanedes-Casey M, Rousseau L, Josephs KA, Parisi JE, Knopman DS, Petersen RC, Boeve BF, Graff-Radford NR, Rademakers R, Lagier-Tourenne C, Edbauer D, Cleveland DW, Dickson DW, Petrucelli L, Boylan KB. Cerebellar c9RAN proteins associate with clinical and neuropathological characteristics of C9ORF72 repeat expansion carriers. Acta Neuropathol. 2015;130:559–573. doi: 10.1007/s00401-015-1474-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chew J, Gendron TF, Prudencio M, Sasaguri H, Zhang YJ, Castanedes-Casey M, Lee CW, Jansen-West K, Kurti A, Murray ME, Bieniek KF, Bauer PO, Whitelaw EC, Rousseau L, Stankowski JN, Stetler C, Daughrity LM, Perkerson EA, Desaro P, Johnston A, Overstreet K, Edbauer D, Rademakers R, Boylan KB, Dickson DW, Fryer JD, Petrucelli L. C9ORF72 repeat expansions in mice cause TDP-43 pathology, neuronal loss, and behavioral deficits. Science. 2015;348:1151–1154. doi: 10.1126/science.aaa9344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van Blitterswijk M, Gendron TF, Baker MC, DeJesus-Hernandez M, Finch NA, Brown PH, Daughrity LM, Murray ME, Heckman MG, Jiang J, Lagier-Tourenne C, Edbauer D, Cleveland DW, Josephs KA, Parisi JE, Knopman DS, Petersen RC, Petrucelli L, Boeve BF, Graff-Radford NR, Boylan KB, Dickson DW, Rademakers R. Novel clinical associations with specific C9ORF72 transcripts in patients with repeat expansions in C9ORF72. Acta Neuropathol. 2015;130:863–876. doi: 10.1007/s00401-015-1480-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lagier-Tourenne C, Baughn M, Rigo F, Sun S, Liu P, Li HR, Jiang J, Watt AT, Chun S, Katz M, Qiu J, Sun Y, Ling SC, Zhu Q, Polymenidou M, Drenner K, Artates JW, McAlonis-Downes M, Markmiller S, Hutt KR, Pizzo DP, Cady J, Harms MB, Baloh RH, Vandenberg SR, Yeo GW, Fu XD, Bennett CF, Cleveland DW, Ravits J. Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc Natl Acad Sci USA. 2013;110:E4530–E4539. doi: 10.1073/pnas.1318835110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kramer NJ, Carlomagno Y, Zhang Y-J, Almeida S, Cook CN, Gendron TF, Prudencio M, Blitterswijk M Van, Belzil V, Couthouis J, Paul JW, III, Goodman LD, Daughrity L, Chew J, Garrett A, Pregent L, Jansen-West K, Tabassian LJ, Rademakers R, Boylan K, Graff-Radford NR, Josephs KA, Parisi JE, Knopman DS, Petersen RC, Boeve BF, Deng N, Feng Y, Cheng T-H, Dickson DW, Cohen SN, Bonini NM, Link CD, Gao F-B, Petrucelli L, Gitler AD. Spt4 selectively regulates the expression of C9orf72 sense and antisense mutant transcripts. Science. 2016;353:708–712. doi: 10.1126/science.aaf7791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vatsavayai SC, Yoon SJ, Gardner RC, Gendron TF, Vargas JNS, Trujillo A, Pribadi M, Phillips JJ, Gaus SE, Hixson JD, Garcia PA, Rabinovici GD, Coppola G, Geschwind DH, Petrucelli L, Miller BL, Seeley WW. Timing and significance of pathological features in C9orf72 expansion-associated frontotemporal dementia. Brain. 2016;139:3202–3216. doi: 10.1093/brain/aww250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Walhout R, Schmidt R, Westeneng HJ, Verstraete E, Seelen M, van Rheenen W, de Reus MA, van Es MA, Hendrikse J, Veldink JH, van den Heuvel MP, van den Berg LH. Brain morphologic changes in asymptomatic C9orf72 repeat expansion carriers. Neurology. 2015;85:1780–1788. doi: 10.1212/WNL.0000000000002135. [DOI] [PubMed] [Google Scholar]
- 26.Rohrer JD, Nicholas JM, Cash DM, Swieten J van, Dopper E, Jiskoot L, Minkelen R van, Rombouts SA, Cardoso MJ, Clegg S, Espak M, Mead S, Thomas DL, De Vita E, Masellis M, Black SE, Freedman M, Keren R, MacIntosh BJ, Rogaeva E, Tang-Wai D, Tartaglia MC, Laforce R, Jr, Tagliavini F, Tiraboschi P, Redaelli V, Prioni S, Grisoli M, Borroni B, Padovani A, Galimberti D, Scarpini E, Arighi A, Fumagalli G, Rowe JB, Coyle-Gilchrist I, Graff C, Fallström M, Jelic V, Ståhlbom AK, Andersson C, Thonberg H, Lilius L, Frisoni GB, Binetti G, Pievani M, Bocchetta M, Benussi L, Ghidoni R, Finger E, Sorbi S, Nacmias B, Lombardi G, Polito C, Warren JD, Ourselin S, Fox NC, Rossor MN. Presymptomatic cognitive and neuroanatomical changes in genetic frontotemporal dementia in the Genetic Frontotemporal dementia Initiative (GENFI) study: A cross-sectional analysis. Lancet Neurol. 2015;14:253–262. doi: 10.1016/S1474-4422(14)70324-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Williams KL, Fifita JA, Vucic S, Durnall JC, Kiernan MC, Blair IP, Nicholson GA. Pathophysiological insights into ALS with C9ORF72 expansions. J Neurol Neurosurg Psychiatry. 2013;84:931–935. doi: 10.1136/jnnp-2012-304529. [DOI] [PubMed] [Google Scholar]
- 28.Le Ber I, Camuzat A, Guillot-Noel L, Hannequin D, Lacomblez L, Golfier V, Puel M, Martinaud O, Deramecourt V, Rivaud-Pechoux S, Millecamps S, Vercelletto M, Couratier P, Sellal F, Pasquier F, Salachas F, Thomas-Antérion C, Didic M, Pariente J, Seilhean D, Ruberg M, Wargon I, Blanc F, Camu W, Michel BF, Berger E, Sauvée M, Thauvin-Robinet C, Mondon K, Tournier-Lasserve E, Goizet C, Fleury M, Viennet G, Verpillat P, Meininger V, Duyckaerts C, Dubois B, Brice A. C9ORF72 repeat expansions in the frontotemporal dementias spectrum of diseases: A flow-chart for genetic testing. J Alzheimers Dis. 2013;34:485–499. doi: 10.3233/JAD-121456. [DOI] [PubMed] [Google Scholar]
- 29.Miller TM, Pestronk A, David W, Rothstein J, Simpson E, Appel SH, Andres PL, Mahoney K, Allred P, Alexander K, Ostrow LW, Schoenfeld D, Macklin EA, Norris DA, Manousakis G, Crisp M, Smith R, Bennett CF, Bishop KM, Cudkowicz ME. An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: A phase 1, randomised, first-in-man study. Lancet Neurol. 2013;12:435–442. doi: 10.1016/S1474-4422(13)70061-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van Elteren PH. On the combination of independent two sample tests of Wilcoxon. Bull Inst Inter Statist. 1960;37:351–361. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Validation of poly(GP) detection in CSF.
Fig. S2. Relationships between CSF poly(GP) and ALSFRS-R score, behavioral impairment, and cognitive impairment.
Fig. S3. Decreases in RNA foci and c9RAN protein pathology are observed in (G4C2)66 mice treated with c9ASO-1.
Fig. S4. Decreases in brain and CSF poly(GP) are observed in (G4C2)66 mice treated with c9ASO-1.
Fig. S5. Validation of poly(GA) Meso Scale Discovery sandwich immunoassay.
Table S1. Distribution of the 254 subjects among the sites that provided CSF.
Table S2. Poly(GP) in CSF from C9ORF72 repeat expansion carriers and noncarriers.
Table S3. Associations of poly(GP) with C9ORF72 repeat expansion and disease status.
Table S4. Characteristics according to C9ORF72 repeat expansion carrier status.
Table S5. Characteristics for asymptomatic C9ORF72 repeat expansion carriers and patients with c9ALS or c9ALS-FTD.
Table S6. Poly(GP) in CSF collected longitudinally from C9ORF72 repeat expansion carriers.
Table S7. Secondary comparisons of CSF poly(GP) in patients with c9ALS or c9ALS-FTD.
Table S8. Neuropsychological test battery for Mayo Clinic and NIH.
Table S9. Characteristics according to behavioral impairment in patients with c9ALS or c9ALS-FTD.
Table S10. Characteristics according to cognitive impairment in patients with c9ALS or c9ALS-FTD.
Table S11. Subject characteristics for PBMCs.
Table S12. Subject characteristics for lymphoblastoid cell lines.
Table S13. Subject characteristics for iPSC lines.