

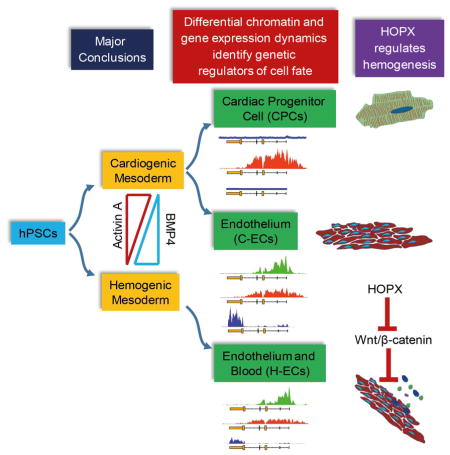
Summary
We analyzed chromatin dynamics and transcriptional activity of human embryonic stem cell (hESC)-derived cardiac progenitor cells (CPCs) and KDR+/CD34+ endothelial cells generated from different mesodermal origins. Using an unbiased algorithm to hierarchically rank genes modulated at the level of chromatin and transcription, we identified candidate regulators of mesodermal lineage determination. HOPX, a non-DNA binding homeodomain protein, was identified as a candidate regulator of blood-forming endothelial cells. Using HOPX reporter and knockout hESCs we show that HOPX regulates blood formation. Loss of HOPX does not impact endothelial fate specification but markedly reduces primitive hematopoiesis, acting at least in part through failure to suppress Wnt/β-catenin signaling. Thus, chromatin state analysis permits identification of regulators of mesodermal specification, including a conserved role for HOPX in governing primitive hematopoiesis.
eTOC
Palpant et al. analyze gene expression and chromatin dynamics in cardiovascular progenitor cells derived from hPSCs to elucidate genes governing cell fate. HOPX is identified as a regulator of primitive hematopoiesis, providing insight into controlling cell lineages from pluripotency for disease modeling or therapeutic applications.
Introduction
During gastrulation, mesoderm is specified from the mid to posterior primitive streak and gives rise to a wide range of derivatives including the heart, skeletal system, vasculature, and blood. An emerging set of studies indicates a significant overlap in the molecules that regulate the bifurcations in lineage between mesodermal derivatives, including MESP1, GATA4, NKX2-5, and TMEM88, among others (Bondue et al., 2008; Chan et al., 2013; Ferdous et al., 2009; Palpant et al., 2013; Peterkin et al., 2009). Furthermore, studies of the developmental origins of mesoderm, particularly the relationship between cardiac and endothelial development, indicate that disrupting expression of chromatin modulators (Delgado-Olguín et al., 2012; Lickert et al., 2004; Wamstad et al., 2012), transcription factors (Van Handel et al., 2012), or signaling molecules (Palpant et al., 2013) critical for specifying cell states can result in alterations in cell fate. As a consequence, tight orchestration of signaling and transcription is critical for determining the wide range of lineages specified in the early embryo. Failure to navigate these formative fate choices often results in miscarriage and/or birth defects (Herion et al., 2014).
Human pluripotent stem cells have provided valuable insights to mechanisms underlying cell fate specification. In a broad range of developmental pathways, lineage regulators show a unique signature in which these loci are repressed (H3K27me3) in cell states in which expression of these genes would be deleterious (Bernstein et al., 2006; Boyer et al., 2006; Cui et al., 2009; Paige et al., 2012). The repressive effects of H3K27me3 are replaced by open chromatin marks (H3K4me3) when expression of the gene is required to mediate a cell fate choice (Paige et al., 2012). We have previously shown that structural and housekeeping genes show less dynamic regulation at the level of chromatin during differentiation, requiring only gain or loss of the activating H3K4me3 to control transcription (Paige et al., 2012). As such, combining chromatin state analysis and mRNA expression provides a powerful means of identifying low abundance regulators of cell fate determination such as transcription factors or signaling molecules. We have successfully used this approach to uncover roles for MEIS2 (Paige et al., 2012) and TMEM88 (Palpant et al., 2013) in heart development.
We recently generated a differentiation method involving modulation of activin A and BMP4 stimulation to direct anterior vs posterior-like mesoderm that gives rise to cell populations reflecting cardiogenic fates vs. blood forming lineages, respectively, from hPSCs (Palpant et al., 2017; Palpant et al., 2015b). Using well established cues from developmental biology we can generate cardiogenic mesoderm, an anterior primitive streak lineage that is directed primarily through stimulation of activin A and gives rise to cardiomyocytes with high efficiency. This induction approach can also give rise to endothelial cells (ECs) that are highly angiogenic, have weak blood-forming activity and express endocardial markers like NFATC1. We can also generate endothelial cells from posterior primitive streak-like mesoderm that is directed primarily through stimulation of BMP4 signaling. This posterior mesodermal lineage shows minimal cardiac specification and gives rise to endothelial cells that are robustly hemogenic and express markers of blood forming mesoderm such as HAND1.
The fundamental goals of this study include first, elucidating the chromatin and gene expression dynamics that delineate distinct but ontologically related mesoderm progenitor populations and, second, identify regulators of cardiovascular fate determination. To accomplish this we analyzed genome-wide chromatin states and gene expression in cardiac progenitor cells (CPCs) and endothelial cells derived from different mesodermal origins. By performing genome editing and loss off function assays in vitro we identified HOPX as a regulator of hemogenesis.
Results
Chromatin states and gene expression of mesodermal lineages
We have developed a differentiation scheme using high density monolayer cultures where hPSCs can be directed into mesodermal fates that give rise to cardiac, endothelial, and blood lineages by varying the concentrations of activin A and BMP4 (Palpant et al., 2015a). High activin A favors cardiogenic fates whereas high BMP4 directs hemogenic fates with minimal cardiac potential. Cardiogenic mesoderm can be directed into cardiac progenitor cells (CPCs) or endothelial cells (cardiogenic mesoderm–derived endothelial cells, C-ECs), while hemogenic mesoderm can be directed into endothelial and blood lineages (hemogenic mesoderm-derived endothelial cells, H-ECs). The specific mechanisms by which blood and endothelial fates are specified has not been described in detail in this protocol. However, we have shown functional blood and endothelial cell types derive coordinately from these conditions. CPC differentiation from mesoderm requires Wnt/β-catenin inhibition, while induction of C-ECs and H-ECs requires treatment with VEGF, BMP4, bFGF, and ascorbic acid (Figure S1) (Palpant et al., 2015a).
In order to identify genomic and transcriptional differences between these closely related mesodermal progenitor populations, we performed RNA-seq and chromatin immunoprecipitation followed by deep sequencing, using antibodies to H3K4me3 and H3K27me3 in day 5 CPCs, C-ECs, and H-ECs (Figure S1 and Figure 1a). The overall distribution of the genome into bins of bivalent, H3K4me3-only, H3K27me3-only and neither chromatin modification was similar amongst the 3 progenitor populations, correlating with previously published distributions of chromatin states in progenitor cells in vitro and in vivo (Paige et al., 2012; Sachs et al., 2013) (Figure 1b). Two dimensional principal component analysis (PCA) of RNA-seq and ChIP-seq data showed that in the eigenvectors of the first component (PC1) endothelial cells clustered together relative to the cardiac progenitor cells (Figure 1c). However, in the second component (PC2) C-ECs clustered more closely to CPCs than H-ECs, supporting a shared phenotype attributable to their developmental ontogeny from cardiogenic mesoderm (Figure 1c).
Figure 1. Gene expression and chromatin dynamics show fidelity to lineage-specific developmental fates.

(a) Schematic diagram showing lineages generated by directed differentiation of hESCs. Day 5 progenitor populations for cardiac and 2 different endothelial lineages were isolated for ChIP-seq and RNA-seq analysis. (b) Genome-wide distribution of chromatin states into non-labeled, H3K4me3, H3K27me3, or bivalently labeled in CPCs, C-EC, and H-EC. (c) Two-dimensional principal component analysis of CPCs, C-EC, and H-EC as determined by expression, H3K4me3, and H3K27me3. The top 5 genes contributing to PCA1 are listed below each graph. (d) Heat maps for expression, H3K4me3, and H3K27me3 for a hand collated list of known regulators of endothelial (left) and cardiac (right) fate showing appropriate lineage specificity of gene sets. (e) Average signal for H3K4me3 and H3K27me3 chromatin dynamics 5kb surrounding the TSS for known lineage specific structure/function genes (blue) vs. lineage regulators (red) for the cardiac lineage (left) and endothelial lineage (right). (f) Raw RNA-seq and ChIP-seq data from all 3 mesodermal progenitor populations showing the myofilament protein TNNI1 representing a structure/function gene for the cardiac lineage and the transcription factors NKX2-5 and TAL1 representing lineage regulators for the cardiac and endothelial lineages, respectively.
Although RNAseq data show clear differences between the three cell populations (Figure 1c–d), RNA analyses are strongly influenced by high abundance transcripts that encode structural or housekeeping functions. Differences in low abundance transcripts encoding transcription factors and other regulatory molecules are more difficult to identify. We analyzed our data on the basis of chromatin states separating structural/functional genes vs. lineage regulators. As with our previous work (Paige et al., 2012), we found that genes encoding proteins with structural functions showed only acquisition of H3K4me3 in the active population with no evidence of H3K27me3 deposited at these loci in the alternate lineages (Figure 1e and f). In contrast, genes encoding proteins involved in mediating cell fate choices such as transcription factors showed markedly increased H3K4me3 in the target lineage coupled with H3K27me3 deposited around the TSS in the alternate lineage(s) (Figure 1e and f, Table S1).
Chromatin state and gene expression reveal regulators of cell fate specification in mesoderm
Compared to in vivo models, pluripotent stem cell differentiation provides a more controlled approach for elucidating mechanisms governing mesoderm differentiation into cardiovascular progenitor populations. Based on previous success using gene expression and chromatin states to identify key regulators of cell fate (Paige et al., 2012; Palpant et al., 2013), we used a similar approach to identify putative regulators of CPCs, C-ECs, and H-ECs.
A schematic outline of the algorithm is show in Figure 2. It was designed as follows: for a given pairwise comparison of lineages, the first list of putative regulators was generated by ranking genes solely on the basis of fold change of transcript abundance. The second round of analysis generated candidate regulator lists incorporating chromatin and gene expression (Figure 2a–b). Genes gained a higher score based on the relative abundance of expression by RNA-seq plus H3K4me3 in the lineage of interest and the relative abundance of H3K27me3 in the alternative lineage (Figure 2c). Data were analyzed in an unbiased manner on a locus-by-locus basis for all population comparisons to generate a score for each gene. If a gene appeared as shared between lineages (e.g. C-ECs and H-ECs), the gene was restricted to the shared regulator list (pan-EC) and removed from the individual regulator lists to enrich for lineage-specific genes in each list. We provide an example for the calculated ranking of TAL1, which is a regulator of C-ECs and H-ECs (Figure 2d).
Figure 2. Lineage-specific molecules involved in cell fate determination are enriched by hierarchical ranking of genes based on chromatin dynamics and gene expression.

(a) Heat map of all genes analyzed which was reduced to (b) those genes (expression and chromatin heat maps shown) that are >2 fold higher in a given population. In this example genes greater than 2 fold higher in C-ECs and H-ECs vs. CPCs are shown. (c) Equations used to generate scoring for regulator list hierarchy. (d) Raw data and correlative score generated based on analysis of the TAL1 gene. (e–f) Regulator lists generated using ranking algorithm outlined in a–d were analyzed by gene ontology analysis with data shown for CPCs (e) and pan-ECs (f). Data are presented as (i) lineage map of population under evaluation, (ii) genes ranked on the basis of cumulative score for regulators based on expression (green) or expression plus chromatin (orange). The number of genes in each regulator list is shown to the side of each graph. (iii–iv) Statistical analysis of gene ontology enrichment categories for Biological Process (iii) and Molecular Function (iv) as a comparison of regulators identified by expression alone (green) or expression plus chromatin (orange). (g) H3K4me3 chromatin breadth 5000 bp upstream and downstream of the TSS for putative regulators identified by expression alone (green) or expression plus chromatin (orange) in CPCs, C-ECs, and H-ECs. Raw H3K4me3 deposition around the TSS for all genes in the list are shown to the right of each graph.
Regulator lists were then analyzed by gene ontology analysis (Figure 2e–f and Figure S2). Data show the lineage of interest in (i) and the cumulative score for each regulator graphed in (ii). Selection for regulators on the basis of chromatin plus expression resulted in a significant attrition of candidate molecules that showed differences in mRNA but minimal difference in chromatin state between the populations (Figure 2e–f, ii). Biological process and molecular function ontologies in genes identified by expression alone predominantly featured structural constituents such as sarcomere organization or actin binding (Figure 2e–f, iii and iv, and Table S2, S5, and S8). Analysis of chromatin alone revealed enrichment for transcriptional regulators and DNA binding molecules but performed poorly in enrichment for cell type-specific biological processes (Table S3, S6, and S9). In contrast, integrating expression and chromatin data identified genes responsible for mediating cell fate choices in a lineage-specific manner, including molecules mediating these cell states with molecular functions involving sequence-specific DNA binding characteristic of transcriptional regulators (Figure 2e–f, iii and iv and Tables S4 and S7–S9).
Recent studies in chromatin dynamics have shown that the breadth of the H3K4me3 mark correlates with genes linked to cell identity (Benayoun et al., 2014). We therefore analyzed H3K4me3 breadth in regulators identified by expression alone or expression plus chromatin as an independent metric for identifying lineage regulators. We found that in all lineages, genes identified by the integrated approach had a significantly broader H3K4me3 domain compared to those identified by expression alone (Figure 2g). Taken together, these data show an essential role for incorporation of open and closed chromatin for enriching genes governing cell identity and function that vastly exceeds the capacity of transcriptional profiling alone.
Identification of the top putative regulators of lineage determination
All putative regulators identified on the basis of expression or chromatin and expression are listed in Table S2–S4. We performed a literature search on the top 10 regulators identified for each cell type to determine whether they had a known role in that lineage (in health or disease) and/or were known developmental regulators (Figure 3a). Many of the top regulators were known to have lineage-specific functions, which validated our overall approach. For example, CELF2 is a known RNA processing molecule involved in regulating splice variants in the embryonic heart (Blech-Hermoni et al., 2013; Singh et al., 2004). The genome-wide analysis also identified 49 known lineage regulators that were shared between CPCs and C-ECs including a preponderance of cardiac and endocardial associated factors including SIX1, FOXC1, RBM20, IRX5, IRX3, TWIST1, and PDGFRα (Table S4). Similarly, canonical mediators of blood forming endothelial cell fate determination including LYL1 (Capron et al., 2006; Giroux et al., 2007) and TAL1 (Gering et al., 1998) were appropriately identified as top regulators in hemogenic endothelium (Figure 3a and Table S4).
Figure 3. Lineage regulators identified by integrated analysis of chromatin dynamics and gene expression and their temporal expression dynamics during directed differentiation.

(a) Table of regulators showing those with known roles in development or disease and those with no known previous role in lineage specification. (b–c) Quantitative RT-PCR analysis between days 2 and 5 of differentiation for all 3 mesodermal lineages. (b) NKX2-5 and TAL1 were assessed to show appropriate regulation of known regulators in a time-dependent and lineage-specific manner. (c) Assessment of the top 5 regulators in each category. All PCR products were confirmed by sequencing. n =4–8 per time point. Values are presented as mean ± sem.
Among the regulators identified through this algorithm, several had no known function in the lineage or only peripheral associations with the lineage without established mechanism. Furthermore, although a large number of genes identified using this approach include transcription factors which are classically defined as governing cell identity, this unbiased approach broadens the scope of gene subtypes as putative regulators of cell identity including those encoding proteins involved in RNA processing, signaling, metabolism, and more. These data show that regulators identified on the basis of chromatin states and expression display a range of candidate molecules for dissecting mechanisms by which mesoderm fates are differentially specified during cardiovascular development.
To gain insights into the dynamics of gene regulation we determined the mRNA expression patterns during differentiation for the top 5 regulators of each of the 3 lineages by qRT-PCR (Figure 3b–c). Two known markers of lineage determination, NKX2-5 and TAL1, were included to confirm that these time course samples have appropriate lineage and temporal specificity (Figure 3b). We found that factors identified as CPC, H-EC, and pan-EC regulators were largely lineage specific and showed significant temporal activation as lineage development progressed (Figure 3c).
HOPX is a regulator of hemogenesis
The litmus test for this analysis was to determine whether this approach could identify regulators of fate decision in mesoderm development. To address this, we focused follow up assays on HOPX as a regulator of endothelial development with the following rationale. HOPX ranks within the top 5 genes regulating endothelial cells, ahead of other known regulators of endothelial development such as TAL1 and no previous studies have elucidated a role for HOPX in the early developmental specification or function of endothelium or blood. HOPX is a non-DNA binding homeodomain protein shown to play a number of roles throughout heart development (Jain et al., 2015b; Trivedi et al., 2010). It is also known to regulate hair follicle, intestinal, and hematopoietic stem cell biology (Takeda et al., 2013; Takeda et al., 2011; Zhang et al., 2013). A recent study has implicated HOPX in adult hematopoietic stem cell function though a potential role in regulating embryonic hematopoietic development during mesodermal patterning has not been studied (Zhou et al., 2015).
We analyzed chromatin and expression dynamics in different mesoderm populations. In our two endothelial lineages, HOPX shows a bivalent chromatin profile biased toward transcriptionally active chromatin (H3K4me3) that correlates with expression as assessed by RNA-seq (Figure 4a). Table 1 provides computational data for the scoring of the HOPX as a regulator of ECs.
Figure 4. HOPX identified as a regulator of cell fate determination in hemato-endothelial differentiation.

(a) Chromatin dynamics and gene expression of the HOPX locus in day 5 CECs, H-ECs, and CPCs. (b) Sashimi isoform analysis of the HOPX locus in endothelial vs. cardiac differentiation showing distinct transcript profiles that differ between these lineages. Orange arrow denotes common translational start site (c) Construct for CRISPR/Cas9 gene targeting of a tdTomato reporter to the HOPX translational start site. (d) FACS analysis of tdTomato mean fluorescence intensity in day 2 mesoderm cells and day 5 KDR+/CD34+ H-ECs normalized to time matched WT cells. Raw FACS plots shown to the right. n≥3 per group. Values are presented as mean ± sem. *P<0.05.
Table 1.
Computational analysis of HOPX RNA-seq and ChIP-seq data generated by ranking algorithm.
| C-EC Expression | 1175.218 |
| H-EC Expression | 709.7971 |
| CPC Expression | 18.98132 |
| C-EC vs CPC log2FoldChange | 5.952204 |
| H-EC vs CPC log2FoldChange | 5.224754 |
| C-EC vs CPC K27me3_log2FoldChange | −2.39 |
| H-EC vs CPC K27me3_log2FoldChange | −2.54 |
| C-EC vs CPC K27me3_padj | 5.99E-05 |
| H-EC vs CPC K27me3_padj | 1.16E-07 |
| C-EC vs CPC K4me3_log2FoldChange | 1.83 |
| H-EC vs CPC K4me3_log2FoldChange | 2.1 |
| C-EC vs CPC K4me3_padj | 4.69E-08 |
| H-EC vs CPC K4me3_padj | 1.94E-08 |
| C-EC vs CPC Rank score | 26.55188 |
| H-EC vs CPC Rank score | 29.64972 |
| Aggregate Score | 56.2016 |
Since HOPX is abundantly expressed in cardiac development, we examined the HOPX transcript from RNAseq databases obtained from one year-old hESC-CMs and fetal human heart versus transcripts identified in ECs focusing on HOPX splice variants between the cell types (Figure 4b). Sashimi plots were used for quantitative visualization of mRNA sequencing reads aligned to gene annotations (Katz et al., 2010). These data show a significant difference in isoform expression between endothelial and cardiac populations with splice variants being consistently expressed in lineage-specific cell types at all stages of development (Figure 4b). Despite retaining a common translational start site, cardiac transcripts showed activation of an extended 5′ UTR compared to transcripts expressed in endothelial differentiation. Thus, endothelial cells use an alternative HOPX transcriptional start compared to cardiomyocytes.
To validate that HOPX is actively expressed in endothelial development we used CRISPR/Cas9 to generate a reporter hESC line in which tdTomato is knocked into the HOPX translational start site (TSS) (Figure S3). This cell line provides a dynamic readout of HOPX expression during differentiation (Figure 4c). HOPX reporter activity was not detected in mesoderm (day 2) above WT control cells but showed significant activation in day 5 CD34+ H-ECs (Figure 4d). While HOPX reporter activity was observed in C-ECs, mean fluorescence was significantly higher in H-ECs (Figure S4a). Previous studies have suggested that blood forming endothelium is confined to the portion of ECs negative for the surface marker CD73 (NT5E) (Choi et al., 2012). HOPX reporter activity was measured in H-ECs separated on the basis of CD73 expression, with the mean fluorescence intensity of HOPX significantly higher in CD73− cells (Figure S4b).
HOPX is expressed during hemato-endothelial lineage specification in vivo
We next assessed HOPX expression during mouse in vivo development from E6.5–E7.75 based on expression profiling work carried out by the Göttgens laboratory (Moignard et al., 2015; Scialdone et al., 2016). Expression profiling of Flk1+ cells during mouse mesoderm lineage development showed HOPX expressed in coordination with ETV2 and TAL1 during hemato-endothelial lineage specification (Moignard et al., 2015) (Figure 5a). At higher resolution, transcriptional profiling of 1,205 individual Flk1+ cells from the epiblast and nascent mesoderm and gastrulating mouse embryo provide single cell-level view of expression changes during mesoderm formation in the mammalian gastrulating embryo (Scialdone et al., 2016). Unsupervised hierarchical clustering of single cell expression profiles indicated HOPX activity in cells also expressing hemato-endothelial genes CDH5, GATA1, Myb, and TAL1 but not in cells expressing early mesodermal lineage specification genes like EOMES and MESP1 (Figure 5b).
Figure 5. HOPX is expressed during hemato-endothelial differentiation in vivo.

(a) Expression profiling of KDR+ cells during mouse development from days 7–7.75 showing up-regulation of HOPX in coordination with TAL1 and ETV2. (b) Heatmap showing key genes representing cells clustered into ten cell groups during the time course of three developmental stages, Head Fold, Neural Plate, and Primitive Streak. (c) tSNE analysis of Neural Plate and Head Fold stage cells excluding E6.5 Epiblast cells. Color coding is as described in panel A for all cell types. HOPX expression profiling across these cell populations is shown to the right. (d–e) The fraction of cells with expression of HOPX (d) and relative expression of HOPX (e) in different subpopulations of cells throughout hemato-endothelial development. Cell populations are color coded as described in panel b. (f) Expression analysis at single cell level ordered by pseudotime showing down-regulated (T) and up-regulated (HBB-BH1) genes relative to HOPX.
Using t-distributed stochastic neighbor embedded analysis (t-SNE) (van der Maaten and Hinton, 2008) to project high dimensional data into two-dimension points, key populations involved in specification of hemogenic endothelium were identified including early uncommitted mesodermal progenitors, posterior mesoderm, endothelium, blood progenitors, and primitive erythrocytes (Figure 5c). Overlaying HOPX expression across populations identified by Scialdone et al showed scattered and rare expression in uncommitted mesoderm with increased expression in endothelium through to blood progenitors and primitive erythrocytes (Figure 5c). Analysis of cell subpopulations showed HOPX positive cells in all cell populations with the highest proportion in endothelium and erythroid cell types where greater than 75% of cells expressed HOPX (Figure 5d–e).
We next analyzed expression of genes using a computational approach to infer location of cells in pseudotime along this developmental trajectory (Ji and Ji, 2016). These data show proper down-regulation of mesoderm marker T followed by up-regulation of HOPX during blood development as marked by fetal hemoglobin expression in erythroid lineage specification (Figure 5f). Taken together, these data show that HOPX expression is activated during hemato-endothelial fate specification in vivo.
HOPX is functionally regulated by SCL in hemato-endothelial differentiation
We analyzed global gene expression data of WT vs. SCL/TAL1 knockout ESC differentiation by Org et al (Org et al., 2015). Loss of SCL/TAL1 during hemato-endothelial differentiation causes cells to undergo a fate shift into the cardiac lineage manifest as ectopic heart formation in the yolk sac (Van Handel et al., 2012). While HOPX is involved in heart development (Jain et al., 2015b), these data show that HOPX is up-regulated during hemato-endothelial differentiation in an SCL/TAL1-dependent manner (Figure S5).
HOPX regulates hematopoiesis at least in part through Wnt inhibition
We next sought to characterize the molecular mechanism by which HOPX regulates blood specification in hESC derivatives. To accomplish this, we generated a HOPX knockout line using CRISPR/Cas9 (Figure S6) and differentiated the cells into H-ECs. No defect was observed in efficiency of lineage specification into endothelium on the basis of CD31 staining in WT and HOPX KO derived ECs (Figure 6a). Endothelial functional assays were carried out to determine HOPX impact on lumen formation (Figure 6b) and angiogenesis (Figure S7), which showed no difference compared to WT cells.
Figure 6. HOPX is functionally required for primitive hematopoiesis.

(a) Fate specification of CD31 endothelial cells is not effected by HOPX KO. (b) HOPX KO does not impact functional endothelial lumen formation assays in collagen. Scale bars = 200 μm. (c–e) Hematopoiesis analysis of CD43+/CD235a+ derivatives in day 5 H-ECs with representative FACS plots (c) and colony forming assays in methylcellulose assaying for Ery-P (d) and macrophage CFUs (e) in WT vs. HOPX KO cells. Representative CFU images are shown to the right for primitive erythroid (Ery-P), and macrophage (Mac) colonies. (f–g) Quantitative RT-PCR analysis of HOPX and genes involved in hematoendothelial cell fate specification (SCL) and function (GATA1 and RUNX1) (f) as well as Wnt/β-catenin target genes AXIN2 and TROY1 in WT vs. HOPX KO cells (g). (h) Wnt-dependent regulation of CD43+/CD235a+ primitive hematopoiesis assessed by treatment of cells with the Wnt agonist CHIR-99021 (1 μM) or the Wnt antagonist XAV-939 (1 μM). (i) Differentiation of HOPX KO and WT cells into H-ECs showing that addition of the Wnt inhibitor XAV-939 (1 μM) partially rescues hematopoietic deficiencies observed in HOPX KO cells. n=4–8 per group. Values are presented as mean ± sem. *P<0.05 vs. WT. # P<0.05 vs. HOPX KO cells.
We next analyzed blood formation by FACS analysis for cells giving rise to primitive hematopoietic lineages (CD43/235a) in addition to colony forming assays (CFU) in methylcellulose, as we have performed previously (Palpant et al., 2015a). Nearly all blood forming progenitor activity at the onset of hESC hematopoiesis occurs in the CD43+ population. Co-expression of CD43 with CD235a delineates the initial wave of primitive hematopoiesis, which contains mostly primitive erythroid and myeloid (primarily macrophage) derivatives (Kennedy et al., 2012; Kennedy et al., 2007; Sturgeon et al., 2014; Vodyanik et al., 2006). In HOPX KO cells there was a marked deficiency in CD43+/CD235a+ blood derivatives vs. control (Figure 6c). This was further corroborated by colony forming assays in methylcellulose, which showed a reduced number of primitive erythroid and macrophage colony forming units in HOPX KO cells vs. control (Figure 6d–e). Following this initial wave of primitive hematopoiesis, a second wave of lineage-restricted hematopoiesis referred to at the erythromyeloid progenitor (EMP) wave gives rise to erythroid progeny expressing both embryonic and adult globins as well as a broader array of myeloid progeny (granulocytes in addition to macrophages) (Palis J. FEBS Lett 2016 Nove 590(22):3965–3974). We previously demonstrated that secondary co-culture of day 5 H-EC with OP9 stroma and hematopoietic cytokines results in the generation of CD45+ progeny with hematopoietic potential resembling the EMP wave (Palpant et al., 2015b). Thus, to determine a role for HOPX in EMP hematopoiesis, day 5 H-ECs were co-cultured with OP9 stroma and assayed for CD45+ cells and colony forming activity. These assays showed no difference between WT and HOPX KO cells (Figure S7b), indicating the defect was most pronounced in primitive hematopoiesis. Multilineage hematopoiesis that includes lymphoid potential was not assayed as our differentiation conditions did not generate progency with robustly detectable lymphoid potential based on our previous studies (Palpant et al., 2015b).
Molecularly, these functional findings were validated by transcriptional analysis of genes required for endothelial and blood development. We found that SCL/TAL1 and GATA1 were markedly down-regulated in HOPX KO cells compared to control (Figure 6f). The hematopoietic transcription factor RUNX1 (Webber et al., 2013), was not different between KO and WT cells (Figure 6f). Taken together, these data suggest a reduced generation or survival of primitive lineage progenitors in the absence of HOPX with no effect on CD31+ endothelial fate specification.
HOPX functions as a transcriptional inhibitor in most contexts (Cheung et al., 2013; Jain et al., 2015b; Trivedi et al., 2010). Chip-seq assays have shown that HOPX binds to Wnt target genes (Jain et al., 2015b), and recent studies have indicated that Wnt/β-catenin signaling is required for definitive hematopoiesis but inhibitory to primitive hematopoiesis (Sturgeon et al., 2014). We therefore analyzed Wnt target genes AXIN2 and TROY1 in day 5 H-ECs. TROY1 was significantly up-regulated in the context of HOPX KO (Figure 6g), and AXIN2 showed a non-significant increase. This is consistent with the observed deficiency in primitive hematopoiesis. We differentiated H-ECs in the presence of Wnt modulators CHIR-99021 and XAV-939 and functionally show Wnt dosage directly regulates efficiency of primitive hematopoiesis from pluripotency in our differentiation protocol (Figure 6h). To determine if HOPX is acting directly through the Wnt pathway to regulate hematopoiesis, we attempted to rescue the differentiation by adding an exogenous small molecule Wnt inhibitor, XAV-939, which stimulates β-catenin degradation by inhibiting tankyrase. Adding 1μM XAV-939 from days 2–5 of differentiation resulted in a partial rescue of the hematopoietic deficiency on the basis of increased CD43+/CD235a+ cells in the HOPX KO cells compared to controls (Figure 6h). We repeated these findings using an independently generated CRISPR/Cas9 HOPX mutant cell line with the same outcomes (Figure S6 and S7c). These data indicate that HOPX is acting at least in part through suppression of Wnt/β catenin signaling to modulate primitive hematopoiesis from earlier mesodermal derivatives.
Discussion
In this study we provide genome-wide analysis of transcriptional activity as well as chromatin to elucidate the molecular states that distinguish the cardiac and two distinct endothelial lineages. This analysis provides a framework with which to study mechanisms of fate determination in mesoderm cell fate specification. Chromatin states in concert with gene expression profiles have previously been used to identify regulators in cardiac development (Paige et al., 2012; Palpant et al., 2013). We applied this approach to identify molecules that regulate mesodermal fates with a specific focus on HOPX. HOPX is a non-DNA binding homeodomain protein that has been shown to modulate cell fate choices in a wide range of contexts (Jain et al., 2015a; Takeda et al., 2013; Takeda et al., 2011; Trivedi et al., 2010; Zhou et al., 2015). In the current study we provide evidence for a mechanism by which HOPX regulates hemato-endothelial development. Previous work has implicated HOPX in modulating cell fate specification through GATA factors and Wnt signaling (Jain et al., 2015b; Trivedi et al., 2010). Wnt signaling is a key pathway that is tightly regulated to mediate cell fate choices in mesoderm (Paige et al., 2010; Palpant et al., 2013; Sturgeon et al., 2014; Ueno et al., 2007), including multiple, stage-specific aspects of hematopoiesis (Chanda et al., 2013; Ruiz-Herguido et al., 2012; Sturgeon et al., 2014). In human ESC models, early Wnt activity is inhibitory to primitive hematopoiesis at the level of the KDR+ mesodermal precursors as well as their subsequent colony forming progeny (Paluru et al., 2014; Sturgeon et al., 2014).
Consistent with this, we identify HOPX as a positive regulator of primitive hematopoiesis that may be acting at least in part through inhibition of the Wnt pathway.. Given that HOPX is expressed between day 3–5 of differentiation (Figure 3c), after mesoderm has been specified (day 2), we hypothesize HOPX is governing emergence of primitive hematopoiesis from specified mesoderm. Future studies, however, will be required to determine whether HOPX functions within KDR+ mesoderm, which is detected by day 3 of differentiation, or during the transition through CD34+ hemogenic endothelium, which occurs shortly thereafter between days 4 and 5. Given studies suggesting Wnt regulation of primitive hematopoiesis at multiple levels, including lineage specification, survival, and expansion (Paluru et al., 2014; Sturgeon et al., 2014); future analysis will also be required to determine whether HOPX regulates primitive hematopoietic progenitor numbers through modulation of proliferation or survival subsequent to their specification from mesoderm.
Following its role in primitive hematopoiesis, Wnt activation has been shown to both positively and negatively regulate subsequent multilineage hematopoiesis, likely reflecting multi-phasic requirements for the Wnt pathway during the transition from hemogenic endothelium to hematopoietic stem/progenitor cells (Chanda et al., 2013; Ruiz-Herguido et al., 2012; Sturgeon et al., 2014). A recent study has identified a role for HOPX in adult hematopoietic stem cell homeostasis and engraftment (Zhou et al 2015), but did not address embryonic HSC formation. Although we did not observe a significant effect on EMP hematopoiesis in the absence of HOPX, further studies will be required to determine whether HOPX also interacts with the Wnt pathway to regulate definitive, multilineage hematopoiesis and particularly embryonic HSC formation, as specification of multilineage hematopoiesis in human PSC models remains inefficient and thus challenging to assess with current differentiation protocols.
Together with our previous work (Paige et al., 2012), this study accentuates the power of identifying regulators of cell fate determination through a combinatorial approach of gene expression and chromatin analysis. These findings provide insights into regulation of mesoderm development and mechanisms controlling hematopoietic lineage specification. Importantly, further understanding of how mesoderm is specified into primitive and definitive blood lineages will be essential for translational applications, including generating hematopoietic stem cells and differentiated blood cells from pluripotent stem cells for disease modelling and cell-based therapeutics. Lastly, this study provides the framework for a wide range of studies on putative regulatory molecules important for specifying mesodermal derivatives in vivo and in vitro.
Experimental Procedures
See supplemental experimental procedures for more details.
Cell Culture
RUES2 human ES cells were maintained as previously described (Paige et al., 2012). In brief, cells were plated on Matrigel (BD) coated plates and maintained in an undifferentiated state with mouse embryonic fibroblast (MEF) conditioned media containing 5 ng/mL hbFGF (Peprotech, 100-18B).
HOPX reporter and KO hESCs
We used CRISPR gene editing technology to genetically modify RUES2 hESCs at the HOPX locus (Figure S3a–e and S6).
Human ESC Directed Differentiation
WT RUES2 human embryonic stem cells were used in this study. Undifferentiated cells were maintained in mouse embryonic fibroblast-conditioned media. Standard cardiomyocyte and endothelial directed differentiation using a monolayer platform was performed with a modified protocol based on previous reports (Palpant et al., 2015a).
Colony forming assays
Day 5 hESC-derived cells were harvested as single cells and plated for colony forming-unit (CFU) progenitors in methylcellulose containing human cytokines (H4034, Stem Cell Technologies). Colonies were scored by morphology after 12–14 days as small, primitive erythroid (CFU-EryP), and macrophage (CFU-Mac).
RNA-seq
Total RNA from bulk cultures of cardiac progenitor cells, C-ECs, and H-ECs were isolated with RNALater (Qiagen, 76104). For each group, 2 biological replicates were submitted for analysis. Samples were submitted to University of Washington High Throughput Genomic Sequencing Center for isolation and analysis. RNA-seq was performed on poly-A enriched samples using Illumina TruSeq. Reads were aligned to version hg19 of the human genome using Gsnap. Transcript abundance was estimated using a Markov chain Monte Carlo algorithm to sample from a hierarchical Bayesian model. Single cell RNA-seq data was originally derived by Scialdone et al (Scialdone et al., 2016). Gene-level read counts data from single cell RNA-seq were downloaded from http://gastrulation.stemcells.cam.ac.uk.
Chromatin immunoprecipitation followed by deep sequencing
Day 5 bulk cultures of cardiac progenitor cells, C-ECs, and H-ECs were fixed in 1% formaldehyde. Crosslinked samples were submitted to the University of Washington High Throughput Genomic Sequencing Center for ChIP-seq analysis for H3K4me3 and H3K27me3 histone modifications. Peaks were called using MACS. Regions and genes with differential chromatin marks were identified using diffReps. All RNA and ChIP-seq data have been curated and approved by Gene Expression Omnibus (GSE 97080).
Generating regulator lists
The basic principle of generating a putative regulator list is to identify genes with significant changes in levels of expression and H3K27me3 or H3K4me3 marks. This includes both filtering and ranking steps. First, only genes with significant changes in levels of expression and at least one histone marks (H3K27me3 or H3K4me3) in a pairwise lineage comparison are kept for further analysis. For each gene g in each data set D in each pairwise comparison of two cell types i and j, score S is defined as:
where i and j = C-ECs, H-ECs or CPCs; di;g and dj;g the values of gene g in data set D (expression, H3K27me3, or H3K4me3), and FDRi:j;g the false discovery rate of gene g in the pairwise comparison between lineages i and j. S is a signed score based on the statistical significance of change for genes with higher values in lineage i.
Then the three data sets are integrated such that genes with consistent changes (higher expression, higher active H3K4me3 marks and lower repressive H3K27me3 marks or vice versa) in either direction will get the highest absolute scores:
To identify regulators for a single lineage i, only genes with positive scores from pairwise comparisons involving lineage i are kept, and the combined scores are defined as:
To identify shared regulators for two lineages i and j, only genes with positive scores from pairwise comparison involving a third lineage k are kept, and the combined scores are defined as:
Genes with FDR values smaller than 1e-15 were truncated at 1e-15 to prevent expression data from dominating the integrative significance score calculation.
The R code to generate integrated, expression only and chromatin only lists are deposited on Github: https://github.com/yuliangwang/regulators_integrating_RNAseq_ChIPseq
Supplementary Material
List of regulators for 3 mesodermal lineages based on expression alone listed in rank order. See separate supplemental file.
List of regulators for 3 mesodermal lineages based on chromatin alone listed in rank order. See separate supplemental file.
List of regulators for 3 mesodermal lineages based on integration of chromatin and expression listed in rank order. See separate supplemental file.
Gene Ontology “Biological Processes” and “Molecular Function” enrichments for regulator lists generated by expression only. See separate supplemental file.
Gene Ontology “Biological Processes” and “Molecular Function” enrichments for regulator lists generated by chromatin only. See separate supplemental file.
Gene Ontology “Biological Processes” and “Molecular Function” enrichments for regulator lists generated by integration of expression and chromatin. See separate supplemental file.
Lists of gene ontology enrichment of Molecular Functions and Biological Process comparing the P-value for shared GO terms for integrated approach vs. expression only. See separate supplemental file.
Lists of gene ontology enrichment of Molecular Functions and Biological Process comparing the P-value for shared GO terms for integrated approach vs. chromatin only. See separate supplemental file.
Quantitative PCR Primers to amplify human genes
Highlights.
Chromatin dynamics reveal genes governing cell identity
HOPX is identified as a regulator of mesoderm lineage determination
HOPX modulates primitive hematopoiesis by inhibition of Wnt signaling
Acknowledgments
We thank the University of Washington high throughput sequencing core (htSEQ) for generating RNA-seq and ChIP-seq data. This work was supported by the following sources: P01 HL094374 (to CEM), R01 HL084642 (to CEM), U01 HL100405 (to CEM), and P01 GM81619 (to CEM), and an award from the Fondation Leducq Transatlantic Network of Excellence (to CEM), and training grant support from T32 HL007312 (to NJP). The authors declare no conflict of interest.
Footnotes
Accession numbers. All RNA and ChIP-seq data have been curated and approved by Gene Expression Omnibus (GSE 97080).
Author Contributions:
NJP: Designed the study, carried out experiments related to hESCs, genome engineering, genomics analysis, and wrote the manuscript
YW: Carried out all computational bioinformatics related to expression profiling and chromatin dynamics
BH: Carried out experiments related to blood development from hESCs
RZ: Carried out transcriptional time course profiling of lineage regulators
DJ: Carried out computational analysis of RNA-seq data
MR: Carried out endothelial lumen formation and angiogenesis assays
LP: Designed the experiments and interpreted the findings
RJ: Assisted with gene targeting of HOPX in hESCs
WR: Supervised analysis of RNA-seq data
JE: Supervised the HOPX gene targeting
YZ: Supervised the endothelial functional assays
IB: Supervised the blood formation assays from hESCs
AM: Supervised the computational work
CEM: Supervised the overall study, designed the experiments, acquired funding, and wrote the manuscript
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Benayoun BA, Pollina EA, Ucar D, Mahmoudi S, Karra K, Wong ED, Devarajan K, Daugherty AC, Kundaje AB, Mancini E, et al. H3K4me3 breadth is linked to cell identity and transcriptional consistency. Cell. 2014;158:673–688. doi: 10.1016/j.cell.2014.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- Blech-Hermoni Y, Stillwagon SJ, Ladd AN. Diversity and conservation of CELF1 and CELF2 RNA and protein expression patterns during embryonic development. Dev Dyn. 2013;242:767–777. doi: 10.1002/dvdy.23959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondue A, Lapouge G, Paulissen C, Semeraro C, Iacovino M, Kyba M, Blanpain C. Mesp1 acts as a master regulator of multipotent cardiovascular progenitor specification. Cell Stem Cell. 2008;3:69–84. doi: 10.1016/j.stem.2008.06.009. [DOI] [PubMed] [Google Scholar]
- Boyer LA, Plath K, Zeitlinger J, Brambrink T, Medeiros LA, Lee TI, Levine SS, Wernig M, Tajonar A, Ray MK, et al. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature. 2006;441:349–353. doi: 10.1038/nature04733. [DOI] [PubMed] [Google Scholar]
- Capron C, Lécluse Y, Kaushik AL, Foudi A, Lacout C, Sekkai D, Godin I, Albagli O, Poullion I, Svinartchouk F, et al. The SCL relative LYL-1 is required for fetal and adult hematopoietic stem cell function and B-cell differentiation. Blood. 2006;107:4678–4686. doi: 10.1182/blood-2005-08-3145. [DOI] [PubMed] [Google Scholar]
- Chan SS, Shi X, Toyama A, Arpke RW, Dandapat A, Iacovino M, Kang J, Le G, Hagen HR, Garry DJ, et al. Mesp1 patterns mesoderm into cardiac, hematopoietic, or skeletal myogenic progenitors in a context-dependent manner. Cell Stem Cell. 2013;12:587–601. doi: 10.1016/j.stem.2013.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanda B, Ditadi A, Iscove NN, Keller G. Retinoic acid signaling is essential for embryonic hematopoietic stem cell development. Cell. 2013;155:215–227. doi: 10.1016/j.cell.2013.08.055. [DOI] [PubMed] [Google Scholar]
- Cheung WK, Zhao M, Liu Z, Stevens LE, Cao PD, Fang JE, Westbrook TF, Nguyen DX. Control of alveolar differentiation by the lineage transcription factors GATA6 and HOPX inhibits lung adenocarcinoma metastasis. Cancer Cell. 2013;23:725–738. doi: 10.1016/j.ccr.2013.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi KD, Vodyanik MA, Togarrati PP, Suknuntha K, Kumar A, Samarjeet F, Probasco MD, Tian S, Stewart R, Thomson JA, et al. Identification of the hemogenic endothelial progenitor and its direct precursor in human pluripotent stem cell differentiation cultures. Cell Rep. 2012;2:553–567. doi: 10.1016/j.celrep.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui K, Zang C, Roh TY, Schones DE, Childs RW, Peng W, Zhao K. Chromatin signatures in multipotent human hematopoietic stem cells indicate the fate of bivalent genes during differentiation. Cell Stem Cell. 2009;4:80–93. doi: 10.1016/j.stem.2008.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado-Olguín P, Huang Y, Li X, Christodoulou D, Seidman CE, Seidman JG, Tarakhovsky A, Bruneau BG. Epigenetic repression of cardiac progenitor gene expression by Ezh2 is required for postnatal cardiac homeostasis. Nat Genet. 2012;44:343–347. doi: 10.1038/ng.1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferdous A, Caprioli A, Iacovino M, Martin CM, Morris J, Richardson JA, Latif S, Hammer RE, Harvey RP, Olson EN, et al. Nkx2-5 transactivates the Ets-related protein 71 gene and specifies an endothelial/endocardial fate in the developing embryo. Proc Natl Acad Sci U S A. 2009;106:814–819. doi: 10.1073/pnas.0807583106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gering M, Rodaway AR, Göttgens B, Patient RK, Green AR. The SCL gene specifies haemangioblast development from early mesoderm. EMBO J. 1998;17:4029–4045. doi: 10.1093/emboj/17.14.4029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giroux S, Kaushik AL, Capron C, Jalil A, Kelaidi C, Sablitzky F, Dumenil D, Albagli O, Godin I. lyl-1 and tal-1/scl, two genes encoding closely related bHLH transcription factors, display highly overlapping expression patterns during cardiovascular and hematopoietic ontogeny. Gene Expr Patterns. 2007;7:215–226. doi: 10.1016/j.modgep.2006.10.004. [DOI] [PubMed] [Google Scholar]
- Herion NJ, Salbaum JM, Kappen C. Traffic jam in the primitive streak: The role of defective mesoderm migration in birth defects. Birth Defects Res A Clin Mol Teratol. 2014 doi: 10.1002/bdra.23283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain R, Barkauskas CE, Takeda N, Bowie EJ, Aghajanian H, Wang Q, Padmanabhan A, Manderfield LJ, Gupta M, Li D, et al. Plasticity of Hopx(+) type I alveolar cells to regenerate type II cells in the lung. Nat Commun. 2015a;6:6727. doi: 10.1038/ncomms7727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain R, Li D, Gupta M, Manderfield LJ, Ifkovits JL, Wang Q, Liu F, Liu Y, Poleshko A, Padmanabhan A, et al. HEART DEVELOPMENT. Integration of Bmp and Wnt signaling by Hopx specifies commitment of cardiomyoblasts. Science. 2015b;348:aaa6071. doi: 10.1126/science.aaa6071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Z, Ji H. TSCAN: Pseudo-time reconstruction and evaluation in single-cell RNA-seq analysis. Nucleic Acids Res. 2016;44:e117. doi: 10.1093/nar/gkw430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz Y, Wang ET, Airoldi EM, Burge CB. Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat Methods. 2010;7:1009–1015. doi: 10.1038/nmeth.1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy M, Awong G, Sturgeon CM, Ditadi A, LaMotte-Mohs R, Zúñiga-Pflücker JC, Keller G. T lymphocyte potential marks the emergence of definitive hematopoietic progenitors in human pluripotent stem cell differentiation cultures. Cell Rep. 2012;2:1722–1735. doi: 10.1016/j.celrep.2012.11.003. [DOI] [PubMed] [Google Scholar]
- Kennedy M, D’Souza SL, Lynch-Kattman M, Schwantz S, Keller G. Development of the hemangioblast defines the onset of hematopoiesis in human ES cell differentiation cultures. Blood. 2007;109:2679–2687. doi: 10.1182/blood-2006-09-047704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lickert H, Takeuchi JK, Von Both I, Walls JR, McAuliffe F, Adamson SL, Henkelman RM, Wrana JL, Rossant J, Bruneau BG. Baf60c is essential for function of BAF chromatin remodelling complexes in heart development. Nature. 2004;432:107–112. doi: 10.1038/nature03071. [DOI] [PubMed] [Google Scholar]
- Moignard V, Woodhouse S, Haghverdi L, Lilly AJ, Tanaka Y, Wilkinson AC, Buettner F, Macaulay IC, Jawaid W, Diamanti E, et al. Decoding the regulatory network of early blood development from single-cell gene expression measurements. Nat Biotechnol. 2015;33:269–276. doi: 10.1038/nbt.3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Org T, Duan D, Ferrari R, Montel-Hagen A, Van Handel B, Kerényi MA, Sasidharan R, Rubbi L, Fujiwara Y, Pellegrini M, et al. Scl binds to primed enhancers in mesoderm to regulate hematopoietic and cardiac fate divergence. EMBO J. 2015;34:759–777. doi: 10.15252/embj.201490542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paige SL, Osugi T, Afanasiev OK, Pabon L, Reinecke H, Murry CE. Endogenous Wnt/beta-catenin signaling is required for cardiac differentiation in human embryonic stem cells. PLoS One. 2010;5:e11134. doi: 10.1371/journal.pone.0011134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paige SL, Thomas S, Stoick-Cooper CL, Wang H, Maves L, Sandstrom R, Pabon L, Reinecke H, Pratt G, Keller G, et al. A temporal chromatin signature in human embryonic stem cells identifies regulators of cardiac development. Cell. 2012;151:221–232. doi: 10.1016/j.cell.2012.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palpant NJ, Pabon L, Friedman CE, Roberts M, Hadland B, Zaunbrecher RJ, Bernstein I, Zheng Y, Murry CE. Generating high-purity cardiac and endothelial derivatives from patterned mesoderm using human pluripotent stem cells. Nat Protoc. 2017;12:15–31. doi: 10.1038/nprot.2016.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palpant NJ, Pabon L, Rabinowitz JS, Hadland BK, Stoick-Cooper CL, Paige SL, Bernstein ID, Moon RT, Murry CE. Transmembrane protein 88: a Wnt regulatory protein that specifies cardiomyocyte development. Development. 2013;140:3799–3808. doi: 10.1242/dev.094789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palpant NJ, Pabon L, Roberts M, Hadland B, Jones D, Jones C, Moon RT, Ruzzo WL, Bernstein I, Zheng Y, et al. Inhibition of β-catenin signaling respecifies anterior-like endothelium into beating human cardiomyocytes. Development. 2015a doi: 10.1242/dev.117010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palpant NJ, Pabon L, Roberts M, Hadland B, Jones D, Jones C, Moon RT, Ruzzo WL, Bernstein I, Zheng Y, et al. Inhibition of β-catenin signaling respecifies anterior-like endothelium into beating human cardiomyocytes. Development. 2015b;142:3198–3209. doi: 10.1242/dev.117010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paluru P, Hudock KM, Cheng X, Mills JA, Ying L, Galvão AM, Lu L, Tiyaboonchai A, Sim X, Sullivan SK, et al. The negative impact of Wnt signaling on megakaryocyte and primitive erythroid progenitors derived from human embryonic stem cells. Stem Cell Res. 2014;12:441–451. doi: 10.1016/j.scr.2013.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterkin T, Gibson A, Patient R. Common genetic control of haemangioblast and cardiac development in zebrafish. Development. 2009;136:1465–1474. doi: 10.1242/dev.032748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Herguido C, Guiu J, D’Altri T, Inglés-Esteve J, Dzierzak E, Espinosa L, Bigas A. Hematopoietic stem cell development requires transient Wnt/β-catenin activity. J Exp Med. 2012;209:1457–1468. doi: 10.1084/jem.20120225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachs M, Onodera C, Blaschke K, Ebata KT, Song JS, Ramalho-Santos M. Bivalent chromatin marks developmental regulatory genes in the mouse embryonic germline in vivo. Cell Rep. 2013;3:1777–1784. doi: 10.1016/j.celrep.2013.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scialdone A, Tanaka Y, Jawaid W, Moignard V, Wilson NK, Macaulay IC, Marioni JC, Göttgens B. Resolving early mesoderm diversification through single-cell expression profiling. Nature. 2016;535:289–293. doi: 10.1038/nature18633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh G, Charlet-B N, Han J, Cooper TA. ETR-3 and CELF4 protein domains required for RNA binding and splicing activity in vivo. Nucleic Acids Res. 2004;32:1232–1241. doi: 10.1093/nar/gkh275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturgeon CM, Ditadi A, Awong G, Kennedy M, Keller G. Wnt signaling controls the specification of definitive and primitive hematopoiesis from human pluripotent stem cells. Nat Biotechnol. 2014;32:554–561. doi: 10.1038/nbt.2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda N, Jain R, Leboeuf MR, Padmanabhan A, Wang Q, Li L, Lu MM, Millar SE, Epstein JA. Hopx expression defines a subset of multipotent hair follicle stem cells and a progenitor population primed to give rise to K6+ niche cells. Development. 2013;140:1655–1664. doi: 10.1242/dev.093005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda N, Jain R, LeBoeuf MR, Wang Q, Lu MM, Epstein JA. Interconversion between intestinal stem cell populations in distinct niches. Science. 2011;334:1420–1424. doi: 10.1126/science.1213214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trivedi CM, Zhu W, Wang Q, Jia C, Kee HJ, Li L, Hannenhalli S, Epstein JA. Hopx and Hdac2 interact to modulate Gata4 acetylation and embryonic cardiac myocyte proliferation. Dev Cell. 2010;19:450–459. doi: 10.1016/j.devcel.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno S, Weidinger G, Osugi T, Kohn AD, Golob JL, Pabon L, Reinecke H, Moon RT, Murry CE. Biphasic role for Wnt/beta-catenin signaling in cardiac specification in zebrafish and embryonic stem cells. Proc Natl Acad Sci U S A. 2007;104:9685–9690. doi: 10.1073/pnas.0702859104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Maaten L, Hinton G. Visualizing data using t-SNE. Journal of Machine Learning Research. 2008;9:2579–2605. [Google Scholar]
- Van Handel B, Montel-Hagen A, Sasidharan R, Nakano H, Ferrari R, Boogerd CJ, Schredelseker J, Wang Y, Hunter S, Org T, et al. Scl represses cardiomyogenesis in prospective hemogenic endothelium and endocardium. Cell. 2012;150:590–605. doi: 10.1016/j.cell.2012.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vodyanik MA, Thomson JA, Slukvin II. Leukosialin (CD43) defines hematopoietic progenitors in human embryonic stem cell differentiation cultures. Blood. 2006;108:2095–2105. doi: 10.1182/blood-2006-02-003327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wamstad JA, Alexander JM, Truty RM, Shrikumar A, Li F, Eilertson KE, Ding H, Wylie JN, Pico AR, Capra JA, et al. Dynamic and coordinated epigenetic regulation of developmental transitions in the cardiac lineage. Cell. 2012;151:206–220. doi: 10.1016/j.cell.2012.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webber BR, Iacovino M, Choi SH, Tolar J, Kyba M, Blazar BR. DNA methylation of Runx1 regulatory regions correlates with transition from primitive to definitive hematopoietic potential in vitro and in vivo. Blood. 2013;122:2978–2986. doi: 10.1182/blood-2013-03-489369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Prak L, Rayon-Estrada V, Thiru P, Flygare J, Lim B, Lodish HF. ZFP36L2 is required for self-renewal of early burst-forming unit erythroid progenitors. Nature. 2013;499:92–96. doi: 10.1038/nature12215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Crow AL, Hartiala J, Spindler TJ, Ghazalpour A, Barsky LW, Bennett BB, Parks BW, Eskin E, Jain R, et al. The Genetic Landscape of Hematopoietic Stem Cell Frequency in Mice. Stem Cell Reports. 2015 doi: 10.1016/j.stemcr.2015.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
List of regulators for 3 mesodermal lineages based on expression alone listed in rank order. See separate supplemental file.
List of regulators for 3 mesodermal lineages based on chromatin alone listed in rank order. See separate supplemental file.
List of regulators for 3 mesodermal lineages based on integration of chromatin and expression listed in rank order. See separate supplemental file.
Gene Ontology “Biological Processes” and “Molecular Function” enrichments for regulator lists generated by expression only. See separate supplemental file.
Gene Ontology “Biological Processes” and “Molecular Function” enrichments for regulator lists generated by chromatin only. See separate supplemental file.
Gene Ontology “Biological Processes” and “Molecular Function” enrichments for regulator lists generated by integration of expression and chromatin. See separate supplemental file.
Lists of gene ontology enrichment of Molecular Functions and Biological Process comparing the P-value for shared GO terms for integrated approach vs. expression only. See separate supplemental file.
Lists of gene ontology enrichment of Molecular Functions and Biological Process comparing the P-value for shared GO terms for integrated approach vs. chromatin only. See separate supplemental file.
Quantitative PCR Primers to amplify human genes