

Abstract
Background
Kallistatin, a serine proteinase inhibitor, has vasodilatory and anti-inflammatory properties and is increased in other inflammatory conditions. We measured kallistatin in HIV for the first time, examined its relationship with inflammation, and determined if statin therapy affected levels.
Methods
Kallistatin levels were measured in subjects from a randomized, double-blinded, placebo-controlled trial.
Results
135 HIV-infected subjects were included. Kallistatin levels were 28.4 μg/mL at baseline and not affected by rosuvastatin. Levels were correlated with hsCRP, interleukin-6, fibrinogen, and insulin resistance.
Conclusions
Kallistatin levels were correlated with some markers of systemic inflammation and should be further explored in the HIV population.
Keywords: HIV, kallistatin, cardiovascular disease, inflammation, immune activation, coagulation, carotid intima-media thickness
INTRODUCTION
Patients with HIV are at increased risk of cardiovascular disease (CVD) compared with the general population (Triant, Lee, Hadigan and Grinspoon, 2007, Obel, Thomsen, Kronborg, Larsen, Hildebrandt, Sorensen and Gerstoft, 2007). The increased risk is due in part to the independent effects of HIV infection despite virologic suppression with antiretroviral therapy (ART) (Grunfeld, Delaney, Wanke, Currier, Scherzer, Biggs, Tien, Shlipak, Sidney, Polak, O’Leary, Bacchetti and Kronmal, 2009, Ross, Rizk, O’Riordan, Dogra, El-Bejjani, Storer, Harrill, Tungsiripat, Adell and McComsey, 2009). It has been hypothesized that increased inflammation, immune activation, and oxidative stress associated with HIV infection contribute to endothelial cell activation and dysfunction, thus leading to atherosclerosis and subsequent CVD (Kuller, Tracy, Belloso, De Wit, Drummond, Lane, Ledergerber, Lundgren, Neuhaus, Nixon, Paton, Neaton and Group, 2008, Triant, Meigs and Grinspoon, 2009, Kaplan, Sinclair, Landay, Lurain, Sharrett, Gange, Xue, Hunt, Karim, Kern, Hodis and Deeks, 2011, Longenecker, Funderburg, Jiang, Debanne, Storer, Labbato, Lederman and McComsey, 2013). In addition, lipid derangements and insulin resistance caused by HIV infection and ART further exacerbate the CVD risk (Riddler, Li, Chu, Kingsley, Dobs, Evans, Palella, Visscher, Chimel and Sharrett, 2007).
Kallistatin, first identified as an inhibitor of a human serine proteinase kallikrein, was subsequently shown to have vasodilatory effects independent of the kallikrein system (Chao, Stallone, Liang, Chen, Wang and Chao, 1997). Also, it was found to reduce inflammatory cytokines and protect tissues from oxidative stress in arthritis (Wang, Chen, Wu, Liu, Jin, Chao and Chao, 2005), heart disease (Chao, Yin, Yao, Shen, Smith and Chao, 2006, Liu, Bledsoe, Hagiwara, Shen, Chao and Chao, 2012), and renal injury (Shen, Hagiwara, Yao, Chao and Chao, 2008). It has been proposed that elevated kallistatin levels may be a result of a compensatory mechanism to mitigate endothelial dysfunction and hypertension (Jenkins, McBride, Januszewski, Karschimkus, Zhang, O’Neal, Nelson, Chung, Harper, Lyons and Ma, 2010).
Studies suggest that kallistatin exerts its effects by two mechanisms. First, kallistatin directly inhibits binding of tumor necrosis factor alpha (TNF-α) to TNF-α receptor (TNFR) on endothelial cells, therefore blocking downstream cascade reactions that lead to nuclear factor κB activation and inflammatory gene expression (Yin, Gao, Shen, Chao and Chao, 2010). Also, kallistatin binds to Kruppel-like factor 4 on endothelial cells and induces nitric oxide synthase (eNOS) expression, thereby increasing nitric oxide levels (Shen, Smith, Hsu, Chao and Chao, 2009, Shen, Gao, Hsu, Bledsoe, Hagiwara, Chao and Chao, 2010). Because of kallistatin’s effects on inflammation and oxidative stress, it is thought to be cardioprotective (Chao, Stallone, Liang, Chen, Wang and Chao, 1997, Liu, Bledsoe, Hagiwara, Shen, Chao and Chao, 2012, Shen, Hagiwara, Yao, Chao and Chao, 2008, Yin, Gao, Shen, Chao and Chao, 2010).
Despite the increased inflammation, oxidative stress, and CVD risk among HIV-infected patients, no studies have investigated kallistatin levels in this population. However, there has been a particular interest in the HIV population in identifying novel circulating biomarkers that could help stratify CVD risk. A study by Jenkins, et al lends support to the idea of utilizing kallistatin as a CVD biomarker. In their cross-sectional study, the authors showed a significant difference in kallistatin levels among Type I diabetic patients with microvascular complications, Type I diabetic patients without microvascular complications, and non-diabetic controls (Jenkins, McBride, Januszewski, Karschimkus, Zhang, O’Neal, Nelson, Chung, Harper, Lyons and Ma, 2010). Thus, exploring kallistatin levels and their associations with inflammation and surrogate markers of CVD could show important relationships, potentially identifying a unique biomarker that could further delineate CVD risk in this population.
Interestingly, kallistatin’s effect on the upregulation of endothelial nitric oxide synthase is one of the same sites of action as 3-hydroxy-3-methylglutarylcoenzyme A reductase inhibitors (statins) (Laufs, La Fata, Plutzky and Liao, 1998, Martinez-Gonzalez, Raposo, Rodriguez and Badimon, 2001, Kureishi, Luo, Shiojima, Bialik, Fulton, Lefer, Sessa and Walsh, 2000), which are well-known to attenuate CVD risk by both their lipid-lowering and anti-inflammatory properties and may have an increasingly important role in the HIV population (Erlandson, Jiang, Debanne and McComsey, 2015, Eckard, Jiang, Debanne, Funderburg and McComsey, 2014). In fact, two recent randomized, controlled trials recently showed that statin therapy produced dramatic reductions in non-calcified plaque volume measured by coronary CT angiography, reductions in the number of plaques with “vulnerable” or high-risk features (Lo, Lu, Ihenachor, Wei, Looby, Fitch, Oh, Zimmerman, Hwang, Abbara, Plutzky, Robbins, Tawakol, Hoffmann and Grinspoon) and halted the progression of carotid intima-media thickness (IMT), a surrogate marker of subclinical atherosclerosis (Longenecker, Jiang, Debanne, Labbato, Kinley, Storer and McComsey, 2015). If kallistatin levels can help delineate CVD risk in HIV and/or are affected by statins, changes in kallistatin levels may offer another way to monitor these patients and/or measure the effects of interventions like statin therapy.
Only one study to date has evaluated the effects of statin therapy on kallistatin levels in the general population (Campbell, Kladis, Zhang, Jenkins, Prior, Yii, Kenny, Black and Kelly, 2010). The authors reported no association between statin use and kallistatin levels in patients with and without Type 2 diabetes. However, this study was not randomized, included a very small number of patients who were undergoing coronary artery bypass graft surgery and were on different doses of various statins.
Thus, the main objectives of this study were to, for the first time, (1) measure kallistatin levels in a group of HIV-infected patients on ART, (2) examine the relationship between kallistatin levels and markers of inflammation and CVD risk in this population, and (3) determine the effect of statin use on kallistatin levels in the context of a placebo-controlled, randomized trial.
METHODS
The Stopping Atherosclerosis and Treating Unhealthy bone with RosuvastatiN in HIV (SATURN-HIV) is an on-going randomized, double-blinded, placebo-controlled trial designed to measure the effect of rosuvastatin on the progression of subclinical CVD in subjects with heightened inflammation or T-cell activation but normal low-density lipoprotein (LDL) cholesterol. Here, we present a post hoc analysis that assessed plasma kallistatin levels in SATURN-HIV participants at baseline and after 24 weeks of statin therapy or placebo.
Complete details of the SATURN-HIV trial can be found elsewhere (Eckard, Jiang, Debanne, Funderburg and McComsey, 2014). Briefly, enrollment occurred between March 2011–August 2012 and included HIV-infected adults ≥18 years old with fasting LDL-cholesterol ≤130 mg/dL and high-sensitivity C-reactive protein (hsCRP) ≥2 mg/L or CD8+ T-cell expression of CD38 and human leukocyte antigen (HLA)-DR antigens ≥19% at screening which occurred ≤30 days prior to enrollment. Additional inclusion criteria included cumulative ART duration ≥6 months, stable ART regimen for ≥12 weeks prior to entry, no plans to change current ART, diet, or exercise regimen, HIV-1 RNA <1,000 copies/mL, fasting triglycerides ≤500 mg/dL, Kamofsky performance score ≥80, and 1 CVD risk factor (i.e. male sex, smoking, hypertension, family history). Main exclusion criteria included a clinically-important illness or laboratory abnormality within 14 days prior to study entry that might place the subject at risk by being exposed to statins or which might confound the interpretation of this investigation, pregnancy/lactation, known CVD, uncontrolled diabetes (hemoglobin A1C ≥8.5%), active or chronic uncontrolled inflammatory condition or opportunistic infection, known underlying myositis/muscle disease, change in lipid or cholesterol modification pharmacotherapy or statin use in the 6 months prior to entry. The study enrolled at the University Hospitals Case Medical Center, Cleveland, OH and was reviewed and approved by the local institutional review board. Written informed consent was provided by all participants. The study was registered on clinicaltrials.gov (NCT01218802).
The intervention consisted of rosuvastatin 10 mg daily or matching placebo. Randomization was done by an investigational pharmacist and stratified by protease inhibitor (PI) use at entry. Participants underwent extensive cardiovascular assessment at pre-determined time points to evaluate for evidence of subclinical vascular disease by measuring carotid IMT, coronary artery calcium (CAC) score and flow-mediated vasodilation (FMD) of the brachial artery (baseline values presented in this current analysis). Plasma levels of inflammation (hsCRP, interleukin-6 (IL-6), soluble tumor necrosis factor receptor-I (sTNFR-I), soluble tumor necrosis factor receptor-II (sTNFR-II)), endothelial adhesion molecules (soluble vascular cell adhesion molecule-1 (sVCAM-1), soluble intracellular adhesion molecule-1 (sICAM-1)), and coagulation markers (D-dimer, fibrinogen) that are associated with CVD in HIV and/or in the general population and elevated in HIV were also measured (baseline and 24-week time point values presented in this current analysis). At each time point, a number of clinical and metabolic variables were also obtained, including fasting lipids, glucose and insulin. Physical examination and weight and height measurements were obtained in all subjects from the same scale, and blood pressure was obtained with an automated, calibrated blood pressure machine. An extensive review of the medical records was conducted and subject-reported data were collected, including demographics, past and current medical diagnoses, concomitant medications, and detailed ART history. HIV-1 RNA level and CD4+ cell count were collected as part of routine clinical care.
SATURN-HIV participants were eligible for this current study if they had completed 24 weeks of intervention at the time of the analysis and had available stored plasma from both baseline and 24 weeks. Plasma was isolated at the time of the initial blood collection (which occurred after a 12-hour fast) and frozen at −70°C without prior thawing. Laboratory personnel were blinded to clinical information. Paired kallistatin levels (from the baseline and 24-week time points) were measured together in duplicate with a specific enzyme-linked immunosorbent (ELISA) assay (R&D Systems, Inc., Minneapolis, MN) as per the manufacturer’s product manual. The calculated means of the measurements were used in the analysis. Inter- and intra-coefficients of variance (CV) were 3.6% and <8.1%, respectively.
Kallistatin levels were first analyzed with all subjects combined at the baseline time point. Continuous measures are described by medians and interquartile ranges (IQR), and nominal variables are described with frequencies and percentages. Differences in baseline kallistatin levels were also determined between several relevant sub-groups (e.g. male vs. female sex) using appropriate two-sample tests.
Between-group and within-group changes in kallistatin levels over 24 weeks in the statin and placebo groups were then considered. Here, variables are described by study group. Nominal variables were compared using χ2 analysis or Fisher’s exact test. For between-group comparisons (baseline and changes from baseline to 24 weeks), kallistatin levels were compared using Wilcoxon rank sum test. For within-group changes from baseline to 24 weeks, kallistatin levels were compared with Wilcoxon signed rank test.
Correlations between kallistatin levels and variables of interest were assessed using Spearman correlation coefficients for continuous variables. Correlations were first performed for variables at baseline for the HIV-infected group combined. Next, correlations were performed between changes in kallistatin levels and changes in other variables over the 24-week time point for the HIV-infected group combined.
The evaluation of kallistatin levels in this study was considered exploratory; no power calculations were determined. All statistical tests were two-sided with a 0.05 nominal significance level. Analyses were performed with SAS version 9.2 (SAS Institute, Cary, NC).
RESULTS
Among the entire study population (N = 147), a total of 135 (92%) HIV-infected subjects were included in this current analysis. Twenty-four, 64, and 47 subjects qualified for the study with hsCRP ≥2 μg/mL, CD8+CD38+HLA-DR+ cells ≥19%, or both, respectively. Table 1 shows demographic and baseline subject characteristics. Overall, the median age was 47 years with 81% male and 67% African American. The median CD4+ cell count was 578 cells/μL with a CD4+ cell count nadir of 177 cells/μL. The median duration of HIV infection was 136 months, with 49% on a PI-containing regimen.
Table 1.
Baseline Characteristics of Subjects by Study Group
| Median (interquartile range) or no. (%) | All Subjects N = 135 |
Statin Group N = 67 |
Placebo Group N = 68 |
|---|---|---|---|
| Age (years) | 47 (39, 53) | 47 (41, 52) | 47 (37, 53) |
| Male sex | 109 (81%) | 55 (82%) | 54 (79%) |
| African American race | 91 (67%) | 46 (69%) | 45 (66%) |
| BMI (kg/m2) | 26.7 (23.3, 30.2) | 26.6 (23.3, 30.7) | 27.0 (23.2, 30.2) |
| Systolic BP (mmHg) | 121 (112, 132) | 121 (111, 132) | 121 (112, 132) |
| Diastolic BP (mmHg) | 80 (72, 84) | 79 (72, 84) | 80 (72, 84) |
| LDL cholesterol (mg/dL) | 96 (77, 113) | 95 (75, 107) | 97 (78, 121) |
| HDL cholesterol (mg/dL) | 45 (37, 57) | 45 (37, 58) | 46 (37, 57) |
| Triglycerides (mg/dL) | 115 (84, 189) | 103 (77, 199) | 122 (87, 187) |
| HOMA-IR | 1.8 (1.1, 3.3) | 1.7 (1.0, 2.8) | 1.9 (1.2, 4.8) |
| Current smoking | 90 (67%) | 40 (60%) | 50 (74%) |
| HIV duration (months) | 136 (70, 213) | 129 (70, 196) | 142 (72, 233) |
| HIV-1 RNA ≤50 copies/mL | 109 (81%) | 54 (81%) | 55 (81%) |
| HIV-1 RNA if >50 copies/mL | 99 (75, 175) | 82 (71, 123) | 131 (81, 178) |
| Current CD4 count (cells/μL) | 578 (432, 836) | 605 (435, 850) | 569 (422, 828) |
| Nadir CD4 count (cells/μL) | 177 (89, 293) | 170 (86, 312) | 190 (90, 279) |
| Cumulative NRTI duration (months) | 62 (38, 115) | 59 (30, 115) | 62 (41, 112) |
| Cumulative PI duration (months) | 39 (7, 89) | 39 (13, 106) | 39 (2, 80) |
| Cumulative NNRTI duration (months) | 24 (6, 54) | 19 (2, 56) | 30 (10, 54) |
| Kallistatin (μg/mL) | 28.4 (19.2, 40.0) | 30.5 (18.4, 39.5) | 26.5 (20.0, 41.1) |
| CD8+CD38+HLA-DR+ (%)* | 12.3 (8.8, 17.4) | 13.4 (9.0, 19.5) | 11.5 (8.5, 16.4) |
| hsCRP (μg/mL) | 1.71 (0.77, 5.02) | 1.66 (0.77, 4.91) | 2.01 (0.72, 5.12) |
| IL-6 (pg/mL) | 2.79 (1.96, 4.74) | 2.89 (1.89, 4.63) | 2.65 (1.97, 5.32) |
| sTNFR-I (pg/mL) | 1610 (1278, 2396) | 1623 (1341, 2200) | 1594 (1236, 2467) |
| sTNFR-II (pg/mL) | 2332 (1625, 2883) | 2474 (1718, 3054) | 2158 (1605, 2629) |
| sVCAM-1 (ng/mL) | 670 (563, 819) | 694 (574, 825) | 643 (544, 750) |
| sICAM-1 (ng/mL) | 237 (185, 308) | 225 (185, 317) | 257 (180, 305) |
| D-dimer (μg/mL) | 0.19 (0.11, 0.32) | 0.20 (0.13, 0.34) | 0.16 (0.09, 0.29) |
| Fibrinogen (mg/dL) | 385 (335, 503) | 388 (339, 513) | 383 (331, 470) |
| CCA IMT (mm) | 0.66 (0.62, 0.76) | 0.66 (0.62, 0.78) | 0.67 (0.60, 0.75) |
| FMD % | 3.9 (2.0, 6.0) | 4.0 (2.0, 6.3) | 3.7 (1.7, 5.3) |
| CAC score >0 | 51 (38%) | 23 (34%) | 28 (41%) |
N.B. There were no significant differences between the statin and placebo groups for any of the variables listed in this table.
% of CD8+ cells expressing CD38 and HLA-DR antigens
BMI, body mass index; HOMA-IR, homeostatic model assessment of insulin resistance; BP, blood pressure; LDL, low-density lipoprotein; HDL, high-density lipoprotein; NRTI, nucleoside/nucleotide analogue reverse transcriptase inhibitor; PI, protease inhibitor; NNRTI, non-nucleoside/nucleotide analogue reverse transcriptase inhibitor; hsCRP, high-sensitivity C-reactive protein; IL-6, interleukin-6; sTNFR-I, soluble tumor necrosis factor receptor-I; sTNFR-II, soluble tumor necrosis factor receptor-II; sVCAM-1, soluble vascular cell adhesion molecule-1; sICAM-1, soluble intracellular adhesion molecule-1; CCA IMT, common carotid artery intima-media thickness; FMD, flow-mediated vasodilation; CAC, coronary artery calcium
The baseline median (IQR) kallistatin level for all the HIV-infected subjects was 28.4 (19.2, 40.0) μg/mL (Table 1). There were no significant differences between kallistatin levels and those subjects with a CAC score of 0 vs. >0, or between those with a detectable vs. undetectable HIV-1 RNA level, male vs. female sex, black vs. non-black race, or current smokers vs. non-smokers (Table 2).
Table 2.
Relationships between Kallistatin Levels and Variables of Interest for All HIV-infected Subjects.
| A. Correlations between Kallistatin Levels and Variables of Interest | ||
|---|---|---|
|
| ||
| Variable | Baseline | Changes over 24 Weeks |
|
| ||
| Markers of Inflammation and Coagulation | ||
|
| ||
| hsCRP | R = −0.29; P = 0.0005 | R = −0.15; P = 0.08 |
| IL-6 | R = −0.23; P = 0.007 | R = −0.19; P = 0.03 |
| sTNFR-I | R = −0.12; P = 0.18 | R = −0.09; P = 0.31 |
| sTNFR-II | R = −0.77; P = 0.38 | R = −0.06; P = 0.49 |
| sVCAM-1 | R = 0.03; P = 0.77 | R = −0.86; P = 0.32 |
| sICAM-1 | R = −0.004; P = 0.96 | R = 0.04; P = 0.65 |
| D-dimer | R = −0.8; P = 0.36 | R = 0.05; P = 0.57 |
| Fibrinogen | R = −0.23; P = 0.007 | R = −0.05; P = 0.54 |
|
| ||
| Markers of Subclinical Vascular Disease | ||
|
| ||
| CCA IMT | R = −0.09; P = 0.31 | N/A |
| FMD | R = 0.09; P = 0.30 | N/A |
|
| ||
| Demographics, Clinical, and Metabolic Parameters | ||
|
| ||
| Age | R = −0.12; P = 0.18 | N/A |
| Body mass index | R = 0.04; P = 0.65 | R = 0.20; P = 0.82 |
| Systolic BP | R = 0.08; P = 0.34 | R = 0.07; P = 0.43 |
| Diastolic BP | R = 0.07; P = 0.39 | R = −0.04; P = 0.64 |
| LDL cholesterol | R = −0.03; P = 0.72 | R = 0.1; P = 0.90 |
| HDL cholesterol | R = −0.06; P = 0.47 | R = −0.90; P = 0.31 |
| Triglycerides | R = 0.13; P = 0.14 | R = 0.18; P = 0.04 |
| HOMA-IR | R = 0.18; P = 0.04 | R = 0.09; P = 0.28 |
|
| ||
| HIV Variables | ||
|
| ||
| CD4+ cell count | R = −0.12; P = 0.16 | N/A |
| Nadir CD4+ cell count | R = −0.01; P = 0.91 | N/A |
| HIV-1 RNA level* | R = 0.07; P = 0.72 | N/A |
| HIV duration | R = −0.03; P = 0.77 | N/A |
| NRTI duration | R = −0.02; P = 0.08 | N/A |
| PI duration | R = 0.08; P = 0.41 | N/A |
| NNRTI duration | R = 0.004; P = 0.97 | N/A |
|
| ||
| B. Differences in Kallistatin Levels in Sub-groups Based on Dichotomous Variables | ||
|
| ||
| Variable | Kallistatin Level, μg/mL | P |
|
| ||
| CAC = 0 | 28.6 (18.7, 42.1) | 0.84 |
|
| ||
| CAC >0 | 28.4 (19.7, 38.0) | |
|
| ||
| Detectable HIV-1 RNA | 28.5 (20.1, 40.4) | 0.58 |
|
| ||
| Undetectable HIV-1 RNA | 28.3 (16.7, 34.0) | |
|
| ||
| Male sex | 28.3 (19.7, 37.5) | 0.31 |
|
| ||
| Female sex | 32.5 (17.0, 53.4) | |
|
| ||
| Black race | 27. (17.9, 39.5) | 0.42 |
|
| ||
| Non-black race | 30.5 (22.7, 40.0) | |
|
| ||
| Current smoker | 26.7 (19.5, 38.8) | 0.72 |
|
| ||
| Current non-smoker | 29.7 (19.2, 41.2) | |
hsCRP, high-sensitivity C-reactive protein; IL-6, interleukin-6; sTNFR-I, soluble tumor necrosis factor receptor-I; sTNFR-II, soluble tumor necrosis factor receptor-II; sVCAM-1, soluble vascular cell adhesion molecule-1; sICAM-1, soluble intracellular adhesion molecule-1; CCA IMT, common carotid artery intima-media thickness; FMD, flow-mediated vasodilation; BP, blood pressure; LDL, low-density lipoprotein; HDL, high-density lipoprotein; HOMA-IR, homeostatic model assessment of insulin resistance; NRTI, nucleoside/nucleotide analogue reverse transcriptase inhibitor; PI, protease inhibitor; NNRTI, non-nucleoside/nucleotide analogue reverse transcriptase inhibitor; CAC, coronary artery calcium
Baseline kallistatin levels were negatively correlated with hsCRP (R = −0.29; P = 0.0005), IL-6 (R = −0.23; P = 0.007) and fibrinogen (R = −0.23; P = 0.007), and positively correlated with homeostasis model assessment of insulin resistance (HOMA-IR) (R = 0.20; P = 0.04) (Table 2 and Figure 1). High-sensitivity C-reactive protein and IL-6 values had one and two extreme outliers, respectively. Thus, these correlations were reconsidered with the outliers removed to ensure that these values were not driving the correlations. When removed, the P values remained significant for both correlations (hsCRP: R = −0.28; P = 0.0009 and IL-6: R = −0.21; P = 0.01). The hsCRP outlier and the highest IL-6 outlier were from the same subject.
Figure 1.
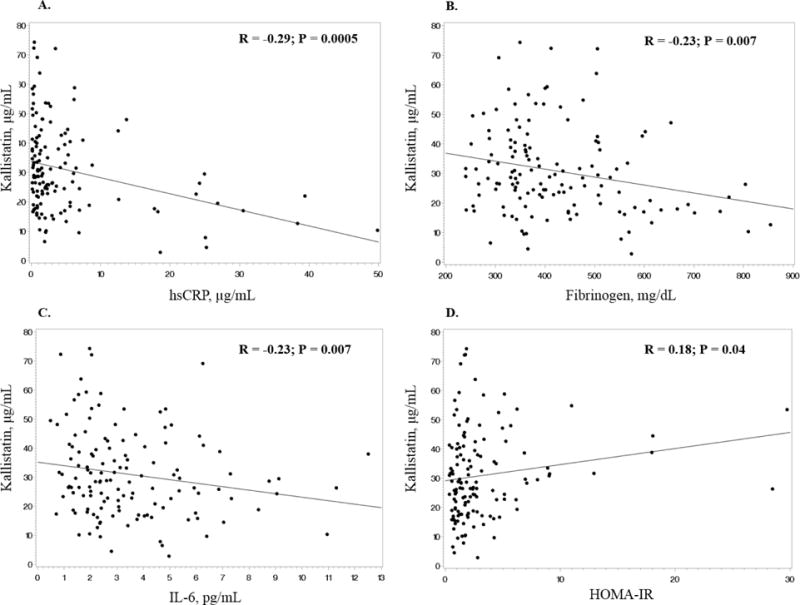
There were no significant correlations between kallistatin levels and the other inflammation markers (sTNFR-I, sTNFR-II), the endothelial adhesion molecules (sVCAM-1, sICAM-1), and the coagulation marker, D-dimer. There were also no significant correlations between kallistatin levels and carotid IMT or FMD, nor any significant correlations between kallistatin and age, body mass index, blood pressure, LDL cholesterol, high-density lipoprotein cholesterol, and triglycerides (Table 2).
Sixty-seven and 68 subjects were randomized to rosuvastatin and placebo, respectively. The statin and placebo groups were well-matched at baseline (Table 1). Median (IQR) kallistatin levels at baseline were 30.5 (18.4, 39.5) μg/mL in the rosuvastatin arm and 26.5 (20.0, 41.1) μg/mL in the placebo arm (P = 1.00). Two subjects changed ART regimens between baseline and 24 weeks: one was started on abacavir instead of didanosine, and one stopped lamivudine/zidovudine and started emtricitabine/tenofovir and maraviroc. One subject stopped ART and had an HIV-1 RNA level of >12,000 copies/mL at 24 weeks, but there was no statistically significant difference in the number of subjects with an undetectable HIV-1 RNA level at baseline vs. 24 weeks in either group.
There was no significant difference between groups in kallistatin changes over the 24-week study period (rosuvastatin group: −0.9 (−12.8, 16.4) μg/mL; placebo group: 1.4 (−9.9, 13.2) μg/mL; P = 0.56). Similarly, at the 24-week time point, kallistatin levels remained unchanged in both groups (rosuvastatin group: 28.6 (23.1, 38.5) μg/mL; placebo group: 28.6 (20.5, 40.9) μg/mL; P = 0.93). As previously reported, there were also no significant differences in changes in inflammation and coagulation markers between subjects on rosuvastatin therapy vs. placebo (Eckard, Jiang, Debanne, Funderburg and McComsey, 2014).
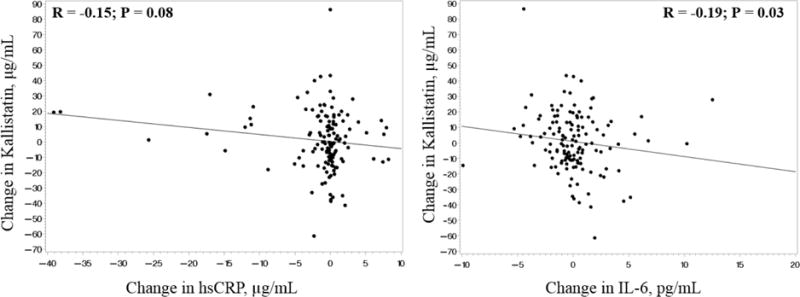
Correlations between changes in kallistatin levels and changes in variables of interest are presented in Table 2. Changes in IL-6 and changes in kallistatin levels were significantly correlated for the HIV-infected group combined, and there was a trend towards significance between changes in hsCRP and changes in kallistatin (Figure 2). The same subject who was the outlier for baseline hsCRP was also the outlier for changes in hsCRP. When the analysis was re-considered this time without the outlier, the P value increased from 0.08 to 0.12 with little change in the correlation coefficient. The only other variable where changes were significantly correlated with changes in kallistatin was triglycerides.
Figure 2.
DISCUSSION
In this study, and for the first time in HIV, we measured the level of the anti-inflammatory marker, kallistatin, in an HIV-infected population on ART with evidence of heightened inflammation and/or immune activation. The median plasma kallistatin level was 28.4 μg/mL for the HIV-infected group combined at baseline, which is comparable to the findings of a previous study in 318 healthy HIV-uninfected African American adolescents (Zhu, Chao, Kotak, Guo, Parikh, Bhagatwala, Dong, Patel, Houk, Chao and Dong, 2013), which reported a mean (standard deviation (SD)) kallistatin level of 27.9 (11.2) μg/mL in males and 26.8 (11.0) μg/mL in females (P = 0.47). Chao, et al also reported a similar mean (SD) of 22.1 (3.5) μg/mL in 30 normal subjects (Chao, Schmaier, Chen, Yang and Chao, 1996).
Interestingly, Chao, et al also found significantly reduced mean kallistatin (SD) levels in subjects with liver disease (7.2 (2.5) μg/mL), sepsis (7.7 (3.5) μg/mL), in women taking oral contraceptives (19.8 (3.8) μg/mL), and during pregnancy (14.9 (3.3) μg/mL) (Chao, Schmaier, Chen, Yang and Chao, 1996). Likewise, in a study of 54 patients with severe community-acquired pneumonia (CAP) who required intensive care unit (ICU) admission in a tertiary center in Taiwan, the median kallistatin level was 8.3 μg/mL on the day 1 of ICU admission and 11.0 μg/mL on the day 4 (Lin, Lu, Lin, Chen, Chao, Chao and Lin, 2013). These studies suggest that acute illness and/or immune compromise may decrease kallistatin levels. One could argue that these subjects are more analogous to our current HIV-infected study group than healthy controls. However, this latter study was performed in a population that was much older (mean age of 77 years), in a setting of severe acute illness, and in a different ethnic group compared to our study population. The authors did include a control group that consisted of 17 healthy individuals who had a higher median (range) than the subjects admitted to the ICU with CAP (17.2 (5.3 – 82.7) μg/mL), but they did not report any demographics or comparison to the study group. Likewise, our HIV-infected subjects were all on ART with well-controlled HIV, perhaps making them more like a healthy uninfected population. On the other hand, given the increased inflammation in this population (which would increase kallistatin levels) coupled with an immunocompromised state (which would decrease kallistatin levels), the sum effect may be “normal” kallistatin levels. Further studies are needed to investigate these potential situations by including additional groups, such as HIV-infected patients not on ART +/− low CD4 counts and patients with clinically-apparent co-morbidities, especially CVD, kidney disease, liver disease, and hypertension.
In this current study, we did demonstrate a significant negative correlation between kallistatin and the inflammatory cytokines, hsCRP and IL-6, which is consistent with findings in other studies (Zhu, Chao, Kotak, Guo, Parikh, Bhagatwala, Dong, Patel, Houk, Chao and Dong, 2013), although data are conflicting (Jenkins, McBride, Januszewski, Karschimkus, Zhang, O’Neal, Nelson, Chung, Harper, Lyons and Ma, 2010). While there was one hsCRP outlier and two IL-6 outliers at baseline, correlations did not change substantially when they were removed. There was also a significant correlation between changes in kallistatin levels and changes in IL-6 over 24 weeks (with no outliers present), indicating that this relationship is likely real. However, further studies should be conducted to verify this relationship as neither variable changed significantly over the 24-week time period.
It is unclear why there was not a significant relationship with either sTNFR-I or sTNFR-II, or if there is a clinical implication to the statistically significant relationship between kallistatin levels and either HOMA-IR or fibrinogen. These findings may suggest that the relationship between kallistatin and proinflammatory cytokines is more complex or altered in the setting of HIV. There was also no significant relationship between kallistatin and markers of subclinical vascular disease, and in particular, FMD, which is a measure of endothelial function. One previous study outside of HIV investigated kallistatin levels and carotid IMT in 60 children and adolescents with Type 1 diabetes (El-Asrar, Andrawes, Ismail and Salem, 2015). Here, the authors showed a significant correlation between kallistatin levels and carotid IMT (left and right IMT, R=0.776 and R=0.881, respectively; both P<0.001). However, 30 of the diabetic subjects had evidence of microvascular complications, and these subjects had higher levels of kallistatin than those without microvascular complications. Thus, the lack of significant correlations between measures of endothelial dysfunction and subclinical atherosclerosis may be due to our chosen HIV population, none of whom had clinical evidence of vascular disease.
Interestingly, in the aforementioned study by El-Asrar, et al, the authors were able to differentiate patients with and without microvascular complications with a kallistatin cut-off value of 6.1 ng/mL with a sensitivity of 96.87% and a specificity of 93.75%. It is difficult to compare levels directly to our study, as the authors used a different method with a much different assay range than ours and other studies. However, this study suggests that kallistatin can be used as a biomarker for detection of microvascular complications in diabetic patients. Likewise, in Jenkins, et al (Jenkins, McBride, Januszewski, Karschimkus, Zhang, O’Neal, Nelson, Chung, Harper, Lyons and Ma, 2010), the authors studied 116 Type 1 diabetic patients (50 with microvascular complications and 66 without microvascular complications) and 29 non-diabetic controls. The mean kallistatin level was 13.4 μg/mL in diabetics with complications, 12.1 μg/mL in diabetics with no complications, and 10.3 μg/mL in controls (ANOVA, P = 0.007). Thus, we believe these studies lend support to the idea of using kallistatin as a biomarker in the HIV-infected population to perhaps differentiate patients with and without micro- and macro-angiopathy. As discussed above, however, further studies are needed with various HIV sub-populations, including those with evidence of vascular disease.
In our current study, we also investigated whether statins affected kallistatin levels. Statins appear to be cardioprotective independent of their effects on LDL cholesterol. One of its non-lipid mechanisms is upregulating eNOS, thereby increasing endothelial nitric oxide levels (Laufs, La Fata, Plutzky and Liao, 1998, Martinez-Gonzalez, Raposo, Rodriguez and Badimon, 2001). Given the overlap in the site of action between statins and kallistatin, it is reasonable to consider the effect of rosuvastatin on plasma kallistatin levels in the HIV-infected population. In our study, however, kallistatin levels were not affected by statin therapy, a finding that mirrors that of the only other available study in the general population (Campbell, Kladis, Zhang, Jenkins, Prior, Yii, Kenny, Black and Kelly, 2010). Given the randomized, placebo-controlled design of this current study, our results provide greater evidence that statins given at normal therapeutic doses do not appear to directly affect plasma kallistatin levels. On the other hand, it is possible that we did not see a change in kallistatin levels because there were no significant changes in measured inflammatory markers in subjects on rosuvastatin therapy compared to those on placebo after 24 weeks. If the primary mechanism by which kallistatin levels change is by changes in inflammation, this could account, at least in part, for the lack of findings. However, statins have a number of pleotropic effects that that may have affected kallistatin levels, independent of changes in inflammatory cytokines.
There are limitations to this study. First, there was no healthy, HIV-negative control group to directly compare plasma kallistatin levels to that of the HIV-infected subjects. This study also investigated a specific HIV population: those on stable ART with low or undetectable HIV-1 RNA and normal LDL cholesterol. Thus, the generalizability to the HIV population as a whole should be determined in other studies. There are also statistical limitations to an exploratory post-hoc analysis, including potential sample size issues, which limits the overall interpretation of the data. This study investigated a number of associations between kallistatin and variables of interest, increasing the risk of Type I errors. Nevertheless, this study offers novel data, which can form the substrate for future research. Given its anti-inflammatory properties, kallistatin should be further explored in the HIV population.
CONCLUSIONS
Kallistatin, a unique serine proteinase inhibitor, with vasodilatory and anti-inflammatory properties, was examined for the first time in HIV-infected patients. The median plasma level in HIV subjects who were virologically-suppressed on ART was comparable to studies in uninfected populations. Levels were negatively correlated with IL-6 and hsCRP, markers of systemic inflammation, but were not correlated with measures of subclinical CVD and were unaffected by 24 weeks of statin therapy. Given our findings with kallistatin and inflammatory cytokines and other studies showing kallistatin as a potential biomarker to detect microvascular complications in diabetics, kallistatin should be further explored in HIV-infected individuals. Studies particularly investigating patients with more co-morbidities would be useful to evaluate kallistatin’s utility as a biomarker in this unique population.
Acknowledgments
This work was supported by the Emory+Children’s Pediatric Research Center Biomarkers Core, Case Western Reserve University Center for AIDS Research (P30 AI36219), National Institutes of Health [R01 NR012642 and R01 HD070490 to GAM; K23 HD069199 to ARE]. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. This study is registered at clinicaltrials.gov (NCT01218802).
Footnotes
Declaration of Interests: ARE has received research funding from Bristol-Myers Squibb, Cubist Pharmaceuticals, and GlaxoSmithKline and has served as an advisor for Gilead. GAM serves as a consultant, speaker, and has received research funding from Bristol-Myers Squibb, ViiV/GlaxoSmithKline, Gilead, ICON, and Merck. All other authors declare no conflicts of interest.
References
- Triant VA, Lee H, Hadigan C, Grinspoon SK. Increased acute myocardial infarction rates and cardiovascular risk factors among patients with human immunodeficiency virus disease. The Journal of clinical endocrinology and metabolism. 2007;92:2506–12. doi: 10.1210/jc.2006-2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obel N, Thomsen H, Kronborg G, Larsen C, Hildebrandt P, Sorensen H, Gerstoft J. Ischemic heart disease in HIV-infected and HIV-uninfected individuals: a population-based cohort study. Clinical Infectious Diseases. 2007;44:1625–31. doi: 10.1086/518285. [DOI] [PubMed] [Google Scholar]
- Grunfeld C, Delaney JA, Wanke C, Currier JS, Scherzer R, Biggs ML, Tien PC, Shlipak MG, Sidney S, Polak JF, O’Leary D, Bacchetti P, Kronmal RA. Preclinical atherosclerosis due to HIV infection: carotid intima-medial thickness measurements from the FRAM study. Aids. 2009;23:1841–9. doi: 10.1097/QAD.0b013e32832d3b85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross AC, Rizk N, O’Riordan MA, Dogra V, El-Bejjani D, Storer N, Harrill D, Tungsiripat M, Adell J, McComsey GA. Relationship between inflammatory markers, endothelial activation markers, and carotid intima-media thickness in HIV-infected patients receiving antiretroviral therapy. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America. 2009;49:1119–27. doi: 10.1086/605578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuller LH, Tracy R, Belloso W, De Wit S, Drummond F, Lane HC, Ledergerber B, Lundgren J, Neuhaus J, Nixon D, Paton NI, Neaton JD, Group ISS Inflammatory and coagulation biomarkers and mortality in patients with HIV infection. PLoS medicine. 2008;5:e203. doi: 10.1371/journal.pmed.0050203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triant VA, Meigs JB, Grinspoon SK. Association of C-reactive protein and HIV infection with acute myocardial infarction. Journal of acquired immune deficiency syndromes. 2009;51:268–73. doi: 10.1097/QAI.0b013e3181a9992c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan RC, Sinclair E, Landay AL, Lurain N, Sharrett AR, Gange SJ, Xue X, Hunt P, Karim R, Kern DM, Hodis HN, Deeks SG. T cell activation and senescence predict subclinical carotid artery disease in HIV-infected women. J Infect Dis. 2011;203:452–63. doi: 10.1093/infdis/jiq071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longenecker C, Funderburg N, Jiang Y, Debanne S, Storer N, Labbato D, Lederman M, McComsey G. Markers of inflammation and CD8 T-cell activation, but not monocyte activation, are associated with subclinical carotid artery disease in HIV-infected individuals. HIV medicine. 2013 doi: 10.1111/hiv.12013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddler S, Li X, Chu A, Kingsley L, Dobs A, Evans R, Palella F, Visscher B, Chimel J, Sharrett A. Longitudinal changes in serum lipids among HIV-infected men on highly active retroviral therapy. HIV Med. 2007;8:280–7. doi: 10.1111/j.1468-1293.2007.00470.x. [DOI] [PubMed] [Google Scholar]
- Chao J, Stallone JN, Liang YM, Chen LM, Wang DZ, Chao L. Kallistatin is a potent new vasodilator. The Journal of clinical investigation. 1997;100:11–7. doi: 10.1172/JCI119502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Chen S, Wu C, Liu M, Jin Y, Chao L, Chao J. Prophylactic adenovirus-mediated human kallistatin gene therapy suppresses rat arthritis by inhibiting angiogenesis and inflammation. Arthritis & Rheumatism. 2005;52:1319–24. doi: 10.1002/art.20991. [DOI] [PubMed] [Google Scholar]
- Chao J, Yin H, Yao YY, Shen B, Smith RJ, Chao L. Novel role of kallistatin in protection against myocardial ischemia-reperfusion injury by preventing apoptosis and inflammation. Hum Gene Ther. 2006;17:1201–13. doi: 10.1089/hum.2006.17.1201. [DOI] [PubMed] [Google Scholar]
- Liu Y, Bledsoe G, Hagiwara M, Shen B, Chao L, Chao J. Depletion of endogenous kallistatin exacerbates renal and cardiovascular oxidative stress, inflammation, and organ remodeling. American journal of physiology Renal physiology. 2012;303:F1230–8. doi: 10.1152/ajprenal.00257.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen B, Hagiwara M, Yao YY, Chao L, Chao J. Salutary effect of kallistatin in salt-induced renal injury, inflammation, and fibrosis via antioxidative stress. Hypertension. 2008;51:1358–65. doi: 10.1161/HYPERTENSIONAHA.107.108514. [DOI] [PubMed] [Google Scholar]
- Jenkins AJ, McBride JD, Januszewski AS, Karschimkus CS, Zhang B, O’Neal DN, Nelson CL, Chung JS, Harper CA, Lyons TJ, Ma JX. Increased serum kallistatin levels in type 1 diabetes patients with vascular complications. Journal of angiogenesis research. 2010;2:19. doi: 10.1186/2040-2384-2-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H, Gao L, Shen B, Chao L, Chao J. Kallistatin inhibits vascular inflammation by antagonizing tumor necrosis factor-alpha-induced nuclear factor kappaB activation. Hypertension. 2010;56:260–7. doi: 10.1161/HYPERTENSIONAHA.110.152330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen B, Smith J, RS, Hsu Y, Chao L, Chao J. Kruppel-like factor 4 is a novel mediator of kallistatin in inhibiting endothelial inflammation via increased endothelial nitric-oxide synthase expression. Journal of Biological Chemistry. 2009;284:35471–78. doi: 10.1074/jbc.M109.046813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen B, Gao L, Hsu Y, Bledsoe G, Hagiwara M, Chao L, Chao J. Kallistatin attenuates endothelial apoptosis through inhibition of oxidative stress and activation of Akt-eNOS signaling. Heart and Circulatory Physiology. 2010;299:H1419–27. doi: 10.1152/ajpheart.00591.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laufs U, La Fata V, Plutzky J, Liao J. Upregulation of endothelial nitric oxide synthase by HMG CoA reductase inhibitors. Circulation. 1998;97:1129–35. doi: 10.1161/01.cir.97.12.1129. [DOI] [PubMed] [Google Scholar]
- Martinez-Gonzalez J, Raposo B, Rodriguez C, Badimon L. 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibition prevents endothelial NO synthase downregulation by aterogenic levels of native LDLs: balance between transcriptional ad posttranscriptional regulation. Arterioscler Thromb Vasc Biol. 2001;21:804–9. doi: 10.1161/01.atv.21.5.804. [DOI] [PubMed] [Google Scholar]
- Kureishi Y, Luo Z, Shiojima I, Bialik A, Fulton D, Lefer D, Sessa W, Walsh K. The HMG-CoA reductase inhibitor simvastatin activates the protein kinase Akt and promotes angiogenesis in normocholesterolemic animals. Nature Medicine. 2000;6:1004–10. doi: 10.1038/79510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erlandson K, Jiang Y, Debanne S, McComsey GA. Effects of randomized rosuvastatin compared with placebo on bone and body composition among HIV-infected adults. Aids. 2015;29:175–82. doi: 10.1097/QAD.0000000000000526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckard AR, Jiang Y, Debanne SM, Funderburg NT, McComsey GA. Effect of 24 weeks of statin therapy on systemic and vascular inflammation in HIV-infected subjects receiving antiretroviral therapy. The Journal of infectious diseases. 2014;209:1156–64. doi: 10.1093/infdis/jiu012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Lo MT, Ihenachor EJ, Wei J, Looby SE, Fitch KV, Oh J, Zimmerman CO, Hwang J, Abbara S, Plutzky J, Robbins G, Tawakol A, Hoffmann U, Grinspoon SK. Effects of statin therapy on coronary artery plaque volume and high-risk plaque morphology in HIV-infected patients with subclinical atherosclerosis: a randomised, double-blind, placebo-controlled trial. The Lancet HIV. 2:e52–e63. doi: 10.1016/S2352-3018(14)00032-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longenecker CT, Jiang Y, Debanne SM, Labbato D, Kinley B, Storer N, McComsey GA. Rosuvastatin Arrests Progression of Carotid Intima-Media Thickness in Treated HIV. 22nd Conference on Retroviruses and Opportunistic Infections (CROI); Seattle, WA, USA. 2015. Editior, editorˆeditors. [Google Scholar]
- Campbell DJ, Kladis A, Zhang Y, Jenkins AJ, Prior DI, Yii M, Kenny JF, Black MJ, Kelly DJ. Increased Tissue Kallikrein Levels in Type 2 Diabetes. Diabetologia. 2010;53:779–85. doi: 10.1007/s00125-009-1645-8. [DOI] [PubMed] [Google Scholar]
- Zhu H, Chao J, Kotak I, Guo D, Parikh SJ, Bhagatwala J, Dong Y, Patel SY, Houk C, Chao L, Dong Y. Plasma kallistatin is associated with adiposity and cardiometabolic risk in apparently healthy African American adolescents. Metabolism. 2013;62:642–6. doi: 10.1016/j.metabol.2012.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao J, Schmaier A, Chen LM, Yang Z, Chao L. Kallistatin, a novel human tissue kallikrein inhibitor: Levels in body fluids, blood cells, and tissues in health and disease. J Lab Clin Med. 1996;127:612–20. doi: 10.1016/s0022-2143(96)90152-3. [DOI] [PubMed] [Google Scholar]
- Lin W-C, Lu S-L, Lin C-F, Chen C-W, Chao L, Chao J, Lin YS. Plasma kallistatin levels in patients with severe community-acquired pneumonia. Critical Care. 2013;17 doi: 10.1186/cc12507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Asrar MA, Andrawes NG, Ismail EA, Salem SM. Kallistatin as a marker of microvascular complications in children and adolescents with type 1 diabetes mellitus: Relation to carotid intima media thickness. Vascular medicine. 2015;20:509–17. doi: 10.1177/1358863X15591089. [DOI] [PubMed] [Google Scholar]