

Abstract
The use of gene panel sequence for diagnostic and prognostic testing is now widespread, but there are so far few objective tests of methods to interpret these data. We describe the design and implementation of a gene panel sequencing data analysis pipeline (VarP) and its assessment in a CAGI4 community experiment. The method was applied to clinical gene panel sequencing data of 106 patients, with the goal of determining which of 14 disease classes each patient has and the corresponding causative variant(s). The disease class was correctly identified for 36 cases, including 10 where the original clinical pipeline did not find causative variants. For a further seven cases, we found strong evidence of an alternative disease to that tested. Many of the potentially causative variants are missense, with no previous association with disease, and these proved the hardest to correctly assign pathogenicity or otherwise. Post analysis showed that three-dimensional structure data could have helped for up to half of these cases. Over-reliance on HGMD annotation led to a number of incorrect disease assignments. We used a largely ad hoc method to assign probabilities of pathogenicity for each variant, and there is much work still to be done in this area.
Keywords: Gene panel sequencing, VarP analysis pipeline, monogenic disease, missense mutations, CAGI
INTRODUCTION
Genetic testing in clinical laboratories is becoming increasingly common: As of March 2017, GeneTests.org contains entries for about 706 labs and 1,083 clinics worldwide performing a total of 67,187 tests on 5,926 genes for 4,963 genetic conditions. So far though, there has been only limited testing of method efficacy (McCarthy et al. 2014; Pirooznia et al. 2014; Cornish and Guda 2015; Hwang et al. 2015). Many of the genetic tests use targeted gene sequencing panels for identifying variants in a set of genes or gene regions that are known to be associated with a disease (Kammermeier et al. 2014; Okazaki et al. 2016). In clinical laboratories specializing in specific diseases or classes of disease, panels provide high coverage data for genes of interest at relatively low cost, and also reduce the issues in reporting incidental findings to patients. A key and challenging step in all these tests is the ability to accurately interpret the genetic variants and assign a likelihood of pathogenicity (Richards et al. 2015).
Potentially pathogenic sequence variants fall into three classes: (a) those almost certain to cause major loss of protein function (LoF), arising from the introduction of premature stop codons, frameshifts caused by small insertions or deletions, and direct hits on splice sites; (b) those that may or may not significantly affect gene regulation (such as regulatory variants at transcription factor binding sites) or protein function, particularly missense variants; and (c) those that are more likely benign, particularly synonymous, UTR, and deep intronic variants. The main challenge lies in understanding the phenotypic consequences of the large fraction of variants falling into the last two classes. Most clinical laboratories follow a semi-automated approach for variant interpretation, first making use of available variant annotation and prioritization tools and then checking the potential causative variants’ association with the disease of interest in databases and the literature. For the first step, there are dozens of annotation and prioritization tools (open-source or commercial) available (for example, Wang et al. 2010; Cingolani et al. 2012; Sifrim et al. 2013; Robinson et al. 2014; McLaren et al. 2016), typically providing potentially causative variants based on inheritance pattern, allele frequency, genomic region of interest, mutation type and in silico analysis of the likely impact of missense mutations. It has been demonstrated that there are substantial discrepancies between existing annotation tools (McCarthy et al. 2014; Pabinger et al. 2014) so that there is a clear need to encourage and monitor advances in this field. In most clinical laboratories, standard guidelines such as those from the American College of Medical Genetics and Genomics (ACMG) (Richards et al. 2015) are followed for variant interpretation and reporting. Although the guidelines accept computational predictions of pathogenicity for variants, these are only considered a ‘supportive’ evidence. Other evidence is required to classify a variant as causative. As a consequence, the overall contribution of computational methods for variant classification is low and this motivates the development and testing of more accurate methods for variant interpretation.
CAGI (Critical Assessment of Genome Interpretation) is an organization that conducts community experiments to objectively assess computational methods for predicting phenotypic impacts of genomic variation (https://genomeinterpretation.org/). The most recent round of experiments (CAGI4) included a challenge to determine which of 14 disease classes each of 106 patients has and the corresponding causal variants, given each patient’s gene panel sequencing data (https://genomeinterpretation.org/content/4-Hopkins_clinical_panel). The gene panel dataset consists of exons with flanking regions and some complete intron sequencing data for 83 genes from each patient. Data were provided by the Johns Hopkins DNA Diagnostic Laboratory. The Laboratory is a CLIA and CAP certified, Maryland, New York, and Pennsylvania licensed clinical genetic testing laboratory specializing in rare, inherited disorder testing (http://www.hopkinsmedicine.org/dnadiagnostic/tests/).
The data were made available to registered CAGI participants, and all were required to deposit disease and variant assignments by a specified deadline. The anonymized submissions were assessed by John-Marc Chandonia (http://enigma.lbl.gov/chandonia-john-marc/) and Shamil R. Sunyaev (http://genetics.bwh.harvard.edu/wiki/sunyaevlab/), and results were later discussed at the CAGI4 conference. A paper on the assessment is part of this CAGI special issue of Human Mutation (ref to Chandonia et al CAGI issue paper when available).
The identification of causal variants requires a number of carefully controlled procedures for assessing the quality of the data, accurate variant annotation, handling of unphased genotypes, and an appropriate probability model that can prioritize primary and secondary disease findings. With these considerations in mind, we developed a new variant prioritization pipeline (implemented in Python) called VarP (https://github.com/kunduk/VarP) using a combination of open-source and in-house software tools for analyzing gene panel sequencing data. This pipeline was the most successful of those used in CAGI, in the sense that it resulted in the correct matching of the highest number of panel exomes to disease class. [https://genomeinterpretation.org/sites/default/files/protected_files/4-Hopkins_clinical_panel_assessor1_AAdhikari_remixable.pptx]. Nevertheless, the results are far from perfect. In this paper, we describe the design and implementation of the variant prioritization pipeline and the results obtained.
MATERIALS AND METHODS
Capture bed files, gene panel sequencing data, and disease class
The Johns Hopkins DNA Diagnostic Laboratory panel sequencing procedure generates sequence for all exons plus a boundary of 50 bases up and down stream and some introns for 83 genes (1350 exonic and 39 intronic regions), covering 14 monogenic disease classes. 73 of these genes are known to harbor mutations for one of the 14 monogenic disease classes. The remaining ten genes are known to harbor mutations for two or more disease classes. Sequences had been captured using one of the two custom probe sets (Agilent SureSelectXT Target Enrichment Kit) and sequenced using Illumina MiSeq to generate paired-end reads (2×100 nt reads). Two capture bed files (v01, v02) describing the two probe sets were provided as part of the challenge. The Hopkins group called sequence variants and produced two VCF files for each patient, one a gVCF for single nucleotide variants (SNVs; using GATK UnifiedGenotyper, v2.7-4) and the other a VCF for insertion-deletion variants (Indels, GATK HaplotypeCaller, v2.7-4). For the challenge, all VCF files from 106 patients had been combined into two files, one each for SNVs and Indels.
Building the gene list for disease classes
All the genes annotated in the two capture bed files (v01 and v02) were extracted to compile a list of genes to examine. The description of 14 disease classes was provided on the challenge webpage (https://genomeinterpretation.org/sites/default/files/protected_files/4-Hopkins_clinical_panel_disorders.pdf). We made extensive use of the Hopkin’s DNA Diagnostic Laboratory website to map genes to disease class (http://www.hopkinsmedicine.org/dnadiagnostic/). The website lists a number of gene panel tests and also gives a detailed description of the genes associated with each disease as well as their inheritance pattern. Using this resource we were able to group 53 of the 83 genes to 12 disease classes and obtain the inheritance pattern. We used literature and the Genetic Home Reference Database (http://ghr.nlm.nih.gov/) to group another 24 genes to some of the disease classes and obtain the inheritance pattern. In total 77 out of 83 genes were grouped among the 14 disease classes as shown in Table 1. The remaining 6 genes (DHODH, TRIM37, EFTUD2, AMACR, AGXT and CAT) are associated with diseases that are not related to any of the 14 disease classes and therefore were excluded from any downstream analysis.
Table 1.
The 14 disease classes and genes identified as relevant to each class. Genes associated with more than one disease class are indicated by an asterisk.
| Disease Class | Gene List |
|---|---|
| Cystic Fibrosis and CF-related disorders | CA12, CFTR, *SCNN1A, *SCNN1B, *SCNN1G |
| Diffuse Lung Disease | ABCA3, AP3B1, CSF2RA, CSF2RB, *DKC1, FOXF1, HPS1, HPS4, NKX2-1, SFTPA2, SFTPB, SFTPC, SLC7A7, *TERC, *TERT, *TINF2 |
| Primary Ciliary Dyskinesia | CCDC103, CCDC39, CCDC40, DNAAF1, DNAAF2, DNAAF3, HEATR2, DNAH11, DNAH5, DNAI1, DNAI2, DNAL1, HYDIN, LRRC6, NME8, RSPH4A, RSPH9 |
| Peroxisomal Beta-Oxidation Defects | ACOX1, HSD17B4, SCP2 |
| Rhizomelic Chondrodysplasia Punctata | AGPS, GNPAT, PEX7, PHYH |
| Zellweger Spectrum Disorders | DNM1L, PEX1, PEX2, PEX3, PEX5, PEX6, PEX10, PEX12, PEX13, PEX14, PEX16, PEX19, PEX26 |
| Loeys-Dietz Syndrome | *TGFBR1, *TGFBR2, TGFB2 |
| Marfan Syndrome | *FBN1, *TGFBR2 |
| Thoracic Aortic Aneurysm and Dissection (TAAD) | *FBN1, ACTA2, MYH11, MYLK, SMAD3, *TGFBR1, *TGFBR2, COL3A1 |
| Ataxia Telangiectasia | ATM |
| Liddle Syndrome | *SCNN1B, *SCNN1G |
| Pseudohypoaldosteronium Type 1 | NR3C2, *SCNN1A, *SCNN1B, *SCNN1G |
| Telomere Shortening Disorders | CTC1, NHP2, NOP10, *TERC, *TERT, *TINF2, WRAP53, *DKC1 |
| Treacher Collins and Related Syndrome | POLR1D, TCOF1, POLR1C, SF3B4 |
Gene Panel Sequencing data analysis pipeline
The method developed for this challenge (VarP – Variant Prioritization) uses open-source and in-house software tools to analyze gene panel sequencing data with respect to rare genetic disorders in an automated manner. The method has four modules – Variant annotation, QC (quality control) analysis, Variant Prioritization, and estimation of the probability of each variant being causative for the disease. The four modules were executed in a sequential manner (Fig. 1). The inputs were the two VCF files and a gene configuration file that contained the genes associated with each disease class and their inheritance pattern (autosomal dominant/recessive, compound heterozygous, pseudoautosomal dominant/recessive, X-linked dominant/recessive).
Figure 1.
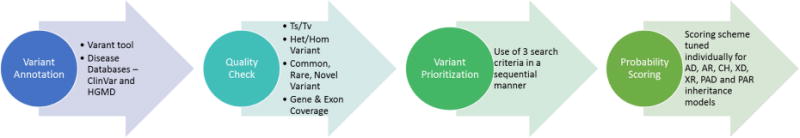
The Variant Prioritization (VarP) Method. Circles represent the four modules. Modules are executed sequentially starting from Variant Annotation and ending with Probability Scoring. The ‘Varant’ tool in step 1 annotates variants with genomic region of occurrence, mutation type, minor allele frequency and prediction of pathogenicity for variants. Ts/Tv=Transition/Transversion, Het/Hom=Heterozygous/Homozygous AD=Autosomal Dominant, AR=Autosomal Recessive, CH=Compound Heterozygous, XD=X-linked dominant, XR=X-linked recessive, PAD=Pseudo Autosomal Dominant, and PAR=Pseudo Autosomal Recessive.
Variant Annotation
The two VCF files (one for SNVs and another for Indels) were annotated using Varant (http://compbio.berkeley.edu/proj/varant, doi:10.5060/D2F47M2C). Details on Varant are provided in the supplementary material. Varant annotated each variant in the VCF files with region of occurrence (intron, exon, splice site or intergenic), observed minor allele frequencies (MAF) from ExAC (Lek et al. 2016) and 1000 Genomes Phase-3 (Auton et al. 2015), mutation type (missense, nonsense, silent, frameshift and non-frameshift indels), predicted impact on protein function, and previously associated phenotypes reported in ClinVar (Landrum et al. 2016). Varant used dbNSFP (v2.9) (Jian et al. 2014) database to fetch the mutation impact predictions from PolyPhen-2 (v2.2.2) (Adzhubei et al. 2013), SIFT (release Jan, 2015) (Kumar et al. 2009) and CADD (v1.2) (Kircher et al. 2014). The RefGene (Pruitt et al. 2014) gene definition file was used for gene and transcript annotations. The principal isoforms of each gene were taken from the APPRIS database (Rodriguez et al. 2013). In addition, the VCF files were annotated with SNPs3D (May, 2015) (Yue et al. 2006) mutation impact predictions, HGMD (version June 2014) (Stenson et al. 2003) disease-related variants and with dbscSNV (Jian et al. 2014) variants that potentially alter splicing.
Quality control Analysis
Three types of QC analysis were run on the Hopkin’s dataset. The first QC analysis is a comparison of Transition vs. Transversion ratio (Ts/Tv), Heterozygous vs. Homozygous variants (Het/Hom), no call sites vs. low quality sites and common vs. rare vs. novel variant counts across all 106 samples and with those in a control variant set from 2,504 samples in 1000 Genomes Phase-3 (Auton et al. 2015). No call sites (sites where neither reference nor alternate allele was called) and low-quality sites (sites not marked PASS and/or genotype quality less than or equal to 30) per sample were computed from the challenge gVCF file. A variant is considered novel if it was not present in the 1000 Genomes and ExAC (Lek et al. 2016) dataset and considered rare if present with an MAF of less than 5% in both of these datasets. Other 1000 Genomes or ExAC variants were considered common. Only SNVs flagged as PASS in the VCF file and with a genotype quality (GQ) greater than 30 were included in the analysis. Scatter plots were generated to represent the results. The QC module also estimated which samples are of African ethnicity, to aid in interpretation of variant count differences. The ethnicity analysis used the population-specific allele frequency (AF) from the 1000 Genomes Phases-3 dataset to identify population enriched variants (i.e. variants that are common (AF > 0.05) in a population but rare (AF <= 0.05) in other populations). Samples whose African population enriched variant count was highest in number compared to other populations in 1000 Genomes (Admix American, South Asian, East Asian and European) were assigned African ethnicity. The second QC analysis is a comparison of the average read depth for 83 genes across 106 samples, using the read depth provided in the gVCF file. The module produced a heat-map of these data, allowing convenient visual inspection for anomalies. The third QC analysis identifies capture regions (exon or intron) with anomalous read depth with respect to other captured regions in the same gene, where the anomaly is found in at least 85% of the samples. Anomalous coverage was identified by first computing the average read depth across the gene (μ) and its standard deviation (σ), and then checking each region for significantly low (< μ − 2σ) or high (> μ + 2σ) coverage. The anomalous coverage regions were then visually inspected using gene coverage plots.
Identification of potentially causative variants
Only rare or novel variants rated high quality (marked PASS and with a GQ > 30 in the VCF files) were considered in the search for causal variants. At this stage, a rare variant was defined as one reported in ExAC (Lek et al. 2016) with a minor allele frequency (MAF) less than or equal to 0.01 and a novel variant was defined as one not found in ExAC. Indels in low complexity regions (LCR) were excluded from the analysis, based on the LCR dataset computed for the human genome by Heng Li (Li 2014). For each sample, each QC qualified variant in each of the 83 genes was assigned to one of four categories, ranked by the likelihood that the variant is causative.
Category 1
Variants reported in HGMD with either DM (disease-causing mutation) or DP (disease-associated polymorphism) status, and/or reported in ClinVar with pathogenic or likely pathogenic clinical significance.
Category 2
Variants annotated as nonsense mutations, direct splicing mutations disrupting either a splice donor or acceptor site, frameshift or non-frameshift causing Indels, splice altering variants predicted in the dbscSNV database, and missense mutations predicted as damaging by one or more of SNPs3D, SIFT, PolyPhen-2 and CADD.
Category 3
Variants annotated as missense but not predicted to be damaging by any of the above methods, and UTR and intronic variants.
Category 4
All other variants (including synonymous and all with MAF > 0.1). These were not considered as potentially causative.
Each variant was also grouped by frequency based on its ExAC MAF: group 1 – novel, 2 – very rare (MAF <= 0.005), or 3 – rare (0.005 < MAF <= 0.01).
For each sample, the variant assigned to the lowest category was taken as the potentially causative variant. If there were two or more variants with the same category, the one in the lowest frequency group was selected. When there were two or more variants with the same category and frequency group, all were selected. Once a selection had been made, no other variants in that sample were considered. Category 1 variants were assumed to be of highest confidence, followed by category 2 and 3 variants and so selection was made in that order: If a suitable variant or variants were found in Category 1, no category 2 ones were considered, and similarly, if suitable variants were found in Category 2, no Category 3 ones were considered. No phase information was available for these data, so for non-homozygous variants where the inheritance model of the gene containing the selected variant required a second allele as part of a compound heterozygous pair, the next ranked variant in that gene was selected.
Thus, for each of the 106 samples, the output from the module was usually one (for dominant or homozygous recessive situations) or two (for compound heterozygous situations) potentially causative variants in a particular gene. Since each gene is associated with one or more of the 14 disease classes (shown in Table 1), identification of a gene implied one or in some cases two possible disease classes. For some samples, no potentially causative variants were found, or for compound heterozygous situations, only a single variant met selection criteria, and so no disease was identified.
Estimating probability for the disease
Table 2 lists the probability of pathogenicity assigned for each category of potentially causative variant. Category 1 variants (based on HGMD or ClinVar entries) were assigned a probability of 1.0, except for some missense variants where prediction methods suggested low impact. Category 2 missense variants were assigned a probability based on the extent of consensus among the four missense impact analysis methods used (SNPs3D, SIFT, PolyPhen-2, and CADD), utilizing a calibration from HGMD data and a control set of inter-species variants. That calibration shows a strong and approximately linear dependence of pathogenic probability on agreement between methods (Supp. Figure S1). Other variant types were subjectively assigned probabilities as shown in Table 2. For autosomal recessive situations, the combined probability of pathogenicity was taken as the product of probabilities for the two contributing variants. Those values were incremented by 0.2 for homozygous cases, as an ad hoc correction for increased confidence, and by 0.1 in compound heterozygous situations. Based on this scoring scheme, a probability of pathogenicity for a disease class was generated for all the samples in which one or more potentially causative variants were identified. For the cases in which a gene was associated with more than one disease class, equal probability was assigned for all the disease classes.
Table 2.
Pathogenicity probability estimates for each variant type.
| Variant Type | Probability Score |
|---|---|
| Reported in HGMD or ClinVar as pathogenic | 1 |
| Missense – Reported in HGMD or ClinVar as pathogenic and predicted damaging by only 2, 1 or 0 out of 4 methods | 0.9 |
| Missense – Predicted damaging by 4/4 methods | 1 |
| Missense – Predicted damaging by 3/4 methods | 0.8 |
| Missense – Predicted damaging by 2/4 methods | 0.5 |
| Missense – Predicted damaging by 1/4 methods | 0.25 |
| Missense – Not predicted damaging by any methods | 0.15 |
| Nonsense | 1 |
| Frameshift/Non-Frameshift Indel | 1 |
| Variant predicted to affect splicing | 0.8 |
| Variant close to Splice Donor site | 0.2 |
| Variant close to Splice Acceptor site | 0.2 |
| UTR Variant | 0.05 |
| Intronic Variant | 0.05 |
| All other variants | 0 |
Post-Challenge Analysis
We performed many post challenge analyses on the results in order to gain insight into the performance, strengths, and weaknesses of the method, and in doing so, made a number of observations. We assessed performance based on the official answer key provided by the Johns Hopkins DNA Diagnostic Laboratory group. For each patient, the key specified the disease class, the possibly causative variants (if any) found in the subset of the 83 genes examined, and a classification of each of these variants (pathogenic, likely pathogenic, VUS (variant of uncertain significance), likely benign and benign). The Hopkins classifications were based on the ACMG evidence rules (Richards et al. 2015).
RESULTS
QC Analysis Summary
Supp. Figures S2 and S3 and Supp. Table S1 together with accompanying text provide details of the QC analysis. Overall, transition/transversion ratios and heterozygosity/homozygosity ratios are consistent with those found in 1000 genome data, with the exception of one sample (P8) with excess homozygosity. There are a maximum of 2000 low quality and 940 no-call calls per sample in the v01 capture data and lower numbers in v02. We expect that any causative variants at these positions would be missed. Common, rare, and novel variant (SNV and Indel) counts for all the samples are consistent with 1000 genome data, except for two outlier samples identified as of African ethnicity which have larger rare Indel counts. The average read depth per gene per sample is high (greater than 100X) with the exception of two capture regions (Exon-53 and Exon-60 of HYDIN gene in Supp. Figure S4) where anomalous coverage could potentially result in causative variants being missed or in false positives.
Missense mutations are amplified in the potentially causative variant set
The VCF files provided for the challenge have a total 2311 unique variants across the 106 patients. This variant set consists of 40% intronic variants, 26% missense variants, 20% synonymous variants, and 14% of variants that are assigned as LoF (frameshift Indels, non-frameshift Indels, and nonsense), UTR, or splicing (Fig. 2A). After applying the PASS (PASS in VCF file), genotype (GQ > 30) and frequency filters (MAF <= 1% in ExAC), the total number of variants was reduced by almost 50% to 1291, with 233 variants filtered because of low quality and 787 further variants filtered because of high MAF. Figure 2B shows that the frameshift and non-frameshift indels decreased the most (by 40% and 27%) on applying the PASS filter and NonSyn, Syn, UTR, CodingIntronic and ‘Close to splice site’ variants decreased the most (by 37 to 42%) on applying the frequency filter. After all filtering, 138 out of the 1291 variants were assigned as potentially causative by the prioritization procedure. In this set, the fraction of LoF variant is 16% and the fraction of missense variants is doubled to more than half (56%), while intronic variants drop to 8% and synonymous to 1%. The high fraction of potentially causative missense variants emphasizes the importance of correctly interpreting this class of mutation.
Figure 2.
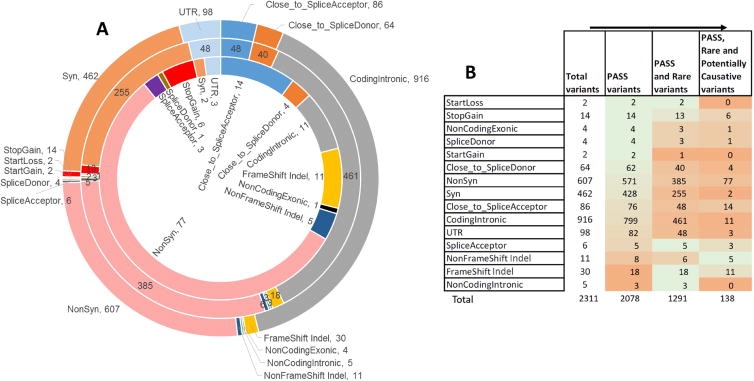
Distribution of variant types for the gene panel sequencing data for 83 genes from 106 patients. Figure 2A: Distribution of variant types. The outer circle shows the distribution for all variants present in the VCF files provided as part of the challenge. The middle circle shows the distribution of high-quality rare variants after applying PASS, GQ and frequency filters using data in the VCF file. The inner circle shows the distribution of potentially causative high quality rare and novel variants in 104 patients after applying the variant selection algorithm. Missense and loss of function variants are substantially enhanced in the latter set. Figure 2B: Changes in the variant type distribution during the filtering process, from total VCF variants, to those annotated as PASS, to those with low frequency, and finally those selected as potentially causative. The heat map indicates the percent decrease in variants on applying each filter (in the direction indicated by the arrows): the larger the decrease, the more orange; the smaller the decrease, the more green. The frameshift and non-frameshift Indel count decreased the most (by 40% and 27%) on applying the PASS filter and NonSyn, Syn, UTR, CodingIntronic, Close to splice site variants decreased the most (by 37 to 42%) on applying the frequency filter.
Matching individuals to disease class
Application of the categorization procedure described in Methods resulted in a non-zero probability for a specific disease class being assigned to 87 of the 106 patients. A further 17 patients were assigned a non-zero probability for two disease classes, as a consequence of a single gene being associated with two of the 14 disease classes. Two patients (P59 and P86) were not assigned to any disease class. P59 had the lowest average read depth for 50 genes out of 83 and next to lowest for the other 33 genes compared to other samples, suggesting that causative variants may have been missed.
Correct disease assignments also made by Hopkins
Overall, the assessors determined that we made correct disease assignments for 36 of 106 cases (Fig. 3A), in the sense that the highest probability was assigned to the disease class specified in the Hopkins answer key. The Hopkins group reported “pathogenic”, “likely pathogenic, or “variant of uncertain significance” (VUS), based on ACMG variant classification, for 43 cases (Fig. 3B). The VarP pipeline assigned the maximum probability to the same disease class for 26 of these 43 cases, with the same variants assigned as causative. There are two primary reasons for our non-identification of the other 17 cases (row 6 and row 10 in Fig. 3A). First, for 10 of these patients, the Hopkins group found only one heterozygous variant in genes known to be associated with disease in a recessive inheritance pattern. Our method considered this insufficient evidence. Second, for the remaining seven patients we found an alternative disease that ranked higher in the variant categorization scheme. As noted in Methods, the selection scheme only considered the disease identified by the highest-ranked variants, and rejected all others. Had we considered diseases identified by lower confidence categorizations, five of these seven cases the Hopkins reported disease would have received 2nd ranking; one 3rd ranking; and one 4th ranking.
Figure 3.

Disease assignment statistics for the 36 patients with correctly identified disease class. Figure 3A: Distribution of the number of patients across prediction performance. The highlighted numbers represent the patients with correct disease class assignment. The reasons for incorrect disease assignment are described in the text. Figure 3B: Distribution of Positive cases (orange) are those found by Hopkins to be carrying pathogenic or likely pathogenic variants, the VUS cases based on ACMG guidelines (yellow) are those carrying variants of uncertain significance and Negative cases (grey) are those in which no causative variant was found by Hopkins. * indicates the number of cases with correct disease class assignment. The VarP pipeline assigned the correct disease class for 26 of the Positive and VUS cases and also correctly assigned disease class (with potentially causative variants) in 10 of the Hopkins Negative cases.
Additional correct disease assignments
Out of the 63 patients for which the Hopkins analysis found no causative variants in the genes ordered as part of the clinical test, our method made 10 correct assignments of disease class and assigned potentially causative variants (row 7 in Fig. 3A). Seven of these patients were found to carry autosomal dominant or homozygous recessive variants and remaining three patients carried compound heterozygous variants. For nine of these 10 cases, the gene hosting the potentially causative variant was not analyzed by the Hopkins group, presumably because coverage was not selected by the requesting physician. For the remaining case, the Hopkins group did not report the potentially causative variant even though they analyzed the relevant gene. For the other 53 patients (row 8 and row 11 in Fig. 3A), neither our method nor the Hopkins group found any causative variants for the expected disease class. However, we found potentially causative variants for a different disease in four of these patients, suggesting alternative diagnoses (see the Alternative Diagnosis section).
Assignment of probability
In order to estimate the accuracy of our probability model, we checked how well the probability of pathogenicity scores correlated with incorrect disease class assignment. The dependence of incorrect disease assignment on assigned probability follows the correct trend, with a high fraction at low probability and a lower fraction at high probability (Fig. 4). However, there are 25 patients with incorrect disease class assignments and a probability greater than 0.8. We found following reasons for this: 1. High confidence given to DM status HGMD variants – 11 of the 25 anomalies are of this type. These are discussed below in the Selection section and listed in Table 4. 2. In five cases, there were pairs of Indels (frameshift or non-frameshift) close together (less than 10 bp apart, Supp. Table S2) in the CCDC40 gene and classified by us as causative compound heterozygous variants. Very likely, these are false Indels arising from alignment errors or errors near perfect repeat regions (Fang et al. 2014). 3. In two cases there are two heterozygous variants predicted damaging by three methods in genes associated with recessive disease. It is possible that these are the same copy of the gene (no phasing information was available). 4. In the remaining seven cases, we found possible alternative diagnoses. These are discussed in detail below.
Figure 4.
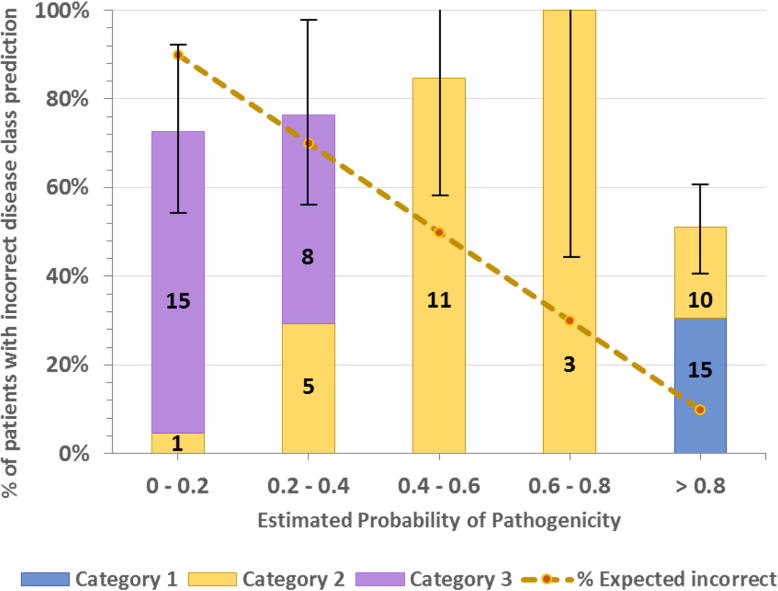
Distribution of patients with incorrectly assigned disease class versus estimated probability of pathogenicity. The dotted line shows the expected value in each bin (e.g. in the 0.8 to 1.0 bin, 10% of disease assignments are expected to be incorrect). Bars show the % of patients in each bin that actually have incorrect assignments. Bar colors show the number of patients with assignments made in each category (Category 1, most confidence). The error bar for each bin is the standard deviation of the number of patients in that bin. As should be the case for a good probability algorithm, patients with a high probability of a correct disease assignment do have a lower rate of incorrect disease classes. However, the plot also shows that there are 25 patients with high probability scores (> 0.8) but incorrect disease class. 15 of these patients carry variant(s) reported as pathogenic (tagged as DM) in the HGMD database. Reasons for this are discussed in the text.
Table 4.
List of variants reported in HGMD with DM or DP status but not supported by other data and leading to an incorrect diagnosis. MAF: Minor Allele Frequency. These variants were present in 11 patients. Green: benign missense prediction, red: deleterious prediction.
| Variant Chr:Pos:Ref:Alt |
Potentially causative variants in # of Patients |
MAF in ExAC |
Mutation Type |
Gene | cDNA Change/Amino Acid |
Impact Predictions B=Benign, D=Damaging, PD=Possibly Damaging |
ClinVar Clinical Significance |
HGMD Status |
HGMD Reported PMID |
Comments |
|---|---|---|---|---|---|---|---|---|---|---|
| 15:48748913:C:T | 2 | 0.0048 | Silent | FBN1 | NM_000138.4:c.5343G>A (p.(V1781=)) |
|
Benign | DM | 17627385 | Reported in a table. No functional study reported. |
| 15:48725102:C:T | 1 | 0.0008 | Missense | FBN1 | NM_000138.4:c.6700G>A (p.(V2234M)) |

|
Other | DM | 17253931 | |
| 3:30733044:T:A | 1 | 0.0014 | Missense | TGFBR2 | NM_003242.5:c.1657T>A (p.(S553T)) |
|
Likely Benign | DM | 16791849 | Reported in a Table. Has a normal transcript, no other data. |
| 5:149740732:C:T | 2 | 0.0023 | Missense | TCOF1 | NM_001135243.1:c.122C>T (p.(A41V)) |

|
– | DM | 12444270 | Unknown Significance. In PMID: 19572402 this variant is reported benign. |
| 7:117305631:A:G | 1 | 0.0034 | Intronic | CFTR | NM_000492.3:c.4242+13A>G |
|
Benign | DM | 15858154 | Variant is not at all reported in the paper. |
| 15:48722907:G:A | 2 | 0.0033 | Missense | FBN1 | NM_000138.4:c.6832C>T (p.(P2278S)) |

|
Benign | DM | 19293843 | Reported as part of a double mutant in the paper. No functional study reported. |
| 15:48818329:A:G | 1 | 0.0023 | Missense | FBN1 | NM_000138.4:c.986T>C (p.(I329T)) |
|
Likely Benign | DM | ||
| 16:23391725:C:T | 1 | 0.0067 | Intronic | SCNN1B | NM_000336.2:c.1543-17C>T |
|
– | DP | 15661075 | Although close (17bp) to a splice site a study showed no splicing effect. |
Variant assignment accuracy for each Selection Category
As described in Methods, we used a work flow to assign variants to one of three categories, ranked by likelihood of pathogenicity. Table 3 shows the percent of correct disease assignments for variants in each category. The highest fraction of cases (42%, 11 out of 26) agreeing with the Hopkins disease class were based on Category 1 variants. The corresponding fractions for Category 2 and Category 3 variants are 38% and 23% respectively. This trend is expected, since assignment confidence decreases with increasing category number.
Table 3.
Percentage of correct disease assignments in each of the three variant selection categories. As expected, accuracy is highest in Category-1, then Category-2, then Category-3. Novel variant assignments are more accurate than for rare variants.
| Category | Variant Considered | Minor Allele Frequency
|
% Correct Assignment | ||
|---|---|---|---|---|---|
| Novel | <= 0.005 | <=0.01 | |||
|
| |||||
| Category-1 | In HGMD with DM, DP status and/or in ClinVar with Pathogenic or Likely pathogenic tag | 4/4 | 7/19 | 0/3 | 11/26: 42% |
|
| |||||
| Category-2 | Missense (Predicted damaging either by SNPs3D, SIFT, PolyPhen2 or CADD) Frameshift/Non-Frameshift Indel NonSense Direct Splicing Any variant predicted damaging by dbscSNVs |
9/14 | 7/28 | 2/5 | 18/47: 38% |
|
| |||||
| Category-3 | All other missense, UTR, and Intronic | 4/17 | 2/12 | 1/2 | 7/31: 23% |
|
| |||||
| 17/35: 49% | 16/59: 27% | 3/10: 30% | |||
As noted earlier, Category 1 variants are those annotated in HGMD and/or ClinVar as disease-causing. Further inspection showed that 11 of the 15 discordant assignments cases had conflicting database annotations and sometimes weak or no supporting evidence (Table 4). In seven cases, the corresponding variant is annotated ’DM’ (disease mutation) in HGMD but is annotated ‘benign’ or ‘likely benign’ in ClinVar. Consistent with the ClinVar annotation, a check of the supporting literature for these showed either no experimental support or no evidence favoring pathogenicity. For the other four cases, ClinVar had no relevant entry and there was no literature support. Seven of these 11 cases involved missense variants, and none of those were rated high confidence pathogenic by our consensus method. With the wisdom of hindsight, we should have factored these considerations into the categorization and probability procedures, and placed less faith in HGMD. The remaining four discordant assignments have either functional validation of the variant as damaging or are annotated as pathogenic in ClinVar as well. As discussed later, these four patients may really have a different disease.
Category 2 variants are those selected because of being a LoF variant, the computational method assigning pathogenicity for missense variants, a direct hit on a splice site, or a prediction of an impact on splicing (Jian et al. 2014). The 18 correct assignment cases include seven compound heterozygous and 11 autosomal dominant or recessive cases. Seven out of these 11 cases carry a LoF (nonsense, frameshift or non-frameshift Indel) or direct splicing variant, and the remaining four carry missense variants (two predicted damaging by two methods and two predicted damaging by one method). The 29 cases with discordant disease class with respect to the Hopkins information in this category include 11 compound heterozygous cases and 18 autosomal dominant or recessive cases. For the 11 discordant compound heterozygous cases, the assumption that the two variants are appropriately phased is a likely cause of misassignment. The 18 other cases include one frameshift Indel and 17 missense variants. Of the 17 missense, only one was high confidence, predicted damaging by all four methods. Four were damaging by three methods (expected accuracy 0.8), 10 were damaging by two methods (expected accuracy 0.5), and two were damaging by only one method (expected accuracy 0.25).
Category 3 variants are missense mutations predicted benign by all four computational methods and those which are intronic or in a UTR. All were assigned low causative probability, ranging from 0.05 to 0.29. There are only seven out of 30 with correct disease class assignments that were assigned based on Category 3 potentially causative variants. Six of these seven cases carried intronic insertions or deletions close to a splice site (within 5 to 30 bases), suggesting proper treatment of this mechanism is important. The remaining case carries a missense mutation predicted benign by the four mutation impact prediction methods.
There is a marked dependence of level of agreement with the Hopkins disease class and the frequency of the potentially causative variants (Table 3): 49% of disease assignments made for novel variants agree with the Hopkins answer key, compared to 27–30% for the other, non-novel variants with less than 1% MAF.
Alternative Diagnoses
There is an important difference between the Hopkins lab procedures and the CAGI challenge. In the lab, in accordance with clinical guidelines, for each patient, variant analysis was performed only on the subset of genes identified by the physician requesting the test, usually those for a single disease, and sometimes only a subset of genes for a single disease. On the other hand, the challenge required analysis of all genes for each patient. That led to a number of findings suggesting that in some cases, causative variants are overlooked in the clinic. Of the 70 cases where our disease assignments and the disease tested by the Hopkins pipeline differ, seven have strong evidence supporting assignment to a different disease (Table 5). In four of these cases, no variants supportive of the tested disease were found by ourselves or by Hopkins. In two further cases, the Hopkins pipeline reported only one variant in a recessive gene and for the remaining case (patient P8 in Table 5), there is evidence that the patient may have two diseases. These seven cases fall into three groups:
Table 5.
Patients carrying putative causative variants for an alternative disease. AD=Autosomal Dominant, AR=Autosomal Recessive, CH=Compound Heterozygous (for AR cases, the listed variant is homozygous). The table is divided into three case types. Green: benign missense prediction, red: deleterious prediction.
| Patient ID |
Variant Chr:Pos:Ref:Alt |
MAF in ExAC |
Inheritance Model |
Mutation Type |
Gene | Amino Acid/ cDNA Change |
Impact Predictions B=Benign, PD=Possibly Damaging, D=Damaging |
ClinVar Clinical Significance |
HGMD Status, PMID |
Predicted Disease Class |
Tested Disease class |
|---|---|---|---|---|---|---|---|---|---|---|---|
| # TYPE-1: Patients carrying variants likely causative of a disease phenotype that has overlapping symptoms with the disease tested at Hopkins. | |||||||||||
| P36 | 16:23200963:G:A | 0.0047 | AD | Missense | SCNN1G | NM_001039.3:c.589G>A (p.(E197K)) |

|
Pathogenic | DM, 18507830 | Bronchiectasis | Diffuse Lung Disease |
| P48 | 16:23200963:G:A | 0.0047 | AD | Missense | SCNN1G | NM_001039.3:c.589G>A (p.(E197K)) |

|
Pathogenic | DM, 18507830 | Bronchiectasis | Pseudohypoaldosteronism type 1 |
| P7 | 5:1293767:G:A | 0.0022 | AD | Missense | TERT | NM_001193376.1:c.1234C>T (p.(H412Y)) |

|
Pathogenic | DM, 15814878 | Pulmonary Fibrosis and/or Bone marrow failure | Cystic Fibrosis and CF-Related |
| # TYPE-2: Patient carrying a variant reported in HGMD and ClinVar with pathogenic clinical significance, for a disease other than that tested at Hopkins. | |||||||||||
| P8 | 7:117230454:G:C | 0.0051 | AR | Missense | CFTR | NM_000492.3:c.1727G>C (p.(G576A)) |

|
Pathogenic | DM, 1545465 | Cystic Fibrosis and CF-related disorders | Peroxisomal Beta-Oxidation Defects |
| # TYPE-3: Patient carrying LoF variant or missense predicted damaging by all reporting computational methods. | |||||||||||
| P40 | 10:13337608:T:C | 0.0001 | CH | Splice Acceptor | PHYH | NM_006214.3:c.135-2A>G |
|
Pathogenic | DM, 9326939 | Zellweger Spectrum Disorders | Cystic Fibrosis and CF-Related |
| 10:13336522:GA:G | Novel | Frame-shift | NM_006214.3:c.319del (p.(S107Rfs*11)) | ||||||||
| P46 | 5:149767634:C:T | 0.0001 | AD | Missense | TCOF1 | NM_001135243.1:c.3029C>T (p.(T1010I)) |

|
Treacher Collins and Related Syndrome | Diffuse Lung Disease | ||
| P75 | 17:78032436:G:A | 0.0018 | CH | Missense | CCDC40 | NM_001243342.1:c.1303G>A (p.(E435K)) |

|
Primary Ciliary Dyskinesia | Diffuse Lung Disease | ||
| 17:78064014: A:ACAACA CGGGACGC GCGCAGGC ACGTGCACG AACAACAC GGGACGC GCGCAGGC ACGTGCAC |
0.0007 | Frame-shift |
NM_001243342.1:c. c.2909_2910ins CAACACGGGACGC GCGCAGGC ACGTGCACG AACAACACGG GACGCGCGC AGGCACGTGCAC (p.(K970Nfs*144)) |
||||||||
Three cases where the patient carried variants likely causative of a disease phenotype that has overlapping symptoms with the disease tested at Hopkins. One of these is a patient (P36) carrying a very rare (AF=0.0047 in ExAC) autosomal dominant missense mutation (rs5738:G>A, NM_001039.3:c.589G>A, p.(E197K)) in exon-3 of the SCNN1G gene. This mutation is reported in HGMD and ClinVar to be causative for Bronchiectasis with pathogenic clinical significance (Fajac et al. 2008). The patient was tested for Diffuse Lung disease and no variants with the required inheritance pattern was found in the relevant genes. Bronchiectasis has been previously shown to be associated with Idiopathic Pulmonary Fibrosis, one of the diseases in the Diffuse Lung disease class. (International Consensus Statement of the American Thoracic Society and the European Respiratory Society. 2000; Bourke 2006).
One case where a patient (P8) carried a variant reported in HGMD and ClinVar to have pathogenic clinical significance for a disease other than that tested for, and where the tested and apparent diseases cannot be easily confused. P8 carries a very rare (AF=0.0051 in ExAC) homozygous recessive mutation (rs1800098:G>C, NM_000492.3:c.1727G>C, p.(G576A)) in the CFTR gene, consistent with the disease class ‘Cystic Fibrosis and CF-related disorders’. A functional study found the mutation causes an increased amount of skipping of exon-12 during splicing (Pagani et al. 2003). This patient was originally tested for Peroxisomal Beta-Oxidation Defects and a homozygous recessive frameshift mutation was found in the relevant gene. We did not report that variant because of finding the CFTR variant which we categorize as higher confidence of pathogenicity. The data are consistent with the patient having both diseases.
Three cases where the patient carried variant(s) predicted damaging by all reporting computational methods or a LoF variant. For example, one of these is a patient (P46) to whom we assigned ‘Treacher Collins and Related Syndromes’ based on a very rare (MAF = 0.0002 in ExAC) missense mutation (rs538401137:C>T, NM_001135243.1:c.3029C>T, p.(T1010I)) in the TCOF1 gene and assigned as damaging by all four computational methods. This patient was tested for the Diffuse Lung disease class in the Hopkins pipeline, and no variants consistent with that phenotype were found by them or us.
Protein structure coverage for potentially causative variants
In principle, information from three-dimensional structure and on the detailed functional roles of residues, motifs, and domains should be of considerable value in evaluating the impact of missense variants. In practice, it is often ignored, and indeed we did not use it in this challenge. What difference might it have made? To investigate this, we considered only potentially causative missense variants that are not included in HGMD or ClinVar. Current ACMG guidelines (Richards et al. 2015) would place a low weight on computational analysis of these, and thus they would likely be reported as VUSs. There are 47 such missense variants distributed over 41 patients. ~50% (23/47) of these are either included in an experimental structure or can be included in a homology model based on 22% or higher sequence identity to an experimental structure (Fig. 5A). Three of these mutations are in proteins with experimental structure (X-Ray structure). We use these three cases to illustrate how protein structure could be used to: (a) supplement the sequence analysis methods to increase confidence in a pathogenic or benign assignment and (b) understand the pathogenicity mechanism at the protein level. Two of these mutations have correctly assigned disease classes and causative variants in our submission. One of those is of a novel mutation (NM_000901.4:c.1807T>A, p.(C603S)) at a highly conserved position in the Mineralocorticoid receptor. This protein is associated with Pseudohypoaldosteronism Type 1. Although we correctly identified this mutation from sequence information, only two of the four (SNPs3D, SIFT, PolyPhen2, CADD) methods assigned it as pathogenic, and the other two did not. Thus additional evidence would have improved confidence in the assignment. Inspection of the structure (PDBID: 4TNT) showed that the wild-type amino acid (CYS-603) is a zinc ligand in a zinc finger domain (Fig. 5B). Many other zinc ligand mutations in these domains cause loss of function of the corresponding proteins (Kambouris et al. 2014; Vincent et al. 2014), providing additional evidence of pathogenicity. The second case with correct disease assignment is of another novel mutation (NM_000492.3:c.3849G>C, p.(R1283S)) at a highly conserved position in the second nucleotide binding domain of the CFTR protein. Mutations in CFTR cause Cystic Fibrosis, one of the disease classes in the Hopkins dataset. This mutation is predicted damaging by three out of four (SNPs3D, SIFT, PolyPhen2, CADD) sequence methods. Inspection of the protein structure (PDBID: 3GD7) hosting this mutation shows the wild type side chain (R1283) makes two charge-dipole interactions with main chain carbonyl groups of L1258 and R1259, providing a helix cap (Hol et al. 1981), consistent with significant destabilization of the structure (Fig. 5C). Loss of protein stability has been shown to be the most common cause of monogenic disease (Wang and Moult 2001; Yue et al. 2005). A different mutation at this position (rs77902683, NM_000492.3:c.3848G>T, p.(R1283M)) has previously been found in CF patients (Cheadle et al. 1992) and has been reported as pathogenic in ClinVar and HGMD.
Figure 5.
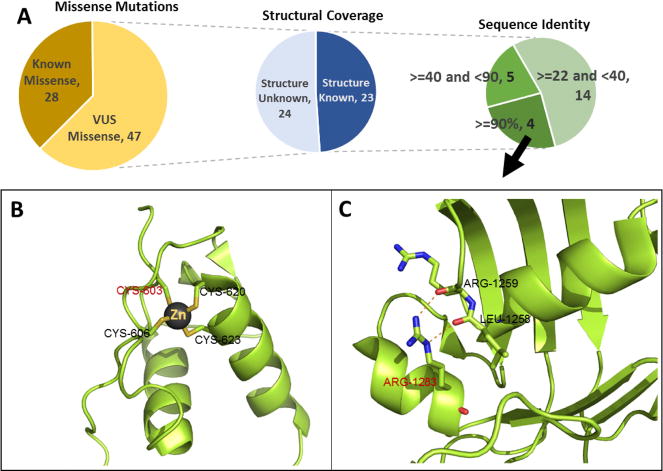
Structural coverage of prioritized missense mutations. Figure 5A: Missense variant distribution: 1) Known (variant reported in HGMD or ClinVar) versus VUS variants, 2) Structural coverage for VUS variants, 3) Number of mutations in different sequence identity ranges between the protein hosting the mutation and the closest available homologous protein in the PDB. Figure 5B and 5C show two examples of structure assisting mutation interpretation. Figure 5B: Part of a zinc finger domain of the Mineralocorticoid receptor protein (PDB: 4TNT) including the mutation C603S found in a Hopkins patient, showing that C603 is one of the Zn ligands. Analogy to other zinc coordinating mutations in zinc fingers provides strong evidence structure and hence function will be disrupted. Figure 5C: Mutation R1283S, found in one of the Hopkins patients, is predicted deleterious by the three out of four computational methods. Inspection of the structure shows disruption of two charge-dipole interactions forming a helix cap, expected to significantly destabilize the structure.
The third mutation with experimental structure coverage is one where we made an incorrect disease assignment on the basis of just one of the four missense analysis methods predicting deleterious. Although that was already a low confidence prediction, further evidence would be useful. This is a very rare (MAF=0.0005 in ExAC) variant (rs147398624:G>A, NM_000901.4: c.2578G>A, p.(V860I)) in the Mineralocorticoid receptor, with an autosomal dominant pattern disease inheritance pattern. The mutation is located on the protein surface (PDBID: 2AA5) and is not part of any known interface, providing further evidence the mutation is benign.
DISCUSSION
The CAGI4 challenge based on panel sequencing data provided by the Johns Hopkins DNA diagnostic laboratory has allowed a blind test of current methods for identifying causative variants in clinical rare disease sequence data. Participants were asked to match each of 106 patients to one of 14 classes of disease. To address this challenge, we developed an analysis pipeline, VarP, designed to identify potentially causative variants. Using this pipeline, we were able to correctly match 36 patients to the reported disease class. The analysis provided a number of insights into issues related to gene panel testing, including the relationship between data quality and success in finding causative variants, variant prioritization procedure limitations, inconsistencies in databases, and cases of possible alternate diagnosis.
Undiagnosed cases
Even with full knowledge of the reported disease class, the Hopkins pipeline could only find potential causative variants for 43 cases, leaving 63 with no causative variants. As discussed below, we were able to find variants correctly matching a further 10, but that still leaves half (53) of the cases where neither we nor Hopkins could find variants. There are three major factors that may contribute to the high fraction of undiagnosed cases. First, a limitation in all studies of this type is data quality. Our QC analysis suggests the Hopkins data are generally of high quality. Read depth per gene per sample is high (between 107X to 983X) and each sample has only about 2000 positions with no call or a low-quality variant call. But there are some particular sample level properties in the data that may affect analysis. For example, sample P8 (tested for an autosomal recessive disease, Peroxisomal Beta-Oxidation defect) has an abnormally high fraction of homozygous variant calls compared with heterozygous ones, increasing the chances of finding an apparently causative homozygous variant. Our pipeline identified a potentially causative homozygous missense variant in CFTR, consistent with cystic fibrosis, and annotated as pathogenic in both HGMD and ClinVar, whereas the Hopkins pipeline found a homozygous frameshift variant in HSD17B4, consistent with the tested disease. There are also some areas of low coverage, for instance 78 samples have zero coverage of Exon-60 in HYDIN. Variants in this gene may cause Primary ciliary dyskinesia. Overall though, sequencing data quality does not appear to make a large contribution to missing diagnostic variants.
A second factor contributing to non-identification of causative variants is that there may be other, unknown, genes where variants cause the disease phenotype. Many new monogenic disease genes are still being discovered (more than 67 genes in a two-year period, Beaulieu et al. 2014). Thirdly, the causative variants may have been not covered in the panel, which consists of mostly exon sequence. Missing variants may include those affecting the expression of a relevant gene, CNVs, and larger scale structural genomic changes. In some rare disease analyses using whole genome sequence (WGS), such as in the SickKids Genome Clinic (http://www.sickkids.ca/CGM/genome-clinic/index.html), the latter type of variant has been found to make a significant contribution (Stavropoulos et al. 2016). However, those patients mostly exhibit major developmental disease phenotypes, and may not be typical of rare disease patients in general.
Correct diagnosis for cases where the Hopkins pipeline did not find causative variants
For 10 cases we were able to identify the reported disease class even though Hopkins reported no potentially causative variants. In nine out of these ten cases, the Hopkins pipeline did not include analysis of the gene carrying the diagnostic variant(s). Apparently this is because the requested test did not include the gene, a choice made by the referring physician. As noted earlier, Hopkins is only permitted to analyze the requested gene set. For the 10th (a compound heterozygous case where one of the variants is missense predicted damaging by four methods and other is an intronic variant close to a splice acceptor), Hopkins did not report the potentially causative variants even though they analyzed the relevant gene.
Missed diagnoses
There are 17 cases where we did not identify the correct disease class, but the Hopkins analysis did find potentially causative variants. For 10 of these, the Hopkins variants are in genes expected to have a recessive inheritance pattern, and only one heterozygous variant was present – not sufficient for our evidence rule. Had we used such a weak criterion for inheritance model filtering many more false positives would have been generated. Thus these should not be regarded as failure of the VarP approach but rather an appropriate filtering strategy used in VarP. In the other seven cases where Hopkins found variants, VarP found stronger evidence for a different disease class. For two of these, as discussed below, we consider the evidence that the patients have the VarP identified disease very strong, and if so, these also are not errors. For the other five, we made two sorts of errors. One was placing too much trust in HGMD that affected three cases – in each of these cases the HGMD annotations were incorrect and contradicted or not supported by ClinVar or experimental data. The other source of error was for two compound heterozygous cases where one of the partner variants was a low impact missense (predicted benign by 1/4 methods) or an intronic variant and so provided very weak evidence. In retrospect, the procedure of taking just the most likely causative variant(s) and ignoring all other variants in a patient was sub-optimal. A better procedure would probably be to use all variants in each gene to assign a probability of pathogenicity and to use those probabilities to infer disease class.
Incorrect diagnoses
For 25 patients VarP made high confidence (probability score > 0.8) incorrect disease class assignments. A primary factor was again over-reliance on HGMD annotation, accounting of 11 of the 25 cases. A further five cases involved pairs of Indels very close to each other (less than 10 base pairs apart), and consistent with a compound heterozygous cause for a recessive disease. In fact, these Indel pairs are probably coupled alignment errors. There are two cases where the assumption that a pair of recessive variants are on different copies of the gene may be incorrect (there was no phasing data available). In seven of the remaining cases, we found high confidence pathogenic variants in genes associated with a different disease from that in the Hopkins answer key. As discussed later, the evidence for some of these is sufficiently strong that they may not be errors.
Distinct potentially causative variants that led to disease classification
VarP identified 105 potentially causative variants each of which occurs once in a total of 78 patients. A further 14 potentially causative variants were seen in two or more of the other 28 patients (Supp. Table S3). We also considered accuracy in terms of the fraction of these 119 distinct variants which led to correct and incorrect disease assignments. By this measure, correct disease identification increases from 34% (36/106) to 36% (33/91). The improvement occurs because the majority of repeat variants are present in cases where an incorrect disease was assigned, and we speculate that some of these may reflect sequencing artifacts.
Reliability of probability for disease assignments
In the clinic, perhaps more important than having an accurate method of determining pathogenicity is having an accurate method for assigning a probability of correctness to a pathogenic assignment. The CAGI challenge required participants to also provide these probabilities, and so it was possible to evaluate how effective our approach was. We used a largely ad hoc probability scale in this analysis. Although there is a reasonable overall correlation between these quantities (Fig. 4), there were a substantial number of variants assigned a high probability that were not in fact pathogenic. There were two primary reasons for that – first, as noted earlier, we misjudged the reliability of HGMD assignments of disease mutations. Had we used a model that included disagreements between HGMD and ClinVar, these cases would have had more appropriate probabilities. Second, as discussed below, in a number of cases we consider the evidence strong that these patients had a different disease.
Reliability of missense probability estimates
As described in the Results, overall, the estimated probabilities of pathogenicity shows qualitative though not quantitatively correct properties. The majority of potentially causative variants are missense, so improved confidence in assigning a probability of pathogenicity to these are of particular importance. As described earlier, we assigned a probability based on the fraction of four different missense analysis methods reporting deleterious. The method was calibrated [Yin el al] using a set of HGMD mutations (all assumed pathogenic) and a set of interspecies variants (assumed benign). There are a number of limitations to this dataset, and so we were interested to see to what extent the estimated probabilities were useful. Interpretation of the results is complicated by the alternative diagnosis cases and by compound heterozygous cases, involving two different variants. Supp. Figure S5 shows the relationship between estimated probabilities and correct disease class assignment, omitting those cases. Counts here are too small to draw firm conclusions. A high proportion of mutations assigned with a probability of less than or equal to 0.5 are incorrect, consistent with expectations. However, more than half of the mutations with probabilities higher than 0.7 are also incorrect, not as expected. Further analysis Yin et al. (ref to Yin et al. CAGI issue paper when available) suggests that a probability method based on more than four missense impact prediction methods would have yielded better results. But clearly a more extensive blind test is needed to evaluate this approach.
Apparent cases of alternative diagnoses
Using quite stringent criteria we identified seven cases where the data are consistent with patients having a different disease class than that provided in the Hopkins answer key. Four of these patients carry variants for the alternative disease class that are reported in HGMD and ClinVar as pathogenic. The remaining cases carry missense variants predicted damaging by all reporting methods, frameshift or non-frameshift indels, or variants directly affecting splicing. In three cases, symptoms of the answer key disease and the alternative overlap, so it is possible that there was a misdiagnosis in the referring clinic. The other cases are more puzzling. Since we have no information as to why a particular test was ordered (and in many cases the Hopkins group may not either), it is difficult to comment further. But it is concerning that in a number of cases there could be confusion of some sort as to what disease patients have. In these seven cases, the Hopkins pipeline did not report any variant for four cases, reported only one variant in a recessive gene for two cases and reported a homozygous frameshift mutation in the remaining case. The pipeline was prevented from discovering the possible alternatives by the current guidelines, which require that only requested genes for a specific disease test be examined. On the basis of these limited data, it is not clear whether on balance this practice is in the patients’ best interest.
VarP performance improves when the patients’ clinical indications are known
Clinical laboratories typically have information on each patient’s disease phenotype, and variants are evaluated with that knowledge. In that aspect, the CAGI Hopkins challenge creates an artificially harder problem, since disease class is not known to participants. If the disease classes were known, would VarP identify the variant(s) reported by Hopkins pipeline? We tested this scenario by searching for potentially causative variant(s) only in genes associated with each patient’s diagnosed disease class, using the VarP pipeline. On this basis, VarP identifies potentially causative variants for 61 patients, 18 more cases than the Hopkins pipeline. However, there are still nine cases where Hopkins identified potentially causative variants and VarP does not. As discussed earlier, these patients each carry only one heterozygous variant in a recessive gene, which we considered insufficient evidence.
Better results would have been obtained not using HGMD
As noted earlier, 11 of the 25 incorrect disease class assignment cases with a probability of pathogenicity higher than 0.8 are a result of accepting HGMD annotations of pathogenicity. Such a high error rate from a single cause suggests that it might be better to ignore HGMD altogether and just use ClinVar for pathogenicity information. We tested this by running the VarP pipeline again, omitting HGMD. The success rate (correct match to disease class) increases from 36 to 40 (Supp. Table S4).
Lessons learned
Going forward, how would we now improve performance of the VarP analysis pipeline? As noted earlier, a suboptimal feature of the procedure was terminating the variant search once a suitable candidate had been found, rather than finding all possible causative variants and assigning each a probability. As also noted earlier, over-reliance on HGMD was a cause of errors and this can be corrected by considering ClinVar and HGMD annotations together, and, where appropriate, include missense impact analysis in assigning a probability to these Category 1 variants. Structure also has the potential for contributing to the discovery of causative variants and providing mechanistic insight. However, full automation of that analysis will require the development of new methods. In general, much more work must be done to provide a reliable probability of pathogenicity, not only for missense but for all types of variants.
Supplementary Material
Acknowledgments
We are grateful to Drs Bethany Buckley, Molly Sheridan, and Garry R. Cutting, The Johns Hopkins University for making the challenge data available.
GRANT SPONSOR
This work was supported in part by NIH R01GM104436 to JM. The CAGI experiment coordination is supported by NIH U41 HG007446 and the CAGI conference by NIH R13 HG006650.
References
- Adzhubei I, Jordan DM, Sunyaev SR. Current Protocols in Human Genetics. Hoboken, NJ, USA: John Wiley & Sons, Inc; 2013. Predicting Functional Effect of Human Missense Mutations Using PolyPhen-2; pp. 7.20.1–7.20.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auton A, Abecasis GR, Altshuler DM, Durbin RM, Abecasis GR, Bentley DR, Chakravarti A, Clark AG, Donnelly P, Eichler EE, Flicek P, Gabriel SB, et al. A global reference for human genetic variation. Nature. 2015;526:68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaulieu CL, Majewski J, Schwartzentruber J, Samuels ME, Fernandez BA, Bernier FP, Brudno M, Knoppers B, Marcadier J, Dyment D, Adam S, Bulman DE, et al. FORGE Canada Consortium: outcomes of a 2-year national rare-disease gene-discovery project. Am J Hum Genet. 2014;94:809–17. doi: 10.1016/j.ajhg.2014.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourke SJ. Interstitial lung disease: progress and problems. Postgrad Med J. 2006;82:494–9. doi: 10.1136/pgmj.2006.046417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan AY, Punwani D, Kadlecek TA, Cowan MJ, Olson JL, Mathes EF, Sunderam U, Man Fu S, Srinivasan R, Kuriyan J, Brenner SE, Weiss A, et al. A novel human autoimmune syndrome caused by combined hypomorphic and activating mutations in ZAP-70. J Exp Med. 2016;213:155–65. doi: 10.1084/jem.20150888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheadle JP, Meredith AL, al-Jader LN. A new missense mutation (R1283M) in exon 20 of the cystic fibrosis transmembrane conductance regulator gene. Hum Mol Genet. 1992;1:123–5. doi: 10.1093/hmg/1.2.123. [DOI] [PubMed] [Google Scholar]
- Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012;6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornish A, Guda C. A Comparison of Variant Calling Pipelines Using Genome in a Bottle as a Reference. Biomed Res Int. 2015;2015:456479. doi: 10.1155/2015/456479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durbin RM, Altshuler DL, Durbin RM, Abecasis GR, Bentley DR, Chakravarti A, Clark AG, Collins FS, De La Vega FM, Donnelly P, Egholm M, Flicek P, et al. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fajac I, Viel M, Sublemontier S, Hubert D, Bienvenu T, Pasteur M, Helliwell S, Houghton S, Webb S, Foreraker J, Coulden R, Flower C, et al. Could a defective epithelial sodium channel lead to bronchiectasis. Respir Res. 2008;9:46. doi: 10.1186/1465-9921-9-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang H, Wu Y, Narzisi G, ORawe JA, Barrón LTJ, Rosenbaum J, Ronemus M, Iossifov I, Schatz MC, Lyon GJ, Gudmundsson J, Sulem P, et al. Reducing INDEL calling errors in whole genome and exome sequencing data. Genome Med. 2014;6:89. doi: 10.1186/s13073-014-0089-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hol WG, Halie LM, Sander C. Dipoles of the alpha-helix and beta-sheet: their role in protein folding. Nature. 1981;294:532–6. doi: 10.1038/294532a0. [DOI] [PubMed] [Google Scholar]
- Hwang S, Kim E, Lee I, Marcotte EM, Church GM, Lunshof JE, Bamshad MJ, Do R, Kathiresan S, Abecasis GR, Pereira PCB, Yang Y, et al. Systematic comparison of variant calling pipelines using gold standard personal exome variants. Sci Rep. 2015;5:17875. doi: 10.1038/srep17875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jian X, Boerwinkle E, Liu X. In silico prediction of splice-altering single nucleotide variants in the human genome. Nucleic Acids Res. 2014;42:13534–44. doi: 10.1093/nar/gku1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kambouris M, Maroun RC, Ben-Omran T, Al-Sarraj Y, Errafii K, Ali R, Boulos H, Curmi PA, El-Shanti H, Seelow D, Schuelke M, Hildebrandt F, et al. Mutations in zinc finger 407 [ZNF407] cause a unique autosomal recessive cognitive impairment syndrome. Orphanet J Rare Dis. 2014;9:80. doi: 10.1186/1750-1172-9-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kammermeier J, Drury S, James CT, Dziubak R, Ocaka L, Elawad M, Beales P, Lench N, Uhlig HH, Bacchelli C, Shah N. Targeted gene panel sequencing in children with very early onset inflammatory bowel disease–evaluation and prospective analysis. J Med Genet. 2014;51:748–55. doi: 10.1136/jmedgenet-2014-102624. [DOI] [PubMed] [Google Scholar]
- Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310–315. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Hoover J, Jang W, Katz K, et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016;44:D862–D868. doi: 10.1093/nar/gkv1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, Tukiainen T, Birnbaum DP, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. Toward better understanding of artifacts in variant calling from high-coverage samples. Bioinformatics. 2014;30:2843–51. doi: 10.1093/bioinformatics/btu356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy DJ, Humburg P, Kanapin A, Rivas MA, Gaulton K, Cazier J-B, Donnelly P, Green E, Guyer M, Schrijver I, Aziz N, Farkas D, et al. Choice of transcripts and software has a large effect on variant annotation. Genome Med. 2014;6:26. doi: 10.1186/gm543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKernan KJ, Peckham HE, Costa GL, McLaughlin SF, Fu Y, Tsung EF, Clouser CR, Duncan C, Ichikawa JK, Lee CC, Zhang Z, Ranade SS, et al. Sequence and structural variation in a human genome uncovered by short-read, massively parallel ligation sequencing using two-base encoding. Genome Res. 2009;19:1527–1541. doi: 10.1101/gr.091868.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GRS, Thormann A, Flicek P, Cunningham F, Eisenstein M, Weil M, Chen A, Visscher P, et al. The Ensembl Variant Effect Predictor. Genome Biol. 2016;17:122. doi: 10.1186/s13059-016-0974-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okazaki T, Murata M, Kai M, Adachi K, Nakagawa N, Kasagi Noriko, Matsumura W, Maegaki Y, Nanba E. Clinical Diagnosis of Mendelian Disorders Using a Comprehensive Gene-Targeted Panel Test for Next-Generation Sequencing. Yonago Acta Med. 2016;59:118–125. [PMC free article] [PubMed] [Google Scholar]
- Pabinger S, Dander A, Fischer M, Snajder R, Sperk M, Efremova M, Krabichler B, Speicher MR, Zschocke J, Trajanoski Z. A survey of tools for variant analysis of next-generation genome sequencing data. Brief Bioinform. 2014;15:256–78. doi: 10.1093/bib/bbs086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagani F, Stuani C, Tzetis M, Kanavakis E, Efthymiadou A, Doudounakis S, Casals T, Baralle FE. New type of disease causing mutations: the example of the composite exonic regulatory elements of splicing in CFTR exon 12. Hum Mol Genet. 2003;12:1111–1120. doi: 10.1093/hmg/ddg131. [DOI] [PubMed] [Google Scholar]
- Patel JP, Puck JM, Srinivasan R, Brown C, Sunderam U, Kundu K, Brenner SE, Gatti RA, Church JA. Nijmegen Breakage Syndrome Detected by Newborn Screening for T Cell Receptor Excision Circles (TRECs) J Clin Immunol. 2015;35:227. doi: 10.1007/s10875-015-0136-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirooznia M, Kramer M, Parla J, Goes FS, Potash JB, McCombie W, Zandi PP, Hodges E, Xuan Z, Balija V, Kramer M, Molla M, et al. Validation and assessment of variant calling pipelines for next-generation sequencing. Hum Genomics. 2014;8:14. doi: 10.1186/1479-7364-8-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruitt KD, Brown GR, Hiatt SM, Thibaud-Nissen F, Astashyn A, Ermolaeva O, Farrell CM, Hart J, Landrum MJ, McGarvey KM, Murphy MR, O’Leary NA, et al. RefSeq: an update on mammalian reference sequences. Nucleic Acids Res. 2014;42:D756–63. doi: 10.1093/nar/gkt1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Punwani D, Zhang Y, Yu J, Cowan MJ, Rana S, Kwan A, Adhikari AN, Lizama CO, Mendelsohn BA, Fahl SP, Chellappan A, Srinivasan R, et al. Multisystem Anomalies in Severe Combined Immunodeficiency with Mutant BCL11B. N Engl J Med. 2016;375:2165–2176. doi: 10.1056/NEJMoa1509164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson PN, Kohler S, Oellrich A, Wang K, Mungall CJ, Lewis SE, Washington N, Bauer S, Seelow D, Krawitz P, Gilissen C, Haendel M, et al. Improved exome prioritization of disease genes through cross-species phenotype comparison. Genome Res. 2014;24:340–348. doi: 10.1101/gr.160325.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez JM, Maietta P, Ezkurdia I, Pietrelli A, Wesselink J-J, Lopez G, Valencia A, Tress ML. APPRIS: annotation of principal and alternative splice isoforms. Nucleic Acids Res. 2013;41:D110–D117. doi: 10.1093/nar/gks1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster SC, Miller W, Ratan A, Tomsho LP, Giardine B, Kasson LR, Harris RS, Petersen DC, Zhao F, Qi J, Alkan C, Kidd JM, et al. Complete Khoisan and Bantu genomes from southern Africa. Nature. 2010;463:943–947. doi: 10.1038/nature08795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sifrim A, Popovic D, Tranchevent L-C, Ardeshirdavani A, Sakai R, Konings P, Vermeesch JR, Aerts J, De Moor B, Moreau Y. eXtasy: variant prioritization by genomic data fusion. Nat Methods. 2013;10:1083–1084. doi: 10.1038/nmeth.2656. [DOI] [PubMed] [Google Scholar]
- Statement IC. Idiopathic Pulmonary Fibrosis: Diagnosis and Treatment. Am J Respir Crit Care Med. 2000;161:646–664. doi: 10.1164/ajrccm.161.2.ats3-00. [DOI] [PubMed] [Google Scholar]
- Stavropoulos DJ, Merico D, Jobling R, Bowdin S, Monfared N, Thiruvahindrapuram B, Nalpathamkalam T, Pellecchia G, Yuen RKC, Szego MJ, Hayeems RZ, Shaul RZ, et al. Whole-genome sequencing expands diagnostic utility and improves clinical management in paediatric medicine. npj. Genomic Med. 2016;1:15012. doi: 10.1038/npjgenmed.2015.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenson PD, Ball EV, Mort M, Phillips AD, Shiel JA, Thomas NST, Abeysinghe S, Krawczak M, Cooper DN. Human Gene Mutation Database (HGMD): 2003 Update. Hum Mutat. 2003;21:577–581. doi: 10.1002/humu.10212. [DOI] [PubMed] [Google Scholar]
- Strom SP, Lee H, Das K, Vilain E, Nelson SF, Grody WW, Deignan JL. Assessing the necessity of confirmatory testing for exome-sequencing results in a clinical molecular diagnostic laboratory. Genet Med. 2014;16:510–515. doi: 10.1038/gim.2013.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent AL, Jordan CA, Cadzow MJ, Merriman TR, McGhee CN, YS R, WJ C, HY P, JM L, CB C, KR D, CA J, et al. Mutations in the Zinc Finger Protein Gene, ZNF469, Contribute to the Pathogenesis of Keratoconus. Investig Opthalmology Vis Sci. 2014;55:5629. doi: 10.1167/iovs.14-14532. [DOI] [PubMed] [Google Scholar]
- Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Moult J. SNPs, protein structure, and disease. Hum Mutat. 2001;17:263–70. doi: 10.1002/humu.22. [DOI] [PubMed] [Google Scholar]
- Yue P, Li Z, Moult J. Loss of protein structure stability as a major causative factor in monogenic disease. J Mol Biol. 2005;353:459–73. doi: 10.1016/j.jmb.2005.08.020. [DOI] [PubMed] [Google Scholar]
- Yue P, Melamud E, Moult J. SNPs3D: Candidate gene and SNP selection for association studies. BMC Bioinformatics. 2006;7:166. doi: 10.1186/1471-2105-7-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zawistowski M, Reppell M, Wegmann D, St Jean PL, Ehm MG, Nelson MR, Novembre J, Zöllner S. Analysis of rare variant population structure in Europeans explains differential stratification of gene-based tests. Eur J Hum Genet. 2014;22:1137–1144. doi: 10.1038/ejhg.2013.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.