

Abstract
Understanding the basis of complex trait disease is a fundamental problem in human genetics. The CAGI Crohn’s Exome challenges are providing insight into the adequacy of current disease models by requiring participants to identify which of a set of individuals has been diagnosed with the disease, given exome data. For the CAGI4 round, we developed a method that used the genotypes from exome sequencing data only to impute the status of Genome Wide Association Studies (GWAS) marker single nucleotide polymorphisms (SNPs). We then used the imputed genotypes as input to several machine learning methods that had been trained to predict disease status from marker SNP information. We achieved the best performance using Naïve Bayes and with a consensus machine learning method, obtaining an area under the curve (AUC) of 0.72, larger than other methods used in CAGI4. We also developed a model that incorporated the contribution from rare missense variants in the exome data, but this performed less well. Future progress is expected to come from the use of whole genome data rather than exomes.
Keywords: Crohn disease, complex disease risk model, Exome Sequencing, GWAS data, Machine Learning model, Naïve Bayes, CAGI
INTRODUCTION
Critical Assessment of Genome Interpretation (CAGI) community experiments provide a platform for assessing computational methods relating genotype to phenotype, where participants are required to make blind predictions (https://genomeinterpretation.org). For complex trait disease, there have been three rounds of Crohn disease exome challenges. In each round, participants were given exome data for a set of individuals, some of whom have been diagnosed with Crohn disease and some of whom have not. The challenge was then to assign a probability of each individual having the disease (https://genomeinterpretation.org/content/4-crohns-exomes). While accurate risk prediction has the potential to facilitate targeted preventive treatments, the broader goal of this type of challenge is to test and improve understanding of the underlying disease mechanism by testing mechanism hypotheses embedded in the prediction models.
Crohn disease, a subtype of inflammatory bowel disease (IBD), is a multifactorial complex disease, where several factors can confound disease risk prediction. It is not possible to identify Crohn patients from DNA sequence alone with complete accuracy for three primary reasons: First, environmental factors play a large role (Petersen et al. 2014) – co-occurrence in monozygotic twins is only 20–55% (Gordon et al. 2015). Second, there is a range of severity of disease, related to disease location (ileal vs. colonic Crohn disease) and age of onset (Cleynen et al. 2016). Several studies have reported rare monogenic forms of IBD for very early onset of the disease (Uhlig et al. 2014; Bianco et al. 2015) and pediatric Crohn patients show enhanced epithelial chemokine production compared with adult patients, suggesting a difference in early immune response between these two groups (Damen et al. 2006; Nieuwenhuis and Escher 2008). In spite of these differences, a high-density immunochip genotyping study did not identify differentiating genetic factors between early and late onset patients (Cutler et al. 2015). Third, some patients diagnosed with Crohn exhibit a wide range of monogenic defects affecting epithelial barrier and response, neutrophil numbers, phagocytosis and bacterial killing, innate hyper and autoinflammatory immune response, T and B cell selection and activation, regulatory T cells and immune regulation (Uhlig 2013). Thus, it is probably unreasonable to expect that one system level model of the disease will be adequate. This is reflected in current disease risk prediction models with ROC AUCs ranging from 0.56 to 0.72 (Ruderfer et al. 2010; Kang et al. 2011). It has been claimed that in theory, given its high heritability, an optimal method for predicting Crohn disease risk should be able to achieve a ROC AUC between 0.96 and 0.98 with the assumption that all Crohn disease risk loci have been identified and all the known genetic variances are explained by the genomic profile (Wray et al. 2010; Jostins and Barrett 2011). However, this theoretical maximum AUC limit is not attainable with linear models based on genetic factors known so far. (Witte et al. 2014) showed that 140 modest-risk variants together with 3 additional high-risk variants from (Jostins et al. 2012) could yield a maximum AUC of 0.77, and explaining about 16.4% of the total Crohn heritability of 72%.
Recently great progress has been made in identifying common variants (markers) associated with complex diseases using microarray based genome wide association studies (GWAS) (Welter et al. 2014). Because of sparse sampling of variants and extensive linkage disequilibrium in the human genome, these marker SNPs are almost always surrogates of the real causative variants, and so do not provide direct insight into the mechanisms underlying the statistical association with disease. As a consequence, they do not provide a basis for directly building a mechanistic model of the disease. Results from GWAS also only capture the contribution of common variants. The declining cost of high-throughput sequencing has begun to make exome sequencing accessible for reasonable sample sizes, and these data hold the promise of identifying both common and rare variants directly contributing to complex disease risk, albeit largely limited to coding region contributions.
The present analysis addresses the challenge of differentiating Crohn patients from healthy individuals with Crohn exome data (https://genomeinterpretation.org/content/4-crohns-exomes) using machine learning approaches. We have previously studied the mechanism landscape underlying seven complex trait diseases in a set of high confidence GWAS loci. In that work, we observed that for Crohn disease, expression plays the biggest role (~50%) compared to high impact missense or auxiliary splicing effects (Pal et al. 2015; Pal and Moult 2015). The variants affecting expression are generally in non-coding regions and it is non-trivial to assess their effects from exome sequencing data alone. So we hypothesized that GWAS markers would provide a better basis for prediction of disease risk than exome sequencing data only, as in principle, the markers associations reflect all underlying mechanisms. Based on this hypothesis, we used machine learning approaches to compare the performance of exome sequencing data vs. imputed marker SNP genotypes in Crohn disease risk prediction.
MATERIALS AND METHODS
Exome sequencing data
Exome sequencing data were available from the three rounds of CAGI Crohn’s exome challenges, held in 2016 (CAGI4) (https://genomeinterpretation.org/content/4-crohns-exomes), in 2013 (https://genomeinterpretation.org/content/crohns-disease-2013) and in 2011 (https://genomeinterpretation.org/content/crohns-disease-2011).
The CAGI4 Crohn challenge provided exome sequence data for 111 individuals. Post-challenge, we learned that these are composed of 64 cases and 47 controls. All exome sequencing data had been generated using the TruSeq exome enrichment kit (Illumina, San Diego, CA, USA) and the Illumina HiSeq2000 instrument and were called together using the Genome Analysis Toolkit (McKenna et al. 2010) (GATK 3.3-0) Haplotype Caller to avoid any data processing artifacts. Human genome build hg19 was used to map the chromosomal positions of the reads. The main challenge was to identify cases and controls among all individuals. Hierarchical clustering by variant similarity shows that all cases were unrelated in CAGI4 set, with two pairs of individuals related in the control set (Supp. Figure S1).
In 2013 data, the 66 exomes contained a total of 28 different pedigrees, including two monozygotic twin pairs, one pair being discordant. There are a total 51 cases and 15 controls in this dataset – this information was provided in the challenge page before prediction submission and the challenge was to identify which 51 individuals are cases and which 15 individuals are controls. All 66 exomes were generated using the TruSeq exome enrichment kit and were called together using the GATK program (McKenna et al. 2010) to avoid data processing artifacts. Chromosomal positions were mapped according to Human genome build hg19. A hierarchical clustering plot by variant similarity shows that all 51 cases were part of one or more pedigrees, while 8 out of 15 controls were unrelated (Supp. Figure S2). These eight controls have a high heterozygosity rate (Supp. Figure S3) compared to the rest of the individuals, making them easily separable from cases, just by clustering.
The earlier 2011 data consists of 56 individuals – 42 cases and 14 controls. The numbers of cases and controls were unknown before submission of predictions and the main challenge was to identify cases and controls from all these 56 exomes. These data contained one trio consisting of two healthy individuals and one with Crohn disease (Supp. Figure S4). For each individual, a separate VCF file was provided, generated using Illumina technology. Base-calling was done using the standard pipeline, consisting of BWA (Li and Durbin 2009), Picard (http://picard.sourceforge.net) and Samtools Pileup (Li et al. 2009). Here the chromosomal positions of the reads were mapped according to Human genome build hg18. In each VCF file, only positions where the observed base differed from the reference genome for one or both copies were reported – no information about missing data or concordance with reference genome bases was provided. We observed batch effects for eight control individuals who had been sequenced separately (Supp. Figure S4).
Compared to earlier data sets, the CAGI4 dataset is of much better quality in terms of both heterozygosity plots (Supp. Figure S5) and distribution of cases and controls within dendrograms (Supp. Figure S1). In some post-prediction analyses, we omitted eight controls from 2013 dataset with very high relative heterozygosity. We did not perform any post-prediction analysis on the 2011 data because of the uncertainty caused by unreported missing data.
Age of onset challenge for CAGI4 data
An additional aspect of the CAGI4 challenge was to identify which individuals had early onset Crohn disease, defined as diagnosis before age 10,. We checked for the presence of rare mutations in the IL10RA gene, which have been shown to be related to early disease onset (Kotlarz et al. 2012), but this approach was not successful. After the challenge was closed, we learned that 39 of the Crohn patients in the CAGI4 set were early onset (<= 10 years) and 25 patients were late onset (> 10 years). In the 2013 set, only four patients were early onset and in the 2011 set, all were late onset using this threshold.
Annotation of VCF files and QC filters
CAGI4 and 2013 exome VCF files were annotated using Varant (doi:10.5060/D2F47M2C), to provide region of occurrence (intron, exon, splice site or intergenic), observed minor allele frequencies (MAF) in the 1000 Genomes Project Phase3 database (Auton et al. 2015) or the ExAC database (Lek et al. 2016), mutation type, and predicted pathogenicity of missense variants. The RefGene (Pruitt et al. 2014) gene definition file was used for gene and transcript annotations in Varant. For the 2013 and CAGI4 data, we only used high quality data (GQ > 30 with ‘PASS’ filter).
Pre-processing of the exome data
PLINK (Purcell et al. 2007) was used to convert the exome VCF files to ped and bed files. These files were then used to calculate the heterozygosity rate per individual and to perform complete linkage agglomerative clustering based on the pairwise identity-by-state (IBS) similarity matrix (where two or more than two individuals share similar exome wide SNPs), providing a basis for hierarchical clustering. The R package ‘hclust’ was used to draw the cluster dendrogram of each set. For most GWAS loci, markers do not fall within the exome sequence, and for these, Impute2 (Howie et al. 2011) was used to impute the genotypes of GWAS marker SNPs for each individual from the exome data and using 1000 Genomes Project Phase3 data (Auton et al. 2015) as a reference. Pre-phasing of the data was performed using ShapeIT (Delaneau et al. 2012).
Choice of GWAS loci
Two sets of genome wide association study (GWAS) loci were used: a set of 90 we had previously compiled (Pal et al. 2015; Pal and Moult 2015) and a set of 140 from (Jostins et al. 2012). We were able to impute marker genotypes for 138 loci out of total 140 Crohn loci from (Jostins et al. 2012). Marker SNPs and corresponding odds ratios were extracted from the GWAS catalog (Welter et al. 2014). We also investigated a set of 573 SNPs (personal communication) from the IBD Genetics Consortium Crohn disease risk assessment, based on ~17,000 Crohn patients and ~22,000 controls typed on the immunochip (Wei et al. 2013). This work reported a ROC AUC of 0.86 for prediction of Crohn status, using machine-learning techniques. We were able to impute the genotypes of 473 of these SNPs from exome data, and these were used for the analysis. Supp. Figure S6 shows the relationships between these three loci sets. The 90 loci set is almost a subset of 138 loci set, with 83 loci common to the two sets. The 473 SNPs occur in 190 loci. Of these, 94 are not present in either of the two sets. 69 loci are common to all three sets.
Identification of high impact missense mutations
A missense variant in any Crohn disease related gene was considered high impact if at least two of five methods assigned a pathogenic score. The methods are SNPs3D profile (Yue et al. 2006), SNPs3d stability (Yue et al. 2005), Polyphen2 (Adzhubei et al. 2010), SIFT (Kumar et al. 2009) and CADD (Kircher et al. 2014). The relatively relaxed threshold of impact prediction (compared to that for monogenic disease missense mutations) was intended to include a higher fraction of true positives, at the expense of a higher fraction of false positives.
Methodology for the CAGI4 challenge
Our methods primarily used GWAS marker genotypes in each individual as a basis for assigning a risk of Crohn disease. Using those data, the relative probability of each individual being diagnosed with Crohn disease was estimated in three different ways (Figure 1):
Figure 1.
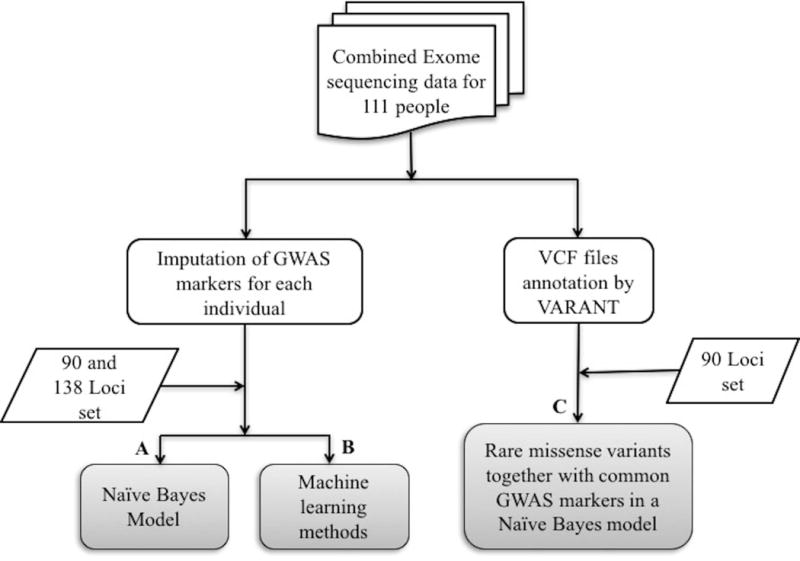
Methodology for CAGI-4 Crohn’s exome challenge.
- (A) With Naïve Bayes related models on GWAS markers: We estimated the relative risk of each individual being diagnosed with Crohn disease based on two untrained Naïve Bayes related models – ‘odds ratio’ based and ‘conditional probability’ based, applied on the 138 loci set and on the 90 loci set. For both the models, we addressed the question: what will be the disease risk for an individual, given the genotypes in a specified GWAS loci set? Now according to Bayes rule,
Where D is the disease risk of an individual with genotypes in ‘n’ GWAS loci. P(D) is the constant prior term which is unaffected by the variation in genotypes in ‘n’ loci. is the probability of genotypes in case individuals and is the probability of genotypes irrespective of the disease status. For both the models, we approximated the relative probabilities of each genotype occurring in case ( ) vs. controls ( ) using genotype frequencies derived from odds ratios and the minor allele frequency of the marker, assuming Hardy Weinberg equilibrium, as described in (Pal and Moult 2015).
For the ‘odds ratio’ model, we calculated heterozygous and minor homozygous odds ratios for each GWAS marker SNP, using the case and control genotype frequencies. The relative disease risk of each individual is then approximated as:
Where the genotypic odds ratio is or , depending on the genotypes of an individual at each of the ‘n’ loci.
For the ‘conditional probability’ based Naïve Bayes model, we approximated the conditional probabilities of an individual belonging to the case and control categories, given the genotypes in the ‘n’ loci set. That is, for each individual we calculated:
and,
Disease risk was taken to be proportional to the ratio / .
For the challenge submissions, each disease risk score from both the models was converted to probability by normalizing within 0 to 1.
(B) With machine learning methods using the GWAS markers: Models were developed for the 138 loci and 90 loci sets using the vector of marker SNP genotypes (values of 0, 1, or 2 minor alleles) for each individual as the input features. We explored four machine learning methods – Logistic Regression, Random Forest, Naïve Bayes (in Weka) and a Neural Network. In general, standard settings in Weka (Witten et al. 2016) were used for all machine learning models. For the Random Forest, we increased the number of trees from default of 100 to 1000 to improve the performance, and for the neural network we adjusted the number of hidden layers to 2. For training purposes, we generated 20 random samples of 1500 cases and 1500 controls, randomly selected from the 2000 cases and 3000 controls in the WTCCC1 Crohn microarray data (Burton et al. 2007). Benchmarking was done in two ways: on the 20 random sets of WTCCC1 500 cases and 500 controls not selected for training and on the 2013 CAGI Crohn exome data set. For each method, the version that performed best on the corresponding WTCCC1 test data was selected for use in CAGI4 challenge.
We also used a consensus machine learning approach – taking the average of the probabilities obtained from the Naïve Bayes (odds ratio based), Logistic Regression, and Random Forest methods. The Neural Network method performed the worst of all the methods, so was not included in this consensus. As both untrained Naïve Bayes models ((A) above) performed better than the Weka based trained Naïve Bayes model on the two benchmarking test sets, we chose one of the untrained Naïve Bayes methods for the consensus set.
(C) Inclusion of contributions from rare high impact missense variants: The ‘odds ratio’ based Naïve Bayes model for GWAS markers described in (A) was combined with contributions from rare predicted high impact missense exome variants (< 5% minor allele frequency in 1000 Genomes Phase3 dataset) in relevant genes. In our previous work, we have analyzed possible underlying mechanisms in each of the 90 Crohn disease loci set, identifying which have common variants in strong linkage disequilibrium with GWAS markers and are predicted high impact missense (Pal and Moult 2015), affect expression, or affect splicing (Pal et al. 2015). In this disease risk prediction model, we checked for rare variant contributions in that previously compiled set of high impact missense mechanism genes. If a GWAS marker risk allele is not present in a locus, we included the contribution of rare variants to the Naïve Bayes model with the same weight as used for common missense variants in that gene. Inclusion of the effect of rare missense variants in loci with putative expression and splicing mechanisms and those where no mechanism was assigned is complicated by not knowing the sign of the relationship between a marker SNP and a putative mechanism variant. For example, the marker SNP may be associated with increased Crohn risk, but a missense variant may have the opposite correlation – its presence is protective. For loci assigned common missense mechanisms, we have found that for about 2/3 of loci there is a positive linkage disequilibrium correlation between the presence of a risk marker allele and missense mechanism variants and in about 1/3 there is an anti-correlation (Pal and Moult 2015). We therefore used a weighted sum of the genotypic odds ratios (as calculated in (A) above) implied by the two genotypes possibilities for the rare predicted high impact missense variants from genes where the putative common variant mechanism is not missense (those with expression or splicing mechanisms), or from loci where the relationship between changed protein activity and the disease risk is unknown. For loci where the GWAS marker risk allele is present, we did not consider additional contributions from rare missense variants, assuming the common variant has a maximal effect.
Altogether, using different combinations of GWAS loci sets and machine learning models, we made six submissions (Table 1) for the Crohn CAGI4 challenge. In the post-challenge analysis of performance, area under the ROC curves (AUC) and confidence intervals from bootstrapping were obtained using the R package ‘fbroc’. The R package ‘pROC’ (Robin et al. 2011) was used to calculate P-values between pairs of ROC AUCs.
Table 1.
Performance of the six CAGI4 submissions
| Method description | GWAS loci used | AUC | 95% CI |
|---|---|---|---|
| Naïve Bayes model (conditional probability based) | 138 | 0.72 | [0.62–0.82] |
| Consensus machine learning method from Naïve Bayes (odds ratio based), Logistic regression and Random Forest (1000 trees) | 138 | 0.72 | [0.63–0.81] |
| Logistic regression | 138 | 0.71 | [0.61–0.80] |
| Logistic regression | 90 | 0.66 | [0.54–0.75] |
| Consensus machine learning method from Naïve Bayes (odds ratio based), Logistic regression and Random Forest (1000 trees) | 90 | 0.65 | [0.55–0.75] |
| Rare predicted high impact missense variant contribution together with common variant contribution in a Naïve Bayes model (odds ratio based) | 90 | 0.63 | [0.54–0.73] |
Methodology for the CAGI 2013 set
In CAGI 2013, we estimated Crohn disease risk in each individual directly from the exome sequencing data: We assumed that in each individual there is a subset of exome variants that directly influence disease risk through alteration of the activity level of relevant genes. The model assumes that the more deleterious missense SNVs an individual carries in Crohn relevant genes, the higher the probability they will have the disease. For each Crohn relevant gene, we identified any SNV in the exome data that is likely to have a significant impact on the function of that gene, based on Annovar annotation (Wang et al. 2010). There are four classes of potential impact SNVs: missense, nonsense, splicing (in a splice junction), and expression. For high impact missense SNVs we used predictions of deleteriousness from SNPs3D profile (Yue et al. 2006), SNPs3d stability (Yue et al. 2005), Polyphen2 (Adzhubei et al. 2010) and SIFT (Kumar et al. 2009). Any missense SNV in a relevant gene for which one or more of the four methods predicts deleterious was considered high impact. Nonsense and splice junctions were identified from Annovar annotations. Expression SNVs are defined as those SNVs that have been found to be associated with gene expression differences in two or more eQTL studies, as compiled in (Yu et al. 2016). Because of many uncertainties in the model, we estimated the relationship between impact SNV load and disease risk in four different ways, leading to four submissions in CAGI 2013 challenge. The most successful of these four, as judged by AUC, calculates relative disease risk as the summation of weighted maximum genotype values over 71 loci (Franke et al. 2010). In a locus containing more than one impact SNV, only the impact SNV with the highest genotype value will contribute to the final sum. Genotype values are 0 for no impact SNVs, 1 for a heterozygous impact SNV and 2 for homozygous impact SNVs. Ad hoc weights were assigned to different classes of impact SNVs as follows: Nonsense SNVs: 0.9; splice SNVs: 0.8; expression SNVs: 04; missense SNVs: 0.5 – 0.7 determined by the number of methods predicting deleteriousness for that SNV.
For comparison purposes, we have now also applied this method to the CAGI4 challenge data. Missense SNVs from 74 of the 138 loci set (Jostins et al. 2012) were included (for other loci, we did not have reliable identification of the genes involved in the disease mechanism). We also applied the most successful CAGI4 method (Naïve Bayes model on GWAS markers) to the CAGI 2013 challenge data to determine how well it performs on that set.
Methodology for the CAGI 2011 set
The model and methodology used for the 2011 CAGI challenge was the same as that for CAGI 2013, with the exception that we used only two missense impact prediction methods, SNPs3D profile (Yue et al. 2006) and SNPs3d stability (Yue et al. 2005). We made six CAGI 2011 submissions, each estimating the relationship between impact SNV load and disease risk in a different way. The most successful of these six, as judged by AUC, was the same genotype summation method as in CAGI 2013, also using the 71 loci in (Franke et al. 2010).
Monogenic Crohn disease data
In the post-challenge analysis, we extracted 22 monogenic Crohn disease like genes (i.e, associated with Crohn disease like immunopathology) from (Uhlig et al. 2014) together with appropriate inheritance patterns. These genes were checked for the presence of rare variants in early onset case individuals.
RESULTS
Imputation of GWAS markers
Only 9% and 7% of all marker SNPs fall within the exome data for 138 loci and 90 loci respectively. 18% of the markers are in intergenic regions for both sets. The remainder are in non-coding regions of genes. The imputation process usually generates three possible genotype probabilities for each position for each individual. If any of these genotype probabilities is greater than 0.8, then we considered that as the imputed genotype, otherwise the marker position was classified as ‘missing data’. Success of the imputation process depends on how close the GWAS marker position is to any SNP in the exome data. Supp. Figure S7 shows the fraction of GWAS marker SNPs that can be reliably imputed as a function of the distance between the marker and the nearest SNP in the exome data (distance range is from 0.0 Mb – 1.65 Mb). For most markers in intergenic regions and a few in UTRs (26 such markers), imputation was unsuccessful, so that there is high fraction of missing data (more than 50%) in these regions. Supp. Figure S8 shows the distribution of missing data across individuals for the 138 loci set. On average there are 22% missing data, with a minimum of 14% and a maximum of 29% and most of the values between 18 and 24%.
Submissions for the CAGI4 challenge
We submitted six prediction sets in the CAGI4 challenge (Table 1). Five of these addressed the contribution of common variants via imputed GWAS marker genotype information using various machine learning models. The sixth submission added contributions from rare variants present in the exome sequencing data to the GWAS marker information, using the odds ratio based Naïve Bayes model (details in Materials and Methods). Table 1 shows the performance of each of the six submitted methods, evaluated in terms of a ROC curve AUC. Performance is consistently better on the 138 loci set than on 90 loci set. Among all the methods, the Naïve Bayes method (conditional probability based) has the largest AUC, but the differences in performance between all the methods are small and not significantly different (based on p-values, Supp. Table S1). Figure 2A shows the ROC curve for the nominally best performing 138 loci Naïve Bayes model with an AUC of 0.72 and a 95% confidence interval of [0.62–0.82]. Figure 2B shows the predicted probabilities of each individual having Crohn, ordered from lowest to highest probability. A perfect model would group all cases at the top of the rankings, and all controls at the bottom. As can be seen in the plot, there is a clear signal in that respect, with the majority of points are in the appropriate part of the curve, but there is also substantial intermingling.
Figure 2.
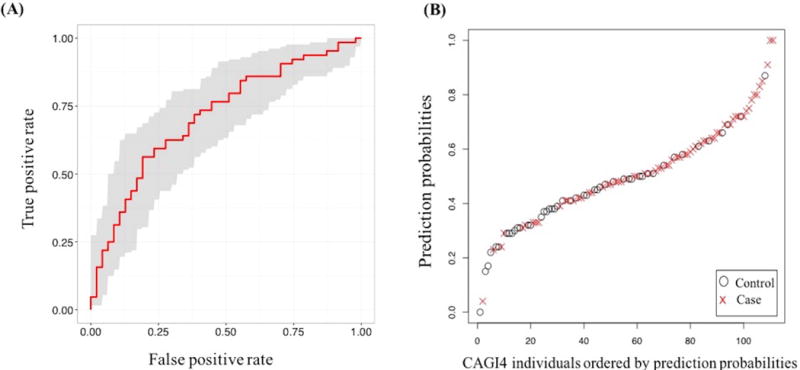
(A) ROC curve for the best performing CAGI4 Naïve Bayes model (conditional probability based) prediction with an AUC of 0.72. The shaded grey area shows the 95% confidence range. (B) CAGI4 Naïve Bayes model (conditional probability based) prediction probabilities, ranked from lowest to the highest probability. Open circles represent controls and red crosses represent cases.
Effect of variation in loci set
As noted above, the performance of predictions based on the 138 loci set is consistently better than that on the 90 loci set. We also investigated the performance of a larger set of marker SNPs, using the same methods, based on a set of 573 SNPs, prioritized for Crohn disease by the IBD Genetics Consortium (Wei et al. 2013). The genotypes of 473 of these were imputable from the exome data. Figure 3 shows AUC values (together with their 95% confidence intervals) for seven different methods for three association sets. In general, the results for the 138 loci set are the best, with the 473 SNPs set AUCs in between the 138 and the 90 loci values. An exception to this pattern is the poor performance of the non-machine learning Naïve Bayes methods (NBCP and NBOR) on the 473 SNPs set. A possible explanation for the generally moderate to low performance of the 473 SNPs set, given its size, is that these SNPs span only 190 loci, with 45% of the loci containing multiple SNPs. The non-machine learning Naïve Bayes methods are strictly additive and so more sensitive to the uneven weighting of loci, while machine learning methods may partly handle this imbalance. Additionally, the 473 SNPs set is not as well established as the two GWAS sets.
Figure 3.
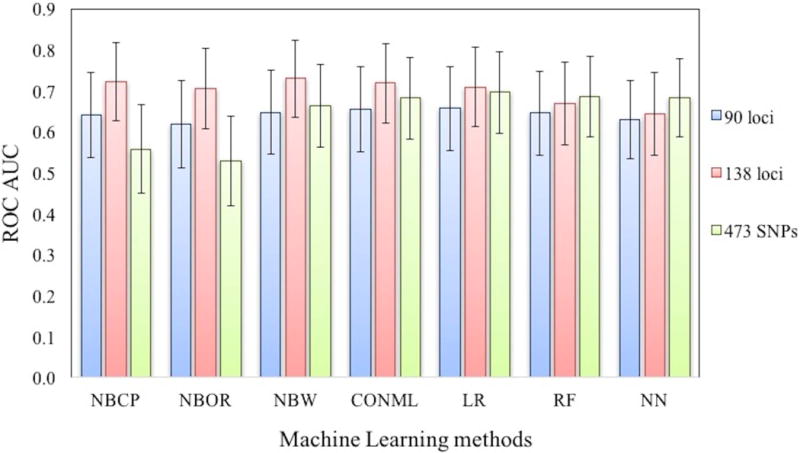
AUC performance of machine learning methods with three Crohn’s association sets (90 loci, 138 loci and 473 SNPs set). 95% confidence intervals of AUCs for each method are shown as error bars in the plot. The methods are conditional probability based Naïve Bayes (NBCP), odds ratio based Naïve Bayes (NBOR), Weka based Naïve Bayes (NBW), Logistic Regression (LR), Neural Net (NN), Random Forest (RF), and consensus machine learning method among NBOR, LR and RF (CONML). The difference in performance between the individual methods is small.
We also examined the sensitivity of the results to the choice of training sample used (details in Methods). Supp. Figure S9 shows the spread of AUCs over the 20 random samples tested in training for all machine learning methods for all three association sets. In the challenge, for each method the sample that gave the best AUC against WTCCC1 test data was selected. While there is some spread of AUC values across training samples (Supp. Table S2), values used in the challenge (Figure 3) are representative of overall relative performance across training samples, except for Logistic Regression on the 473 SNPs set. For that method, the best performing test sample turned out to be by far the best performing on the challenge data (outlier high point in Supp. Figure S9).
Robustness of the methods
Among the machine learning methods used, Neural Nets and Random Forests are capable of capturing nonlinear relationships present in the data, while Naïve Bayes and Logistic Regression are not. We do not observe any significant difference in the AUC values obtained with the different classes of methods (Table 1, Supp. Table S1), implying either that there is not much non-linear information in the data or, more likely, that the Neural Net or Random Forest structures used here were not capable of capturing that information.
We also examined the effect of training set size on the accuracy of the four machine learning methods (Supp. Figure S10). The median AUC values of all four methods plateau at training sizes up to half that used, so that training set size did not limit performance. However, for the neural net, fluctuations in the interquartile range are relatively high even with data sets more than 25% larger than that used. This instability suggests the neural net model is less reliable with limited training data.
A limitation of using the GWAS marker SNPs as the basis for predicting disease risk is that it is not possible to reliably impute all the genotypes from the exome data. We examined whether or not the resulting average of 22% missing data had a significant effect on accuracy of the best result (conditional probability Naïve Bayes on the 138 loci set) as follows. The CAGI4 individuals were divided into eight subsets, four with relatively low missing data and four with relatively high missing data (Supp. Table S3). The subsets show no obvious trend for higher accuracy with less missing data, and pairwise P values between AUCs also show no significant effect. A possible confounding factor in these comparisons is that samples with low missing data may tend to contain more erroneous genotypes. To control for this effect, we increased the threshold P value for accepted genotypes from 0.8 to 0.9 (increasing the average missing data to 29%) and repeated the analysis. Here too, there is no significant difference in accuracy as a function of % missing data. Although this analysis is somewhat limited by moderate sample sizes, the results are consistent with the amount of missing data not being a limiting factor on accuracy.
Rare variant model
Addition of the contribution of predicted high impact rare missense variants to that from imputed marker SNPs in the Naïve Bayes model (odds ratio based) on the 90 loci set (details in Methods) produced an AUC of 0.63 (Table 1), similar to that from using imputed marker SNPs alone. Thus, this rare variant model did not improve the disease risk prediction performance. Supp. Figure S11 shows the distribution of predicted high impact rare missense variants found in for those loci where the GWAS marker risk allele is absent. For missense loci where the risk marker allele is present we did not include any contribution from rare missense variants, assuming the common variant had a maximal effect. Counts of such rare high impact missense variants are low, with no variants in an individual the most usual situation. Thus it is not surprising that our rare variant model made no difference to the results. However, we do find that for missense loci where the GWAS marker risk allele is absent, there is a weak signal for a higher level of rare high impact missense variants in cases than controls, in partial support of the model used (P value = 0.0343, chi-square test).
We also searched the CAGI4 data for predicted high impact rare variants in 22 monogenic Crohn disease like genes (Uhlig et al. 2014), as early onset cases have been found to sometimes carry very rare variants with large effect sizes. In the CAGI4 data, we found only two early onset cases (2 and 7 years) carrying autosomal dominant mutations in GUCY2C, with symptoms of familial diarrhea, a monogenic form of IBD. However, one of these mutations (p.Q784K) is deleterious only by CADD (Kircher et al. 2014) and the other mutation (I514T) is deleterious only by both polyphen-2 (Adzhubei et al. 2010) and CADD (Kircher et al. 2014). Also, (Uhlig et al. 2014) mention that symptoms for mutations in this gene are not usually early onset, but develop during adulthood, mainly after 20 years of age. More detailed information about clinical symptoms of these patients are needed to resolve whether these rare variants are really causative variants.
Application of the best CAGI4 method to the 2013 CAGI Crohn challenge dataset
We also tested how the CAGI4 methods perform on the earlier 2013 CAGI challenge set (Supp. Figure S12). The best performing method in CAGI4 (conditional probability Naïve Bayes) returns a high AUC of 0.81, compared with the CAGI4 0.72. The four machine learning methods do not perform as well however. The distorted data architecture in the 2013 challenge makes these results hard to interpret.
As there is no major homozygous genotype information in the 2011 CAGI Crohn dataset, we were unable to apply all methods there.
Application of the exome variant based method to the CAGI4 data
We also investigated the performance of the model we developed for the 2013 Crohn challenge on the CAGI4 138 loci data. That method is based on the exome impact variant load in each individual (details in Methods). Inclusion of impact variants (both common or rare) in the 74 loci where exome impact variants were found, results in a low AUC of 0.59.
Using that method on the CAGI 2013 dataset with 71 loci (Franke et al. 2010) (excluding the eight controls which are easily separable by clustering, Supp. Figure S2), we obtained a higher AUC of 0.71 (the best method of four submissions). For the CAGI 2011 set on the same 71 loci, an exome based method (details in Methods) generated an even higher AUC of 0.81, excluding the eight controls which are easily separable by clustering (Supp. Figure S4). But performance on these datasets is not comparable with that on the CAGI4 set, as both earlier sets have case/control substantial biases.
DISCUSSION
Microarray versus exome data for estimating Crohn disease risk
At first glance, it seems obvious that since exome data is so much richer than microarray results, it should help to interpret disease mechanism, and by implication improve our ability to predict who is at most risk for particular diseases. Microarray data only provide associations between the presence of specific SNPs represented on the chip and disease phenotypes. Correlation is not cause, and because of the extensive linkage disequilibrium in the human genome, and the sparse representation of variants on a microarray, it is unlikely that the marker SNPs are themselves involved in disease mechanism. Yet our approach of only using the exome data to impute the status of microarray derived marker SNPs proved the best predictor of Crohn risk in the CAGI4 challenge, both in comparison with those of other CAGI participants and with our own current and previous exome oriented methods. The result is even more surprising, given the difficulties of reliably imputing marker status from exome sequence, resulting in substantial missing data. There are two primary reasons for the better performance of the marker SNP method. First, unlike monogenic disease, many of the variants involved in complex trait disease mechanism are not in exons. In our earlier analysis (Pal et al. 2015) we found that about 50% of Crohn loci involve mechanisms affecting gene expression, and often the corresponding variant is not included in exome data. Second, and more significantly, even if the mechanism variant is included in exome capture, it is necessary to know which one it is and the relationship between the presence of the variant and disease risk –does the presence of the variant increase or decrease risk, and by how much. Our previous work allowed us to derive these data fairly completely for the approximately 1/3 of Crohn loci where a missense variant mechanism is involved. In about 1/3 of those loci, the mechanism missense SNP is protective – its presence reduces the risk of disease, so assuming a variant always increases risk is a poor approximation. Thus knowing the sign of the risk/SNP relationship is critical. For expression and splicing mechanisms in non-missense loci, in most cases we were not able to identify which SNP is directly involved in mechanism or whether its presence represented increased or decreased disease risk. These uncertainties about underlying disease mechanism and the limited coverage of exome data result in microarray data currently providing a more accurate approach to estimating Crohn disease risk. In future, whole genome data rather than just exome sequence, together with more extensive data on which SNPs are associated with expression changes (Melé et al. 2015), will make a more detailed mechanism model more tractable. Issues of which cell type expression changes are relevant will still remain, however.
Inclusion of more loci improves prediction accuracy
We considered three disease association sets one with 90 GWAS loci, one with a 138 GWAS loci, and a set of 473 SNPs spanning 190 loci. For all methods, accuracy is higher on the 138 disease loci than on 90 loci. (As noted earlier, for 473 SNPs set, issues with the varying number of SNPs in each loci complicate the analysis, so it is not possible to draw conclusions on the relationship between number of loci and accuracy). Up to now, as study sizes have become larger, the number of disease associated loci has increased for Crohn and other complex traits (Park et al. 2010). That suggests there is an approximately power law relationship between the effect size of loci and the number of loci – a long tail of yet to be discovered loci that nevertheless contribute a significant amount of disease risk signal. Thus, study size is one of the factors limiting accuracy in predicting Crohn disease risk.
Role of rare variants
A second widely discussed limitation on accuracy of predicting disease risk is the role of rare variants (Kiezun et al. 2012; Maher et al. 2012). We attempted to add this effect by including the contribution of rare missense variants present in relevant genes in GWAS loci. In practice, there are multiple limitations in doing this. First, in many loci it is not yet clear which gene or genes are involved in the mechanism. We adopted a conservative approach of only including contributions from reasonably well established mechanism genes, thus omitting many possible contributions. Second, as also discussed above, it is necessary to know the sign of the relationship between the presence of the variant and the disease risk. For loci where we had previously found a common missense SNP mechanism, we assumed the sign is same for all missense variants. For other loci, we used a weighted sum of the two possibilities, a poor approximation. Third, there is an issue of which rare variants will contribute. In loci where a large change in protein activity is necessary to significantly affect risk (those where there are common, high impact, missense mechanism SNPs), it is reasonable to only include rare predicted high impact missense variants, and that is what we did. But in loci where more subtle effects significantly contribute to disease risk, such as those where a SNP related expression mechanism is implied, it would be necessary to estimate the effect of all rare variants present – missense and those affecting expression. At present, there is no reliable way of doing that, and we did not include this contribution. Fourth, at best GWAS studies only identify mechanism genes where there happens to be a common SNP that affects disease risk. It is likely there are many more genes, sometimes in the same pathways, sometimes not, where there are no significant common SNP, but rare variants play a role (https://doi.org/10.1101/077180), (Rivas et al. 2011). Indeed, we have shown that genes where activity is most tightly coupled to disease risk, such as drug targets, are less likely to contain common disease related SNPs (Cao and Moult 2014). At present, there is no effective way of identifying these non-GWAS detectable mechanism genes, and so their contribution is also omitted from our model. Given all these caveats, it is not surprising that our inclusion of rare variants did not improve prediction accuracy, and that should certainly not be taken as evidence that rare variants are not important.
Future prospects
As discussed above, the obstacles to developing really effective mechanism based methods for predicting disease risk are formidable. There are additional difficulties, such as unidentified epistatic effects (Maki-Tanila and Hill 2014). Nevertheless, larger GWAS study sizes, fine grained follow-up investigations (for example (Gorlatova et al. 2011; Kauder et al. 2013) for the MSP locus), and improved computational models will make a difference, and so it is important that CAGI continue to include complex trait disease challenges of this type.
Supplementary Material
Acknowledgments
We wish to express our thanks and appreciate to the Crohn challenge data providers, Prof. Dr. Andre Franke & Dr Britt-Sabina Petersen, for making these challenges possible.
GRANT SPONSOR
This work was supported by NIH R01GM104436 to JM and R01GM102810 to JM and Osnat Herzberg. The CAGI experiment coordination is supported by NIH U41 HG007446 and the CAGI conference by NIH R13 HG006650.
References
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–9. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auton A, Abecasis GR, Altshuler DM, Durbin RM, Abecasis GR, Bentley DR, Chakravarti A, Clark AG, Donnelly P, Eichler EE, Flicek P, Gabriel SB, et al. A global reference for human genetic variation. Nature. 2015;526:68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianco AM, Girardelli M, Tommasini A. Genetics of inflammatory bowel disease from multifactorial to monogenic forms. World J Gastroenterol. 2015;21:12296–310. doi: 10.3748/wjg.v21.i43.12296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton PR, Clayton DG, Cardon LR, Craddock N, Deloukas P, Duncanson A, Kwiatkowski DP, McCarthy MI, Ouwehand WH, Samani NJ, Todd JA, Donnelly P, et al. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao C, Moult J. GWAS and drug targets. BMC Genomics. 2014;15(Suppl 4):S5. doi: 10.1186/1471-2164-15-S4-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleynen I, Boucher G, Jostins L, Schumm LP, Zeissig S, Ahmad T, Andersen V, Andrews JM, Annese V, Brand S, Brant SR, Cho JH, et al. Inherited determinants of Crohn’s disease and ulcerative colitis phenotypes: a genetic association study. Lancet. 2016;387:156–167. doi: 10.1016/S0140-6736(15)00465-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutler DJ, Zwick ME, Okou DT, Prahalad S, Walters T, Guthery SL, Dubinsky M, Baldassano R, Crandall WV, Rosh J, Markowitz J, Stephens M, et al. Dissecting Allele Architecture of Early Onset IBD Using High-Density Genotyping. PLoS One. 2015;10:e0128074. doi: 10.1371/journal.pone.0128074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damen GM, Hol J, de Ruiter L, Bouquet J, Sinaasappel M, van der Woude J, Laman JD, Hop WCJ, Büller HA, Escher JC, Nieuwenhuis EES. Chemokine production by buccal epithelium as a distinctive feature of pediatric Crohn disease. J Pediatr Gastroenterol Nutr. 2006;42:142–9. doi: 10.1097/01.mpg.0000189336.70021.8a. [DOI] [PubMed] [Google Scholar]
- Delaneau O, Marchini J, Zagury J-F. A linear complexity phasing method for thousands of genomes. Nat Methods. 2012;9:179–81. doi: 10.1038/nmeth.1785. [DOI] [PubMed] [Google Scholar]
- Franke A, McGovern DPB, Barrett JC, Wang K, Radford-Smith GL, Ahmad T, Lees CW, Balschun T, Lee J, Roberts R, Anderson CA, Bis JC, et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat Genet. 2010;42:1118–25. doi: 10.1038/ng.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon H, Trier Moller F, Andersen V, Harbord M. Heritability in Inflammatory Bowel Disease. Inflamm Bowel Dis. 2015;21:1428–1434. doi: 10.1097/MIB.0000000000000393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorlatova N, Chao K, Pal LR, Araj RH, Galkin A, Turko I, Moult J, Herzberg O. Protein characterization of a candidate mechanism SNP for Crohn’s disease: the macrophage stimulating protein R689C substitution. PLoS One. 2011;6:e27269. doi: 10.1371/journal.pone.0027269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howie B, Marchini J, Stephens M. Genotype imputation with thousands of genomes. G3 (Bethesda) 2011;1:457–70. doi: 10.1534/g3.111.001198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jostins L, Barrett JC. Genetic risk prediction in complex disease. Hum Mol Genet. 2011;20:R182–8. doi: 10.1093/hmg/ddr378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, Lee JC, Schumm LP, Sharma Y, Anderson CA, Essers J, Mitrovic M, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491:119–24. doi: 10.1038/nature11582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang J, Kugathasan S, Georges M, Zhao H, Cho JH, NIDDK IBD Genetics Consortium Improved risk prediction for Crohn’s disease with a multi-locus approach. Hum Mol Genet. 2011;20:2435–42. doi: 10.1093/hmg/ddr116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauder SE, Santell L, Mai E, Wright LY, Luis E, N’Diaye EN, Lutman J, Ratti N, Sa SM, Maun HR, Stefanich E, Gonzalez LC, et al. Functional consequences of the macrophage stimulating protein 689C inflammatory bowel disease risk allele. PLoS One. 2013;8:e83958. doi: 10.1371/journal.pone.0083958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiezun A, Garimella K, Do R, Stitziel NO, Neale BM, McLaren PJ, Gupta N, Sklar P, Sullivan PF, Moran JL, Hultman CM, Lichtenstein P, et al. Exome sequencing and the genetic basis of complex traits. Nat Genet. 2012;44:623–30. doi: 10.1038/ng.2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310–315. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotlarz D, Beier R, Murugan D, Diestelhorst J, Jensen O, Boztug K, Pfeifer D, Kreipe H, Pfister E, Baumann U, Puchalka J, Bohne J, et al. Loss of Interleukin-10 Signaling and Infantile Inflammatory Bowel Disease: Implications for Diagnosis and Therapy. Gastroenterology. 2012;143:347–355. doi: 10.1053/j.gastro.2012.04.045. [DOI] [PubMed] [Google Scholar]
- Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–81. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, Oanne H, Ware J, Hill andrew J, Cummings BB, Tukiainen T, Birnbaum DP, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher MC, Uricchio LH, Torgerson DG, Hernandez RD. Population Genetics of Rare Variants and Complex Diseases. Hum Hered. 2012;74:118–128. doi: 10.1159/000346826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maki-Tanila A, Hill WG. Influence of Gene Interaction on Complex Trait Variation with Multilocus Models. Genetics. 2014;198:355–367. doi: 10.1534/genetics.114.165282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melé M, Ferreira PG, Reverter F, DeLuca DS, Monlong J, Sammeth M, Young TR, Goldmann JM, Pervouchine DD, Sullivan TJ, Johnson R, Segrè AV, et al. Human genomics. The human transcriptome across tissues and individuals. Science. 2015;348:660–5. doi: 10.1126/science.aaa0355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieuwenhuis EES, Escher JC. Early onset IBD: what’s the difference? Dig Liver Dis. 2008;40:12–5. doi: 10.1016/j.dld.2007.07.166. [DOI] [PubMed] [Google Scholar]
- Pal LR, Moult J. Genetic basis of common human disease: Insight into the role of missense SNPs from genome-wide association studies. J Mol Biol. 2015;427:2271–2289. doi: 10.1016/j.jmb.2015.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal LR, Yu C-H, Mount SM, Moult J. Insights from GWAS: emerging landscape of mechanisms underlying complex trait disease. BMC Genomics. 2015;16(Suppl 8):S4. doi: 10.1186/1471-2164-16-S8-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J-H, Wacholder S, Gail MH, Peters U, Jacobs KB, Chanock SJ, Chatterjee N. Estimation of effect size distribution from genome-wide association studies and implications for future discoveries. Nat Genet. 2010;42:570–5. doi: 10.1038/ng.610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen B-S, Spehlmann ME, Raedler A, Stade B, Thomsen I, Rabionet R, Rosenstiel P, Schreiber S, Franke A. Whole genome and exome sequencing of monozygotic twins discordant for Crohn’s disease. BMC Genomics. 2014;15:564. doi: 10.1186/1471-2164-15-564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruitt KD, Brown GR, Hiatt SM, Thibaud-Nissen F, Astashyn A, Ermolaeva O, Farrell CM, Hart J, Landrum MJ, McGarvey KM, Murphy MR, O’Leary NA, et al. RefSeq: an update on mammalian reference sequences. Nucleic Acids Res. 2014;42:D756–63. doi: 10.1093/nar/gkt1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, Maller J, Sklar P, de Bakker PIW, Daly MJ, Sham PC. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivas MA, Beaudoin M, Gardet A, Stevens C, Sharma Y, Zhang CK, Boucher G, Ripke S, Ellinghaus D, Burtt N, Fennell T, Kirby A, et al. Deep resequencing of GWAS loci identifies independent rare variants associated with inflammatory bowel disease. Nat Genet. 2011;43:1066–1073. doi: 10.1038/ng.952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robin X, Turck N, Hainard A, Tiberti N, Lisacek F, Sanchez J-C, Müller M. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics. 2011;12:77. doi: 10.1186/1471-2105-12-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruderfer DM, Korn J, Purcell SM. Family-based genetic risk prediction of multifactorial disease. Genome Med. 2010;2:2. doi: 10.1186/gm123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlig HH. Monogenic diseases associated with intestinal inflammation: implications for the understanding of inflammatory bowel disease. Gut. 2013;62:1795–805. doi: 10.1136/gutjnl-2012-303956. [DOI] [PubMed] [Google Scholar]
- Uhlig HH, Schwerd T, Koletzko S, Shah N, Kammermeier J, Elkadri A, Ouahed J, Wilson DC, Travis SP, Turner D, Klein C, Snapper SB, et al. The diagnostic approach to monogenic very early onset inflammatory bowel disease. Gastroenterology. 2014;147:990–1007.e3. doi: 10.1053/j.gastro.2014.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164–e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Z, Wang W, Bradfield J, Li J, Cardinale C, Frackelton E, Kim C, Mentch F, Van Steen K, Visscher PM, Baldassano RN, Hakonarson H, et al. Large Sample Size, Wide Variant Spectrum, and Advanced Machine-Learning Technique Boost Risk Prediction for Inflammatory Bowel Disease. Am J Hum Genet. 2013;92:1008–1012. doi: 10.1016/j.ajhg.2013.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welter D, MacArthur J, Morales J, Burdett T, Hall P, Junkins H, Klemm A, Flicek P, Manolio T, Hindorff L, Parkinson H. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2014;42:D1001–6. doi: 10.1093/nar/gkt1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witte JS, Visscher PM, Wray NR. The contribution of genetic variants to disease depends on the ruler. Nat Rev Genet. 2014;15:765–776. doi: 10.1038/nrg3786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witten IH, Frank EHMP. Data Mining. Fourth Morgan Kaufmann; 2016. Practical Machine Learning Tools and Techniques. [Google Scholar]
- Wray NR, Yang J, Goddard ME, Visscher PM. The genetic interpretation of area under the ROC curve in genomic profiling. PLoS Genet. 2010;6:e1000864. doi: 10.1371/journal.pgen.1000864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu C-H, Pal LR, Moult J. Consensus Genome-Wide Expression Quantitative Trait Loci and Their Relationship with Human Complex Trait Disease. Omi A J Integr Biol. 2016;20:400–414. doi: 10.1089/omi.2016.0063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue P, Li Z, Moult J. Loss of protein structure stability as a major causative factor in monogenic disease. J Mol Biol. 2005;353:459–73. doi: 10.1016/j.jmb.2005.08.020. [DOI] [PubMed] [Google Scholar]
- Yue P, Melamud E, Moult J. SNPs3D: candidate gene and SNP selection for association studies. BMC Bioinformatics. 2006;7:166. doi: 10.1186/1471-2105-7-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.