

Significance
Myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) devastates the lives of millions of people and has remained a mystery illness despite decades of research. It has long been suspected that inflammation is central to its pathogenesis. Although only two cytokines were found to be different (TGF-β higher and resistin lower) in ME/CFS patients compared with controls, 17 cytokines correlated with ME/CFS severity. Thirteen of these cytokines are proinflammatory and may contribute to many of the symptoms these patients experience for several years. Only CXCL9 (MIG) inversely correlated with fatigue duration.
Keywords: cytokines, chronic fatigue syndrome, immune monitoring, severity, myalgic encephalomyelitis
Abstract
Although some signs of inflammation have been reported previously in patients with myalgic encephalomyelitis or chronic fatigue syndrome (ME/CFS), the data are limited and contradictory. High-throughput methods now allow us to interrogate the human immune system for multiple markers of inflammation at a scale that was not previously possible. To determine whether a signature of serum cytokines could be associated with ME/CFS and correlated with disease severity and fatigue duration, cytokines of 192 ME/CFS patients and 392 healthy controls were measured using a 51-multiplex array on a Luminex system. Each cytokine’s preprocessed data were regressed on ME/CFS severity plus covariates for age, sex, race, and an assay property of newly discovered importance: nonspecific binding. On average, TGF-β was elevated (P = 0.0052) and resistin was lower (P = 0.0052) in patients compared with controls. Seventeen cytokines had a statistically significant upward linear trend that correlated with ME/CFS severity: CCL11 (Eotaxin-1), CXCL1 (GROα), CXCL10 (IP-10), IFN-γ, IL-4, IL-5, IL-7, IL-12p70, IL-13, IL-17F, leptin, G-CSF, GM-CSF, LIF, NGF, SCF, and TGF-α. Of the 17 cytokines that correlated with severity, 13 are proinflammatory, likely contributing to many of the symptoms experienced by patients and establishing a strong immune system component of the disease. Only CXCL9 (MIG) inversely correlated with fatigue duration.
Myalgic encephalomyelitis or chronic fatigue syndrome (ME/CFS) is a complex and debilitating disease of unknown etiology affecting more than one million Americans and millions of individuals worldwide (1, 2). ME/CFS is characterized by persistent or relapsing unexplained fatigue of at least 6-mo duration that is not alleviated by rest and results in a substantial reduction in previous levels of occupational, educational, social, and personal activities (2–5). In ME/CFS patients, fatigue is just one of multiple incapacitating symptoms that include cognitive impairment, postexertional malaise, unrefreshing sleep, headaches, myalgias, arthralgias, sore throats, lymphadenopathy, hypersensitivity to noise, light, or certain food items, and autonomic disturbances (4). These symptoms often cluster in each patient in varying combinations and intensity. “ME/CFS” has been the term generally preferred by researchers, but the terms “myalgic encephalomyelitis” (ME) or “chronic fatigue and immune dysfunction syndrome” (CFIDS) are favored by clinicians and patients, given the wide range of complaints and the heterogeneity of the illness (1, 4, 6, 7). In a recent report, the Institute of Medicine proposed a new definition for ME/CFS and a new name: “systemic exertion intolerance disease” (SEID) (2).
The presence of ongoing or fluctuating flu-like symptoms, arthralgias, myalgias, autonomic disturbances, and a striking hypersensitivity to stimuli in many patients with this illness has led to the suspicion that ME/CFS is an inflammatory or immunological disorder (8). Surprisingly, conventional markers of inflammation commonly used in the daily practice of medicine (e.g., erythrocyte sedimentation rate, C-reactive protein) are seldom elevated in ME/CFS patients (9). Tests measuring innate and adaptive immune responses have been reported as abnormal but often yield negative or conflicting results (8, 10–12). However, in a longitudinal study, fatigue severity was associated with daily fluctuations of the inflammatory adipokine leptin (13). Also, many studies have found increased numbers of circulating cytotoxic CD8+ cells bearing activation antigens (8, 14–16). In addition, in a cross-sectional study, Hornig et al. (17) reported a distinct cytokine inflammatory signature associated with early disease.
One large epidemiological study reported a higher risk of non-Hodgkin’s lymphoma (NHL) [odds ratio (OR) = 1.29, 95% CI = 1.16–1.43, P value < 0.0001], marginal zone lymphoma (MZL) (OR = 1.88, 95% CI = 1.38–2.57), and diffuse large B-cell lymphoma (DLBCL) (OR = 1.34, 95% CI = 1.12–1.61) in patients older than 65 y of age with ME/CFS (15). The state of chronically activated cellular immunity that has been reported by several laboratories might plausibly explain this association.
The purpose of the present study was to use a comprehensive immune-profiling approach to determine whether an abnormal profile of circulating cytokines could be identified in ME/CFS patients and whether this profile correlated with disease severity and/or fatigue duration.
Results
Basic Demographics.
ME/CFS patients and healthy controls had a comparable age (49.9 and 50.1 y, respectively) and sex distribution (76.6 and 77.3% female, respectively) as expected from the age and sex-matched design (Table 1). The ME/CFS patient group had a higher proportion of Caucasian individuals (91.7%) compared with healthy controls (71.2%; P < 0.0001). Race data were missing for 10 participants. Six of the ten participants for whom race data were lacking were ME/CFS cases, and four were controls. The six ME/CFS patients did not differ from the 186 ME/CFS cases that were included in the study. The four controls did not differ from the 388 controls included in the study. Because of their missing race data, these 10 participants were excluded from the cytokine analysis, yielding a final sample size of 574 individuals.
Table 1.
Study population demographics
| Cases | Healthy controls | ||||
| Characteristics | N | % | N | % | P value |
| Total number of participants | 192 | 100.0 | 392 | 100.0 | |
| Age, mean ± SD | 49.9 ± 12.7 | 50.1 ± 12.5 | 0.8576* | ||
| Sex | 0.8349† | ||||
| Female | 147 | 76.6 | 303 | 77.3 | |
| Male | 45 | 23.4 | 89 | 22.7 | |
| Race | <0.0001‡ | ||||
| Asian | 5 | 2.6 | 38 | 9.7 | |
| Hispanic | 3 | 1.6 | 21 | 5.4 | |
| Black | 1 | 0.5 | 32 | 8.2 | |
| White | 176 | 91.7 | 279 | 71.2 | |
| All other | 1 | 0.5 | 18 | 4.6 | |
| No data | 6 | 3.1 | 4 | 1.0 | |
| 1994 CDC case definition | |||||
| Impaired memory | 184 | 95.8 | 4 | 1.0 | <0.0001‡ |
| Sore throat | 117 | 60.9 | 1 | 0.3 | <0.0001‡ |
| Tender lymph nodes | 118 | 61.5 | 2 | 0.5 | <0.0001‡ |
| Muscle pain | 175 | 91.2 | 10 | 2.6 | <0.0001‡ |
| Multijoint pain | 132 | 68.8 | 22 | 5.6 | <0.0001‡ |
| New headaches | 137 | 71.4 | 30 | 7.7 | <0.0001‡ |
| Unrefreshing sleep | 186 | 96.9 | 8 | 2.0 | <0.0001‡ |
| Postexertional malaise | 186 | 96.9 | 3 | 0.8 | <0.0001‡ |
Note: 1994 CDC Case Definition P values did not change when excluding observations in which the participant’s response was unknown or in which no data were provided. Race data were missing for 10 participants. These 10 participants were excluded from the cytokine analysis to adjust for race. Thus, the sample size for the cytokine analysis is 574 participants.
Case vs. control comparison used t test for unequal variances.
Case vs. control comparison used Fisher’s exact test.
Case vs. control comparison used Freeman–Halton exact test with Monte Carlo approximation.
Cytokine findings presented in this section are only those following appropriate adjustment for nonspecific binding. These analyses were performed separately by cytokine. Cytokine preprocessed median fluorescence intensity (pMFI) was regressed on age, sex, race, and nonspecific binding. Additionally, only findings that were statistically significant following adjustment for multiple comparisons [controlling false discovery rate (FDR) at 5%] are reported in this section.
Analysis of ME/CFS Cases vs. Healthy Controls.
The average pMFI of two cytokines was found to be significantly different in ME/CFS patients and in healthy controls. TGF-β (P = 0.0052) was elevated in ME/CFS patients, and resistin (P = 0.0052) was lower (Table 2). In addition, three cytokines were significantly different in cases and controls when stratified by severity, as shown in Fig. 1 (red brackets). The average pMFI of IL-13 was significantly higher in the severe group (P = 0.0250) than in controls; leptin was significantly lower in the mild group (P = 0.0495), and resistin was significantly lower in the mild (P = 0.0370) and severe (P = 0.0208) groups (Table 3).
Table 2.
Comparison of cytokine levels (pMFI) in ME/CFS patients and in healthy controls
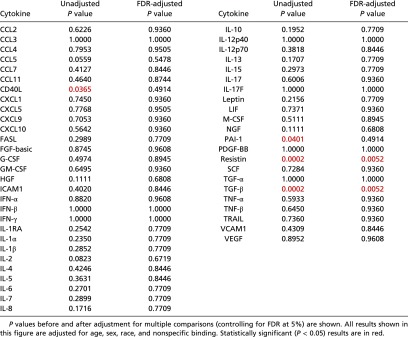 |
Fig. 1.

Mean cytokine levels in healthy controls (Con) and ME/CFS patients grouped by mild, moderate (Mod), and severe (Sev) disease. Means for pMFI ± 1 SE for each cytokine are shown within vertical brackets. The dotted horizontal line within each cytokine panel represents the average value for healthy controls. Statistically significant comparisons of disease severity level vs. healthy controls (adjusted P < 0.05, Table 3) are in red. Results were adjusted for multiple comparisons, and covariates of age, sex, race, and nonspecific binding. Depression of resistin and elevation of TGF-β in cases overall are each evident (Table 2) as are nonlinear trends across disease severity levels in ICAM1 and resistin (Table 4).
Table 3.
Comparison of mean cytokine levels (pMFI) in ME/CFS patients, grouped by mild, moderate, and severe disease, and in healthy controls
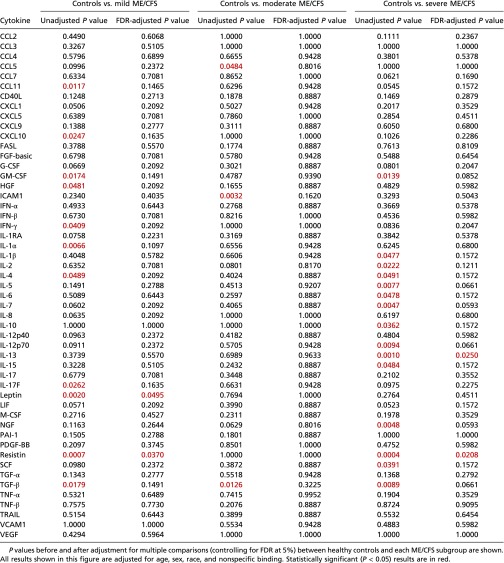 |
Analysis of ME/CFS Cases by Disease Severity.
Overall, 17 cytokines were found to have a statistically significant upward linear trend across the sequence of mild, moderate, and severe ME/CFS severity: CCL11 (P = 0.0069), CXCL1 (P = 0.0266), CXCL10 (P = 0.0100), G-CSF (P = 0.0110), GM-CSF (P = 0.0063), IFN-γ (P = 0.0101), IL-4 (P = 0.0103), IL-5 (P = 0.0073), IL-7 (P = 0.0063), IL-12p70 (P = 0.0069), IL-13 (P = 0.0069), IL-17F (P = 0.0103), leptin (P = 0.0100), leukemia inhibitory factor (LIF) (P = 0.0100), nerve growth factor (NGF) (P = 0.0069), stem cell factor (SCF) (P = 0.0145), and TGF-α (P = 0.0367) (Fig. 2 and Table 4). Intercellular adhesion molecule 1 (ICAM1) and resistin exhibited a statistically significant, nonlinear, inverted trend (P = 0.0334 for each) (Fig. 1).
Fig. 2.

Mean cytokine levels with statistically significant linear trends in ME/CFS patients grouped by mild, moderate (Mod), and severe (Sev) disease compared with healthy controls (Con). Mean pMFI ± 1 SE are shown as vertical brackets for each cytokine. P values shown are for the significance of the linear trend. Only statistically significant linear trends adjusted for multiple comparisons (P < 0.05) are shown. All results shown in this figure were also adjusted for age, sex, race, and nonspecific binding. The dotted horizontal line within each cytokine panel represents the average value for healthy controls.
Table 4.
Trend analysis for mean cytokine levels (pMFI) in ME/CFS patients across the sequence of mild, moderate, and severe disease
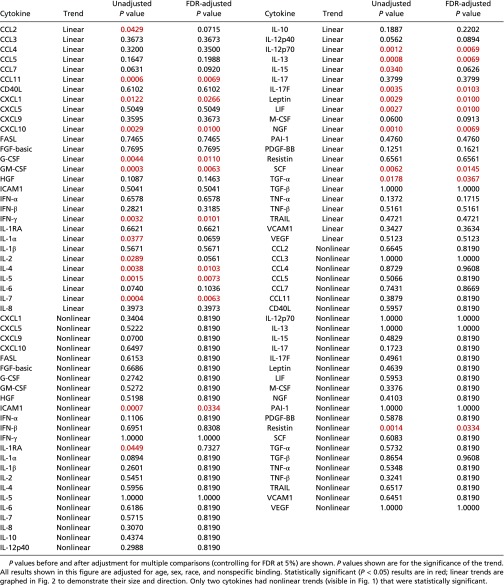 |
Analysis of ME/CFS Cases by Fatigue Duration.
In Spearman correlation analyses, before controlling for potentially confounding variables, several cytokines were found to correlate inversely with fatigue duration: Growth related oncogene-α (GRO-α or CXCL1), IFN-β, IL-1α, IL-1RA, IL-2, IL-8, IL-15, TGF-α, and TGF-β (SI Appendix, SI Materials and Methods and Table S1). Following correction for age, nonspecific binding, race, and sex, only one cytokine, CXCL9 (monokine induced by interferon-γ, MIG), was found to correlate inversely with fatigue duration (unadjusted for multiple comparisons, P = 0.0123) (SI Appendix, Table S1). Although the inclusion of fatigue duration as an additional covariate was expected to result in a loss of statistical power, regression analysis by both disease severity and fatigue duration revealed that the upward linear trend across disease severity remained statistically significant for CCL11, CXCL10, G-CSF, GM-CSF, IFN-γ, IL-4, IL-5, IL-7, IL-12p70, IL-13, IL-17F, leptin, LIF, NGF, ICAM1, and resistin. The findings by severity for CXCL1, SCF, and TGF-α shifted slightly above the threshold for statistical significance (CXCL1, adjusted P = 0.0536; SCF, adjusted P = 0.0531; TGF-α, adjusted P = 0.0797), possibly because of variance inflation (18) by the independent covariate of fatigue duration. Only CXCL9 and IL-1α inversely correlated with fatigue duration but lost statistical significance after correction for multiple comparisons (SI Appendix, Table S2). We also compared mean cytokine levels in cases with ≤3-y fatigue duration (n = 30) and those with >3-y fatigue duration (n = 156), as per Hornig et al. (17). We did not find any cytokine to be significantly different between these two groups (SI Appendix, Table S3).
Discussion
Fifty-one serum cytokines were measured in a cross-sectional study of 186 ME/CFS patients and 388 healthy controls matched by age and sex. A single serum sample was obtained at baseline without any physical, emotional, or neurocognitive stimulation. Only two cytokines were found to be significantly different on average in ME/CFS patients when compared as a group with healthy controls: TGF-β was elevated, and resistin was lower.
TGF-β has been found to be elevated in ME/CFS patients in five of eight studies (63%), as highlighted in a meta-analysis by Blundell et al. (10). TGF-β is a 112-amino acid protein that provides cells with the pleiotropic capacity to affect cell-developmental programs and behavior, including cell proliferation, differentiation, morphogenesis, tissue homeostasis, and regeneration (19). The principal cell sources of TGF-β include monocytes, macrophages, T cells (primarily regulatory T cells), chondrocytes, and intestinal epithelial cells, involving both innate and adaptive immune responses. Given the pleiotropism of TGF-β and its wide availability, its implication in the pathogenesis of apparently dissimilar conditions such as Marfan syndrome (20), cancer (in both control and development) (21), renal fibrosis (22), chronic pulmonary diseases (23), liver disease (24), and inflammatory bowel disease (IBD) (25) is unsurprising. Because TGF-β has been implicated in the development of cancer, elevation of this cytokine in ME/CFS patients older than 65 y of age could contribute to their possibly increased risk of NHL and two defined NHL subtypes, MZL and DLBCL, following their ME/CFS diagnosis (26). Moreover, apart from ME/CFS, other studies have found and proposed a direct link between circulating levels of TGF-β and the risk of NHL (27, 28).
TGF-β, along with IL-10, is primarily viewed as an anti-inflammatory cytokine. The TGF-β elevation in ME/CFS patients may represent down-regulatory activity by these patients’ immune systems against unremitting inflammation; if so, however, one would expect TGF-β levels to correlate with ME/CFS severity, as was observed with several proinflammatory cytokines in this study. Alternatively, it appears that TGF-β does not always function to counteract inflammation. Despite the overarching view of TGF-β as an immunosuppressive (anti-inflammatory) cytokine, its net effect may depend on the local immunological milieu at target tissues and the overall levels of TGF-β (29). For instance, in patients with active IBD, TGF-β has been found at increased levels in the inflamed gut compared with mucosa unaffected by the disease (25). Thus, elevated levels of TGF-β in ME/CFS patients may actually be detrimental and may be a major factor in promoting relentless inflammation and a “fibrotic” milieu resistant to therapeutic interventions in some ME/CFS patients.
Resistin is a cytokine produced primarily by peripheral blood mononuclear cells (PBMCs) in humans and by adipocytes (i.e., as an adipocytokine) in rodents (30). Resistin in humans appears to have a significant proinflammatory role by targeting PBMCs, endothelial cells, smooth muscle cells, platelets (30), and chondrocytes (31) and by increasing the release of IL-1β, IL-6, and TNF-α via the NF-κB pathway (32). Resistin has been reported to be a marker of inflammation in systemic lupus erythematosus (SLE) and Crohn’s disease in humans (33). It is unclear at this time why resistin had this unusual behavior in our study, increasing with mild to moderate disease severity but decreasing with moderate to severe disease. A similar trend was observed in other cytokines including ICAM1 (Fig. 1). An analogous biological behavior is observed in other disease processes such as hepatitis, in which transaminases increase with the severity of inflammation in the liver but actually decline after a certain level of severity is reached as a result of exhaustion and the inability of the hepatocytes to produce these enzymes.
Remarkably, 17 cytokines were associated with severity in ME/CFS patients. Thirteen of these 17 cytokines are primarily proinflammatory: CCL11, CXCL1, CXCL10, IFN-γ, IL-4, IL-5, IL-7, IL-12, IL-13, IL-17, leptin, G-CSF, and GM-CSF. Interestingly, 11 (65%) of the 17 cytokines and 9 (69%) of the 13 proinflammatory cytokines are classified as “type I” by sharing a similar 3D structure, i.e., a four α-helical bundle structure. Their linear relationship with severity was statistically significant even after correction for multiple comparisons, even though these 17 cytokines did not distinguish cases from controls overall. This apparent paradox is explained by the levels of these cytokines in patients with mild disease being below or in the lower range for healthy controls and the levels in patients with severe disease being in the higher or upper range for healthy controls (Figs. 1 and 2). This dysregulation to extremes of normative range also may explain why several studies, including ours, have reported few or no cytokine levels that distinguish ME/CFS cases from controls (10). Above all, it suggests that severity may be a key variable for subgrouping ME/CFS. Furthermore, the levels of circulating cytokines in response to inflammatory triggers such as infection have been reported by various groups to be lower than in controls for some cytokines and higher for others (34–36). A response with lower levels of cytokines may represent a down-regulatory effort by the immune system in an attempt to attenuate more severe immunopathology, resulting in milder or even no symptomatology. A response with higher levels may indicate that the immune system is dealing with a greater challenge that is more likely to result in immunopathology and symptoms. ME/CFS patients in the mild category (with cytokine levels in the lower range) would be protected from more severe disease through this mechanism, whereas those in the severe category would suffer on the opposite side of the spectrum. In addition to a response to an inflammatory trigger, these cytokine findings associated with severity also suggest a dose–response defect in the metabolism or excretion of cytokines. With the lack of large and long-term longitudinal studies in ME/CFS patients, it is not possible at this time to establish whether patients evolve over time as a continuum from mild, to moderate, to severe disease or if a patient is set to stay within a range of a specific category of severity for the duration of the illness.
A second apparent paradox is harder to explain: The two cytokines that did distinguish cases from controls, TGF-β and resistin, did not exhibit a linear relationship with disease severity. It may be that TGF-β and resistin contribute to ME/CFS pathogenesis independent of disease severity.
One of the challenging clinical features of ME/CFS is the capacity of the illness to persist for several years. Thus, the inflammatory state described here might persist, unabated, for decades. Adipokines have been proposed as mediators and perpetuators of chronic inflammatory diseases (37). In this study, two adipokines were identified as being important: leptin and resistin. Thus, it is biologically plausible that changes in adipocyte tissue (in the bone marrow and/or peripheral tissues) linked to increased production of these adipokines may be a factor in the propagation of an inflammatory state in ME/CFS.
In the data presented here, leptin was found to correlate with disease severity. It also was found to correlate significantly with fatigue severity in a longitudinal study led by Younger et al. (13). In addition to playing an important role in regulating body weight by promoting satiety and increasing energy consumption, leptin has been identified as a major proinflammatory cytokine involved in innate and adaptive immune responses (38). In addition, leptin has been shown to be involved in neutrophil recruitment, macrophage activation, phagocytosis, activation of NK cells, dendritic cell survival, skewing T cells toward a proinflammatory and Th1 phenotype, and acting as a negative regulator of regulatory T cells (38, 39). Two additional clinical features in ME/CFS could be explained by the leptin levels in our patients. ME/CFS occurs more frequently in women than in men (2). Leptin levels are higher in females than males, even when corrected for confounding variables such as body mass index (BMI) or level of adiposity (40, 41), and therefore some have suggested that leptin may play a role in the influence of sex on the development of diseases including multiple sclerosis and SLE, which predominantly affect females (37); ME/CFS may be another such disease. In addition, ME/CFS patients often complain of significant cognitive and neurological symptoms, and a recent study suggested that neuroinflammation could be a central feature of the disease (42, 43). Recently, adipokines have been invoked as mediators of an ongoing crosstalk between adipose tissue and the CNS that on occasion, following an unknown trigger, can result in neuroinflammation and neurodegenerative diseases (44). Leptin also has been reported recently to up-regulate the recruitment of neutrophils into the brain in a murine model of sepsis induced by systemic administration of LPS, providing another plausible mechanism for its ability to cause neuroinflammation (45). Thus, it is possible that the CNS abnormalities observed in ME/CFS patients can be explained, at least in part, by the ability of leptin and resistin to cross and/or disrupt the blood–brain barrier (46). Moreover, systemic inflammation, such as that found in our patients, has been invoked as a mechanism for neuroinflammation in other neurodegenerative disease models (47–49). In animal models, systemic administration of LPS alone has been shown to result in neuroinflammation (45, 50).
Recently, Hornig et al. (17) reported a group of cytokines that inversely correlated with fatigue duration, and in our cytokine assay we investigated the same 51 cytokines. In our study, several cytokines [GRO-α (CXCL-1), IFN-β, IL-15, IL-1A, IL-1RA, IL-2, IL-8, TGF-α, and TGF-β] also inversely correlated with fatigue duration before controlling for potentially confounding variables (age, nonspecific binding, race, and sex). After correction for age (SI Appendix, Fig. S3), nonspecific binding, and race, only one cytokine, CXCL9, inversely correlated with fatigue duration. When analysis was performed for both disease severity and fatigue duration, the findings for disease severity remained statistically significant for most cytokines, whereas those for fatigue duration did not. It is possible that disease duration and severity interact in their association with cytokine expression. To assess that possibility for each cytokine separately, we fit a regression model that allows mean cytokine expression levels to vary flexibly over the 2D distribution of total scores from the Multidimensional Fatigue Inventory (MFI-20) assessment of disease severity (51) and fatigue duration in years (52). This analysis found no evidence that the relationship between mean cytokine expression and disease severity changes with duration of disease (SI Appendix, Table S4). The importance of analyzing ME/CFS data by severity is further supported by the lack of correlation between fatigue duration and disease severity (SI Appendix, Fig. S4). However, the lack of observed correlation between disease (fatigue) duration and cytokine levels may result from the lower statistical power of the current study, because the sizes of both our overall sample of cases (n = 186) and, especially, of cases with ≤3-y fatigue duration (n = 30), were smaller than those studied by Hornig et al. Thus, our failure to discover any differences between short and long duration does not allow us to rule out definitively the possibility that mean serum levels might differ between these two specific fatigue-duration categories (≤3 y vs. >3 y). A study by Landi et al. (53) measured 31 cytokines in 100 ME/CFS patients with disease of “long duration” and in 79 healthy controls and observed reductions in IL-16, IL-7, and VEGF-A levels in ME/CFS patients. Theirs is the first ME/CFS study to measure IL-16. Although the Landi et al. study did not adjust for disease severity, the authors concluded through multivariate data analysis techniques that IL-16 could be an important candidate for a biomarker profile in ME/CFS (53). Unfortunately, our cytokine assay did not include IL-16.
A nonspecific binding in the Luminex 200 IS system potentially impacting pMFI signals of some cytokines was found in our study (SI Appendix, SI Materials and Methods). This discovery was possible through analysis of residual data that usually are considered inconsequential in most studies. The adjustment for nonspecific binding was applied to the 51 cytokines of all participants and was significant for some of the cytokines (e.g., IFN-β) (SI Appendix, Fig. S2). This adjustment allowed our study to decrease noise and, we believe, to produce data that may be closer to the actual biological underpinnings of ME/CFS. Multiplex assays that allow the measurement of a significant number of analytes, in contrast to assays that measure only a few analytes, are an efficient tool for advancing our understanding of disease processes. However, pitfalls such as this one for nonspecific binding should be anticipated and, if found, corrected.
Limitations of our study include the cross-sectional design. Future longitudinal studies are necessary to address whether ME/CFS patients remain within their cytokine signature and disease severity category over time or fluctuate among them. We sampled only peripheral blood and not other compartments such as cerebrospinal fluid (CSF). Cytokine studies in the CSF of ME/CFS patients have reported abnormalities even though the sample size has been modest and simultaneous serum samples were not analyzed (54, 55). In one study, a diathesis to allergic and inflammatory responses in CSF, similar to some of our findings, were found in these patients (54, 55). In addition, because resistin and leptin are important adipokines, correction for patients’ BMI would have been ideal. However, this correction was not possible with the current dataset. Findings in this study provide further evidence that ME/CFS likely involves a systemic inflammatory process (17). These findings also support biological plausibility for the propensity of these patients to experience several major and ongoing clinical manifestations, offer a mechanism for the disease’s predilection to affect women and the increased risk for development of NHL, and support the suitability of exploring immunomodulation as a primary or adjuvant therapy (56, 57). Future cytokine research in the peripheral blood of ME/CFS patients should embrace longitudinal designs and seek correlations with neuroradiology, neuroinflammation, and CSF studies. If using multiplex array-based technologies, investigators should pay attention to residual data in regression analysis to identify and correct unrecognized confounders such as nonspecific binding.
Materials and Methods
Study Design.
An age- and sex-matched case-control cross-sectional study was conducted at Stanford University in 2009 to investigate the role of immune responses, genetic predisposition, and infection in ME/CFS. A total of 192 ME/CFS cases and 392 healthy controls were included in this analysis. However, the final sample size was 186 ME/CFS cases and 388 healthy controls because of missing race information in six ME/CFS cases and four healthy controls. In addition to the serum analyzed in this report, other peripheral blood specimens were collected from these individuals and are stored for ongoing and future immune, genetic, and pathogen-discovery studies. Each case participant was matched to two control participants by sex and age (±6 mo). To be included in the study, participants had to be 14 y of age or older, reside in Northern California, and provide written informed consent and Health Insurance Portability and Accountability Act of 1996 authorization as required by the Stanford University Institutional Review Board (protocol numbers 18068 and 18155).Participants were classified as cases if they met the 1994 CDC CFS case definition (3). Of note, symptoms such as unrefreshing sleep, postexertional malaise, and impaired memory (also referred to by patients as “brain fog”) were present in 96.9, 96.9, and 95.8% of ME/CFS patients, respectively (Table 1). Controls in the study were eligible if they did not have history of fatigue and did not meet the ME/CFS case definition. Exclusion criteria for both groups included active or uncontrolled morbidities that would have interfered with the patient’s ability to participate in the study, particularly conditions or medications causing immunosuppression or immunodeficiency (additional exclusion criteria are given in SI Appendix, SI Materials and Methods).
Participants were recruited from March 2, 2010 to September 1, 2011, and their peripheral blood was drawn between 8:30 AM and 3:30 PM on the day of enrollment. Blood specimens including serum, plasma, whole-blood DNA and whole-blood RNA (collected in PAXgene tubes), and PBMCs were obtained and processed on the day of enrollment by the Stanford Center for Clinical and Translational Research and Education (spectrum.stanford.edu/accordions/clinical-and-translational-research-unit) and were stored on the same day by the Stanford Human Immune Monitoring Center (HIMC: iti.stanford.edu/himc.html).
Participants’ age, sex, and age of onset of ME/CFS were recorded at baseline. The MFI-20, a 20-item questionnaire (51), was administered to each participant on the day of blood sample collection. A higher score indicates greater severity. This instrument has been validated in the ME/CFS population (58).
Cytokine Assay.
Cytokines were measured for each participant in serum using a 51-multiplex array on the Luminex 200 IS system (Affymetrix) performed at the Stanford HIMC. The manufacturer’s protocol was followed, with variations as described by Brodin, et al. (59).
A total of 19 plates were used. Each participant’s sample was entered in two replicate wells, and matched sets of ME/CFS cases and healthy controls were always mixed in all plates to reduce confounding case status with plate artifacts. Results were accepted as final (569 samples) if more than 95% of data had a coefficient of variation (CV) <10%. When the CV exceeded 30% (15 samples), the averaging over duplicate wells reduced the technical variance in median fluorescence intensities (FIs) by twofold.
Each plate also contained two wells of internal control and wells to account for generic binding to the beads (CHEX1, CHEX2, CHEX3, CHEX4) unrelated to the target cytokine. Assay CheX beads (Radix BioSolutions) are a mixture of four quality-control beads that are spiked into each well of a Luminex immunoassay. Each of the four beads monitors a part of the assay process: instrument performance, application of detection antibody, application of fluorescent reporter, and nonspecific binding. The last parameter is monitored by the CHEX4 beads, which have very low intrinsic fluorescence. Elevated CHEX4 fluorescence is indicative of samples containing high levels of nonspecific binding activity. The fact that nonspecific binding was affecting our results was discovered by statistical analysis of residual data (variation in observed data not explained by fit of the regression line, as per graphical explanation of residuals in SI Appendix, Fig. S1) from regression analysis. T.H.H. discovered that these residual data contained structure retrievable by multivariate statistical methods. He subsequently discovered that this “residual structure” was strongly correlated with nonspecific binding and that nonspecific binding was correlated with pMFI in many cytokines and with case status. Therefore, nonspecific binding was included as a covariate to prevent introduction of bias (60).
Luminex measures the FIs of the cytokines and produces a distribution of typically 200–300 FIs per well. We computed the median FIs for each distribution per well. Because every participant’s sample was entered in two wells, two median FIs per cytokine were computed for each participant. For the median FIs that had bead counts of at least 90, we then computed the mean value from the two median FIs.
Statistical Analysis.
Preprocessing.
Consistent preprocessing across cytokines facilitated their comparison and biological interpretation. MFI data were preprocessed (pMFI) for each cytokine through a sequence of averaging over duplicate wells, natural-logarithm transformation to reduce variance heterogeneity, isolation and removal of plate effects, and centering and scaling. Use of population marginal means (61) adjusted for covariates of age, sex, and race (white vs. nonwhite) permitted estimation and removal by subtraction of plate effects that were balanced (i.e., 1:1 rather than 2:1) with respect to control vs. case status. Centering and scaling entailed subtracting the sample mean and dividing by the sample SD.
Primary analysis: Disease severity.
A priori, cases were classified into tertiles for ME/CFS severity: MFI-20 scores from 51–75 were classified as mild disease, scores from 76–85 as moderate disease, and scores from 86–100 as severe disease. For each cytokine, generalized maximum entropy estimation (GME) (62) was used to fit a regression model to test hypotheses regarding the four pMFI means (control and three severities). Specifically, we regressed pMFI on disease severity category (control, mild, moderate, and severe), sex, race, age, pMFI of the nonspecific binding control (CHEX4) (for further information on nonspecific binding, see SI Appendix, SI Materials and Methods), and, to allow for the possibility that covariate effects differ between cases and controls, interaction terms between case status and each covariate (sex, race, age, and pMFI of the nonspecific binding control). [We also performed a linear mixed model regression analysis that, in addition to these covariates, adjusted for matched set as a random coefficient to account for any remaining structure caused by the matching process. Matched set explained ∼0% of the variance in cytokine levels for nearly all cytokines (in the presence of other covariates) and so was not retained in the results presented here.] Using the fit of the pMFI data to this regression model, hypothesis testing was used to compare covariate-adjusted pMFI means between (i) each disease severity group versus control and (ii) the equally weighted average across all three severity groups versus control. To compare controls to cases overall, covariates were held at their sample mean values for cases. To compare controls to each specific case, severity level, covariates were held at their sample mean values for the severity level of that specific case. In addition, in post hoc analyses, the fit of the pMFI data to this regression model was used to examine the association between mean cytokine response and severity level within cases. Specifically, we tested for linear and curvilinear trends in pMFI means across the sequence of mild, moderate, and severe disease. Shapes of sample distributions of pMFI values across individuals in this sample varied widely among cytokines. This variety of distributions made GME especially suitable because this estimation method does not require that the regression response variable (here the pMFI) follow any particular parametric distribution (e.g., normal distribution) (62). Further technical details on application of GME are provided in SI Appendix, SI Materials and Methods.
Secondary analysis: Fatigue duration.
In an analysis limited to cases, we regressed pMFI on disease severity category (mild, moderate, and severe), sex, race, age, pMFI of the nonspecific binding control (CHEX4), and the additional covariate of fatigue duration (in years). Because we allowed covariate effects to differ between cases and controls in the primary analysis, regression models for cases are identical for primary and secondary analyses with the exception of fatigue duration serving as a covariate in secondary analyses. With primary and secondary regression models otherwise identical, the secondary analysis was able to isolate the effect of fatigue duration on pMFI. [To examine the robustness of findings to the method of parameter estimation, in addition to GME, we also fit regression models to the pMFI data using ordinary least squares (OLS). An OLS (or closely allied) method was used in a previous report by Hornig et al. (17). Findings reported here are similar for the GME and OLS regression methods and are available upon request. Further technical details about the application of OLS are provided in SI Appendix, SI Materials and Methods.] To permit direct comparison with another recent report (17), we calculated estimates of Spearman rank correlation coefficients between each cytokine’s pMFI and fatigue duration, and we took this analysis a step further by adjusting estimates of Spearman rank correlation coefficients for age, nonspecific binding, race, and sex. Further technical details are provided in SI Appendix, SI Materials and Methods.
Type I error control.
Throughout, P values have been adjusted to account for the accumulation of type 1 error across multiple hypothesis tests. Specifically, P value adjustments used an adaptive two-stage linear step-up procedure to control the FDR at 5% (60, 63) across the 51 cytokines. FDR control was performed separately by group (e.g., severity level) to allow for group differences in the proportions of truly null hypotheses. All analyses were performed in SAS 9.4 (SAS Institute) and R 3.2.2 through 3.3.2 (https://www.R-project.org/).
Supplementary Material
Acknowledgments
We thank the ME/CFS patients who volunteered and participated in our studies; the Stanford Institute for Immunity, Transplantation and Infection and the Stanford Human Immune Monitoring Center for support; Dr. Manisha Desai and Aya Mitani in the Quantitative Sciences Unit, Department of Medicine, Stanford University School of Medicine, for their initial work and analysis of our data, reinforcing the robustness of our findings; Donn W. Garvert, MS, Statistical Programmer (Stanford ME/CFS Initiative); and Ben B. Varasteh (The Stanford Center for Clinical and Translational Research and Education) and Jane Norris, PA, for patient recruitment efforts.
Footnotes
Conflict of interest statement: M.M.D. is a member of the Scientific Advisory Board of the Open Medicine Foundation. A.L.K. and J.G.M. have published together, most recently in 2017.
See Commentary on page 8914.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1710519114/-/DCSupplemental.
References
- 1.Prins JB, van der Meer JW, Bleijenberg G. Chronic fatigue syndrome. Lancet. 2006;367:346–355. doi: 10.1016/S0140-6736(06)68073-2. [DOI] [PubMed] [Google Scholar]
- 2.Clayton EW. Beyond myalgic encephalomyelitis/chronic fatigue syndrome: An IOM report on redefining an illness. JAMA. 2015;313:1101–1102. doi: 10.1001/jama.2015.1346. [DOI] [PubMed] [Google Scholar]
- 3.Fukuda K, et al. International Chronic Fatigue Syndrome Study Group The chronic fatigue syndrome: A comprehensive approach to its definition and study. Ann Intern Med. 1994;121:953–959. doi: 10.7326/0003-4819-121-12-199412150-00009. [DOI] [PubMed] [Google Scholar]
- 4.Carruthers BM, et al. Myalgic encephalomyelitis/chronic fatigue syndrome: Clinical working case definition, diagnostic and treatment protocols. J Chronic Fatigue Syndr. 2003;11:7–36. [Google Scholar]
- 5.Carruthers BM, et al. Myalgic encephalomyelitis: International consensus criteria. J Intern Med. 2011;270:327–338. doi: 10.1111/j.1365-2796.2011.02428.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jason LA, Brown A, Evans M, Sunnquist M, Newton JL. Contrasting chronic fatigue syndrome versus myalgic encephalomyelitis/chronic fatigue syndrome. Fatigue. 2013;1:168–183. doi: 10.1080/21641846.2013.774556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Komaroff AL. Myalgic encephalomyelitis/chronic fatigue syndrome: A real illness. Ann Intern Med. 2015;162:871–872. doi: 10.7326/M15-0647. [DOI] [PubMed] [Google Scholar]
- 8.Lorusso L, et al. Immunological aspects of chronic fatigue syndrome. Autoimmun Rev. 2009;8:287–291. doi: 10.1016/j.autrev.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 9.Raison CL, Lin JM, Reeves WC. Association of peripheral inflammatory markers with chronic fatigue in a population-based sample. Brain Behav Immun. 2009;23:327–337. doi: 10.1016/j.bbi.2008.11.005. [DOI] [PubMed] [Google Scholar]
- 10.Blundell S, Ray KK, Buckland M, White PD. Chronic fatigue syndrome and circulating cytokines: A systematic review. Brain Behav Immun. 2015;50:186–195. doi: 10.1016/j.bbi.2015.07.004. [DOI] [PubMed] [Google Scholar]
- 11.Russell L, et al. Illness progression in chronic fatigue syndrome: A shifting immune baseline. BMC Immunol. 2016;17:3. doi: 10.1186/s12865-016-0142-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Klimas NG, Broderick G, Fletcher MA. Biomarkers for chronic fatigue. Brain Behav Immun. 2012;26:1202–1210. doi: 10.1016/j.bbi.2012.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stringer EA, et al. Daily cytokine fluctuations, driven by leptin, are associated with fatigue severity in chronic fatigue syndrome: Evidence of inflammatory pathology. J Transl Med. 2013;11:93. doi: 10.1186/1479-5876-11-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Curriu M, et al. Screening NK-, B- and T-cell phenotype and function in patients suffering from chronic fatigue syndrome. J Transl Med. 2013;11:68. doi: 10.1186/1479-5876-11-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ford B, Bradley AS, Bansal AS. Altered functional T cell subset populations and cytokine profile in patients with chronic fatigue syndrome: A pilot study. J Chronic Dis Manag. 2016;1:1004. [Google Scholar]
- 16.Tirelli U, Marotta G, Improta S, Pinto A. Immunological abnormalities in patients with chronic fatigue syndrome. Scand J Immunol. 1994;40:601–608. doi: 10.1111/j.1365-3083.1994.tb03511.x. [DOI] [PubMed] [Google Scholar]
- 17.Hornig M, et al. Distinct plasma immune signatures in ME/CFS are present early in the course of illness. Sci Adv. 2015;1:e1400121. doi: 10.1126/sciadv.1400121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hsieh FY, Bloch DA, Larsen MD. A simple method of sample size calculation for linear and logistic regression. Stat Med. 1998;17:1623–1634. doi: 10.1002/(sici)1097-0258(19980730)17:14<1623::aid-sim871>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 19.Massagué J. TGFβ signalling in context. Nat Rev Mol Cell Biol. 2012;13:616–630. doi: 10.1038/nrm3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cannaerts E, van de Beek G, Verstraeten A, Van Laer L, Loeys B. TGF-β signalopathies as a paradigm for translational medicine. Eur J Med Genet. 2015;58:695–703. doi: 10.1016/j.ejmg.2015.10.010. [DOI] [PubMed] [Google Scholar]
- 21.Massagué J. TGFbeta in cancer. Cell. 2008;134:215–230. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meng XM, Nikolic-Paterson DJ, Lan HY. TGF-β: The master regulator of fibrosis. Nat Rev Nephrol. 2016;12:325–338. doi: 10.1038/nrneph.2016.48. [DOI] [PubMed] [Google Scholar]
- 23.Aschner Y, Downey GP. Transforming growth factor-β: Master regulator of the respiratory system in health and disease. Am J Respir Cell Mol Biol. 2016;54:647–655. doi: 10.1165/rcmb.2015-0391TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fabregat I, et al. IT-LIVER Consortium TGF-β signalling and liver disease. FEBS J. 2016;283:2219–2232. doi: 10.1111/febs.13665. [DOI] [PubMed] [Google Scholar]
- 25.Shen Y, Zhang C, Chen Y. TGF-β in inflammatory bowel diseases: A tale of the Janus-like cytokine. Crit Rev Eukaryot Gene Expr. 2015;25:335–347. doi: 10.1615/critreveukaryotgeneexpr.2015013974. [DOI] [PubMed] [Google Scholar]
- 26.Chang CM, Warren JL, Engels EA. Chronic fatigue syndrome and subsequent risk of cancer among elderly US adults. Cancer. 2012;118:5929–5936. doi: 10.1002/cncr.27612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mazur G, Bogunia-Kubik K, Wrobel T, Kuliczkowski K, Lange A. TGF-beta1 gene polymorphisms influence the course of the disease in non-Hodgkin’s lymphoma patients. Cytokine. 2006;33:145–149. doi: 10.1016/j.cyto.2005.12.010. [DOI] [PubMed] [Google Scholar]
- 28.Yang ZZ, et al. TGF-β upregulates CD70 expression and induces exhaustion of effector memory T cells in B-cell non-Hodgkin’s lymphoma. Leukemia. 2014;28:1872–1884. doi: 10.1038/leu.2014.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morikawa M, Derynck R, Miyazono K. TGF-β and the TGF-β family: Context-dependent roles in cell and tissue physiology. Cold Spring Harb Perspect Biol. 2016;8:a021873. doi: 10.1101/cshperspect.a021873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang X, Yang Z. Resistin’s, obesity and insulin resistance: The continuing disconnect between rodents and humans. J Endocrinol Invest. 2016;39:607–615. doi: 10.1007/s40618-015-0408-2. [DOI] [PubMed] [Google Scholar]
- 31.Zhang Z, et al. Resistin induces expression of proinflammatory cytokines and chemokines in human articular chondrocytes via transcription and messenger RNA stabilization. Arthritis Rheum. 2010;62:1993–2003. doi: 10.1002/art.27473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Almehed K, d’Elia HF, Bokarewa M, Carlsten H. Role of resistin as a marker of inflammation in systemic lupus erythematosus. Arthritis Res Ther. 2008;10:R15. doi: 10.1186/ar2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Konrad A, et al. Resistin is an inflammatory marker of inflammatory bowel disease in humans. Eur J Gastroenterol Hepatol. 2007;19:1070–1074. doi: 10.1097/MEG.0b013e3282f16251. [DOI] [PubMed] [Google Scholar]
- 34.Meira CS, et al. Toxoplasma Groups Cerebral and ocular toxoplasmosis related with IFN-γ, TNF-α, and IL-10 levels. Front Microbiol. 2014;5:492. doi: 10.3389/fmicb.2014.00492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pernas L, Ramirez R, Holmes TH, Montoya JG, Boothroyd JC. Immune profiling of pregnant Toxoplasma-infected US and Colombia patients reveals surprising impacts of infection on peripheral blood cytokines. J Infect Dis. 2014;210:923–931. doi: 10.1093/infdis/jiu189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rey A, et al. Cytokine profiling reveals decreased serum levels of CCL2 in active ocular toxoplasmosis. Br J Ophthalmol. 2013;97:1338–1342. doi: 10.1136/bjophthalmol-2013-303092. [DOI] [PubMed] [Google Scholar]
- 37.Hutcheson J. Adipokines influence the inflammatory balance in autoimmunity. Cytokine. 2015;75:272–279. doi: 10.1016/j.cyto.2015.04.004. [DOI] [PubMed] [Google Scholar]
- 38.Procaccini C, et al. Leptin as immune mediator: Interaction between neuroendocrine and immune system. Dev Comp Immunol. 2017;66:120–129. doi: 10.1016/j.dci.2016.06.006. [DOI] [PubMed] [Google Scholar]
- 39.Procaccini C, Jirillo E, Matarese G. Leptin as an immunomodulator. Mol Aspects Med. 2012;33:35–45. doi: 10.1016/j.mam.2011.10.012. [DOI] [PubMed] [Google Scholar]
- 40.Ostlund RE, Jr, Yang JW, Klein S, Gingerich R. Relation between plasma leptin concentration and body fat, gender, diet, age, and metabolic covariates. J Clin Endocrinol Metab. 1996;81:3909–3913. doi: 10.1210/jcem.81.11.8923837. [DOI] [PubMed] [Google Scholar]
- 41.Saad MF, et al. Sexual dimorphism in plasma leptin concentration. J Clin Endocrinol Metab. 1997;82:579–584. doi: 10.1210/jcem.82.2.3739. [DOI] [PubMed] [Google Scholar]
- 42.Nakatomi Y, et al. Neuroinflammation in patients with chronic fatigue syndrome/myalgic encephalomyelitis: An 11C-(R)-PK11195 PET study. J Nucl Med. 2014;55:945–950. doi: 10.2967/jnumed.113.131045. [DOI] [PubMed] [Google Scholar]
- 43.Natelson BH, Weaver SA, Tseng CL, Ottenweller JE. Spinal fluid abnormalities in patients with chronic fatigue syndrome. Clin Diagn Lab Immunol. 2005;12:52–55. doi: 10.1128/CDLI.12.1.52-55.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Parimisetty A, et al. Secret talk between adipose tissue and central nervous system via secreted factors-an emerging frontier in the neurodegenerative research. J Neuroinflammation. 2016;13:67. doi: 10.1186/s12974-016-0530-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rummel C, Inoue W, Poole S, Luheshi GN. Leptin regulates leukocyte recruitment into the brain following systemic LPS-induced inflammation. Mol Psychiatry. 2010;15:523–534. doi: 10.1038/mp.2009.98. [DOI] [PubMed] [Google Scholar]
- 46.Mauro C, De Rosa V, Marelli-Berg F, Solito E. Metabolic syndrome and the immunological affair with the blood-brain barrier. Front Immunol. 2015;5:677. doi: 10.3389/fimmu.2014.00677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Takeda S, Sato N, Morishita R. Systemic inflammation, blood-brain barrier vulnerability and cognitive/non-cognitive symptoms in Alzheimer disease: Relevance to pathogenesis and therapy. Front Aging Neurosci. 2014;6:171. doi: 10.3389/fnagi.2014.00171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wardill HR, et al. Cytokine-mediated blood brain barrier disruption as a conduit for cancer/chemotherapy-associated neurotoxicity and cognitive dysfunction. Int J Cancer. 2016;139:2635–2645. doi: 10.1002/ijc.30252. [DOI] [PubMed] [Google Scholar]
- 49.Morris G, Berk M, Walder K, Maes M. Central pathways causing fatigue in neuro-inflammatory and autoimmune illnesses. BMC Med. 2015;13:28. doi: 10.1186/s12916-014-0259-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schweighöfer H, Rummel C, Roth J, Rosengarten B. Modulatory effects of vagal stimulation on neurophysiological parameters and the cellular immune response in the rat brain during systemic inflammation. Intensive Care Med Exp. 2016;4:19. doi: 10.1186/s40635-016-0091-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lin JM, et al. Further validation of the Multidimensional Fatigue Inventory in a US adult population sample. Popul Health Metr. 2009;7:18. doi: 10.1186/1478-7954-7-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Long JS, Ervin LH. Using heteroscedasticity consistent standard errors in the linear regression model. Am Stat. 2000;54:217–224. [Google Scholar]
- 53.Landi A, Broadhurst D, Vernon SD, Tyrrell DL, Houghton M. Reductions in circulating levels of IL-16, IL-7 and VEGF-A in myalgic encephalomyelitis/chronic fatigue syndrome. Cytokine. 2016;78:27–36. doi: 10.1016/j.cyto.2015.11.018. [DOI] [PubMed] [Google Scholar]
- 54.Hornig M, et al. Cytokine network analysis of cerebrospinal fluid in myalgic encephalomyelitis/chronic fatigue syndrome. Mol Psychiatry. 2016;21:261–269. doi: 10.1038/mp.2015.29. [DOI] [PubMed] [Google Scholar]
- 55.Peterson D, et al. Cytokines in the cerebrospinal fluids of patients with chronic fatigue syndrome/myalgic encephalomyelitis. Mediators Inflamm. 2015;2015:929720. doi: 10.1155/2015/929720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fluge Ø, et al. Benefit from B-lymphocyte depletion using the anti-CD20 antibody rituximab in chronic fatigue syndrome. A double-blind and placebo-controlled study. PLoS One. 2011;6:e26358. doi: 10.1371/journal.pone.0026358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Montoya JG, et al. Randomized clinical trial to evaluate the efficacy and safety of valganciclovir in a subset of patients with chronic fatigue syndrome. J Med Virol. 2013;85:2101–2109. doi: 10.1002/jmv.23713. [DOI] [PubMed] [Google Scholar]
- 58.Smets EM, Garssen B, Bonke B, De Haes JC. The Multidimensional Fatigue Inventory (MFI) psychometric qualities of an instrument to assess fatigue. J Psychosom Res. 1995;39:315–325. doi: 10.1016/0022-3999(94)00125-o. [DOI] [PubMed] [Google Scholar]
- 59.Brodin P, et al. Variation in the human immune system is largely driven by non-heritable influences. Cell. 2015;160:37–47. doi: 10.1016/j.cell.2014.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kim KI, van de Wiel MA. Effects of dependence in high-dimensional multiple testing problems. BMC Bioinformatics. 2008;9:114. doi: 10.1186/1471-2105-9-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Milliken GA, Johnson DE. 1992. Analysis of Messy Data (Van Nostrand Reinhold, New York) Vol 1: Designed Experiments.
- 62.Golan A, Judge GG, Miller DJ. Maximum Entropy Econometrics: Robust Estimation with Limited Data. John Wiley & Sons; Chichester, UK: 1996. [Google Scholar]
- 63.Benjamini Y, Krieger AM, Yekutieli D. Adaptive linear step-up procedures that control the false discovery rate. Biometrika. 2006;93:491–507. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.