

Abstract
Human pluripotent stem cells (hpscs) provide a valuable model for the study of human development and a means to generate a scalable source of cells for therapeutic applications. This protocol specifies cell fate efficiently into cardiac and endothelial lineages from hPSCs. The protocol takes 2 weeks to complete and requires experience in hPSC culture and differentiation techniques. Building on lessons taken from early development, this monolayer-directed differentiation protocol uses different concentrations of activin A and bone morphogenetic protein 4 (BMP4) to polarize cells into mesodermal subtypes that reflect mid-primitive-streak cardiogenic mesoderm and posterior-primitive-streak hemogenic mesoderm. this differentiation platform provides a basis for generating distinct cardiovascular progenitor populations that enable the derivation of cardiomyocytes and functionally distinct endothelial cell (EC) subtypes from cardiogenic versus hemogenic mesoderm with high efficiency without cell sorting. ecs derived from cardiogenic and hemogenic mesoderm can be matured into >90% CD31+/VE-cadherin+ definitive ECs. To test the functionality of ECs at different stages of differentiation, we provide methods for assaying the blood-forming potential and de novo lumen-forming activity of ECs. To our knowledge, this is the first protocol that provides a common platform for directed differentiation of cardiomyocytes and endothelial subtypes from hPSCs. This protocol yields endothelial differentiation efficiencies exceeding those of previously published protocols. Derivation of these cell types is a critical step toward understanding the basis of disease and generating cells with therapeutic potential.
Introduction
The heart is the earliest organ to form in the developing embryo, providing the basis for a functional circulation as other organ systems develop. Emerging bioengineering and biotechnology approaches for studying the formation of the mesoderm and its cellular lineages provide us with a great opportunity to develop new insights into this complex developmental process. In particular, hPSCs provide an ideal system with which to study these questions because they (i) are of human origin, (ii) are scalable, (iii) allow for the use of advanced molecular biology tools for analysis, and (iv) provide a simplified system for studying cell-fate choices in early development.
During embryogenesis, cell-fate decisions are coordinated by gradients of cytokines and morphogens that allow for differentiation and organization of multiple cell types into complex tissues1. The capacity to direct these complex fate choices is mediated by critical spatiotemporally orchestrated cues required to direct specific cell fates and cell subtypes. Well-described anterior–posterior morphogen gradients principally involving activin/nodal and BMP4 are required for developing a polarized axis during gastrulation2–5. This polarization of mesoderm gives rise to the heterogeneous cell types of the cardiovascular system, including cardiomyocytes and those of the endocardium, vascular endothelium, and the hematopoietic system (Fig. 1). These three lineages are differentiated by lineage-specific modulation of key signaling pathways, including the vascular endothelial growth factor (VEGF) signaling pathway for ECs and Wnt signaling inhibition for cardiomyocytes.
Figure 1.
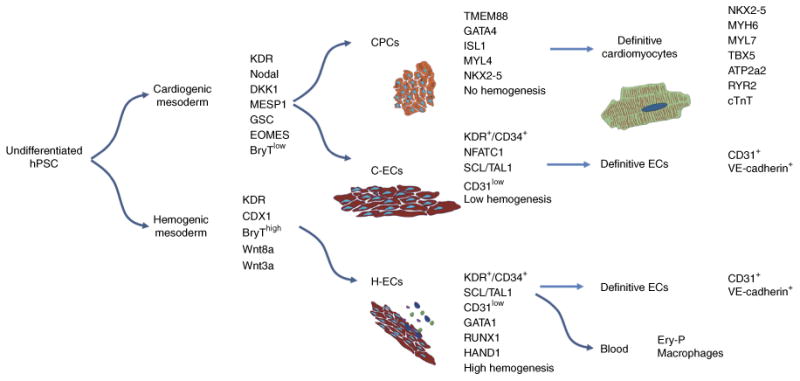
Lineage fate choices in cardiovascular development. Schematic outlining major cell-fate decisions from pluripotency to definitive cardiac and vascular cell types. Molecular markers and functional characteristics are noted for each population. C-ECs, cardiogenic-mesoderm-derived endothelial cells; CPCs, cardiac progenitor cells; ECs, endothelial cells; Ery-P, primitive erythroid; H-ECs, hemogenic-mesoderm-derived endothelial cells; hPSCs, human pluripotent stem cells.
Advantages, limitations, and alternative methods
Studies using hPSCs, by our laboratory6,7 and others8,9, have contributed substantially to knowledge about mechanisms of human mesodermal patterning. The protocol presented here enables the polarization of hPSC mesoderm such that closely related yet distinct cardiovascular populations can be generated efficiently without the need of post facto enrichment, which can disturb cell regulatory states and reduce viability and yield. hPSC polarization toward cardiogenic mesoderm allows for the derivation of both cardiomyocytes and ECs, whereas hPSC polarization toward hemogenic mesoderm gives rise to only blood-forming ECs.
Although efforts have been made to define conditions for polarization of lateral plate mesoderm from embryonic stem cells in vitro3–5,10,11, there has been limited success in translating these observations into protocols to generate high-purity definitive populations. Studies have shown that polarization of hPSCs during specification of mesoderm provides developmental cues that are deterministic for generating definitive cell types6,10. This is in keeping with a history of studies showing that developmental specification of definitive lineages occurs as early as gastrulation, at which point gene programs required to direct one lineage also simultaneously actively repress gene programs for other lineages10–13.
hPSCs have been used extensively in efforts to derive human endothelium14–17. Despite substantial effort, attempts to generate high-purity endothelium from hPSCs have met with modest success, with efficiencies ranging from 5 to 30% cells positive for lineage markers, including VE-cadherin, kinase insert domain receptor (KDR; also known as vascular endothelial growth factor receptor 2), and CD34 (refs. 16,18,19). As such, most widely implemented protocols currently require cell sorting to enrich ECs to purity16,17. This is problematic because it shows that control of cell-fate choices directed toward the endothelial lineage—despite being well understood during embryogenesis—are not efficiently controlled in hPSC-directed differentiation.
The current protocol provides important strides forward for hPSC-directed differentiation into the cardiovascular lineages. First, this protocol provides a common differentiation platform for deriving cardiac and endothelial lineages through simple modulation of activin A and BMP4 concentrations. Second, developmental studies have shown distinct origins and mechanisms for specifying endothelial subtypes such as endocardial, vascular, and lymphatic ECs20–25. To this end—using an approach of polarizing hPSC mesoderm at the onset of differentiation—this protocol provides the first approach for controlling EC differentiation from cardiogenic versus hemogenic mesoderm. Third, unlike other protocols reported so far, we have identified conditions for directing differentiation of all lineages to >90% purity without any sorting steps. We have previously provided extensive characterization of these mesoderm populations using gene expression, proteomics, and analysis of endogenous Wnt/β-catenin signaling activity6.
Despite these advances, the current approach sustains ongoing limitations with existing reagents for differentiation. In the first place, the protocol requires the use of Matrigel, B-27 supplement, and recombinant proteins, which are prone to batch effects, making differentiation variable, requiring lot testing. Second, this protocol is limited in its impact on the cardiac differentiation protocols, given the extensiveness of simple small-molecule-based approaches that have been optimized26–28. This component of the protocol is helpful for studies that aim to decipher how cardiomyocyte versus EC lineage potential is specified from cardiogenic mesoderm.
Although this protocol is an advancement in the field for studying developmental lineage decisions in cardiac and endothelial differentiation, other protocols provide important complementary methods for using these cell types in a range of applications that include primary vascular plexus formation using hPSC-derived pericytes16, vasculature xenografts in zebrafish in vivo16, and the generation of definitive cell types in a scalable manner for therapeutics29. Other protocols also provide methods for using hPSCs to derive pericytes, smooth muscle cells, arterial versus venous ECs, and cardiomyocytes that can be used in coordination with the methods outlined here16,17,27,28,30.
Applications
This protocol provides opportunities to study fundamental questions about lineage choices, advance approaches for tissue engineering, and derive a broader scope of cell types for cardiovascular therapeutic applications. The common platform for deriving meso-dermal subtypes provides an opportunity to study mechanisms underlying polarization of mesoderm and fate choices that govern lineage specification from common progenitor populations. The generation of ECs from cardiogenic mesoderm also provides an opportunity to study the process of endocardial development and valve formation that could lead to commercial applications for therapeutic outcomes. In an effort to re-create the lineages of the heart from hPSCs, this method for derivation of ECs from cardiogenic mesoderm couples well with existing protocols for generating vascular ECs, pericytes, smooth muscle cells, cardiomyocytes, epicardial cells, and fibroblasts16,17,30,31. This protocol's high efficiency of differentiation is a major advance, providing a scalable source of cells for applications such as tissue engineering, cell therapy, disease modeling, and drug discovery.
Overview of the procedure and experimental design
In this report, we use developmental cues in a monolayer differentiation approach to generate different cardiovascular subtypes, reproducibly achieving >90% purity of all lineages without sorting (an overview of this protocol is provided in Fig. 2). The efficient control of differentiation of diverse cardiovascular subtypes from hPSCs opens opportunities to explore genomics, epigenetics, functional biology, drug screening, and therapeutic applications.
Figure 2.
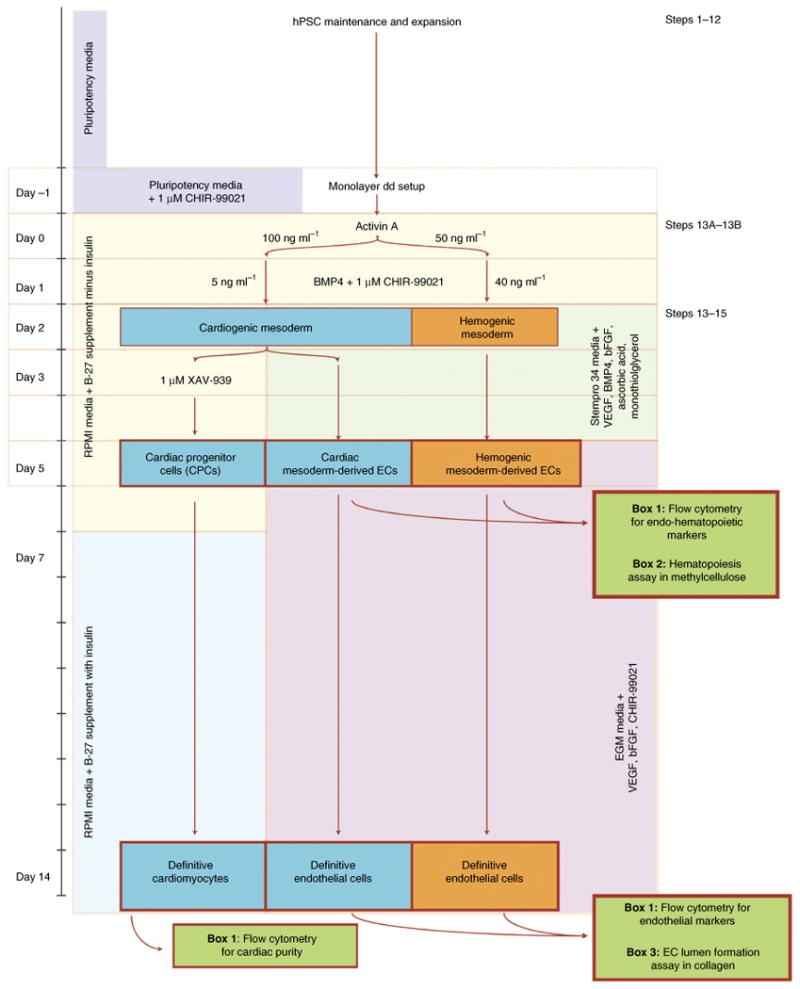
Outline of protocol for the derivation of cardiac and vascular lineages from hPSCs. The overall approach is outlined with lineage fate choices demarcated by arrows mediated by stage-specific changes in cytokine and media conditions. dd, directed differentiation.
hPSC maintenance, passaging, and setup (Steps 1–12)
We have used this protocol to derive distinct cardiovascular populations from both human embryonic stem cell (hESC; RUES2 cells) and human induced pluripotent stem cell (hiPSC) lines (WTC and IMR90 cells). Successful results have been obtained when the pluripotent stem cells are maintained in either mouse embryonic-fibroblast-conditioned medium or defined mTeSR medium. This protocol provides detailed steps for thawing hPSCs, standard maintenance, passaging, directed differentiation, and phenotyping.
During setup of differentiation, cells are dispersed to single cells using the chelating agent Versene (PBS with EDTA) and plated in pluripotency medium containing the Wnt/β-catenin agonist CHIR-99021 for 24 h before induction with activin A. Plating density of hPSCs at the onset of differentiation is a well-known variable that influences differentiation efficiency. We therefore tested three different plating densities (Fig. 3a) and determined the impact on lineage specification into cardiac and EC types, as shown throughout this protocol.
Figure 3.
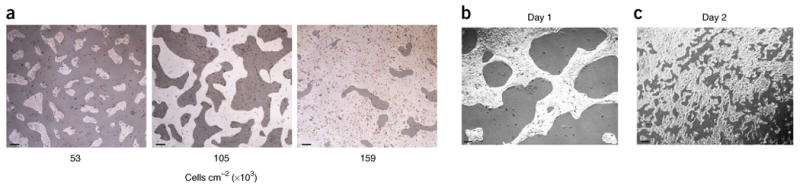
Cell culture density and phenotype from pluripotency to mesoderm. (a) Different cell densities tested throughout this protocol to assess density dependency of differentiation into cardiac and endothelial lineages. (b,c) Phenotypic changes that are observed as cells transition from pluripotency to day 1, remaining in close contact during exposure to activin A, (b) and then undergo near single-cell dispersion across the plate during exposure to BMP4 and progression to mesoderm on day 2 (c). Scale bars, 500 μm.
Polarization of lineages into cardiogenic versus hemogenic mesoderm (Step 13A and B)
This protocol provides a method for generating polarized mesoderm by induction of differentiation under varying concentrations of activin A (day 0) and BMP4 (day 1) in RPMI medium with B-27 supplement minus insulin. We also make use of the approach optimized by Zhang et al.32, in which Matrigel is added to the induction medium in an effort to facilitate signaling cues for epithelial-to-mesenchymal transition, which occurs during gastrulation. However, not all cell lines require Matrigel for efficient differentiation. In keeping with developmental gradients of activin A that direct polarization of mesoderm, cardiogenic mesoderm (mid-primitive streak) is derived under conditions of 100 ng ml−1 activin A, whereas hemogenic mesoderm (posterior-primitive streak) is derived under conditions of 50 ng ml−1 activin A. RPMI medium with B-27 supplement minus insulin is used in both cases6. We have previously shown that similar results are achieved using doses <50 ng ml−1(ref. 6). During exposure to activin A (17–18 h), cells appear to ‘tighten’, leaving acellular regions throughout the plate (Fig. 3b).
On day 1, the medium is changed to RPMI medium with B-27 supplement minus insulin, containing the Wnt/β-catenin signaling agonist CHIR-99021. Cardiogenic mesoderm is derived from cultures exposed to 5 ng ml−1 BMP4, whereas hemogenic mesoderm is derived from cultures exposed to 40 ng ml−1 BMP4. We have empirically shown that mesoderm is formed on day 2, based on time-course assays of gene expression6,7. Under the conditions described, the cells transition from a compact state on day 1 to a dispersed monolayer throughout the plate on day 2 (Fig. 3c). Table 1 provides primers for quantitative RT–PCR for analysis of pan-mesoderm genes, as well as those that show lineage-specific expression in cardiogenic versus hemogenic mesoderm. Cardiomyocyte purity is assessed at day 14 using FACS to detect cardiac Troponin T-positive (cTnT+) cells. KDR+/CD34+ expression at day 5 is used as an indicator of purity during early stages of pan-endothelial differentiation.
Table 1.
Quantitative RT–PCR primers for human genes.
| Gene name | Forward primer | Reverse primer |
|---|---|---|
| Housekeeping | ||
| HPRT | TGACACTGGCAAAACAATGCA | GGTCCTTTTCACCAGCAAGCT |
| Pan mesoderm | ||
| T | CAAATCCTCATCCTCAGTTTG | GTCAGAATAGGTTGGAGAATTG |
| KDR | ATGCACGGCATCTGGGAATC | GTCACTGTCCTGCAAGTTGCTGTC |
| Cardiogenic mesoderm | ||
| NODAL | TGGAGGTGGGATGAAGTCACCTAT | AACCCAGCCTGAGGCAATGAGATT |
| DKK1 | AACAGCTATCCAAATGCAG | TCACAGGGGAGTTCCATAAA |
| MESP1 | TCGAAGTGGTTCCTTGGCAGAC | CCTCCTGCTTGCCTACAAAGTGTC |
| GSC | GAGGAGAAAGTGGAGGTCTGGTT | CTCTGATGAGGACCGCTTCTG |
| Hemogenic mesoderm | ||
| CDX1 | GGTGGCAGCGGTAAGACTC | TGTAACGGCTGTAATGAAACTCC |
| WNT8a | GCAGAGGCGGAACTGATCTT | CGACCCTCTGTGCCATAGATG |
| WNT3a | AACTACGTGGAGATCATGCCC | GACTCCCTGGTAGCTTTGTC |
| Cardiac progenitor cells | ||
| TMEM88 | GCTGCCTTCAATCTTCTCCTG | ATAAAGGGCTCGGCTGTAGG |
| GATA4 | ACACCCCAATCTCGATATGTTTG | GTTGCACAGATAGTGACCCGT |
| ISL1 | ATTTCCCTATGTGTTGGTTGC | CGTTCTTGCTGAAGCCGATG |
| NKX2.5 | CCAAGGACCCTAGAGCCGAA | ATAGGCGGGGTAGGCGTTAT |
| MYL4 | TCAAAGAGGCCTTTTCATTG | CGTCTCAAAGTCCAGCATCT |
| Cardiomyocytes | ||
| TNNT2 | TTCACCAAAGATCTGCTCCTCGCT | TTATTACTGGTGTGGAGTGGGTGTGG |
| MYH6 | CAAGTTGGAAGACGAGTGCT | ATGGGCCTCTTGTAGAGCTT |
| MYL7 | TCCAACGTCTTTTCCATGTT | TCTGTCCCATTGAGCTTCTC |
| TBX5 | GAACCACAAGATCACGCAATTA | ACACCATTCTCACACTGGTAT |
| ATP2A2 | ATGACAACCCACTGAGAAGAGAA | CGAAGGTCAGATTGGTCTCATATTT |
| RYR2 | AGAACTTACACACACGCGACCTG | CATCTCTAACCGGACCATACTGC |
| Pan endothelium | ||
| SCL | AAGGGCACAGCATCTGTAGTCA | AAGTCTTCAGCAGAGGGTCACGTA |
| CD34 | AAATCCTCTTCCTCTGAGGCTGGA | AAGAGGCAGCTGGTGATAAGGGTT |
| CD31 | ATCATTTCTAGCGCATGGCCTGGT | ATTTGTGGAGGGCGAGGTCATAGA |
| Endocardial endothelium | ||
| NFATC1 | GCATTTTCCTTGATCCCTGT | AGCAGCTTTAGGGTGCAAAT |
| Hemogenic endothelium | ||
| HAND1 | TCAAAGACGCACTCTTCCAC | GTGCAGCGACAAAAAGAAAA |
| G ATA 1 | CTTTCAGGTGTACCCATTGC | AAAGTCTCCAGGAAGCTGGT |
| RUNX1 | ATGTGGTCCTATTTAAGCCAGCCC | TCATCTGGCTGAAGACACCAGCTT |
Cardiac differentiation (Steps 13A–14A)
The efficiency of cardiac differentiation from pluripotency is directly related to the ability to specify mid-streak cardiogenic mesoderm as opposed to the more anterior-streak endoderm or posterior-streak hemogenic mesoderm. As we have shown previously, deviating from precise specification of cardiogenic mesoderm has marked effects on the efficiency of cardiomyocyte differentiation6. We also show that the density of cells has major implications for proper specification of high-purity derivatives (Fig. 4a; Supplementary Methods).
Figure 4.
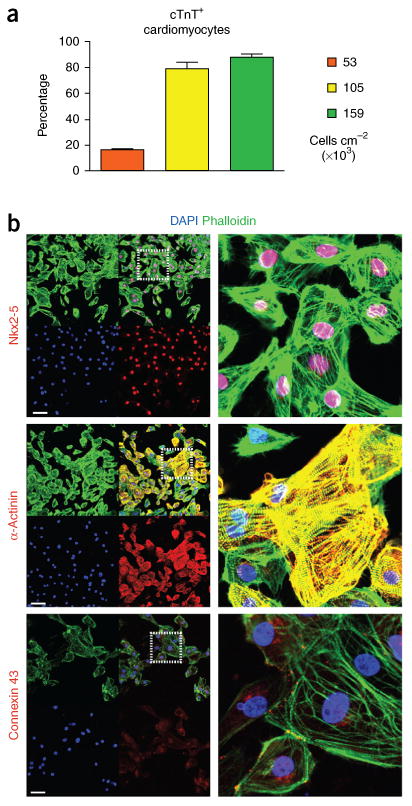
Cardiac directed differentiation. (a) Effect of cell density on cardiomyocyte differentiation efficiency based on FACS analyses of cTnT+ cells on day 14. (b) Immunohistochemistry of cardiomyocytes generated from high-density differentiation (159 cells cm−2) at day 14 based on staining for the transcription factor Nkx2-5, the myofilament protein α-actinin, and the intercalated disk protein connexin 43. n = 6 biological replicates per sample. The antobodies used are listed in the Materials section. Data are represented as mean ± sem. Scale bars, 100 μm.
Proper dosage of Wnt/β-catenin signaling has been shown to be one of the major determinants of specification into the cardiac lineage6,7,9,28,33,34. Although Wnts are required for mesoderm specification, their signaling subsequently must be inhibited to direct cells into the cardiac lineage7,9,28,34. We therefore add an exogenous tankyrase inhibitor, XAV-939, on day 3 of differentiation to reinforce this key step during cardiac progenitor cell specification. Previous work has suggested that cardiac progenitor cells, which emerge on day 5 of differentiation, can be assessed for purity on the basis of KDR/PDGFRα expression3,35. Using our protocol, we have found a statistically significant (P < 0.05) negative correlation with KDR+/PDGFRα+ cells at day 5 versus the efficiency of cardiac differentiation at day 14, and therefore we do not advocate for this phenotyping approach6. Table 1 provides primers for amplifying genes involved in specification of the cardiac progenitor cell, including NKX2-5, GATA4, TMEM88, ISL1, and MYL4.
As the protocol progresses past the progenitor cell stage, we make use of insulin in a stage-dependent manner during cardiac differentiation. This approach has been supported by studies showing that insulin is inhibitory before cardiac progenitor cell specification but is required for definitive cardiomyocyte development36. Accordingly, RPMI medium is changed on day 7 to include the B-27 supplement containing insulin. Beating cardiomyocytes can be seen as early as day 7. Cells progressively mature to definitive cardiomyocytes by day 14, at which point they are ∼90% cTnT+ as ascertained by FACS analysis and express high levels of Nkx2-5, α-actinin, and the junctional protein connexin 43 (Fig. 4b). Box 1 describes methods for staining cardiomyocytes for quantification of cTnT and smooth muscle actin (SMA) by FACS analysis. SMA is expressed in immature cardiomyocytes but is replaced by cardiac α-actin (ACTC1), as cardiomyocytes become more mature. Table 1 provides primers for amplifying genes expressed in definitive cardiomyocytes, including the myofilament genes TNNT2 and MYH6, the transcription factor TBX5, and the calcium-handling genes ATP2a2 and RYR2. For further phenotyping, we recommend previous protocols that have provided detailed methods for characterization of cardiomyocytes by immunohistochemistry27. As with other lineages outlined in the protocol, this protocol does not require purification (e.g., using selection or sorting37–41) at any point to achieve high-purity cardiomyocytes.
Box 1. Flow cytometry analysis of hPSC-derived cardiomyocytes and ECs.
Additional Materials
96-Well round-bottom plate (Costar, cat. no. 3799)
Procedure
Wash the cells with PBS. Aspirate the PBS and add 10% (vol/vol) trypsin-Versene solution (0.25% (vol/vol) trypsin) and incubate the cells at 37 °C.
-
Monitor the cells under a microscope until they are single cells—roughly 2-5 min.
▲ CRITICAL STEP For all cells, triturate no more than three to five times to resuspend. To transition the colonies to single cells,keeping cells longer in trypsin solution is better than over-triturating the cells. Stop the trypsin-Versene reaction by transferring cellsfrom step 1 to a tube with an equal volume of stop solution (FBS:DMEM 1:1 + 200 units ml−1 of DNase 1).
Centrifuge the cells at 300g for 5 min at 22–24 °C and aspirate the medium.
-
Stain the cardiomyocytes as described in option A and stain the endothelial cells as described in option B.
-
Staining of cardiomyocytes
-
If performing cTnT/SMA staining, cells must be fixed before staining. Resuspend cells from step 3 in 200 μl of 4% (wt/wt) paraformaldehyde solution and incubate at 4 °C for 10 min. Centrifuge the cells at 300g for 5 min at 22-24 °C, pipette the paraformaldehyde into an appropriate waste container, and resuspend the cell pellets in 300-500 μl of 5% (vol/vol) FBS–PBS solution.
■ PAUSE POINT The cells can be kept fixed at 4 °C for up to 7 d before staining.
Count cells with a hemocytometer and transfer 1.0–5.0 × 105 cells from each sample to one well of a 96-well round-bottom plate. Centrifuge the plate at 250g for 5 min at 22–24 °C and decant the supernatant.
Resuspend the pellet with 50 μl of 0.75% (wt/vol) saponin in 5% (vol/vol) FBS–PBS with the appropriate dilution of primary antibody according to Table 3. Antibody combinations of cTnT and SMA are recommended for double staining. Incubate the mixture at room temperature for at least 30 min.
Add 150 μl of 0.75% saponin in 5% (vol/vol) FBS–PBS to each well and triturate five times. Centrifuge the plate at 300g for 5 min at 22–24 °C and decant the supernatant.
Repeat Step 4A(iv) of the PROCEDURE using 200 μl of 0.75% saponin in 5% (vol/vol) FBS–PBS.
Resuspend the pellet with 50 μl of 5% (vol/vol) FBS–PBS with the appropriate dilution of secondary antibody according to Table 3. Incubate at room temperature for at least 30 min in darkness.
Repeat Steps 4A(iv and v) of the PROCEDURE using 5% (vol/vol) FBS–PBS.
-
Add 200 μl of 5% (vol/vol) FBS–PBS to each well and triturate five times. Transfer each sample to a separate 5-ml FACS tube. Add an additional 200 μl of 5% (vol/vol) FBS-PBS and 100 μl of 4% (wt/wt) paraformaldehyde.
■ PAUSE POINT Cells can be kept at 4 °C for up to 7 d before flow cytometric analysis.
-
-
Staining of endothelial cells
This protocol is useful for analyzing day 5 ECs or later-stage definitive ECs.
▲ CRITICAL ECs and hematopoietic derivatives are stained and analyzed live. Do not fix cells at any point in the protocol.
Resuspend the cells in 200 μl of DMEM + 10% FBS and transfer them to one well of a 96-well round-bottom plate. Centrifuge the plate at 250g for 5 min at 22–24 °C, and decant the supernatant.
Resuspend the cells in DMEM containing appropriate dilutions of the desired antibody, with a total volume of 50 μl (Table 3).
Incubate the cells at 4 °C for 30–45 min.
Add 150 μl of 5% (vol/vol) FBS–PBS to each well and triturate five times. Centrifuge the plate at 250g for 5 min at 22–24 °C and decant the supernatant.
Repeat Step 4B(iv) of the PROCEDURE using 200 μl of 5% (vol/vol) FBS–PBS.
Analyze the cells immediately by flow cytometry.
-
Differentiation of ECs from mesodermal subtypes (Steps 13 and 14B)
Previous work has shown a requirement for VEGF and active Wnt/β-catenin signaling in the development of endothelium42,43. We tested a wide range of culture conditions adapted from previous differentiation protocols14,18,19,44,45 to determine optimal parameters for differentiation of the endothelium, building from our monolayer-directed mesodermal differentiation platform (Supplementary Fig. 1). This yielded some interesting observations. First, as compared with the marked effects observed with cardiac differentiation, we found that the seeding densities tested at the onset of differentiation did not markedly influence the purity of ECs generated by this protocol (Fig. 5). Second, in contrast to current embryoid body protocols14,18,19,44, high levels of VEGF stimulation are required to direct endothelial differentiation in a monolayer format (Supplemental Fig. 1; Fig. 5). Third, we determined that cells remain responsive to VEGF stimulation between days 2 and 3 of differentiation but lack responsiveness before or after that window of time (Supplementary Fig. 2). Last, we found that augmenting Wnt/β-catenin pathway activity after mesoderm induction using the agonist CHIR-99021 was inhibitory to endothelial differentiation at all doses tested (Supplementary Fig. 1). Among a wide range of conditions vetted, we describe here a unique protocol that generates >90% pure ECs from cardiogenic and hemogenic mesoderm. ECs generated from cardiogenic mesoderm have poor blood-forming capacity and express markers of endocardial endothelium, whereas ECs generated from hemogenic mesoderm (e.g., those expressing NFATC1) have robust blood-forming potential and express markers of the extraembryonic mesoderm (e.g., HAND1)6.
Figure 5.
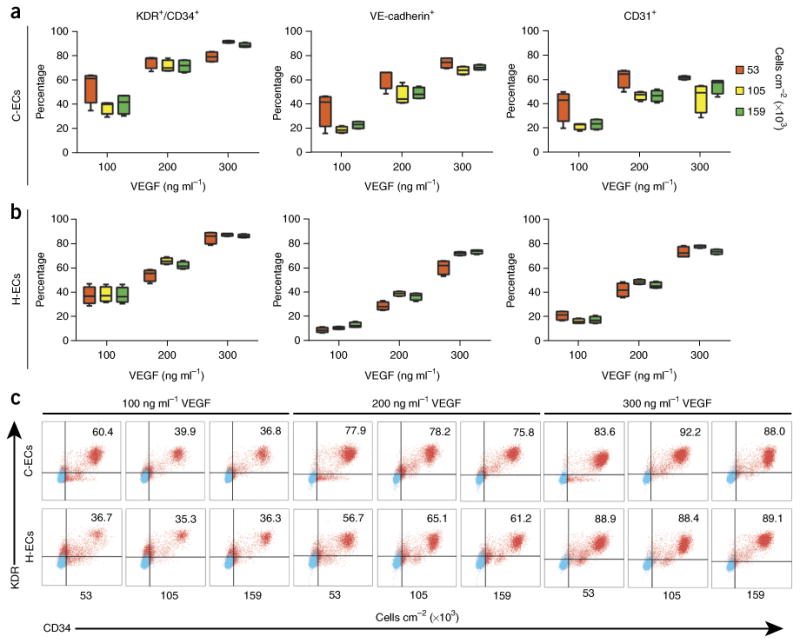
Variables influencing endothelial differentiation efficiency. (a,b) Cells were differentiated into C-ECs (a) and H-ECs (b), and tested for efficiency on the basis of density at the onset of differentiation and amount of VEGF exposure at day 2. Cells were phenotyped on day 5 of differentiation for KDR/CD34 (c), VE-cadherin, and CD31 by FACS analysis. (c) Raw FACS plots for KDR/CD34 analysis. These data show a clear dependency of differentiation on the basis of VEGF stimulation but much less effect of density at the onset of differentiation. n = 6 biological replicates per sample. Data are represented as mean ± sem.
In contrast to cardiomyocyte differentiation, which is highly sensitive to specification of hPSCs through cardiogenic mesoderm, endothelium is known to form from all mesodermal origins1,46. We have similarly shown this with hPSC endothelial differentiation6. On day 2, when hPSCs have formed mesoderm subtypes (cardiogenic or hemogenic), EC differentiation is directed by changing the medium to StemPro-34, containing VEGF, BMP4, bFGF, ascorbic acid, and monothioglycerol. Cells remain in this medium until day 5. Endothelial purity is assayed at day 5 by FACS analysis for KDR+/CD34+ cells; however, markers of definitive endothelium, including VE-cadherin and CD31, are also evident at this time point (Fig. 5; Supplementary Fig. 3; Supplementary Methods). Box 1 describes methods for staining ECs for analysis of KDR and CD34 by FACS analysis. Table 1 outlines primers for quantitative RT–PCR analysis of pan-endothelial markers (e.g., SCL) versus those that specifically identify endocardial endothelium (e.g., NFATC1) versus blood-forming endothelium (e.g. GATA1, RUNX1, and HAND1).
Findings during mouse development have shown that endocardial endothelium has very limited, transient hematopoietic activity, whereas the posterior-streak-derived hemogenic endothelium is the primary source for blood formation in development47. We have shown that the hPSC endothelium derived from cardiogenic mesoderm exhibits modest blood-forming activity, largely giving rise to primitive erythroid derivatives, whereas hPSC–ECs derived from hemogenic mesoderm have the capacity for both the erythroid and myeloid lineages6. By contrast, the hPSC cardiac progenitor cells have no blood-forming activity7. As described in Box 1, we use phenotyping of day 5 cells initially to characterize primitive erythroid cell types based on the percentage of CD43+/CD235a+ cells (Fig. 6a; Supplementary Fig. 3; Supplementary Methods)6. As a secondary assay, we provide a brief description in Box 2 for the blood colony-forming assay in methylcellulose (Fig. 6b). Methods for differentiation into later-stage hematopoietic derivatives have been described elsewhere6,7,48.
Figure 6.
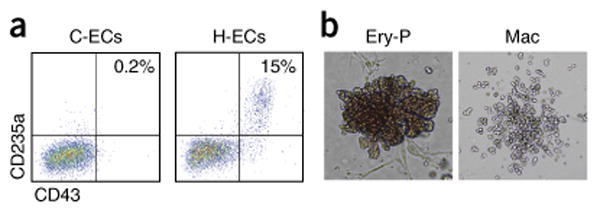
Hematopoiesis assays from hESC-ECs. (a) Representative FACS plot of day 5 C-ECs and H-ECs showing differences in primitive hematopoietic activity, indicated by CD43/CD235a double-positive cells. (b) Representative images of primitive erythroid (Ery-P) and macrophage (Mac) colonies in methylcellulose colony-forming assays. Images are magnified for Ery-P (100×) and Mac (50×) to show details of colony morphology.
Box 2. Primitive hematopoietic colony-forming assays.
Additional Materials
Iscove's Modified Dulbecco's Medium (Gibco, cat. no. 12440)
Methylcellulose medium with human cytokines (H4034, cat. no. StemCell Technologies)
35-mm Suspension culture dishes (Corning, cat. no. 430588)
Procedure
Harvest and count cells with a hemocytometer. Centrifuge at 250g for 5 min at 22–24 °C and resuspend cells at a concentration of 1 × 106 cells per ml in Iscove's Modified Dulbecco's Medium.
Add 300 μl of cells to 3 ml of methylcellulose medium containing recombinant human stem cell factor, granulocyte-macrophage colony-stimulating factor (GM-CSF), granulocyte colony–stimulating factor (G-CSF), interleukin-3, and erythropoietin. Vortex vigorously to evenly distribute cells.
Use a 3-ml syringe and a 16-gauge 1.5-inch needle to add 1 ml of methylcellulose medium with cells to each of three 35-mm suspension culture dishes. Rotate the plates to evenly distribute the methylcellulose medium.
Place two plates with methylcellulose medium into a 10-cm tissue culture dish with one additional 35-mm dish without a lid, containing sterile water, to maintain proper humidity for the methylcellulose. Incubate at 37 °C in 5% CO2.
After 12–14 d, hematopoietic colonies can be scored visually. At this early stage of hematopoietic differentiation, colonies consist predominantly of primitive erythroid (Ery-P) and macrophage (Mac) lineages (Fig. 5).
To mature cells into definitive ECs, day 5 cells are passaged for expansion in endothelial growth medium (EGM) containing VEGF, bFGF, and CHIR-99021 for an additional 5–10 d. We tested a number of matrix substrates to determine their effect on maturation of ECs, including Matrigel, fibronectin, and gelatin, and found no difference (Fig. 7). We found that differentiation under these conditions led to near 99% CD31+ ECs from both cardiogenic and hemogenic mesoderm without requiring sorting (Fig. 7a,b).
Figure 7.
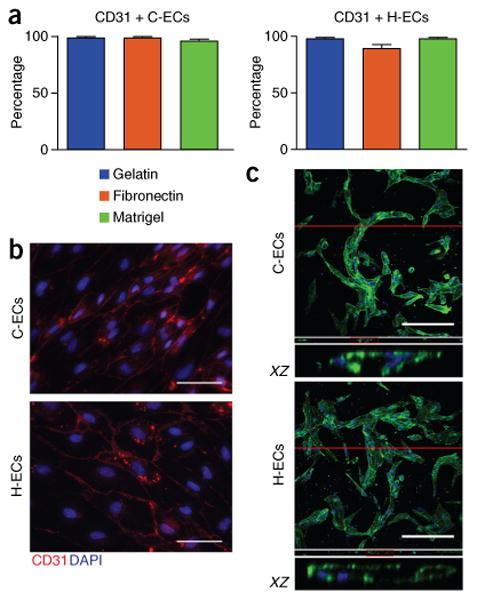
Differentiation into definitive endothelial cells. (a) Analysis of EC differentiation using different matrix substrates, showing no effect on differentiation efficiency using gelatin, fibronectin, or Matrigel. (b) Staining of C-ECs and H-ECs in a monolayer for CD31. (c) Confocal z stack showing de novo lumen formation in collagen gel, as seen in both C-ECs and H-ECs stained with phalloidin (green) and DAPI (blue). Orthogonal slices (XZ) below each z stack show lumen formation. n = 5 biological replicates per sample for C-ECs and 9 replicates for H-ECs. Data are represented as mean ± sem. Scale bars, 100 μm.
In our previous work, we have shown marked differences in the functionality of EC subtypes in the context of engineered microvascular networks6. Given the technically specialized nature of this method49, we provide a more classic approach for assaying EC functionality on the basis of lumen formation in collagen gels (Box 3). Previous protocols have also provided extensive details for performing immunohistochemistry for endothelial markers, as well as a variety of additional functional end points for assaying ECs in vitro and in vivo16,17.
Box 3. Collagen-based tubulogenesis assay.
Additional Materials
Rat tail collagen, type I (Life Technologies, cat. no. A1048301)
10× M199 (Sigma-Aldrich, cat. no. M0650)
1N NaOH (Sigma, cat. no. S2770)
μ-Slide Angiogenesis (Ibidi, cat. no. 81506)
Alexa Fluor 488 Phalloidin (Invitrogen, cat. no. A12379)
Alexa Fluor goat anti-rabbit 568 (Invitrogen, cat. no. A11011)
Rb pAb CD31 (Abcam, cat. no. 28364)
Triton X-100 (Sigma, cat. no. Sigma X100)
Procedure
Harvest day 14 ECs with standard enzymatic digestion methods (0.05% (vol/vol) trypsin). Count the cells using a hemocytometer and set aside on ice during collagen preparation.
-
Dilute and neutralize the collagen gel to a final concentration of 2 mg ml−1. Calculate the volume of reagents needed (table 4). Combine 10× M199 medium, NaOH, and half of the total EGM. Mix together and combine with the collagen. Mix well by pipetting slowly until a homogeneous solution is obtained.
▲ CRITICAL STEP Keep the collagen on ice while mixing to prevent gelation until ready. Mix slowly and carefully to avoid bubbles.
▲ CRITICAL STEP Monitor the pH of the collagen mixture to ensure that it is properly neutralized. If the mixture is too acidic, continue to bring the pH up with NaOH until a pH of 7.2 is obtained. Adjust the volume of EGM used in step 2 as needed to maintain constant final volume.
Determine the number of cells required to achieve a final cell density of 2 × 106 per ml (no. cells = 2 × 106 × VFINAL). Resuspend the cells in the remaining EGM volume and add to the neutralized collagen mixture. Mix well by pipetting until the cells are evenly distributed.
Pipette the collagen + cell mixture into wells of angiogenesis μ-slides (4-mm-diameter wells, 10-μl volume per well). Allow the samples to gel at 37 °C for 20 min.
After gelation, feed the cells with EGM supplemented with 1 μM CHIR-99021, 20 ng ml−1 bFGF, and 20 ng ml−1 VEGF and replace with fresh medium every day.
After 2 d, fix the samples in 3.7% (wt/wt) formaldehyde for 10 min and wash them in PBS.
Stain in situ for CD31 and phalloidin (using standard immunohistochemistry methods)
-
7
First, block fixed samples in PBS with 2% (wt/vol) BSA and 0.5% (vol/vol) Triton X-100.
-
8
Next, incubate the cells overnight with primary antibody at 4 °C: 1:30 Rb pAb to hCD31.
-
9
Wash the cells with PBS three times, 15 min each.
-
10
Incubate with secondary antibodies for 1 h at room temperature: 1:100 Alexa Fluor 488 Phalloidin and 1:100 Alexa Fluor goat anti-rabbit 568.
-
11
Image the samples using confocal microscopy to obtain 3D z stacks.
Materials
Reagents
1-Thioglycerol (MTG; Sigma-Aldrich, cat. no. M6145-25 ml)
1× StemPro-34 SFM (Life Technologies, cat. no. 10639-011)
Activin A (R&D Systems, cat. no. 338-AC)
Allophycocyanin (APC)-conjugated donkey anti-rabbit IgG antibody (Jackson, cat. no. 711-136-152)
APC-conjugated mouse anti-human CD43 antibody (BD, cat. no. 560198)
B-27 supplement (Life Technologies, cat. no. 17504-044)
B-27 supplement minus insulin (Life Technologies, cat. no. A18956-01)
bFGF (PeproTech, cat. no. AF-100-18B)
BMP4 (R&D Systems, cat. no. 314-BP)
BSA (Sigma-Aldrich, cat. no. A9418-50G)
CHIR-99021(Cayman Chemical, cat. no. 13122)
DMEM (Life Technologies, cat. no. 10313-021)
DMEM/F12 (Life Technologies, cat. no. 11320-033)
DMSO (Sigma-Aldrich, cat. no. D2650)
DNase 1, bovine pancreas (Calbiochem, cat. no. 260913)
EGM BulletKit (Lonza, cat. no. CC-3124)
FBS (HyClone, cat. no. SH30396.03)
FITC mouse anti-human CD31 antibody (BD Pharmingen, cat. no. 555445)
5% (vol/vol) Gelatin solution (Sigma-Aldrich, cat. no. G1393-100ml)
12 M Hydrochloric acid (HCl; Fisher, cat. no. A14451-212)
l-Ascorbic acid 2-phosphate sesquimagnesium salt hydrate (Sigma-Aldrich, cat. no. A8960-5G)
l-Glutamine (Life Technologies, cat. no. 25030-081)
Matrigel (Corning, cat. no. 356231)
Mouse IgG (FITC) isotype control (Abcam, cat. no. ab37356)
Mouse IgG1 Isotype Control (eBioscience, cat. no. 14-4714-85)
mTeSR1 Complete Kit (StemCell Technologies, cat. no. 05850)
4% (wt/wt) Paraformaldehyde solution (Affymetrix, cat. no. 199943)
PBS (Life Technologies, cat. no. 14190-144)
Phycoerythrin (PE)-conjugated anti-human VEGF R2/KDR mouse IgG1 antibody (R&D, cat. no. FAB357P)
PE-conjugated goat anti-mouse IgG antibody (Jackson, cat. no. 115-116-146)
PE/Cy7-conjugated mouse IgG1 isotype control (BioLegend, cat. no. 400125)
PE/Cy7-conjugated mouse anti-human CD235a antibody (BD Pharmingen, cat. no. 563666)
PE/Cy7 mouse IgG1 isotype control (BioLegend, cat. no. 400125)
Penicillin–streptomycin (Life Technologies, cat. no. 15140-122)
Peridinin chlorophyll (PerCP)-conjugated anti-human CD34 antibody (BD, cat. no. 340430)
PerCP-conjugated mouse IgG1 isotype control (BD Pharmingen, cat. no. 559425)
Rabbit anti-alpha smooth muscle actin antibody [E184] (Abcam, cat. no. ab32575)
Rabbit IgG isotype control (Abcam, cat. no. ab171870)
RPMI medium (Life Technologies, cat. no. 11875-093)
Human pluripotent stem cells ! CAUTION Appropriate national laws and institutional regulatory board guidelines must be followed, and informed consent should be obtained from human subjects. The cell lines used should be regularly checked to ensure that they are authentic and that they are not infected with mycoplasma. The cell lines that we have successfully used with this protocol include RUES2 (Rockefeller University), IMR90 (WiCell), and WTC11 (University of San Francisco).
Saponin, from Quillaja Bark (Sigma-Aldrich, cat. no. S7900-100G)
Tris base (Fisher, cat. no. BP152-500)
Troponin T Cardiac Isoform antibody (Thermo Scientific, cat. no. MA5-12960)
2.5% (vol/vol) trypsin (Life Technologies, cat. no. 15090-046)
VE-cadherin antibody (Abcam, cat. no. 7047)
VEGF (PeproTech, cat. no. 100-20)
Versene (Life Technologies, cat. no. 15040-066)
XAV-939 (Tocris Bioscience, cat. no. 3748)
Y-27632 (Tocris Bioscience, cat. no. 1254)
Y-27632 (Tocris Bioscience, cat. no. 1254)
EQUIPMENT
Plates (100 mM; Corning, cat. no. 353003)
24-Well plates (Corning, cat. no. 353047)
Polystyrene round-bottom tube (5 ml; Corning, cat. no. 352054)
CL2 centrifuge (Thermo Scientific, cat. no. 66001 BA)
Conical tubes (15 and 50 ml; Thermo Scientific, cat. nos. 339651 and 339653)
FACS machine (2-laser, 6-color; BD FACSCanto II)
FACS acquisition software (BD FACSDiva)
FACS analysis software (FlowJo v9.8.5)
Hemocytometer (Fisher Scientific, cat. no. 0267110)
Humidified tissue culture incubator (37 °C, 5% CO2; Heraeus HERA Cell, cat. no. 51013669)
Inverted contrasting microscope (Leica DMIL, cat. no. 090-135-001)
Serological pipettes (5, 10, 25, and 50 ml; Costar, cat. nos. 4487, 4488, 4489, and 4490)
Shel Lab H2O bath incubator (Sheldon Manufacturing, cat. no. W14M)
Sterile biological safety cabinets (Nuaire Class II Type A2)
Sterile filters (125, 250, 500, and 1,000 ml; Thermo Scientific, cat. nos. 565-0020, 568-0020, 569-0020, and 567-0020)
Sterilized Pasteur pipettes (VWR, cat. no. 14672-200)
Reagent reservoir (50 ml; Costar, cat. no. 4870)
Reagent Setup
Activin A (10 mg ml−1)
In a sterile hood, on ice, add 5 ml of 4 mM HCl in 0.1% (wt/vol) BSA–PBS to a 50-μg vial of activin A and dissolve. Divide the solution into 300-μl aliquots and store them at −20 °C for up to 6 months.
BMP4 (10 mg ml−1)
In a sterile hood, on ice, add 5 ml of 4 mM HCl in 0.1% (wt/vol) BSA–PBS to a 50-μg vial of BMP4 and dissolve. Divide the solution into 20-μl aliquots and store them at −80 °C for up to 6 months.
bFGF (10 mg ml−1)
In a sterile hood, on ice, reconstitute a 1-mg vial of bFGF using 100 ml of 10 mM Tris (pH 7.6) in 0.1% (wt/vol) BSA–H2O. Divide the solution into 800-μl and 400-μl aliquots in 1.5-ml tubes and store them at −80 °C for up to 1 year.
VEGF (500 mg ml−1)
In a sterile hood, on ice, reconstitute one vial (50 μg) of VEGF powder in 100 μl of 0.05% (wt/vol) BSA–H2O. Divide the solution into 5-μl single-use aliquots and store them at −80 °C for up to 1 year. Use the aliquots immediately after thawing.
CHIR-99021 (25 mM)
In a sterile hood, add 860 μl of DMSO to 10 mg of CHIR-99021. Divide the solution into 10-μl aliquots and store them at −20 °C for up to 1 year.
XAV-939 (10 mM)
In a sterile hood, add 3.2 ml of room-temperature (22–24 °C) DMSO to one 10-mg vial of XAV-939 and resuspend. Divide the solution into 100-μl aliquots in 1.5-ml tubes and store them at −20 °C for up to 1 month.
Y-27632 (10 mM)
In a sterile hood, add the appropriate volume of DMSO to a 50-mg vial and dissolve it to a concentration of 10 mM. Divide the solution into 100-μl aliquots and store them at −20 °C for up to 1 year.
1 M Tris (pH 7.6) (500 ml)
In a sterile hood, add 60.57 g of Tris base to 350 ml of Millipore water. Adjust the volume of the solution to 500 ml and adjust the pH to 7.6 ± 0.1. This solution can be stored at room temperature for up to 1 year.
10 mM Tris (pH 7.6) in 0.1% (wt/vol) BSA–H2O (500 ml)
In a sterile hood, add 5 ml of 1 M Tris (pH 7.6) solution to 495 ml of 0.1% (wt/vol) BSA–H2O solution and filter-sterilize using a 500-ml sterile filter. Store the solution at 4 °C for up to 1 year.
0.05% (wt/vol) BSA–H2O (100 ml)
In a sterile hood, add 50 mg of BSA to 100 ml of Millipore H2O and filter using a 125-ml sterile filter. Store this solution at 4 °C for up to 1 year.
0.1% (wt/vol) BSA–PBS (1,000 ml)
In a sterile hood, add 1 g of BSA to 1,000 ml of PBS and filter using a 1,000-ml sterile filter. Store this solution at 4 °C for up to 1 year.
0.4 mM HCl in 0.1% (wt/vol) BSA–PBS (1,000 ml)
In a sterile hood, add 33.33 μl of 12 M HCl solution to 1,000 ml of 0.1% (wt/vol) BSA–PBS and mix thoroughly. Store this solution at 4 °C for up to 6 months.
5% (vol/vol) FBS–PBS (500 ml)
In a sterile hood, mix 25 ml of FBS with 475 ml of PBS. The solution can be stored at 4 °C for up to 6 months.
0.1% (vol/vol) Gelatin–H2O(500 ml)
In a sterile hood, mix 10 ml of 5% (vol/vol) Gelatin solution with 490 ml of Millipore water. Filter using a 500-ml sterile filter. If the 5% (vol/vol) stock gelatin solution is congealed, warm it in a 37 °C water bath until it has dissolved. The solution can be stored at 4 °C for up to 6 months.
30× Matrigel–DMEM/F12 (20 ml)
In a sterile hood, add 10 ml of 4° C DMEM/F12 to one vial (10 ml) of 60× Matrigel at 4 °C and resuspend.
! CAUTION Matrigel is very sensitive to changes in temperature and may gel if it is not aliquotted quickly. Divide the solution into (3 ml) aliquots and freeze them at −20° C. The solution can be stored at 4 °C for up to 6 months.
10% (vol/vol) trypsin–Versene solution(10 ml)
In a sterile hood, gently mix 1 ml of 2.5% (vol/vol) trypsin at 4 °C with 9 ml of Versene at 37 °C in a 15-ml conical tube. We do not recommend storing this solution; use it fresh.
Stop solution
In a sterile hood, gently mix equal volumes of FBS and DMEM/F12 with 200 units ml−1 of DNase 1. The solution can be stored at 4 °C for up to 1 month.
0.75% Saponin in 5% (wt/vol) FBS–PBS (500 ml)
In a sterile hood, mix 3.75 g of dry saponin with 500 ml of 5% (wt/vol) FBS–PBS.
! CAUTION Saponin is a very dangerous, light chemical that must be weighed in a fume hood. After thoroughly mixing, filter the solution using a 500-ml sterile filter. The solution can be stored at 4 °C for up to 6 months.
mTeSR1 basal medium + 1× mTeSR1 supplement + 1 mg ml−1 penicillin–streptomycin (500 ml)
In a sterile hood, add the contents of one 5× mTeSR1 supplement vial (100 ml) to 400 ml of mTeSR1 basal medium and mix thoroughly to make mTeSR1 medium. Then, mix 5 ml of 10 mg ml−1 penicillin–streptomycin with 495 ml of the mTeSR1 medium. The medium can be stored at 4 °C for up to 4 weeks. The mTeSR1 medium used in this protocol includes the supplement.
RPMI + 100 ng (or 50 ng) ml−1 activin A + 1× B-27 without insulin + 1× Matrigel
In a sterile hood, add appropriate volumes (see the PROCEDURE for protocols) of 10 μg ml−1 activin A, 50× B-27 without insulin, and 30× Matrigel-DMEM/F12 solution to RPMI to achieve your total desired volume (see Table 2 for respective plate format volumes). We do not recommend storing this solution.
Table 2.
Media volumes (ml) for cell feeding during differentiation based on plate format.
| plate format | |||||
|---|---|---|---|---|---|
|
|
|||||
| ml per well added: | 24 wells | 12 wells | 6 wells | 10 cm | 15 cm |
| Step 13A(i) (day 0) | 0.5 | 1 | 1.5 | 10 | 30 |
| Steps 13A(ii)–14AB | 1 | 2 | 4 | 20 | 60 |
RPMI + 5 ng (or 40 ng) ml−1 BMP4 + 1 mM CHIR-99021 + 1× B-27 without insulin
In a sterile hood, add appropriate volumes (see the PROCEDURE for protocols) of 10 μg ml−1 BMP4, 25 mM CHIR-99021, and 50× B-27 minus insulin to RPMI to achieve your total desired volume (see Table 2 for respective plate format volumes). We do not recommend storing this solution.
RPMI + 1 μM XAV 939 + 1× B-27 without insulin
In a sterile hood, add appropriate volumes (see the PROCEDURE for protocols) of 10 mM XAV 939 and 50× B-27 minus insulin to RPMI to achieve your total desired volume (see Table 2 for respective plate format volumes). We do not recommend storing this solution.
RPMI + 1× B-27 minus (or plus) insulin (510 ml)
In a sterile hood, add one vial (10 ml) of 50× B-27 minus (or plus) insulin to 500 ml of RPMI. We do not recommend storing this solution.
StemPro + 4 × 10−4 M MTG + 2 mM L-glutamine + 50 μg ml−1 ascorbic Acid + 10 ng ml−1 BMP4 + 5 ng ml−1 bFGF + 300 ng ml−1 VEGF (25 ml)
In a sterile hood, add appropriate volumes (see the PROCEDURE for protocols) of 1 M MTG, 200 mM l-glutamine, 5 mg ml−1 ascorbic acid, 10 μg ml−1 BMP4, 10 μg ml−1 bFGF, and 500 μg ml−1 VEGF to StemPro medium to achieve your total desired volume (see Table 2 for respective plate format volumes). We do not recommend storing this solution.
EBM + EGM supplements
In a sterile hood, thoroughly mix the EGM supplements from the BulletKit with EBM (500 ml). The medium can be stored at 4 °C for up to 2 weeks. The EGM referred to in the PROCEDURE is EBM with EGM supplements already added.
EGM + 20 ng ml−1 VEGF + 20 ng ml−1 bFGF + 1 μM CHIR-99021
In a sterile hood, add appropriate volumes (see the PROCEDURE for protocols) of 500 μg ml−1 VEGF, 10 μg ml−1 bFGF, and 25 mM CHIR-99021 to EGM medium to achieve your total desired volume (see Table 2 for respective plate format volumes). We do not recommend storing this solution.
Equipment Setup
Matrigel-coated plates (25 ml)
In a sterile hood, gently mix 0.83 ml of Matrigel-DMEM/F12 30× with 24.17 ml of DMEM/F12 in a 50-ml conical tube. Immediately add Matrigel in DMEM/F12 at a volume of 6 ml per 10-cm plate or 500 μl per well of a 24-well plate. Allow the Matrigel to set overnight at 4 °C. The Matrigel-coated plates can be stored at 4 °C for up to 3 weeks.
0.1% (vol/vol) Gelatin–H2O-coated plates
In a sterile hood, add 6 ml of gelatin–H2O solution per 10-cm plate or 500 μl per well of a 24-well plate. Allow the gelatin-–H2O solution to set at room temperature for 10 min. Use gelatin–H2O-coated plates immediately. We do not recommend storing any gelatin–H2O-coated plates for later use.
Procedure
Feeder-free culture of hPSCs ● TIMING 30 min, plus 4 d for maintenance
Take a Matrigel-coated plate from 4 °C and place it in a biological safety cabinet at room temperature for 15 min to warm.
-
Remove hPSCs, such as RUES2s (Rockefeller University), from liquid N2 and thaw rapidly.
! CAUTION Appropriate national laws and institutional regulatory board guidelines must be followed, and informed consent should be obtained from human subjects. All necessary consent forms were acquired for hPSCs used in this study.
Transfer the cells to a 50-ml conical tube containing 5 ml of mTeSR1 medium with 1 μg ml−1 penicillin–streptomycin.
Centrifuge the cell suspension at 200g for 5 min at room temperature. Aspirate and discard the supernatant with a sterilized Pasteur pipette.
-
Using a 5-ml pipette, gently resuspend the cell pellet in mTeSR1 medium + 10 μM Y27632 + 1 μg ml−1 penicillin-streptomycin. Limit the number of triturations. Count the cells. After aspirating the liquid from the warmed Matrigel-coated plate, slowly add cells to the medium for a plating density of ∼1.2 × 104 cells cm−2. Put the plate into a 37 °C, 5% CO2 incubator. Move the plate in quick, short, to-and-fro and side-to-side motions to evenly distribute the cells across the surface and then leave them undisturbed overnight.
▲ CRITICAL STEP Including the Rho kinase inhibitor Y-27632 is very important for high hPSC recovery after freezing and thawing.
The next day, aspirate the medium and replace it with fresh 37 °C mTeSR1 medium containing 1 μg ml-1 penicillin–streptomycin. Repeat this medium replacement daily until the cells are ready for passage (Step 7).
Passaging of hPSCs using Versene ● TIMING 20 min, plus 4 d for maintenance
-
7
When the cells are 75–85% confluent or if individual colonies are wider than the diameter of the field with a 10× objective, aspirate the old medium and add room-temperature PBS. Aspirate the PBS and add Versene prewarmed to 37 °C.
? TROUBLESHOOTING
-
8
Incubate the plate at 37 °C, 5% CO2, and wait for 2 min. Monitor the progress of cell detachment by viewing with a microscope. Continue until the cells in the center of colonies begin to round up.
-
9
Aspirate the Versene. Resuspend single cells using mTeSR1 + 10 μM Y27632 by washing the plate starting at the bottom and working side-to-side until reaching the top. Continue this process until all of the cells are detached. Be sure to limit the number of triturations. After the cells are removed from the surface of the well, add the contents of the well to a sterile conical tube.
-
10
Aspirate the liquid from a room-temperature Matrigel-coated plate. Add mTeSR1 + 10 μM Y27632 + 1 μg ml−1 penicillin–streptomycin to the plate. Next, slowly add a fraction of the cell suspension from Step 9 to the plate to achieve a plating density of ∼1.2 × 104 cells cm−2. Place the plate in the incubator. Move the plate in quick, short, to-and-fro and side-to-side motions to evenly distribute the cells across the surface of the plate. Leave the plate undisturbed overnight.
-
11
The next day, aspirate the medium and replace it with fresh 37 °C mTeSR1 + 1 μg ml−1 penicillin–streptomycin. Repeat this medium replacement daily until the cells are 75–85% confluent or any one individual colony is wider than the diameter of the viewing field with a 10× objective (the amount of time this takes depends on the split ratio); daily monitoring is necessary. Pluripotent cell culture can be maintained by repeating Steps 7–11 before moving to the next steps for monolayer differentiations of hPSCs.
Monolayer differentiation setup for hPSCs ● TIMING 20 min
-
12
For cells prepared for differentiation, add an appropriate volume of cells from Step 9 to a volume of 37 °C mTeSR1 + 1 μM CHIR-99021 + 10 μM Y27632, resulting in an appropriate concentration of cells to achieve proper seeding density (see Figs. 5 and 6 for more information). For differentiation into cardiomyocytes, plate at ∼1.6 × 105 cells cm−2. The plating density for endothelial differentiation is less critical. Differentiation conditions have been optimized for a 24-well plate but can be expanded to accommodate any format.
? TROUNLESHOOTING
Directed differentiation into polarized mesodermal lineages
-
13
Both cardiogenic and hemogenic mesoderm can readily be derived from hPSCs by varying activin A and BMP4 concentrations on days 0 and 1, respectively. Follow option A for differentiation into cardiogenic mesoderm. Follow option B for hemogenic mesoderm differentiation.
-
cardiogenic mesoderm differentiation ● TIMING 2 d
Aspirate the medium and wash the wells with an equal volume of PBS. Aspirate PBS and add the appropriate volume (Table 2) of 37 °C RPMI + 100 ng ml−1 activin A + 1× B-27 without insulin + 1× Matrigel to each well of the plate. Incubate the plate for 17 h at 37 °C, 5% CO2. This is designated day 0.
-
On day 1, aspirate the activin-A-containing medium and add the appropriate volume (Table 2) of 37 °C RPMI + 5 ng ml−1 BMP4 + 1 μM CHIR-99021 + 1× B-27 without insulin to each well of the plate. Place the plate in the incubator.
▲ CRITICAL STEP To generate cardiogenic-mesoderm-derived C-ECs, leave the medium unchanged for 24 h (until day 2) and then go to Step 14B(i). To generate cardiomyoctes, leave the medium unchanged for 48 h (until day 3) and then go to Step 14A(i).
? TROUBLESHOOTING
-
Hemogenic mesoderm differentiation ● TIMING 2 d
Aspirate the medium and wash the wells with an equal volume of PBS. Aspirate the PBS and add the appropriate volume (Table 2) of 37 °C RPMI + 50 ng ml−1 activin A + 1× B-27 without insulin + 1× Matrigel to each well of the plate. Incubate the plate for 17 h at 37 °C, 5% CO2. This is designated day 0.
-
On day 1, aspirate the activin-A-containing medium and add the appropriate volume (Table 2) of 37 °C RPMI + 40 ng ml−1 BMP4 + 1 μM CHIR-99021 + 1× B-27 without insulin to each well of the plate. Place the plate in the incubator. To generate hemogenic-mesoderm-derived ECs (H-ECs), leave the medium unchanged for 24 h (until day 2) and then go to Step 14B(i).
? TROUBLESHOOTING
-
Further directed differentiation
-
14
If you performed Step 13A and you wish to proceed with differentiation into cardiomyocytes, follow option A. Endothelial subtypes can be generated by directed differentiation (option B) following differentiation into cardiogenic mesoderm (obtained by following Step 13A) or hemogenic mesoderm (obtained by following Step 13B).
-
Directed differentiation into cardiomyocytes ● TIMING 12 d
Starting with day 3 cardiogenic mesoderm (from Step 13A(ii)), aspirate the BMP4-containing medium and add the appropriate volume (Table 2) of 37 °C RPMI + 1 μM XAV 939 + 1× B-27 without insulin to each well of the plate. Perform this step no later than 48 h after Step 13A(ii) media addition. Incubate the plate for 48 h at 37 °C, 5% CO2.
On day 5, aspirate the XAV-containing medium and add the appropriate volume (Table 2) of 37 °C RPMI + 1× B-27 minus insulin to each well. Incubate the plate for 48 h at 37 °C, 5% CO2.
On day 7, aspirate the medium and add the appropriate volume (Table 2) of 37 °C RPMI + 1× B-27 plus insulin to each well. Incubate the plate for 48 h at 37 °C, 5% CO2. Spontaneous beating can be observed beginning around day 7.
-
From day 7 on, after aspirating the old medium, add 37 °C RPMI + 1× B-27 plus insulin medium to the wells every other day. Cells will differentiate into definitive cardiomyocytes by day 14. Flow cytometry analysis of day 14 hPSC-derived cardiomyocytes is outlined in Box 1. More information about antibodies can be found in Table 3.
? TROUBLESHOOTING
-
Directed differentiation of endothelial subtypes ● TIMING 3–12 d
-
On day 2, aspirate the BMP4-containing medium and add the appropriate volume (Table 2) of 37 °C StemPro + 4 × 10−4 M MTG + 2 mM L-glutamine + 50 ug ml−1 ascorbic acid + 10 ng ml−1 BMP4 + 5 ng ml−1 bFGF + 300 ng ml−1 VEGF to each well. Incubate the mixture for 72 h at 37 °C, 5% CO2.
▲ CRITICAL STEP After thawing, VEGF can be used only once.
-
On day 5, cells can be analyzed by FACS staining for the endothelial markers KDR, CD34, CD31, and VE-cadherin. These markers do not distinguish C-ECs from H-ECs. However, blood formation potential is a phenotype that distinguishes C-ECs from H-ECs. We provide detailed methods for analyzing blood-forming activity by FACS analysis for markers of primitive hematopoiesis, including CD43 and CD235a, as well as for colony-forming activity in methylcellulose. See Box 1 for FACS analysis methods and Box 2 for the methylcellulose CFU assay.
? TROUBLESHOOTING
-
On day 5, split cells using 37 °C Versene and 0.25% (vol/vol) trypsin and replate with the appropriate volume in each well (Table 2) of 37 °C EGM + 20 ng ml−1 VEGF + 20 ng ml−1 bFGF + 1 μM CHIR-99021 at a density of 9.0 × 103 cells cm−2 on 0.1% (vol/vol) gelatin-coated plates.
▲ CRITICAL STEP Cells are highly sensitive to shear stress. When replating, triturate minimally to avoid cell death.
? TROUBLESHOOTING
Split cultures when they achieve 80–90% confluence.
-
Perform FACS analysis to determine the purity of CD31+/VE-cadherin+ cells, as described in Box 1. Mature ECs will show de novo lumen formation potential as a key phenotype for functional endothelium. We provide a detailed protocol for assaying tubulogenesis in collagen, as outlined in Box 3 and Table 4.
? TROUBLESHOOTING
-
-
Table 3.
Antibody information.
| Antibody | Application | Stage | Vendor (cat. no.) | Dilution | Conjugate |
|---|---|---|---|---|---|
| cTnT | IHC/FACS | Primary | Thermo | 1:100 | NA |
| SMA | FACS | Primary | Abcam | 1:50 | NA |
| KDR | FACS | NA | R&D | 1:6 | PE |
| CD34 | FACS | NA | BD | 1:5 | PerCP |
| CD43 | FACS | NA | BD | 1:5 | APC |
| CD235a | FACS | NA | BD | 1:37 | PECy-7 |
| CD31 | IHC | Primary | Abcam (28364) | 1:30 | NA |
| CD31 | FACS | NA | BD | 1:5 | FITC |
| VE-cadherin | IHC/FACS | NA | Abcam (7047) | 1:5 | PE |
| Phalloidin | IHC | NA | Invitrogen (A12379) | 1:100 | Alexa Fluor 488 |
| Mouse anti-goat | IHC/FACS | Secondary | Jackson | 1:200 | PE |
| Rabbit anti-donkey | IHC/FACS | Secondary | Jackson | 1:500 | APC |
| Goat anti-rabbit | IHC | Secondary | Invitrogen (A11011) | 1:100 | Alexa Fluor 568 |
| Isotypes | |||||
| Rabbit IgG | IHC/FACS | NA | Abcam | 1:100 | NA |
| Mouse IgG1 | IHC/FACS | NA | eBioscience | 1:100 | NA |
| PerCP IgG1 | FACS | NA | BD | 1:50 | NA |
| PECy-7 IgG1 | FACS | NA | BioLegend | 1:50 | NA |
| FITC IgG | FACS | NA | Abcam | 1:50 | NA |
NA, not applicable.
Table 4.
Volume calculations for collagen gel.
| Reagent | Stock concentration | Final concentration | Volume | |
|---|---|---|---|---|
| Rat tail collagen, type I | 3 mg ml−1 | 2 mg ml−1 |
|
|
| 10× M199 | 10× | 1× |
|
|
| 1 N NaOH | 1 N | — | VNaOH = 0.025 × VCOLLAGEN | |
| EGM | — | — | VEGM = VFINAL − VM199 − VNaOH − VCOLLAGEN |
? TROUBLESHOOTING
Troubleshooting advice can be found in Table 5.
Table 5. Troubleshooting table.
| Step(s) | Problem | Possible reason | Solution |
|---|---|---|---|
| 7 | Undifferentiated cells do not have homogeneous morphology | Cells are growing too densely or are not being split at a proper ratio Spontaneous differentiation can occur if hPSCs grow too confluent | The split ratio is variable, although generally a ratio between 1:4 and 1:15 is appropriate when using Versene for passaging. Avoid passaging more than once every 3–4 d |
| 12 | Suboptimal cell seeding | Improper handling of the plate during seeding or excessive cell death. Cell death and uneven cell distribution | Cells are highly sensitive to trituration during passaging. Keep trituration to a minimum. Use Y-27632 during plating to increase survival. Careful handling of the plate after seeding is necessary for even distribution of cells |
| 13–14 | Poor differentiation in all lineages | Quality of undifferentiated cells | If undifferentiated cells are not maintained properly, this will cause poor differentiation. Keep large stocks of karyotypically normal cells. We recommend not passaging more than ten times before thawing a new batch of cells |
| Cytokine and small-molecule bioactivity | For cytokines and small molecules, bioactivity is markedly compromised through repeat freeze–thaw cycles. Optimally, prepare aliquots of cytokines for single-use purposes and use immediately after thawing. Use small molecules according to manufacturer instructions | ||
| Media quality | Media pH will become more basic with repeated opening. Use media within 2 weeks of first use. B-27 should be aliquotted and frozen for single use | ||
| Timing of media changes and technique | Cell-fate decisions, particularly during the early stages of differentiation, are highly sensitive to timing. We therefore try to make media changes ±30 min around the time designated in this protocol. As a matter of technique, also try to add medium very slowly at every change so as not to disrupt the cell monolayer | ||
| 14A(iv) | Low cardiomyocyte purity | Incorrect density at the time of activin A induction | Some cell lines require very high density seeding (e.g., WTC cells), whereas others are more flexible and can generate cardiomyocytes in a wide range of densities (e.g., RUES2 cells). Optimal results should be determined empirically |
| Poor timing of Wnt manipulation | The timing of Wnt inhibition on day 3 can be sensitive. Optimize the timing of XAV-939 addition in Step 14(i) | ||
| 14B(ii) | Low KDR+/CD34+ cell yield at day 5 | Seeding density is too high | C-EC formation is more sensitive to high seeding density compared with that of H-ECs. Test different seeding densities in Step 12 |
| VEGF concentration is not optimal | Cell lines show variable requirements for VEGF during EC induction. Optimize VEGF concentrations in Step 14B(i,iii) | ||
| Reagent bioactivity is low | Some of the reagents should be frozen for long-term storage and thawed for one-time use only. VEGF aliquots must be stored at −80 °C and then thawed for immediate use. Do not refreeze aliquots or store them at 4 °C for future use. Ascorbic acid is also a one-time-use reagent | ||
| 14B(iii) | Cell death after replating C-ECs | Too much trituration during passaging | C-ECs are highly sensitive to shear stress as compared with H-ECs. Pretreat C-ECs with fresh StemPro-34 medium with Y-27632 2 h before passaging and use minimal trituration in Step 9 |
| 14B(v) | Low CD31+ endothelial cell yield at day 14 | Medium is not fresh | The efficiency of generating definitive ECs requires fresh EGM medium. We recommend using EGM within 2 weeks after supplements have been added |
● TIMING
Steps 1–5, thawing hPSCs: 30 min
Step 6, daily maintenance of hPSCs: 4 d
Steps 7–11, passaging hPSCs using Versene: 20 min, plus 4 d for maintenance
Step 12, monolayer differentiation setup for hPSCs: 20 min
Step 13A, cardiogenic mesoderm differentiation: 2 d
Step 13B, hemogenic mesoderm differentiation: 2 d
Step 14A, directed differentiation into cardiomyocytes: 12 d
Step 14B, directed differentiation of endothelium subtypes: 3–12 d
Anticipated results
This protocol provides an effcient methodology for the generation of high-purity definitive mesodermal subtypes from hPSCs, including cardiomyocytes and endothelial cells. Upon induction of differentiation with activin A and BMP4, cells should show marked morphological changes as they transit from pluripotency to mesoderm (Fig. 4b), with marked differences in gene expression (Tables 1 and 3) consistent with lineage specifcation into cardiogenic versus hemogenic mesoderm. Under conditions of Wnt inhibition from cardiogenic mesoderm, differentiation should show time-dependent activation of cardiac transcription factor and myoflament genes by day 5 of differentiation. Beating should be observed by days 7–10, with differentiation achieving ∼90% cTnT+ cells by day 14 (Fig. 4).
For endothelial differentiation, cells should progress to day 5 of differentiation, achieving ∼90% KDR+/CD34+ cells with gene expression patterns that distinguish cardiogenic versus hemogenic EC lineages. Evidence of primitive hematopoiesis should be seen almost entirely in ECs derived from hemogenic mesoderm, based on the presence of CD43+/CD235a+ cells (Fig. 6a). Similarly, functional blood-forming assays in methylcellulose should show very little blood-forming potential in ECs derived from cardiogenic mesoderm, whereas ECs derived from hemogenic mesoderm should show robust primitive erythroid and macrophage colony-forming units (Fig. 6b). As cells mature into definitive endothelial cells, they should become >90% CD31+/VE-cadherin+ by day 14 of differentiation and show robust lumen formation in collagen (Fig. 7). This protocol has been found to be versatile, with high-efficiency differentiation from hESC and hiPSC lines (Fig. 8).
Figure 8.
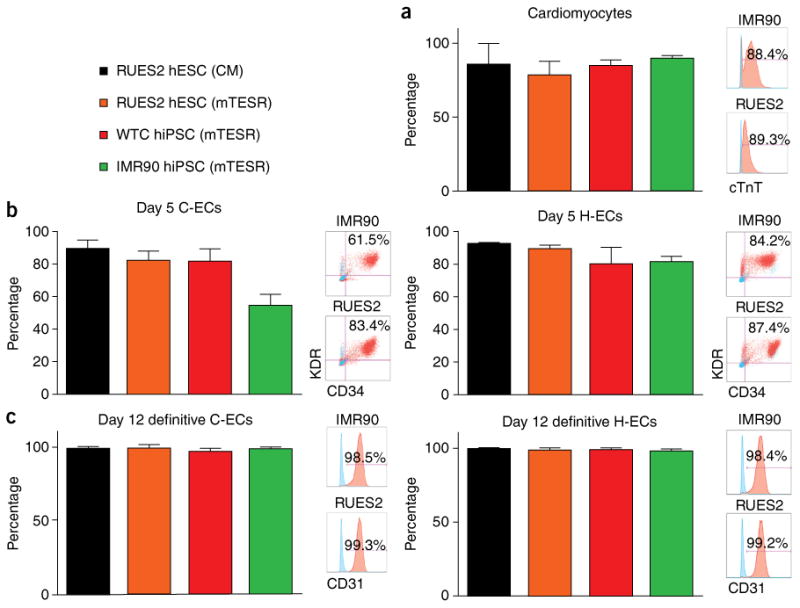
Efficiency of differentiation as assessed in multiple hPSC lines. The efficiency of differentiation was assessed in RUES2 cells cultured in condition medium (CM) and defined medium (mTESR), WTC hiPSCs, and IMR90 hiPSCs. (a–c) Cells were differentiated into cardiomyocytes based on cTnT+ cells on day 14 of differentiation (a), C-ECs and H-ECs based KDR+/CD34+ cells on day 5 (b), and definitive C-ECs and H-ECs based on CD31+ cells on day 14 of differentiation (c). n = 6 biological replicates for a and 8 replicates for b and c. Data are represented as mean ± sem.
Supplementary Material
Acknowledgments
This work was supported by the following grants: PO1 GM081619, PO1 HL094374, RO1 HL084642, and UO1 HL100405 (C.E.M.); NIH DP2DK102258-01 (Y.Z.); T32 EB001650 (R.J.Z.); NIH T32EB001650 and NIH T32HL007312 (M.R.); NIH UO1 HL100395 and U01 HL099997 (I.B.); and Alex's Lemonade Stand Young Investigator Award and the Hyundai Hope Scholar Grant (B.H.).
Footnotes
Note: Any Supplementary Information and Source Data files are available in the online version of the paper.
Competing Financial Interests The authors declare no competing financial interests.
Author Contributions N.J.P. conceptually developed protocols, designed and performed experiments, analyzed data, and wrote the manuscript. L.P. conceptually developed protocols, designed experiments, and assisted in preparation of the manuscript. C.E.F., M.R., B.H., and R.J.Z. performed experiments, analyzed data, and helped prepare protocol details. Y.Z. and I.B. designed experiments and assisted in preparation of the manuscript. C.E.M. supervised the project, obtained funding for the research, and wrote and approved the manuscript.
References
- 1.Murry CE, Keller G. Differentiation of embryonic stem cells to clinically relevant populations: lessons from embryonic development. Cell. 2008;132:661–680. doi: 10.1016/j.cell.2008.02.008. [DOI] [PubMed] [Google Scholar]
- 2.Xu PF, Houssin N, Ferri-Lagneau KF, Thisse B, Thisse C. Construction of a vertebrate embryo from two opposing morphogen gradients. Science. 2014;344:87–89. doi: 10.1126/science.1248252. [DOI] [PubMed] [Google Scholar]
- 3.Kattman SJ, et al. Stage-specific optimization of activin/nodal and BMP signaling promotes cardiac differentiation of mouse and human pluripotent stem cell lines. Cell Stem Cell. 2011;8:228–240. doi: 10.1016/j.stem.2010.12.008. [DOI] [PubMed] [Google Scholar]
- 4.Nostro MC, Cheng X, Keller GM, Gadue P. Wnt, activin, and BMP signaling regulate distinct stages in the developmental pathway from embryonic stem cells to blood. Cell Stem Cell. 2008;2:60–71. doi: 10.1016/j.stem.2007.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sumi T, Tsuneyoshi N, Nakatsuji N, Suemori H. Defining early lineage specification of human embryonic stem cells by the orchestrated balance of canonical Wnt/beta-catenin, Activin/Nodal and BMP signaling. Development. 2008;135:2969–2979. doi: 10.1242/dev.021121. [DOI] [PubMed] [Google Scholar]
- 6.Palpant NJ, et al. Inhibition of β-catenin signaling respecifies anterior-like endothelium into beating human cardiomyocytes. Development. 2015;142:3198–209. doi: 10.1242/dev.117010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Palpant NJ, et al. Transmembrane protein 88: a Wnt regulatory protein that specifies cardiomyocyte development. Development. 2013;140:3799–3808. doi: 10.1242/dev.094789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wamstad JA, et al. Dynamic and coordinated epigenetic regulation of developmental transitions in the cardiac lineage. Cell. 2012;151:206–220. doi: 10.1016/j.cell.2012.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Willems E, et al. Small-molecule inhibitors of the Wnt pathway potently promote cardiomyocytes from human embryonic stem cell-derived mesoderm. Circ Res. 2011;109:360–364. doi: 10.1161/CIRCRESAHA.111.249540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mendjan S, et al. NANOG and CDX2 pattern distinct subtypes of human mesoderm during exit from pluripotency. Cell Stem Cell. 2014;15:310–325. doi: 10.1016/j.stem.2014.06.006. [DOI] [PubMed] [Google Scholar]
- 11.Faial T, et al. Brachyury and SMAD signalling collaboratively orchestrate distinct mesoderm and endoderm gene regulatory networks in differentiating human embryonic stem cells. Development. 2015;142:2121–2135. doi: 10.1242/dev.117838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yutzey KE, Bader D. Diversification of cardiomyogenic cell lineages during early heart development. Circ Res. 1995;77:216–219. doi: 10.1161/01.res.77.2.216. [DOI] [PubMed] [Google Scholar]
- 13.Van Handel B, et al. Scl represses cardiomyogenesis in prospective hemogenic endothelium and endocardium. Cell. 2012;150:590–605. doi: 10.1016/j.cell.2012.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rafii S, et al. Human ESC-derived hemogenic endothelial cells undergo distinct waves of endothelial to hematopoietic transition. Blood. 2013;121:770–780. doi: 10.1182/blood-2012-07-444208. [DOI] [PubMed] [Google Scholar]
- 15.Nourse MB, et al. VEGF induces differentiation of functional endothelium from human embryonic stem cells: implications for tissue engineering. Arterioscler Thromb Vasc Biol. 2010;30:80–89. doi: 10.1161/ATVBAHA.109.194233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Orlova VV, et al. Generation, expansion and functional analysis of endothelial cells and pericytes derived from human pluripotent stem cells. Nat Protoc. 2014;9:1514–1531. doi: 10.1038/nprot.2014.102. [DOI] [PubMed] [Google Scholar]
- 17.Levenberg S, Ferreira LS, Chen-Konak L, Kraehenbuehl TP, Langer R. Isolation, differentiation and characterization of vascular cells derived from human embryonic stem cells. Nat Protoc. 2010;5:1115–1126. doi: 10.1038/nprot.2010.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kennedy M, D'Souza SL, Lynch-Kattman M, Schwantz S, Keller G. Development of the hemangioblast defines the onset of hematopoiesis in human ES cell differentiation cultures. Blood. 2007;109:2679–2687. doi: 10.1182/blood-2006-09-047704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.White MP, et al. Limited gene expression variation in human embryonic stem cell and induced pluripotent stem cell-derived endothelial cells. Stem Cells. 2013;31:92–103. doi: 10.1002/stem.1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Misfeldt AM, et al. Endocardial cells are a distinct endothelial lineage derived from Flk1+ multipotent cardiovascular progenitors. Dev Biol. 2009;333:78–89. doi: 10.1016/j.ydbio.2009.06.033. [DOI] [PubMed] [Google Scholar]
- 21.Peterkin T, Gibson A, Patient R. Common genetic control of haemangioblast and cardiac development in zebrafish. Development. 2009;136:1465–1474. doi: 10.1242/dev.032748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de la Pompa JL, et al. Role of the NF-ATc transcription factor in morphogenesis of cardiac valves and septum. Nature. 1998;392:182–186. doi: 10.1038/32419. [DOI] [PubMed] [Google Scholar]
- 23.Ranger AM, et al. The transcription factor NF-ATc is essential for cardiac valve formation. Nature. 1998;392:186–190. doi: 10.1038/32426. [DOI] [PubMed] [Google Scholar]
- 24.Morikawa Y, Cserjesi P. Extra-embryonic vasculature development is regulated by the transcription factor HAND1. Development. 2004;131:2195–2204. doi: 10.1242/dev.01091. [DOI] [PubMed] [Google Scholar]
- 25.Barnes RM, Firulli BA, Conway SJ, Vincentz JW, Firulli AB. Analysis of the Hand1 cell lineage reveals novel contributions to cardiovascular, neural crest, extra-embryonic, and lateral mesoderm derivatives. Dev Dyn. 2010;239:3086–3097. doi: 10.1002/dvdy.22428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burridge PW, et al. Chemically defined generation of human cardiomyocytes. Nat Methods. 2014;11:855–860. doi: 10.1038/nmeth.2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lian X, et al. Directed cardiomyocyte differentiation from human pluripotent stem cells by modulating Wnt/β-catenin signaling under fully defined conditions. Nat Protoc. 2013;8:162–175. doi: 10.1038/nprot.2012.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lian X, et al. Robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical Wnt signaling. Proc Natl Acad Sci USA. 2012;109:E1848–E1857. doi: 10.1073/pnas.1200250109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kempf H, Kropp C, Olmer R, Martin U, Zweigerdt R. Cardiac differentiation of human pluripotent stem cells in scalable suspension culture. Nat Protoc. 2015;10:1345–1361. doi: 10.1038/nprot.2015.089. [DOI] [PubMed] [Google Scholar]
- 30.Ditadi A, et al. Human definitive haemogenic endothelium and arterial vascular endothelium represent distinct lineages. Nat Cell Biol. 2015;17:580–591. doi: 10.1038/ncb3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Witty AD, et al. Generation of the epicardial lineage from human pluripotent stem cells. Nat Biotechnol. 2014;32:1026–1035. doi: 10.1038/nbt.3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang J, et al. Extracellular matrix promotes highly efficient cardiac differentiation of human pluripotent stem cells: the matrix sandwich method. Circ Res. 2012;111:1125–1136. doi: 10.1161/CIRCRESAHA.112.273144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Paige SL, et al. Endogenous Wnt/beta-catenin signaling is required for cardiac differentiation in human embryonic stem cells. PLoS One. 2010;5:e11134. doi: 10.1371/journal.pone.0011134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ueno S, et al. Biphasic role for Wnt/beta-catenin signaling in cardiac specification in zebrafish and embryonic stem cells. Proc Natl Acad Sci USA. 2007;104:9685–9690. doi: 10.1073/pnas.0702859104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mummery CL, et al. Differentiation of human embryonic stem cells and induced pluripotent stem cells to cardiomyocytes: a methods overview. Circ Res. 2012;111:344–358. doi: 10.1161/CIRCRESAHA.110.227512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lian X, Zhang J, Zhu K, Kamp TJ, Palecek SP. Insulin inhibits cardiac mesoderm, not mesendoderm, formation during cardiac differentiation of human pluripotent stem cells and modulation of canonical Wnt signaling can rescue this inhibition. Stem Cells. 2013;31:447–457. doi: 10.1002/stem.1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dubois NC, et al. SIRPA is a specific cell-surface marker for isolating cardiomyocytes derived from human pluripotent stem cells. Nat Biotechnol. 2011;29:1011–1018. doi: 10.1038/nbt.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gantz JA, et al. Targeted genomic integration of a selectable floxed dual fluorescence reporter in human embryonic stem cells. PLoS One. 2012;7:e46971. doi: 10.1371/journal.pone.0046971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ovchinnikov DA, et al. Isolation of contractile cardiomyocytes from human pluripotent stem-cell-derived cardiomyogenic cultures using a human NCX1-EGFP reporter. Stem Cells Dev. 2015;24:11–20. doi: 10.1089/scd.2014.0195. [DOI] [PubMed] [Google Scholar]
- 40.Elliott DA, et al. NKX2-5(eGFP/w) hESCs for isolation of human cardiac progenitors and cardiomyocytes. Nat Methods. 2011;8:1037–1040. doi: 10.1038/nmeth.1740. [DOI] [PubMed] [Google Scholar]
- 41.Tohyama S, et al. Distinct metabolic flow enables large-scale purification of mouse and human pluripotent stem cell-derived cardiomyocytes. Cell Stem Cell. 2013;12:127–137. doi: 10.1016/j.stem.2012.09.013. [DOI] [PubMed] [Google Scholar]
- 42.Ciau-Uitz A, Pinheiro P, Gupta R, Enver T, Patient R. Tel1/ETV6 specifies blood stem cells through the agency of VEGF signaling. Dev Cell. 2010;18:569–578. doi: 10.1016/j.devcel.2010.02.009. [DOI] [PubMed] [Google Scholar]
- 43.Ciau-Uitz A, Pinheiro P, Kirmizitas A, Zuo J, Patient R. VEGFA-dependent and -independent pathways synergise to drive Scl expression and initiate programming of the blood stem cell lineage in Xenopus. Development. 2013;140:2632–2642. doi: 10.1242/dev.090829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kennedy M, et al. T lymphocyte potential marks the emergence of definitive hematopoietic progenitors in human pluripotent stem cell differentiation cultures. Cell Rep. 2012;2:1722–1735. doi: 10.1016/j.celrep.2012.11.003. [DOI] [PubMed] [Google Scholar]
- 45.Choi KD, et al. Identification of the hemogenic endothelial progenitor and its direct precursor in human pluripotent stem cell differentiation cultures. Cell Rep. 2012;2:553–567. doi: 10.1016/j.celrep.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nakano A, Nakano H, Smith K, Palpant NJ. The developmental origins and lineage contributions of endocardial endothelium. Biochim Biophys Acta. 2016;1863:1937–1947. doi: 10.1016/j.bbamcr.2016.01.022. [DOI] [PubMed] [Google Scholar]
- 47.Nakano H, et al. Haemogenic endocardium contributes to transient definitive haematopoiesis. Nat Commun. 2013;4:1564. doi: 10.1038/ncomms2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sturgeon CM, Ditadi A, Awong G, Kennedy M, Keller G. Wnt signaling controls the specification of definitive and primitive hematopoiesis from human pluripotent stem cells. Nat Biotechnol. 2014;32:554–561. doi: 10.1038/nbt.2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zheng Y, et al. In vitro microvessels for the study of angiogenesis and thrombosis. Proc Natl Acad Sci USA. 2012;109:9342–9347. doi: 10.1073/pnas.1201240109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.