

Summary
Central regulation of metabolic physiology is mediated critically through neuronal functions; however, if astrocytes are also essential remains unclear. Here we show that the high-order processes of astrocytes in the mediobasal hypothalamus displayed shortening in fasting while elongation in fed status. Chronic overnutrition and astrocytic IKKβ/NF-κB upregulation similarly impaired astrocytic plasticity leading to sustained shortening of high-order processes. In physiology, astrocytic IKKβ/NF-κB upregulation resulted in early-onset effects including glucose intolerance and blood pressure rise and late-onset effects including body weight and fat gain. Appropriate inhibition in astrocytic IKKβ/NF-κB protected against chronic overnutrition from impairing astrocytic plasticity and these physiological functions. Mechanistically, astrocytic regulation of hypothalamic extracellular GABA level and therefore BDNF expression were found partly accountable. Hence, astrocytic process plasticity and IKKβ/NF-κB play significant roles in central control of blood glucose, blood pressure and body weight as well as the central induction of these physiological disorders leading to disease.
Keywords: Astrocyte, hypothalamus, IKKβ, NF-κB, inflammation, blood glucose, blood pressure, obesity, diabetes
eTOC blurb
XXX et al report that astrocytes can actively respond to metabolic information such as fasting/refeeding and chronic over-nutrition by dynamically changing their process length and density. Chronic overnutrition inflames astrocytes, thus impairing their plasticity and resulting in changes in extracellular neurotransmitter levels and downstream neuropeptides, which affect metabolism.
Introduction
Hypothalamic neurons are important for the regulation of feeding behavior and related metabolic physiology (Cone, 2005; Dietrich and Horvath, 2011; Flier and Maratos-Flier, 1998; Munzberg and Myers, 2005; Schwartz et al., 2000; Tschop et al., 2006). To understand the regulatory functions of hypothalamic neurons, for example, through secretion of various neuropeptides or formation of various neural circuits, has been a major focus of research including many studies during recent years (Chiappini et al., 2014; Cohen et al., 2001; Coppari et al., 2005; Flak et al., 2014; Ghamari-Langroudi et al., 2015; Kievit et al., 2006; Kitamura et al., 2006; Kleinridders et al., 2013; Lam et al., 2005; Zeltser et al., 2012; Zhang et al., 2008). In contrast to neurons, astrocytes have not received adequate attention, despite that they are not only the most abundant neural cells but display remarkably high diversities in morphology and functions (Eroglu and Barres, 2010; Fields and Stevens-Graham, 2002; Garcia-Caceres et al., 2016; Ishibashi et al., 2006; Muroyama et al., 2005; Muthukumar et al., 2014; Nedergaard et al., 2003; Perea et al., 2014; Rangroo Thrane et al., 2013; Shigetomi et al., 2012; Theodosis et al., 2008; Wyss-Coray et al., 2003). Astrocytes and neurons are strongly interconnected and the astrocyte-to-neuron ratio surges with increased complexity of the brain (Nedergaard et al., 2003). Previous studies have shown that astrocytes secret cytokines and neurotrophic factors to promote neuronal development and plasticity, to modulate synaptic excitation through uptake and release of neurotransmitters, and to maintain various types of homeostasis in brain tissues (Eroglu and Barres, 2010; Jo et al., 2014; Otsu et al., 2015; Rangroo Thrane et al., 2013; Schachtrup et al., 2015; Shigetomi et al., 2012). Also, astrocytes can influence the brain’s metabolic condition by controlling glycogen storages and supplying fuel to neurons (Belanger et al., 2011; Choi et al., 2012), and it has been reported that hypothalamic astrocytes can sense glucose via glucose transporter-2, and register leptin signaling via leptin receptor (Fuente-Martin et al., 2012; Marty et al., 2005). Recently, hypothalamic astrocyte processes were found to change in response to leptin (Kim et al., 2014), and lack of insulin signaling in astrocytes can alter the processes and brain glucose uptake (Garcia-Caceres et al., 2016). From disease perspective, high-fat diet (HFD)-induced obesity in mice has been shown to be associated with hypothalamic astrogliosis (Thaler et al., 2012). Despite these observations, the role of astrocytes in hypothalamic control of metabolic physiology or disease still remains undetermined.
As known, the majority of astrocytic regulatory functions rely on their astrocytic processes (Haydon and Carmignoto, 2006; Kimelberg and Nedergaard, 2010). Indeed, the processes of astrocytes are interspersed between neurons, shrouding neuronal somata, synapses and dendrites (Theodosis et al., 2008). Astrocytes also express numerous transporters and receptors that are mainly localized in these processes (Chung et al., 2013; Minelli et al., 1996; Muthukumar et al., 2014; Shigetomi et al., 2012). The coating of neurons by astrocyte processes facilitates the containment of ions and neurotransmitters within neurons; such a close connection is necessary for proper functioning of neurons (Sykova, 2004). Notably, the astrocyte coat is sensitive to changing conditions, rendering astrocyte plasticity to play as a key part in regulating neuronal function (Theodosis et al., 2008). Rapid astrocyte process remodeling in certain areas of the hypothalamus has been observed in the context of reproductive physiology, for instance, changing estrogen levels in the arcuate nucleus (ARC) affect the coating of neurons by astrocyte processes (Garcia-Segura et al., 1994). Moreover, in the primate preoptic hypothalamic area, the astrocyte coating of neurons that secret gonadotropin-releasing hormone is closely connected to plasma steroid levels (Garcia-Segura et al., 1994; Witkin et al., 1991). In this work, we studied if hypothalamic astrocytic process plasticity could be involved in metabolic regulation and/or disease development, and if so, how.
Results
Hypothalamic astrocytes change high-order processes according to nutrition
Grounded at the critical role by the hypothalamus in regulation of metabolic physiology, several years ago we started to study if hypothalamic astrocytes might dynamically alter their morphology and particularly their detailed processes according to nutritional changes. Because conventional approaches, such as immunostaining of glial fibrillary acid protein (GFAP), hardly reveal high-order details of astrocytic processes, we resorted to Golgi staining which has been used to study the detailed branches of neural cells including astrocytes (Alvarez et al., 2015; Grosche et al., 2013; Hama et al., 2004; Marin-Padilla, 1995; Ogata and Kosaka, 2002; Sullivan et al., 2010). In 2012, Ranjan et al modified Golgi staining protocol so that it preferentially stains glial cells, predominantly astrocytes (Ranjan and Mallick, 2012). We further optimized this protocol and were able to stain a significant number of astrocytes which were easily identifiable among other cell types based on their distinct characteristics in morphology (Fig. S1). Astrocytes were sporadically and randomly stained by this chemical method, usually without being staggered together, which made it technically feasible to track individual astrocytes and their own processes. In our experiment, adult chow-fed C57BL/6 mice under ad-libitum feeding condition were compared to overnight fasting. Astrocytes in the mediobasal hypothalamus (MBH) which comprises the ARC and the ventral medial hypothalamic nucleus (VMH) were intensively examined. Despite that the domain sizes of astrocytes in the ARC were smaller than those in the VMH, high-order processes of these cells in both subregions from fasting condition were shorter and less dense compared to those from ad-libitum fed condition (Fig. 1A–D, Fig. S2A). Following fasting, astrocytes showed significant decreases in the length of branches from order 4 to 8 in the ARC and from order 4 to 11 in the VMH, and decreases in the numbers of intersections across the Scholl radii of 6–14 μm in the ARC and 20–52 μm in the VMH. Such nutritional condition-dependent plastic changes in astrocytes were observed in the MBH but negligibly found in other brain regions such as the cortex in the same sections (Fig. 1E, F). Along this line, we examined mice which were maintained chronically under a normal chow versus an HFD, the latter resulting in obesity and associated metabolic disorders. Of interest, shortening of hypothalamic astrocytic high-order processes was sustained in HFD-fed mice while reversed in chow-fed mice when examined under ad-libitum feeding (Fig. 2A, B, Fig. S2B). Shortened processes of MBH astrocytes in HFD-fed mice were associated with an increased number of GFAP-positive cells (which predominantly reflected astrocytes) in the MBH (Fig. 2C). Altogether, hypothalamic astrocytes adjust the size of their high-order processes according to nutritional condition, and the development of HFD-induced obesity is associated with altered process plasticity of astrocytes in the mediobasal hypothalamic region.
Figure 1. Hypothalamic and brain astrocytic processes in different nutritional conditions.
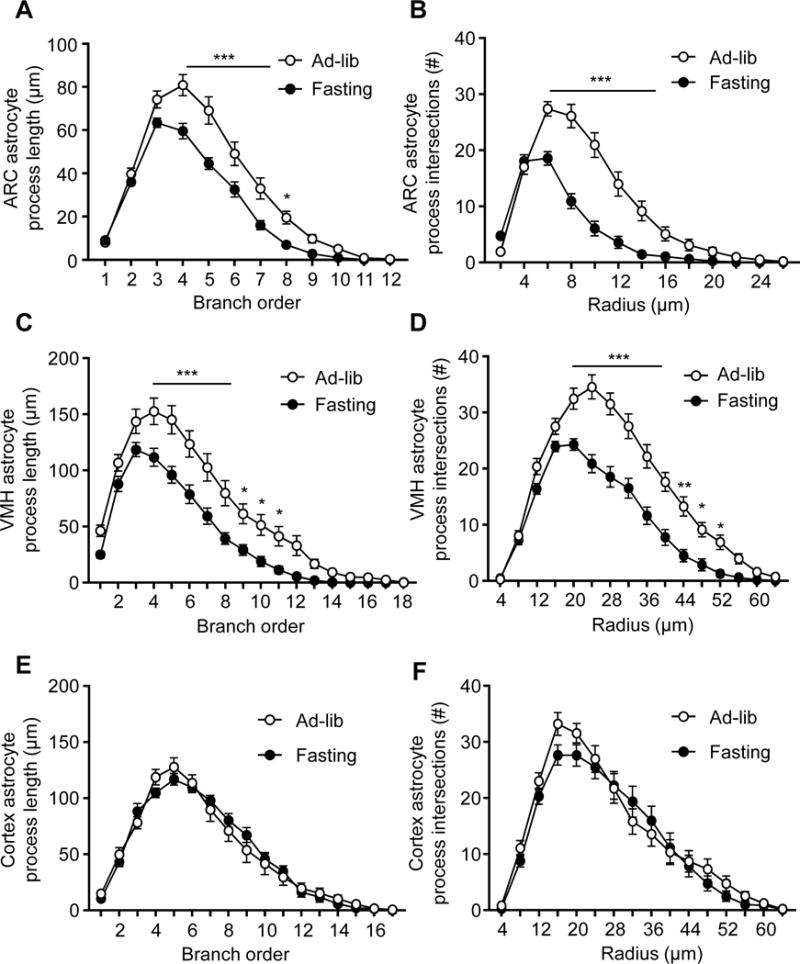
Brain sections from standard C57BL/6 mice (3-month-old males) under ad-libitum chow feeding (Ad-lib) vs. 16-hour overnight fasting were processed for Golgi staining. Astrocytic processes in the ARC (A, B), VMH (C, D) and cortex (E, F) were quantified according to the process lengths of indicated branch orders (A, C, E) and the number of intersections across the indicated radius from the center point of cell body (B, D, F). A total number of 70–100 representative astrocytes from 5 mice per nutritional condition were randomly traced for all processes revealed by Golgi staining using computerized program and plotted according to subregions. * p < 0.05, ** p < 0.01, *** p < 0.001; n = 20–35 data sets per point, compared between ad-libitum feeding and fasting at underlined or indicated points. Error bars reflect mean ± s.e.m. See also Figure S1–S2.
Figure 2. Effects of HFD feeding or IKKβ/NF-κB on hypothalamic astrocytic processes.
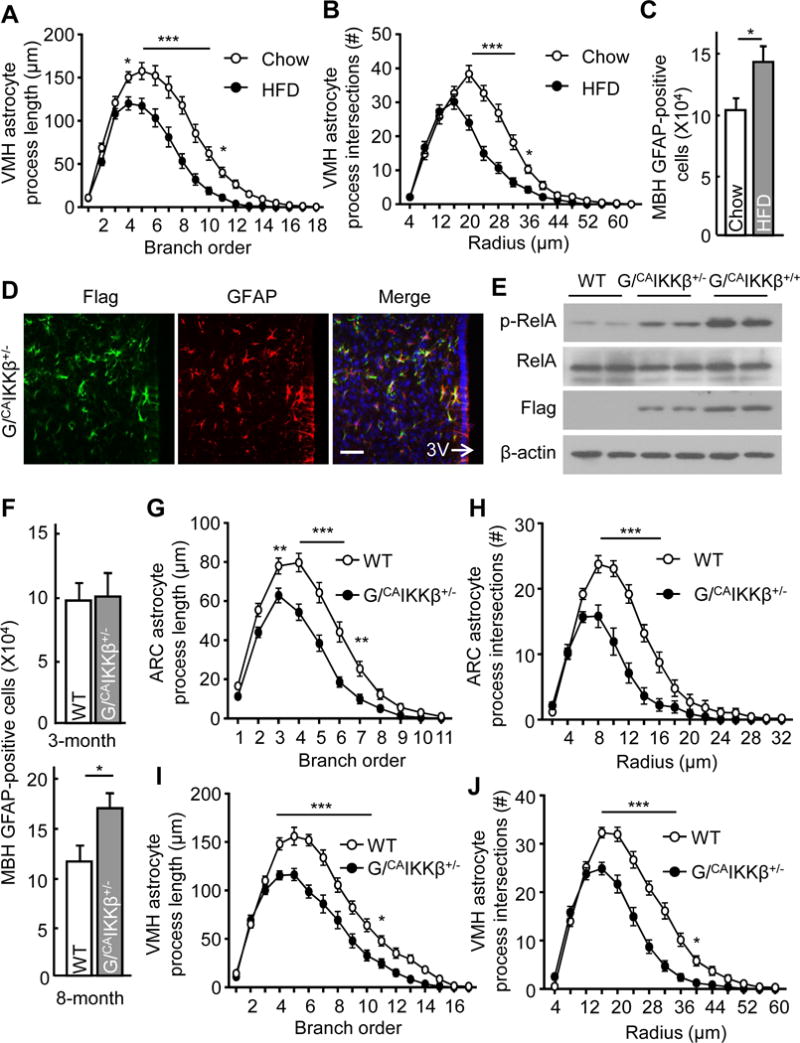
A–C Brains from ad-libitum fed mice following 3-month HFD vs. chow feeding were processed for (A, B) Golgi staining and process tracing as described in Figure 1 and (C) immunostaining for GFAP followed by counting of GFAP-positive cells in the MBH. D, E Immunostaining of hypothalamus sections (D) and Western blot of cultured astrocytes (E) obtained from the indicated mice. G/CAIKKβ+/−: GFAP/CAIKKβ+/− mice; G/CAIKKβ+/+: GFAP/CAIKKβ+/+ mice; WT: genotype-matched (lox-STOP-lox-CAIKKβ+/−) wildtype mice; 3V: third ventricle. Scale bar, 50 μm. F–J MBH sections from ad-libitum fed GFAP/CAIKKβ+/− and WT mice were used for GFAP immunostaining followed by counting of GFAP-positive cells (F) and Golgi staining followed by astrocytic process analyses (G–J). Experiments were based on 3- vs. 8-month-old (F) or 3-month-old (G–J) male mice, all maintained on a normal chow. * p < 0.05, ** p < 0.01, *** p < 0.001, n = 25–30 data sets from 5 mice per point (A, B, G–J) and n = 4 mice per group (C, F), compared between HFD and chow (A–C) and between G/CAIKKβ+/− and WT (F–J) at underlined or indicated points. Error bars reflect mean ± s.e.m. See also Figure S1–S3.
Shortening of astrocytic processes by moderate-level IKKβ/NF-κB upregulation
We have recently postulated that nutritional surplus can acutely and chronically upregulate hypothalamic IKKβ/NF-κB pathway and mediate the physiological and pathological response, respectively. Here, we designed to study if IKKβ/NF-κB might take part in the control of hypothalamic astrocytic process changes. Supportively, we observed that increased IKKβ/NF-κB activation by expression of constitutively-active IKKβ (CAIKKβ) in cultured hypothalamic astrocytes greatly blunted their processes and even the primary-order branches revealed by GFAP immunostaining (Fig. S3). To focus on the in vivo conditions, we employed a cell-specific transgenic mouse model in which CAIKKβ was expressed in the astrocytes to activate NF-κB in these cells. To generate these mice, we crossed Rosa 26-lox-STOP-lox-CAIKKβ (flag-tagged) mice with GFAP-Cre mice having Cre expression controlled by GFAP promoter – which is known to predominantly (although not exclusively) target astrocytes. The offspring compound mice were termed GFAP/CAIKKβ and compared to littermate Rosa 26-lox-STOP-lox-CAIKKβ mice which represented genotype-matched wildtype (WT) controls. Immunostaining confirmed that flag-tagged CAIKKβ was expressed in GFAP-positive cells (Fig. 2D). As noted, while homozygous GFAP/CAIKKβ+/+ mice suffered from brain damage and sickness due to severe neuroinflammation, heterozygous GFAP/CAIKKβ+/− mice were indistinguishable from the littermate control mice. Using NF-κB subunit RelA phosphorylation to report NF-κB activation, we confirmed that NF-κB was slightly or moderately upregulated in GFAP/CAIKKβ+/− astrocytes but strongly upregulated in GFAP/CAIKKβ+/+ astrocytes, compared to control astrocytes (Fig. 2E). The number of GFAP-positive cells (which mainly reflect astrocytes) in the MBH of these mice of young ages did not change until the mice were older (Fig. 2F). However, young GFAP/CAIKKβ+/− mice already displayed shortening of high-order processes of MBH astrocytes compared to the control group (Fig. 2G–J). Such altered astrocytic processes in GFAP/CAIKKβ+/− mice were already observable when they were young. Hence, moderate-level upregulation in IKKβ/NF-κB causes shortening of hypothalamic astrocytic high-order processes prior to a longer-term effect in increasing the number of astrocytes in this hypothalamic region.
Early-onset physiological effects of moderate-level astrocytic IKKβ/NF-κB upregulation
Since astrocytic IKKβ/NF-κB can acutely affect astrocytic process plasticity, but obesity represents a chronic status, we decided to examine if it might affect obesity-associated metabolic parameters which are known to be acutely regulated. These included glucose tolerance, insulin secretion, energy expenditure and blood pressure, as each of these functions can be acutely altered in normal physiology, and chronic impairments in these functions include glucose intolerance, insulin resistance and high blood pressure which collectively reflect the so-called “metabolic syndrome”. While homozygous GFAP/CAIKKβ+/+ mice were not suitable as many of them were developmentally unhealthy due to severe neuroinflammation, we employed heterozygous GFAP/CAIKKβ+/− mice which appeared normal in general. Indeed, young GFAP/CAIKKβ+/− mice and genotype-matched WT controls were comparable in terms of body weight, fat and lean mass and food intake (Fig. S4A–D). In contrast, these young GFAP/CAIKKβ+/− mice already demonstrated several early-onset metabolic changes, including glucose intolerance (Fig. 3A) and increased plasma insulin level (Fig. 3B). The basal metabolic rate of GFAP/CAIKKβ+/− mice, indicated by oxygen consumption, tended to be higher than that of control mice, but the difference did not reach a statistical significance (Fig. S4E). In a separate experiment, we profiled blood pressure of these mice using telemetric approach, results showing that blood pressure was higher in GFAP/CAIKKβ+/− mice than WT control mice during the resting-stage daytime (Fig. 3C–F). Thus, in association with hypothalamic astrocytic process shortening, moderate-level astrocytic IKKβ/NF-κB upregulation led to early-onset physiological changes including glucose intolerance and blood pressure increase.
Figure 3. Early-onset physiological effects of astrocytic IKKβ/NF-κB manipulation.
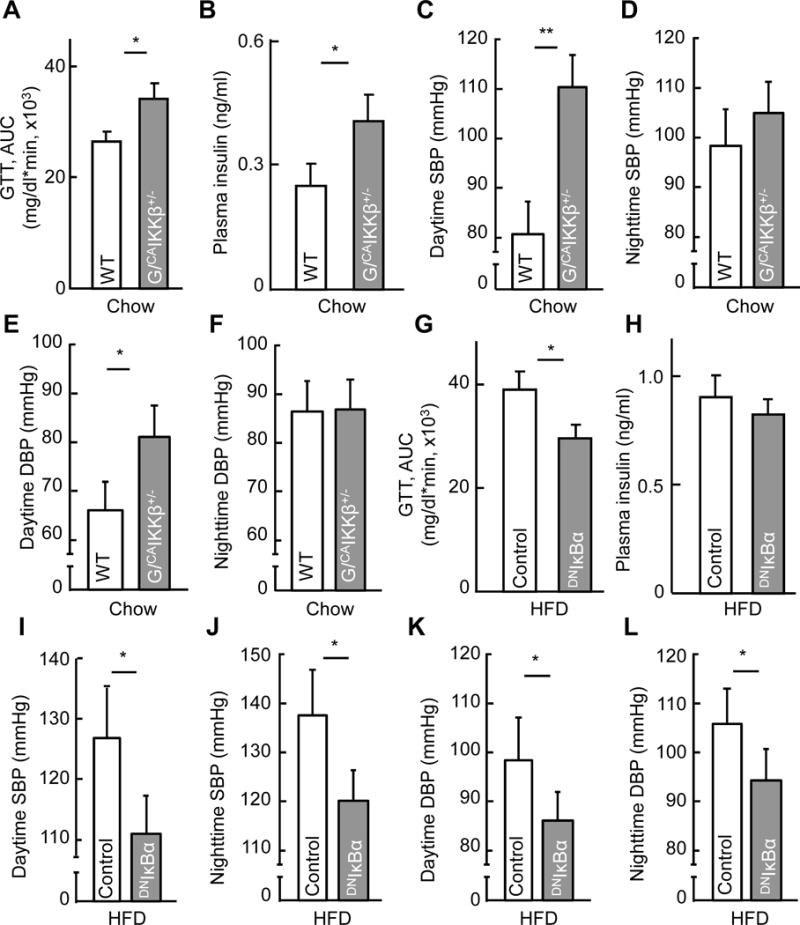
Chow-fed GFAP/CAIKKβ+/− mice vs. littermate genotype-matched (lox-STOP-lox-CAIKKβ+/−) WT mice (~4-month-old males) (A–F), and HFD-fed male C57BL/6 mice (5-month HFD feeding) following intra-MBH (mainly the VMH) injections of GFAP promoter-driven DNIκBα vs. control lentivirus (G–L), were profiled for glucose tolerance via GTT (A, G) and fasting plasma insulin level (B, H). Subgroups of these mice received aortic radio-telemetry implantation for blood pressure (BP) measurement (C–F, I–L). G/CAIKKβ+/−: GFAP/CAIKKβ+/− mice; GTT: glucose tolerance test; AUC: area under curve of GTT; SBP: systolic BP; DBP: diastolic BP. * p < 0.05, ** p < 0.01, n = 9–10 mice per group (A, B, G, H) and n = 4 mice per group (C–F, I–L). Error bars reflect mean ± s.e.m. See also Figure S4–S5.
Early-onset physiological effects of hypothalamic astrocytic NF-κB inhibition
To further develop this study, we asked if HFD-induced astrocytic process shortening could be acutely reversed by astrocytic IKKβ/NF-κB inhibition, and if so, whether this cellular effect might precede its physiological effect in reducing obesity. To answer this question, we designed an experiment in which astrocytic IKKβ/NF-κB was acutely inhibited in C57BL/6 mice that already developed obesity through HFD feeding. Acute inhibition of hypothalamic astrocytic IKKβ/NF-κB in these mice was achieved by intra-hypothalamic (largely the VMH subregion) injection of lentivirus expressing dominant-negative IκBα (DNIκBα) controlled by GFAP promoter (Fig. S5A). As shown in Fig. S5B–D, lentiviral DNIκBα led to a rapid reversal of HFD-induced astrocytic process shortening, but HFD-induced astrogliosis (indicated by increases in the cell number of astrocytes) did not rapidly reverse. Then, we analyzed the mice with HFD-induced obesity but hypothalamic astrocytic NF-κB was acutely inhibited through lentiviral DNIκBα injection. At an early time when the extent of obesity did not drop (Fig. S5E–H), lentiviral DNIκBα-injected mice demonstrated an improvement in glucose tolerance (Fig. 3G) as well as insulin sensitivity (Fig. 3H). Besides, obesity-related hypertension was found to be reversed during both daytime and nighttime (Fig. 3I–L). Thus, based on the gain- and loss-of-functional models, astrocytic IKKβ/NF-κB can acutely regulate astrocytic process plasticity and physiological control of glucose metabolism and blood pressure.
Late-onset effect on body weight by moderate astrocytic IKKβ/NF-κB upregulation
In our previous studies, we have established the role of IKKβ/NF-κB-dependent molecular inflammation in the development of HFD-induced weight gain and obesity (Li et al., 2012; Yan et al., 2014; Zhang et al., 2008; Zhang et al., 2015). Here, we continued to employ GFAP/CAIKKβ+/− mice to test if astrocytic IKKβ/NF-κB could be involved in obesity development. According to Western blot results, increased NF-κB activity in the hypothalamus of chow-fed GFAP/CAIKKβ+/− mice was comparable with the increase induced by chronic HFD feeding in standard C57BL/6 mice (Fig. 4A), but we should mention that the affected cell types probably differed between these two experimental conditions. To dissociate the long-term effect of astrocytic IKKβ/NF-κB from that of chronic HFD feeding, GFAP/CAIKKβ+/− mice and their controls were maintained on a normal chow. Over a longitudinal follow-up, we noted that GFAP/CAIKKβ+/− mice tended to eat more than did WT controls, but this effect was hardly statistically significant until animals were half a year old (Fig. 4B). Similarly, GFAP/CAIKKβ+/− mice weighed similarly as did WT controls when they were young but were found heavier than control mice at older ages (Fig. 4C). To check if body weight increase in these GFAP/CAIKKβ+/− mice could be suggestive of pro-obesity development, we examined the lean and fat composition of these mice, showing that GFAP/CAIKKβ+/− mice had similar lean mass but doubled fat mass compared to controls (Fig. 4D, E). Thus, in addition to the early-onset effects on glucose and blood pressure, astrocytic IKKβ/NF-κB activation exerts a late-onset effect on body weight and fat mass.
Figure 4. Late-onset effect of astrocytic IKKβ/NF-κB manipulation on body weight.
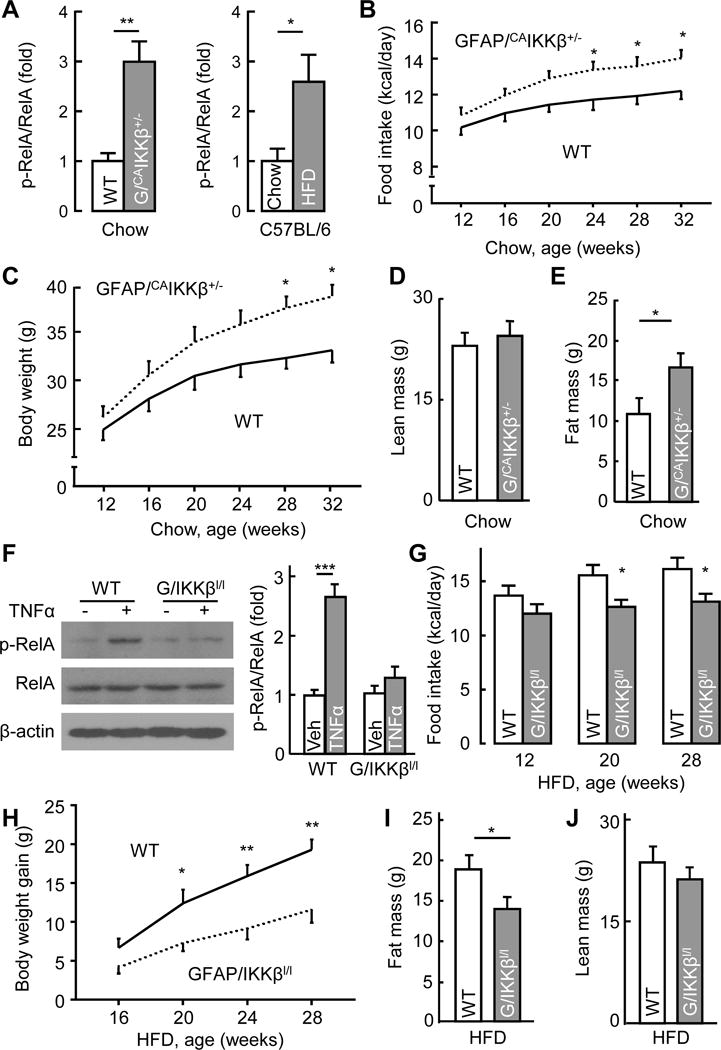
A Hypothalamic samples from chow-fed GFAP/CAIKKβ+/− and littermate WT (lox-STOP-lox-CAIKKβ+/−) mice or from C57BL/6 mice following 5-month HFD vs. chow feeding were analyzed via Western blotting for p-RelA levels and presented following normalization with RelA levels. B–E Chow-fed GFAP/CAIKKβ+/− mice vs. littermate WT (lox-STOP-lox-CAIKKβ+/−) mice were longitudinally profiled for food intake (B) and body weight (C), and measured for lean mass (D) and fat mass (E) when mice were half a year old. F Astrocytes from new-born GFAP/IKKβlox/lox mice vs. littermate WT (IKKβlox/lox) controls were cultured and treated with TNFα (30 ng/ml) vs. vehicle for 30 min before cell lysates were analyzed via Western blotting with the indicated antibodies. Bar graph: intensity of p-RelA blots normalized by RelA blots. G–J GFAP/IKKβlox/lox mice and littermate WT (IKKβlox/lox) controls were maintained on a HFD since 12 weeks of age and profiled for food intake (G) and body weight gain (H) at the indicated ages, and measured for fat mass (I) and lean mass (J) at the age of half a year. G/CAIKKβ+/−: GFAP/CAIKKβ+/− mice; G/IKKβl/l: GFAP/IKKβlox/lox mice; Veh: vehicle. * p < 0.05, ** p < 0.01, *** p < 0.001, n = 7–8 mice per group (B–E, G–J) and n = 4 mice per group (A, F). Error bars reflect mean ± s.e.m. See also Figure S4 and S6.
Counteraction against dietary obesity through astrocytic IKKβ/NF-κB inhibition
Subsequently we studied if an inhibition of IKKβ/NF-κB in the astrocytes could reduce the effects of chronic overnutrition on body weight and fat mass. To do so, we developed a loss-of-function genetic mouse model with suppression of IKKβ in the astrocytes. To achieve this, we crossed IKKβlox/lox mice with GFAP-Cre mice, both lines were bred on a C57BL/6 background, leading to offspring GFAP/IKKβlox/lox and the littermate IKKβlox/lox controls. Because GFAP-Cre did not lead to a complete gene knockout, GFAP/IKKβlox/lox mice maintained the basal NF-κB activity which appeared to be important for normal biology, since these mice were completely normal in terms of development and general physiology. On the other hand, the partial ablation of IKKβ substantially prevented NF-κB activation in response to pathological challenges, for example, TNFα failed to elevate NF-κB subunit RelA phosphorylation in cultured astrocytes which were isolated from GFAP/IKKβlox/lox mice (Fig. 4F). Under chow feeding, food intake of GFAP/IKKβlox/lox mice was noticeably lower than the control mice, but their body weight levels did not remarkably differ. Starting at 12 weeks of age, we placed a group of GFAP/IKKβlox/lox and littermate genotype-matched WT controls on HFD feeding. Compared to WT controls, GFAP/IKKβlox/lox mice almost immediately showed a reduction in HFD intake which maintained throughout the follow-up (Fig 4G). Body weight profiling revealed that GFAP/IKKβlox/lox mice significantly resisted the development of HFD-induced body weight and fat gain (Fig 4H–J). In addition, we generated a mouse model through injecting GFAP promoter-driven Cre lentiviruses into the MBH of IKKβlox/lox mice; these mice also demonstrated a protection against chronic HFD feeding-induced body weight excess as a result of a counteractive effect against chronic HFD feeding-associated overeating (Fig. S6). Altogether, IKKβ/NF-κB in the hypothalamus is critically involved in the astrocytic mechanism of obesity development.
Astrocytic IKKβ/NF-κB in extracellular GABA-induced neuronal activation
Since nutritional status as well as inflammatory condition affect both the morphology of astrocytes and energy balance, we conceived that a change in the length of astrocytic process could affect hypothalamic neuronal activity and therefore mediate energy balance regulation. Since astrocytes are crucial for the homeostasis of neurotransmitters, we paid attention to a few and quickly noted that GABA was importantly relevant. In research, GABAergic neurons have been reported to regulate feeding, body weight and metabolic functions (Kim et al., 2012; Tong et al., 2008; Wu et al., 2009; Wu et al., 2015); however, it is still unclear if synaptic or non-synaptic extracellular GABA is mostly responsible. Using c-Fos as a neuronal activation marker, we found that compared to chow-fed WT control mice, many cells in the paraventricular nucleus (PVN) as well as the VMH of chow-fed GFAP/CAIKKβ+/− mice were more sensitive to GABA agonist Baclofen-induced neuronal activation (Fig. 5A, B). Astrocytic IKKβ/NF-κB loss-of-function model was also studied, using HFD-fed mice which received MBH injection of DNIκBα vs. control lentivirus as described in Fig. 3G–L. At an early time point when HFD-induced obesity was still comparable between two groups, these HFD-fed mice received an administration of Baclofen through an intra-hypothalamic injection. Compared to chow-fed WT control mice in Figure 5A–B, much more cells became c-Fos positive in the VMH and PVN of HFD-fed control mice following Baclofen injection (Fig. 5C). This increase largely represented an effect of chronic HFD feeding (data not shown), although the underlying basis still remains unclear. On the other hand, despite HFD feeding, Baclofen-induced neuronal activation was markedly prevented in DNIκBα-injected mice (Fig. 5C). Thus, a moderate level of IKKβ/NF-κB activation can turn up the hypothalamic response to GABA through astrocytes.
Figure 5. Effect of astrocytic IKKβ/NF-κB manipulation on neuronal activation.

GFAP/CAIKKβ+/− mice versus littermate WT (lox-STOP-lox-CAIKKβ+/− ) mice (3 months old, chow fed males) (A, B) and HFD-fed C57BL/6 mice with MBH injections of GFAP promoter driven DNIκBα or control lentiviruses described in Figures 3I–L (C) were injected in the MBH with baclofen (20 ng) versus vehicle, and brains were dissected at 2 hours post-injection for c-Fos immunostaining and counting analysis. Images show the PVN from WT versus GFAP/CAIKKβ+/− mice following the injection as indicated. Scale bar, 100 μm. Bar graphs present the average numbers of cells positive for c-Fos in median sections of the PVN and VMH of the indicated mice. *p < 0.05, **p < 0.01, n = 4 mice per group (B, C). Error bars reflect mean ± SEM.
Control of hypothalamic extracellular GABA level by astrocytes and IKKβ/NF-κB
An important function of astrocytes in regulating GABA neural circuit is to clear GABA from synaptic cleft and the extracellular space through GABA transporters that are located on processes (Belanger et al., 2011; Minelli et al., 1996). Therefore, we measured extracellular GABA content in the MBH of mice under different feeding conditions via microdialysis. According to previous research, GABA transporter-3 (GAT3) is abundant in astrocytes and specifically localized in astrocytic process (Minelli et al., 1996). We found that GAT3 protein levels were higher in the hypothalamus than many other brain regions such as the cortex, hippocampus and cerebellum (Fig. S7A), indicating that this transporter may play a role in regulating GABA homeostasis in the hypothalamus. Since astrocytic process length was found sensitive to IKKβ/NF-κB manipulation, several experiments were performed using IKKβ/NF-κB models. Primary astrocytes from the hypothalamus were infected with lentiviruses expressing CAIKKβ (Fig. S7B). The results showed that CAIKKβ expression was sufficient to impair GABA uptake of astrocytes from culture medium. Next, we measured GABA concentrations in the MBH of various mouse models with chronic HFD feeding or IKKβ/NF-κB manipulations. The results showed that chronic HFD feeding led to an increase in extracellular GABA content within the MBH of C57BL/6 mice (Fig. 6A). Similarly, MBH extracellular GABA levels were significantly higher in GFAP/CAIKKβ+/− mice than in controls (Fig. 6B). GFAP/IKKβlox/lox mice were also studied, showing that IKKβ knockout prevented HFD feeding from increasing MBH extracellular GABA content (Fig. 6C). These results suggest that astrocytic IKKβ/NF-κB activity has an influence on extracellular GABA-induced neuronal activation, and this effect could be partially related to astrocytic regulation of extracellular GABA which therefore changes the sensitivity of GABA-dependent neuronal excitation.
Figure 6. Effects of astrocytic IKKβ/NF-κB on extracellular GABA and BDNF.
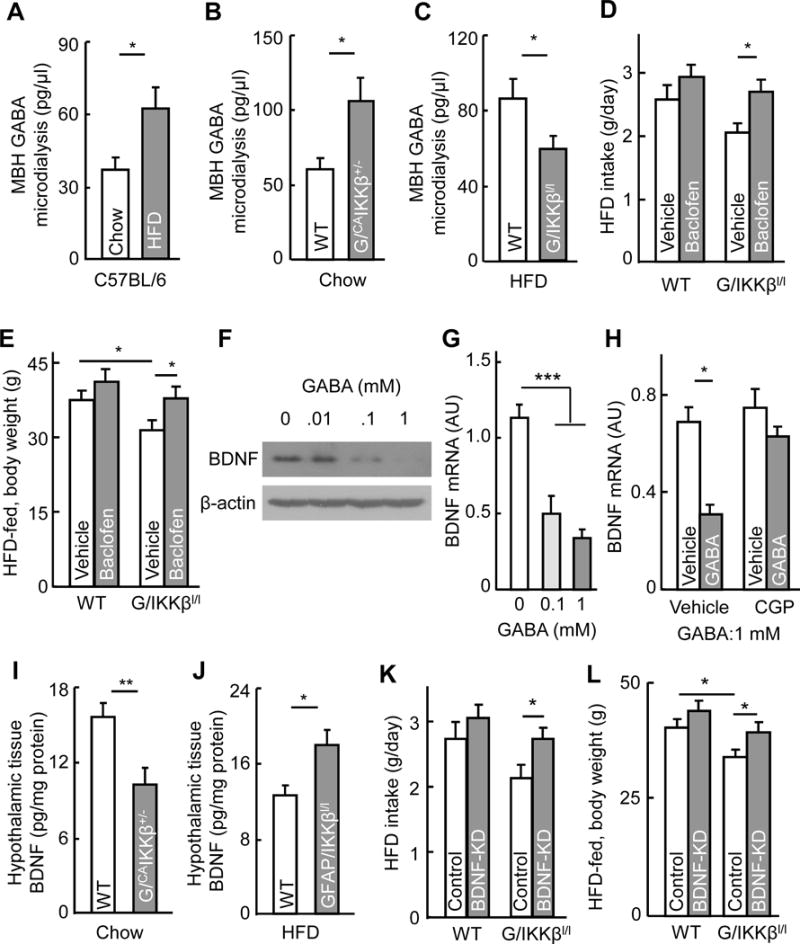
A–C Extracellular GABA contents in the MBH were measured through microdialysis for (A) C57BL/6 mice which received 3-month HFD vs. chow feeding, (B) GFAP/CAIKKβ+/− mice vs. littermate WT controls, and (C) GFAP/IKKβlox/lox mice vs. littermate WT controls. D–E HFD-fed GFAP/IKKβlox/lox mice and littermate WT (IKKβlox/lox) mice received intra-third ventricle administration of Baclofen (20 ng) twice per week for 4 weeks and subsequently measured for HFD intake (D) and body weight (E). F–H Cultured hypothalamus neurons were treated with GABA at indicated concentrations for 4 hours (F–H), together with pre-treatment of CGP54626 (10 μM) for 30 min prior to GABA (1 mM) treatment (H), and measured for BDNF protein (F) and mRNA (G, H) levels. I–J Hypothalamic samples collected from overnight-fasted GFAP/CAIKKβ+/− mice (I) and GFAP/IKKβlox/lox mice (J) vs. their respective littermate WT controls (I, J) were lysed and measured for BDNF via ELISA. K–L HFD-fed GFAP/IKKβlox/lox mice vs. littermate WT (IKKβlox/lox) controls were injected in the MBH with BDNF shRNA or scramble shRNA lentiviruses and measured for HFD intake (K) and body weight (L) after 4-week lentiviral treatment. * p < 0.05, ** p < 0.01, *** p < 0.001, n = 15 mice per group and 3 mice pooled per sample (A–C), n = 6–8 mice per group (D, E, I–L); n = 6 samples per group (G, H). Error bars reflect mean ± s.e.m. See also Figure S7.
Astrocytic IKKβ/NF-κB-controlled extracellular GABA affects feeding and body weight
Presumably, changes in astrocytic process plasticity can affect the uptake of various neurotransmitters, for example, glutamate, if neurotransmitter uptake system is significantly present in the processes of astrocytes. In this study, we have shown that astrocytic IKKβ/NF-κB manipulation led to early-term effects on glucose and blood pressure vs. late-term effects on feeding and body weight, which might involve different neurotransmitters. Given that hypothalamic GABAergic neurons are known to strikingly affect feeding and body weight (Kim et al., 2012; Tong et al., 2008; Wu et al., 2009; Wu et al., 2015), we wondered if extracellular GABA might be particularly important for feeding and body weight phenotypes in our IKKβ/NF-κB mouse models. To gain an insight into this possibility, we studied HFD-fed GFAP/IKKβlox/lox mice which greatly benefited from feeding control and obesity prevention. These knockout mice and WT controls were maintained on HFD feeding, and during this feeding intervention, both mice were centrally treated for 4 weeks via intra-hypothalamic third ventricle injection with GABA receptor agonist Baclofen. Baclofen partially restored HFD intake GFAP/IKKβlox/lox mice to the level of HFD intake experienced in vehicle-treated control mice (Fig. 6D). Consistently, the anti-obesity effect in GFAP/IKKβlox/lox mice was significantly abolished by the treatment (Fig. 6E). Thus, while the neurotransmitter mechanism for early-onset and body weight-independent effects of astrocytic IKKβ/NF-κB remains to be studied, these results here support a role of GABA pathway in linking astrocytic IKKβ/NF-κB in association with astrocytic process plasticity to feeding and body weight control.
GABA-controlled BDNF in feeding and body weight effect of astrocytic IKKβ/NF-κB
Through controlling extracellular GABA, astrocytes might modulate many GABA receptor-expressing neurons, such as POMC and AGRP neurons, since some of these neurons are reported to be responsive to GABA (Cowley et al., 2001; Smith et al., 2013). While future investigation on these neurons is needed, we directed a research effort in this work to BDNF-expressing neurons, because previous reports have pointed out that administration of GABA receptor antagonist can increase BDNF in several brain regions (Enna et al., 2006; Heese et al., 2000), and besides, BDNF is expressed in the VMH and PVN of hypothalamus and masterly regulates feeding behavior and energy homeostasis (An et al., 2015; Cordeira et al., 2014), while our results in Figure 5 revealed that astrocytic IKKβ/NF-κB activation remarkably rendered many neurons in these two subregions to be more responsible to GABA. We questioned if BDNF neurons in these subregions might be partially responsible for feeding and body weight in the same line as the effect of GABA manipulation. In cultured hypothalamic neurons, we confirmed that increases in GABA concentration led to reduction in BDNF protein and mRNA levels (Fig. 6F, G). To test whether the decrease of BDNF expression by GABA is induced by GABAB receptor, cultured hypothalamic neurons were pretreated with GABAB receptor antagonist CGP54626 before GABA was added. Our results showed that this pre-treatment blunted the inhibition on BDNF expression by GABA (Fig. 6H). By measuring BDNF in hypothalamic tissues, we found that BDNF content in the MBH was lower in chow-fed GFAP/CAIKKβ+/− mice than chow-fed WT control mice (Fig. 6I) and was higher in HFD-fed GFAP/IKKβlox/lox mice compared to HFD-fed WT control mice (Fig. 6J). To assess if BDNF could be physiologically involved in the anti-obesity effect of this astrocytic IKKβ knock model, we injected lentiviral BDNF shRNA to the MBH area of GFAP/IKKβlox/lox and control mice under HFD feeding. The results showed that the protective effects of IKKβ knockout on food intake and body weight were diminished by BDNF shRNA (Fig. 6K, L). Thus, while we predict that there are additional types of neurons which are accountable, BDNF neurons represent a link between extracellular GABA and the feeding/body weight changes induced by astrocytic IKKβ/NF-κB.
Discussion
Astrocytic process plasticity in hypothalamic control of physiology
The central nervous system and in particular the hypothalamus are important for the regulation of metabolic physiology, and multiple neuronal circuits in the hypothalamus and other brain regions have been implicated in this regulation (Andrews et al., 2008; Chiappini et al., 2014; Coppari et al., 2005; Dodd et al., 2015; Flak et al., 2014; Kleinridders et al., 2013; Koch et al., 2015; Thaler et al., 2012; Zeltser et al., 2012). Over recent years astrocytes have increasingly been appreciated for the roles in brain’s functioning such as synaptic plasticity, neural excitation, and development (Chung et al., 2015; Eroglu and Barres, 2010; Jo et al., 2014; Otsu et al., 2015; Rangroo Thrane et al., 2013; Shigetomi et al., 2012). Very recently it was reported that the field size and morphology of hypothalamic astrocytes are influenced by leptin signaling (Kim et al., 2014) and that insulin receptor ablation can also affect the primary processes of astrocytes as well as brain glucose update and metabolism (Garcia-Caceres et al., 2016). Here our study revealed that the astrocytic processes in the mediobasal hypothalamic region undergo plastic changes according to nutritional states such as fasting vs. feeding. Of note, these changes occur at high-order processes rather than cell bodies or primary trunks low-order branches. Hence together with recent literature, we suggest that astrocytes can actively receive and react to metabolic information, resulting in dynamic changes of process length and density which adjust the hypothalamic condition and regulate the physiological response. Based on our results, this regulation by astrocytic processes involves feeding-associated functions such as blood glucose and blood pressure and perhaps appetite as well. Yet, the underlying mediators for changing astrocyte processes are still unclear, which, however, could be related to intracellular calcium since it determines cell morphology (Fawthrop and Evans, 1987; Molotkov et al., 2013; Otsu et al., 2015) and/or autophagy since it is more activated in the brain during fasting (Alirezaei et al., 2010). Despite these questions, IKKβ/NF-κB appears important, given that IKKβ/NF-κB activation or inhibition can both rapidly alter astrocytic process plasticity along with or prior to the accordant induction of physiological effects.
Metabolic regulation of astrocytes via extracellular neurotransmitter changes
Besides the prototypical role of hypothalamic neuropeptides in central control of metabolic physiology, lately synaptic neurotransmitters such as GABA have been related to metabolic regulation (Dietrich and Horvath, 2009; Ghamari-Langroudi et al., 2015; Kim et al., 2015a; Kim et al., 2012; Tong et al., 2008; Vong et al., 2011; Wu et al., 2015). However, as synaptic regulation occurs very fast, conceivably it should require additional modulations to process a slower-process regulation on metabolic physiology. The control of food intake via GABAergic synaptic transmission has been shown to occur within 20 milliseconds (Vong et al., 2011), but this feature differs from the physiological conditions in which feeding and metabolic responses to changing nutrient signals take much longer (up to hours), implicating that an extra-synaptic GABA action might exist. In this study, using microdialysis which mostly reflects non-synaptic GABA, we provided data showing that nutritional changes as well as astrocytic IKKβ/NF-κB activation or inhibition consistently affect hypothalamic extracellular GABA levels. The levels of extracellular GABA were negatively correlated with the sizes of hypothalamic astrocytic processes consistently in all models that we studied. Given that astrocytic processes importantly regulate the update of extracellular GABA (Belanger et al., 2011; Minelli et al., 1996), a cause-effect relationship between them can be reasoned although not exactly proven in this study. Along this line, we identified a role of extracellular GABA-controlled BDNF in affecting food intake and body weight, providing an example of extracellular neurotransmitter-dependent neuropeptide in metabolic regulation. While this study focused on studying GABA-BDNF axis, we should point out that other neurotransmitters (such as glutamate) could also be involved, if astrocytic processes contain the update systems for these neurotransmitters. Indeed, early-onset vs. late-onset effects of astrocytic IKKβ/NF-κB in different physiology also suggest involvements of different neurotransmitters which influence different downstream neuropeptides.
Disease relevance of astrocytic IKKβ/NF-κB and process plasticity impairment
Under chronic overnutrition, hypothalamic dysfunctions are important for the development of dietary obesity and associated metabolic syndrome (such as glucose intolerance, insulin resistance and hypertension). The responsible mechanisms have multiple levels, one of which was recently related to a sustained, moderate level of IKKβ/NF-κB activation leading to hypothalamic molecular inflammatory changes (Li et al., 2012; Purkayastha et al., 2011; Yan et al., 2014; Zhang et al., 2008). This set of hypothalamic molecular changes, appreciably termed “hypothalamic inflammation” although still vaguely defined, can substantially influence the normal functions of hypothalamic cells including neurons. In recent literature, it has been reported that overnutrition-induced hypothalamic inflammation is associated with astrogliosis (Thaler et al., 2012); however, it remains unknown if astrocytes are causally important for the development of obesity and related metabolic syndrome. In this study, we found that astrocyic process plasticity is impaired similarly by chronic overnutrition and by moderate-level IKKβ/NF-κB upregulation. Using genetic models, we revealed that IKKβ/NF-κB activation led to astrocytic process reduction while IKKβ/NF-κB inhibition rapidly reversed this process defect in mice with obesity, and these effects on astrocytic processes occurred before changes in cell numbers in both experimental conditions. Thus, we dissected out an event of process plasticity change which is independent of cell number. Altogether, as elucidated in Figure 7, a conceptual diagram is postulated, that is, when physiology is under changes from fed status to fasting, hypothalamic astrocytic processes are dynamically reduced, extracellular neurotransmitters such as GABA levels become elevated, which employ neuropeptides (such as GABA-controlled BDNF) to physiologically mediate different metabolic responses; on the other hand, some of these regulatory mechanisms and actions are enhanced under the condition of chronic overnutrition, critically through sustained IKKβ/NF-κB upregulation, which contribute to the loss of physiological homeostasis and eventually disease.
Figure 7. Concluding points and proposed conceptual model.
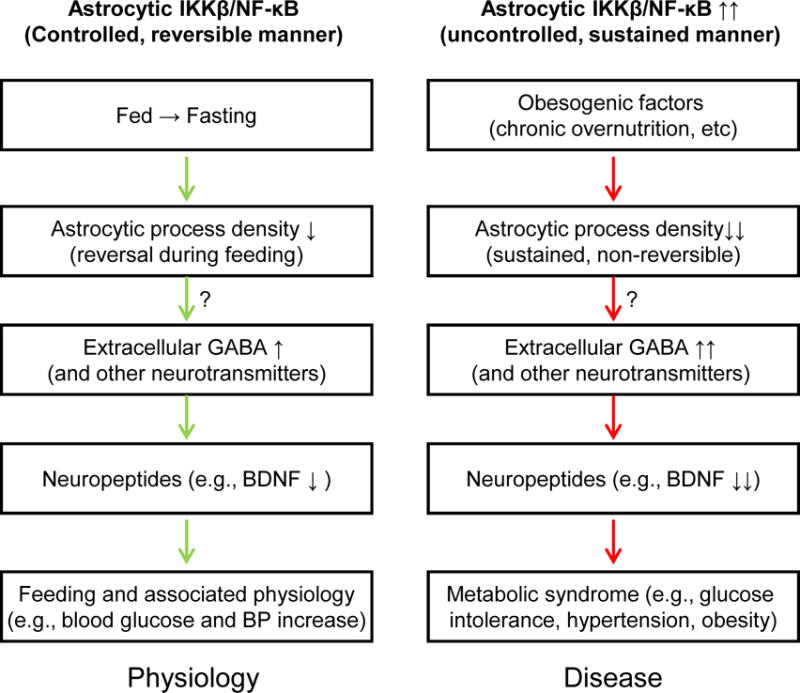
Physiological changes from fed status to fasting lead to decreases in hypothalamic astrocytic process density; accordingly hypothalamic extracellular neurotransmitters (such as GABA) levels are adjusted to modulate downstream neuropeptides (such as BDNF) and promote physiological switch from fasting to feeding which is associated with increased glucose and BP. Chronic overnutrition or sustained astrocytic IKKβ/NF-κB activation can similarly maintain astrocytic process shortening and alterations in extracellular GABA and BDNF expression, thus contributing to the development of metabolic syndrome. Potential connections indicated with question markers can be suggested from this work but remain to be established.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents should be directed to and will be fulfilled by the corresponding author Dongsheng Cai (dongsheng.cai@einstein.yu.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animal study
Standard C57BL/6 mice were purchased from Jackson Laboratory. Rosa 26-lox-STOP-lox-CAIKKβ mice and GFAP-Cre mice obtained from Jackson Laboratory were then maintained on C57BL/6 strain background. IKKβlox/lox mice were also maintained on C57BL/6 strain background (Zhang et al., 2008). Mice were housed in standard, pathogen-free animal facility, 3–5 mice per cage, 12h/12h light and darkness cycles and maintained on a standard normal chow since weaning. As male mice in C57BL/6 background are prone to the development of obesity and associated disorders and it is standard procedure to use males to study these diseases, we used male mice in all studies. For obesity-related studies, mice were fed a HFD (60% kcal fat, Research Diets, Inc.) for indicated durations. Body weights were regularly measured and food intake was determined periodically through individual housing mice. Energy expenditure was determined using metabolic chambers (Columbus Instrument, Inc.), lean and fat mass of mice were measured via MRI, both performed through the physiology core at Albert Einstein College of Medicine. Glucose tolerance test (GTT) was performed in mice through intraperitoneal injection of glucose at 2g/kg body weight. Blood glucose levels during GTT were measured with LifeScan® blood glucose monitoring system. Blood insulin was measured using ELISA Kit (ALPCO). All procedures were approved by the Institutional Animal Care and Use Committee of Albert Einstein College of Medicine.
Cell cultures
Hypothalamus tissues were dissected from C57BL/6 mice on postnatal Day 4 for the isolation of astrocytes, the meninges were removed, and cells were dissociated in 0.05% trypsin-EDTA. Mixed cells were cultured at 37 °C in 5% CO2. Non-adherent cells were removed by changing the media every 3 days. After 7–10 days, cells were shaken rigorously in an orbital incubator at 0.23 g at 37 °C for 2 hours to detach microglia, then shaken again at 0.23 g at 37 °C overnight to remove oligodendrocytes. Remaining cells were astrocytes and were trypsinized and reseeded for further culture. Hypothalamus neurons were collected mouse embryos on embryonic Day 19. Hypothalamus was dissected using a stereo microscope, and then placed in 0.05% trypsin-EDTA for 30 min at 37°C. After trituration in Neurobasal media, cells were passed through a 70-μm cell strainer to remove tissue debris followed with a 40-μm cell strainer. After centrifuging, cells were resuspended in Neurobasal media containing 2% B27 and 2 mM glutamine and plated in 6-well plates pre-coated with 25 μg/ml poly-D-lysine for further culture. GABA contents in culture medium were detected via ELISA kits (Biotang). For immunostaining, astrocytes on coverslips were fixed with 4% PFA, blocked with the serum, and treated with mouse anti-GFAP (MAB3402, Millipore) primary antibody followed by reaction with Alexa Fluor 488 secondary antibody (Invitrogen). Images were taken using a confocal microscope.
METHOD DETAILS
Cannulation and brain injection
As previously described (Kim et al., 2015b), using an ultra-precise (10 μm resolution) small animal stereotactic apparatus (David Kopf Instruments), a 26 gauge guide cannula (Plastics One, Inc.) was implanted into third ventricle of anesthetized mice at the midline coordinates of 1.7 mm posterior to bregma and 5.0 mm below the surface of skull. Intra-third ventricular injection was carried out with a 33-gauge internal cannula (Plastics One) connected to a 5-μl Hamilton Syringe. All drugs were dissolved in 0.5-μl artificial cerebrospinal fluid (aCSF) for injection. Injection of aCSF was used as vehicle control. For the bilateral lentivirus injection, an ultra-precise stereotactic apparatus were located to the hypothalamic area centering at the VMH at coordinates of 1.7 mm posterior to bregma, 5.6–5.8 mm below the surface of skull, and 0.25 mm lateral to midline. Purified lentiviruses suspended in 0.2 μl aCSF was injected over ~10-min period via a 26-gauge guide cannula and a 33-gauge internal injector (Plastics One) connected to a 5-μl Hamilton Syringe and infusion pump (WPI Instruments). Lentiviruses injection accuracy for each individual mouse was assessed post experiment through immunostaining of GFP which was contained in the lentiviral vector, and mice at least with 80% of GFP expression within the target region were considered qualified while mice which did not meet this criterion (<20% failure rate) were excluded from the experiment.
Microdialysis and ELISA
A guide cannula for microdialysis (CMA) was implanted into the MBH area of anesthetized mice at coordinates of 1.7 mm posterior to bregma, 5.8 mm below the surface of skull, and 0.25 mm lateral to midline. As described in the literature (Gobrogge et al., 2009), after one-week recovery, a dialysis probe (CMA) with 1.0-mm membrane was inserted through the guide cannula and perfused continuously at 0.5 μl/min with aCSF using a glass Hamilton syringe connected to an infusion pump (WPI Instruments). Samples were collected through a tube connected to the probe. During sampling, mice were conscious with free movement. GABA contents in microdialysis samples were analyzed in Stable Isotope and Metabolomics Core Facility of the Albert Einstein College of Medicine. In brief, microdialysis samples were added with methanol, vortexed, and after centrifugation, supernatant was transferred and dried under gentle air flow. Linearity curve samples using a series of GABA quantity were prepared in the same procedure. Samples were analyzed by gas chromatography time-of-flight mass spectrometry (GC-TOFMS premier, Waters, USA). Metabolites separation was performed on a 30m DB-5MS column coupled with 10m guard column (Agilent, USA). Standard curve samples were run twice (before and after the microdialysis samples). The data was collected under a positive chemical ionization (PCI) mode. The mass range was set to 100 to 1200, and the lock mass was set to 286.0027. Data was processed with Masslynx software (Waters, MA, USA). To measure tissue BDNF content, hypothalamic tissue was homogenized in the lysis buffer (20 mM Tris-HCl, 50 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM β-glycerolphosphate, protease inhibitor cocktail (Thermo), the homogenates were centrifuged at 13,000 g for 30 min, and the supernatant was used for BDNF assay via ELISA kit (Biotang).
Golgi staining
Golgi staining was performed as previously described (Giusti et al., 2014; Reichel et al., 2016), and to shift the staining towards glia cells instead of neurons, we adapted a modified protocol described by Ranjan and Mallick (Ranjan and Mallick, 2012). Mice were sacrificed under anesthesia, the brains carefully dissected out and briefly rinsed in dH2O, and the brains were incubated in 4% PFA-PBS at room temperature for 72 h. Subsequently the protocol of the FD Rapid GolgiStain™ Kit (FD Neurotechnologies, INC) was followed. A sufficient number of astrocytes across the ARC and VMH of each mouse was randomly traced using 100× objective and the Neurolucida 5 software (MBF Bioscience, Williston, VT), and an adequate number of cells per condition were independently traced and analyzed quantitatively. Scholl analyses and dendritic length were performed using the Neurolucida Explorer (MBF Bioscience) software. Concentric Scholl circles around the center points of cell bodies were determined at various intervals. Cell types were identified according to literature-derived criteria (Bradl and Lassmann, 2010; Kimelberg and Nedergaard, 2010; Ranjan and Mallick, 2012) and our own observations (as exemplified in Fig. S1). In brief, astrocytes under Golgi staining were recognized by distinct characteristics including a star-like cellular shape, the domain area of which typically ranged from 25 to 65 μm in diameter, with a dense cell body staining (almost fused with the initial portions of cell processes) and highly-ramified thick processes which differ from processes of other glial cells. Microglia under Golgi staining characteristically presented a small cell body (usually <5 μm in diameter) with less densely-stained, low-degree ramified and thin processes, the domain of which ranged from 10 to 20 μm in diameter. Oligodendrocytes under Golgi staining were identified by the presence of a round-oval distinctive cell body with the presence of highly densely-stained but few thick un-ramified short processes (often 1~3 densely stained processes) typically with a domain of 25–40 μm in diameter.
Immunohistology
Brain histology was analyzed using brain sections and immunostaining. Mice under anesthesia were transcardially perfused with 4% PFA and brains were removed, post-fixed in 4% PFA for 24 hours, and infiltrated with 20–30% sucrose. Brain sections (20 μm-thick) were cut using cryostat, blocked with serum of appropriate species, treated with primary antibodies: mouse antic-Fos (sc-166940, Santa Cruz), mouse anti-GFP (A-11120, Thermo Fisher), mouse anti-FLAG (F1804, Sigma) and chicken anti-GFAP (ab4674, Abcam), subsequently reacted with Alexa Fluor 488 or 555 secondary antibodies (Invitrogen). Naïve IgGs of appropriate species were used as negative controls. DAPI staining was used to reveal all cells in the section. Images were taken using a confocal microscope.
Telemetric probe implantation and recording
Radiotelemetric catheters (model TA11PA–C10, Data Sciences International, DSI) were implanted in the carotid artery, using the procedure we described previously (Purkayastha et al., 2011). Briefly, after mice were anesthetized, a ventral midline skin incision was made and the left common carotid artery was isolated under a surgical microscope. The proximal end of the artery was ligated below the carotid bifurcation, and the distal end was occluded with a microclip. A small incision was made near the proximal end, and pressure transmission catheter was guided into the artery, advanced to the aorta, and secured in place with sutures. The transmitter device was placed subcutaneously on the right flank, as close to the hind limb as possible. Subsequently, neck incision was closed using 5–0 sutures (Ethicon), and mice were allowed for 1~2 weeks of post-surgical recovery, and the pressure signals of each animal under conscious, free-moving conditions were recorded using the computerized method (DSI). BP data were sampled continuously with a sampling rate of 1,000 Hz with 1-minute segment duration.
Recombinant lentiviruses
The GFAP promoter was a gift kindly provided by Dr. Michael Brenner. Lentiviral vector of GFAP promoter-driven DNIκBα was generated by subcloning GFAP promoter into DNIκBα expressed lentiviral vector. Mouse BDNF shRNA and control scramble shRNA were constructed using pSico (Addgene). BDNF shRNA sequence: 5′-GAATTGGCTGGCGATTCAT-3′. Lentiviruses were produced by co-transfecting the viral expression vector with the packaging plasmids into HEK293T cells. At 24–36 hours after transfection, culture media were collected and filtered through a 0.45-μm filter to remove cell debris and followed by ultracentrifugation. After centrifugation, supernatant was removed, and lentiviruses in the pellet were re-suspended, and lentivirus titer was determined via ELISA kit (Clontech Laboratories).
mRNA analysis and western blot
Total RNA was extracted from tissues or cultured cells using TRIzol® (Invitrogen), and cDNA was synthesized using M-MLV RT System (Promega) followed by PCR amplification and quantification using SYBR® Green PCR Master Mix (Applied Biosystems). House-keeping β-actin mRNA was used for PCR normalization. Animal tissues were homogenized, and proteins were dissolved in a lysis buffer. Proteins dissolved in lysis buffer were separated by SDS-PAGE and identified by immunoblotting. Primary antibodies included rabbit anti-p-RelA (3033, Cell Signaling), rabbit anti-RelA (8242, Cell Signaling), rabbit anti-β-actin (4970, Cell Signaling), mouse anti-flag (F1804, Sigma), rabbit anti-BDNF (ab72439, Abcam) and rabbit anti-GAT3 (ab431, Abcam). Secondary antibodies included HRP-conjugated anti-mouse and anti-rabbit antibodies (Pierce).
QUANTIFICATION AND STATISTICAL ANALYSIS
Two-tailed Student’s t test was used for comparisons between two groups, and ANOVA and appropriate (Tukey) post-hoc analyses were used for comparisons among more than two groups. Data presented met normal distribution, data variance among comparable experimental groups was similar, and statistical tests for each figure were justified appropriate. Adequate sample sizes were chosen based on our previous research; animal number was 7–10 mice per group for metabolic physiological analysis and 4–6 independent samples per group for molecular analysis. The variances of each pair of data sets being compared were similar to each other and consistent with their being normally distributed. Mice were randomized into different groups with approximately equal numbers of animals in each group. None of qualified samples or data points were excluded from the statistical analysis. Experimental performers were not usually blind to animal treatments but outcomes were subjected to blind analysis whenever possible. Statistical analyses were performed using GraphPad Prism 7. Data were presented as mean ± s.e.m. P < 0.05 was considered statistically significant.
Supplementary Material
Highlights.
Astrocytic process plasticity is vital for the central control of metabolic balance
Astrocytes control extracellular neurotransmitter level to regulate metabolic balance
Astrocytic plasticity is impaired by chronic overnutrition or metabolic inflammation
Inflamed astrocytes lead to metabolic imbalance and thus metabolic syndrome
Acknowledgments
The authors sincerely thank Cai’s laboratory members for technical assistance. This study was supported by NIH R01 DK078750, R01 AG031774, R01 HL113180, and R01 DK 099136 (all to D. Cai).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
COMPETING INTERESTS STATEMENT
The authors state that they have no competing financial interests.
Author contributions: DC conceived and designed the study and organized experimentation. YZ co-designed and performed majority of experiments, JR initiated and co-developed Golgi staining experiments, CH performed animal surgeries and involved physiological recordings. JZ did imaging of Golgi staining. All authors did data analysis and interpretation. YZ prepared all figures, YZ and JR assisted writing, and DC wrote the paper.
References
- Alirezaei M, Kemball CC, Flynn CT, Wood MR, Whitton JL, Kiosses WB. Short-term fasting induces profound neuronal autophagy. Autophagy. 2010;6:702–710. doi: 10.4161/auto.6.6.12376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez MI, Rivas L, Lacruz C, Toledano A. Astroglial cell subtypes in the cerebella of normal adults, elderly adults, and patients with Alzheimer’s disease: a histological and immunohistochemical comparison. Glia. 2015;63:287–312. doi: 10.1002/glia.22751. [DOI] [PubMed] [Google Scholar]
- An JJ, Liao GY, Kinney CE, Sahibzada N, Xu B. Discrete BDNF Neurons in the Paraventricular Hypothalamus Control Feeding and Energy Expenditure. Cell Metab. 2015;22:175–188. doi: 10.1016/j.cmet.2015.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews ZB, Liu ZW, Walllingford N, Erion DM, Borok E, Friedman JM, Tschop MH, Shanabrough M, Cline G, Shulman GI, et al. UCP2 mediates ghrelin’s action on NPY/AgRP neurons by lowering free radicals. Nature. 2008;454:846–851. doi: 10.1038/nature07181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belanger M, Allaman I, Magistretti PJ. Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metab. 2011;14:724–738. doi: 10.1016/j.cmet.2011.08.016. [DOI] [PubMed] [Google Scholar]
- Bradl M, Lassmann H. Oligodendrocytes: biology and pathology. Acta neuropathologica. 2010;119:37–53. doi: 10.1007/s00401-009-0601-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiappini F, Catalano KJ, Lee J, Peroni OD, Lynch J, Dhaneshwar AS, Wellenstein K, Sontheimer A, Neel BG, Kahn BB. Ventromedial hypothalamus-specific Ptpn1 deletion exacerbates diet-induced obesity in female mice. J Clin Invest. 2014;124:3781–3792. doi: 10.1172/JCI68585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi HB, Gordon GR, Zhou N, Tai C, Rungta RL, Martinez J, Milner TA, Ryu JK, McLarnon JG, Tresguerres M, et al. Metabolic communication between astrocytes and neurons via bicarbonate-responsive soluble adenylyl cyclase. Neuron. 2012;75:1094–1104. doi: 10.1016/j.neuron.2012.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung WS, Allen NJ, Eroglu C. Astrocytes Control Synapse Formation, Function, and Elimination. Cold Spring Harbor Perspectives in Biology. 2015;7:a020370. doi: 10.1101/cshperspect.a020370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung WS, Clarke LE, Wang GX, Stafford BK, Sher A, Chakraborty C, Joung J, Foo LC, Thompson A, Chen C, et al. Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature. 2013;504:394–400. doi: 10.1038/nature12776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen P, Zhao C, Cai X, Montez JM, Rohani SC, Feinstein P, Mombaerts P, Friedman JM. Selective deletion of leptin receptor in neurons leads to obesity. J Clin Invest. 2001;108:1113–1121. doi: 10.1172/JCI13914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cone RD. Anatomy and regulation of the central melanocortin system. Nat Neurosci. 2005;8:571–578. doi: 10.1038/nn1455. [DOI] [PubMed] [Google Scholar]
- Coppari R, Ichinose M, Lee CE, Pullen AE, Kenny CD, McGovern RA, Tang V, Liu SM, Ludwig T, Chua SC, Jr, et al. The hypothalamic arcuate nucleus: a key site for mediating leptin’s effects on glucose homeostasis and locomotor activity. Cell Metab. 2005;1:63–72. doi: 10.1016/j.cmet.2004.12.004. [DOI] [PubMed] [Google Scholar]
- Cordeira JW, Felsted JA, Teillon S, Daftary S, Panessiti M, Wirth J, Sena-Esteves M, Rios M. Hypothalamic dysfunction of the thrombospondin receptor alpha2delta-1 underlies the overeating and obesity triggered by brain-derived neurotrophic factor deficiency. J Neurosci. 2014;34:554–565. doi: 10.1523/JNEUROSCI.1572-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowley MA, Smart JL, Rubinstein M, Cerdan MG, Diano S, Horvath TL, Cone RD, Low MJ. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature. 2001;411:480–484. doi: 10.1038/35078085. [DOI] [PubMed] [Google Scholar]
- Dietrich MO, Horvath TL. GABA Keeps Up an Appetite for Life. Cell. 2009;137:1177–1179. doi: 10.1016/j.cell.2009.06.002. [DOI] [PubMed] [Google Scholar]
- Dietrich MO, Horvath TL. Synaptic plasticity of feeding circuits: hormones and hysteresis. Cell. 2011;146:863–865. doi: 10.1016/j.cell.2011.08.031. [DOI] [PubMed] [Google Scholar]
- Dodd GT, Decherf S, Loh K, Simonds SE, Wiede F, Balland E, Merry TL, Munzberg H, Zhang ZY, Kahn BB, et al. Leptin and insulin act on POMC neurons to promote the browning of white fat. Cell. 2015;160:88–104. doi: 10.1016/j.cell.2014.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enna SJ, Reisman SA, Stanford JA. CGP 56999A, a GABA(B) receptor antagonist, enhances expression of brain-derived neurotrophic factor and attenuates dopamine depletion in the rat corpus striatum following a 6-hydroxydopamine lesion of the nigrostriatal pathway. Neurosci Lett. 2006;406:102–106. doi: 10.1016/j.neulet.2006.07.004. [DOI] [PubMed] [Google Scholar]
- Eroglu C, Barres BA. Regulation of synaptic connectivity by glia. Nature. 2010;468:223–231. doi: 10.1038/nature09612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fawthrop DJ, Evans RJ. Morphological changes in cultured astrocytes following exposure to calcium ionophores. Neurosci Lett. 1987;81:250–256. doi: 10.1016/0304-3940(87)90391-0. [DOI] [PubMed] [Google Scholar]
- Fields RD, Stevens-Graham B. New insights into neuron-glia communication. Science. 2002;298:556–562. doi: 10.1126/science.298.5593.556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flak JN, Patterson CM, Garfield AS, D’Agostino G, Goforth PB, Sutton AK, Malec PA, Wong JM, Germani M, Jones JC, et al. Leptin-inhibited PBN neurons enhance responses to hypoglycemia in negative energy balance. Nat Neurosci. 2014;17:1744–1750. doi: 10.1038/nn.3861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flier JS, Maratos-Flier E. Obesity and the hypothalamus: novel peptides for new pathways. Cell. 1998;92:437–440. doi: 10.1016/s0092-8674(00)80937-x. [DOI] [PubMed] [Google Scholar]
- Fuente-Martin E, Garcia-Caceres C, Granado M, de Ceballos ML, Sanchez-Garrido MA, Sarman B, Liu ZW, Dietrich MO, Tena-Sempere M, Argente-Arizon P, et al. Leptin regulates glutamate and glucose transporters in hypothalamic astrocytes. J Clin Invest. 2012;122:3900–3913. doi: 10.1172/JCI64102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Caceres C, Quarta C, Varela L, Gao Y, Gruber T, Legutko B, Jastroch M, Johansson P, Ninkovic J, Yi CX, et al. Astrocytic Insulin Signaling Couples Brain Glucose Uptake with Nutrient Availability. Cell. 2016;166:867–880. doi: 10.1016/j.cell.2016.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Segura LM, Chowen JA, Parducz A, Naftolin F. Gonadal hormones as promoters of structural synaptic plasticity: cellular mechanisms. Progress in neurobiology. 1994;44:279–307. doi: 10.1016/0301-0082(94)90042-6. [DOI] [PubMed] [Google Scholar]
- Ghamari-Langroudi M, Digby GJ, Sebag JA, Millhauser GL, Palomino R, Matthews R, Gillyard T, Panaro BL, Tough IR, Cox HM, et al. G-protein-independent coupling of MC4R to Kir7.1 in hypothalamic neurons. Nature. 2015;520:94–98. doi: 10.1038/nature14051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giusti SA, Vogl AM, Brockmann MM, Vercelli CA, Rein ML, Trumbach D, Wurst W, Cazalla D, Stein V, Deussing JM, et al. MicroRNA-9 controls dendritic development by targeting REST. eLife. 2014;3 doi: 10.7554/eLife.02755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gobrogge KL, Liu Y, Young LJ, Wang Z. Anterior hypothalamic vasopressin regulates pair-bonding and drug-induced aggression in a monogamous rodent. Proc Natl Acad Sci U S A. 2009;106:19144–19149. doi: 10.1073/pnas.0908620106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosche A, Grosche J, Tackenberg M, Scheller D, Gerstner G, Gumprecht A, Pannicke T, Hirrlinger PG, Wilhelmsson U, Huttmann K, et al. Versatile and simple approach to determine astrocyte territories in mouse neocortex and hippocampus. PLoS One. 2013;8:e69143. doi: 10.1371/journal.pone.0069143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hama K, Arii T, Katayama E, Marton M, Ellisman MH. Tri-dimensional morphometric analysis of astrocytic processes with high voltage electron microscopy of thick Golgi preparations. Journal of neurocytology. 2004;33:277–285. doi: 10.1023/B:NEUR.0000044189.08240.a2. [DOI] [PubMed] [Google Scholar]
- Haydon PG, Carmignoto G. Astrocyte control of synaptic transmission and neurovascular coupling. Physiol Rev. 2006;86:1009–1031. doi: 10.1152/physrev.00049.2005. [DOI] [PubMed] [Google Scholar]
- Heese K, Otten U, Mathivet P, Raiteri M, Marescaux C, Bernasconi R. GABA(B) receptor antagonists elevate both mRNA and protein levels of the neurotrophins nerve growth factor (NGF) and brain-derived neurotrophic factor (BDNF) but not neurotrophin-3 (NT-3) in brain and spinal cord of rats. Neuropharmacology. 2000;39:449–462. doi: 10.1016/s0028-3908(99)00166-5. [DOI] [PubMed] [Google Scholar]
- Ishibashi T, Dakin KA, Stevens B, Lee PR, Kozlov SV, Stewart CL, Fields RD. Astrocytes promote myelination in response to electrical impulses. Neuron. 2006;49:823–832. doi: 10.1016/j.neuron.2006.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo S, Yarishkin O, Hwang YJ, Chun YE, Park M, Woo DH, Bae JY, Kim T, Lee J, Chun H, et al. GABA from reactive astrocytes impairs memory in mouse models of Alzheimer’s disease. Nat Med. 2014;20:886–896. doi: 10.1038/nm.3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kievit P, Howard JK, Badman MK, Balthasar N, Coppari R, Mori H, Lee CE, Elmquist JK, Yoshimura A, Flier JS. Enhanced leptin sensitivity and improved glucose homeostasis in mice lacking suppressor of cytokine signaling-3 in POMC-expressing cells. Cell Metab. 2006;4:123–132. doi: 10.1016/j.cmet.2006.06.010. [DOI] [PubMed] [Google Scholar]
- Kim ER, Wu Z, Sun H, Xu Y, Mangieri LR, Xu Y, Tong Q. Hypothalamic Non-AgRP, Non-POMC GABAergic Neurons Are Required for Postweaning Feeding and NPY Hyperphagia. J Neurosci. 2015a;35:10440–10450. doi: 10.1523/JNEUROSCI.1110-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JG, Suyama S, Koch M, Jin S, Argente-Arizon P, Argente J, Liu ZW, Zimmer MR, Jeong JK, Szigeti-Buck K, et al. Leptin signaling in astrocytes regulates hypothalamic neuronal circuits and feeding. Nat Neurosci. 2014;17:908–910. doi: 10.1038/nn.3725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KW, Donato J, Jr, Berglund ED, Choi YH, Kohno D, Elias CF, DePinho RA, Elmquist JK. FOXO1 in the ventromedial hypothalamus regulates energy balance. J Clin Invest. 2012;122:2578–2589. doi: 10.1172/JCI62848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MS, Yan J, Wu W, Zhang G, Zhang Y, Cai D. Rapid linkage of innate immunological signals to adaptive immunity by the brain-fat axis. Nat Immunol. 2015b;16:525–533. doi: 10.1038/ni.3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimelberg HK, Nedergaard M. Functions of astrocytes and their potential as therapeutic targets. Neurotherapeutics : the journal of the American Society for Experimental NeuroTherapeutics. 2010;7:338–353. doi: 10.1016/j.nurt.2010.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura T, Feng Y, Kitamura YI, Chua SC, Jr, Xu AW, Barsh GS, Rossetti L, Accili D. Forkhead protein FoxO1 mediates Agrp-dependent effects of leptin on food intake. Nat Med. 2006;12:534–540. doi: 10.1038/nm1392. [DOI] [PubMed] [Google Scholar]
- Kleinridders A, Lauritzen HP, Ussar S, Christensen JH, Mori MA, Bross P, Kahn CR. Leptin regulation of Hsp60 impacts hypothalamic insulin signaling. J Clin Invest. 2013;123:4667–4680. doi: 10.1172/JCI67615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch M, Varela L, Kim JG, Kim JD, Hernandez-Nuno F, Simonds SE, Castorena CM, Vianna CR, Elmquist JK, Morozov YM, et al. Hypothalamic POMC neurons promote cannabinoid-induced feeding. Nature. 2015;519:45–50. doi: 10.1038/nature14260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam TK, Pocai A, Gutierrez-Juarez R, Obici S, Bryan J, Guilar-Bryan L, Schwartz GJ, Rossetti L. Hypothalamic sensing of circulating fatty acids is required for glucose homeostasis. Nat Med. 2005;11:320–327. doi: 10.1038/nm1201. [DOI] [PubMed] [Google Scholar]
- Li J, Tang Y, Cai D. IKKbeta/NF-kappaB disrupts adult hypothalamic neural stem cells to mediate a neurodegenerative mechanism of dietary obesity and pre-diabetes. Nat Cell Biol. 2012;14:999–1012. doi: 10.1038/ncb2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin-Padilla M. Prenatal development of fibrous (white matter), protoplasmic (gray matter), and layer I astrocytes in the human cerebral cortex: a Golgi study. J Comp Neurol. 1995;357:554–572. doi: 10.1002/cne.903570407. [DOI] [PubMed] [Google Scholar]
- Marty N, Dallaporta M, Foretz M, Emery M, Tarussio D, Bady I, Binnert C, Beermann F, Thorens B. Regulation of glucagon secretion by glucose transporter type 2 (glut2) and astrocyte-dependent glucose sensors. J Clin Invest. 2005;115:3545–3553. doi: 10.1172/JCI26309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minelli A, DeBiasi S, Brecha NC, Zuccarello LV, Conti F. GAT-3, a high-affinity GABA plasma membrane transporter, is localized to astrocytic processes, and it is not confined to the vicinity of GABAergic synapses in the cerebral cortex. J Neurosci. 1996;16:6255–6264. doi: 10.1523/JNEUROSCI.16-19-06255.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molotkov D, Zobova S, Arcas JM, Khiroug L. Calcium-induced outgrowth of astrocytic peripheral processes requires actin binding by Profilin-1. Cell calcium. 2013;53:338–348. doi: 10.1016/j.ceca.2013.03.001. [DOI] [PubMed] [Google Scholar]
- Munzberg H, Myers MG., Jr Molecular and anatomical determinants of central leptin resistance. Nat Neurosci. 2005;8:566–570. doi: 10.1038/nn1454. [DOI] [PubMed] [Google Scholar]
- Muroyama Y, Fujiwara Y, Orkin SH, Rowitch DH. Specification of astrocytes by bHLH protein SCL in a restricted region of the neural tube. Nature. 2005;438:360–363. doi: 10.1038/nature04139. [DOI] [PubMed] [Google Scholar]
- Muthukumar AK, Stork T, Freeman MR. Activity-dependent regulation of astrocyte GAT levels during synaptogenesis. Nat Neurosci. 2014;17:1340–1350. doi: 10.1038/nn.3791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedergaard M, Ransom B, Goldman SA. New roles for astrocytes: redefining the functional architecture of the brain. Trends Neurosci. 2003;26:523–530. doi: 10.1016/j.tins.2003.08.008. [DOI] [PubMed] [Google Scholar]
- Ogata K, Kosaka T. Structural and quantitative analysis of astrocytes in the mouse hippocampus. Neuroscience. 2002;113:221–233. doi: 10.1016/s0306-4522(02)00041-6. [DOI] [PubMed] [Google Scholar]
- Otsu Y, Couchman K, Lyons DG, Collot M, Agarwal A, Mallet JM, Pfrieger FW, Bergles DE, Charpak S. Calcium dynamics in astrocyte processes during neurovascular coupling. Nat Neurosci. 2015;18:210–218. doi: 10.1038/nn.3906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perea G, Yang A, Boyden ES, Sur M. Optogenetic astrocyte activation modulates response selectivity of visual cortex neurons in vivo. Nat Commun. 2014;5:3262. doi: 10.1038/ncomms4262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purkayastha S, Zhang G, Cai D. Uncoupling the mechanisms of obesity and hypertension by targeting hypothalamic IKK-beta and NF-kappaB. Nat Med. 2011;17:883–887. doi: 10.1038/nm.2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangroo Thrane V, Thrane AS, Wang F, Cotrina ML, Smith NA, Chen M, Xu Q, Kang N, Fujita T, Nagelhus EA, et al. Ammonia triggers neuronal disinhibition and seizures by impairing astrocyte potassium buffering. Nat Med. 2013;19:1643–1648. doi: 10.1038/nm.3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranjan A, Mallick BN. Differential staining of glia and neurons by modified Golgi-Cox method. Journal of Neuroscience Methods. 2012;209:269–279. doi: 10.1016/j.jneumeth.2012.06.023. [DOI] [PubMed] [Google Scholar]
- Reichel JM, Bedenk BT, Gassen NC, Hafner K, Bura SA, Almeida-Correa S, Genewsky A, Dedic N, Giesert F, Agarwal A, et al. Beware of your Cre-ation: lacZ expression impairs neuronal integrity and hippocampus-dependent memory. Hippocampus. 2016 doi: 10.1002/hipo.22601. [DOI] [PubMed] [Google Scholar]
- Schachtrup C, Ryu JK, Mammadzada K, Khan AS, Carlton PM, Perez A, Christian F, Le Moan N, Vagena E, Baeza-Raja B, et al. Nuclear pore complex remodeling by p75(NTR) cleavage controls TGF-beta signaling and astrocyte functions. Nat Neurosci. 2015;18:1077–1080. doi: 10.1038/nn.4054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz MW, Woods SC, Porte D, Jr, Seeley RJ, Baskin DG. Central nervous system control of food intake. Nature. 2000;404:661–671. doi: 10.1038/35007534. [DOI] [PubMed] [Google Scholar]
- Shigetomi E, Tong X, Kwan KY, Corey DP, Khakh BS. TRPA1 channels regulate astrocyte resting calcium and inhibitory synapse efficacy through GAT-3. Nat Neurosci. 2012;15:70–80. doi: 10.1038/nn.3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AW, Bosch MA, Wagner EJ, Ronnekleiv OK, Kelly MJ. The membrane estrogen receptor ligand STX rapidly enhances GABAergic signaling in NPY/AgRP neurons: role in mediating the anorexigenic effects of 17beta-estradiol. American journal of physiology. Endocrinology and metabolism. 2013;305:E632–640. doi: 10.1152/ajpendo.00281.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan SM, Bjorkman ST, Miller SM, Colditz PB, Pow DV. Morphological changes in white matter astrocytes in response to hypoxia/ischemia in the neonatal pig. Brain research. 2010;1319:164–174. doi: 10.1016/j.brainres.2010.01.010. [DOI] [PubMed] [Google Scholar]
- Sykova E. Extrasynaptic volume transmission and diffusion parameters of the extracellular space. Neuroscience. 2004;129:861–876. doi: 10.1016/j.neuroscience.2004.06.077. [DOI] [PubMed] [Google Scholar]
- Thaler JP, Yi CX, Schur EA, Guyenet SJ, Hwang BH, Dietrich MO, Zhao X, Sarruf DA, Izgur V, Maravilla KR, et al. Obesity is associated with hypothalamic injury in rodents and humans. J Clin Invest. 2012;122:153–162. doi: 10.1172/JCI59660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theodosis DT, Poulain DA, Oliet SH. Activity-dependent structural and functional plasticity of astrocyte-neuron interactions. Physiol Rev. 2008;88:983–1008. doi: 10.1152/physrev.00036.2007. [DOI] [PubMed] [Google Scholar]
- Tong Q, Ye CP, Jones JE, Elmquist JK, Lowell BB. Synaptic release of GABA by AgRP neurons is required for normal regulation of energy balance. Nat Neurosci. 2008;11:998–1000. doi: 10.1038/nn.2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tschop MH, Castaneda TR, Woods SC. The brain is getting ready for dinner. Cell Metab. 2006;4:257–258. doi: 10.1016/j.cmet.2006.09.001. [DOI] [PubMed] [Google Scholar]
- Vong L, Ye C, Yang Z, Choi B, Chua S, Jr, Lowell BB. Leptin action on GABAergic neurons prevents obesity and reduces inhibitory tone to POMC neurons. Neuron. 2011;71:142–154. doi: 10.1016/j.neuron.2011.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witkin JW, Ferin M, Popilskis SJ, Silverman AJ. Effects of gonadal steroids on the ultrastructure of GnRH neurons in the rhesus monkey: synaptic input and glial apposition. Endocrinology. 1991;129:1083–1092. doi: 10.1210/endo-129-2-1083. [DOI] [PubMed] [Google Scholar]
- Wu Q, Boyle MP, Palmiter RD. Loss of GABAergic signaling by AgRP neurons to the parabrachial nucleus leads to starvation. Cell. 2009;137:1225–1234. doi: 10.1016/j.cell.2009.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Kim ER, Sun H, Xu Y, Mangieri LR, Li DP, Pan HL, Xu Y, Arenkiel BR, Tong Q. GABAergic projections from lateral hypothalamus to paraventricular hypothalamic nucleus promote feeding. J Neurosci. 2015;35:3312–3318. doi: 10.1523/JNEUROSCI.3720-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyss-Coray T, Loike JD, Brionne TC, Lu E, Anankov R, Yan F, Silverstein SC, Husemann J. Adult mouse astrocytes degrade amyloid-beta in vitro and in situ. Nat Med. 2003;9:453–457. doi: 10.1038/nm838. [DOI] [PubMed] [Google Scholar]
- Yan J, Zhang H, Yin Y, Li J, Tang Y, Purkayastha S, Li L, Cai D. Obesity-and aging-induced excess of central transforming growth factor-beta potentiates diabetic development via an RNA stress response. Nat Med. 2014;20:1001–1008. doi: 10.1038/nm.3616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeltser LM, Seeley RJ, Tschop MH. Synaptic plasticity in neuronal circuits regulating energy balance. Nat Neurosci. 2012;15:1336–1342. doi: 10.1038/nn.3219. [DOI] [PubMed] [Google Scholar]
- Zhang X, Zhang G, Zhang H, Karin M, Bai H, Cai D. Hypothalamic IKKbeta/NF-kappaB and ER stress link overnutrition to energy imbalance and obesity. Cell. 2008;135:61–73. doi: 10.1016/j.cell.2008.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu G, Yan J, Zhang Y, Li B, Cai D. Metabolic learning and memory formation by the brain influence systemic metabolic homeostasis. Nat Commun. 2015;6:6704. doi: 10.1038/ncomms7704. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.