

Abstract
Neurons are terminally differentiated cells with a high rate of metabolism and multiple biological properties distinct from their undifferentiated precursors. Previous studies showed that nucleotide excision DNA repair is down-regulated in post-mitotic muscle cells and neurons. Here, we characterize DNA damage susceptibility and base excision DNA repair (BER) capacity in undifferentiated and differentiated human neural cells. The results show that undifferentiated human SH-SY5Y neuroblastoma cells are less sensitive to oxidative damage than their differentiated counterparts, in part due to having robust BER capacity, which is heavily attenuated in the post-mitotic neurons. The reduction in BER activity in the differentiated cells correlates with diminished protein levels of key long patch BER components FEN-1, PCNA and Ligase I. Thus, due to their higher BER capacity, proliferative neural progenitor cells are more efficient at repairing DNA damage compared to their neuronally differentiated progeny.
Keywords: BER, differentiation, DNA repair, post-mitotic
1. Introduction
Neurons are highly differentiated non-dividing cells that exhibit electrical excitability, a high metabolic rate and abundant transcriptional activity. The high metabolic activity in these cells generates relatively large amounts of reactive oxygen species (ROS) that can damage lipids, proteins and DNA. Unrepaired ROS-induced oxidative DNA damage in the nuclear and mitochondrial DNA (mtDNA) of neurons can lead to cell death and contribute to aging and neurodegeneration.
Base excision DNA repair (BER) is the major pathway that repairs oxidative lesions, alkylated bases, single-strand breaks (SSBs) and abasic sites in nuclear and mtDNA [8, 19]. BER involves the sequential action of multiple enzymes, typically starting with the recognition and removal of an inappropriate base by a DNA glycosylase [15]. Glycosylases excise the damaged base forming an abasic site, which is primarily processed by an apurinic/apyrimidinic endonuclease (APE1). APE1 incises the sugar phosphate backbone at the abasic site generating a one-nucleotide SSB. In short patch BER (SP-BER), the gap is filled by DNA polymerase β (Pol β) and, following 5’-terminal processing by Polβ, the nick is ligated by DNA ligase III . Other 3’ and 5’ terminal blocking groups are also resolved prior to ligation by BER-related factors such as aprataxin, polynucleotide kinase phosphatase (PNKP) and tyrosyl DNA phosphodiesterase 1 (TDP1) [9, 28, 31, 36]. Long patch BER (LP-BER), a sub-pathway of BER that engages several replication-associated proteins, involves strand-displacement synthesis and 5’-flap processing, often necessary to cope with complex 5’-termini. In this process, Pol β or DNA polymerase delta/epsilon (Pol δ/ε), flap endonuclease-1 (FEN-1), proliferating cell nuclear antigen (PCNA) and DNA ligase I coordinately resynthesize 2 -10 nucleotides, displace the downstream strand, cleave away the 5’-flap that harbors the blocking group, and seal the remaining nick [9].
Previous findings suggest that terminally differentiated muscle cells express very low levels of DNA ligase I and ligase III/ XRCC1 [25],leading to an impairment of both short and long patch BER in the myotubes . Hildrestrand et al. suggested that 8-oxoguanine glycoslyase (OGG1) protein levels were lower in fully differentiated neurospheres than in undifferentiated neurospheres [12], whereas Linn et al. reported that APE1 activity, but not Poḷ and uracil DNA glycosylase (UDG) activities, were higher in differentiated neuroblastoma cells [13]. Additional studies suggest that expression of DNA repair proteins may vary in both a species- and tissue-specific manner [3, 11].
In the present study, we investigated BER capacity in undifferentiated and neuronally differentiated human SH-SY5Y neuroblastoma cells. DNA damage and cell viability were examined in differentiated and undifferentiated cells treated with hydrogen peroxide or methyl methane sulfonate (MMS), and the protein levels and enzyme activities of several BER factors were quantified. We report here that differentiated cells are more susceptible to DNA damage induced by oxidative stress, but not alkylating agents. This heightened sensitivity correlates with a dramatic decrease in BER components, potentially leading to the accumulation of a subset of oxidative DNA lesions in differentiated cells.
2. Materials and Methods
2.1 Cell culture and differentiation
The human neuroblastoma cell line SH-SY5Y (obtained from ATCC) was maintained in regular DMEM medium (Invitrogen) with 10% fetal bovine serum (FBS) and 1% Penicillin-Streptomycin in 5% CO2 at 37°C. Mitotic SH-SY5Y cells are able to differentiate into post-mitotic neuronal-like cells when cultured in medium with reduced serum levels and treatment with retinoic acid [17]. In the present study the post-mitotic state was achieved by treating cells with 10 μM retinoic acid for 5 days followed by reseeding of the cells to polyethyleneimine (PEI)-coated Petri dishes in serum-free DMEM containing 10 ng/ml brain derived neurotrophic factor (BDNF, Chemicon) for another 5 days.
2.2 Immunocytochemistry
The cells were fixed in 4% paraformaldehyde (in phosphate buffer saline (PBS)) and incubated with lysis buffer (0.5% Triton X-100, 100 mM glycine, 1% BSA, 0.7 mM EDTA) for 10 min on ice. After lysis the cells were incubated with blocking buffer (10% donkey serum, 0.01% sodium azide in PBS). The blocked cells were incubated with primary antibodies, Tuj1 (1:250, Sigma-Aldrich) or MAP2 (1:500, Chemicon.), for 60 minutes at 37 °C. The samples were then briefly rinsed with wash buffer (0.05% Triton-X 100 in PBS) three times for 5 minutes at room temperature before incubation with conjugated secondary antibody (Alexa 488, 1: 1000, donkey anti-mouse, Invitrogen) for 60 minutes at 37 °C. After the incubation the cells were washed with PBS (3 × 5 minutes) and mounted with Prolong Gold containing 4`,6`- diamidino-2-phenylindole (DAPI, nuclear marker, Invitrogen). Images of immunostained cells were captured using a Nikon Eclipse TE2000 confocal microscope system running Volocity software (Perkin Elmer).
2.3 Measurement of cell viability
Cells (10,000/well in a 96-well plate coated with PEI) were treated in triplicate with MMS (0.1 - 4 mM) for 60 minutes or hydrogen peroxide (0.01-1 mM) for 40 minutes in standard growth medium for replicating SH-SY5Y cells or in serum-free/BDNF medium for differentiated cells. Following treatment, cells were refreshed with 200μl of respective medium and incubated at 37°C for 24 hours. Viability was quantified using the lactate dehydrogenase (LDH) cytotoxicity detection kit (Roche Applied Science, Indianapolis, Indiana, USA) by incubating 100 μl media from each well with 100 μl reaction mixture for 15 minutes at room temperature and reading on a Bio-Rad microplate spectrophotometer. Cytotoxicity was proportional to the absorbance (OD) at 492 nm (reference wavelength 690 nm) as a measure of LDH activity released from damaged cells into the media and was calculated against that measured for untreated controls. Maximal cellular LDH release was determined by treating cells with 1% Triton X-100 for 60 minutes; OD values above this level were excluded.
The WST-8 assay was also used to assess cytotoxicity. Cells (20,000/well in a 96-well plate coated with PEI) were treated in triplicate with MMS (0.02 - 2 mM) or hydrogen peroxide (0.01-1 mM) for 4 hours at 37°C in 100 μl standard growth medium for replicating SH-SY5Y cells or serum-free/BDNF medium for differentiated cells. Viability was quantified using the WST-8 cytotoxicity assay kit (Dojindo Molecular Technologies, Rockville, Maryland, USA) by incubating wells in 10 μl CCK-8 solution for 2 hours at 37°C and reading on a Bio-Rad microplate spectrophotometer. Cell viability was directly proportional to the absorbance (OD) at 450 nm as a measure of dehydrogenase activity in viable cells and was calculated against that measured for untreated controls.
2.4 Alkali single-cell gel electrophoresis (comet assay)
Alkali single-cell gel electrophoresis was performed as described previously [22]. Cells were washed with PBS before medium containing 50 μM H2O2 was added and cultures were incubated for 40 minutes at 37°C or treated with 100 μM MMS for 60 minutes at 37°C. After the chemical treatment, cells were washed once with fresh DMEM. Cells were prepared at 0 hours, with and without treatment, 6 hours and 24 hours after treatment. Briefly, cells were rinsed in PBS, and approximately 10,000 cells were mixed with 75 μl of low melt point agarose (LMPA) and spread onto an agarose-coated glass slide. After the LMPA had set, the coverslip was removed, another 75 μl of low melt point agarose was added and the slide was placed on a chilled aluminum tray for 5 minutes. The slide was incubated in lysis buffer (2.5 M NaCl, 100 mM EDTA, 10 mM Tris, and 1% Triton X-100) for at least 4 hours then washed with neutralization buffer (0.4 M Tris, pH 7.4), followed by a 10 minute incubation with REC buffer (10 mM HEPES-KOH, 100 mM KCl, 10 mM EDTA, 0.1 mg/ml BSA; pH 7.4). For enzymatic treatments, slides were incubated with formamidopyrimidine DNA glycosylase (Fpg, New England Biolabs) or mouse 3-methyladenine DNA glycosylase (Aag, Trevigen) at 37°C for 60 minutes. Fpg treatment was used on samples exposed to hydrogen peroxide and related controls, and Aag treatment on MMS-exposed sets and related controls. The enzyme-treated slides were rinsed with alkali buffer (300 mM NaOH and 1 mM EDTA; pH 12.1) for 20 minutes to denature DNA. Electrophoresis was performed and the slides dehydrated in 100% ethanol for 5 minutes before staining with ethidium bromide (10 ng/ml). Images of nuclei were acquired under fluorescence illumination using a Zeiss Axiovert microscope, and images were analyzed using Komet 5.5 software (Kinetic Imagine). Statistical analysis was carried out using Graph Pad Prism software, statistical significance was assessed by comparing corresponding undifferentiated and differentiated data sets using student’s t-test.
2.5 BER activities
Cells were washed with PBS, centrifuged, and the cell pellets were used immediately for extraction or stored at -80°C for future use. Total protein was extracted by incubating the cell pellet in RIPA buffer with protease inhibitors for 30 minutes at 4°C , the lysate was then briefly sonicated on ice (3x 10s, 10% power) using a Branson sonicator then spun down at 10,000g at 4°C for 30 minutes to remove cellular debris and DNA. Protein levels were quantified using the bicinchoninic acid method (BCA) using bovine serum albmin as a known concentration standard. Extracts were used fresh or aliquoted and stored at -80°C. All oligonucleotide DNA substrates used to measure various BER activities have been used extensively by our group and others in the field and are as listed in Table 1. The SP-BER activity assay was used to measure both polymerase incorporation and total ligation activity. The assay was initially performed using varying concentrations of protein extract. This preliminary step was conducted to identify the amount of protein that gave the least substrate degradation but still had detectable activity (not shown). The quantitative assay was then run using increasing incubation times, with only time points determined to be in the linear phase of the experiment subsequently used for further quantitative analysis. Experiments to assess the rate limiting step included the enzyme T4 ligase (Roche) and Pol β was also used as a positive control. More detailed methods regarding the activity assays are described in previous studies [34, 35].
Table 1.
Combinations of oligonucleotides used to test the various BER activities.
| Activity | Substrate |
|---|---|
|
| |
| 8-oxo-G incision | 5′-GAA CTG T(O)A GAC TGA T, |
| 3′- GCT GAC A C T GAA CTG ACG ACT A; | |
|
| |
| Uracil incision | 5′-ATA GG(U) CGG ATC AAG ATT, |
| 3′-TAT CC (G or A) GCC GAA TAA | |
|
| |
| AP incision | 5′-GAA CTG T (F) A GAC TGA T, |
| 3′- GCT GAC A C T GAA CTG ACG ACT A | |
|
| |
| Incorporation | 5′-CTG CTG ( )GT ACG GAT CGG C, |
| 3′-GAC GTC GAC GCA GCC G. | |
|
| |
| Nick | 5`-TCC GTT GAA GCC TGC TTT_GAC ATA CTA ACT TGA GCG AAA CGG-T- 3` |
| (Ligation) | 3`- T- ACG CAA CTT CGG ACG AAA CTG TAT GAT TGA ACT CGC TTT GCC-5` |
|
| |
| BER | 5`-TCC GTT GAA GCC TGC TTT_()AC ATA CTA ACT TGA GCG AAA CGG-T- 3` |
| (Incorporation and Ligation) | 3`- T- ACG CAA CTT CGG ACG AAA CTG TAT GAT TGA ACT CGC TTT GCC-5` |
|
| |
| LP-BER | 5`-GTC CAC CCG ACG CCA CCT CCT GCC TTC AAT GTG CTG ( ) GAT CCT ACA ACC AAG ACG AAT TCC GGA TAC GAC GGC CAG TGC CGA CCG TGC CAG CCT AAA TTT CAA TCC ACC C- 3` |
| 3`- CAG GTG GGC TGC GGT GGA GGA CGG AAG TTA CAC GAC C CTA GGA TGT TGG TTC TGC TTA AGG CCT ATG CTG CCG GTC ACG GCT GGC ACG GTC GGA TTT AAA GTT AGG TGG G- 5` | |
2.6 Immunoblot analysis
Proteins were separated by SDS-polyacrylamide gel electrophoresis (SDS-PAGE), and transferred to a (PVDF) membrane. After blocking in 5% non-fat milk protein in TBST (Tris buffer saline with 0.01% Tween-20), the membrane was exposed to one of the following primary antibodies; XRCC1 (1:500, Abcam), UDG (1: 200, Santa Cruz), OGG1 (1: 200, Santa Cruz), DNA ligase III (LIGIII) (1:500, BD Bioscience Technology), APE1 (1: 1000, Imgenex), Pol β (1: 500, Abcam), PCNA (1: 2000, Santa Cruz), DNA ligase I (LIGI) (1: 500, Santa Cruz), FEN-1 (1: 250, Abcam), (Pol ε) ( 1: 250, Abcam), GAPDH (1:1000, Abcam) and HSP60 (1:500, Abcam). β-actin was used as a loading and transfer control (1: 5000, Sigma-Aldrich). Secondary antibodies utilized were as follows; anti-mouse (1: 5000) (Vector Laboratories), anti-goat (1: 10000) (Vector Laboratories), and anti-rabbit (1: 5000) (Perkin Elmer).
2.7 LP-BER assay
Long patch BER was measured using a substrate specific for only this repair activity. The substrate utilized a 110mer backbone that has a biotin group on both ends, (gift from Dr. Bambara, University of Rochester, NY). Annealed to the backbone is an unlabelled 36mer and a 73mer that has a 5’- tetrahydrofuran blocking group and a 3’ -P32 end label. Before use, the annealed substrate was incubated with streptavidin for 30 minutes at 4°C; this molecule forms a strong non-covalent bond with the biotin blocking both ends of the substrate. When the substrate is in this blocked conformation, PCNA must be loaded onto the substrate and cannot simply slide on or off [14]. LP-BER activity was measured in total protein extracts (described above) from differentiated and undifferentiated cells.
The repair reactions were assembled in a 20 μl volume. Depending on the assay conducted, either the protein concentration or incubation time was variedFor protein concentration dependent reactions, between 0.5- 2 μg of total protein extract was added to a reaction containing 50 fmol end-labeled substrate, 10 mM ATP, 10 μM dNTPs, 20 ng DNA (λ), in reaction buffer (50 mM Tris-HCL, pH 8, 10 mM KCl, 10 mM MgCl2, 1 mM dithiothreitol, and 1% glycerol) as per [33]. The reaction was carried out at 37°C for 30 minutes, terminated with stopping buffer (30 mM Tris-HCL pH 8, 30 mM EDTA pH 8, 30% glycerol, 0.9% SDS, 0.05% bromophenol blue) and heating at 95°C for 5 minutes. Reaction products were separated in a 15% urea-acrylamide gel, at 15W for 120 minutes. The gel products were quantitated using a GE Typhoon phosphoimager. The above reaction was also carried out using varied incubation times between 15 minutes and 90 minutes under identical conditions described above. As previously described for the SPBER assay, this approach allowed us to first identify an amount of protein extract that gave clean reproducible results, and then using this amount and varying incubation time we could accurately determine the rate of repair ensuring that the assay was always in the linear phase when we did quantitative analysis.
3. Results
3.1 Differentiating mitotic SH-SY5Y cells into neuron-like post-mitotic cells
For this research, it was critical to distinguish between neuroblasts that are simply non-proliferating and differentiated neurons that are post-mitotic. Human SH-SY5Y neuroblastoma cells can be induced to differentiate into post-mitotic neurons by manipulating culture conditions [17]. When grown in DMEM with 10% FBS, SH-SY5Y cells are morphologically similar to fibroblasts (Figure 1A, left panels). After incubation for 5 days in medium containing 10 μM retinoic acid, the cells begin to form neurites, although some cells continued to replicate. Subsequent use of medium containing BDNF resulted in at least 95% of the cells being post-mitotic and exhibiting outgrowth of long neurites as seen by standard phase contrast microscopy (Figure 1A, right panels).
Figure 1. Morphological change of SH-SY5Y cells differentiated from mitotic into post-mitotic (neuron-like) cells.
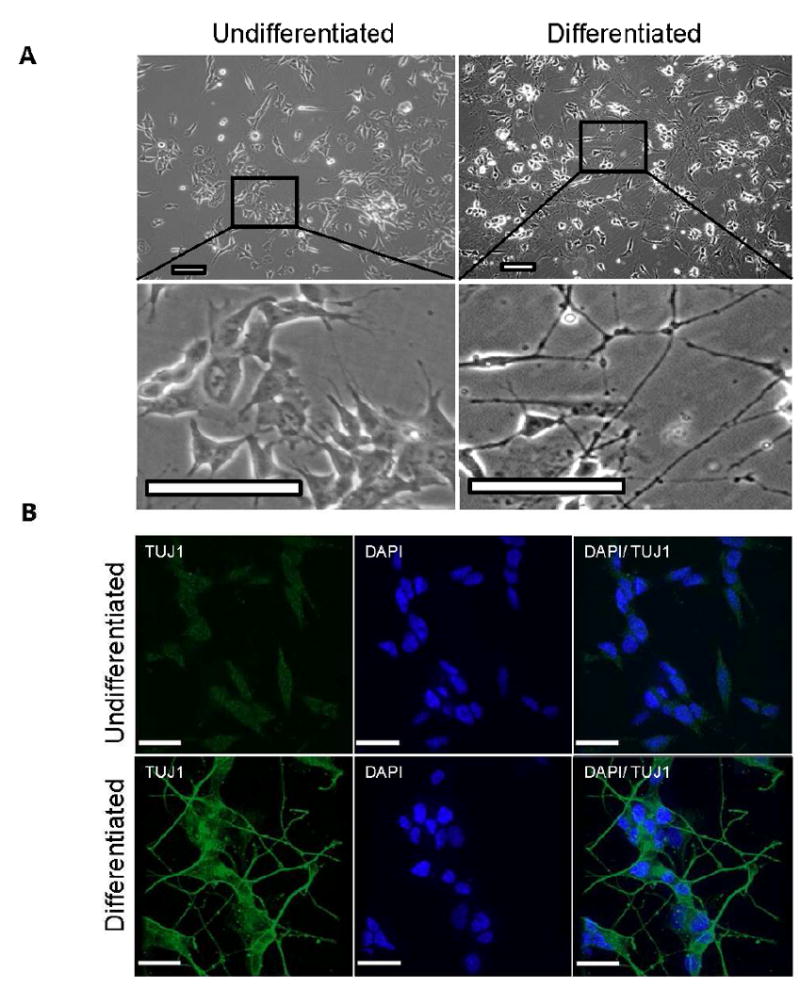
A. (Panels left) Neuroblastoma cells before differentiation. Cells are treated with 10 μM retinoic acid for 5 days; at this time point some cells have extended short neurites but many cells are still dividing. (Panel right) Cells after a further 5 days of culture in serum-free medium containing 10 ng/ml brainderived growth factor (BDNF); at this time point the majority of cells are post-mitotic and exhibit long neurite formation B. The neuron-specific class III beta-tubulin (Tuj1) marker was present in the outgrowth axons of neurons after retinoic acid and BDNF treatment. In comparison untreated SH-SY5Y cells only showed light staining of Tuj1. (Scale bar = 25 μM)
To confirm that the morphologically differentiated cells have neuronal characteristics, we tested for expression of neuron-specific markers, including neuron-specific class III beta-tubulin (Tuj1), by standard immunocytochemistry techniques. Tuj1 stabilizes microtubules in neuronal cell bodies and axons, and plays a role in axonal transport. The fully differentiated SH-SY5Y cells had Tuj1 immuno reactive axons, whereas this antibody labeling was weak or absent in undifferentiated cells (Figure 1B). The Tuj1 image shows points of bright staining along the axons which we propose may be areas of protein aggregation. We also tested for the presence of an additional neuronal marker, microtubule associated protein 2 (MAP2). MAP2 is believed to protect and stabilize newly formed microtubules and is a pertinent factor in neuronal development. Seen in Supplementary Figure 1, only the post mitotic cells show any degree of MAP2 immunoreactivity. Using these well-established neuronal markers, we thus verified that the protocol used here with human SH-SY5Y neuroblastoma cells resulted in differentiation into post-mitotic neuron-like cells.
3.2 Neurons can adequately repair damage caused by an alkylating agent
Previous research investigating the toxic effect of alkylating agents in the central nervous system (CNS) has been largely restricted to murine and non-isogenic models. To establish whether the differentiation status of human neuroblastoma cells could influence sensitivity to DNA damaging agents, cells were exposed to the alkylating chemical MMS. As graphed in Figure 2A and B, we tested the vulnerability of undifferentiated and differentiated SH-SY5Y cells to a range of MMS concentrations. The cytotoxicity results presented in Figure 2A show that after four hours of exposure to concentrations of MMS above 1 mM, there is a modest difference in survival between the two groups, significant at 1mM and 1.5mM but not at 2 mM. Similar survival trends were also observed using an alternate treatment regime involving a 1 hour exposure at the indicated concentrations of MMS, followed by 24 hours of recovery (Figure 2B).
Figure 2. Methyl Methane Sulfonate (MMS) induced DNA damage is repaired efficiently in both differentiated and undifferentiated cells.
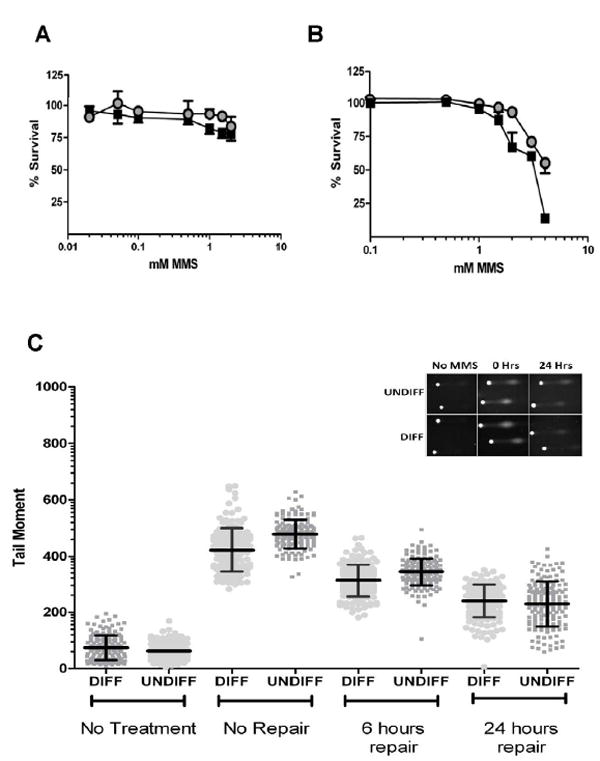
A. Cell survival after extended MMS exposure (4 hours). Cells were exposed to the indicated concentrations of MMS with survival measured using the WST-8 assay. B. Cellular survival after 1 hour MMS exposure and a 24 hour repair period using a LDH based assay. ■ denotes undifferentiated cells, ● denotes differentiated cells. All survival assays repeated in triplicate. C. Quantitative analysis of the tail moment after exposure to MMS. After 24 hours repair both groups had comparable amounts of remaining DNA damage. Middle line in scatter plot represents the mean ± SD. Comet repeated in duplicate with consistent results. Graph inserts of representative comets images.
We next used the alkali single-cell gel electrophoresis (comet) assay to determine the rate of DNA repair after MMS exposure. The assay here employed treatment with the DNA glycosylase Aag, which excises alkylated base modifications creating alkaline labile AP sites. Using this assay, the number of Aag-induced and also endogenous SSBs can be estimated from the length of the comet “tail” or moment (Figure 2C, insert and Supplementary Figure 2. Initially, we determined the amount of endogenous damage present in both sets of cells (Figure 2C, far left). Differentiated and undifferentiated cells without treatment had only low levels of endogenous DNA damage. (Figure 2C; Table 2).
Table 2.
Numerical analysis of comet moment.
| No Treatment | No Repair | 6 Hours repair | 24 hours repair | |||||
|---|---|---|---|---|---|---|---|---|
| DIFF | UNDIFF | DIFF | UNDIFF | DIFF | UNDIFF | DIFF | UNDIFF | |
| MMS | ||||||||
| Mean | 74 | 63 | 422 | 479 | 313 | 344 | 240 | 229 |
| Std. Dev. | 44 | 28 | 78 | 51 | 57 | 49 | 58 | 80 |
| Std. Error | 3 | 2 | 6 | 4 | 5 | 4 | 5 | 6 |
| Number of values (n) | 161 | 159 | 146 | 147 | 144 | 169 | 151 | 158 |
| H2O2 | ||||||||
| Mean | 69 | 55 | 703 | 752 | 487 | 563 | 346 | 270 |
| Std. Dev. | 48 | 49 | 76 | 92 | 124 | 116 | 135 | 131 |
| Std. Error | 5 | 5 | 10 | 12 | 11 | 12 | 12 | 14 |
| Number of values (n) | 110 | 102 | 56 | 61 | 125 | 90 | 126 | 90 |
To compare the rate of repair, both groups of cells were treated with 100 μM MMS for 1 hour, with comet tail length measured immediately post-treatment and after periods of 6 or 24 hours of repair; all data points gathered are graphed in Figure 2C and are summarized in Table 2. Immediately after MMS treatment, based on the results obtained from comparing the comet tails of 100 or more cells from both groups, undifferentiated cells (479±51, arbitrary units) had accumulated a statistically higher (p <0.01) amount of damage compared to differentiated cells (422±78); we note that this analysis does not account for repair that may have taken place during the short period of exposure to MMS. DNA damage remained higher in undifferentiated cells after 6 hours of repair (344±49 undifferentiated versus 313±57 differentiated, p<0.01), consistent with both groups repairing MMS-induced damage at a similar rate (28% of damage repaired in undifferentiated versus 26% in differentiated). Despite this, after 24 hours of repair the undifferentiated cells had repaired 52% of damage compared to only 44% for the differentiated group, resulting in both groups having comparable amounts of remaining damage (p=0.16). Thus, the undifferentiated cells were more susceptible to the formation of MMS-induced lesions, yet appeared to repair this damage slightly more efficiently, resulting in similar amounts of remaining damage after 24 hours.
3.3 Neurons do not repair hydrogen peroxide-induced DNA damage efficiently
The above comet and survival results support the notion that alkylating DNA damage is repaired effectively in both cycling neuroblasts and post mitotic neuronal cells. To determine if neurons are particularly susceptible to oxidative DNA damage, we treated undifferentiated and differentiated cells with hydrogen peroxide at concentrations that cause oxidative DNA damage, including SSBs. Cellular survival after hydrogen peroxide exposure was tested as described earlier for MMS. After 4 hours of exposure, the differentiated cells were significantly more sensitive to hydrogen peroxide at concentrations above 100 μM, as compared to undifferentiated cells (Figure 3A). Further, after a 40 minute exposure and 24 hours of repair, low concentrations of hydrogen peroxide (50μM) resulted in a noticeable difference in cellular survival, with the differentiated cells being significantly more sensitive to hydrogen peroxide damage than undifferentiated cells (Figure 3B).
Figure 3. Neuronal cells are more sensitive to oxidative DNA damage.
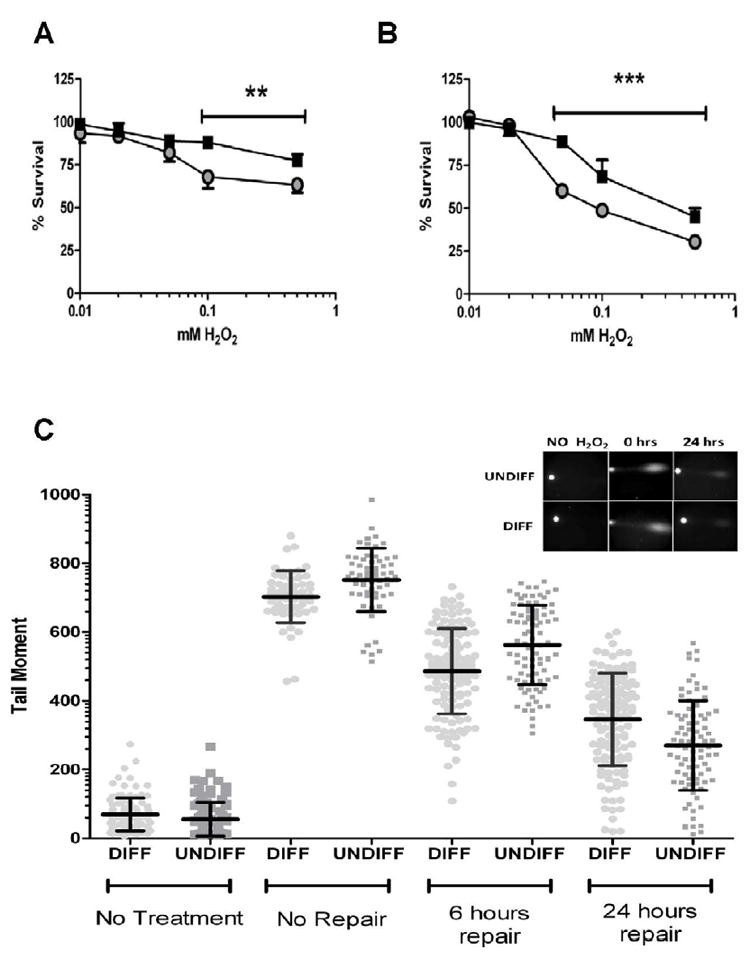
A. Cellular survival after extended (4 hours) exposure to H2O2 treatment. Survival was measured by WST-8 assay. B. Cellular survival 24 hours after acute (40 minute) hydrogen peroxide treatment measured using an LDH based assay. All survival assays have been repeated in triplicate. ■ denotes undifferentiated cells, ● denotes differentiated cells. (**p<0.05, *** p<0.01) C. Analysis of the comet moment determined that undifferentiated cells had more DNA damage compared to differentiated cells after exposure to H2O2. However, 24 hours after exposure to H2O2 the undifferentiated cells had repaired more of the damage than differentiated cells. Middle line in scatter plot represents the mean ± SD. Comet experiments have been performed in duplicate. Graph inserts of representative comets images.
We used the comet assay to determine whether the increased sensitivity of the differentiated cells was due to reduced DNA repair capacity. For this assay, we used a low concentration of hydrogen peroxide, which caused significantly more cell death after 24 hours in the differentiated cells (50 μM). This time, the DNA glycoslyase fpg was used to digest sites of oxidative damage and generate SSBs before comet analysis. Levels of endogenous DNA damage in both groups were found to be comparably low (Figure 3C, far left; Table 2).
The DNA damage immediately after treatment caused by hydrogen peroxide was significantly higher (p<0.01) in undifferentiated cells (752±92 versus 703±76 in differentiated cells). After 6 hours of repair, the undifferentiated group had repaired ~25% of the initial damage, whereas the differentiated group had repaired 31%, resulting in the undifferentiated group still having significantly more remaining DNA damage (p<0.01) (Table 2). In the 6 to 24 hour time frame, however, the undifferentiated cells repaired twice the amount of DNA damage as the differentiated cells. Hence, despite the undifferentiated cells accumulating more damage initially and having comparable levels of repair after 6 hours, these cells had a significantly lower (p<0.01) amount of remaining DNA damage after 24 hours of repair. These data are consistent with the toxicity results showing that differentiated cells are more vulnerable to sustained DNA damage induced by 50 μM of hydrogen peroxide (Figure 3B).
3.4 Modulation of SP-BER components in neuronally differentiated cells
SP-BER is regarded as the primary DNA repair mechanism for most forms of alkylative, oxidative, and SSB damage. Differences in BER capacity could reflect different levels of expression or differential activation of BER enzymes in undifferentiated and differentiated SH-SY5Y cells. This possibility was tested by performing immunoblot and enzyme activity assays on extracts from undifferentiated and differentiated cells. We consulted previous research using the SHSY-5Y cells from Castano and colleagues, [7] to carefully select the western loading controls used in this study. We reproduced and then expand on the previously published results and report that it is the retinoic acid treatments that affected the loading controls in the aforementioned study not necessarily the process of differentiation (Supplementary Figure 3). Here we find that both β-Actin and GAPDH loading controls gave comparable, reproducible results using extracts from undifferentiated and differentiated cells. We initially investigated BER activity and protein levels (Figure 4A). There was no significant difference in levels of the DNA glycoslyases OGG1 and UDG. However, core BER components APE1 (p<0.05) and LIGIII (p<0.05) were significantly decreased in differentiated neurons. XRCC1 protein was undetectable in the differentiated extracts Previous preliminary studies using microarray analysis had indicated no significant change in XRCC1 transcript levels following differentiation of SY5Y neuroblastoma cells (unpublished observation of [17]); however in this study, our studies recapitulate the findings of Narciso and colleagues [25], where XRCC1 protein levels are significantly reduced upon differentiation. To more accurately determine XRCC1 transcript levels real time RT-PCR was conducted using RNA extracted from differentiated and undifferentiated cells. In Supplementary Figure 4 we show that there is a significant down regulation of XRCC1 gene expression, however this did not correlate completely with the dramatic reduction in XRCC1 protein levels. These changes in BER enzyme levels between undifferentiated and differentiated cells caused a reduction in the SPBER repair activity in the extracts from differentiated cells, measured using a substrate requiring both incorporation and ligation activity (p<0.05) ( Figure 4B). The reduced SP-BER activity appeared independent of Pol β protein levels, however, did correlate with the reduction in XRCC1 protein levels. Further, we used the same substrate with the addition of T4 ligase to determine if there was an accumulation of ligatable intermediates indicating that ligase activity was potentially rate limiting (Supplementary Figure 5). Indeed in both differentiated and undifferentiated extracts the addition of T4 ligase resulted in an increase of DNA repair product. However, the amount of increase was far greater in the differentiated extracts suggesting that that these cells may be particularly deficient in ligation activity (Supplementary Figure 5B). We expanded our investigation and measured individual enzymatic activities to more completely determine which activities associated with BER may be modulated by neural differentiation. In Figure 4C, we show that total 8-oxoguanine incision activity (mainly a function of OGG1) is significantly decreased in the differentiated cells. UDG activity on a U:A substrate is moderately increased, whereas the comparable activity on a U:G substrate is slightly decreased (Figure 4C). We also investigated the effect of differentiation on the three core enzymatic activities associated with SP-BER: AP site incision, singlenucleotide incorporation, and nick ligation (Figure 4D). The decrease in total AP endonuclease activity in differentiated cells correlated with the reduction in APE1 protein levels. A reproducible decrease in incorporation activity was also observed in the differentiated group, a result that was surprising in light of the slightly increased Pol β protein level. Ligation activity in differentiated cells was reduced by approximately 90% (Figure 4D), despite LIGIII protein levels decreasing by less than 25% (Figure 4A).
Figure 4. Changes in SP-BER components and activity levels in differentiated cells.
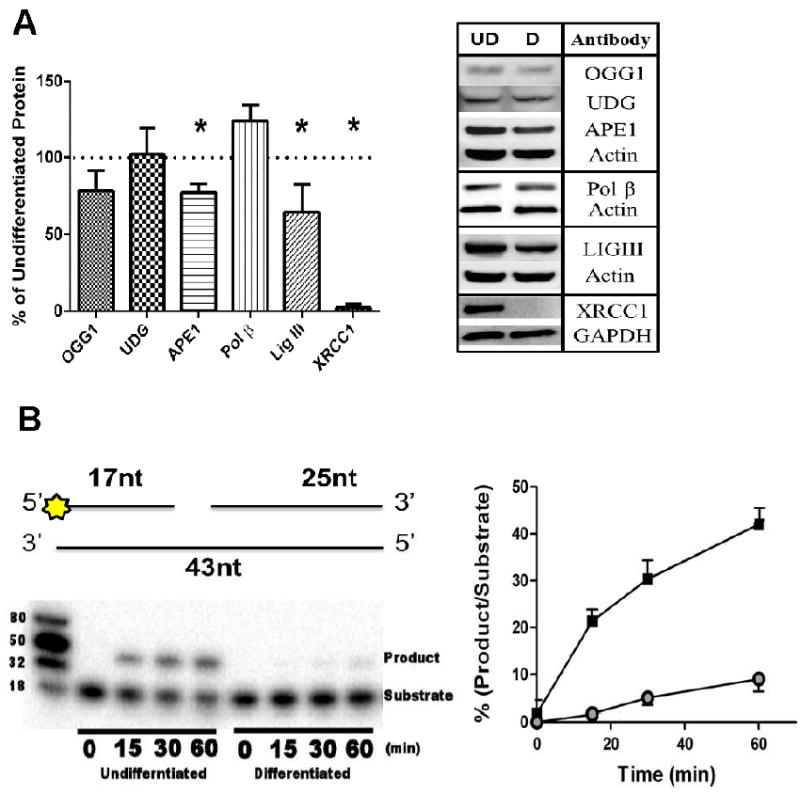
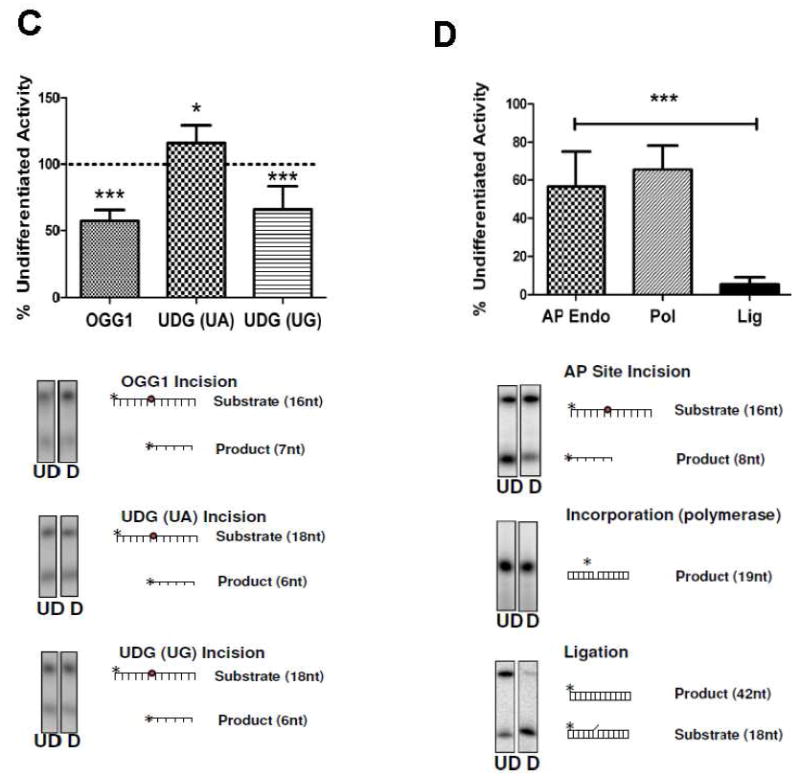
(A) (Right panel) Representative images from western analysis (UD= undifferentiated, D= differentiated). β actin was used as a loading control (*, p<0.05, n=4), Protein levels ofAPE1 and LIGIII were moderately yet significantly decreased after differentiation with only XRCC1 heavily decreased. (Left panel) (B) (Right panel) SP- BER assay (top) Diagrammatic representation of oligonucleotide combination used in the assay. Star denotes 5’ radiolabeling site (refer to materials and methods for additional information).(bottom) Representative gel image from the SP-BER assay shows that there is robust SP-BER activity in the undifferentiated extracts in contrast differentiated extracts had greatly reduced activity (Left panel). Undifferentiated cells have significantly higher levels of repair activity (p<0.01) measured using a BER substrate incubated with 2 μg cell extract (■ denotes undifferentiated cells, ● denotes differentiated cells). Assay conducted in triplicate. Data is mean±SD. (C) Activities of 8- oxoguanine glycosylase 1 (OGG1), uracil DNA glycosylase (UDG) (U:G) were significantly decreased after cellular differentiation (***, p<0.01). (D) Core BER activities are decreased in differentiated cells. Incision and incorporation activity was moderately decreased. In contrast, ligation activity is heavily reduced in neurons. (***, p<0.01, conducted at least in triplicate). In all panels the mean is plotted ±SD.
3.5 Evidence that long patch BER activity is reduced in differentiated neurons
While the results of the ligation assay did not reflect the LIGIII protein levels the reduction in activity may be a result of reduced XRCC1 protein levels. Alternatively LIGI (which is primarily involved in LP-BER) would also be active on the nick substrate and may also influence the reduction in activity. Using immunoblot analysis, we determined that the LIGI protein level was substantially decreased (~90%) in differentiated neurons (Figure 5A). Considering that many other LP-BER proteins are involved in DNA replication, we investigated whether the non- replicating status of the differentiated cells would modulate LP-BER protein levels. Indeed, FEN-1, Pol ε, and PCNA were all significantly reduced in post mitotic neurons compared to proliferating undifferentiated cells (Figure 5A).
Figure 5. Protein levels of LP-BER associated proteins are heavily reduced in neurons resulting in a decrease in LP-BER capacity.
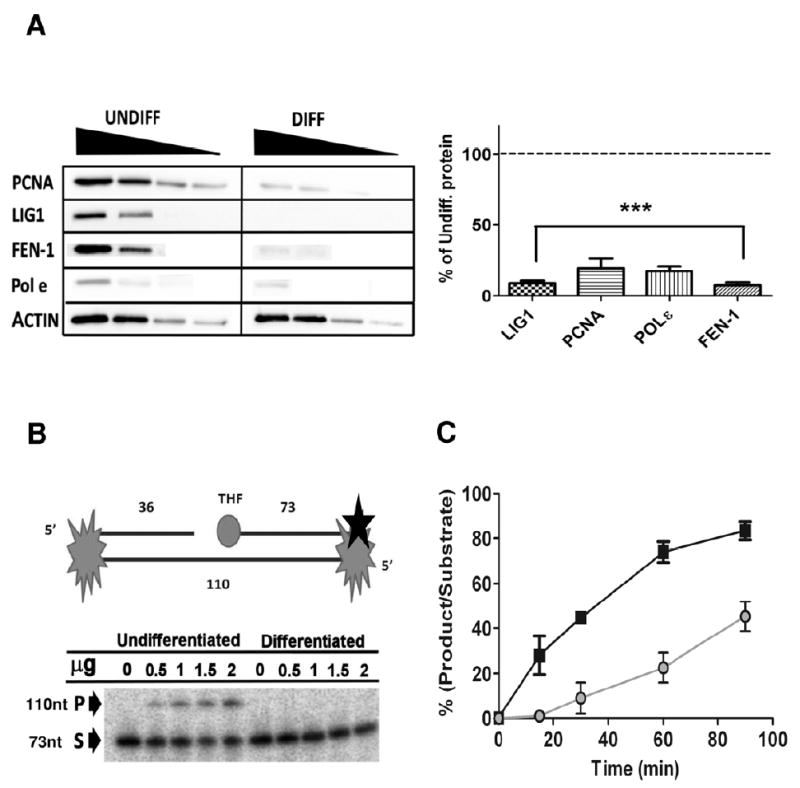
(A) (Left panel) Representative western blot using 80, 40, 20 and 10 μg of total protein extracts. (Right panel) All LPBER proteins investigated were significantly reduced after differentiation. (***, p<0.01, n=3) (B) (Upper) Diagrammatic representation of LP-BER substrate (refer to materials and methods for additional information) Black star denotes 3’ radiolabeling site. (Lower panel) Representative gel of LP-BER substrate using increasing total protein concentrations (0-2 μg) shows no LP-BER activity in differentiated extracts after 15 minutes incubation (P= product, S= substrate). (C) Time course for LP-BER assay demonstrates that LP-BER activity in undifferentiated total cell extracts is significantly (p<0.01, n=3) higher at all-time points investigated (■ denotes differentiated cells, ● denotes undifferentiated cells). Data is mean±SD.
Next, we measured what effect the dramatic decrease in LP-BER components had on LP-BER activity. The LP-BER substrate we used contains a 5’ THF group refractory to the deoxyribophosphodiesterase (dRpase) activity of Pol β and requires processing by LP-BER. The previously reported weak retention of PCNA on linear substrates was circumvented by blocking both ends with a biotin- strepavidin conjugate, requiring PCNA to be loaded on and off the substrate (Figure 5B, top). Since preliminary experiments showed that a high level of substrate degradation was present in the extracts from undifferentiated cells, we used lower protein concentrations to compare activity between the two groups. As seen as Figure 5B, after 30 minutes of incubation at a range of protein concentrations, LP-BER activity was only detected in extracts from the undifferentiated cells, consistent with the low protein levels seen for the integral LP-BER components in the differentiated cells (Figure 5A). As shown in Figure 5C, we also measured LP-BER activity over an extended period of 90 minutes, and our results indicate that LP-BER is approximately four times higher in undifferentiated cells compared to differentiated cells.
4. Discussion
The goal of this study was to evaluate how neuronal cell differentiation affects BER efficiency and the response to DNA damage. Using SH-SY5Y neuroblastoma cells as a model, we observed that replicating cells treated with hydrogen peroxide or MMS show a higher capacity to repair DNA damage and also, by and large, a higher level of expression of BER proteins and activities. One of the central assertions of this study is that BER components are modulated according to the differentiation status of the neuron. We make this assertion based on the results of eight distinct quantitative biochemical assays, measuring a total of twelve different DNA repair. In each case the assay produced highly reproducible results which, when coupled with extensively western analysis on the proteins involved in both SP and LP-BER allowed us to confidently determine that after neuronal differentiation SP-BER repair is reduced due to a decrease in XRCC1 but also Ligase I. Despite this reduction, LP-BER does not compensate, decreasing dramatically due to a reduction in LP-BER components associated with replication. We find that despite the post mitotic cells not being more sensitive to alkylating damage, they are prone to cellular toxicity induced by oxidative stress, apparently because of an impaired ability to repair the associated DNA damage.
One possible explanation for the observed increased vulnerability of genomic DNA in undifferentiated cells is that actively replicating chromatin is especially susceptible to DNA damage. Consistent with this hypothesis, previous studies from our group demonstrated that Chinese hamster ovary cells were hyper-sensitive to UV-irradiation during S-phase [26, 27]. Thus, chromatin of undifferentiated cells, which is maintained in an open conformation to facilitate active DNA replication and transcription, is more available for modification by exogenous DNA damaging agents [6]. Notably, Bill et al. reported results that are consistent with the present study, including that undifferentiated mesenchymal stem cells have a higher DNA repair rate in actively transcribed genes and a higher DNA repair efficiency than terminally differentiated cells [5]. Moreover, two recent studies reported that the speed of DNA repair depends on the relaxed or compacted state of chromatin [2, 23]. Of note, the work presented here focusses primarily on repair in the nucleus, however, many of the enzymes involved in nuclear DNA repair are also associated with mitochondrial DNA repair (reviewed ([30])). It remains to be tested whether the repair of mitochondrial damage is also decreased after differentiation and how this impacts cellular survival after genotoxic stress.
Our data indicate that ligation activity is particularly affected in post-mitotic neurons and suggest that the reduced ligation is a direct effect of a pronounced decrease in LIGI levels. A decrease in LIGI protein levels has also been reported in non-neural human cell models, including terminally differentiated muscle cells [25] and non-proliferating keratinocytes [15], suggesting that the result may be indicative of a non-proliferating cell regardless of differentiation status. Importantly, in all these reports, including our own, LIGI protein is not completely absent, but heavily reduced. This corroborates early reports [20] describing that ligase I mRNA was detected at low levels in a number of non-proliferating cell types, including mature rat neurons, further supporting the involvement of this enzyme in DNA repair in differentiated cells.
The functional redundancy of ligases in DNA repair is of particular interest in neurons considering the low levels of LIGI. In recent work by Gao et al., LIGIII was shown to be dispensable in the nucleus, but not in the mitochondria, in cells from the murine nervous system [10]. This exchangeability is also supported by work conducted by Sleeth et al. [29], who demonstrated the ability of LIGI to participate in SP- and LPBER, while LIGIII participated exclusively in SP-BER. Here, we show that human neuronal cells are not particularly sensitive to MMS. Our data suggest that alkylating damage is largely unaffected by the reduced LIGI protein levels. This notion is strengthened by data from Bentley et al. [4] showing that the ethyl methane sulfonate (EMS) survival curve for ligase I null mouse fibroblasts was indistinguishable from the wild type mouse control. Despite previous reports that LIGI participates in SP-BER, it does not appear to be required for the repair of alkylating damage in neuronal cells.
An important contribution of this work is that it clearly shows that neurons are particularly susceptible to agents that induce oxidative DNA damage. This may be in part attributed to reduced levels of OGG1-related activity, because neurons from OGG1 deficient mice exhibit increased vulnerability to oxidative stress [18]. However, this heightened sensitivity is unlikely to be caused by a breakdown in any of the core SP-BER components (APE1, Pol β, LIGIII), considering these cells efficiently repair alkylating damage. Instead, it may be related to the spectrum of DNA damage produced by oxidizing agents.
Oxidative damage can result in direct DNA damage, such as 8-oxo-G lesions, but can also generate significant amounts of SSBs. While SSBs with simple 3’-hydroxyl and 5’-phosphate ends are substrates for SP-BER, many 5’ modifications that are refractory to Pol β dRP lyase activity require LP-BER. We show that neuronal differentiation is coupled with a decrease in LP-BER proteins FEN-1, PCNA, Pol ε and LIGI, resulting in a severely reduced BER capacity. In support of our data, Wei and Englander [33] recently looked at differences in BER processing in mitotic and proliferating tissue and found that LP-BER capacity for a 5’ blocking group was nearly six fold lower in brain samples compared to proliferating intestinal mucosa. While comparing DNA repair capacity across different tissue types should be approached with caution, these results are similar to some in this study and suggest that repair of 5’ blocking groups in the brain is particularly poor. Furthermore, the severe progressive cerebellar ataxia associated with ataxia with ocular motor apraxia 1 (AOA1) is evidence that reduced repair of 5’ blocking groups is particularly deleterious in the brain. AOA1 is caused by mutations in aprataxin (APTX), a protein responsible for resolving 5’-AMP ligation intermediates. The predominant neural specificity of the disorder may stem from a lack of back up DNA repair mechanisms available to replicating cells. Here we report that LP-BER, a potential back up repair mechanism for the 5’-AMP in the absence of aprataxin, is severely reduced in neurons and this may contribute to the neural specificity of AOA1.
The brain requires large amounts of oxygen to sustain high levels of metabolic activity in neuronal and associated cells. This activity invariably produces ROS with the potential to cause oxidative damage. Normally, however, BER can be up-regulated in response to excitation of neurons, as was recently shown in cultured rat cerebral cortical neurons where activation of glutamate receptors induced the expression of APE1 and enhanced BER [37]. Many neurodegenerative disorders are associated with an increase in oxidative stress and impaired DNA repair. Loss of BER capacity may contribute to the accumulation of DNA damage reported to occur in vulnerable populations of neurons in several neurodegenerative disorders, including Alzheimer disease [21, 35], Parkinson disease [24, 38], Huntington disease [32] and amyotrophic lateral sclerosis [1]. Interestingly, brain cells of Alzheimer disease patients show significant BER dysfunction and reduced expression of UDG and OGG1 activities [35]. We propose that loss of BER capacity in terminally differentiated neurons could cause persistent DNA damage to accumulate over time. This higher level of DNA damage might be more readily tolerated in a non-replicating cell than in a replicating cell, in which persistent DNA damage activates cell cycle checkpoints, such as those mediated by ATM/ATR and P53, and triggers cell death [16]. Without this cell cycle signaling in differentiated neurons, DNA damage can accumulate and impair function, a process observed in normal aging and this may be more severe in the dysfunctional BER background of neurodegenerative diseases.
Supplementary Material
Acknowledgments
We thank Dr. Robert Bambara for reagents provided. We also thank Dr. Misiak and Dr. Illuzzi for critical reading of the manuscript. This research is supported by intramural research program of National Institute on Aging, National Institutes of Health.
Footnotes
Disclosure statement: The authors report no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Aguirre N, Beal MF, Matson WR, Bogdanov MB. Increased oxidative damage to DNA in an animal model of amyotrophic lateral sclerosis. Free Radic Res. 2005;39(4):383–8. doi: 10.1080/10715760400027979. [DOI] [PubMed] [Google Scholar]
- 2.Amouroux R, Campalans A, Epe B, Radicella JP. Oxidative stress triggers the preferential assembly of base excision repair complexes on open chromatin regions. Nucleic Acids Res. 2010;38(9):2878–90. doi: 10.1093/nar/gkp1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aprelikova ON, Tomilin NV. Activity of uracil-DNA glycosylase in different rat tissues and in regenerating rat liver. FEBS Lett. 1982;137(2):193–5. doi: 10.1016/0014-5793(82)80347-5. [DOI] [PubMed] [Google Scholar]
- 4.Bentley DJ, Harrison C, Ketchen AM, Redhead NJ, Samuel K, Waterfall M, Ansell JD, Melton DW. DNA ligase I null mouse cells show normal DNA repair activity but altered DNA replication and reduced genome stability. J Cell Sci. 2002;115(Pt 7):1551–61. doi: 10.1242/jcs.115.7.1551. [DOI] [PubMed] [Google Scholar]
- 5.Bill CA, Grochan BM, Meyn RE, Bohr VA, Tofilon PJ. Loss of intragenomic DNA repair heterogeneity with cellular differentiation. J Biol Chem. 1991;266(32):21821–6. [PubMed] [Google Scholar]
- 6.Bubley GJ, Xu J, Kupiec N, Sanders D, Foss F, O’Brien M, Emi Y, Teicher BA, Patierno SR. Effect of DNA conformation on cisplatin adduct formation. Biochem Pharmacol. 1996;51(5):717–21. doi: 10.1016/s0006-2952(95)02256-2. [DOI] [PubMed] [Google Scholar]
- 7.Castano Z, Kypt R. Housekeeping Proteins: Limitations as references during neuronal differntiation. The Open Neuroscience Journal. 2008;2:36–40. [Google Scholar]
- 8.Englander EW. Brain capacity for repair of oxidatively damaged DNA and preservation of neuronal function. Mech Ageing Dev. 2008;129(7-8):475–82. doi: 10.1016/j.mad.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Frosina G, Fortini P, Rossi O, Carrozzino F, Raspaglio G, Cox LS, Lane DP, Abbondandolo A, Dogliotti E. Two pathways for base excision repair in mammalian cells. J Biol Chem. 1996;271(16):9573–8. doi: 10.1074/jbc.271.16.9573. [DOI] [PubMed] [Google Scholar]
- 10.Gao Y, Katyal S, Lee Y, Zhao J, Rehg JE, Russell HR, McKinnon PJ. DNA ligase III is critical for mtDNA integrity but not Xrcc1-mediated nuclear DNA repair. Nature. 2011;471(7337):240–4. doi: 10.1038/nature09773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haug T, Skorpen F, Aas PA, Malm V, Skjelbred C, Krokan HE. Regulation of expression of nuclear and mitochondrial forms of human uracil-DNA glycosylase. Nucleic Acids Res. 1998;26(6):1449–57. doi: 10.1093/nar/26.6.1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hildrestrand GA, Diep DB, Kunke D, Bolstad N, Bjoras M, Krauss S, Luna L. The capacity to remove 8-oxoG is enhanced in newborn neural stem/progenitor cells and decreases in juvenile mice and upon cell differentiation. DNA Repair (Amst) 2007;6(6):723–32. doi: 10.1016/j.dnarep.2006.12.008. [DOI] [PubMed] [Google Scholar]
- 13.Jensen L, Linn S. A reduced rate of bulky DNA adduct removal is coincident with differentiation of human neuroblastoma cells induced by nerve growth factor. Mol Cell Biol. 1988;8(9):3964–8. doi: 10.1128/mcb.8.9.3964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jonsson ZO, Hindges R, Hubscher U. Regulation of DNA replication and repair proteins through interaction with the front side of proliferating cell nuclear antigen. EMBO J. 1998;17(8):2412–25. doi: 10.1093/emboj/17.8.2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Krokan HE, Standal R, Slupphaug G. DNA glycosylases in the base excision repair of DNA. Biochem J. 1997;325(Pt 1):1–16. doi: 10.1042/bj3250001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kruman II, Wersto RP, Cardozo-Pelaez F, Smilenov L, Chan SL, Chrest FJ, Emokpae R, Jr, Gorospe M, Mattson MP. Cell cycle activation linked to neuronal cell death initiated by DNA damage. Neuron. 2004;41(4):549–61. doi: 10.1016/s0896-6273(04)00017-0. [DOI] [PubMed] [Google Scholar]
- 17.Kulkarni A, McNeill DR, Gleichmann M, Mattson MP, Wilson DM., III XRCC1 protects against the lethality of induced oxidative DNA damage in nondividing neural cells. Nucleic Acids Res. 2008;36(15):5111–21. doi: 10.1093/nar/gkn480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu D, Croteau DL, Souza-Pinto N, Pitta M, Tian J, Wu C, Jiang H, Mustafa K, Keijzers G, Bohr VA, Mattson MP. Evidence that OGG1 glycosylase protects neurons against oxidative DNA damage and cell death under ischemic conditions. J Cereb Blood Flow Metab. 2011;31(2):680–92. doi: 10.1038/jcbfm.2010.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martin LJ. DNA damage and repair: relevance to mechanisms of neurodegeneration. J Neuropathol Exp Neurol. 2008;67(5):377–87. doi: 10.1097/NEN.0b013e31816ff780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Montecucco A, Biamonti G, Savini E, Focher F, Spadari S, Ciarrocchi G. DNA ligase I gene expression during differentiation and cell proliferation. Nucleic Acids Res. 1992;20(23):6209–14. doi: 10.1093/nar/20.23.6209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moreira PI, Nunomura A, Nakamura M, Takeda A, Shenk JC, Aliev G, Smith MA, Perry G. Nucleic acid oxidation in Alzheimer disease. Free Radic Biol Med. 2008;44(8):1493–505. doi: 10.1016/j.freeradbiomed.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 22.Morris EJ, Dreixler JC, Cheng KY, Wilson PM, Gin RM, Geller HM. Optimization of single-cell gel electrophoresis (SCGE) for quantitative analysis of neuronal DNA damage. Biotechniques. 1999;26(2):282–9. doi: 10.2144/99262st02. [DOI] [PubMed] [Google Scholar]
- 23.Mosesso P, Palitti F, Pepe G, Pinero J, Bellacima R, Ahnstrom G, Natarajan AT. Relationship between chromatin structure, DNA damage and repair following Xirradiation of human lymphocytes. Mutat Res. 2010 doi: 10.1016/j.mrgentox.2010.03.005. [DOI] [PubMed] [Google Scholar]
- 24.Nakabeppu Y, Tsuchimoto D, Yamaguchi H, Sakumi K. Oxidative damage in nucleic acids and Parkinson’s disease. J Neurosci Res. 2007;85(5):919–34. doi: 10.1002/jnr.21191. [DOI] [PubMed] [Google Scholar]
- 25.Narciso L, Fortini P, Pajalunga D, Franchitto A, Liu P, Degan P, Frechet M, Demple B, Crescenzi M, Dogliotti E. Terminally differentiated muscle cells are defective in base excision DNA repair and hypersensitive to oxygen injury. Proc Natl Acad Sci U S A. 2007;104(43):17010–5. doi: 10.1073/pnas.0701743104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Orren DK, Petersen LN, Bohr VA. Persistent DNA damage inhibits S-phase and G2 progression, and results in apoptosis. Mol Biol Cell. 1997;8(6):1129–42. doi: 10.1091/mbc.8.6.1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Petersen LN, Orren DK, Bohr VA. Gene-specific and strand-specific DNA repair in the G1 and G2 phases of the cell cycle. Mol Cell Biol. 1995;15(7):3731–7. doi: 10.1128/mcb.15.7.3731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Raymond AC, Staker BL, Burgin AB., Jr Substrate specificity of tyrosyl-DNA phosphodiesterase I (Tdp1) J Biol Chem. 2005;280(23):22029–35. doi: 10.1074/jbc.M502148200. [DOI] [PubMed] [Google Scholar]
- 29.Sleeth KM, Robson RL, Dianov GL. Exchangeability of mammalian DNA ligases between base excision repair pathways. Biochemistry. 2004;43(40):12924–30. doi: 10.1021/bi0492612. [DOI] [PubMed] [Google Scholar]
- 30.Sykora P, Wilson DM, III, Bohr VA. Repair of persistent strand breaks in the mitochondrial genome. Mech Ageing Dev. 2012;133(4):169–75. doi: 10.1016/j.mad.2011.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Takahashi T, Tada M, Igarashi S, Koyama A, Date H, Yokoseki A, Shiga A, Yoshida Y, Tsuji S, Nishizawa M, Onodera O. Aprataxin, causative gene product for EAOH/AOA1, repairs DNA single-strand breaks with damaged 3’-phosphate and 3’-phosphoglycolate ends. Nucleic Acids Res. 2007;35(11):3797–809. doi: 10.1093/nar/gkm158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Trushina E, McMurray CT. Oxidative stress and mitochondrial dysfunction in neurodegenerative diseases. Neuroscience. 2007;145(4):1233–48. doi: 10.1016/j.neuroscience.2006.10.056. [DOI] [PubMed] [Google Scholar]
- 33.Wei W, Englander EW. DNA polymerase beta-catalyzed-PCNA independent long patch base excision repair synthesis: a mechanism for repair of oxidatively damaged DNA ends in post-mitotic brain. J Neurochem. 2008;107(3):734–44. doi: 10.1111/j.1471-4159.2008.05644.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weissman L, de Souza-Pinto NC, Mattson MP, Bohr VA. DNA base excision repair activities in mouse models of Alzheimer’s disease. Neurobiol Aging. 2009;30(12):2080–1. doi: 10.1016/j.neurobiolaging.2008.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weissman L, Jo DG, Sorensen MM, de Souza-Pinto NC, Markesbery WR, Mattson MP, Bohr VA. Defective DNA base excision repair in brain from individuals with Alzheimer’s disease and amnestic mild cognitive impairment. Nucleic Acids Res. 2007;35(16):5545–55. doi: 10.1093/nar/gkm605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Whitehouse CJ, Taylor RM, Thistlethwaite A, Zhang H, Karimi-Busheri F, Lasko DD, Weinfeld M, Caldecott KW. XRCC1 stimulates human polynucleotide kinase activity at damaged DNA termini and accelerates DNA single-strand break repair. Cell. 2001;104(1):107–17. doi: 10.1016/s0092-8674(01)00195-7. [DOI] [PubMed] [Google Scholar]
- 37.Yang JL, Tadokoro T, Keijzers G, Mattson MP, Bohr VA. Neurons efficiently repair glutamate-induced oxidative DNA damage by a process involving CREBmediated up-regulation of apurinic endonuclease 1. J Biol Chem. 2010;285(36):28191–9. doi: 10.1074/jbc.M109.082883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang JL, Weissman L, Bohr VA, Mattson MP. Mitochondrial DNA damage and repair in neurodegenerative disorders. DNA Repair (Amst) 2008;7(7):1110–20. doi: 10.1016/j.dnarep.2008.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.