

SUMMARY
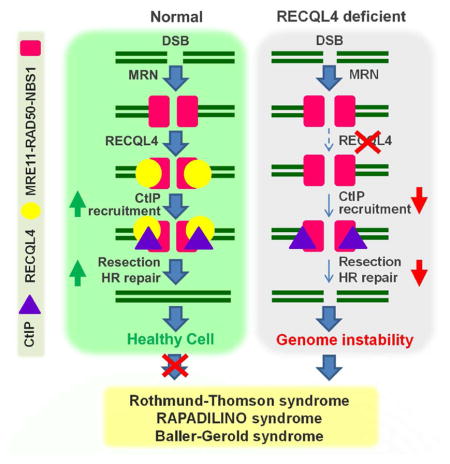
The RecQ helicase RECQL4, mutated in Rothmund-Thomson syndrome, regulates genome stability, aging and cancer. Here, we identify a crucial role for RECQL4 in DNA end resection, which is the initial and an essential step of homologous recombination (HR)-dependent DNA double-strand break repair (DSBR). Depletion of RECQL4 severely reduces HR-mediated repair and 5’ end resection in vivo. RECQL4 physically interacts with MRE11-RAD50-NBS1 (MRN), which senses DSBs and initiates DNA end resection with CtIP. The MRE11 exonuclease regulates the retention of RECQL4 at laser-induced DSBs. RECQL4 also directly interacts with CtIP via its N-terminal domain and promotes CtIP recruitment to the MRN complex at DSBs. Moreover, inactivation of RECQL4’s helicase activity impairs DNA end processing and HR-dependent DSBR without affecting its interaction with MRE11 and CtIP, suggesting an important role for RECQL4’s unwinding activity in the process. Thus, we report here that RECQL4 is an important participant in HR-dependent DSBR.
Keywords: RECQL4, Rothmund-Thomson syndrome, DNA resection, homologous recombination, DNA repair, RecQ-like helicase
ETOC BLURB
RECQL4, a RecQ helicase mutated in Rothmund-Thomson syndrome, is a guardian of genome stability and repairs DNA, but the underlying mechanisms remain unclear. Lu et al show that RECQL4 plays a role in homologous recombination repair of DNA double-strand breaks (DSBs). RECQL4 promotes 5’ DNA end resection through the MRE11-RAD50-NBS1 and CtIP complexes.
INTRODUCTION
DNA double-strand breaks (DSBs) are generated by exogenous stress, endogenous replication and programmed recombination events. Improperly repaired DSBs can lead to genome instability, chromosomal rearrangements and/or cell death (Symington, 2014). DSBs are usually repaired by one of two major pathways: homologous recombination (HR) and non-homologous end joining (NHEJ) (Aparicio et al., 2014).
HR-dependent DSBR is mostly error-free, but it requires a sister or non-sister chromatid as template, and is only active during the S and G2 phases of the cell cycle. In contrast, NHEJ-dependent DSBR is error-prone, DNA template-independent and active during all phases of the cell cycle. HR-dependent DSBR is initiated by 5' end resection of the DSBs, which generates 3’ protruding single-strand DNA (ssDNA) tails (Chen et al., 2013; Zhu et al., 2008). RPA coats the ssDNA, and then RAD51 replaces RPA to promote strand invasion. This is followed by repair synthesis, dissolution and resolution of Holliday junctions and ligation of the ends (Prakash et al., 2015). It is generally considered that DNA end resection occurs in two steps (Cejka et al., 2010; Gravel et al., 2008; Mimitou and Symington, 2008; Nimonkar et al., 2011; Niu et al., 2010; Zhu et al., 2008). The first step is the initial resection by Mre11-Rad50-Xrs2 (MRX) and Sae2 at the DSB in yeast (Cannavo and Cejka, 2014; Mimitou and Symington, 2008) or by MRE11-RAD50-NBS1 (MRN) and CtIP (CtBP-interacting protein) in human cells (Sartori et al., 2007; You et al., 2009). This is followed by extensive resection by either exonuclease1 (EXO1) or DNA2/BLM/TOP3/RMI1/2 (Dna2/Sgs1/Top3/Rmi1 in yeast) (Cejka et al., 2010; Gravel et al., 2008; Mimitou and Symington, 2008; Nimonkar et al., 2011; Nimonkar et al., 2008; Niu et al., 2010; Zhu et al., 2008). RECQL4 is one of five RecQ helicase proteins in mammalian cells. Defects in human RECQL4 are associated with three genetic diseases: Rothmund-Thomson Syndrome (RTS), RAPADILINO and Baller-Gerold Syndrome (Siitonen et al., 2009) as well as several cancers (Fang et al., 2013; Lu et al., 2014b; Su et al., 2010). It is well established that RECQL4 is required for the assembly of the DNA replication initiation machinery (Im et al., 2009; Sangrithi et al., 2005; Xu et al., 2009). However, the role of RECQL4 in DNA repair is less clear (Croteau et al., 2014). Lack of RECQL4 increases persistent DNA damage and triggers cellular senescence in human and mouse primary fibroblasts (Lu et al., 2014a).
RECQL4 is recruited to laser-induced DSBs and RTS fibroblasts are sensitive to ionizing radiation (IR), suggesting that RECQL4 plays a role in DSBR (Singh et al., 2010). Recently, we showed that depletion of RECQL4 inhibits NHEJ in U2OS cells (Shamanna et al., 2014). Nevertheless, RECQL4 is highly expressed during S phase (Singh et al., 2012; Xu et al., 2009), when HR-dependent DSBR dominates. Thus, here we explore the possibility that RECQL4 also plays a role in HR-dependent DSBR. We find that RECQL4 promotes DNA end resection and HR-dependent DSBR by stimulating the association of CtIP with MRN at DSBs and that the helicase activity of RECQL4 is necessary for DNA end resection. Together, these findings suggest that RECQL4 plays an important role in the DNA end resection step of HR-mediated DSBR in human cells.
RESULTS
RECQL4 promotes DNA end resection during HR-dependent DSBR
Endogenous RECQL4 co-localized with γH2AX at laser-induced DSBs in U2OS cells (Fig.1A) and depletion of RECQL4 caused U2OS and HeLa cells to be significantly more sensitive to IR (Fig. 1B and Fig. S1A). Since DSB repair pathway choice is cell cycle regulated (Aparicio et al., 2014), we first examined the effect of RECQL4 depletion on cell cycle progression. Knockdown of RECQL4 did not perturb cell cycle progression significantly in U2OS or HEK293T cells and did not alter expression of cell cycle marker proteins Cyclin A and Cyclin D1 (Fig. S2), which is consistent with a previous finding in HEK293 cells (Park et al., 2006).
Figure 1.

RECQL4 is required for HR-mediated repair and DNA end resection. A. Co-localization of endogenous RECQL4 and γH2AX at laser-induced DSB tracks. Bar, 10μm. B. Clonogenic survival of siRQ4-transfected U2OS cells treated with γ radiation. C. Representative dot plot images of DR-GFP U2OS cells showing in vivo HR. D. Quantification of HR repair. E. RPA foci in U2OS cells treated with control or RECQL4 siRNA 1 hour after 10 Gy IR. Bar, 10 μm. F. IR-induced RPA32 phosphorylation on serine 4 and 8. U2OS cells expressing control or RECQL4 shRNA were exposed to 10 Gy of IR then allowed to recover for the indicated time. UT, untreated. G. Quantification of ssDNA generated by 5’ end resection at two AsiSI-induced DSBs in AID-DIvA U2OS cells. All graphs show mean± SEM from at least three biological repeats. P values (*, P<0.05; **, P<0.01) was determined by Student’s t-test. See also Figures S1 and S2.
The role of RECQL4 in HR-dependent DSBR was then investigated in DR-GFP U2OS cells, which can be scored for efficiency of HR-mediated repair of an I-SceI endonuclease-induced DSB by measuring the fraction of GFP-positive cells (Pierce et al., 1999; Wang et al., 2014). DR-GFP U2OS cells were transfected with one of three RECQL4-targeted siRNAs. The efficiency of RECQL4 knockdown was about 90%, 60% and 50% for siRQ4, siRQ4-2 and siRQ4-3, respectively (Fig. 1D). Since siRQ4 produced the greatest knockdown it was used in all subsequent experiments. Depletion of RECQL4 by siRQ4 significantly reduced the proportion of GFP-positive cells by 73%, from 5.5% in control cells to 1.47% in knockdown cells (Fig.1C, D), suggesting that RECQL4 plays a crucial role in HR-dependent DSBR. The other two siRNAs, siRQ4-2 and siRQ4-3, also significantly reduced the proportions of GFP-positive cells to 3.71% and 4.0%, respectively (Fig.1C, D). These data show that knockdown efficiency of RECQL4 correlates with a decrease of HR-mediated DSBR.
RECQL4 is rapidly recruited to laser-induced DSBs where it is retained for a short time (Singh et al., 2010). Thus, we speculated that it plays a role in the early stages of HR repair. As mentioned above, the first step of HR-mediated DSBR is 5’-3’ end resection of the DSBs to generate 3’ protruding ssDNA tails, which are rapidly coated by RPA to form a nuclease-resistant protective protein-DNA filament (Chen et al., 2013). We then examined RPA foci formation in RECQL4-depleted cells. After exposure to 10 Gy of IR, the fraction of cells with > 20 RPA foci was ~33.6% in the siRQ4-treated U2OS cells, significantly less than 75.8% in the control cells (Fig.1E). Depletion of RECQL4 also repressed RPA foci formation after IR in HeLa cells (Fig. S1B). Consistent with these results, the abundance of phosphorylated RPA32 on serine 4 and serine 8, a maker of ssDNA-bound RPA (Shao et al., 1999), was increased in IR-treated control U2OS and HEK293T cells, but not in RECQL4-knockdown cells (Fig. 1F and Fig. S1C).
AID-DIvA U2OS cells have been used to directly quantify ssDNA generated by 5’ end resection at two AsiSI-induced DSBs (Aymard et al., 2014). With addition of 4-hydroxytamoxifen (4-OHT), the AsiSI endonuclease fused to an estrogen receptor ligand binding domain translocates from the cytoplasm to the nucleus to induce DSBs (Aymard et al., 2014). Genomic DNA from these cells was prepared and analyzed for ssDNA at two DSBs by TaqMan qPCR, as previously described (Aymard et al., 2014; Zhou et al., 2014). Consistent with a previous report (Zhou et al., 2014), we observed notably lower ssDNA in cells treated with CtIP siRNA because CtIP stimulates DNA end resection (Sartori et al., 2007). On the contrary, depletion of 53BP1 increased the amount of ssDNA (Fig.1G), since 53BP1 inhibits resection (Bunting et al., 2010). To investigate the role of RECQL4 in 5’ end resection, ssDNA content was measured in siRQ4-transfected AID-DIvA U2OS cells. Interestingly, the amount of ssDNA generated at position 335nt from DSB1 was 40.2% lower than that in siCtrl-treated cells, and similar to the reduction caused by CtIP depletion. At DSB2, depletion of RECQL4 reduced ssDNA content by 57.2%, 70.1% and 75.2% at positions 364 nt, 1754 nt and 3574 nt, respectively. Together, these results demonstrate that RECQL4 is important for HR by promoting 5’ end resection of DSBs.
Retention of RECQL4 at DSBs depends on MRE11
To explore the function of RECQL4 in 5' end resection, we used mass spectroscopy to analyze proteins captured by co-immunoprecipitation (IP) with RECQL4-3XFLAG from irradiated HEK293T cells in the presence of benzonase. MRN components MRE11 and RAD50 and other DNA resection proteins BLM, EXO1 and DNA2 were identified (Fig. S3A and Table S1), and their interactions with RECQL4 were independently confirmed by IP with GFP-RECQL4 in the presence of benzonase (Fig. S3B) or ethidium bromide (Fig. S3C). We found that RECQL4 co-localized with MRE11 at DSBs and that the RECQL4-MRN interaction is stimulated by IR (Fig. 2A, B). Purified recombinant RECQL4 also immunoprecipitated recombinant MRE11, RAD50 and NBS1 (Fig. 2C), indicating complex formation between RECQL4 and MRN. To map the interaction region of RECQL4 with MRE11, purified RECQL4-3XFLAG and truncation fragments were incubated with purified YFP-MRE11 bound to GFP agarose beads. YFP-MRE11 pulled down full length and the N-terminal domain of RECQL4 (Fig. 2D), indicating that the N-terminal fragment of RECQL4 is responsible for the interaction with MRE11. To determine whether the interaction between RECQL4 and MRN is functional, we measured the nuclease activity of MRN on closed-circular single-strand PhiX174 DNA in the presence of RECQL4 in vitro as previously reported (Sartori et al., 2007). Wild type MRN and nuclease-dead MRN-ND (H129L/D130V) (Stracker et al., 2002) as well as RECQL4 and its helicase-dead mutant RQ4KM were used (Fig. S4A). We found that RECQL4 slightly stimulated the nuclease activity of MRN on closed-circular single-strand PhiX174 DNA (Fig. 2E).
Figure 2.

MRE11 mediates recruitment of RECQL4 to DSBs to promote HR repair. A. Co-localization of endogenous RECQL4 and MRE11 at laser-induced DSB tracks in U2OS cells. Bar, 10 μm. B. RECQL4 interacts with MRN complex in vivo. FLAG-IP was carried out using extracts prepared from vector and RQ4-3xFLAG expressing HEK293T cells treated with a 10 Gy IR and recovered for 10 min. C. Co-IP of recombinant MRE11, RAD50 and NBS1 with RECQL4. D. N-terminal domain of RECQL4 interacts with MRE11. E. Nuclease of MRN is stimulated by RECQL4 on closed circular single strand PhiX174 DNA. MRN (20 nM) or the nuclease-dead mutant, MRN-ND, was incubated with 20 nM RECQL4, boiled RECQL4, or BSA. Buffer, nuclease reaction buffer. F. MRE11 promotes RECQL4 recruitment to DSBs in U2OS cells. The recruitment of GFP-RECQL4 to DSB tracks, generated with 435 nm laser, was monitored in the control and MRE11-depleted U2OS cells and the fluorescence intensity were quantified. n=27. Bar, 10 μm. G. Retention of RECQL4 at DSBs depends on the exonuclease activity of MRE11. U2OS cells were treated with100 μM mirin. Graphic quantification below images, n=21. Bar, 10 μm. H. RECQL4 and MRE11 regulate resection at DSBs. Bar graph showing percent of ssDNA content generated at DSB1 in siRNA- or mirin-treated cells. Error bars represent SEM from four biological repeats. I. HR repair assay and western blots from RECQL4 and MRE11 knockdown DR-GFP cells. All data are presented as mean ± SEM from at least three independent experiments with P-value calculated with Student’s t-test. *, P<0.05. See also Figures S3, Figure S4 and Table S1.
Both RECQL4 and MRE11 are rapidly recruited to DSB (Haince et al., 2008; Singh et al., 2010), and thus we evaluated whether RECQL4 and MRE11 affected each other’s recruitment to DNA damage. GFP-RECQL4 was recruited significantly less to laser-induced DSBs in siMRE11-treated U2OS cells than in control cells (Fig. 2F), and there is less chromatin-bound RECQL4 in siMRE11-treated U2OS cells than in control cells after IR (Fig. S4B). However, depletion of RECQL4 did not affect the recruitment of YFP-MRE11 to laser-induced DSB (Fig. S4C), suggesting that recruitment of RECQL4 to DSBs requires MRE11, but not vice versa.
Since MRE11 nuclease regulates the pathway choice between NHEJ and HR (Shibata et al., 2014), the dynamics of GFP-RECQL4 recruitment was also evaluated in cells exposed to mirin, which specifically inhibits the MRE11 exonuclease but does not inhibit MRN complex formation (Dupre et al., 2008). RECQL4 was still rapidly recruited to laser-induced DSBs in mirin-treated cells (Fig. 2G), indicating that the recruitment of RECQL4 does not depend on the exonuclease activity of MRE11. However, GFP-RECQL4 was retained at DSBs for a significantly shorter time after mirin treatment (Fig.2G). This suggests that retention of RECQL4 at DSBs is regulated by MRE11 nuclease activity.
When ssDNA was measured at AsiSI-induced DSB in AID-DIvA U2OS cells pre-treated with mirin, siRQ4 or siMRE11, a lower amount of ssDNA was detected (Fig. 2H). However, the effect was not additive (Fig. 2H). Using the DR-GFP reporter system, it was observed that pre-treatment with siRQ4 or siMRE11 significantly reduced HR-mediated DSBR, but the effect was also not additive (Fig. 2I). These results suggest that RECQL4 functions downstream of MRN to promote DNA 5' end resection and HR-dependent DSBR.
RECQL4 promotes recruitment of CtIP to DSBs
CtIP is required for initiation of MRN-catalyzed 5' end resection at DSBs (Chen et al., 2008; Sartori et al., 2007; Yuan and Chen, 2009). Here we found that RECQL4 co-localized with CtIP at laser-induced DSBs (Fig. 3A) and interacted with CtIP in irradiated HEK293T cells (Fig. 3B and Fig. S3B, C). The interaction between CtIP and RECQL4 appeared to be stronger in IR-treated cells (Fig. 3B). Co-IP of recombinant RECQL4 and CtIP suggests that RECQL4 interacts directly with CtIP (Fig. 3C), and the N-terminus of RECQL4 was mapped as the interacting region with CtIP (Fig. 3D).
Figure 3.

RECQL4 promotes CtIP recruitment to DSBs for DNA end resection and HR repair. A. Co-localization of endogenous RECQL4 and CtIP at laser-induced DSB tracks. Bar, 5 μm B. Co-IP of CtIP with RECQL4 in response to IR. C. In vitro co-IP analysis of recombinant RECQL4 and CtIP. D. The N-terminus of RECQL4 interacts with CtIP. E. Subcellular distribution of CtIP in control and RECQL4-depleted U2OS cells 10 min after IR. F. GFP-CtIP foci in the control and RECQL4-depeted U2OS cells 30 min after IR. Bar, 10μm. G. Recruitment of GFP-CtIP to DSB tracks in control and RECQL4-depeted U2OS cells. n=29. Bar, 10 μm. H. RECQL4 supports the interaction between MRN and CtIP. Western analysis of indicated proteins pulled-down with YFP-MRE11 or GFP-CtIP from control and RECQL4 knockdown HEK293T cells 10 min after IR. I. Quantification of ssDNA generated at DSB1 in AID-DIvA U2OS cells after knockdown of RECQL4 and CtIP. J. HR repair assay after knockdown of RECQL4 and CtIP in DR-GFP U2OS cells. Error bars for I and J represent SEM from three independent experiments. The IR dose is 10Gy. See also Figure S2, Figure S3 and Table S1.
Recruitment of RECQL4 reaches its peak about one minute after laser damage (Fig. 2G), while CtIP needs much longer (Wang et al., 2013). Considering the direct interaction between RECQL4 and CtIP, it is possible that RECQL4 promotes CtIP recruitment to DSBs. To test this hypothesis, we first measured the abundance of chromatin-bound CtIP in control and RECQL4-knockdown U2OS cells after IR and found that IR increased chromatin-bound CtIP in the control cells but not in RECQL4-depleted cells (Fig. 3E). Interestingly, more mobility shift of chromatin-bound CtIP was detected in control cells that in RECQL4-depleted cells after IR (Fig. 3E), indicating that RECQL4 promotes IR-induced posttranslational modification of CtIP. Also, IR-treated control U2OS cells had an average of 22.6 GFP-CtIP foci per cell, significantly higher than that in siRQ4-treated cells (Fig. 3F). Furthermore, in the RECQL4 knockdown U2OS cells, recruitment of GFP-CtIP was significantly slower and less efficient than that in control cells (Fig. 3G). Together these data suggest that RECQL4 promotes stable CtIP recruitment to DSBs.
Since RECQL4 promotes recruitment of CtIP to DSBs, we asked whether RECQL4 is required for MRN-CtIP complex formation after IR. Pulldown assays were conducted in control- and RECQL4-knockdown HEK293T cells expressing YFP-MRE11 or GFP-CtIP. Expression levels of MRE11, RAD50, NBS1 and CtIP proteins were similar in control and RECQL4 knockdown cells (Input of Fig. 3H). Cell cycle status was not significantly different between RECQL4-depleted and control HEK293T cells (Fig. S2D, E). IP of YFP-MRE11 efficiently pulled down similar amounts of RAD50 and NBS1 from control and RECQL4 knockdown cells. In contrast, the interaction between MRE11 and CtIP was inhibited by knockdown of RECQL4 (Fig. 3H). In the reverse experiments using GFP-CtIP expressing cells, GFP-CtIP efficiently co-immunoprecipitated MRE11, RAD50 and NBS1 from control cells but much less from RECQL4 knockdown cells (Fig. 3H). These data are consistent with the idea that RECQL4 promotes the interaction between MRN and CtIP in human cells.
Depletion of RECQL4 or CtIP significantly reduced ssDNA generation at DSB1 in AID-DIvA cells (Fig. 3I). However, there were no differences among RECQL4 or CtIP-depleted cells and RECQL4/CtIP double knockdown cells. A similar result was obtained from the experiments measuring the HR efficiency (Fig. 3J). These results imply that RECQL4 and CtIP both play a role in HR-dependent DSBR and that RECQL4 promotes recruitment of CtIP to DSBs.
BLM and EXO1 act downstream of RECQL4 during HR-mediated DSBR
5’ resection, initiated by the MRN-CtIP complex, is extended by BLM/DNA2 and EXO1 via two alternative pathways (Cejka, 2015; Symington, 2014). Recruitment of both BLM and EXO1 to DSBs requires CtIP (Wang et al., 2013). Since RECQL4 promotes CtIP recruitment to DSBs, we tested whether removal of RECQL4 could result in failure of the two extensive resection pathways. We found that BLM, DNA2 and EXO1 interact with RECQL4 in irradiated HEK293T cells (Fig. S3). In addition, the retention of GFP-BLM at DSBs was reduced in U2OS cells after RECQL4 knockdown (Fig. 4A), suggesting that RECQL4 stimulates retention of BLM at IR-induced DSBs. In addition, knockdown of BLM, RECQL4 or both inhibited 5' resection to a similar extent at DSB1 in AID-DIvA U2OS cells (Fig. 4B). In the HR assay, knockdown of BLM, RECQL4, or both significantly reduced HR by 40%, 60% or 60%, respectively (Fig. 4C). These findings suggest that RECQL4 promotes retention of BLM at DSBs to stimulate HR.
Figure 4.

RECQL4 promotes recruitment of BLM and EXO1 to laser-induced DSBs. A. Recruitment of GFP-BLM in control and RECQL4-depeted U2OS cells. n= 22. Bar, 10 μm. B. Quantification of ssDNA content generated at DSB1 in AID-DIvA U2OS cells depleted for RECQL4 and BLM. C. Quantification of GFP-positive cells from the HR repair in control, RECQL4 and BLM knockdown cells. D. Recruitment of GFP-EXO1 in control and RECQL4-knockdown U2OS cells. n=27. Bar, 10 μm. E. Quantification of the ssDNA generated from resection at DSB1 in cells with knockdown for RECQL4 and EXO1. F. HR assay from EXO1 and RECQL4 knockdown DR-GFP cells. Data presented as mean ± SEM from three biological repeats. See also Figure S3 and Table S1.
We also evaluated the impact of RECQL4 loss on EXO1-mediated resection. RECQL4 co-localized with EXO1 at laser-induced DSBs in U2OS cells (Fig. S3E) and co-immunoprecipitated endogenous EXO1 in irradiated U2OS cells but not in untreated cells (Fig. S3F). GFP-EXO1 was rapidly recruited to DSBs in U2OS cells, but significantly more slowly in siRQ4-treated U2OS cells (Fig. 4D), indicating that RECQL4 also promotes EXO1 function at DSBs. Knockdown of EXO1 reduced the amount of 5’ end-resected DSBs in AID-DIvA U2OS cells (Fig. 4E), which is consistent with previous findings (Zhou et al., 2014). However, EXO1 and RECQL4 double knockdown was not additive in resection of DSBs (Fig. 4E). EXO1 knockdown reduced HR by about 31.4% but did not significantly exacerbate the reduction of HR in combination with RECQL4 depletion (Fig. 4F). These data support a model in which RECQL4 acts upstream of DNA2/BLM and EXO1 in HR-dependent DSBR.
RECQL4 helicase activity is required for DNA end resection during HR-mediated repair
The RecQ proteins share a conserved RecQ helicase domain and possess 3’-5’ DNA unwinding activity (Croteau et al., 2014). The helicase activity of human RECQL4 is weak compared to the others in vitro (Rossi et al., 2010; Xu and Liu, 2009). However, mutations in the helicase domain have been identified in many reported RECQL4-asscociated syndrome patients (Siitonen et al., 2009), indicating the importance of the helicase domain in vivo. To explore whether RECQL4 helicase activity is involved in DNA end resection and HR repair, we transfected siRQ4-resistant plasmids to ectopically express 3XFLAG-tagged WT RECQL4 or the helicase-dead mutant RECQL4-KM in siRQ4-treated AID-DIvA U2OS or DR-GFP U2OS cells. Western blots showed that endogenous RECQL4 was depleted by siRQ4 and that 3XFLAG-tagged RECQL4 and RECQL4-KM were expressed in AID-DIvA U2OS cells (Fig. 5A). Depletion of RECQL4 resulted in a reduction of ssDNA generated by DNA resection at the DSB1 site, and overexpression of RECQL4-3XFLAG completely restored the loss of 5’ end resection in siRQ4-transfected cells, whereas overexpression of RECQL4-KM-3XFLAG did not (Fig. 5A). These results indicate that the RECQL4 helicase activity is required for DNA end resection.
Figure 5.

Helicase activity of RECQL4 is required for RECQL4 to function in DNA resection and HR repair. A. Quantification of the ssDNA generated from resection at DSB1 after endogenous RECQL4-depletion of AID-DIvA U2OS cells but complementation with RECQL4-WT-3XFLAG or RECQL4-KM-3XFLAG. Data presented are mean ± SEM from three biological repeats. B. HR repair assay after endogenous RECLQ4-depletion in DR-GFP U2OS cells expressing wild type or helicase dead mutant RECQL4. Error bars represent SEM from four independent experiments. N.S., no significance. Pull-down assay using YFP-MRE11 (C) or GFP-CtIP (D) with RQ4Wt- 3XFLAG and RQ4KM-3XFLAG in vitro. E. Both WT and helicase-dead mutant RECQL4 significantly stimulate nuclease of MRN on closed circular single strand PhiX174 DNA. The concentration of MRN, RECQL4 and RQ4KM were 20 nM. Error bars represent SEM from three repeats with P-value by Student’s t-test. F. RPA displaces RECQL4 from ssDNA. Various substrates as shown were pre-incubated with RECQL4 then RPA or BSA were added to compete off RECQL4. Detection of displaced RECQL4 and DNA-bound RPA were visualized by Western Blotting. G. Model showing RECQL4’s role in DNA end resection of HR-mediated repair. MRN complex recognizes and binds to DSBs and recruits RECQL4 to the sites of damage. In turn RECQL4 promotes the stable recruitment of CtIP to DSBs and performs unwinding at the DNA ends thereby promoting resection. See also Figure S5.
In the HR repair assay, DSBs were generated by transfection of I-SceI-expressing plasmid into DR-GFP U2OS cells (Pierce et al., 1999). To reduce competition with I-SceI-expressing plasmid, the amount of pCMVtag4A-RQ4-siR, pCMVtag4A-RQ4KM-siR or vector was reduced to 0.5 μg for 2X106 cells, which resulted in a low expression level of 3XFLAG tagged RECQL4 and the mutant. However, this level of RECQL4-3xFLAG still significantly increased the percentage of GFP-positive cells, depleted for endogenous RECQL4 (Fig. 5B). This is consistent with our observation that RECQL4 levels correlate with HR repair (Fig. 1C, D). The RECQL4-KM-3XFLAG expression was higher than that of WT RECQL4, but did not significantly rescue the loss of HR repair after RECQL4-depletion. These data suggest that the RECQL4’s helicase activity is important for DNA end resection and HR-dependent DSBR.
Since RECQL4 promotes complex formation between CtIP and MRN by interacting with these proteins, we then measured whether inactivation of the helicase impairs RECQL4’s ability to interact with CtIP and MRE11. RECQL4-KM-3XFLAG was pulled down with YFP-MRE11 to the same extent as RECQL4-3XFLAG (Fig. 5C). Similarly, helicase-dead RECQL4 also interacted with CtIP as well as WT RECQL4 did (Fig. 5D). These finding suggests that inactivation of the helicase domain of RECQL4 does not affect the interaction between RECQL4 and MRE11 or CtIP. Additionally, we found that both helicase-dead and WT RECQL4 proteins were able to stimulate the nuclease activity of MRN on closed circle single strand PhiX174 DNA (Fig. 5E). Taken together, the DNA unwinding activity of RECQL4 is required to promote DNA end resection and HR repair.
RPA-mediated displacement of RECQL4 from ssDNA
Unlike BLM and WRN, RECQL4 remains at DSB sites for only a short time (Singh et al., 2010), suggesting that it falls off or is displaced. After DNA end resection, RPA coats the ssDNA tails for protection, which supported by the observation that RPA recruitment to DSBs increases continuously in 1 hour (Fig. S5A), as previously reported (Costelloe et al., 2012). Thus, we used an in vitro RECQL4 displacement assay to determine whether RPA could remove RECQL4 from ssDNA. Biotin-labeled ssDNA, dsDNA or 3’ tailed dsDNA substrates were first incubated with RECQL4 and then with either RPA or BSA. RPA-mediated RECQL4 displacement was detected by visualization of RECQL4 in the supernatant (Fig. 5F). Consistent with previous findings (Jensen et al., 2012; Keller et al., 2014), RECQL4 binds to ssDNA, dsDNA and 3’ tailed dsDNA substrates (Fig. 5F). RPA prefers to bind DNA substrates with longer ssDNA (Fig. S5C, D). When RPA was added to the RECQL4 coated 80-nt long ssDNA G80, RECQL4 was displaced as RPA bound to this substrate (Fig. 5F). A similar phenomenon was observed using GC40 and GC60 DNA substrates, which contain 40 or 20 nucleotide 3’ tails, respectively (Fig. 5F). However, very little RECQL4 was replaced by RPA from the blunt-ended dsDNA GC80 or from the 6nt-tailed dsDNA G80/C74 (Fig. 5F). BSA, our negative control, did not displace RECQL4 from the tested DNA substrates (Fig. 5F). Together, these data imply that RPA can displace RECQL4 from ssDNA or 3’ tailed dsDNA in vitro but not duplex DNA.
DISCUSSION
DNA end resection generates 3’ tailed ssDNA which is critical for launching HR repair. The MRN complex initiates 5’ end resection with CtIP, and then extensive resection is carried out by the nucleases EXO1 or DNA2 in two alternative pathways (Cejka, 2015; Symington, 2014). In the present work, we establish that RECQL4 is required for robust DNA end resection by regulating the interaction between MRN and CtIP and further that the helicase activity of RECQL4 is required for the process. We show that depletion of RECQL4 results in loss of HR repair as a result of diminished 5’ resection. IR enhances the physical interaction of RECQL4 with MRN and CtIP. The nuclease activity of MRE11 regulates the retention of RECQL4 at DSBs, and RECQL4 promotes recruitment of CtIP, as well as downstream players like BLM, DNA2 and EXO1, which participate in the extensive resection step of HR. Thus this work ascribes a hitherto unrecognized role for RECQL4 as an important regulator of DNA end resection in HR repair.
The data presented here indicate that rapid recruitment of RECQL4 to laser-induced DSBs depends on prior recruitment of MRN while RECQL4 is not required for recruitment of MRN to IR-induced DSBs. Additionally, RECQL4 interacts physically with the MRN complex in living cells and under cell-free conditions in vitro (Fig. 2A-D). RECQL4 and MRN function in the same pathway during 5’ end resection in HR repair (Fig. 2H, I). These findings suggest that RECQL4 functions downstream of MRN in HR DSBR. Combining these findings with the previous reports that both MRE11 and RECQL4 immediately gather at laser-induced DSB (Haince et al., 2008; Singh et al., 2010), it is likely that MRN and RECQL4 act sequentially and cooperatively at DSBs to promote 5’ end resection at DNA DSBs.
Retention of RECQL4 at DSBs is dramatically reduced in the presence of mirin (Fig. 2G). Mirin inhibits MRE11 exonuclease and also represses MRN-dependent ATM activation (Dupre et al., 2008). After recruitment by MRN, RECQL4 is likely retained at DSBs by the resected DNA, which depends on the exonuclease activity of MRE11. Meanwhile, RECQL4 stimulates the nuclease activity of MRN in vitro and promotes MRN-CtIP complex formation after IR, which is required for initiation of DNA resection (Sartori et al., 2007; You et al., 2009). Therefore, it is possible that RECQL4 promotes MRE11-mediated resection at a limited level, which further stimulates retention of RECQL4 at DSBs. Meanwhile, initially resected DNA also leads to limited ATR activation and ATR-dependent phosphorylation of CtIP, and in turn promotes stable chromatin association of CtIP for robust resection (Peterson et al., 2013). With 5’ resection ongoing, RPA binds to long ssDNA and disassociates RECQL4. Therefore, MRN, RECQL4, CtIP and checkpoint kinases can function in a feedback loop during DNA end processing at DSBs.
Recruitment of CtIP to DSBs depends on MRN and ATM (You et al., 2009). MRN is recruited earlier than CtIP to DSBs, therefore MRN may not directly recruit CtIP (You et al., 2009). This study reports that RECQL4 physically interacts with CtIP and promotes stable recruitment of CtIP to DSBs. However, the kinetics of the accumulation of CtIP differs from that of RECQL4. CtIP reaches its peak of abundance at laser-induced DSBs around 15 minutes after microirradation (Fig. 3G), as previously reported (Wang et al., 2013; You et al., 2009). However, RECQL4 only needs about one minute to reach its recruitment peak (Fig. 2G). Therefore, it is possible that RECQL4 recruits CtIP directly to initiate DNA resection, which further promotes more recruitment of CtIP due to checkpoint activation. RECQL4 also facilitates formation of an MRN-CtIP complex in vivo after IR (Fig. 3H). In summary, the data are consistent with the model that RECQL4 promotes CtIP recruitment to DSBs and thereby directly promotes end processing and HR-dependent DSBR.
BLM/DNA2 and EXO1 are required for extensive 5' end-resection step during HR-dependent DSBR (Symington, 2014). In contrast to RECQL4, BLM appears to play a somewhat more complex role during HR in human cells, possibly at two steps, once during end processing and a second time during dissolution of Holliday junctions together with TopoIIIα/RMI1/2 (Croteau et al., 2014). Here we show that RECQL4 plays a role in recruiting and retaining BLM and EXO1 at DSBs and that RECQL4 acts in the same pathway as BLM and EXO1 (Fig.4). Since MRE11 and CtIP are required to recruit/retain BLM/EXO1 at DSBs (Eid et al., 2010; Truong et al., 2013; Wang et al., 2013), this lack of retention of BLM/EXO1 on DSBs in RECQL4-depleted cells is probably a consequence of the inhibition of CtIP recruitment caused by loss of RECQL4. Another possibility is that RECQL4 directly recruits BLM and EXO1 in order to switch from initial resection to extensive resection.
RECQL4 is unlikely to be directly involved in the extensive resection step with DNA2/BLM or EXO1, although it interacts with the all of them. Short retention of RECQL4 at DSBs reduces the possibility that RECQL4 works together with DNA2/BLM or EXO1 in extensive resection. Secondly, BLM, but not RECQL4 specifically stimulates the nuclease activities of both DNA2 and EXO1 in vitro (Nimonkar et al., 2011; Nimonkar et al., 2008). Moreover, RPA displaces RECQL4 from ssDNA (Fig. 5F). However, it is possible that RECQL4 after interacting with MRN and CtIP remains bound to the ssDNA long enough to interact with DNA2/BLM and EXO1 and promotes their recruitment and chromatin association, which may activate the switch from initial resection to extensive resection.
Here, we found that depletion of either BLM or EXO1 reduces DNA resection and HR (Fig. 4). In yeast, absence of Exo1 reduced resection 1–5 kb from the DSB (Llorente and Symington, 2004; Mimitou and Symington, 2008). Dysfunction of sgs1 also markedly reduced the DNA resection rate and efficiency (Zhu et al., 2008) However, co-depletion of sgs1 and exo1 caused more dramatic loss of DNA resection (Mimitou and Symington, 2008; Zhu et al., 2008). In human cells, depletion of EXO1, DNA2 or BLM reduced 5’ DNA resection in U2OS cells (Grabarz et al., 2013; Gravel et al., 2008; Myler et al., 2016; Tomimatsu et al., 2012; Zhou et al., 2014). DNA2/BLM (Dna2/sgs1) and EXO1 can compensate for each other in extensive resection (Symington, 2014). However, dysfunction of either pathway could affect efficiency of DNA resection in vivo.
The helicase-dead mutant RECQL4-KM did not rescue the loss of either DNA end resection or HR repair (Fig. 5), demonstrating that the helicase activity of RECQL4 is required for DNA end resection and HR repair. RECQL4-KM was generated by replacing lysine 508 in the Walker A motif of the SFII helicase domain with methionine, and the mutation eliminates 3’-5’ DNA unwinding activity and ATPase of RECQL4 but not its annealing activity (Rossi et al., 2010). The expression of RECQL4-KM only partially restored the ability of RECQL4 to prevent cellular senescence in primary human fibroblasts with depletion of the endogenous RECQL4, indicating the importance of helicase activity in vivo (Lu et al., 2014a). However, the helicase activity is neither involved in the physical interaction between RECQL4 and MRN and CtIP, nor in the stimulation of the nuclease activity of MRN. In vivo DSBs are complex due to chromatin organization and DSB binding proteins competing at the site. RECQL4 has several activities. The failure of RECQL4-KM to rescue the resection and HR in endogenous RECQL4-depleted cells may reflect that it acts as a dominant negative. RECQL4 can unwind dsDNA, dsDNA with 3’ overhang (not 5’ overhang), bubble-structured dsDNA, Y-structured duplex and D-loop (Ghosh et al., 2012; Rossi et al., 2010; Xu and Liu, 2009). Thus, these activities might also help resolve secondary structures near the DNA ends to facilitate the initiation or extensive steps of DNA end resection.
A model for 5’ DNA resection is presented in Figure 5G, which highlights the role of RECQL4 in initiating 5' end resection at a nascent DSB. DSB arise due to endo- or exogenous insults, which are first sensed by the MRN complex. RECQL4 is recruited by MRN, and RECQL4 possibly promotes a limited resection with MRN, which also promotes retention of RECQL4 at DSB. The MRN-RECQL4 complex then promotes recruitment of CtIP to DSBs. After CtIP enters the complex, the nuclease activity of MRN is greatly stimulated (Sartori et al., 2007). The resultant short ssDNA strands then may also promote greater retention of RECQL4 and CtIP. This feedback loop would then facilitate recruitment of proteins involved in the extensive end resection step like BLM/DNA2 and EXO1. We are proposing that an activity of RECQL4 may remove DNA secondary structure barriers near the ends of the DNA to promote MRN-mediated DNA resection. After some length of resection, RPA binds the ssDNA and promotes displacement of RECQL4 allowing it to be recycled for use at other dsDNA break sites.
EXPERIMENTAL PROCEDURES
Cell culture, knockdown, DNA transfection, γ radiation and survival assay
U2OS, HEK293T and HeLa cell lines were cultured in DMEM medium with 10% fetal bovine serum (Sigma), 1X penicillin/streptomycin (Gibco). All cells were cultured in an atmosphere of 5% CO2 at 37°C. Lentivirus-mediated shRNA knockdown and siRNA knockdown were performed as reported (Lu et al., 2014a). The sequences of siRNA and shRNA are listed in Table S2. Polyplus JetPrime® was used for DNA transfection. γ rays were generated using a cesium-137 source (Gammacell Exactor 40, Best Theratronics). Radiation dose is 10 Gy, and post-irradiation recovery time is indicated in the figure legends. For the colony formation assay, cells were irradiated and then stained with 2% methylene blue in 5% ethanol 10 days after IR. The colonies with over 50 cells were counted. The results are presented as mean ± SEM from three independent experiments with P value by Student’s t-test.
Laser induced DNA damage and real time recruitment of fluorescence proteins
Laser-induced DSB and the recruitment of GFP-RECQL4, GFP-CtIP, GFP-BLM, GFP-EXO1, and GFP-RPA were performed as described (Singh et al., 2010). For mirin treatment, U2OS cells expressing GFP-RECQL4 were pre-incubated with 100 μM mirin for 4 hrs before laser microirradation. The results are presented as mean ± SEM with P value by Student’s t-test.
HR assay
RECQL4 and other target proteins were knocked down by siRNA in the DR-GFP U2OS cells (a gift from Prof. Xiaofan Wang), and 72 hrs after siRNA transfection, the HR assay was performed as reported (Pierce et al., 1999; Wang et al., 2014). The siRQ4-resistant plasmids, pCMVTag4A-RQ4-siR and pCMVTag4A-RQ4KM-siR, expressing 3XFLAG tagged wildtype RECQL4 and helicase-dead mutant RECQL4KM, respectively, were generated by PCR with primers RQ4-siR-PF and RQ4-siR-PR (Supplementary Table 2). 0.5 μg vector, pCMVTag4A-RQ4-siR or pCMVTag4A-RQ4KM-siR, were transfected into 5X105 siRQ4-treated DR-GFP U2OS cells. 24 hr later, cells were then processed for HR assay. The results are presented as mean ± SEM from three independent experiments with P value by Student’s t-test.
5’ resection assay
In vivo 5’ end resection was measured in AID-DIvA U2OS cells (a gift from Dr. Gaëlle Legube), as previously described (Zhou, Caron et al. 2014). For the rescue assay, 5 μg pCMVTag4A-RQ4-siR, pCMVTag4A-RQ4KM-siR or control vector was transfected into 2X106 RECQL4 siRQ4-treated cells. 45 hr later, cells were treated with 4-OHT and then processed for the resection assay. At least three biological repeats were performed and data are presented as the mean ± SEM.
Subcellular fractionation, Western blotting and immunofluorescence microscopy
Subcellular fractions were isolated using a subcellular protein fractionation kit (Thermo Fisher) according to the manufacturer’s instructions and the resultant fractions were analyzed with Western blotting. Western blot and immunofluorescence microscopy were performed as previously described (Lu et al., 2014a). Antibodies used in this study are listed in the extended experimental procedures.
Protein purification
The MRE11-RAD50-NBS1 complex was purified from insect cells as previously described (Cheng et al., 2004). Purification of recombinant RECQL4 and helicase-dead mutant RECQL4-KM were performed as described (Rossi et al., 2010). RPA Purification was performed as described (Henricksen et al., 1994). The details of CtIP purification and 3XFLAG-tagged RECQL4 and its truncation fragments are provided in the extended experimental procedure.
IP, pull down assay, silver staining and protein identification
Control and γ-irradiated cells were incubated for 10 minutes, and then sonicated on ice in IP Lysis Buffer 2 containing 40 mM Tris-HCl pH 7.4, 150 mM NaCl, 2 mM MgCl2, 0.2% NP-40, 0.4% Triton 100, 1X protease inhibitor cocktail (Thermo fisher), 1X phosphatase inhibitor cocktail 2 and 3 (Sigma-Aldrich), 20 U/mL benzonase (Novagen). 50μg/mL ethidium bromide was added in lysate where it was indicated. For co-IP with RECQL4 antibody, 2 mg protein was incubated with 2 μg of RECQL4 antibody (Lu et al., 2014a) or normal Rabbit IgG (Thermo Fisher). For FLAG IP, the cell lysate was incubated with M2 FLAG-magnetic beads (Sigma-Aldrich). For GFP IP, GFP-TRAP beads (ChromoTek) were used to capture GFP-RECQL4 or YFP-MRE11. The beads were washed with cold Washing buffer 4 (20 mM Tris-HCl, pH7.4, 150 mM NaCl, 0.2% Triton X100) for 5 times and then subjected to Western Blotting, Silver staining or mass spectrometry analysis by Harvard Taplin Mass Spectrometry Facility. The details of mass spectrometry are listed in Table S1.
For in vitro IP, purified RECQL4 was incubated with recombinant MRN complex or CtIP in Binding Buffer 1 (20 mM Tris-HCl pH7.4, 100 mM NaCl, 0.2% Triton X100) for 2 hrs at 4 °C, and then was incubated with anti-RECQL4 antibody or normal rabbit IgG. After washing with Binding Buffer, the proteins remained on the beads were analyzed by Western blotting.
Nuclease assay
Nuclease assays were carried out with 20 nM MRN or nuclease-dead MRN-ND in the presence of 20 nM wildtype RECQL4 or helicase-dead RECQL4 RQ4KM on 50 ng closed circular single stranded PhiX174 DNA in the resection buffer containing 20 mM MOPS, pH 7.2, 1 mM DTT, 5 mM MgCl2, 5 mM MnCl2, 1 mM ATP, as previously described (Sartori et al., 2007). After 3 hour incubation at 37 °C, DNA was separated in 0.8% agarose gel, further stained with SYBR Gold, visualized with Chemidoc™ XRS+ system (Bio-Rad) and quantified with Bio-Rad Image Lab™ (Version 3.0). Data were presented as mean ± SEM from three repeats.
Displacement assay
A biotin-labeled oligonucleotide (G80) was annealed with C80, C74, C60 and C40 and resulted in dsDNA GC80 and 3’ tailed dsDNA GC74, GC60, and GC40, respectively (See the sequences in Table S2). RECQL4 (200 nM) was incubated with 20 nM DNA substrates bound to M280-streptavidin beads (Life Technologies) in the binding buffer (20 mM HEPES pH7.4, 100 mM NaCl, 1 mM MgCl2, 0.1% Triton X100, 200 ng/mL BSA) at RT for 15 min. After washing with the binding buffer, the beads were incubated with 200 nM RPA or 200 ng/mL BSA at RT for 15 min. The supernatants and beads were then collected for detecting displaced RECQL4 and DNA-bound RPA by Western blotting.
Supplementary Material
HIGHLIGHTS.
RECQL4 promotes DNA double-strand breaks 5’ end resection
RECQL4 recruitment to DSBs depends on MRE11.
RECQL4 recruits CtIP to DSBs.
RECQL4 helicase activity is required for 5’ DNA end resection.
Acknowledgments
AID-DIVA U2OS is under MTA between Dr. Gaëlle Legube and NIA. We thank Dr. Xiaofan Wang for DR-GFP U2OS, Drs. Dik van Gent and Roland Kanaar for YFP-MRE11 plasmid, Dr. Marc Wold for the GFP-RPA plasmids, Dr. Binghui Shen for FLAG-DNA2 plasmids, Dr. Tomasz Kulikowicz and Christopher Dunn for RECQL4 protein, Tomasz Kulikowicz and Alfred May for IR operation, and Drs. Morten Scheibye-Knudsen and Jaya Sarkar for their critical comments. This research was supported entirely by the Intramural Research Program of the NIH, National Institute on Aging.
Footnotes
COMPETING FINANCIAL INTEREST The authors declare no conflict of interest.
AUTHOR CONTRIBUTIONS Conceptualization: H.L., R.A.S., D.L.C., and V.A.B.; Methodology: H.L. and R.A.S.; Investigation: H.L. and R.A.S.; Writing-Original Draft, H.L. and R.A.S.; Review & Editing, H.L., R.A.S., L.J.R., P.C., D.L.C. and V.A.B.; Funding Acquisition, V.A.B.; Resources, G.K., R.A., L.J.R. and P.C.; Supervision, D.L.C. and V.A.B.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aparicio T, Baer R, Gautier J. DNA double-strand break repair pathway choice and cancer. DNA Repair (Amst) 2014;19:169–175. doi: 10.1016/j.dnarep.2014.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aymard F, Bugler B, Schmidt CK, Guillou E, Caron P, Briois S, Iacovoni JS, Daburon V, Miller KM, Jackson SP, et al. Transcriptionally active chromatin recruits homologous recombination at DNA double-strand breaks. Nat Struct Mol Biol. 2014;21:366–374. doi: 10.1038/nsmb.2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunting SF, Callen E, Wong N, Chen HT, Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez-Capetillo O, Cao L, et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 2010;141:243–254. doi: 10.1016/j.cell.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannavo E, Cejka P. Sae2 promotes dsDNA endonuclease activity within Mre11-Rad50-Xrs2 to resect DNA breaks. Nature. 2014;514:122–125. doi: 10.1038/nature13771. [DOI] [PubMed] [Google Scholar]
- Cejka P. DNA end resection: nucleases team up with the right partners to initiate homologous recombination. J Biol Chem. 2015;290:22931–22938. doi: 10.1074/jbc.R115.675942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cejka P, Cannavo E, Polaczek P, Masuda-Sasa T, Pokharel S, Campbell JL, Kowalczykowski SC. DNA end resection by Dna2-Sgs1-RPA and its stimulation by Top3-Rmi1 and Mre11-Rad50-Xrs2. Nature. 2010;467:112–116. doi: 10.1038/nature09355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Lisby M, Symington LS. RPA coordinates DNA end resection and prevents formation of DNA hairpins. Mol Cell. 2013;50:589–600. doi: 10.1016/j.molcel.2013.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Nievera CJ, Lee AY, Wu X. Cell cycle-dependent complex formation of BRCA1.CtIP.MRN is important for DNA double-strand break repair. J Biol Chem. 2008;283:7713–7720. doi: 10.1074/jbc.M710245200. [DOI] [PubMed] [Google Scholar]
- Cheng WH, von Kobbe C, Opresko PL, Arthur LM, Komatsu K, Seidman MM, Carney JP, Bohr VA. Linkage between Werner syndrome protein and the Mre11 complex via Nbs1. J Biol Chem. 2004;279:21169–21176. doi: 10.1074/jbc.M312770200. [DOI] [PubMed] [Google Scholar]
- Costelloe T, Louge R, Tomimatsu N, Mukherjee B, Martini E, Khadaroo B, Dubois K, Wiegant WW, Thierry A, Burma S, et al. The yeast Fun30 and human SMARCAD1 chromatin remodellers promote DNA end resection. Nature. 2012;489:581–584. doi: 10.1038/nature11353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croteau DL, Popuri V, Opresko PL, Bohr VA. Human RecQ helicases in DNA repair, recombination, and replication. Annu Rev Biochem. 2014;83:519–552. doi: 10.1146/annurev-biochem-060713-035428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupre A, Boyer-Chatenet L, Sattler RM, Modi AP, Lee JH, Nicolette ML, Kopelovich L, Jasin M, Baer R, Paull TT, et al. A forward chemical genetic screen reveals an inhibitor of the Mre11-Rad50-Nbs1 complex. Nat Chem Biol. 2008;4:119–125. doi: 10.1038/nchembio.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eid W, Steger M, El-Shemerly M, Ferretti LP, Pena-Diaz J, Konig C, Valtorta E, Sartori AA, Ferrari S. DNA end resection by CtIP and exonuclease 1 prevents genomic instability. EMBO Rep. 2010;11:962–968. doi: 10.1038/embor.2010.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang H, Nie L, Chi Z, Liu J, Guo D, Lu X, Hei TK, Balajee AS, Zhao Y. RecQL4 helicase amplification is involved in human breast tumorigenesis. PLoS One. 2013;8:e69600. doi: 10.1371/journal.pone.0069600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh AK, Rossi ML, Singh DK, Dunn C, Ramamoorthy M, Croteau DL, Liu Y, Bohr VA. RECQL4, the protein mutated in Rothmund-Thomson syndrome, functions in telomere maintenance. J Biol Chem. 2012;287:196–209. doi: 10.1074/jbc.M111.295063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabarz A, Guirouilh-Barbat J, Barascu A, Pennarun G, Genet D, Rass E, Germann SM, Bertrand P, Hickson ID, Lopez BS. A role for BLM in double-strand break repair pathway choice: prevention of CtIP/Mre11-mediated alternative nonhomologous end-joining. Cell Rep. 2013;5:21–28. doi: 10.1016/j.celrep.2013.08.034. [DOI] [PubMed] [Google Scholar]
- Gravel S, Chapman JR, Magill C, Jackson SP. DNA helicases Sgs1 and BLM promote DNA double-strand break resection. Genes Dev. 2008;22:2767–2772. doi: 10.1101/gad.503108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haince JF, McDonald D, Rodrigue A, Dery U, Masson JY, Hendzel MJ, Poirier GG. PARP1-dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites. J Biol Chem. 2008;283:1197–1208. doi: 10.1074/jbc.M706734200. [DOI] [PubMed] [Google Scholar]
- Henricksen LA, Umbricht CB, Wold MS. Recombinant replication protein A: expression, complex formation, and functional characterization. J Biol Chem. 1994;269:11121–11132. [PubMed] [Google Scholar]
- Im JS, Ki SH, Farina A, Jung DS, Hurwitz J, Lee JK. Assembly of the Cdc45-Mcm2-7-GINS complex in human cells requires the Ctf4/And-1, RecQL4, and Mcm10 proteins. Proc Natl Acad Sci U S A. 2009;106:15628–15632. doi: 10.1073/pnas.0908039106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen MB, Dunn CA, Keijzers G, Kulikowicz T, Rasmussen LJ, Croteau DL, Bohr VA. The helicase and ATPase activities of RECQL4 are compromised by mutations reported in three human patients. Aging (Albany NY) 2012;4:790–802. doi: 10.18632/aging.100506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller H, Kiosze K, Sachsenweger J, Haumann S, Ohlenschlager O, Nuutinen T, Syvaoja JE, Gorlach M, Grosse F, Pospiech H. The intrinsically disordered amino-terminal region of human RecQL4: multiple DNA-binding domains confer annealing, strand exchange and G4 DNA binding. Nucleic Acids Res. 2014;42:12614–12627. doi: 10.1093/nar/gku993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llorente B, Symington LS. The Mre11 nuclease is not required for 5' to 3' resection at multiple HO-induced double-strand breaks. Mol Cell Biol. 2004;24:9682–9694. doi: 10.1128/MCB.24.21.9682-9694.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Fang EF, Sykora P, Kulikowicz T, Zhang Y, Becker KG, Croteau DL, Bohr VA. Senescence induced by RECQL4 dysfunction contributes to Rothmund-Thomson syndrome features in mice. Cell Death Dis. 2014a;5:e1226. doi: 10.1038/cddis.2014.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu L, Jin W, Liu H, Wang LL. RECQ DNA helicases and osteosarcoma. Adv Exp Med Biol. 2014b;804:129–145. doi: 10.1007/978-3-319-04843-7_7. [DOI] [PubMed] [Google Scholar]
- Mimitou EP, Symington LS. Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature. 2008;455:770–774. doi: 10.1038/nature07312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myler LR, Gallardo IF, Zhou Y, Gong F, Yang SH, Wold MS, Miller KM, Paull TT, Finkelstein IJ. Single-molecule imaging reveals the mechanism of Exo1 regulation by single-stranded DNA binding proteins. Proc Natl Acad Sci U S A. 2016;113:E1170–1179. doi: 10.1073/pnas.1516674113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimonkar AV, Genschel J, Kinoshita E, Polaczek P, Campbell JL, Wyman C, Modrich P, Kowalczykowski SC. BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev. 2011;25:350–362. doi: 10.1101/gad.2003811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimonkar AV, Ozsoy AZ, Genschel J, Modrich P, Kowalczykowski SC. Human exonuclease 1 and BLM helicase interact to resect DNA and initiate DNA repair. Proc Natl Acad Sci U S A. 2008;105:16906–16911. doi: 10.1073/pnas.0809380105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu H, Chung WH, Zhu Z, Kwon Y, Zhao W, Chi P, Prakash R, Seong C, Liu D, Lu L, et al. Mechanism of the ATP-dependent DNA end-resection machinery from Saccharomyces cerevisiae. Nature. 2010;467:108–111. doi: 10.1038/nature09318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SJ, Lee YJ, Beck BD, Lee SH. A positive involvement of RecQL4 in UV-induced S-phase arrest. DNA Cell Biol. 2006;25:696–703. doi: 10.1089/dna.2006.25.696. [DOI] [PubMed] [Google Scholar]
- Peterson SE, Li Y, Wu-Baer F, Chait BT, Baer R, Yan H, Gottesman ME, Gautier J. Activation of DSB processing requires phosphorylation of CtIP by ATR. Mol Cell. 2013;49:657–667. doi: 10.1016/j.molcel.2012.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce AJ, Johnson RD, Thompson LH, Jasin M. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev. 1999;13:2633–2638. doi: 10.1101/gad.13.20.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash R, Zhang Y, Feng W, Jasin M. Homologous recombination and human health: the roles of BRCA1, BRCA2, and associated proteins. Cold Spring Harb Perspect Biol. 2015;7:a016600. doi: 10.1101/cshperspect.a016600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi ML, Ghosh AK, Kulikowicz T, Croteau DL, Bohr VA. Conserved helicase domain of human RecQ4 is required for strand annealing-independent DNA unwinding. DNA Repair (Amst) 2010;9:796–804. doi: 10.1016/j.dnarep.2010.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sangrithi MN, Bernal JA, Madine M, Philpott A, Lee J, Dunphy WG, Venkitaraman AR. Initiation of DNA replication requires the RECQL4 protein mutated in Rothmund-Thomson syndrome. Cell. 2005;121:887–898. doi: 10.1016/j.cell.2005.05.015. [DOI] [PubMed] [Google Scholar]
- Sartori AA, Lukas C, Coates J, Mistrik M, Fu S, Bartek J, Baer R, Lukas J, Jackson SP. Human CtIP promotes DNA end resection. Nature. 2007;450:509–514. doi: 10.1038/nature06337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamanna RA, Singh DK, Lu H, Mirey G, Keijzers G, Salles B, Croteau DL, Bohr VA. RECQ helicase RECQL4 participates in non-homologous end joining and interacts with the Ku complex. Carcinogenesis. 2014;35:2415–2424. doi: 10.1093/carcin/bgu137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao RG, Cao CX, Zhang H, Kohn KW, Wold MS, Pommier Y. Replication-mediated DNA damage by camptothecin induces phosphorylation of RPA by DNA-dependent protein kinase and dissociates RPA:DNA-PK complexes. EMBO J. 1999;18:1397–1406. doi: 10.1093/emboj/18.5.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata A, Moiani D, Arvai AS, Perry J, Harding SM, Genois MM, Maity R, van Rossum-Fikkert S, Kertokalio A, Romoli F, et al. DNA double-strand break repair pathway choice is directed by distinct MRE11 nuclease activities. Mol Cell. 2014;53:7–18. doi: 10.1016/j.molcel.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siitonen HA, Sotkasiira J, Biervliet M, Benmansour A, Capri Y, Cormier-Daire V, Crandall B, Hannula-Jouppi K, Hennekam R, Herzog D, et al. The mutation spectrum in RECQL4 diseases. Eur J Hum Genet. 2009;17:151–158. doi: 10.1038/ejhg.2008.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh DK, Karmakar P, Aamann M, Schurman SH, May A, Croteau DL, Burks L, Plon SE, Bohr VA. The involvement of human RECQL4 in DNA double-strand break repair. Aging Cell. 2010;9:358–371. doi: 10.1111/j.1474-9726.2010.00562.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh DK, Popuri V, Kulikowicz T, Shevelev I, Ghosh AK, Ramamoorthy M, Rossi ML, Janscak P, Croteau DL, Bohr VA. The human RecQ helicases BLM and RECQL4 cooperate to preserve genome stability. Nucleic Acids Res. 2012;40:6632–6648. doi: 10.1093/nar/gks349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stracker TH, Carson CT, Weitzman MD. Adenovirus oncoproteins inactivate the Mre11-Rad50-NBS1 DNA repair complex. Nature. 2002;418:348–352. doi: 10.1038/nature00863. [DOI] [PubMed] [Google Scholar]
- Su Y, Meador JA, Calaf GM, Proietti De-Santis L, Zhao Y, Bohr VA, Balajee AS. Human RecQL4 helicase plays critical roles in prostate carcinogenesis. Cancer Res. 2010;70:9207–9217. doi: 10.1158/0008-5472.CAN-10-1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symington LS. End resection at double-strand breaks: mechanism and regulation. Cold Spring Harb Perspect Biol. 2014;6 doi: 10.1101/cshperspect.a016436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomimatsu N, Mukherjee B, Deland K, Kurimasa A, Bolderson E, Khanna KK, Burma S. Exo1 plays a major role in DNA end resection in humans and influences double-strand break repair and damage signaling decisions. DNA Repair (Amst) 2012;11:441–448. doi: 10.1016/j.dnarep.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truong LN, Li Y, Shi LZ, Hwang PY, He J, Wang H, Razavian N, Berns MW, Wu X. Microhomology-mediated End Joining and Homologous Recombination share the initial end resection step to repair DNA double-strand breaks in mammalian cells. Proc Natl Acad Sci U S A. 2013;110:7720–7725. doi: 10.1073/pnas.1213431110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Shi LZ, Wong CC, Han X, Hwang PY, Truong LN, Zhu Q, Shao Z, Chen DJ, Berns MW, et al. The interaction of CtIP and Nbs1 connects CDK and ATM to regulate HR-mediated double-strand break repair. PLoS Genet. 2013;9:e1003277. doi: 10.1371/journal.pgen.1003277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Goldstein M, Alexander P, Wakeman TP, Sun T, Feng J, Lou Z, Kastan MB, Wang XF. Rad17 recruits the MRE11-RAD50-NBS1 complex to regulate the cellular response to DNA double-strand breaks. EMBO J. 2014;33:862–877. doi: 10.1002/embj.201386064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Liu Y. Dual DNA unwinding activities of the Rothmund-Thomson syndrome protein, RECQ4. EMBO J. 2009;28:568–577. doi: 10.1038/emboj.2009.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Rochette PJ, Feyissa EA, Su TV, Liu Y. MCM10 mediates RECQ4 association with MCM2-7 helicase complex during DNA replication. EMBO J. 2009;28:3005–3014. doi: 10.1038/emboj.2009.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You Z, Shi LZ, Zhu Q, Wu P, Zhang YW, Basilio A, Tonnu N, Verma IM, Berns MW, Hunter T. CtIP links DNA double-strand break sensing to resection. Mol Cell. 2009;36:954–969. doi: 10.1016/j.molcel.2009.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J, Chen J. N terminus of CtIP is critical for homologous recombination-mediated double-strand break repair. J Biol Chem. 2009;284:31746–31752. doi: 10.1074/jbc.M109.023424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Caron P, Legube G, Paull TT. Quantitation of DNA double-strand break resection intermediates in human cells. Nucleic Acids Res. 2014;42:e19. doi: 10.1093/nar/gkt1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z, Chung WH, Shim EY, Lee SE, Ira G. Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell. 2008;134:981–994. doi: 10.1016/j.cell.2008.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.