


Abstract
The ends of human chromosomes (telomeres) lose up to 200 bp of DNA per cell division. Chromosomal shortening ultimately leads to senescence and death in normal cells. Many human carcinoma lines are immortal in vitro, suggesting that these cells have a mechanism for maintaining the ends of their chromosomes. Telomerase is a ribonucleoprotein complex that synthesizes telomeric DNA onto chromosomes using its RNA component as template. Telomerase activity is found in most tumor cells, but is absent from normal cells. Little is known about how normal human cells repress telomerase (hTERT) gene expression. Mice carrying an E2F-1 null mutation develop a variety of malignant tumors, suggesting that this transcription factor has a tumor suppressor function. To determine mechanisms by which E2F-1 suppresses tumor formation, we examined the role of this transcription factor in regulation of the hTERT promoter in human cells. We identified two putative E2F-1-binding sites proximal to the transcriptional start site of the hTERT promoter. Mutation of these sites produced dramatic increases in promoter activity. Overexpression of E2F-1 but not a mutant E2F-1 repressed hTERT promoter activity in reporter gene assays. This repression was abolished by mutation of the E2F-1-binding sites in the hTERT promoter. Human cancer cell lines stably overexpressing E2F-1 exhibited decreased hTERT mRNA expression and telomerase activity. We conclude that E2F-1 has an atypical function as a transcriptional repressor of the hTERT gene in human cells.
INTRODUCTION
The ends of human chromosomes are protected from degradation and fusion by telomeres (1). Telomeres consist of tandem repeats of the sequence TTAGGG (2). Chromosomal analysis has shown that telomeres lose up to 200 bp of DNA per cell division in vivo and in vitro (3). This is due to the inability of DNA polymerase to completely replicate the chromosomal ends (4). Chromosomal shortening ultimately leads to senescence and death in normal cells (5). Therefore, progressive telomere shortening is considered to be a mitotic clock that limits the lifespan of normal cells (6).
In contrast to normal somatic cells, many immortal lines do not exhibit telomere shortening during DNA replication, suggesting that maintenance of these structures is required to escape replicative senescence (7). Telomere shortening is frequently arrested in immortal and tumor cell lines (8,9). Telomere maintenance in these cells is the result of the activity of a ribonucleoprotein complex known as telomerase (1). Telomerase synthesizes telomeric DNA onto chromosome ends using its RNA component as template (10,11). Mutations in the telomerase RNA template result in greatly increased telomere length in yeast (12) and nullizygous mice exhibit a decreased lifespan, a diminished stress response and increased tumor formation, likely due to chromosomal abnormalities (13,14). Components of the telomeric protein complex have been cloned (15–18) and the telomerase catalytic subunit has been identified (16,19,20). Overexpression of the catalytic subunit extends the lifespan of mouse embryo fibroblasts (21), but inactivation of retinoblastoma (Rb) and p16INK4A in addition to telomerase activity was required to immortalize human epithelial cells (21).
Telomerase activity has been localized to regenerative and stem cell populations (22) and lines immortalized by viral oncogenes (23). Terminal differentiation of these cells results in inhibition of telomerase activity (24). Telomerase activity is a common feature of tumor tissue and cell lines (25). Telomerase activation has been suggested to be a late event in tumorigenesis (26) and high levels of activity result in an unfavorable clinical prognosis (27). Regulation of telomerase activity has been controversial. Some studies have shown that telomerase activity is expressed throughout the cell cycle (28), while others have demonstrated phase-specific regulation (29), suggesting that telomerase regulation may be a cell type-specific process. Buchkovich and Greider (30) demonstrated that cycling but not quiescent leukocytes express telomerase activity, indicating that telomerase activity may be a proliferation marker. Recent experiments have demonstrated that telomerase activity may be associated with specific G1 phase defects in breast cancer cells (31).
Progression from G1 to S phase is regulated by cyclin-dependent kinase (cdk) phosphorylation of Rb family proteins (for a review see 32). The activity of cdks is regulated by two groups of proteins, the cyclins and cdk inhibitors. Phosphorylation of Rb releases E2F transcription factors which regulate target genes involved in cell division. However, E2F-1 null mutant mice develop a variety of malignant tumors, suggesting that this transcription factor has a tumor suppressor function (33). We hypothesized that repression of telomerase activity would select against the immortalized phenotype of human cancer cells. In order to determine if the tumor suppressor function of E2F-1 included repression of telomerase, we examined the human telomerase promoter for potential E2F-binding sites. Two non-canonical sites were found to mediate repression of the telomerase promoter by E2F-1. This study suggests that repression of the telomerase gene by E2F-1 may be an important tumor suppressor function of this transcription factor.
MATERIALS AND METHODS
Cell culture
The SCC25 cell line used in this study has been described previously (34). Cells were cultured in Dulbecco’s modified Eagle’s medium, 10% fetal bovine serum and 40 µg/ml gentamicin. All cells were grown at 37°C in a humidified atmosphere of 5% CO2.
hTERT promoter constructs and transient transfection
Cloning of the human telomerase promoter has been reported previously (35). Deletion of the 5′-end of the construct to –200 bp was performed by PCR and the resulting downstream fragment was subcloned into the luciferase reporter vector pGL3. Mutation of the putative E2F sites at –174 and –98 bp to 5′-CGCct-3′ in the full-length and deletion constructs was performed using a Gene Editor site-directed mutagenesis kit (Promega). Triplicate cultures of SCC25 cells were transiently transfected with 5 µg of the indicated hTERT promoter/reporter vectors along with 2 µg E2F-1 expression plasmid or blank vector using LipofectAMINE according to the manufacturer’s recommendations (Life Technologies). The E2F-1 expression vectors were provided by Dr Karen Vousden (36). An aliquot of 1 µg β-galactosidase expression plasmid (Vical) was used to normalize for transfection efficiency. Cells were harvested and reporter gene activity determined using a commercially available kit (Dual-light; Tropix). Luciferase activity was normalized to β-galactosidase levels for each sample.
Electrophoretic mobility shift assay
SCC25 nuclei (107) were extracted in 20 mM HEPES, pH 7.9, 25% glycerol, 1.5 mM MgCl2, 1.2 M KCl, 0.2 mM EDTA, 0.2 mM PMSF and 0.5 mM DTT for 30 min at 4°C. Following centrifugation at 10 000 g for 30 min at 4°C, the supernatant was removed and dialyzed against 20 mM HEPES, pH 7.9, 20% glycerol, 0.1 M KCl, 0.2 mM EDTA, 0.2 mM PMSF and 0.5 mM DTT for 1 h at 4°C. An aliquot of 15 µg of dialyzed nuclear extract was incubated in binding reactions containing 2 µg poly(dI-dC)·poly(dI-dC) and 10 000 c.p.m. 32P-end-labeled double-stranded oligonucleotide corresponding to –184 to –161 bp of the hTERT promoter containing the putative E2F site (5′-CGCCCAGGACCGCGCTCCCCACGT-3′). For binding competition analysis, a 10- to 1000-fold molar excess of unlabeled probe or 5′-CGCCCAGGACCGCctTCCCCACGT-3′ mutated oligonucleotide was included in the reactions. To determine if E2F-1 was present in the shifted complexes, 1 µl of anti-human E2F-1 antibody (Santa Cruz Biotechnology) or control IgG was included in the binding reactions. Reactions were incubated at room temperature for 15 min and subjected to native polyacrylamide gel electrophoresis using 0.5× Tris/borate/EDTA running buffer. Gels were dried and exposed to Kodak XAR5 autoradiographic film for 16 h at –80°C.
Northern blotting
An aliquot of 30 µg total cellular RNA was electrophoresed in 1% agarose gels containing 2.2 M formaldehyde using 1× MOPS running buffer. RNA was capillary transferred to nylon membranes (Nytran; Schleicher and Schuell) and crosslinked using a UV Stratalinker (Stratagene). Membranes were prehybridized in 50% formamide, 5× SSPE, 1× Denhardt’s solution and 0.2% SDS at 42°C for 8 h followed by addition of 6 × 106 c.p.m. 32P-labeled hTERT cDNA probe (19). Blots were incubated at 42°C for 16 h followed by two washes in 2× SSC, 0.1% SDS at 50°C for 45 min each and one wash in 0.2× SSC, 0.1% SDS at 65°C for 45 min. Blots were exposed to Kodak XAR5 autoradiographic film for 16 h at –80°C. Blots were stripped and hybridized with an 18S rRNA probe to normalize for the amount of RNA in each lane. Bands were quantitated using a Molecular Dynamics PhosphorImager.
Telomeric repeat amplification protocol
The telomeric repeat amplification protocol (TRAP) has been described previously (25). SCC25 cells were lysed in buffer containing 10 mM Tris–HCl, pH 7.5, 1 mM MgCl2, 1 mM EGTA, 0.1 mM benzamidine, 5 mM β-mercaptoethanol, 0.5% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate and 10% glycerol. After a 30 min incubation on ice, the lysates were centrifuged for 30 min at 12 000 g and the supernatant stored at –80°C. Protein concentrations were determined by the Bradford method using Bio-Rad protein dye reagent according to the manufacturer’s recommendations. Extracts were diluted in lysis buffer and between 5 ng and 5 µg protein was incubated with 0.1 µg TS primer (5′-AATCCGTCGAGCAGAGTT-3′) and 0.1 µg CX primer (5′-CCCTACCCTACCCTACCCTAA-3′). The 50 µl reaction mixture also contained 50 µM each deoxynucleotide triphosphate and 5 µCi [α-32P]dCTP in 20 mM Tris–HCl, pH 8.3, 1.5 mM MgCl2, 63 mM KCl, 0.05% Tween-20, 1 mM EGTA and 2.5 U Taq DNA polymerase (Boehringer Mannheim). Following a 30 min incubation at 30°C, samples were subjected to 20 cycles of PCR at 95°C for 30 s, 50°C for 30 s and 72°C for 1 min. The PCR products were separated on 10% non-denaturing polyacrylamide gels using 0.5× Tris/borate/EDTA running buffer. Gels were dried and exposed to autoradiographic film (Kodak XAR5) at –80°C for 16 h. Heat-inactivated extracts were used as the negative control.
RESULTS
Sequence analysis of the hTERT promoter revealed two potential E2F-binding sites located at –174 and –98 bp relative to the transcription start site (Fig. 1A). To eliminate potential contributions of upstream divergent sites, a second construct containing only the proximal 200 bp of the hTERT promoter was created (–200 hTERT, Fig. 1B). To evaluate the contributions of the putative E2F sites at –174 and –98 bp, these sequences were each mutated to CGCct (m174 hTERT and m98 hTERT). A fifth construct in which both sites were mutated was also created (m174m98 hTERT). These constructs were individually introduced into SCC25 cells by transient transfection. As shown in Figure 2, deletion of the 5′-end of the hTERT promoter to –200 bp only marginally reduced luciferase activity. However, when the putative E2F site at –174 bp was mutated, a dramatic increase in promoter activity was observed. The activity of the m174 hTERT construct was ∼4-fold higher than the intact promoter or the –200 hTERT vector. Similarly, mutation of the putative E2F site at –98 bp also induced promoter activity, although to a lesser extent than the m174 hTERT construct (3-fold increase, Fig. 2). When both sites were mutated, an even greater induction of promoter activity was observed (>5-fold for m174m98 hTERT). These data indicate that the putative E2F sites at –174 and –98 bp in the hTERT promoter act as potent repressors of transcriptional activity.
Figure 1.


(A) Sequence of the hTERT promoter flanking the two putative E2F-binding sites. Numbering is relative to the transcription start site. The two putative E2F-binding sites are underlined. (B) The hTERT promoter constructs containing mutations in the two putative E2F-binding sites. The full-length promoter construct (hTERT) contains two putative E2F-binding sites at –174 and –98 (CGCGC) relative to the start of transcription. This sequence was cloned upstream of the luciferase reporter gene in pGL3 (Promega). The second construct contains only the 200 bp containing the putative E2F sites 5′ to the transcription start (–200 hTERT). The third construct contains the 200 bp 5′ to the transcription start and a mutation (CGCct) in the –174 E2F-binding site (m174 hTERT). The fourth construct contains the 200 bp 5′ to the transcription start and a mutation (CGCct) in the –98 E2F-binding site (m98 hTERT). The fifth construct contains the 200 bp 5′ to the transcription start and mutations in both the –174 and –98 E2F-binding sites (m174m98 hTERT).
Figure 2.

Mutation of the E2F sites increase transcription from the hTERT promoter. Luciferase activity measured in relative light units from each of the hTERT promoter constructs is shown. These experiments were performed three times with similar results. Error bars indicate SEM.
In order to determine if E2F-1 could bind the canonical sites in the hTERT promoter, electrophoretic mobility shift analysis using the sequence flanking the –174 bp site was performed. As shown in Figure 3, incubation of the radiolabeled probe with SCC25 nuclear extract produced a shifted complex. Inclusion of anti-E2F-1 antibody in the binding reactions produced a slight but consistent supershift, indicating the presence of this transcription factor in the complex. Incubation of the nuclear extracts with up to a 1000-fold molar excess of unlabeled probe effectively competed for binding to the radiolabeled oligonucleotide. However, incubation with unlabeled probe in which the putative E2F-binding site was mutated did not compete with the radiolabeled wild-type oligonucleotide. No binding to the radiolabeled mutant probe alone was observed nor was a shifted complex evident in the absence of nuclear extract. Similar results were obtained using a probe corresponding to the –98 bp binding site (data not shown). These results indicate that E2F-1 can specifically bind to the putative E2F sites located at –174 and –98 bp of the hTERT promoter.
Figure 3.

E2F-1 specifically binds to the E2F sites in the hTERT promoter. Nuclear extract from SCC25 cells was incubated with radiolabeled probe as described in Materials and Methods. Anti-E2F-1 antibody produced a supershifted band indicating the presence of E2F-1 in the complex. Some binding reactions were incubated with a 10- to 1000-fold molar excess of unlabeled probe (comp.) or a mutant probe (mut.) to determine binding specificity. No binding to the mutant probe alone was observed. The position of the free probe is shown. These experiments were performed three times with similar results. A representative gel is shown.
To determine the effects of E2F-1 overexpression on the activity of the hTERT promoter constructs, an E2F-1 expression vector or one lacking DNA-binding activity (E132) (36) was co-transfected along with the reporter plasmids. Blank expression vector was used for comparison. As shown in Figure 4, E2F-1 overexpression decreased the activity of the hTERT and –200 hTERT constructs by 70%, suggesting that E2F-1 acts as a transcriptional repressor of this promoter. E2F-1 also decreased the activity of the m174 hTERT construct by 50%. However, E2F-1 repressed transcription from the m98 hTERT construct by only 30% and the double mutant m174m98 hTERT construct was inhibited by only 10%. These results suggest that the putative E2F sites of the hTERT promoter mediate transcriptional repression by E2F-1. The E132 mutant E2F-1 expression vector lacking DNA-binding activity was ineffective at mediating transcriptional repression of the reporter vectors (Fig. 4). These data indicate that both of the putative E2F sites mediate transcriptional repression of the hTERT promoter, with the –98 site being somewhat more effective. E2F-1-mediated repression was also dependent on its DNA-binding ability.
Figure 4.

E2F-1 but not mutant E2F-1 represses transcription from the hTERT promoter. The hTERT promoter constructs described in Materials and Methods were transiently transfected into SCC25 cells with blank vector, an E2F-1 expression plasmid or a mutant E2F-1 lacking DNA-binding activity (E132). Luciferase activity from triplicate cultures was measured as described in Materials and Methods. These experiments were performed three times with similar results. Error bars indicate SEM.
In order to determine if E2F-1 could mediate repression of the endogenous telomerase gene, we stably transfected the E2F-1 expression plasmid or blank vector into SCC25 cells. hTERT mRNA expression and enzymatic activity were determined by northern blot and TRAP assay. As shown in Figure 5A, E2F-1 overexpression reduced hTERT mRNA levels by 4-fold. This reduction in hTERT mRNA expression was accompanied by decreased telomerase activity as determined by TRAP assay (Fig. 5B). These data indicate that E2F-1 inhibits expression of the endogenous hTERT gene at the level of transcription.
Figure 5.
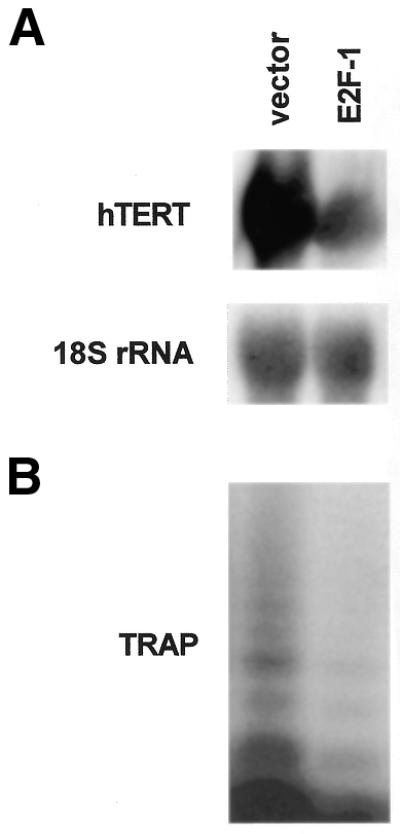
E2F-1 represses hTERT mRNA expression and telomerase activity in SCC25 cells. Cultures were stably transfected with an E2F-1 expression plasmid or blank vector. (A) hTERT mRNA expression was determined by northern blotting. Blots were stripped and probed with an 18S rRNA cDNA to ensure equal loading in each lane. (B) Telomerase activity was determined by the TRAP as described in Materials and Methods. These experiments were performed three times with similar results. Representative blots and gels are shown.
DISCUSSION
The key finding of this study is that E2F-1 represses transcription of the human telomerase gene. This repression was mediated by two non-canonical E2F-binding sites in the proximal portion of the hTERT promoter. E2F-1 bound specifically to these sites and its repressive effect was dependent on an intact DNA-binding domain. E2F-1 has been shown to promote cell cycle progression in numerous studies (for a review see 32). However, E2F-1 null mutant mice develop a number of malignant tumors, including lymphoma, lung adenocarcinoma and uterine sarcoma among others (33). The mechanisms by which inactivation of this transcription factor leads to this wide variety of cancers in mice are unclear. This study provides a potential insight into this paradox. If one of the normal functions of E2F-1 is inhibition of telomerase activity, functional inactivation of this factor may allow activation of the enzyme, thereby predisposing potential cancer cells to immortalization. However, it is likely that other genetic events occur during tumorigenesis in these mice which are independent of E2F-1. While our promoter experiments, northern blot data and TRAP assays all confirm inhibition of telomerase activity by E2F-1, a direct demonstration of transcriptional repression by nuclear run-off analysis will be required in future experiments. Additionally, our results do not rule out the indirect effects of loss of E2F-1 function on downstream target genes.
Historically, E2F proteins have been characterized as transcriptional activators (37). Few studies have demonstrated transcriptional repression by these proteins (38). Our characterization of E2F-1 as a transcriptional repressor of human telomerase gene expression represents a unique new role for these factors. Whether other E2F proteins can also repress the telomerase promoter will be the subject of future experiments. The application of large-scale expression analyses will likely reveal additional genes which are repressed by E2F transcription factors.
The role of other tumor suppressors in regulating transcription of the hTERT gene has been investigated. The Wilms’ tumor suppressor (WT1) has been shown to repress the activity of hTERT promoter constructs (39). The effects of WT1 on the hTERT promoter were mediated by a canonical site between –307 and –423 bp. This repression was not observed in HeLa cells, which lack functional WT1. These results suggest that hTERT activation is at least in part cell specific.
Conversely, activation of the hTERT promoter and the endogenous gene by c-Myc has been the focus of several studies (40–42). Myc overexpression significantly increases activity of the core promoter. Myc/Max heterodimers are believed to be the activating complex whereas Mad/Max acts as a transcriptional repressor. Sp1 has also been shown to activate the hTERT promoter in cooperation with Myc. These two proteins are induced when fibroblasts overcome replicative senescence, which correlates with telomerase activation. In breast cancer cells, estrogen acting via its nuclear receptor has been shown to activate the hTERT promoter via an imperfect palindrome, but also in conjunction with Myc/Max (43).
In summary, E2F-1 represses transcription via consensus binding sites in the hTERT promoter, suggesting a potential mechanism for tumor formation in the absence of this factor. Recently, adenoviral transduction of E2F-1 into human squamous cell carcinoma lines was shown to inhibit telomerase activity (44). Regulation of telomerase gene expression remains a complex issue. Future experiments will be aimed at dissecting the cell cycle progression and tumor suppression functions of E2F-1 with regard to its effects on telomerase expression.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Dr Robert Weinberg for the hTERT cDNA and Dr Karen Vousden for E2F-1 expression vectors. This study was supported by American Cancer Society grant IRG-58-007-41-02.
References
- 1.Blackburn E.H. (1991) Structure and function of telomeres. Nature, 350, 569–573. [DOI] [PubMed] [Google Scholar]
- 2.Zakian V.H. (1995) Telomeres: beginning to understand the end. Science, 270, 1601–1606. [DOI] [PubMed] [Google Scholar]
- 3.Chang E. and Harley,C.B. (1995) Telomere length and replicative aging in human vascular tissues. Proc. Natl Acad. Sci. USA, 92, 11190–11194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blackburn E.H. (1994) Telomeres: no end in sight. Cell, 77, 621–623. [DOI] [PubMed] [Google Scholar]
- 5.Wright W.E., Brasiskyte,D., Piatyszek,M.A. and Shay,J.W. (1996) Experimental elongation of telomeres extends the lifespan of immortal × normal cell hybrids. EMBO J., 15, 1734–1741. [PMC free article] [PubMed] [Google Scholar]
- 6.Harley C.B. (1991) Telomere loss: mitotic clock or genetic time bomb? Mutat. Res., 256, 271–282. [DOI] [PubMed] [Google Scholar]
- 7.Axelrod N. (1995) Of telomeres and tumors. Nature Med., 2, 158–159. [DOI] [PubMed] [Google Scholar]
- 8.Counter C.M., Avilion,A.A., LeFeuvre,C.E., Stewart,N.G., Greider,C.W., Harley,C.B. and Bacchetti,S. (1992) Telomere shortening associated with chromosome instability is arrested in immortal cells which express telomerase activity. EMBO J., 11, 1921–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bryan T.M., Englezou,A., Gupta,J., Bacchetti,S. and Reddel,R.R. (1995) Telomere elongation in immortal human cells without detectable telomerase activity. EMBO J., 14, 4240–4248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blasco M.A., Funk,W., Villeponteau,B. and Greider,C.W. (1995) Functional characterization and developmental regulation of mouse telomerase RNA. Science, 269, 1267–1270. [DOI] [PubMed] [Google Scholar]
- 11.Feng J., Funk,W.D., Wang,S.S., Weinrich,S.L., Avilion,A.A., Chiu,C.P., Adams,R.R., Chang,E., Allsopp,R.C., Yu,J., Le,S., West,M.D., Harley,C.B., Andrews,A.W., Greider,C.W. and Villeponteau,B. (1995) The RNA component of human telomerase. Science, 269, 1236–1241. [DOI] [PubMed] [Google Scholar]
- 12.McEachern M.J. and Blackburn,E.H. (1995) Runaway telomere elongation caused by telomerase RNA gene mutations. Nature, 376, 403–409. [DOI] [PubMed] [Google Scholar]
- 13.Blasco M.A., Lee,H.W., Hande,M.P., Samper,E., Lansdorp,P.M., DePinho,R.A. and Greider,C.W. (1997) Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell, 91, 25–34. [DOI] [PubMed] [Google Scholar]
- 14.Rudolph K.L., Chang,S., Lee,H.W., Blasco,M., Gottlieb,G.J., Greider,C. and DePinho,R.A. (1999) Longevity, stress response and cancer in aging telomerase deficient mice. Cell, 96, 701–712. [DOI] [PubMed] [Google Scholar]
- 15.Chong L., van Steensel,B., Broccoli,D., Erdjument-Bromage,H., Hanish,J., Tempst,P. and de Lange,T. (1995) A human telomeric protein. Science, 270, 1663–1667. [DOI] [PubMed] [Google Scholar]
- 16.Counter C.M., Meyerson,M., Eaton,E.N. and Weinberg,R.A. (1997) The catalytic subunit of yeast telomerase. Proc. Natl Acad. Sci. USA, 94, 9202–9207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harrington L., McPhail,T., Mar,V., Zhou,W., Oulton,R., Amgen EST Program, Bass,M.B., Arruda,I. and Robinson,M.O. (1997) A mammalian telomerase associated protein. Science, 275, 973–977. [DOI] [PubMed] [Google Scholar]
- 18.Van Steensel B. and de Lange,T. (1997) Control of telomere length by the human telomeric protein TRF-1. Nature, 385, 740–743. [DOI] [PubMed] [Google Scholar]
- 19.Meyerson M., Counter,C.M., Eaton,E.N., Ellisen,L.W., Steiner,P., Caddle,S.D., Ziaugra,L., Beijersbergen,R.L., Davidoff,M.J., Liu,Q., Bacchetti,S., Haber,D.A. and Weinberg,R.A. (1997) hEST2, the putative human telomerase catalytic subunit gene, is upregulated in tumor cells and during immortalization. Cell, 90, 785–795. [DOI] [PubMed] [Google Scholar]
- 20.Nakamura T.M., Morin,G.B., Chapman,K.B., Weinrich,S.L., Andrews,W.H., Lingner,J., Harley,C.B. and Cech,T.R. (1997) Telomerase catalytic subunit homologs from fission yeast and human. Science, 277, 955–959. [DOI] [PubMed] [Google Scholar]
- 21.Bodnar A.G., Ouellette,M., Frolkis,M., Holt,S.E., Chiu,C.P., Morin,G.B., Harley,C.B., Shay,J.W., Lichtsteiner,S. and Wright,W.E. (1998) Extension of lifespan by introduction of telomerase into normal human cells. Science, 279, 349–352. [DOI] [PubMed] [Google Scholar]
- 22.Harle-Bachor C. and Boukamp,P. (1996) Telomerase activity in the regenerative basal layer of epidermis in human skin and in immortal and carcinoma derived skin keratinocytes. Proc. Natl Acad. Sci. USA, 93, 6476–6481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klingelhutz A.J., Foster,S.A. and McDougall,J.K. (1996) Telomerase activation by the E6 gene product of human papillomavirus type 16. Nature, 380, 79–82. [DOI] [PubMed] [Google Scholar]
- 24.Bestilny L.J., Brown,C.B., Miura,Y., Robertson,L.D. and Riabowol,K.T. (1996) Selective inhibition of telomerase activity during terminal differentiation of immortal cell lines. Cancer Res., 56, 1796–1802. [PubMed] [Google Scholar]
- 25.Kim N.W., Piatyszek,M.A., Prowse,K.R., Harley,C.B., West,M.D., Ho,P.L.C., Coviello,G.M., Wright,W.E., Weinrich,S.L. and Shay,J.W. (1994) Specific association of human telomerase activity with immortal cells and cancer. Science, 266, 2011–2014. [DOI] [PubMed] [Google Scholar]
- 26.Blasco M.A., Rizen,M., Greider,C.W. and Hanahan,D. (1996) Differential regulation of telomerase activity and telomerase RNA during multistage tumorigenesis. Nature Genet., 12, 200–204. [DOI] [PubMed] [Google Scholar]
- 27.Hiyama E., Hiyama,K., Yokoyama,T., Matsuura,Y., Piatyszek,M.A. and Shay,J.W. (1995) Correlating telomerase activity levels with human neuroblastoma outcomes. Nature Med., 1, 249–255. [DOI] [PubMed] [Google Scholar]
- 28.Holt S.E., Wright,W.E. and Shay,J.W. (1996) Regulation of telomerase activity in immortal cell lines. Mol. Cell. Biol., 16, 2932–2939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhu X., Kumar,R., Mandal,M., Sharma,N., Sharma,H.W., Dhingra,U., Sokoloski,J.A., Hsiao,R. and Narayanan,R. (1996) Cell cycle dependent modulation of telomerase activity in tumor cells. Proc. Natl Acad. Sci. USA, 93, 6091–6095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Buchkovich K.J. and Greider,C.W. (1996) Telomerase regulation during entry into the cell cycle in normal human T cells. Mol. Biol. Cell, 7, 1443–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Landberg G., Nielsen,N.H., Nilsson,P., Emdin,S.O., Cajander,J. and Roos,G. (1997) Telomerase activity is associated with cell cycle deregulation in human breast cancer. Cancer Res., 57, 549–554. [PubMed] [Google Scholar]
- 32.Sherr C.J. (1996) Cancer cell cycles. Science, 274, 1672–1677. [DOI] [PubMed] [Google Scholar]
- 33.Yamasaki L., Jacks,T., Bronson,R., Goillot,E., Harlow,E. and Dyson,N.J. (1996) Tumor induction and tissue atrophy in mice lacking E2F-1. Cell, 85, 537–548. [DOI] [PubMed] [Google Scholar]
- 34.Nguyen D.C. and Crowe,D.L. (1999) Intact functional domains of the retinoblastoma gene product (pRb) are required for downregulation of telomerase activity. Biochim. Biophys. Acta, 1445, 207–215. [DOI] [PubMed] [Google Scholar]
- 35.Takakura M., Kyo,S., Kanaya,T., Hirano,H., Takeda,J., Yutsudo,M. and Inoue,M. (1999) Cloning of human telomerase catalytic subunit (hTERT) gene promoter and indentification of proximal core promoter sequences essential for transcriptional activation in immortalized and cancer cells. Cancer Res., 59, 551–557. [PubMed] [Google Scholar]
- 36.Phillips A.C., Bates,S., Ryan,K.M., Helin,K. and Vousden,K.H. (1997) Induction of DNA synthesis and apoptosis are separable functions of E2F-1. Genes Dev., 11, 1853–1863. [DOI] [PubMed] [Google Scholar]
- 37.Muller H., Bracken,A.P., Vernell,R., Moroni,M.C., Christians,F., Grassilli,E., Prosperini,E., Vigo,E., Oliner,J.D. and Helin,K. (2001) E2Fs regulate the expression of genes involved in differentiation, development, proliferation and apoptosis. Genes Dev., 15, 267–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Koziczak M., Krek,W. and Nagamine,Y. (2000) Pocket protein independent repression of urokinase type plasminogen activator and plasminogen activator inhibitor 1 gene expression by E2F1. Mol. Cell. Biol., 20, 2014–2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Oh S., Song,Y., Yim,J. and Kim,T.K. (1999) The Wilms’ tumor 1 tumor suppressor gene represses transcription of the human telomerase reverse transcriptase gene. J. Biol. Chem., 274, 37473–37478. [DOI] [PubMed] [Google Scholar]
- 40.Wang J., Xie,L.Y., Allan,S., Beach,D. and Hannon,G.J. (1998) Myc activates telomerase. Genes Dev., 12, 1769–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Horikawa I., Cable,P.L., Afshari,C. and Barrett,J.C. (1999) Cloning and characterization of the promoter region of human telomerase reverse transcriptase gene. Cancer Res., 59, 826–830. [PubMed] [Google Scholar]
- 42.Kyo S., Takakura,M., Taira,T., Kanaya,T., Itoh,H., Yutsudo,M., Ariga,H. and Inoue,M. (2000) Sp1 cooperates with c-myc to activate transcription of the human telomerase reverse transcriptase gene (hTERT). Nucleic Acids Res., 28, 669–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kyo S., Takakura,M., Kanaya,T., Zhuo,W., Fujimoto,K., Nishio,Y., Orimo,A. and Inoue,M. (1999) Estrogen activates telomerase. Cancer Res., 59, 5917–5921. [PubMed] [Google Scholar]
- 44.Henderson Y.C., Breau,R.L., Liu,T.J. and Clayman,G.L. (2000) Telomerase activity in head and neck tumors after introduction of wild type p53, p21, p16 and E2F-1 genes by means of recombinant adenovirus. Head Neck, 22, 347–354. [DOI] [PubMed] [Google Scholar]