

Abstract
Thoracic Aortic Aneurysm and Dissection (TAAD) is a life-threatening pathology and remains challenging worldwide. Up to 40% of TAAD are hereditary with complex heterogeneous genetic backgrounds. Recently, next-generation sequencing (NGS) has been successfully applied to identify genetic variants in an efficient and cost-effective manner. In our study, NGS coupled with DNA target-capture array was used to screen 11 known causative genes of TAAD in 70 patients from Southern China. All the identified variants were confirmed by Sanger sequencing. We identified forty variants in 36 patients (51.4%), including three known pathogenic (7.5%), 10 likely pathogenic variants (25%, 9 in FBN1, 1 in ACTA2), and 27 variants with uncertain significance (VUS) (67.5%). Among the 27 VUS, 14 (51.9%) were in the FBN1 gene, 3 in Col5A2, 2 in ACTA2, 2 in MYH11, 2 in MYLK, 2 in SLC2A10, 1 in MSTN and 1 in SMAD3 respectively. Based on the segregation data and independent reports, five known likely pathogenic variants and four VUS were upgraded to pathogenic variant and likely pathogenic variant respectively. Our data indicate that NGS is a highly efficient genetic method for identification of pathogenic variants in TAAD patients.
Introduction
Thoracic aortic aneurysm (TAA) is life-threatening disease with significant morbidity and mortality. The actual incidence of TAA is hard to estimate due to the fact that it can be non-symptomatic until the onset of acute aortic dissection (AD) or rupture. Thoracic aortic aneurysm and dissection (TAAD) may develop into a highly lethal situation. The incidence of acute AD has been reported to be approximately 2–3.5/100,000 inhabitants per year in the US1. It is estimated that individual lifetime risk of rupture or dissection reaches 34% by the time aorta achieves a diameter of 6 cm2. Since TAAD at the early stage is silent and non-symptomatic, early detection and evaluation of the risk of onset and disease progression are crucial for avoiding life-threatening situations.
Previous studies have described the complex and heterogeneous genetic background of TAAD which is often associated with connective tissue diseases including Marfan syndrome (MFS), Ehlers-Danlos syndrome (EDS), Shprintzen-Goldberg syndrome (SGS) and Loeys–Dietz syndrome (LDS). The term symptomatic TAAD is used for characterizing this special group of TAAD patients3. However, it was reported that TAAD patients with family history took up approximately 20–40% of the TAAD population4. Since identification of the very first mutation in the FBN1 in MFS5, 6, causative mutations have subsequently been identified in a dozen others including COL3A1 and COL5A2 mutations associated with EDS pathogenesis7, 8, TGFBR2 mutations associated with Marfan-like syndrome9, and TGFBR1, TGFBR2 and SMAD3 associated with LDS10–12. Gene mutations including ACTA2, MSTN, MYH11, MYLK, and SLC2A10 8, 11, 13–17 are associated with vascular diseases and are responsible for TAAD.
The high throughput, multiplexing next-generation sequencing (NGS) is currently regarded as the most powerful technology for genetic testing in clinical settings. NGS application as a screening method for the diagnosis of TAAD has been reported in Caucasian population. Proost et al. screened 14 genes from 55 patients and identified 15 pathogenic mutation and six variants of uncertain significance (VUS)18; Ziganshin et al. recruited a group of 102 patients with 21 genes examined and found that 4% of the patients had likely pathogenic variants and 22% had VUS19; In a cohort of 175 patients, Wooderchak-Donahue et al. found 51 rare variants in 10 selected genes20, in which pathogenic variants presented in 10% patients and VUS in 18% patients.
In the current study, 11 TAAD-associated genes including ACTA2, Col3A1, Col5A2, FBN1, MSTN, MYH11, MYLK, SLC2A10, SMAD3, TGFBR1, and TGFBR2 were analyzed by NGS among 70 TAAD subjects enrolled from southern China. Forty variants were identified in 36 TAAD patients. Among all the variants, 12 pathogenic/likely pathogenic variants were in FBN1 gene, one likely pathogenic variant was in ACTA2 gene, and the other 27 VUS presented in eight genes.
Results
Clinical findings
The clinical characteristics of TAAD patients were summarized in Table 1. The mean age at the time of genetic testing was 45.7 years. Age of the patients ranged from 18 to 66 years. Sixty (85.7%) patients were male and 10 (14.3%) patients were female. Thirteen (18.6%) patients had a family history of TAAD. Twenty-four (34.3%) patients revealed a history of tobacco use, while 11 (15.7%) patients revealed a history of alcohol use.
Table 1.
Clinical characteristics of the study group.
| Clinical characteristics of the study group, n = 70 | |
|---|---|
| Male (n,%) | 60(85.7) |
| Female (n,%) | 10(14.3) |
| Age(mean, SD) | 45.7(8.5) |
| Tobacco use (n,%) | 24(34.3) |
| Alcohol use (n,%) | 11(15.7) |
| Family history (n,%) | 13(18.6) |
| Suspected MFS (n,%) | 15(21.4) |
| CAD (n,%) | 5(7.1) |
| Hypertension (n,%) | 31(44.3) |
| Hyperlipidemia (n,%) | 18(25.7) |
| Associated structural abnormalities | |
| Thoracic AD (n,%) | 43(61.4) |
| TAA (n,%) | 27(38.6) |
| BAV (n,%) | 4(5.7) |
| Other CV* | 28(40.0) |
AD: aortic dissection, BAV: Bicuspid aortic valve, CAD: coronary artery disease, CV: cardiovascular, Other CV*: includes findings such as mitral valve prolapse, tricuspid valve prolapse, mitral regurgitation or congenital heart disease, thick aortic valve, TAA: thoracic aortic aneurysm.
A variety of structural abnormalities associated with TAAD were identified among several patients: 43(61.4%) patients had thoracic AD, TAA was present in 27 (38.6%) patients. Bicuspid aortic valve (BAV) was present in 4 (5.7%) patients. Twenty-eight (40%) patients had other cardiovascular diseases. Five (7.1%) patients had coronary artery disease (CAD). Thirty-one (44.3%) patients had hypertension. Eighteen (25.7%) patients were hyperlipidemic. Fifteen (21.4%) patients were suspected of MFS, with their clinical features and identified variants shown in Table 2. Eight (53.3%) had a positive family history, all included patients have aortic root dilatation z score ≥2, eight included patients have systemic score >=7 points. Cardiovascular, skeletal, ectopia lentis, dural, skin and lung problems were identified in 15(100%), 15(100%), 9(60%), 1(6.7%), 1(6.7%) and 3(20.0%) of patients, respectively. Therefore, there are 12 patients diagnosed MFS based on clinical data, and 3 (TAAD003, TAAD024, TAAD038) based on genetic examinations according to the revised Ghent criteria.
Table 2.
Clinical Data and Variants identified in suspected Marfan syndrome patients.
| Family | Cardiova-scular | Skeletal | ectopia lentis | Dural ectasia | skin | lung | Z score | System-ic score | Family History (Y/N) | Fulfill Ghent Criteria before genetic test (Y/N) | Gene | Classification |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TAAD003 | + | + | − | − | − | − | 12.91 | 5 | N | N | FBN1 | Likely patho |
| TAAD005 | + | + | + | − | − | − | 11.11 | ≥7 | N | Y | FBN1 | VUS |
| TAAD013 | + | + | + | − | − | − | 8.87 | ≥7 | N | Y | FBN1 | Likely patho |
| TAAD020 | + | + | − | − | + | − | 24.44 | ≥7 | Y | Y | FBN1 | VUS |
| TAAD023 | + | + | − | − | − | + | 22.06 | ≥7 | N | Y | FBN1 | VUS |
| TAAD024 | + | + | − | − | − | + | 18.11 | 4 | N | N | FBN1 | Patho |
| TAAD036 | + | + | + | − | − | − | 15.83 | ≥7 | Y | Y | FBN1 | VUS |
| TAAD038 | + | + | − | − | − | − | 4.92 | 5 | N | N | FBN1 | Likely patho |
| TAAD039 | + | + | + | + | − | − | 4.47 | ≥7 | Y | Y | FBN1 | VUS |
| TAAD045 | + | + | − | − | − | − | 14.19 | 4 | Y | Y | FBN1 | Likely patho |
| TAAD046 | + | + | + | − | − | + | 4.01 | ≥7 | Y | Y | FBN1 | VUS |
| TAAD056 | + | + | + | − | − | − | 8.54 | ≥7 | N | Y | FBN1 | Patho |
| TAAD068 | + | + | + | − | − | − | 9.48 | 6 | Y | Y | FBN1 | Likely patho |
| TAAD069 | + | + | + | − | − | − | 6.16 | 5 | Y | Y | FBN1 | Likely patho |
| TAAD070 | + | + | + | − | − | − | 7.02 | 6 | Y | Y | FBN1 | VUS |
+:positive, −;negative; Patho: pathogenic; VUS: variants of uncertain significance.
Mutation analysis
In a total of 70 analyzed TAAD patients, 40 rare variants were identified in 36 patients (36/70, 51.4%) (Tables 2 and 3). Twenty-seven of these 40 identified rare variants (67.5%) were novel findings. Variants were classified in line with recommendations from the American College of Medical Genetics (ACMG)21 based on the following information: (I) published data including functional and clinical information, (II) variant frequency in the dbSNP and Exome Variant Server and presence in any public variant databases, (III) conservation of the altered residue, (IV) computational prediction programs for variant causality including splicing effects: SIFT22 and PolyPhen23, and (V) family segregation studies. Available evidences for each new variant were evaluated by two independent reviewers.
Table 3.
Variants identified in FBN1 genes.
| Family | Disease | Family History(Y/N) | Nucleotide | Protein | Type | Classification | Reference |
|---|---|---|---|---|---|---|---|
| TAAD001 | AD + AR | N | c.7204+6T > G | — | Splicing | VUS | * |
| TAAD003 | MFS + AD + AR | N | c.1098G > T | p.Trp366Cys | Missense | Likely patho | 25 |
| TAAD005 | MFS + AD + AR | N | c.8123A > G | p.Asn2708Ser | Missense | VUS | 44 |
| TAAD011 | TAA + AR | N | c.1129T > A | p.Cys377Ser | Missense | VUS | * |
| TAAD013 | MFS + TAA + AR | N | c.7471 delA | p.Thr2491ProfsX191 | Frameshift | Likely patho | * |
| TAAD014 | TAA + AR | N | c.1496G > C | p.Cys499Ser | Missense | Likely patho | 27 |
| TAAD017 | AD | N | c.7754T > C | p.Ile2585Thr | Missense | patho | 45 |
| TAAD020 | MFS + TAA + AR | Y | c.4049G > A | p.Cys1350Tyr | Missense | VUS | 46 |
| TAAD021 | TAA + AR | Y | c.5084G > T | p.Cys1695Phe | Missense | VUS | * |
| TAAD023 | MFS + TAA + AR | N | c.7988G > A | p.Cys2663Tyr | Missense | VUS | * |
| TAAD024 | MFS + AD | N | c.8147_8148insA | p.Tyr2716Ter | Frameshift | patho | 18 |
| TAAD026 | AD | N | c.911G > T | p.Cys304Phe | Missense | VUS | * |
| TAAD030 | AD | N | c.5742C > A | p.Cys1914Ter | Nonsense | Likely patho | * |
| TAAD034 | AD | N | c.8069T > G | p.Met2690Arg | Missense | VUS | * |
| TAAD036 | MFS + MR + TR | Y | c.5579G > T | p.Cys1860Phe | Missense | VUS | 46 |
| TAAD038 | MFS + AD + AR | N | c.4454G > A | p.Cys1485Tyr | Missense | Likely patho | 28 |
| TAAD039 | MFS + AD + AR | N | c.2627G > T | p.Cys876Phe | Missense | VUS | * |
| TAAD045 | MFS + TAA + AR | Y | EX5_54 DEL | — | Exon deletion | Likely patho | * |
| TAAD046 | MFS + AD + AR | Y | c.2216G > A | p.Cys739Tyr | Missense | VUS | * |
| TAAD050 | TAA + AR + MR | Y | EX4_53 DEL | — | Exon deletion | Likely patho | * |
| TAAD051 | AD | Y | c.2056G > A | p.Ala686Thr | Missense | VUS | 29 |
| TAAD053 | AD | Y | c.7567A > C | p.Ile2523Leu | Missense | VUS | * |
| TAAD056 | MFS + AR + MR | N | c.247 + 1G > A | — | Splicing | patho | 47 |
| TAAD068 | MFS + TAA + AR | Y | c.4292G > A | p.Cys1431Tyr | Missense | Likely patho | 32 |
| TAAD069 | MFS + AR + MR | Y | c.8292_8293insT | p.Phe2764PhefsX10 | Frameshift | Likely patho | * |
| TAAD070 | MFS + TAA + AR | Y | c.6801C > A | p.Asn2267Lys | Missense | VUS | 18 |
*This study.
The underline patients were with concomitant mutations in different genes.
AD: aortic dissection; AR: aortic regurgitation; MFS: Marfan syndrome; MR: mitral regurgitation; TAA: thoracic aortic aneurysm; TR: tricuspid regurgitation; Patho: pathogenic; VUS: variants of uncertain significance.
Based on software estimations, functional analysis and the segregation data, it was assumed that among all the 40 variants, 3 were pathogenic (7.5%, all in FBN1 gene), 10 were likely pathogenic (25.0%, 9 for the FBN1 gene, 1 for the ACTA2 gene), and 27 were VUS (67.5%). Among the 27 VUS, 14 (51.8%) were in the FBN1 gene, 3 in Col5A2, 2 in ACTA2, 2 in MYH11, 2 in MYLK, 2 in SLC2A10, 1 in MSTN and 1 in SMAD3 respectively. The distribution of identified variants in TAAD associated genes were shown in Fig. 1.
Figure 1.
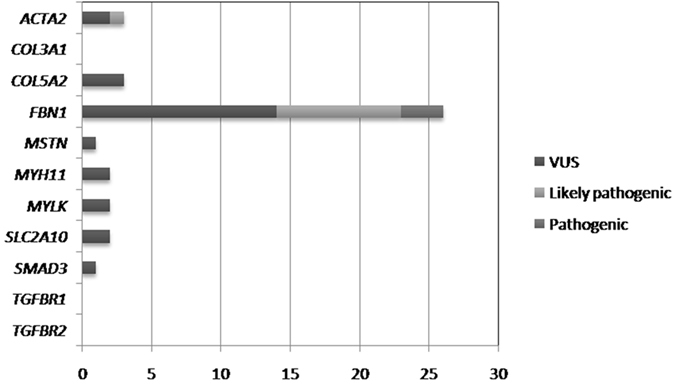
Showed the distribution of identified variants in TAAD associated genes in the current study. Among all the 40 variants, 3 were pathogenic (all in FBN1 gene), 10 were likely pathogenic (9 for the FBN1 gene, 1 for the ACTA2 gene), and 27 were VUS (14 in FBN1 gene, 3 in Col5A2, 2 in ACTA2, 2 in MYH11,2 in MYLK, 2 in SLC2A10,1 in MSTN and 1 in SMAD3 respectively).
FBN1
Similar to results reported by other studies18, 20, 24, the present study again confirmed that most of the variants were found in FBN1 gene, which is the primary causative gene in TAAD. As listed in Table 3, three pathogenic mutations, c.7754T > C missense mutation (TAAD017), c.8147_8148insA frameshift mutation (TAAD024), and c.247 + 1G > A splicing mutation (TAAD056) were identified. In addition, we detected four reported likely pathogenic mutation, including c.1098G > T (TAAD003), c.1496G > C (TAAD014), c.4454G > A (TAAD038) and c4292G > A (TAAD068). According to the latest ACMG recommendations for interpreting and reporting sequence variations21, these four likely pathogenic variants could be reclassified into pathogenic variants due to previous independent reports25–32. Five out of these seven probands were diagnosed as MFS, and four of them were with family history.
Importantly, five novel variants, including 2 frameshift (c.7471delAand c.8292_8293insT), 2 exon deletion (EX5_54 DEL and EX4_53 DEL)and a missense mutation (c.5742C > A) in FBN1 gene were classified as likely pathogenic variants. Mutation c.7471delA in case TAAD013 without family history located in the Von Willebrand factor type A (vWA) domain, which is a hallmark of blood coagulation protein von Willebrand Factor. Truncation of the vWA domain may affect function of protein Fibrillin 133. Another frameshift mutation c.8292_8293insT identified in case TAAD069 with MFS is located in the Asprosin domain and was carried by the patient’s sister without any symptom yet, whose long term follow-up was scheduled. The EX5_54 DEL mutation was identified in a 52-year-old female with MFS (TAAD045). A family history of the disease was found in her father (42 y) and son (16 y), both of whom died due to rupturing of dissecting aneurysm developed from MFS. The second Exon deletion mutation EX4_53 DEL (TAAD050) was found in a 21-year male patient diagnosed with TAA as well as severe aortic valve and mitral valve regurgitation. Family history revealed that his mother died of AD at the age of 38.The remaining novel nonsense mutation c.5742C > A was detected in a 41-year-old male with Stanford A AD (TAAD030) located in the calcium-binding epidermal growth factor (ebEGF) modules, contributing to early termination of encoded protein for the amino acid, producing truncated protein and eventually causing great changes on the structure and function of the protein.
Additionally, fourteen VUS in FBN1 were found. Seven out of them have confirmed TAAD family history (TAAD020, TAAD021, TAAD036, TAAD046, TAAD051, TAAD053, TAAD070). Nine were reported for the first time while the other 5 had been reported by other studies. A large majority of them were missense mutation (13/14, 92.9%). Notably, two novel VUS, c.5084G > T, p.Cys1695Phe (TAAD021) and c.7988G > A, p.Cys2663Tyr (TAAD023), were similar to pathogenic mutations of c.5084G > A, p.Cys1695Tyr26 and c.8121G > C, p.Cys2663Ser27 described in previous studies in terms of mutation site. Aside from that, SIFT and PolyPhen protein function had both been proved to be detrimental. Thus, these variants were highly likely to be pathogenic. Although 4 novel VUS for FBN1, c.1129T > A (TAAD011), c.911G > T (TAAD026), c.2627G > T(TAAD039) and c.2216G > A (TAAD046) were predicted to be detrimental by SIFT and PolyPhen protein function prediction, their pathogenicity is still not well established.
ACTA2
A previously reported missense mutation variant c.635G > A (p.Arg212Gln) in the ACTIN domain of ACTA2 34–36 was carried by a 48-year-old male (TAAD027) with diagnosis of Stanford A AD, ascending aortic aneurysm, and severe aortic valve insufficiency in the current study. This mutation was originally classified as likely pathogenic and elevated to pathogenic with regard to the fact that it was proved by two dependent studies and predicted harmful by SIFT and PolyPhen software. No family history was found in this case, and family verification were negative.
In addition, 2 novel missense VUS in ACTA2 (TAAD041, TAAD065) were found in the current study in absence of family history. Their clinical significance and pathogenesis have yet to be elucidated.
Other TAAD associated genes
Besides 14 VUS found in FBN1, 2 VUS in ACTA2, as well as 11 other VUS, including 3 in Col5A2, 2 in SLC2A10, 2 in MYH11, 2 in MYLK, 1 in MSTN and 1 in SMAD3 were identified in the current study (Table 4). Almost all VUS (9/11, 81.8%) were missense mutation. Detailed information was listed in Table 4.
Table 4.
Thirteen novel VUS identified in other TAAD associated genes.
| Gene | Family | Disease | Family History(Y/N) | Nucleotide | Protein | Type |
|---|---|---|---|---|---|---|
| ACTA2 | TAAD041 | AD | N | c.401T > C | p.Met134Thr | Missense |
| ACTA2 | TAAD064 | AD | N | c.574A > G | p.Met192Val | Missense |
| COL5A2 | TAAD046 | MFS + AD + AR | Y | c.2846C > G | p.Ser949Cys | Missense |
| COL5A2 | TAAD054 | AD + AR | N | c.3388A > G | p.Lys1130Glu | Missense |
| COL5A2 | TAAD061 | AD | N | c.3856G > A | p.Asp1286Asn | Missense |
| MSTN | TAAD022 | AD | N | c.747 + 8A > G | — | Splicing |
| MYH11 | TAAD034 | AD | N | c.1523G > A | p.Arg508His | Missense |
| MYH11 | TAAD040 | AD | N | c.1907A > G | p.Asp636Gly | Missense |
| MYLK | TAAD019 | AD + AR | N | c.4882G > A | p.Val1628Met | Missense |
| MYLK | TAAD065 | AD | N | c.3923A > G | p.Tyr1308Cys | Missense |
| SLC2A10 | TAAD016 | AD + AR | N | c.1154C > T | p.Ala385Val | Missense |
| SLC2A10 | TAAD051 | AD | Y | c.1456G > T | p.Ala486Ser | Missense |
| SMAD3 | TAAD051 | AD | Y | c.532 + 9G > A | — | Splicing |
The underline patients were with concomitant mutations in different genes.
To be noted, more than one VUS in different genes were found in 3 probands. Case TAAD034 with AD had two novel heterozygous variants: c.8069T > G in FBN1 and c.1523G > A in MYH11.Although no family history was confirmed in this patient, c.1523G > A in MYH11was predicted to be pathogenic by two programs (SIFT and PolyPhen) while c.8069T > G in FBN1 showed a nonpathogenic result. Thus, MYH11c.1523G > A was more likely than FBN1c.8069T > G to be pathogenic. TAAD046 was diagnosed MFS and AD with confirmed family history. His mother died of AD (<40 y), and his two brothers were both diagnosed as MFS. The old brother received Betall operation but died of cerebral hemorrhage. The young brother died due to rupture of AD. This proband had two novel VUS (FBN1 c.2216G > A and COL5A2 c.2846C > G), both of which were missense variants. Further prediction on the protein function was performed indicating that FBN1 c.2216 G > A was harmful both by SIFT and Polyphen while COL5A2c.2846C > G was pathogenic by SIFT and non-pathogenic by Polyphen, respectively. However, COL5A2 c.2846C > G was downgraded to likely benign after familial targeted sequencing revealed that the variant was present in his unaffected grandson. At this point, we believe that FBN1 c.2216G > A was much likely to be the pathogenic mutation. Case TAAD051 with Stanford A AD had two missense and one splicing mutation (FBN1 c.2056G > A, SLC2A10 c.1456G > T and SMAD3 c.532 + 9G > A). FBN1 c.2056G > A has been reported in a previous study with unknown clinical significance29, 37, while SLC2A10 c.1456G > T and SMAD3 c.532 + 9G > A were novel identified variants in this study. All of them were similarly predicted to be pathogenic and non-pathogenic by SIFT and PolyPhen protein function prediction, respectively. Except for the pathology of aortic artery, further investigation of this patient showed no clinical presentation of MFS, EDS or LDSIII such as abnormality of skin, crystalline, skeleton as well as osteoarthritis and so on so forth. Family history also revealed that the patient had an affected father, but his DNA was unavailable. Therefore, the pathogenicity of these mutations remained uncertain.
Family confirmation
Furthermore, 21 of 36 probands were further verified in the family, including 13 with pathogenic/likely pathogenic and 8 VUS with family history. Among the 21 verified families, 5 members from different families were found to have same pathogenic variants with probands (TAAD030, TAAD053, TAAD068, TAAD069, TAAD070). Three of them (TAAD053-brother, TAAD068-sister, TAAD070-daugther) had been previously diagnosed with TAAD. Therefore, the pathogenicity of 2 VUS, FBN1 c.7567A > C in TAAD053 and FBN1 c.6801C > A in TAAD070 respectively, were validated and upgraded in accordance of ACMG guideline. The other two members (TAAD030-son, TAAD069-sister) were identified as pathogenic mutation carrier in our study. Both of them received imaging test and laboratory examination for TAAD subsequently. Results revealed abnormality of skeleton and crystalline and systemic score ≥ 7 in TAAD030-son meeting the diagnostic criteria of MFS. Detailed clinical characteristics of genotype positive probands and family members were shown in Table 5, pedigrees of these families were shown in Fig. 2.
Table 5.
Clinical characteristics of probands with pathogenic/likely pathogenic mutation and related mutation carriers.
| Family | Position on pedigree-status | Sex | Age | Type Of syndr-ome | CVS involve-ment of the aorta | Aortic root | AR | LV-EF | Extensi-on | Other CVS involve-ment | Type of surgery | Age at surg-ery | Gene | Mutation |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TAAD 003 | II:1-prob-And | M | 28 | MFS | AD | 56 | 3 | 60 | R, As, Ar, D, Ab | NO | B + TAR + DASI | 28 | FBN1 | c.1098G > T |
| TAAD 013 | II:1-prob-and | M | 35 | MFS | AD | 65 | 3 | 45 | R, As, Ar, D, Ab | MR(1), TR(1) | B + TAR + DASI | 35 | FBN1 | c.7471 delA |
| TAAD 014 | II:2-prob-and | M | 42 | N | TAA | 68 | 3 | 44 | NO | MR(1), TR(1) | B | 42 | FBN1 | c.1496G > C |
| TAAD 017 | II:3-prob-and | F | 43 | N | AD | 42 | 2 | 55 | As, Ar, D, Ab | MR(1), CAD | B + TAR + DASI + CABG | 43 | FBN1 | c.7754T > C |
| TAAD 024 | II:1-prob-and | M | 33 | MFS | AD | 82 | 3 | 71 | R, As, Ar, D, Ab | TR(1) | B + TAR + DASI | 33 | FBN1 | c.8147_8148insA |
| TAAD 027 | II:2-prob-and | M | 48 | N | AD | 67 | 3 | 42 | R, As, | MR(1), TR(1) | B | 48 | ACTA2 | c.635G > A |
| TAAD 030 | II:2-prob-and | M | 41 | N | AD | 49 | 2 | 75 | Ar, D, Ab | MR(1) | B + TAR + DASI | 41 | FBN1 | c.5742C > A |
| TAAD 030 | III:1-son | M | 18 | MFS | N | 32 | 0 | 67 | NO | NO | NA | NA | FBN1 | c.5742C > A |
| TAAD 038 | II:2-prob-and | M | 25 | MFS | AD | 90 | 3 | 70 | As, Ar, D, Ab | MR(1) | B + TAR + DASI | 25 | FBN1 | c.4454G > A |
| TAAD 045 | II:2-prob-and | F | 52 | MFS | TAA | 54 | 2 | 67 | No | MR(1), TR(1) | B | 52 | FBN1 | EX5_54 DEL |
| TAAD 046 | II:1-prob-and | M | 66 | MFS | AD | 65 | 2 | 55 | As, Ar, D, Ab | NO | B + TAR + DASI | 66 | FBN1 | c.2216G > A |
| COL5A2 | c.2846C > G | |||||||||||||
| TAAD 046 | IV:1-gran-dson | M | 22 | N | N | 31 | 0 | 72 | NO | NO | NA | NA | COL5A2 | c.2846C > G |
| TAAD 050 | II:1-prob-and | M | 21 | N | TAA | 45 | 0 | 58 | NO | MR(3), TR(1) | B + MVR | 21 | FBN1 | EX4_53 DEL |
| TAAD 053 | II:2-prob-and | M | 55 | N | AD | 38 | 2 | 68 | As, Ar, D, Ab | NO | DAV + TAR + DASI | 55 | FBN1 | c.7567A > C |
| TAAD 053 | II:3-broth-er | M | 56 | N | AD | Surge-ry | Sur-gery | 65 | AS | NO | B | 51 | FBN1 | c.7567A > C |
| TAAD 056 | II:1-prob-and | M | 24 | MFS | AD | 72 | 3 | 61 | As, Ar, D, Ab | MR(3), TI(3) | B + TAR + DASI + MVR + TVP | 24 | FBN1 | c.247 + 1G > A |
| TAAD 068 | II:1-prob-and | M | 18 | MFS | TAA | 51 | 2 | 65 | NO | NO | B | 18 | FBN1 | c.4292G > A |
| TAAD 068 | II:2-sister | F | 21 | MFS mutation carrier | N | 33 | 0 | 60 | NO | NO | NA | NA | FBN1 | c.4292G > A |
| TAAD 069 | II:1-prob-and | M | 23 | MFS | TAA | 48 | 1 | 72 | NO | MR(3), TR(1) | B + MVR + TVP | 23 | FBN1 | c.8292_8293insT |
| TAAD 069 | II:2-sister | F | 21 | MFS mutate on carrier | N | 29 | 0 | 68 | NO | NO | NA | NA | FBN1 | c.8292_8293insT |
| TAAD 070 | II:2-prob-and | M | 45 | MFS | TAA | 52 | 3 | 55 | NO | TR(3) | B + TVP | 45 | FBN1 | c.6801C > A |
| TAAD 070 | III:3-daug-hter | F | 20 | MFS | TAA | 45 | 2 | 60 | NO | NO | NA | NA | FBN1 | c.6801C > A |
The underline families were with VUS and have positive family history and verification.
Ab: abdominal aorta, AD: aortic dissection, AR: aortic regurgitation(0 none,1 mild, 2 moderate, 3 severe), Ar: aortic arch, As: thoracic ascending aorta, B: Bentall procedure, CABG: coronary artery bypass grafting, CVS: cardiovascular system, D: thoracic descending aorta, DAV: David procedure, DASI: descending aorta stent implantation, F: female, LVEF: left ventricular ejection fraction, M: male, MFS: Marfan syndrome, MR: mitral regurgitation (0 none, 1 mild, 2 moderate, 3 severe), MVR: mitral valve replacement, N: none, NA: not applicable, TAA: thoracic aortic aneurysm, TAR: total aortic arch replacement, TR: tricuspid regurgitation (0 none, 1 mild, 2 moderate, 3 severe), TVP: tricuspid valve plastic.
Figure 2.

Provided the pedigrees of families with pathogenic and likely pathogenic mutations as well as 3 VUS cases with positive family verifications. Five members from different families were found to have same pathogenic variants with probands (TAAD030, TAAD053, TAAD068, TAAD069, TAAD070).
Discussion
NGS for molecular diagnosis of TAAD has recently become a practical screening method to identify disease-related gene mutations, which offers the patient and physician an opportunity to intervene and prevent emergency events for patients and their families. In the current study, NGS was performed to determine mutations in 11 candidate gene associated with TAAD in 70 patients from Southern China. Forty variants, were identified in eight genes in 36 TAAD patients (36/70, 51.4%), among whom 66.7% (24/36) patients had novel variants. The total pathogenic/likely pathogenic variants were 13, in which 5 pathogenic were reclassified from likely pathogenic in this study. Only 10 out of these 36 patients (10/36, 27.8%) showed hypertension and 3(3/36, 8.3%) showed hyperlipidemia, which supports the genetic origin of TAAD. There were 13 cases (13/70, 18.6%) with family history in the current study, which approached the range of 20–40% family history as reported previously5. When family history was taken into consideration, the mutation detection rate was 92.3% (12/13) compared with 45.6% (26/57) in non-family history cases. This result implies a greater chance of taking genetic tests among TAAD patients with positive family history.
FBN1 was first documented as an associated gene with MFS38–40. More and more studies have shown that patient with a pathogenic FBN1 mutation is at risk for developing Marfan-like syndromes such as severe cardiovascular, skeletal, and ophthalmologic complications5, 6, 20 and et al. Faivre L et al.40 pointed out that exons 24–32 represented a hotspot for neonatal MFS and severe forms of MFS. Some recent researches also indicated that variants of FBN1 was strongly related to the developing of TAAD18, 24 aside from MFS. Similarly, most variants identified in our TAAD patients located in FBN1 gene, including 12 pathogenic/likely pathogenic variants and 14 VUS. The majority of them were missense mutations. In the 26 patients with FBN1 mutations, 15 were diagnosed as MFS, the remaining were TAAD. The data imply that FBN1 is the primary disease-causative gene for TAAD in the population of Southern China.
Besides pathogenic mutation discussed above, there were a total of 27 variants found in the current study, which were considered as VUS, owe to insufficient data from segregation study and ambiguous results by software estimation. Most of them were missense mutation, which lead to the difficulties to verify their pathogenicity. Nevertheless, in accordance with recommendations from ACMG, pathogenicity of 3 novel and 1 reported VUS in FBN1 were further established through family validation or previous data. Missense mutation c.7567A > C in TAAD053 and c.6801C > A in TAAD070 can be upgraded because the same mutation was detected in their affected family members. The other two novel VUS, c.5084G > T (p.Cys1695Phe) from TAAD021 and c.7988G > A (p.Cys2663Tyr) from TAAD023, were considered to be highly pathogenic due to the fact that they were same at the site and characteristic to the previous-reported pathogenic mutation. Based on that, we believed these 4 VUS could be reclassified to the likely pathogenic category.
Interestingly, three of 70 patients (4.3%) were found to carry more than one VUS in different genes in this study, c.8069T > Gin FBN1 and c.1523G > A in MYH11 (TAAD034); c.2216G > A in FBN1 and c.2846 C > G in COL5A2 (TAAD046); c.2056G > A in FBN1, c.1456G > T in SLC2A10 and c.532 + G > A in SMAD3 (TAAD051). Concomitant mutations in different genes in TAAD patients have been reported in previous studies41, thus resulting in the complexity and difficulty of discovering the pathogenicity for TAAD. Therefore, family verification on the pathogenicity of these variants is strongly recommended and results must be carefully evaluated and defined by comparing to other independent studies in combination with prediction outcomes of protein function. As in the current study, the pathogenicity of c.2216G > A in FBN1 was confirmed and c.2846C > G in COL5A2 was proved to be benign after family verification of the proband TAAD046. Notably, the three patients with concomitant mutation in different genes were both diagnosed as AD and suffered a severe situation. Therefore, it remained yet to be investigated whether concomitant multiple variants in different genes predict disease severity. Further investigation is needed in order to address this question.
Finally, to translate our findings to clinical practice, all patients carrying pathogenic/likely pathogenic variants and with family history of TAAD were verified and further examined by imaging study. Close follow-ups were scheduled for all these patients. Until now, five out of 21 families were with positive validation, and two members (TAAD030-son, TAAD069-sister) from these 5 families had never received clinical examinations and no previous histories of disease reported. Further clinical examination confirmed their diagnosis of TAAD and they were treated accordingly, suggesting that use of NGS might be particularly useful in determining underlying genetic predisposition for TAAD. Efficient molecular findings combined with NGS can be used to guide optimal management, surveillance, and timely treatment in order to alter the natural course of TAAD.
This study had several limitations. Firstly, the sample size was not large enough. Even though most subjects recruited in the present study were typical and comparatively young, often complicated with MFS diagnoses, and with little to no history of hypertension, further study with a large sample is needed to verify the findings in the present study. Secondly, for patients with VUS, family validation was only performed when family history existed, which might lead to omission of some potential positive information. Thirdly, out of the 11 candidate genes, there could be other mutations among the unselected genes, which could hardly be detected due to the limitation of current methodology and technology. A whole exome sequencing by our team is currently being pursued to overcome these shortcomings.
Our findings broaden the spectrum of genetic backgrounds for thoracic aneurysms and dissections, introducing genetic background as a potential prognostic factor for clinical evaluation of patients with TAAD. Our data has established the FBN1 gene as the most common causative gene in a TAAD patient population from Southern China.
Materials and Methods
Patients
The present study was approved by the ethics committee of Guangdong General Hospital, Guangzhou, China. All experiments were performed in accordance with relevant guidelines and regulations. The study cohort included 70 unrelated patients with TAAD hospitalized in the department of cardiac surgery at Guangdong General Hospital from April 2015 to March 2016. The inclusion criteria were age above 18 years old, born and raised in southern China with only southern China family members, diagnosed as TAAD. For the diagnosis of TAAD, the patients meet the following standards according to the AFFC/AHA Guidelines for the diagnosis and treatment of thoracic aortic disease (2010)1. (1) True aneurysm and dissection involving the thoracic aorta. (2) Aneurysm (or true aneurysm): a permanent localized dilatation of an artery, having at least a 50% increase in diameter compared to the expected normal diameter of the artery in question. (3) Aortic dissection: disruption of the media layer of the aorta with bleeding within and along the wall of the aorta. Rupture of thoracic aortic artery caused by trauma and pseudoaortic aneurysm were excluded in this study. Age at diagnosis, gender, tobacco use, alcohol use, hypertension history, hyperlipidemia history, the status of the cardiovascular system, history of AD and surgeries were recorded. A family history of TAAD and other diseases was collected. A generation pedigree was drawn for every individual patient and family. Revised Ghent criteria42 was used to define MFS for the suspected and a detailed questionnaire was applied to define the involvement of other systems and organs. A Doppler echocardiographic study and CT scan of the entire aorta were performed for all included patients. The presence of mitral valve prolapses (MVP) and mitral regurgitation (MR) was determined using echocardiography and data concerning the mitral valve recorded. Family history was defined as the presence of more than one patient with TAAD in the family. All participants were informed about the study procedures and informed consent for genetic testing and permission to results publication was signed.
Genetic testing
Genetic testing was performed using NGS coupled with a DNA target-capture array on an IlluminaHiSeq. 2500 platform by BGI (Shenzhen, China) as previously reported43. Briefly, eleven genes (ACTA2, Col3A1, Col5A2, FBN1, MSTN, MYH11, MYLK, SLC2A10, SMAD3, TGFBR1, and TGFBR2) (Table 6) relevant to TAAD were selected for one capture array (NimbleGen, Roche, Madison, WI, USA), which was designed mainly to capture the CDS of 2,181 known pathogenic genes associated with 561 Mendelian diseases based on the GeneReviews (NCBI) and Genetics Home Reference. Genomic DNA from peripheral blood or abortion tissues were fragmented into lengths ranging from 200 bp to 250 bp. The primers, adapters and indexes were then ligated to the DNA fragments to construct libraries. The DNA fragments were pooled and hybridized to the capture array. After hybridization and enrichment, the DNA sample was sequenced on IlluminaHiSeq. 2500 Analyzers to generate paired-end reads (90 bps).
Table 6.
List of analyzed genes.
| Gene | GenBank accession no | Chromosomal locus | Exons | Protein name | Disease | Reference |
|---|---|---|---|---|---|---|
| ACTA2 | NM_001613 | 10q22–q24 | 9 | Smooth muscle actin, alpha 2 | TAAD | 11 |
| COL3A1 | NM_000090 | 2q31 | 51 | Collagen type III alpha 1 | Ehlers–Danlos type IV | 7 |
| COL5A2 | NM_000393 | 2q14-q32 | 55 | Collagen type V alpha 2 | Ehlers–Danlos syndrome | 8 |
| FBN1 | NM_000138 | 15q21.1 | 66 | Fibrillin 1 | Marfan/MASS/TAAD | 5 |
| MSTN | NM_005259 | 2q32.2 | 3 | myostatin | myodystrophy | 8 |
| MYH11 | NM_002474 | 16p13.13–p13.12 | 41 | Myosin heavy chain 11 | TAAD-patent ductus arteriosus | 14 |
| MYLK | NM_053025 | 3q21 | 34 | Myosin light chain kinase | TAAD | 16 |
| SLC2A10 | NM_030777 | 20q13.1 | 5 | Solute carrier family 2 (facilitated glucose transporter) member 10 | Arterial tortuosity | 13 |
| SMAD3 | NM_005902 | 15q22.33 | 9 | Mothers against decapentaplegic homolog (SMAD) 3 | Loeys–Dietz syndrome type III | 12 |
| TGFBR1 | NM_004612 | 9q33–q34 | 9 | Transforming growth factor, beta receptor 1 | Loeys–Dietz syndrome TAAD Marfan type II | 10 |
| TGFBR2 | NM_003242 | 3p22 | 7 | Transforming growth factor, beta receptor 2 | Loeys–Dietz TAAD Marfan Type II | 9 |
MASS: Mitral valve prolapse, TAAD: Thoracic aortic aneurysm and dissection.
Short reads mapping, alignment were performed using BWA software (Burrows Wheeler Aligner). SNPs and indels were detected using the SOAPsnp software and GATK IndelGenotyper (http://www.broadinstitute.org/gsa/wiki/index.php/, The Genome Analysis Toolkit) respectively. All reference sequences were based on the NCBI37/hg19 assembly of the human genome (a novel mutation of IDS gene in a Chinese patient with mucopolysaccharidosis II by NGS).
Interpretation of all variants referred to the latest version of ACMG standards and guidelines for the interpretation of sequence variants (PMID:25741868). Normal population frequencies information was from dbSNP, HapMap databases, one thousand genome project database, 100 Chinese healthy adults, EXAC database, NHLBI GO Exome Sequencing Project (ESP). Other information was from HGMD database, published literatures, OMIM database, NCBI-books. In silico deleterious effect was evaluated using PolyPhen and SIFT programs.
Statistical analysis
All results for categorical variables were presented as numbers and percentages and for continuous variables as the mean and standard deviation (SD). Calculations were performed using SPSS v 20.0.
Acknowledgements
We thank Prof. Yafang Hu and Prof. Hui Liu for their technical advice to this study. This work was supported by the Science and Technology Planning Project of Guangdong Province, China (Grant number: 2015A020214017); And the Medical Scientific Research Foundation of Guangdong Province, China (Grant number: A2015121).
Author Contributions
Miaoxian Fang and Ruixin Fan designed and coordinated the study; Changjiang Yu and Xin Li made and controlled the sample collections; Miaoxian Fang and Changjiang Yu carried out NGS measurements, Weiping Xiong and Rong Zeng collected the patient’s clinical information; Miaoxian Fang, Changjiang Yu, Ruixin Fan and Xin Li contributed to data mining, and the evaluation of the results, Miaoxian Fang, Siyao Chen, Ruixin Fan and Jian Zhuang prepared the manuscript. All authors reviewed the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Hiratzka LF, et al. ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM guidelines for the diagnosis and management of patients with thoracic aortic disease: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, American Association for Thoracic Surgery, American College of Radiology, American Stroke Association, Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, Society of Interventional Radiology, Society of Thoracic Surgeons, and Society for Vascular Medicine. Circulation. 2010;121:e266–369. doi: 10.1161/CIR.0b013e3181d4739e. [DOI] [PubMed] [Google Scholar]
- 2.Elefteriades JA, Farkas EA. Thoracic aortic aneurysm clinically pertinent controversies and uncertainties. J Am Coll Cardiol. 2010;55:841–857. doi: 10.1016/j.jacc.2009.08.084. [DOI] [PubMed] [Google Scholar]
- 3.Milewicz DM, Carlson AA, Regalado ES. Genetic testing in aortic aneurysm disease: PRO. Cardiol Clin. 2010;28:191–197. doi: 10.1016/j.ccl.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Disabella E, et al. Risk of dissection in thoracic aneurysms associated with mutations of smooth muscle alpha-actin 2 (ACTA2) Heart. 2011;97:321–326. doi: 10.1136/hrt.2010.204388. [DOI] [PubMed] [Google Scholar]
- 5.Dietz HC, et al. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature. 1991;352:337–339. doi: 10.1038/352337a0. [DOI] [PubMed] [Google Scholar]
- 6.Maslen CL, Corson GM, Maddox BK, Glanville RW, Sakai LY. Partial sequence of a candidate gene for the Marfan syndrome. Nature. 1991;352:334–337. doi: 10.1038/352334a0. [DOI] [PubMed] [Google Scholar]
- 7.Kroes HY, Pals G, van Essen AJ. Ehlers-Danlos syndrome type IV: unusual congenital anomalies in a mother and son with a COL3A1 mutation and a normal collagen III protein profile. Clin Genet. 2003;63:224–227. doi: 10.1034/j.1399-0004.2003.00047.x. [DOI] [PubMed] [Google Scholar]
- 8.Meienberg J, et al. Hemizygous deletion of COL3A1, COL5A2, and MSTN causes a complex phenotype with aortic dissection: a lesson for and from true haploinsufficiency. Eur J Hum Genet. 2010;18:1315–1321. doi: 10.1038/ejhg.2010.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mizuguchi T, et al. Heterozygous TGFBR2 mutations in Marfan syndrome. Nat Genet. 2004;36:855–860. doi: 10.1038/ng1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Loeys BL, et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet. 2005;37:275–281. doi: 10.1038/ng1511. [DOI] [PubMed] [Google Scholar]
- 11.Guo DC, et al. Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat Genet. 2007;39:1488–1493. doi: 10.1038/ng.2007.6. [DOI] [PubMed] [Google Scholar]
- 12.Regalado ES, et al. Exome sequencing identifies SMAD3 mutations as a cause of familial thoracic aortic aneurysm and dissection with intracranial and other arterial aneurysms. Circ Res. 2011;109:680–686. doi: 10.1161/CIRCRESAHA.111.248161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Coucke PJ, et al. Mutations in the facilitative glucose transporter GLUT10 alter angiogenesis and cause arterial tortuosity syndrome. Nat Genet. 2006;38:452–457. doi: 10.1038/ng1764. [DOI] [PubMed] [Google Scholar]
- 14.Zhu L, et al. Mutations in myosin heavy chain11 cause a syndrome associating thoracic aortic aneurysm/aortic dissection and patent ductusarteriosus. Nat Genet. 2006;38:343–349. doi: 10.1038/ng1721. [DOI] [PubMed] [Google Scholar]
- 15.El-Hamamsy L, Yacoub MH. Cellular and molecular mechanisms of thoracic aortic aneurysms. Nat Rev Cardiol. 2009;6:771–786. doi: 10.1038/nrcardio.2009.191. [DOI] [PubMed] [Google Scholar]
- 16.Wang L, et al. Mutations in Myosin Light Chain KinaseCause Familial Aortic Dissections. Am J Hum Genet. 2010;87:701–707. doi: 10.1016/j.ajhg.2010.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Regalado E, et al. Autosomal dominant inheritance of a predisposition to thoracic aortic aneurysms and dissections and intracranial saccular aneurysms. Am J Med Genet A. 2011;155A:2125–2130. doi: 10.1002/ajmg.a.34050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Proost D, Vandeweyer G, Meester JA. Performant Mutation Identification Using Targeted Next-Generation Sequencing of 14 Thoracic Aortic Aneurysm Genes. Hum Mutat. 2015;36:808–814. doi: 10.1002/humu.22802. [DOI] [PubMed] [Google Scholar]
- 19.Ziganshin BA, Bailey AE, Coons C. Routine Genetic Testing for Thoracic Aortic Aneurysm and Dissection in a Clinical Setting. Ann Thorac Surg. 2015;100:1604–1611. doi: 10.1016/j.athoracsur.2015.04.106. [DOI] [PubMed] [Google Scholar]
- 20.Wooderchak-Donahue W, VanSant-Webb C, Tvrdik T. Clinical utility of a next generation sequencing panel assay for Marfan and Marfan-like syndromes featuring aortopathy. Am J Med Genet A. 2015;167A:1747–1757. doi: 10.1002/ajmg.a.37085. [DOI] [PubMed] [Google Scholar]
- 21.Richards S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–524. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding nonsynonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 23.Adzhubei IA, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Poninska JK, Bilinska ZT, Franaszczyk M. Next-generation sequencing for diagnosis of thoracic aortic aneurysms and dissections: diagnostic yield, novel mutations and genotype phenotype correlations. J Transl Med. 2016;14 doi: 10.1186/s12967-016-0870-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Loeys BL, Nuytinck L, Delvaux I, De Bie S, De Paepe A. Genotype and phenotype analysis of 171 patients referred for molecular study of the fibrillin-1 gene FBN1 because of suspected Marfan syndrome. Arch Intern Med. 2001;161:2447–2454. doi: 10.1001/archinte.161.20.2447. [DOI] [PubMed] [Google Scholar]
- 26.Robinson PN, et al. Mutations of FBN1 and genotype-phenotype correlations in Marfan syndrome and related fibrillinopathies. Hum Mutat. 2002;20:153–161. doi: 10.1002/humu.10113. [DOI] [PubMed] [Google Scholar]
- 27.Uyeda T, et al. Three novel mutations of the fibrillin-1 gene and ten single nucleotide polymorphisms of the fibrillin-3 gene in Marfan syndrome patients. J Hum Genet. 2004;49:404–407. doi: 10.1007/s10038-004-0168-x. [DOI] [PubMed] [Google Scholar]
- 28.Howarth R, Yearwood C, Harvey JF. Application of dHPLC for mutation detection of the fibrillin-1 gene for the diagnosis of Marfan syndrome in a National Health Service Laboratory. Genet Test. 2007;11:146–152. doi: 10.1089/gte.2006.0514. [DOI] [PubMed] [Google Scholar]
- 29.Hung CC, et al. Mutation spectrum of the fibrillin-1 (FBN1) gene in Taiwanese patients with Marfan syndrome. Ann Hum Genet. 2009;73:559–567. doi: 10.1111/j.1469-1809.2009.00545.x. [DOI] [PubMed] [Google Scholar]
- 30.Turner CL, et al. Detection of 53 FBN1 mutations (41 novel and 12 recurrent) and genotype–phenotype correlations in 113 unrelated probands referred with Marfan syndrome, or a related fibrillin opathy. Am J Med Genet A. 2009;149A:161–170. doi: 10.1002/ajmg.a.32593. [DOI] [PubMed] [Google Scholar]
- 31.Yoo EH, et al. Clinical and genetic analysis of Korean patients with Marfan syndrome: possible ethnic differences in clinical manifestation. Clinl Genet. 2010;77:177–182. doi: 10.1111/j.1399-0004.2009.01287.x. [DOI] [PubMed] [Google Scholar]
- 32.Baetens M, et al. Applying massive parallel sequencing to molecular diagnosis of Marfan and Loeys-Dietz syndromes. Hum Mutat. 2011;32:1053–1062. doi: 10.1002/humu.21525. [DOI] [PubMed] [Google Scholar]
- 33.Ross JM, et al. Fibrillin containing elastic microfibrils support platelet adhesion under dynamic shear conditions. Thromb Haemost. 1998;79:155–161. [PubMed] [Google Scholar]
- 34.Guo DC, et al. Mutations in smooth muscle alpha-actin (ACTA2) cause coronary artery disease, stroke, and Moyamoya disease, along with thoracic aortic disease. Am J Hum Genet. 2009;84:617–627. doi: 10.1016/j.ajhg.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Morisaki H, et al. Mutation of ACTA2 gene as an important cause of familial and nonfamilial nonsyndromatic thoracic aortic aneurysm and/or dissection (TAAD) Hum Mutat. 2009;30:1406–1411. doi: 10.1002/humu.21081. [DOI] [PubMed] [Google Scholar]
- 36.Shimojima K, Yamamoto T. ACTA2 is not a major disease-causing gene for moyamoya disease. J Hum Genet. 2009;54:687–688. doi: 10.1038/jhg.2009.91. [DOI] [PubMed] [Google Scholar]
- 37.Groth KA, et al. Difficulties in diagnosing Marfan syndrome using current FBN1 databases. Genet Med. 2016;18:98–102. doi: 10.1038/gim.2015.32. [DOI] [PubMed] [Google Scholar]
- 38.Loeys BL, et al. Comprehensive molecular screening of the FBN1 gene favors locus homogeneityof classical Marfan syndrome. Hum Mutat. 2004;24:140–146. doi: 10.1002/humu.20070. [DOI] [PubMed] [Google Scholar]
- 39.Comeglio P, et al. The importance of mutation detection in Marfan syndrome and Marfan-related disorders: report of 193 FBN1 mutations. Hum Mutat. 2007;28 doi: 10.1002/humu.9505. [DOI] [PubMed] [Google Scholar]
- 40.Faivre L, et al. Effect of mutation type and location on clinical outcome in 1,013 probands with Marfan syndrome or related phenotypes and FBN1 mutations: an international study. Am J Hum Genet. 2007;81:454–466. doi: 10.1086/520125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Haruya H, et al. Rapid detection of gene mutations responsible for non-syndromic aortic aneurysm and dissection using two different methods: resequencing microarray technology and next-generation sequencing. Hum Genet. 2012;131:591–599. doi: 10.1007/s00439-011-1105-7. [DOI] [PubMed] [Google Scholar]
- 42.Loeys BL, et al. The revised Ghent nosology for the Marfan syndrome. J MedGenet. 2010;47:476–485. doi: 10.1136/jmg.2009.072785. [DOI] [PubMed] [Google Scholar]
- 43.Wei X, et al. Identification of sequence variants in genetic disease-causing genes using targeted next-generation sequencing. Plos One. 2011;6 doi: 10.1371/journal.pone.0029500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chung BH, et al. Identification of novel FBN1 and TGFBR2 mutations in 65 probands with Marfan syndrome or Marfan‐like phenotypes. Am J Med Genet A. 2009;149:1452–1459. doi: 10.1002/ajmg.a.32918. [DOI] [PubMed] [Google Scholar]
- 45.Liu WO, Oefner PJ, Qian C, Odom RS, Francke U. Denaturing HPLC-identified novel FBN1 mutations, polymorphisms, and sequence variants in Marfan syndrome and related connective tissue disorders. Genet Test. 1997;1:237–242. doi: 10.1089/gte.1997.1.237. [DOI] [PubMed] [Google Scholar]
- 46.Stheneur C, et al. Identification of the minimal combination of clinical features in probands for efficient mutation detection in the FBN1 gene. Eur J Hum Genet. 2009;17:1121–1128. doi: 10.1038/ejhg.2009.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang WJ, et al. Exon 47 skipping of fibrillin-1 leads preferentially to cardiovascular defects in patients with thoracic aortic aneurysms and dissections. J Mol Med (Berl). 2013;91:37–47. doi: 10.1007/s00109-012-0931-y. [DOI] [PubMed] [Google Scholar]