

Abstract
ALS is a devastating and debilitating human disease characterized by the progressive death of upper and lower motor neurons. Although much effort has been made to elucidate molecular determinants underlying the onset and progression of the disorder, the causes of ALS remain largely unknown. In the present work, we have deeply sequenced whole transcriptome from spinal cord ventral horns of post-mortem ALS human donors affected by the sporadic form of the disease (which comprises ~90% of the cases but which is less investigated than the inherited form of the disease). We observe 1160 deregulated genes including 18 miRNAs and show that down regulated genes are mainly of neuronal derivation while up regulated genes have glial origin and tend to be involved in neuroinflammation or cell death. Remarkably, we find strong deregulation of SNAP25 and STX1B at both mRNA and protein levels suggesting impaired synaptic function through SNAP25 reduction as a possible cause of calcium elevation and glutamate excitotoxicity. We also note aberrant alternative splicing but not disrupted RNA editing.
Subject terms: Gene expression profiling, Amyotrophic lateral sclerosis
Introduction
Amyotrophic lateral sclerosis (ALS) is a fatal and devastating neurodegenerative disorder that causes the progressive death of upper and lower motor neurons1. Symptoms and clinical signs, including muscle weakness and atrophy, fasciculations, hyperreflexia, dysarthria and dysphagia emerge when axonal connections fail, leading to denervation1. The worldwide incidence of ALS is approximately two per 100,000 individuals and the mean age of onset is 55–60 years, with a higher incidence among men than women1.
The vast majority of documented cases are sporadic (~90%), without any associated family history. Sporadic and inherited (usually through an autosomal dominant pattern) forms of ALS show common clinical and pathological traits, suggesting equivalent pathogenic mechanisms1.
Although much effort has been done in elucidating molecular factors underlying the onset and progression of the disorder, the causes of ALS remain unknown2. However, numerous investigations in humans and animal models have associated nucleotide variants in a small group of genes (including SOD1, UBQLN2, VCP, OPTN, TDP43, FUS, MATR3 and a hexanucleotide repeat expansion within c9orf72) with the familial form of ALS2.
Microarray-based transcriptome studies have revealed multiple perturbations of motor neuron function in both forms of ALS3, 4, consistent with the hypothesis that various cellular events, including mitochondrial dysfunction, enhanced apoptosis, glutamate-mediated excitotoxicity, free radical injury, protein misfolding, abnormal calcium metabolism, altered axonal transport and activation of proteases and nucleases contribute to the pathobiology of the disease5.
Since motor neurons cannot be sampled during life, investigations into ALS neurodegenerative process are carried out in neuronal-like cells in culture, animal models carrying mutant transgenes or post-mortem material from patients. In the last case, the spinal cord represents the elective tissue and a pivotal source of RNA for gene expression profiling in ALS. While mixed cell populations in which astrocytes, microglia and oligodendrocytes may obscure the distinctive expression profile of motor neurons, which are the most affected in ALS, the co-culture of motor neurons and glial cells has shown an intricate interplay, suggesting that non-neuronal cells play an active role in neurodegeneration6. Accordingly, transcriptomic studies using high throughput sequencing approaches (RNA-Seq) to target specific tissues, such as ventral horns of the spinal cord, may provide new insights into genes and pathways involved in the degeneration of motor neurons. Indeed, RNA-Seq allows more accurate and complete expression profiling than classical microarrays, overcoming their reliance on pre-designed probes. Recently, two studies exploring transcriptome alterations in ALS by the RNAseq technology have been reported7, 8. The first one investigated transcriptome changes in cerebellum and frontal cortex from familiar (fALS) (with mutated c9orf72 gene) and sporadic ALS (sALS) donors, showing extensive dysregulation of alternative splicing and alternative polyadenylation in both ALS groups7. The second one, based on RNAseq technology, investigated transcriptome changes in cervical spinal cord tissues from sALS donors showing that neuroinflammation mediated by TNF (tumor necrosis factor) is the main transcriptome abnormality in sALS8.
In the current work, we also report the analysis of whole transcriptome sequencing data from matched control and sALS post-mortem donors but, differently from previous studies, we focus on the detection and characterization of significant changes in ventral horns of the human lumbar spinal cord, a tissue never investigated at single nucleotide resolution. While massive sequencing of the low molecular weight RNA fraction showed 18 dysregulated miRNAs involved in TGF-beta signaling, axon survival and outgrowth, RNA-Seq data revealed altered expression of 1142 genes belonging to a restricted range of ALS deregulated molecular pathways including an impaired synaptic function. In particular, we focused our attention on two downregulated neuronal t-SNARE proteins, SNAP25 and Syntaxin-1B (STX1B), as they form with the v-SNARE protein synaptobrevin, a ternary complex mediating the neurotransmitter release at the presynaptic membrane. Notably, SNAP25 negatively regulates native voltage-gated calcium channels (VGCCs) in glutamatergic neurons9, and its reduced expression enhances glutamatergic neurotransmission10 and causes neurodegeneration11.
Taken together, our results may represent a remarkable breakthrough to unveil sALS, supporting a primary role of t-SNARE protein expression as possible cause of intracellular calcium elevation and glutamate excitotoxicity12 and suggesting SNAP25 and related SNARE proteins as candidate novel indicators and signatures of motor neuron death to be monitored in ALS patients.
Results
RNA-Seq data analysis
Our study includes ventral horns of the lumbar spinal cord from six human donors affected by sporadic ALS and five age, sex and ethnicity matched controls (Table 1). ALS and control transcriptomes were sequenced employing the Illumina technology on the NextSeq 500 platform. We generated 1.9 billion 100 bp strand-specific paired end reads with, on average, 179 million pairs of reads per sample (Table 2). About 73% of these reads were uniquely aligned to the reference human genome (hg19 assembly) after a cleaning step to remove low quality regions (Table 2). Using RefSeq annotations we verified that about 98% of mapped reads maintained the correct strand orientation and, on average, 46% of mapped bases corresponded to UTR and coding regions of known mRNA transcripts (Table 2).
Table 1.
Control and ALS samples used in this study.
| Bank | ID | BID | Age | Sex | Race | Diagnosis | PMI | RNA-Seq | miRNA-Seq | qRT-PCR | WB | IF |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NICHD | A1 | 5388 | 61 | M | Ca | sALS | 8 | ✓ | ||||
| NICHD | A2 | 925 | 54 | M | Ca | sALS | 3 | ✓ | ||||
| NICHD | A3 | 4762 | 59 | M | Ca | sALS | 6 | ✓ | ||||
| NICHD | A4 | 5595 | 56 | M | Ca | sALS | 16 | ✓ | ✓ | ✓ | ✓ | |
| NICHD | A5 | 1292 | 45 | M | Ca | sALS | 15 | ✓ | ✓ | ✓ | ✓ | ✓ |
| HBSFRC | A6 | 5215 | 59 | M | Ca | sALS | 12,5 | ✓ | ✓ | ✓ | ✓ | |
| HBSFRC | A7 | 5187 | 69 | M | Ca | sALS | 13,1 | ✓ | ✓ | ✓ | ✓ | |
| HBSFRC | A8 | 5165 | 55 | M | Ca | sALS | 18,3 | ✓ | ✓ | ✓ | ✓ | ✓ |
| HBSFRC | A9 | 5223 | 47 | M | Ca | sALS | 16 | ✓ | ✓ | ✓ | ||
| HBSFRC | A10 | 5216 | 53 | M | Ca | sALS | 16,8 | ✓ | ✓ | ✓ | ||
| NICHD | A11 | 5678 | 55 | M | Ca | sALS | 6 | ✓ | ||||
| NICHD | H1 | 5456 | 57 | M | Ca | ACD | 16 | ✓ | ✓ | ✓ | ✓ | ✓ |
| NICHD | H2 | 5663 | 42 | M | Ca | UK | 16 | ✓ | ✓ | ✓ | ✓ | |
| NICHD | H3 | 5458 | 64 | M | Ca | MI | 13 | ✓ | ✓ | ✓ | ✓ | |
| NICHD | H4 | 5361 | 42 | M | Ca | CT | 19 | ✓ | ✓ | ✓ | ✓ | ✓ |
| NICHD | H5 | 5617 | 59 | M | Ca | MI | 10 | ✓ | ✓ | ✓ | ✓ | ✓ |
| NICHD | H6 | 5393 | 56 | M | Ca | ACD | 22 | ✓ | ||||
| NICHD | H7 | 5353 | 43 | M | Ca | CD | 9 | ✓ | ✓ | ✓ |
For each sample we report: Bank (Tissue bank: NICHD or HBSFRC), ID (Sample ID used in the manuscript, Figures and Tables), BID (unique Bank ID from tissue bank), age, sex, race (Ca = Caucasian), main diagnosis (sALS = Sporadic ALS; ACD = Atherosclerosis Cardiovascular Disease; HEM = Hemopericardium; UK = Unknown; CD = Combined Drugs; MI = Multiple injuries; CT = Cardiac tamponade), post-mortem delay (in hours), flags indicating if the sample has been assayed for RNA-Seq, miRNA-Seq, qRT-PCR, immunoblotting (WB) or immunofluorescence (IF).
Table 2.
Main statistics for RNA sequencing.
| ID | #FR | #CFR | #AFR | % AFR | % RB | % COD | % UTR | % INTR | % INTER | % MRNA | % COR_STR |
|---|---|---|---|---|---|---|---|---|---|---|---|
| A4 | 54680251 | 45068732 | 40934057 | 90,83 | 1,00E-06 | 0,153 | 0,135 | 0,433 | 0,279 | 0,288 | 0,972 |
| A5 | 85144930 | 66912832 | 58474545 | 87,39 | 2,00E-06 | 0,217 | 0,162 | 0,440 | 0,182 | 0,378 | 0,984 |
| A6 | 76440384 | 61373970 | 53373122 | 86,96 | 2,00E-06 | 0,277 | 0,210 | 0,386 | 0,127 | 0,487 | 0,992 |
| A7 | 83340776 | 74094106 | 64311655 | 86,80 | 2,00E-06 | 0,292 | 0,226 | 0,347 | 0,134 | 0,518 | 0,993 |
| A8 | 74664234 | 55858789 | 48680941 | 87,15 | 2,00E-06 | 0,299 | 0,219 | 0,358 | 0,123 | 0,519 | 0,993 |
| A9 | 96273069 | 85995872 | 75818167 | 88,16 | 2,00E-06 | 0,234 | 0,203 | 0,428 | 0,135 | 0,437 | 0,990 |
| H1 | 109502733 | 88454014 | 77268812 | 87,35 | 2,00E-06 | 0,237 | 0,197 | 0,432 | 0,133 | 0,434 | 0,990 |
| H2 | 106819322 | 91146575 | 80112958 | 87,89 | 2,00E-06 | 0,243 | 0,193 | 0,423 | 0,142 | 0,435 | 0,989 |
| H3 | 103962426 | 88206006 | 77528076 | 87,89 | 2,00E-06 | 0,302 | 0,233 | 0,342 | 0,122 | 0,536 | 0,993 |
| H4 | 92670547 | 81318295 | 73329651 | 90,18 | 1,00E-06 | 0,297 | 0,230 | 0,338 | 0,134 | 0,528 | 0,992 |
| H5 | 101742727 | 79106426 | 69660162 | 88,06 | 2,00E-06 | 0,267 | 0,216 | 0,384 | 0,133 | 0,483 | 0,992 |
| Mean | 89567400 | 74321420 | 65408377 | 88,06 | 1,82E-06 | 0,256 | 0,202 | 0,392 | 0,149 | 0,459 | 0,989 |
Here we report main statistics about RNA sequencing calculated by in house python scripts and Picard tools. RefSeq gene annotations were downloaded from UCSC genome browser. Column headers are: ID (Unique Sample ID); #FR (Number of sequenced Fragments); #CFR (Number of cleaned Fragments); #AFR (Number of uniquely aligned Fragments); % AFR (Fraction of uniquely aligned Fragments); % RB (Fraction of Ribosomal Bases); % COD (Fraction of Protein Coding Bases); % UTR (Fraction of UTR Bases); % INTR (Fraction of Intronic Bases); % INTER (Fraction of Intergenic Bases); % MRNA (Fraction of mRNA Bases); % COR_STR (Fraction of Fragments in the correct orientation). The effective number of sequenced read is equal to the double of the number of fragments.
To confirm the sporadic disease nature of our samples, we checked for the presence of known ALS mutations in aligned RNAseq data using REDItools13 and did not find any potentially fALS- associated variant in the causative genes annotated in the ALS Online Genetic Database (ALSoD http://alsod.iop.kcl.ac.uk/)14.
We calculated RNAseq based gene expression levels in FPKM and performed unsupervised hierarchical clustering to detect similarities in gene expression profiles between ALS and control samples. We observed a clear separation between diseased and normal groups, suggesting specific transcriptome signatures for ALS (Fig. 1A). Principal component analysis (PCA) of variant genes re-marked such separation (Fig. 1B).
Figure 1.

Hierarchical clustering and PCA analysis of RNAseq data. (A) Hierarchical clustering conducted on CuffDiff2 expression values (in FPKM). ALS and control samples are clearly separated in two distinct groups. (B) PCA analysis of variant genes in which input samples are clustered in diseased (red circles) and normal (blue circles) groups.
Using cell type specific genes for neurons, motorneurons, astrocytes, oligodendrocytes and microglia collected from the literature6, 15, we verified the cell type composition of our spinal cord samples (Fig. 2 and Supplementary Table 1). Interestingly, we found statistically significant changes in cell populations composed by neurons, motorneurons, oligodendrocytes and microglia. No substantial differences were detected in the astrocyte population (Fig. 2). While neuron, motorneuron and oligodendrocyte populations appeared depleted in ALS samples, microglial population was over-represented suggesting neuroinflammation, that is thought to be critically important in the neurodegenerative process16 (Fig. 2).
Figure 2.

Cell type composition analysis of human spinal cord samples. For each cell type we show the geometric mean of the FPKM values of the cell type marker genes (Supplementary Table 1). Statistically different cell type composition is calculated by Mann-Whitney U test (two-tails). *P < 0.05; **P < 0.01; −Not significant.
Differentially expressed genes in ALS and controls
Differential gene expression between ALS and control samples was performed using only unique and concordant RNA-Seq reads. To improve the computational detection of differentially expressed (DE) genes, we applied two independent tools: CuffDiff2 based on FPKM metric that accounts for both uncertainty resulting from read mapping ambiguity and cross-replicate variability17, and DESeq2 based on counts of reads in genes18. The final list of DE genes was selected by the intersection between CuffDiff2 and DEseq2 lists of significant DE genes and taking into account only genes with |log2(FC)| higher than 1. On the whole, we identified 1142 deregulated genes, for some of them a specific cellular origin is known from the literature (Supplementary Table 2), with log2(FC) values highly correlated between CuffDiff2 and DEseq2 (Pearson r = 0.98) (Fig. 3 and Supplementary Table 2). Of these, 559 (49%) were upregulated in ALS samples and mainly involved in neuroinflammation or activation of immune response (Fig. 4, Supplementary Table 3). Fifty-seven upregulated genes were also found deregulated in a previous study based on peripheral blood of ALS patients19 and, thus, they may represent a precious source of potential biomarkers.
Figure 3.

Gene expression analysis of ALS and controls samples using RNAseq. (A) Correlation between log2(FC) values of differently expressed genes detected by CuffDiff2 and DEseq2 (Pearson r = 0.98). (B) Heat map of 1142 genes differentially expressed between ALS and control samples. Color bars above the heat map indicate the disease status: red, ALS; blue, control. Each row of the heat map represents the Z-score transformed FPKM values of one differentially expressed gene across all samples.
Figure 4.

Gene Ontology (GO) analysis of differentially expressed genes. Diagram of GO (Biological Process) terms that are significantly enriched in differentially expressed genes. Categories in the upper part of the figure are enriched in down regulated genes in ALS. Categories in lower part of the figure are enriched in up regulated genes in ALS. The Enrich combined score is reported for each category66.
Downregulated genes account for the 51% (583) of all DE genes and were significantly enriched (P ≪ 10−4 hypergeometric test) in neuronal-specific genes (Fig. 4 and Supplementary Tables 4 and 5). Consistent with past investigations, down expressed genes were involved in several biological processes of nervous system as impulse transmission (SLC1A2, CHAT, SLC12A5, HTR2C) or synapse function (SNPH, SYT4, SNAP25, STX1B) or calcium metabolism (GRIN1, GRIN2A, CACNA1G) (Fig. 4 and Supplementary Tables 2 and 4). RNA-Seq analysis revealed also downregulation of key enzymes in cholesterol biosynthesis including HMGCR (3-hydroxy-3-methylglutaryl-CoA reductase), HMGCS1 (3-hydroxy-3-methylglutaryl-CoA synthase 1), MSMO1 (methylsterol monooxygenase 1) and SQLE (squalene epoxidase), which may affect the organization of cellular membranes and, in turn, modify axon guidance and synaptic transmission (Fig. 4 and Supplementary Tables 2 and 4).
Within the group of down-regulated genes we found a set of 15 synaptic proteins related to the synaptic vesicle cycle (P ≪ 0.0001 hypergeometric test) that consists of docking and fusion of the vesicles, and subsequent exocytosis and neurotransmission (Table 3). Changes in synaptic transmission have been investigated in Alzheimer’s disease20 and may have a primary role in ALS for which little is known. Our results indicate deregulation of key genes for synaptic function including the two neuronal t-SNARE proteins, SNAP25 and STX1B. Recent evidence suggests novel functions for SNAP25 in controlling calcium dynamics and enhancing glutamate release in neurons9, 10, consistent with a role for SNAP25 in ALS pathogenesis.
Table 3.
DE genes linked to synaptic function.
| Gene | CuffDiff2 | DESeq2 | ||||||
|---|---|---|---|---|---|---|---|---|
| CTRL | ALS | log2(fc) | pvalue | padj | log2(fc) | pvalue | padj | |
| CACNA1B | 4,755 | 2,003 | −1,247 | 5E-05 | 0,00071 | −1,070 | 0,00013 | 0,00175 |
| CPLX1 | 33,535 | 5,542 | −2,597 | 5E-05 | 0,00071 | −2,111 | 0,00000 | 0,00000 |
| CPLX2 | 26,962 | 6,223 | −2,115 | 5E-05 | 0,00071 | −1,528 | 0,00064 | 0,00611 |
| DNM1 | 32,800 | 7,355 | −2,157 | 7E-04 | 0,00650 | −1,614 | 0,00007 | 0,00116 |
| NSF | 26,828 | 8,603 | −1,641 | 5E-05 | 0,00071 | −1,379 | 0,00016 | 0,00208 |
| RAB3A | 33,164 | 6,083 | −2,447 | 5E-05 | 0,00071 | −1,849 | 0,00001 | 0,00027 |
| RIMS1 | 11,579 | 3,910 | −1,566 | 5E-05 | 0,00071 | −1,408 | 0,00000 | 0,00000 |
| SLC17A6 | 4,820 | 1,057 | −2,190 | 5E-05 | 0,00071 | −1,432 | 0,00261 | 0,01776 |
| SLC18A3 | 17,019 | 1,829 | −3,218 | 5E-05 | 0,00071 | −2,409 | 0,00000 | 0,00000 |
| SLC32A1 | 6,263 | 1,280 | −2,290 | 5E-05 | 0,00071 | −1,674 | 0,00015 | 0,00197 |
| SNAP25 | 232,315 | 36,663 | −2,664 | 5E-05 | 0,00071 | −1,741 | 0,00031 | 0,00352 |
| STX1B | 20,025 | 4,881 | −2,037 | 5E-05 | 0,00071 | −1,665 | 0,00002 | 0,00032 |
| STXBP1 | 42,523 | 16,587 | −1,358 | 5E-05 | 0,00071 | −1,225 | 0,00001 | 0,00025 |
| UNC13A | 3,875 | 1,350 | −1,521 | 5E-05 | 0,00071 | −1,318 | 0,00000 | 0,00002 |
| UNC13C | 5,286 | 2,190 | −1,271 | 5E-05 | 0,00071 | −1,187 | 0,00000 | 0,00012 |
List of DE genes linked to synaptic function. Per each gene we report: gene name, mean expression levels (in FPKM) in control and ALS groups (from CuffDiff2), log2(fc), P-value and corrected P-value calculated by CuffDiff2 and log2(fc), P-value and corrected P-value calculated by DESeq2.
SLC1A2 which encodes a glutamate transporter essential for terminating the postsynaptic action through the glutamate clearance, appeared negatively regulated in ALS. This finding was also consistent with previous investigations showing down regulation of human SLC1A2 protein in ALS spinal cord21.
Fourteen out of 126 genes associated with ALS (mostly by mutation or GWAS studies) from the specialized ALSoD database14 showed differential expression in our ALS samples, 10 were down-regulated (including PRPH, UNC13A, SLC1A2, SNCG and NEFH) and 4 were up-regulated (SOD2, APOE, LOX and RNASE2).
Mitochondrial dysfunction is common in ALS pathogenesis since mitochondria have a central role in intracellular energy production, calcium homeostasis and control of apoptosis22. Indeed, we found several differentially expressed genes correlated with mitochondrial functions. However, the expression levels of genes transcribed by the mitochondrial genome were unchanged, indicating that mitochondrial transcription may not be impaired in ALS (Supplementary Table 6). Nonetheless, mutations in the mitochondrial genome may occur due to oxidative stress - a hallmark of motor neuron injury22.
Pathway analyses
Differentially expressed genes were further analyzed through the IPA tool (Ingenuity® Pathway Analysis, www.ingenuity.com) in order to identify the most significantly up-regulated and down-regulated canonical pathways associated to neurodegeneration in ALS.
Consistent with other transcriptome investigations in transgenic SOD1 mice and human samples, the most highly activated pathways were related to neuroinflammation and immune response (Fig. 5 and Supplementary Table 7). Indeed, our dataset included many up-regulated genes encoding inflammatory mediators such as chemokines and interleukins and several components of the complement system, oncostatin M signaling and STAT3 pathways (Supplementary Table 7).
Figure 5.

Pathway analysis of differentially expressed genes. IPA pathways that are significantly enriched in differentially expressed genes. Pathways in the upper part of the figure are enriched in down regulated genes in ALS. Pathways in the lower part of the figure are enriched in up regulated genes in ALS. The ratio between the number of differentially expressed genes and the number of genes in the specific pathway is reported on x axis.
Significantly down-regulated pathways, instead, included cholesterol biosynthesis, calcium signaling, glutamate receptor signaling and GABA receptor signaling (Fig. 5 and Supplementary Table 7). In addition, several under expressed genes in ALS samples were associated with neurological and neurodegenerative human disorders such as bipolar disorder, schizophrenia, Huntington’s disease or Alzheimer’s disease (Supplementary Table 8). IPA analysis revealed also a significant enrichment for amyotrophic lateral sclerosis signaling, involving nine genes encoding mainly for calcium channels and glutamate receptors (Supplementary Table 8).
Our results were further corroborated by GO-PCA software implementing an unsupervised method to explore gene expression data using prior knowledge in the form of gene ontology (GO) annotations23. GO-PCA is able to detect specific signatures, consisting of small set of genes that are strongly correlated by their expression as well as functionally related by GO annotations23.
We found 10 significant signatures (P ≪ 0.001) containing between 5 and 10 genes (Table 4). Five signatures, including down-regulated genes, were derived from GO terms related to cholesterol biosynthesis, glutamate secretion, motor activity or axon functionality (Table 4). Remaining five signatures, instead, were associated to inflammation and comprised up-regulated genes (Table 4).
Table 4.
GO-PCA analysis.
| Label | Status | Gene set ID | P-value | Median correlation | Genes |
|---|---|---|---|---|---|
| MF: motor activity [5/5] | UP | GO:0003774 | 0,0000012 | 0,98 | DYNC1I1, KIF3A, KIF3C, KIF5A, KIF5C |
| CC: microtubule [7/10] | UP | GO:0005874 | 0,0000073 | 0,99 | KCNAB2, KIF3A, MAPRE3, REEP2, TUBA4A, TUBB2A, TUBB3 |
| BP: glutamate secretion [5/8] | UP | GO:0014047 | 0,00016 | 0,96 | CPLX1, RAB3A, SLC1A2, SNAP25, STXBP1 |
| BP: cholesterol biosynthetic process [7/10] | UP | GO:0006695 | 0,000026 | 0,92 | ACLY, CYP51A1, DHCR24, HMGCS1, IDI1, MSMO1, SQLE |
| CC: axon [10/21] | UP | GO:0030424 | 0,0001 | 0,99 | CPNE6, EPB41L3, KCNA1, KCNAB2, NEFH, NEFL, NEFM, PTPRN, RAB3A, SCN1B |
| BP: neg. regulation of viral process [5/8] | DW | GO:0048525 | 0,0000011 | 0,43 | FBLN1, IFITM1, IFITM2, IFITM3, SLPI |
| CC: blood microparticle [8/16] | DW | GO:0072562 | 3,9E-09 | 0,7 | ANGPTL4, APOE, C1QB, C1QC, C3, CP, SERPINA3, STOM |
| BP: reactive oxygen species metabolic process [6/9] | DW | GO:0072593 | 0,000013 | 0,49 | CYBA, CYBB, GPX1, GPX3, PDK4, SOD2 |
| BP: receptor-mediated endocytosis [5/17] | DW | GO:0006898 | 0,00042 | 0,82 | APOE, CD163, FCER1G, ITGB2, MSR1 |
| BP: pos. regulation of inflammatory response [6/10] | DW | GO:0050729 | 0,00061 | 0,7 | HYAL2, OSMR, S100A9, TLR2, TNFRSF1A, VAMP8 |
Significant GO signatures detected by GO-PCA tool.
qRT-PCR validation
We validated RNA-Seq results performing quantitative Reverse Transcription PCR (qRT-PCR) assay using RNA extracted from the ventral horns of the lumbar spinal cord in an enlarged sample group (Table 1). We included donors assayed by RNA-Seq (5 controls and 6 ALSs) and an independent group of 2 controls and 3 ALSs (Table 1). From the list of differentially expressed genes, we selected 16 candidates including genes known to be deregulated in ALS such as INA, HECW1 and SLC1A2 (representing a sort of internal control) and genes not yet described as altered in ALS but associated to significantly disrupted pathways such as the cholesterol biosynthesis, the complement system or the synaptic transmission (Table 5). All genes belonging to deregulated pathways were randomly selected (with |log2FC| ≥ 1), except for SNAP25 and STX1B involved in the t-SNARE complex mediating the neurotransmitter release at the presynaptic membrane.
Table 5.
List of qRT-PCR validated genes and TaqMan Assay IDs.
| Gene Symbol | Description | Assay ID |
|---|---|---|
| APOC1 | apolipoprotein C-I | Hs00155790_m1 |
| ATP2B3 | ATPase, Ca++ transporting, plasma membrane 3 | Hs00222625_m1 |
| CHD5 | chromodomain helicase DNA binding protein 5 | Hs01086457_m1 |
| MYT1L | myelin transcription factor 1-like | Hs00903951_m1 |
| STX1B | syntaxin 1B | Hs01041315_m1 |
| SNAP25 | synaptosomal-associated protein, 25 kDa | Hs00938962_m1 |
| CTSS | cathepsin S | Hs00175407_m1 |
| SOD2 | superoxide dismutase 2, mitochondrial | Hs00167309_m1 |
| SLC1A2 | solute carrier family 1 (glial high affinity glutamate transporter), member 2 | Hs01102423_m1 |
| GPX3 | glutathione peroxidase 3 | Hs01078668_m1 |
| HMGCR | 3-hydroxy-3-methylglutaryl-CoA reductase | Hs00168352_m1 |
| HMGCS1 | 3-hydroxy-3-methylglutaryl-CoA synthase 1 (soluble) | Hs00940429_m1 |
| INA | internexin neuronal intermediate filament protein, alpha | Hs00190771_m1 |
| SERPINA3 | serpin peptidase inhibitor, clade A (alpha-1 antiproteinase, antitrypsin), member 3 | Hs00153674_m1 |
| HECW1 | HECT, C2 and WW domain containing E3 ubiquitin protein ligase 1 | Hs00389648_m1 |
| CD48 | CD48 molecule | Hs00914738_m1 |
| GAPDH* | glyceraldehyde-3-phosphate dehydrogenase | Hs99999905_m1 |
| HPRT1* | hypoxanthine phosphoribosyltransferase 1 | Hs99999909_m1 |
| PGK1* | phosphoglycerate kinase 1 | Hs99999906_m1 |
| POLR2A* | polymerase (RNA) II (DNA directed) polypeptide A, 220 kDa | Hs00172187_m1 |
List of genes selected for qRT-PCR validation reporting the TaqMan Assay ID per gene symbol and the corresponding description. Genes indicated by * have been used as internal controls.
The qRT-PCR analysis confirmed the differential expression emerged in RNA-seq analyses for all 16 genes (Fig. 6A) and the estimates of fold change in expression level were highly consistent with those from RNA-Seq (r = 0.98, P = 8.3 × 10−11 for CuffDiff2 and r = 0.96, P = 4.2 × 10−9 for DESeq2) (Fig. 6B).
Figure 6.

Results from qRT-PCR validation. (A) Results from qRT-PCR validation of 16 selected DE genes (Table 3) in the input sample group of 6 controls and 5 Als patients (blue bars), and in an independent group of 2 controls and 3 Als (red bars). Up and down regulation are in respect to ALS donors. (B) Linear correlation between log2(FC) values computed by CuffDiff2 (blue circles) or DESEq2 (red circles) on RNA-Seq data and log2(FC) values detected by qRT-PCR for 16 selected genes.
Aberrant Alternative Splicing
RNA-Seq read alignments were further investigated to identify differential splicing between control and ALS samples. Using MATS24, a software tools implementing Bayesian multivariate analysis of transcript splicing, we discovered 794 aberrant splicing events (Table 6). Of these, 98% involved exon skipping (including mutually exclusive exons) and 2% altered donor or acceptor splice sites and intron retention (Table 6).
Table 6.
Differential alternative splicing detected in RNA-Seq data by MATS program.
| Event Type | N | S |
|---|---|---|
| SE | 72689 | 601 |
| MXE | 18275 | 178 |
| A5SS | 1004 | 5 |
| A3SS | 1205 | 2 |
| RI | 452 | 8 |
| Total | 93625 | 794 |
For each alternative splicing type we repot the number of events in UCSC database (N) and the number of significant events (S). SE: skipped exon; MXE: mutually exclusive exon; A5SS: alternative 5′ splice site; A3SS: alternative 3′ splice site; RI: retained intron.
IPA analysis of gene targets for differential splicing revealed perturbations in genes with important neuronal functions (Supplementary Table 9). In particular, we found genes involved in the actin cytoskeleton signaling, which plays central roles in dynamic processes such as cell motility or axon guidance. GNRH signaling, which coordinates the levels of hormones in the hypothalamic-pituitary-gonadal axis and promotes the activation of G-Proteins, PLC, PKC or the mobilization and influx of calcium was also enriched in genes showing differential splicing as well as genes involved in triacylglycerol biosynthesis and mitochondrial functions (Supplementary Table 9).
Twenty-one of the genes exhibiting differential exon skipping were previously identified in an exon array analysis of motorneurons in sporadic ALS samples25.
Gene Ontology terms highly enriched in differentially spliced genes included: axon guidance, neuron projection guidance, anchoring junction and actin binding (Supplementary Table 10).
miRNA-Seq analysis
An emerging hallmark of neurodegenerative diseases is the defective RNA metabolism. It involves aberrant alternative splicing as demonstrated also by our RNA-Seq results and deregulated gene expression due to malfunction of miRNA activity26. Several previous investigations have documented a crucial role of miRNA gene regulation in the development and function of neurons. Consequently, alterations in the function of miRNA may contribute to neurodegenerative processes. Indeed, specific miRNA alterations have been reported for ALS microglia27 and affecting the stability of the low molecular weight neurofilament mRNA28.
We performed massive sequencing of the low molecular weight RNA fraction extracted from the same control and ALS donor tissues using in the RNA-seq analysis. Between 2.8 and 4.2 million reads were generated per sample and adaptors were removed using a custom script (adapters were detected in between 94.7–95.7% of reads for each sample) (Supplementary Figure 1). Reads were mapped to the human genome and miRNAs (reads mapping perfectly to annotated miRNAs and miRNA* were counted and assigned to non-redundant annotated miRNA sequences).
The vast majority of reads in the peak from 30–34 nt in length corresponded to the 5′ portions of annotated tRNA genes (consistently from nt1 to the anticodon loop) as observed previously29. The peak at 18 nt was comprised, predominantly (>95%), of tRNA derived small RNAs, almost exclusively consisting of few fragments representing positions 1–18 of tRNA-gly and tRNA-ala molecules. None of the aforementioned reads demonstrated statistically significant differential expression between ALS patients and healthy controls.
The peak at 21 and 22 nucleotides was strongly enriched from known miRNAs that were subjected to differential expression analysis. After correction for multiple hypothesis testing, we found 18 dysregulated miRNAs (Padj ≤ 0.05) (Table 7). Of these, 3 were up-regulated and 15 down-regulated in ALS samples.
Table 7.
MiRNAs differentially expressed in healthy controls and ALS patients.
| miRNA | log2FoldChange | P-value | Corrected P-value |
|---|---|---|---|
| hsa-mir-577 | −3,28 | 1,46E-10 | 3,87E-08 |
| hsa-mir-182-5p | −2,70 | 6,50E-15 | 3,44E-12 |
| hsa-mir-485-5p | −2,12 | 8,79E-05 | 8,45E-03 |
| hsa-mir-124-3p | −1,83 | 1,05E-05 | 1,85E-03 |
| hsa-mir-218-5p | −1,68 | 1,54E-04 | 1,16E-02 |
| hsa-mir-183-5p | −1,62 | 3,14E-04 | 2,08E-02 |
| hsa-mir-873-5p | −1,58 | 9,80E-04 | 3,25E-02 |
| hsa-mir-133a | −1,53 | 6,05E-04 | 2,29E-02 |
| hsa-mir-487b | −1,45 | 1,56E-03 | 4,59E-02 |
| hsa-mir-219-5p | −1,44 | 9,55E-04 | 3,25E-02 |
| hsa-mir-409-3p | −1,34 | 1,12E-03 | 3,48E-02 |
| hsa-mir-889 | −1,23 | 5,71E-04 | 2,29E-02 |
| hsa-mir-136-3p | −1,15 | 1,75E-03 | 4,87E-02 |
| hsa-mir-410 | −1,12 | 4,02E-04 | 2,29E-02 |
| hsa-mir-127-3p | −0,95 | 5,61E-04 | 2,29E-02 |
| hsa-mir-148a-3p | 1,23 | 4,99E-04 | 2,29E-02 |
| hsa-mir-155-5p | 1,82 | 9,56E-05 | 8,45E-03 |
| hsa-mir-221-3p | 2,10 | 4,82E-04 | 2,29E-02 |
For each differently expressed miRNA we report the standard name according to MirBase database, the log2 fold-change, the P-value and the corresponding corrected P-value as calculated by DESeq.
MiR-155 has previously been shown to be overexpressed in ALS microglial cells30 and inhibition of its expression has been shown to increase survival time in a murine ALS model31. MiR-485 has been implicated in synaptic formation and maintenance and has been reported to be dysregulated in Alzheimer’s disease and Huntington’s disease32, miR-124 is known as a neurodevelopmental regulator33, miR-219 required for neural precursor differentiation in zebrafish34 and promotes myelination in rats35, miR-127 regulates cell proliferation and senescence by targeting BCL636 while miR-136, miR-873 and miR-410 have all been studied in the context of glioma growth, invasiveness and apoptosis37–39.
Uncertainties regarding both the reliability of target predictions and the extent of degradation of individual targeted mRNAs in vivo, as well as the mixed nature of the tissue samples complicate direct reconciliation of miRNA and putative target expression profiles. The Diana-miRPath tool40 recovered a striking set of 6 pathways (p ≤ 10−16) enriched in genes with predicted targets for differentially expressed miRNAs: the PI3K-Akt signaling pathway, Axon guidance, Neurotrophin signaling pathway, Focal adhesion, TGF-beta signaling pathway and Insulin signaling pathway (Supplementary Table 11).
RNA editing analysis
To further characterize the transcriptome of ALS spinal cord samples, we investigated RNA editing limiting our search to A-to-I changes, as these account for over 99% of editing events in recent large-scale human investigations41, 42. In particular, for each RNA-Seq sample we explored known A-to-I events stored in the REDIportal database43, selecting only genomic positions supported by at least 30 reads and showing a RNA editing level higher than 0.1. Comparing positions in common between healthy and ALS donors, we found a strong correlation of 0,88 (Pval < 10e−14 by Spearman correlation test) in RNA editing levels. We also performed the non-metric multidimensional scaling (NMD) analysis on detected A-to-I changes in order to verify the grouping of input samples by RNA editing. Interestingly, we did not find specific ALS and control clusters as expected in case of global RNA editing alteration (Supplementary Figure 2), suggesting that it should not be impaired in ALS spinal cord tissues.
We observed reduction of RNA editing levels at the recoding GRIA2 Q/R site, known to be edited at nearly 100% in human brain44 and under-edited in motor neurons of ALS donors and, thus, implicated in cell death by excitotoxicity45. However, reduced RNA editing levels at the recoding GRIA2 Q/R site were also observed in control samples and the difference between ALS and control groups was not significant (Mann-Whitney P = 0.43). Only one ALS sample showed strong A-to-I reduction level of 0.19 at GRIA2 Q/R site. Additionally, we compared our RNA editing levels of GRIA2 Q/R site with corresponding values from spinal cord and other brain locations of an independent group of healthy individuals from GTEx project and stored in our REDIportal database. Interestingly, GRIA2 Q/R site appeared highly edited in almost all brain locations (average editing levels from 0.82 to 0.99), while editing levels of spinal cord samples (n = 15, average editing levels of 0.66) were quite similar to our values for both ALS (average editing levels of 0.62) and control (average editing levels of 0.66) groups, meaning that RNA editing levels at GRIA2 Q/R site are not always edited at nearly 100% in spinal cord (Fig. 7A and Supplementary Table 12). Editing frequencies of GRIA2 Q/R site in spinal cord samples were always significantly lower than corresponding values from all tested brain locations (Mann-Whitney P < 0.05) (Table 8).
Figure 7.
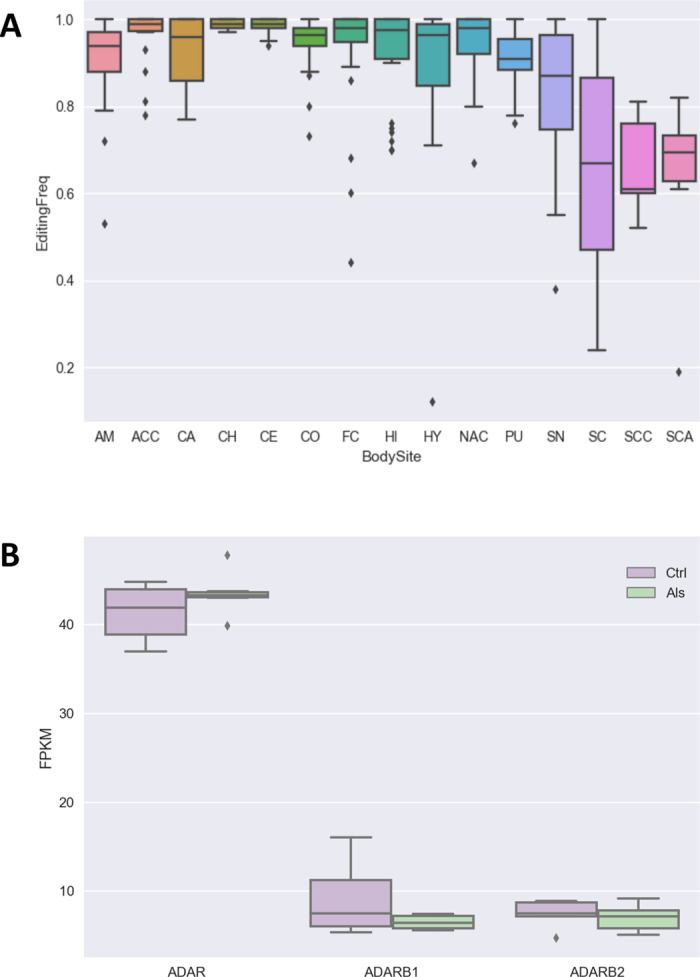
RNA editing levels of GRIA2 Q/R site and the expression of ADAR genes. (A) RNA editing levels of GRIA2 Q/R site in spinal cord and other brain locations. Abbreviated body site names are as follow: SC, spinal cord; AM, Amygdala; ACC, Anterior cingulate cortex; CA, Caudate; CH, Cerebellar Hemisphere; CE, Cerebellum; CO, Cortex; FC, Frontal Cortex; HI, Hippocampus; HY, Hypothalamus; NAC, Nucleus accumbens; PU, Putamen; SN, Substantia nigra; SCC, Spinal cord control group; SCA, Spinal cord ALS group. (B) Expression levels of ADAR genes from RNA-Seq experiments using the Cuffdiff2 tool. FPKM values for controls and ALS donors are shown as box plots.
Table 8.
Comparison between editing frequencies at GRIA2 Q/R site in spinal cord and other brain locations.
| SpinalCord | BrainBodySites | N. SC | N. BodySites | U-Stat | Pval |
|---|---|---|---|---|---|
| SC | ACC | 15 | 22 | 56,5 | 0,00031 |
| SC | NAC | 15 | 27 | 79,5 | 0,00057 |
| SC | CH | 15 | 28 | 64,5 | 0,00007 |
| SC | CO | 15 | 30 | 94,5 | 0,00085 |
| SC | PU | 15 | 19 | 70,5 | 0,00647 |
| SC | CA | 15 | 29 | 88 | 0,00058 |
| SC | AM | 15 | 21 | 80,5 | 0,00699 |
| SC | CE | 15 | 32 | 76,5 | 0,00007 |
| SC | FC | 15 | 28 | 87,5 | 0,00073 |
| SC | HI | 15 | 26 | 82 | 0,00098 |
| SC | SN | 15 | 18 | 86,5 | 0,04087 |
| SC | HY | 15 | 24 | 91 | 0,00513 |
| SC | SCC | 15 | 5 | 35 | 0,43058 |
| SC | SCA | 15 | 6 | 43 | 0,45344 |
Statistical significance was assessed by Mann-Whitney U Test (U-Stat and Pval columns). N. SC indicates the number of samples in spinal cord from GTEx project, N. BodySites report the number of samples per each brain body site from GTEx project. Abbreviated body site names in BrainBodySites are as follow: SC, spinal cord; AM, Amygdala; ACC, Anterior cingulate cortex; CA, Caudate; CH, Cerebellar Hemisphere; CE, Cerebellum; CO, Cortex; FC, Frontal Cortex; HI, Hippocampus; HY, Hypothalamus; NAC, Nucleus accumbens; PU, Putamen; SN, Substantia nigra; SCC, Spinal cord control group; SCA, Spinal cord ALS group.
Further, we explored RNA editing in miRNAs employing massive sequencing of the low molecular weight RNA. We found 12 A-to-I changes in 12 miRNAs (Supplementary Table 13). Of these, 9 were known as edited in the literature, while 3 appeared as novel RNA editing events. All detected changes were observed in both control and diseased donors, even though RNA editing levels were not significantly different (Supplementary Table 13).
Finally, we explored expression values of ADAR genes but no significant differences were detected between healthy and ALS tissues (Fig. 7B).
Immunoblotting and Immunofluorescence of synaptic SNARE proteins in ventral horns of ALS donors
We corroborated our RNA-Seq and qRT-PCR results performing immunoblotting of two synaptic SNARE proteins, SNAP25 and STX1B, since they might provide new insights into neurotransmission and calcium homeostasis alterations in ALS. Western blot analysis was performed on ventral horn of six patients with sporadic ALS and six controls to quantify the protein expression levels of SNAP25 and STX1B. Results obtained showed that SNAP25 and STX1B were significantly downregulated at protein level in ALS patients, in line with the mRNA data (Fig. 8 and Supplementary Figure 3). Since a functional redundancy between STX1B and its homologous STX1A has been reported46, STX1A protein expression levels were also analyzed in parallel. Results revealed an opposite trend for STX1 A being significantly upregulated in ALS patients (Fig. 8 and Supplementary Figure 3), as observed in FUS-silenced motor neurons47. Such upregulation appeared more prominent in five out of six ALS patients.
Figure 8.
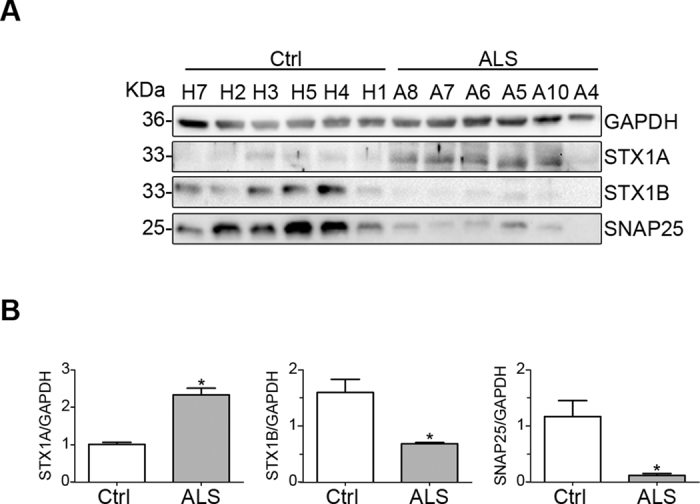
Immunoblot analysis of SNARE proteins in control (Ctrl) and ALS patient ventral horns. (A) Ventral horn samples were examined for STX1A, STX1B and SNAP25 expression by immunoblotting. GAPDH was used as internal control for protein loading. (B) Histograms summarizing the densitometry analysis of the protein bands shown in A, calculated as each SNARE/GAPDH expression ratio, as indicated (n = 6 for Ctrl and n = 6 for ALS; *P < 0.002). Samples are indicated by their Bank ID as reported in Table 1. The image in A was cropped for clarity from the original in Supplementary Figure 3.
Additionally, we investigated in more detail the SNAP25 and STX1A deregulation by immunofluorescence followed by quantification analysis (Figs 9 and 10, Supplementary Figure 4) using confocal microscopy and β3 tubulin as motor neuron marker. The intensity of SNAP25 and STX1A signal was normalized on the size of motor neuron cell body that we found to be reduced by almost 50% in ALS patients (Fig. 9B). In agreement with the SNARE-complex deregulation detected by Western blot (Fig. 8), we found that SNAP25 signal was almost completely absent in ALS patients (Fig. 9), whereas STX1A was significantly upregulated (Fig. 10).
Figure 9.

Confocal microscopy analysis of SNAP25 and β3-tubulin in spinal cord ventral horns of control and ALS patients. (A) Confocal microscopy images of SNAP25 (green) and β3-tubulin (red) of a typical motor neuron in control and ALS patient. (B,C) Histograms showing the motor neuron body size expressed in µm2 (B) and the quantitative analysis of SNAP25 signal normalized on motor neuron size (C) of control and ALS patients (*P < 0.05). (D) Confocal 3D reconstruction of SNAP25 and β3-tubulin staining
Figure 10.

Confocal microscopy analysis of STX1A and β3-tubulin in spinal cord ventral horn of control and ALS patients. (A) Confocal microscopy images of STX1A (green) and β3-tubulin (red) of a typical motor neuron in control and ALS patient. (B) Histograms showing the quantitative analysis of STX1A signal normalized on motor neuron size of control and ALS patients (*P < 0.05). (C) Confocal 3D reconstruction of STX1A and β3-tubulin staining.
Together, these results validate the RNA-Seq data on the altered expression pattern of genes in ALS and provide new information on the molecular basis of synapsis and calcium handling abnormalities associated with the motor neuron degeneration typical of this pathology.
Discussion
ALS is a rare and devastating neurodegenerative disease for which no effective therapies are available1. Large-scale genetic investigations, at least in the familial form of ALS, have identified several mutations in key genes for motor neuron survival2. Nonetheless, the causes of ALS onset and progression are yet elusive. Whole transcriptome analyses have revealed the disruption of a variety of cellular pathways contributing to the elucidation of molecular dynamics characterizing the pathobiology of ALS3.
The main obstacle complicating ALS investigations is that motor neurons cannot be sampled during life. As a consequence, animal models and spinal cord tissue from post-mortem donors represent the elective materials for the study of gene expression in ALS. In humans, previous transcriptome studies have been conducted on laser microdissected motor neurons, in order to avoid biases from mixed cell populations. Although motor neurons are the most affected in ALS, other cell types such as astrocytes, microglia and oligodendrocytes may contribute to disease progression. Indeed, co-culture of motor neurons and glial cells has shown an intricate interplay and an active role of non-neuronal cells in neurodegeneration6.
In order to characterize whole transcriptome modifications occurring in sALS, we conducted a study in which total RNA from the lumbar spinal cord of ALS donors were sequenced using the RNA-Seq technology.
Our results demonstrate the suitability of deep transcriptome sequencing for capturing molecular changes in the lumbar spinal cord and provide new exciting insights still not revealed by recent whole transcriptome studies based on RNAseq in brain7 and cervical spinal cord8 from fALS and sALS samples.
Using a dataset of cell type-specific genes derived from various cell types in the CNS15 as astrocytes, oligodendrocytes and neurons, our data show a depletion of neuron, motorneuron and oligodendrocyte populations with down regulated genes of neuronal derivation and linked to relevant neurological functions as impulse transmission, synaptic transmission, calcium ion transport or neurotransmitter secretion. Up regulated genes, instead, are generally involved in neuroinflammation, immune responses or cell death and tend to have microglial origin.
RNA-Seq of ALS lumbar spinal cord therefore provides a snapshot of transcriptomic events characterizing the disease, suggesting that neuronal function is compromised through the down regulation of many genes. As a consequence of neurological injury, surrounding cells are activated leading to a cascade of inflammatory events. Although our data do not provide evidence regarding early molecular events causing the death of motor neurons, transcriptomic results would seem to support these speculations. In addition, comparing our results with those by Brohawn et al.8 in cervical spinal cord of sALS donors using similar technology and methodology, it emerges that neuroinflammation is common in all sections of spinal cord but molecular signatures of neurodegeneration are more consistent in the lumbar rather than cervical spinal region. Our down regulated genes actually include genes already known to be associated to ALS motor neuron death such as INA, HECW1 or SLC1A2.
As a remarkable result, RNA-Seq data support the down-regulation in ALS donors of SNAP25 and STX1B, neuronal t-SNAREs involved in vesicle trafficking and calcium dynamics. Since a redundant role between STX1B and its homologous STX1A has been reported48 and being STX1A the isoform interacting with SNAP25 and Voltage Gated Calcium Channels (VGCCs) to control calcium signaling, we have explored the t-SNARE pattern as a whole. In particular, we show that SNAP25 and STX1B are strongly downregulated also at protein level. STX1A, instead, was upregulated in five out of six ALS patients. Through immunofluorescence followed by quantification analysis (Figs 9 and 10) using confocal microscopy and β3 tubulin as motor neuron marker, we confirm the deregulation of SNAP25 and STX1A in ALS patients and demonstrate that the down expression of SNAP25 is mainly due to the reduced size of motor neurons.
Ikemoto et al.49 have investigated SNAP25 and STX1 expression in ALS spinal cord finding no apparent decrease or only a mild reduction for both proteins in ALS patients. We believe that the immunolocalization approach performed in the study by Ikemoto49, without quantification analysis, has most likely led to an underestimation of the t-SNARE reduction in ALS patients. Moreover, the antibody used to detect STX1 was not isoform specific and therefore STX1 unaltered expression reported by Ikemoto49 is consistent with the data here reported where STX1A is upregulated and STX1B downregulated. Additionally, upregulation of STX1A and deregulation of SNAP25 splicing were observed also in FUS-silenced motor neurons47.
Experiments performed in animal models and primary cultures have shown that loss of SNAP25 impairs calcium-evoked exocytosis leaving almost unaffected spontaneous vesicle release50, 51 while deletion of STX1A has no effect on both types of synaptic transmission probably due to compensation by STX1B. Finally, STX1B deletion impairs both spontaneous and evoked fast synaptic vesicle exocytosis48.
According to our results on altered t-SNARE protein pattern, SNAP25 and STX1B reduction seem to strictly relate with the motor neuron dysfunction characteristics of ALS ventral horns in which STX1A up regulation may compensate for STX1B loss. Notably, the observed SNAP25 reduction highlights a potentially novel and relevant role of t-SNARE pattern in elevating intracellular calcium concentration and in triggering the glutamate excitotoxicity. Indeed, SNAP25 silencing in glutamatergic neurons has been reported to increase the concentration of intracellular calcium evoked by depolarization52. Therefore an initial accident altering the t-SNARE pattern causing SNAP25 reduction should lead to the increased intracellular calcium levels found in ALS motorneurons12. In turn, elevated intracellular calcium concentration causes motor neuron injury leading to increased glutamate release and excitotoxicity. In this context, STX1A may compensate for SNAP25 reduction in order to restore calcium homeostasis.
Another remarkable finding of our work is the dysregulation of cholesterol synthesis, which has been linked to chronic neurodegenerative disorders53. This alteration could contribute to the impaired axon guidance and synaptic transmission observed in ALS, as cholesterol is essential for the organization and physiological fluidity of cellular membranes.
RNA-Seq also confirms the disruption of RNA metabolism through aberrant alternative splicing potentially resulting from neuronal stress.
Additionally, we investigated RNA editing since its deregulation has been linked to several neurodegenerative diseases including ALS45. Indeed, it has been shown that reduced levels of recoding A-to-I editing at GRIA2 Q/R site may be involved in motor neuronal death through an increased Ca2+ permeability of AMPA receptor45. Our results, based on known RNA editing events, do not support significant alterations. A closer investigation of RNA editing levels at the recoding GRIA2 Q/R site reveals that A-to-I frequencies of GRIA2 Q/R site are not impaired in spinal cord tissue of ALS donors, rather their values are similar to those observed in physiological conditions without altered expression of ADAR enzymes. However, our study was conducted on entire tissues and, thus, we cannot exclude a compensating effect from non-neural cells obscuring specific motor neuron RNA editing alterations.
Small RNA expression analyses suggest important roles for miRNAs regulating signaling pathways determining cell survival and axon guidance during ALS progression. Numerous lines of evidence implicate TGF-beta signaling in motor neuron diseases. It participates in the interplay between neuronal and glial cells during ALS progression6 and increased levels of TGF-beta provoke an acute improvement in the motor performance of SOD1 mice54. Participants in the TGF-beta signaling pathway predicted to be targets of DE miRNAs include Smad2,4,5 and 7. TDP-43 and phosphorylated Smad2 are co-localized within cytoplasmic inclusions in the anterior horn cells of sporadic ALS patients55. Activation of the TGFβ/Smad signaling system is protective against aggregate formation of cytoplasmically mislocalized TDP-4355.
The Axon Guidance pathway has recently been proposed to represent a prime effector in ALS pathobiology56 and is intimately linked to the regulation of actin cytoskeleton and Focal Adhesion circuits.
The phosphatidylinositol 3-kinase (PI3K) /ATK prosurvival pathway has been implicated as a mediator of the protective effect of vascular endothelial growth factor in G93A-SOD1 neurons in vitro and induction of ATK3 is neuroprotective in SOD1_G93A mice57. The pathway contributes, in conjunction with mTOR family proteins, to the regulation of the activity of the glutamate transporter GLT1 in astrocytes58.
Neurotrophins, including Nerve Growth Factor (NGF), brain-derived neurotrophic factor (BDNF), and Neurotrophins 3 and 4 (NTF3 and NTF4) lie upstream of the Neurotrophin pathway which regulates the axon cytoskeleton (and in turn axon outgrowth/guidance and synapse formation) as well as cell differentiation and survival. NFT3, NTF4, and BDNF, as well as their cellular receptors and numerous downstream genes are all predicted targets of miRNAs dysregulated in ALS. NGF, BDNF, NT-3 are neuroprotective on axotomized extraocular motoneurons in neonatal rats59, while 7,8-dihydroxyflavone (7,8-DHF) a small molecule tyrosine kinase receptor B (TrkB) agonist that mimics the effects of BDNF, is neuroprotective in SOD1 mice60.
In conclusion, our study remarks the use of massive RNA sequencing technologies to improve deregulated pathways of ALS neurodegeneration and provide new exiting insights as the SNAP25 reduction as possible cause of calcium elevation and glutamate excitotoxicity, suggesting t-SNARE protein expression as novel indicators and potential biomarkers for sporadic ALS.
Methods
Post mortem tissues
Frozen lumbar spinal cord samples from 11 male human donors affected by sporadic ALS and 7 age, sex and ethnicity matched controls were obtained from the NICHD Brain & Tissue Bank for Developmental Disorders (University of Maryland - http://medschool.umaryland.edu/btbank/) and the Human Brain and Spinal Fluid Resource Center (Los Angeles, CA). Additionally, we required as an additional criterion, the presence of clinical symptoms associated to the altered function of lower motor neurons.
Each tissue was longitudinally dissected and ventral horns were collected for downstream analyses.
Detailed information regarding samples used in RNA-seq, miRNA-seq, qRT-PCR and western blot analyses are provided in Table 1, also including age, gender, ethnicity and post-mortem interval as well as donor bank IDs to access to further clinical info.
RNA extraction
RNA was extracted from the ventral horns of the lumbar spinal cord after grinding with mortar and pestle using the mirVana™ miRNA Isolation Kits (Life Technologies Inc., Carlsbad, CA, USA) according to the manufacturer’s instructions to separate total and small RNA fractions. Each RNA sample was treated with RNAse-free DNAse (Life Technologies) and qualitatively and quantitatively checked on Agilent 2100 Bioanalyzer RNA Nano Chip (Agilent, Santa Clara, CA, USA).
RNA sequencing
Directional RNA-Seq libraries were prepared from 1 μg of total RNA using the TruSeq Stranded Total RNA Sample Prep Kit (Illumina) according to the manufacturer’s protocol. Sequencing was performed on an Illumina NextSeq 500 platform (Illumina, San Diego, CA), generating for each sample from 109 to 219 millions of 100pb × 2 paired-end reads. RNA-Seq statistics are reported in Table 2.
miRNA sequencing
Indexed cDNA libraries from the RNA fraction at low molecular were prepared using the TruSeq small RNA sample Preparation kit (Illumina, San Diego, CA) according to the manufacturer’s protocol and recommendations. Single end sequencing (1 × 50b), after fluorimetric quantification, was performed on an Illumina Miseq platform.
Preprocessing and analysis of RNA-Seq reads
RNA-Seq reads in FASTQ format were initially inspected using FASTQC program (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Adaptors and low quality regions (phred cutoff of 20) were trimmed using Trim Galore (http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/), excluding reads with final length less than 50 bases.
Cleaned reads were aligned onto the complete human genome (assembly hg19) by means of GSNAP61 version 2013-11-27 (using as parameters: -B 5 -d hg19 -t5 -s splicesites -E1000 -N1 -n1 -Q -O-nofails -A sam–force-xs-dir -a paired) providing a list of exon-exon junctions from Ensembl, UCSC and RefSeq databases. Unique and concordant alignments in SAM format were converted in the binary BAM format by SAMtools62 and basic statistics were calculated using the CollectRnaSeqMetrics tool of Picard package (https://broadinstitute.github.io/picard/index.html) (Table 2). The sporadic disease nature of our samples was checked using REDItools13, screening known ALS mutations from ALSoD database in aligned RNAseq data14. Only positions showing a frequency variation higher than 0.1 and supported by more than 10 reads we taken into account.
Differential expression was performed using two independent tools: CuffDiff2 version 2.1.117 (using as main parameters:–library-type fr-firststrand–labels Ctl,Als -u -b hg19.fa refgenes.gtf) and DESeq2 version 1.10.118. Read counts on known human genes were calculated by featureCounts version 1.5.163. Reference human transcriptome was obtained from iGenomes repository (http://cole-trapnell-lab.github.io/cufflinks/igenome_table/) and annotations for rRNA genes were downloaded from UCSC genome browser selecting the RepeatMask table.
DE genes was selected intersecting lists of significant DE genes (corrected P value < 0.05) obtained by CuffDiff2 and DEseq2. We took into account only genes with |log2(FC)| higher than 1.
Changes in cell type composition of spinal cord samples was detected according to the method described in Lin et al.64 and using cell type specific genes for neurons, motorneurons, astrocytes, oligodendrocytes and microglia collected from the literature6, 15. The majority of cell type specific genes were obtained from Cahoy et al.15 taking into account only genes showing >10-fold enrichment in this study. Since Cahoy et al. performed their investigations in mouse, we selected only genes with corresponding human orthologues and showing FPKM values > 0. Motor neuron specific genes, instead, were collected from Phatnani et al.6.
Differential alternative splicing was detected with MATS version 3.0.924 using default parameters except for the read length (fixed to 90). Events were selected at 0.05 significant level, corrected for multiple testing. Heat-maps and PCA plots were generated using ClustVis65.
IPA system was used for pathway analysis. Enrich66, instead, was used to generate lists of Gene Ontology terms enriched in the upregulated, downregulated or differentially spliced gene lists, with default setting. To identify sets of genes that are both strongly correlated to expression data and functionally related to their GO annotations, we used the GO-PCA method23. In brief, it adopts a two-step approach in which PCA is performed first. Then, each principal component is tested for whether it is driven by functionally related genes (in the form of gene ontology annotations).
Preprocessing and analysis of miRNA-Seq reads
Raw sequence data were processed with a custom Python script to remove adapters from inserts of between 14 and 38 nt in length and subsequently mapped onto the human genome (version hg19) using Bowtie67. Counts of reads exactly matching human miRNAs and (where annotated in miRBase) miRNA* sequences were submitted to the DESeq software68 for statistical analysis of differential expression using default parameters and treating different ALS and healthy control patients as biological replicates. Pathway analysis was conducted with miRPath v2.040 with default settings.
RNA editing detection and analysis
RNA editing candidates per each sample were detected using the REDItools package (https://sourceforge.net/projects/reditools/)13. In particular, REDItoolKnown.py was used to explore known RNA editing sites stored in REDIPortal database43. Non-Metric Multidimensional Scaling analysis of RNA editing sites was performed in R using the vegan package. RNA editing levels at the GRIA2 Q/R site in different brain regions were extracted from REDIportal43. Custom python scripts were used to calculate RNA editing correlation between control and ALS samples.
RNA editing detection in miRNAs was performed according to the protocol developed by Alon et al.69, while RNA editing levels were calculated by REDItools13.
qRT-PCR validation
Gene expression analyses were carried out on custom TaqMan Array Fast Plates (Life Technologies), configured in order to include four potential human endogenous control genes (GAPDH, HPRT1, PGK1 and POLR2A), and sixteen genes selected for validation (Table 5), each of them in duplicate per sample. Reverse transcription of 500 ng of total RNA was performed using “iScript Reverse Transcription Supermix for RT-qPCR” (Bio-Rad, Hercules, CA, USA). qPCR reactions were set up according to the manufacturer’s instruction of “TaqMan Fast Universal PCR Master Mix” kit (Life Technologies), starting from 1 µl of diluted cDNA (1:3) as template and performed using the ABI PRISM 7900HT platform (Applied Biosystem, Life Technologies) with the following amplification conditions: hot start at 95 °C for 10 min; 45 amplification cycles (95° for 15 sec, 60° for 1 min).
Control reverse transcription reactions (RT-) were also prepared and PCR reactions, using primers for HPRT1 transcript (Forward: 5′-TGACACTGGCAAAACAATGCA-3′; Reverse: 5′-GGTCCTTTTCACCAGCAAGCT-3′), were carried out as control for genomic DNA contamination (data not shown).
Fluorescence raw data were exported from the SDS 2.2.1 software and analyzed using the comparative quantification cycle (Cq) method to determine the relative ratio of transcripts. First, the ΔCt values were obtained by calculating, for each sample, the difference of the Cq of each target compared to the arithmetic mean of the endogenous Cq genes (GAPDH and HPRT1). Then, the relative expression ratio (rER) was expressed as Log2(2−ΔCt); rER for each type of samples were averaged and calibrated respect to normal samples. Analyses were carried out using data related to two independent experiments.
Antibodies
The following primary antibodies were used: goat anti-SNAP25 (SC-7538, Santa Cruz, CA, USA), rabbit anti-Syntaxin-1A (110302, Synaptic System, Goettingen, Germany), rabbit anti-Syntaxin-1B (110402, Synaptic System, Goettingen, Germany), mouse anti-GAPDH (MAB 374, Millipore, Billerica, MA, USA) and mouse anti-β3 tubulin (SC-80016, Santa Cruz Biotechnology, Santa Cruz, CA, USA).
The following secondary antibodies were used for Western blot: HorseRadishPeroxidase (HRP)-conjugated donkey anti-goat, goat anti-rabbit and goat anti-mouse IgG (Santa Cruz Biotechnology, Santa Cruz, CA, USA).
The following secondary antibodies were used for Immunofluorescence: donkey anti-rabbit and donkey anti-goat AlexaFluor488-conjugate IgG (A-21206, Thermofisher, Milan, Italy), and donkey anti-mouse AlexaFluor594-conjugate IgG (A-27027, Thermofisher, Milan, Italy).
Protein sample preparation and immunoblotting
1 mm slices of Ctrl and ALS ventral horn samples, were collected, cut into small pieces with a razor blade and immediately solubilized in 8 volumes of RIPA buffer (10 mM Tris-HCl, pH 7.4; 140 mM NaCl; 1% Triton X-100; 1% Na deoxycholate; 0.1% SDS; 1 mM Na3VO4; 1 mM NaF and 1 mM EDTA) added with a cocktail of protease inhibitors (Roche, Milan, Italy). The lysis was performed as previously described70. Briefly, on ice for 1 h and the samples were then centrifuged at 22,000xg for 1 h. The protein content of the supernatant was measured with a bicinchoninic acid (BCA) Protein Assay Kit (Rockford, IL, USA).
30 ug of protein samples were separated by 13% Tris-Glycine-SDS-PAGE and transferred to polyvinylidene difluoride membranes (Millipore, Milan, Italy) as described previously71. Membranes with blotted proteins were incubated with primary antibodies, washed, and incubated with peroxidase-conjugated secondary antibodies. Reactive proteins were revealed with an enhanced chemiluminescent detection system (ECL Plus; GE Healthcare, Buckinghamshire, UK) and visualized on a Versadoc imaging system (BioRad, Milan, Italy). Densitometry analysis was performed using Scion Image software (Frederick, MD, USA).
All data are reported as mean ± SEM. We used the Student’s t test for unpaired data and analysis of variance for multiple statistical comparisons between groups, the significance level being set at P < 0.05. The number of samples is indicated in the figure legend.
Immunofluorescence
Fresh sections of control and ALS spinal cord samples were rehydrated with PBS and fixed for 10 min with 4% paraformaldehyde in PBS (HT5014, Sigma, Milan Italy). Sections were washed in PBS and incubated with 1:200 diluted anti-SNAP25 and 1:1000 diluted anti-β3 tubulin or 1:200 diluted anti-Syntaxin-1A and 1:1000 diluted anti-β3 tubulin in 0.3% Triton X-100 in PBS overnight at 4 °C. After washings with PBS, sections were saturated with 0.1% Gelatin (Sigma, Milan, Italy) in PBS, and incubated with 1:1000 diluted AlexaFluor488 and AlexaFluor594 conjugated secondary antibodies for 1 hour at room temperature. After washings, sections were washed in PBS and mounted with Mowiol (Sima, Milan, Italy) and acquired using Leica TCS SP2 confocal microscope (Leica Microsystems, Heidelberg GmbH) for the quantitative analysis described in the next paragraph. Confocal 3D reconstruction was obtained using Leica TCS SP8 STED 3X and Leica HC PL APO 100x/1.40 Oil white objective with Type F Immersion liquid with 1.5 refractive index (Pisani et al., 2016). Excitation of AlexaFluor488 and AlexaFluor594 dyes was performed with a continuous 488 nm and 594 nm wavelength diode laser with a maxim light output in focal plane of 10 mW (NKT Photonics sign supercontinuum laser). To obtain the confocal 3D projection, 30 planes were acquired in a Z-stack of 6–12 μm, and the series processed by Leica LASX software (Leica Microsystems, Mannheim, Germany).
Quantitative analysis of motor neuron size and SNAP25 and STX1A expression by immunofluorescence in ventral horn sections
Single confocal planes of randomly chosen areas of ventral horn sections from three control donors and three ALS patients, stained as described above, were acquired by Leica TCS SP2 confocal microscope (Leica Microsystems, Heidelberg GmbH). Leica Confocal Software Version 2.61 was used to measure motor neuron size and to quantify SNAP25 and STX1A expression levels. Motor neuron cell bodies were drawn based on the β3 tubulin staining, and the surface quantified and expressed in µm2. The average signal value relative to SNAP25 and STX1A for each motor neuron was also calculated and normalized on the motor neuron surface. Results were expressed as means ± standard error. Statistical significance was evaluated using unpaired Student’s t test. Difference with p < 0.05 were considered statistically significant.
Data availability
All sequencing data produced in the present work have been submitted to dbGaP database under the accession phs000747.
Electronic supplementary material
Acknowledgements
We kindly thank J. Cottrell at NICHD Brain Bank for assistance with spinal cord tissues. Authors are grateful to the NICHD Brain & Tissue Bank for Developmental Disorders (University of Maryland - USA) and the Human Brain and Spinal Fluid Resource Center (Los Angeles, CA) for providing lumbar spinal cord tissues used in this study. This work was supported by the Agency for Research on Amyotrophic Lateral Sclerosis (AriSLA); Italian Ministero dell’Istruzione, Università e Ricerca (MIUR): PRIN 2009 and 2010; Consiglio Nazionale delle Ricerche: Flagship Project Epigen, Medicina Personalizzata, Aging Program 2012–2014 and Interomics.
Author Contributions
A.M.D. performed RNAseq sequencing and supervised all laboratory activities. A.G., S.R. and F.L. performed RNA editing analyses and contributed to the manuscript draft. D.S.H. and M.C. carried out miRNA-Seq analyses and revised the manuscript. C.M. performed miRNA sequencing. A.V., I.A., F.M. supported C.M. in miRNA sequencing, carried out qRT-PCR validations and handled tissue samples. L.C. carried out RNA editing detection in miRNAs. F.P., G.P.N. and M.S. performed immunoblotting and immunostaining analyses. E.P. performed bioinformatics analyses. GP and EP conceived the study and wrote the manuscript. All authors approved the final version of the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Graziano Pesole, Email: graziano.pesole@uniba.it.
Ernesto Picardi, Email: ernesto.picardi@uniba.it.
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-10488-7
References
- 1.Kiernan MC, et al. Amyotrophic lateral sclerosis. Lancet. 2011;377:942–955. doi: 10.1016/S0140-6736(10)61156-7. [DOI] [PubMed] [Google Scholar]
- 2.Renton AE, Chio A, Traynor BJ. State of play in amyotrophic lateral sclerosis genetics. Nature neuroscience. 2014;17:17–23. doi: 10.1038/nn.3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Heath PR, Kirby J, Shaw PJ. Investigating cell death mechanisms in amyotrophic lateral sclerosis using transcriptomics. Front Cell Neurosci. 2013;7:259. doi: 10.3389/fncel.2013.00259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cooper-Knock J, et al. Gene expression profiling in human neurodegenerative disease. Nature reviews. Neurology. 2012;8:518–530. doi: 10.1038/nrneurol.2012.156. [DOI] [PubMed] [Google Scholar]
- 5.Ferraiuolo L, Kirby J, Grierson AJ, Sendtner M, Shaw PJ. Molecular pathways of motor neuron injury in amyotrophic lateral sclerosis. Nat Rev Neurol. 2011;7:616–630. doi: 10.1038/nrneurol.2011.152. [DOI] [PubMed] [Google Scholar]
- 6.Phatnani HP, et al. Intricate interplay between astrocytes and motor neurons in ALS. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:E756–765. doi: 10.1073/pnas.1222361110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Prudencio M, et al. Distinct brain transcriptome profiles in C9orf72-associated and sporadic ALS. Nature neuroscience. 2015;18:1175–1182. doi: 10.1038/nn.4065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brohawn DG, O’Brien LC, Bennett JP., Jr. RNAseq Analyses Identify Tumor Necrosis Factor-Mediated Inflammation as a Major Abnormality in ALS Spinal Cord. PloS one. 2016;11:e0160520. doi: 10.1371/journal.pone.0160520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Condliffe SB, Corradini I, Pozzi D, Verderio C, Matteoli M. Endogenous SNAP-25 regulates native voltage-gated calcium channels in glutamatergic neurons. The Journal of biological chemistry. 2010;285:24968–24976. doi: 10.1074/jbc.M110.145813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Antonucci F, et al. Reduced SNAP-25 alters short-term plasticity at developing glutamatergic synapses. EMBO reports. 2013;14:645–651. doi: 10.1038/embor.2013.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sharma M, et al. CSPalpha knockout causes neurodegeneration by impairing SNAP-25 function. The EMBO journal. 2012;31:829–841. doi: 10.1038/emboj.2011.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Appel SH, Beers D, Siklos L, Engelhardt JI, Mosier DR. Calcium: the Darth Vader of ALS. Amyotrophic Lateral Sclerosis. 2001;2:47–54. [PubMed] [Google Scholar]
- 13.Picardi E, Pesole G. REDItools: high-throughput RNA editing detection made easy. Bioinformatics. 2013;29:1813–1814. doi: 10.1093/bioinformatics/btt287. [DOI] [PubMed] [Google Scholar]
- 14.Abel O, Powell JF, Andersen PM, Al-Chalabi A. ALSoD: A user-friendly online bioinformatics tool for amyotrophic lateral sclerosis genetics. Human mutation. 2012;33:1345–1351. doi: 10.1002/humu.22157. [DOI] [PubMed] [Google Scholar]
- 15.Cahoy JD, et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2008;28:264–278. doi: 10.1523/JNEUROSCI.4178-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Streit WJ, Mrak RE, Griffin WS. Microglia and neuroinflammation: a pathological perspective. J Neuroinflammation. 2004;1:14. doi: 10.1186/1742-2094-1-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Trapnell C, et al. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nature biotechnology. 2013;31:46–53. doi: 10.1038/nbt.2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq 2. Genome biology. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saris CG, et al. Weighted gene co-expression network analysis of the peripheral blood from Amyotrophic Lateral Sclerosis patients. BMC genomics. 2009;10:405. doi: 10.1186/1471-2164-10-405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shankar GM, Walsh DM. Alzheimer’s disease: synaptic dysfunction and Abeta. Mol Neurodegener. 2009;4:48. doi: 10.1186/1750-1326-4-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bruijn LI, Miller TM, Cleveland DW. Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu Rev Neurosci. 2004;27:723–749. doi: 10.1146/annurev.neuro.27.070203.144244. [DOI] [PubMed] [Google Scholar]
- 22.Tan W, Pasinelli P, Trotti D. Role of mitochondria in mutant SOD1 linked amyotrophic lateral sclerosis. Biochimica et biophysica acta. 2014;1842:1295–1301. doi: 10.1016/j.bbadis.2014.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wagner F. GO-PCA: An Unsupervised Method to Explore Gene Expression Data Using Prior Knowledge. PloS one. 2015;10:e0143196. doi: 10.1371/journal.pone.0143196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shen S, et al. MATS: a Bayesian framework for flexible detection of differential alternative splicing from RNA-Seq data. Nucleic acids research. 2012;40:e61. doi: 10.1093/nar/gkr1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rabin SJ, et al. Sporadic ALS has compartment-specific aberrant exon splicing and altered cell-matrix adhesion biology. Human molecular genetics. 2010;19:313–328. doi: 10.1093/hmg/ddp498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haramati S, et al. miRNA malfunction causes spinal motor neuron disease. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:13111–13116. doi: 10.1073/pnas.1006151107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Parisi C, et al. Dysregulated microRNAs in amyotrophic lateral sclerosis microglia modulate genes linked to neuroinflammation. Cell Death Dis. 2013;4:e959. doi: 10.1038/cddis.2013.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Campos-Melo D, Droppelmann CA, He Z, Volkening K, Strong MJ. Altered microRNA expression profile in Amyotrophic Lateral Sclerosis: a role in the regulation of NFL mRNA levels. Mol Brain. 2013;6:26. doi: 10.1186/1756-6606-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cole C, et al. Filtering of deep sequencing data reveals the existence of abundant Dicer-dependent small RNAs derived from tRNAs. Rna. 2009;15:2147–2160. doi: 10.1261/rna.1738409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dhahbi JM, et al. 5′ tRNA halves are present as abundant complexes in serum, concentrated in blood cells, and modulated by aging and calorie restriction. BMC genomics. 2013;14:298. doi: 10.1186/1471-2164-14-298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koval ED, et al. Method for widespread microRNA-155 inhibition prolongs survival in ALS-model mice. Human molecular genetics. 2013;22:4127–4135. doi: 10.1093/hmg/ddt261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cohen JE, Lee PR, Chen S, Li W, Fields RD. MicroRNA regulation of homeostatic synaptic plasticity. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:11650–11655. doi: 10.1073/pnas.1017576108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Neo, W. H. et al. MicroRNA miR-124 controls the choice between neuronal and astrocyte differentiation by fine-tuning Ezh2 expression. The Journal of biological chemistry, doi:10.1074/jbc.M113.525493 (2014). [DOI] [PMC free article] [PubMed]
- 34.Hudish LI, Blasky AJ, Appel B. miR-219 regulates neural precursor differentiation by direct inhibition of apical par polarity proteins. Developmental cell. 2013;27:387–398. doi: 10.1016/j.devcel.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pusic AD, Kraig RP. Youth and environmental enrichment generate serum exosomes containing miR-219 that promote CNS myelination. Glia. 2014;62:284–299. doi: 10.1002/glia.22606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen J, Wang M, Guo M, Xie Y, Cong Y. S. miR-127 regulates cell proliferation and senescence by targeting BCL6. PloS one. 2013;8:e80266. doi: 10.1371/journal.pone.0080266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang Y, et al. MiR-136 promotes apoptosis of glioma cells by targeting AEG-1 and Bcl-2. FEBS letters. 2012;586:3608–3612. doi: 10.1016/j.febslet.2012.08.003. [DOI] [PubMed] [Google Scholar]
- 38.Skalsky RL, Cullen BR. Reduced expression of brain-enriched microRNAs in glioblastomas permits targeted regulation of a cell death gene. PloS one. 2011;6:e24248. doi: 10.1371/journal.pone.0024248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen L, et al. MiR-410 regulates MET to influence the proliferation and invasion of glioma. Int J Biochem Cell Biol. 2012;44:1711–1717. doi: 10.1016/j.biocel.2012.06.027. [DOI] [PubMed] [Google Scholar]
- 40.Vlachos IS, et al. DIANA miRPath v.2.0: investigating the combinatorial effect of microRNAs in pathways. Nucleic acids research. 2012;40:W498–504. doi: 10.1093/nar/gks494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bazak L, et al. A-to-I RNA editing occurs at over a hundred million genomic sites, located in a majority of human genes. Genome research. 2014;24:365–376. doi: 10.1101/gr.164749.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Picardi E, et al. Profiling RNA editing in human tissues: towards the inosinome Atlas. Sci Rep. 2015;5:14941. doi: 10.1038/srep14941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Picardi, E., D’Erchia, A. M., Lo Giudice, C. & Pesole, G. REDIportal: a comprehensive database of A-to-I RNA editing events in humans. Nucleic acids research, gkw767, doi:10.1093/nar/gkw767 (2016). [DOI] [PMC free article] [PubMed]
- 44.Seeburg PH, Higuchi M, Sprengel R. RNA editing of brain glutamate receptor channels: mechanism and physiology. Brain Res Brain Res Rev. 1998;26:217–229. doi: 10.1016/s0165-0173(97)00062-3. [DOI] [PubMed] [Google Scholar]
- 45.Hideyama T, et al. Induced loss of ADAR2 engenders slow death of motor neurons from Q/R site-unedited GluR2. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2010;30:11917–11925. doi: 10.1523/JNEUROSCI.2021-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhou P, et al. Syntaxin-1 N-peptide and Habc-domain perform distinct essential functions in synaptic vesicle fusion. The EMBO journal. 2013;32:159–171. doi: 10.1038/emboj.2012.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fujioka Y, et al. FUS-regulated region- and cell-type-specific transcriptome is associated with cell selectivity in ALS/FTLD. Sci Rep. 2013;3:2388. doi: 10.1038/srep02388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mishima T, et al. Syntaxin 1B, but not syntaxin 1A, is necessary for the regulation of synaptic vesicle exocytosis and of the readily releasable pool at central synapses. PloS one. 2014;9:e90004. doi: 10.1371/journal.pone.0090004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ikemoto A, Nakamura S, Akiguchi I, Hirano A. Differential expression between synaptic vesicle proteins and presynaptic plasma membrane proteins in the anterior horn of amyotrophic lateral sclerosis. Acta neuropathologica. 2002;103:179–187. doi: 10.1007/s004010100449. [DOI] [PubMed] [Google Scholar]
- 50.Washbourne P, et al. Genetic ablation of the t-SNARE SNAP-25 distinguishes mechanisms of neuroexocytosis. Nature neuroscience. 2002;5:19–26. doi: 10.1038/nn783. [DOI] [PubMed] [Google Scholar]
- 51.Schoch S, et al. SNARE function analyzed in synaptobrevin/VAMP knockout mice. Science. 2001;294:1117–1122. doi: 10.1126/science.1064335. [DOI] [PubMed] [Google Scholar]
- 52.Verderio C, et al. SNAP-25 modulation of calcium dynamics underlies differences in GABAergic and glutamatergic responsiveness to depolarization. Neuron. 2004;41:599–610. doi: 10.1016/s0896-6273(04)00077-7. [DOI] [PubMed] [Google Scholar]
- 53.Vance JE. Dysregulation of cholesterol balance in the brain: contribution to neurodegenerative diseases. Dis Model Mech. 2012;5:746–755. doi: 10.1242/dmm.010124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Day WA, Koishi K, Nukuda H, McLennan IS. Transforming growth factor-beta 2 causes an acute improvement in the motor performance of transgenic ALS mice. Neurobiology of disease. 2005;19:323–330. doi: 10.1016/j.nbd.2005.01.010. [DOI] [PubMed] [Google Scholar]
- 55.Nakamura M, et al. Activation of transforming growth factor-beta/Smad signaling reduces aggregate formation of mislocalized TAR DNA-binding protein-43. Neuro-degenerative diseases. 2013;11:182–193. doi: 10.1159/000338151. [DOI] [PubMed] [Google Scholar]
- 56.Schmidt ER, Pasterkamp RJ, van den Berg LH. Axon guidance proteins: novel therapeutic targets for ALS? Prog Neurobiol. 2009;88:286–301. doi: 10.1016/j.pneurobio.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 57.Peviani M, Tortarolo M, Battaglia E, Piva R, Bendotti C. Specific induction of Akt3 in spinal cord motor neurons is neuroprotective in a mouse model of familial amyotrophic lateral sclerosis. Molecular neurobiology. 2014;49:136–148. doi: 10.1007/s12035-013-8507-6. [DOI] [PubMed] [Google Scholar]
- 58.Wu X, Kihara T, Akaike A, Niidome T, Sugimoto H. PI3K/Akt/mTOR signaling regulates glutamate transporter 1 in astrocytes. Biochemical and biophysical research communications. 2010;393:514–518. doi: 10.1016/j.bbrc.2010.02.038. [DOI] [PubMed] [Google Scholar]
- 59.Morcuende S, Munoz-Hernandez R, Benitez-Temino B, Pastor AM, de la Cruz RR. Neuroprotective effects of NGF, BDNF, NT-3 and GDNF on axotomized extraocular motoneurons in neonatal rats. Neuroscience. 2013;250:31–48. doi: 10.1016/j.neuroscience.2013.06.050. [DOI] [PubMed] [Google Scholar]
- 60.Korkmaz OT, et al. 7,8-Dihydroxyflavone improves motor performance and enhances lower motor neuronal survival in a mouse model of amyotrophic lateral sclerosis. Neuroscience letters. 2014;566:286–291. doi: 10.1016/j.neulet.2014.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wu TD, Nacu S. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics. 2011;26:873–881. doi: 10.1093/bioinformatics/btq057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li H, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 64.Lin L, et al. Transcriptome sequencing reveals aberrant alternative splicing in Huntington’s disease. Human molecular genetics. 2016;25:3454–3466. doi: 10.1093/hmg/ddw187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Metsalu T, Vilo J. ClustVis: a web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic acids research. 2015;43:W566–570. doi: 10.1093/nar/gkv468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kuleshov MV, et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic acids research. 2016;44:W90–97. doi: 10.1093/nar/gkw377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome biology. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Anders S, Huber W. Differential expression analysis for sequence count data. Genome biology. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Alon S, et al. Systematic identification of edited microRNAs in the human brain. Genome research. 2012;22:1533–1540. doi: 10.1101/gr.131573.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mola MG, et al. The speed of swelling kinetics modulates cell volume regulation and calcium signaling in astrocytes: A different point of view on the role of aquaporins. Glia. 2016;64:139–154. doi: 10.1002/glia.22921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pisani F, et al. Identification of a point mutation impairing the binding between aquaporin-4 and neuromyelitis optica autoantibodies. The Journal of biological chemistry. 2014;289:30578–30589. doi: 10.1074/jbc.M114.582221. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequencing data produced in the present work have been submitted to dbGaP database under the accession phs000747.