


Abstract
The human MutY homolog, hMYH, is an adenine-specific DNA glycosylase that removes adenines or 2-hydroxyadenines mispaired with guanines or 8-oxoguanines. In order to prevent mutations, this activity must be directed to the newly synthesized strand and not the template strand during DNA synthesis. The subcellular localization and expression of hMYH has been studied in serum-stimulated, proliferating MRC5 cells. Using specific antibodies, we demonstrate that endogenous hMYH protein localized both to nuclei and mitochondria. hMYH in the nuclei is distinctly distributed and co-localized with BrdU at replication foci and with proliferating cell nuclear antigen (PCNA). The levels of hMYH in the nucleus increased 3- to 4-fold during progression of the cell cycle and reached maximum levels in S phase compared to early G1. Similar results were obtained for PCNA, while there were no notable changes in expression of 8-oxoguanine glycosylase or the human MutT homolog, MTH1, throughout the cell cycle. The cell cycle-dependent expression and localization of hMYH at sites of DNA replication suggest a role for this glycosylase in immediate post-replication DNA base excision repair.
INTRODUCTION
Oxidative DNA damage is primarily repaired by several enzymes that are components of the base excision repair (BER) pathway (reviewed in 1,2). In Escherichia coli there are complex mechanisms that limit mutagenesis due to the relatively abundant oxidative lesion, 8-oxo-7,8-dihydroguanine (8-oxoG). These pathways are initiated via either: (i) MutM at sites of formation of 8-oxoG; (ii) MutT by hydrolyzing d8-oxoGTP and preventing incorporation of 8-oxoG in DNA; or (iii) MutY repairing post-replicative adenines mispaired with 8-oxoG lesions (3). Although the mechanistic details have not been clearly elucidated, it is believed that similar pathways function in mammalian cells. The human MutT, hMTH1, functions as an 8-oxoGTPase and degrades the mutagenic 8-oxoG substrate from the nucleotide pool, preventing incorporation during DNA replication (4). During DNA replication, dCMP or dAMP (depending on the DNA polymerase involved) can be selectively incorporated opposite 8-oxoG (5,6). When the daughter strand contains a C opposite 8-oxoG, human 8-oxoguanine glycosylase (hOGG1) removes the 8-oxoG and allows the BER machinery to fill the gap with G and thus restore the original sequence (reviewed in 7). However, when A is incorporated opposite 8-oxoG, the human MutY homolog (hMYH) is responsible for removal of the mispaired adenine, thus avoiding G:C→T:A transversion mutations (8). Interestingly, a second 8-oxoG specific activity has been identified that specifically removes 8-oxoG opposite adenines (9). Unlike hMYH, this activity must be directed towards the nascent DNA strand to prevent mutation. In vitro and in vivo experiments utilizing recombinantly expressed hMYH have demonstrated adenine-specific glycosylase activity when adenine is paired with either G or 8-oxoG (10–13). Activity for the removal of 2-hydroxyadenine has also been reported (14).
In order to counteract the mutagenic effects of replication of 8-oxoG lesions in mitochondrial and nuclear DNAs, hMYH must localize in these organelles. The hMYH sequence contains both a nuclear localization signal (NLS) and a mitochondrial targeting signal (MTS) and appears to exist in multiple forms both in the mitochondria and the nucleus (12–15). A calf liver mitochondrial MutY homolog of smaller size has also been reported (16).
Due to the nature of the hMYH mismatch substrate, it is expected that hMYH will function in a post-replication repair pathway and, as such, be coupled to DNA replication and potentially be regulated as a function of the cell cycle. Several DNA repair proteins have been shown to have cell cycle-dependent expression and subcellular localization, including UNG2 (human uracil DNA glycosylase), RAD51, RAD52, FEN-1, APE (human AP endonuclease), NTH (human endonuclease III) and MPG (17–23).
In addition to a close association with DNA replication, the adenine-specific glycosylase activity of hMYH must be specifically targeted to the newly synthesized strand, suggesting that there may be some protein–protein interactions between hMYH and replicative proteins. hMYH contains a consensus proliferating cell nuclear antigen (PCNA)-binding site motif [QXX(I/L/M)XX(F)F/X] at the C-terminus which may serve to direct hMYH to the replication machinery (24). This putative interaction may also serve to orient hMYH to the daughter strand during DNA replication, enabling strand discrimination and proper removal of the adenine. This interaction has recently been demonstrated and suggests that hMYH may initiate adenine excision via the PCNA-dependent long patch BER pathway (25).
Here we demonstrate that the human MutY homolog, hMYH, an adenine-specific glycosylase, localizes to the nucleus during G1/S phase and associates specifically with the replication foci and PCNA. In addition, the levels of hMYH protein are increased 3- to 4-fold during S phase and remain elevated throughout mitosis. These data suggest a replication-coupled BER mechanism for the removal of adenines, possibly coordinated via PCNA interactions with hMYH.
MATERIALS AND METHODS
Cell culture
MRC5 and WI-38 normal diploid and tumor-derived cell lines HeLa S3, HT-116, NH3T3, HepG2, U937, A2780 and V79 were obtained from ATCC. Cells were maintained in the medium recommended by ATCC containing 10% fetal bovine serum (FBS), 100 U/ml penicillin and 100 µg/ml streptomycin. All reagents were from Gibco BRL (Gaithersburg, MD).
Antibodies
Rabbit polyclonal antibodies were raised against oligopeptides identical to hMYH residues 32–51 (no. 32) and 167–181 (no. 167). Antibody no. 32 was a generous gift provided by S. Mitra (University of Texas Medical Branch, Galveston, TX). Antibody no. 167 was custom made and affinity purified by Alpha Diagnostic (San Antonio, TX). Polyclonal antibodies to residues 65–79 of hOgg1 and C-terminal residues 143–156 of human MTH1 were purchased from Novus Biologicals (Littleton, CO). Anti-rabbit PCNA polyclonal antibody against residues 1–261 of human PCNA was purchased from Santa Cruz Biotechnology (Santa Cruz, CA).
Whole cell lysis
HeLa cells were pelleted using a Sorvall centrifuge at 800 g for 10 min. Cells were resuspended in lysis buffer [50 mM Tris–HCl, pH 7.4, 150 mM NaCl, 0.5% NP-40, 5 mM EDTA, 50 mM NaF, 1 mM PMSF and a protease inhibitor (PI) cocktail: 25 µg/ml pepstatin A, 50 µg/ml leupeptin and 0.2% aprotinin] and allowed to swell on ice for 30 min. Cells were then frozen in a dry ice bath and thawed on ice. Cells were spun at 10 000 r.p.m. for 10 min and the supernatant was frozen at –80°C. Bradford reagent (Bio-Rad) was used to quantitate the protein concentration per sample.
Purification of nuclei
Nuclei were purified as previously described (26). Cells were sedimented at 800 g for 10 min and then washed with 50 vol of phosphate-buffered saline (PBS). The pellet was resuspended in a 5× packed cell volume of a hypotonic buffer (10 mM HEPES, pH 7.9, 0.75 mM spermidine, 0.15 mM spermine, 0.1 mM EDTA and 0.1 mM EGTA). Cells were allowed to swell at 4°C for 10 min then sedimented at 300 g for 10 min. The supernatant was removed and replaced with 2 vol of fresh hypotonic buffer plus PI cocktail. Cells were homogenized by 3–5 strokes in a Dounce homogenizer and sucrose restoration buffer was added (1 vol of 500 mM HEPES, pH 7.9, 7.5 mM spermidine, 1.5 mM spermine, 2 mM EDTA, 2 mM EGTA and 10 mM DTT in 9 vol of 7.5% sucrose). Nuclei were sedimented in a Sorvall centrifuge for 30 s at 14 000 r.p.m. The pellet was resuspended in lysis buffer and stored in small aliquots at –80°C. The supernatant was saved and used for mitochondrial purification.
Purification of mitochondria
The procedure for mitochondrial purification was carried out at 4°C as previously described (27). Mitochondria were sedimented using a Sorvall centrifuge at 12 500 r.p.m. for 30 min. The supernatant was discarded and the pellet was resuspended in 25 mM sucrose (in TE, pH 7.4, with PI cocktail). Mitochondria were purified on a step gradient consisting of 1.5 M sucrose (in TE, pH 7.4) and 1.0 M sucrose (in TE, pH 7.4) spun in a SW28 rotor at 25 000 r.p.m. for 1 h. Mitochondria were collected in the 1 M sucrose layer and dialyzed against 10 mM HEPES, pH 7.9, 20% glycerol, 100 mM KCl, 0.2 mM EDTA, 0.2 mM EGTA and 2 mM DTT. Mitochondrial suspensions were sedimented at 14 000 r.p.m. for 20 min in a Sorvall centrifuge and the pellet was resuspended in lysis buffer. Centifugation at 14 000 r.p.m. for 20 min clarified the mitochondrial lysate and the supernatant was used for western blot analyses. Protein concentrations were quantified with Bradford reagent (Bio-Rad) and lysates were stored at –80°C in small aliquots.
Western blot analysis
Cells were harvested, washed in PBS, and lysed as described above. The cell lysates were clarified by high speed centrifugation (15 000 g for 10 min at 4°C). The protein content was determined by Bradford analysis (Bio-Rad) and equal amounts of protein per lane were separated by electrophoresis in a 10% SDS–PAGE gel. The gel was equilibrated in CAPS transfer buffer [10 mM cyclohexylamino-1-propanesulfonic acid (CAPS), pH 11, and 20% methanol] or Tris buffer (10 mM Tris, 20 mM glycine and 20% methanol, pH 8.3) for 30 min prior to transfer overnight at 15 V at 4°C onto a pre-equilibrated PVDF membrane (Amersham). Bound primary antibody was immunochemically detected according to the Enhanced Chemiluminescence (ECL) western blotting protocol (Amersham) using horseradish peroxidase-conjugated secondary antibody (Amersham). The chemiluminescence was visualized with luminescence detection film (Hyperfilm-ECL; Amersham).
Analysis of cell cycle
MRC5 cells (5 × 106 cells/T75 flask) were grown to 50% confluency at 5% CO2 and 37°C in minimal essential Eagle’s medium supplemented with 10% FBS, 100 U/ml penicillin and 100 µg/ml streptomycin. After 120 h in serum-free medium, cells were stimulated to proliferate with complete 10% FCS-containing medium and triplicate flasks of cells were harvested at 0, 6, 12, 18, 21, 24, 27 and 30 h and processed for cell cycle analysis (28). Briefly, cells were suspended in a low salt buffer containing 3% polyethylene glycol, 5 µg/ml propidium iodide, 0.1% Triton X-100, 4 mM Na citrate and 100 µg/ml RNase and incubated at 37°C for 30 min. An equal volume of high salt buffer (3% polyethylene glycol, 5 µg/ml propidium iodide, 0.1% Triton X-100 and 400 mM NaCl) was added and the cells were kept at 4°C overnight. The cellular DNA content was evaluated by flow cytometry using a FACScan flow cytometer (Becton Dickinson). Histograms were analyzed using ModFit LT cell cycle analysis software (Verity Software House) to determine the percent of cells in various stages of the cell cycle. Instrument calibration was performed daily using Calibrate Beads according to the recommendations of the manufacturer (Becton Dickinson). A total of 10 000 events were collected in all samples.
Immunostaining
MRC5 cells (5 × 105 cells/Petri dish) were grown to 50% confluency on glass coverslips (25 mm) in 5% CO2 at 37°C and synchronized by serum starvation for 5 days. In some experiments, actively replicating cultures of MRC5 cells on coverslips were used for immunostaining. Briefly, at 0, 6, 12, 18, 21, 24 and 30 h after serum addition cells were washed with PBS, dried, and fixed in acetone–methanol (1:1 v/v). After exposure to heterologous preimmune serum [0.1 µg in PBS containing 0.05% Tween 20 and 0.5% BSA (PBS-T)], cells were incubated (30 min at 37°C) with primary antibody. A fluorescein-conjugated secondary antibody (Santa Cruz Biotechnology) was then applied to cells for 30 min at 37°C and washed in PBS-T. Nuclei of cells were stained for 15 min with 10 ng/ml DAPI (4′,6-diamidino-2-phenylindole dihydrochloride). Mitotracker staining was performed following the manufacturer’s suggested protocol using 10 ng/ml MitoFluor Red stain (Molecular Probes). Cells were then mounted in anti-fade medium (Dako, Carpinteria, CA) on microscope slides and fluorescent images were evaluated and photographed at a magnification of 400× using a Zeiss UV microscope (Axiophot2; Zeiss, Jena, Germany). Final composite figures were made using an HP Scanjet 6100C scanner and Adobe Photoshop software.
Identification of DNA replication foci
Cells grown on glass coverslips were pulsed for 1 h with 10 µM 5-bromodeoxyuridine (BrdU) in the dark to prevent photolysis of BrdU-substituted DNA. Cells were quickly washed with PBS, fixed in 4% paraformaldehyde at 4°C for 15 min and placed in 0.1 N HCl containing 100 µg/ml pepsin for 30 min at 37°C. For denaturation of the DNA in situ, cells were treated with 1.5 N HCl for 15 min and then treated with Na borate for 5 min. After washing, the incorporated BrdU was detected by immunostaining with monoclonal antibody to BrdU (Roche Molecular Biochemicals). For the co-localization studies, serum-arrested MRC5 cells were serum stimulated and fixed at 21 h (S phase) in paraformaldehyde and processed for immunostaining.
RESULTS
Subcellular localization of hMYH
The subcellular localization of hMYH was studied by indirect immunofluorescence using two independent rabbit antisera against an N-terminal and a central peptide of the protein. Antibody specific to hMYH showed that the majority of this repair enzyme was distributed in the cytoplasm of asynchronous HeLa cell cultures (Fig. 1A). These data are in agreement with a mitochondrial association of hMYH (15). However, a small percentage (3–5%) of the cells demonstrated that hMYH-specific fluorescence was also localized to the nuclei (Fig. 1A, inset).
Figure 1.
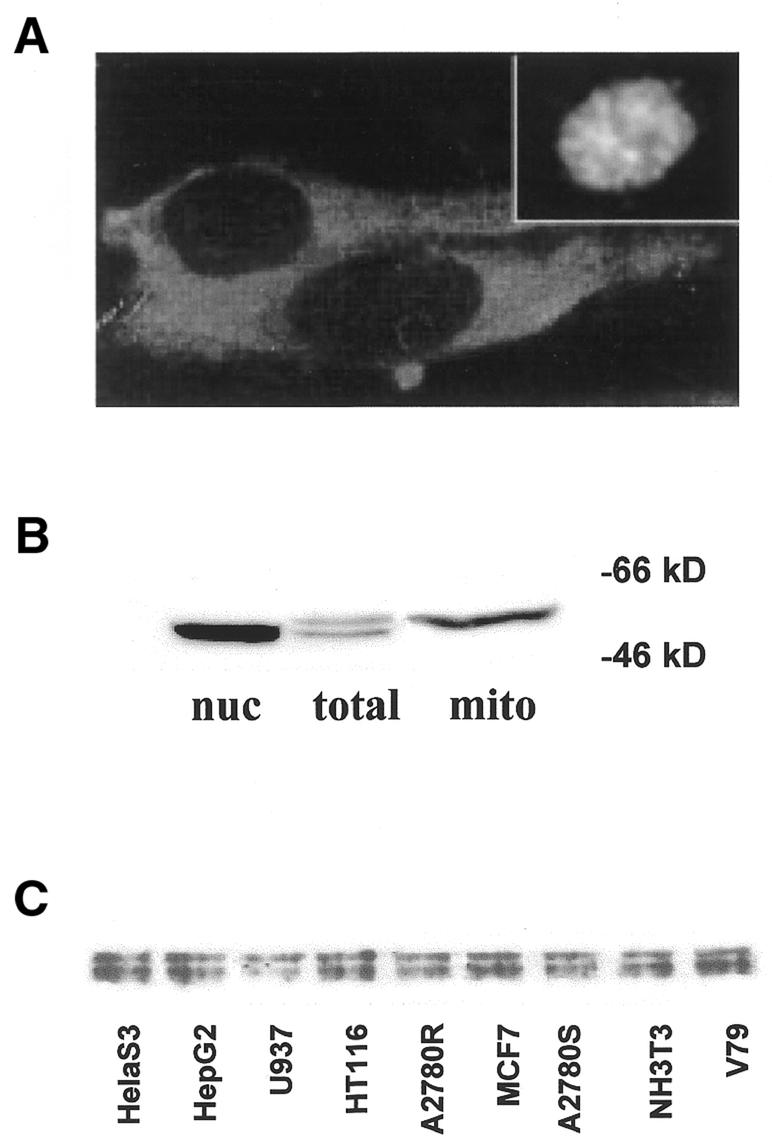
Localization and expression of endogenous hMYH. (A) Subcellular localization of hMYH. Asynchronous HeLa cells were cultured on coverslips, fixed and incubated with anti-hMYH antibody (no. 32) and FITC-conjugated anti-rabbit IgG. (Inset) Example of nuclear-specific hMYH staining. 400× magnification. (B) Multiple forms of hMYH protein. Mitochondrial and nuclear lysates were prepared and proteins separated on a 10% SDS–PAGE gel. Proteins were transferred to PVDF membrane and immunoblotted with anti-hMYH antibody (no. 167). (C) Ubiquitous expression of multiple forms of hMYH in various cell lines. The indicated cell lines were examined for expression of hMYH in asynchronous log phase cultures.
To determine the nature of the fluorescence in the nucleus, extracts were prepared from isolated mitochondria and nuclei. As shown by the western blot analyses in Figure 1B, two electrophoretically distinct forms of hMYH are observed in the total extract preparation. Upon cell fractionation, the lower molecular weight (mol. wt) band corresponding to the 52 and 53 kDa forms is distributed in the nuclei and the higher mol. wt band (corresponding to ∼57 kDa) is localized to the mitochondria. These findings are consistent with data previously reported in Jurkat cells (14), but in contrast to the data of Tsai-Wu et al. which suggest that the larger form is nuclear specific (13). These differences may be due to the cell type examined or the method of sample preparation. The specific banding patterns observed by western analysis were consistent in various cell types examined (Fig. 1C).
To study the extent of expression and subcellular localization of hMYH in individual cells, actively replicating MRC5 cells were fixed and prepared for immunochemistry and fluorescent or confocal microscopy. Similar to HeLa cells, the hMYH-specific fluorescence showed a considerable variation among individual cells. A bright nuclear fluorescence was observed in ∼20% of the cells (80% of cells showed faint nuclear staining) and all cells exhibited cytoplasmic staining (Fig. 2A and B). In the nuclei, fluorescence appeared granulated or punctate and showed apparent nucleoli exclusion (Fig. 2B). In the cytoplasm, distinct granulated staining was observed, as well as some diffuse staining. Superimposition of hMYH-mediated green fluorescence with Mitotracker Red staining (red fluorescence) resulted in yellow images (Fig. 2B), indicating that most of the cytoplasmic hMYH was located in the mitochondria.
Figure 2.

Subcellular localization of hMYH in actively replicating MRC5 cells. (A) Actively replicating MRC5 cells were fixed and stained with anti-hMYH antibody (no. 32). Specific hMYH peptide (competitor) abolishes nuclear and granulated cytoplasmic staining. (B) MitoTracker Red staining demonstrating that hMYH-specific cytoplasmic fluorescence is localized to mitochondria.
These results are consistent with multiple forms of hMYH that are believed to be localized both in nuclei and mitochondria (12–14). Antibodies nos 32 and 167 to endogenous hMYH both demonstrated similar staining patterns. The distinctive nuclear staining pattern and all the granulated cytoplasmic staining (presumably mitochondria) were abolished by incubation with the specific peptides (Fig. 2A, right).
Expression of hMYH as a function of cell cycle
Since the biological role of hMYH is post-replicative removal of adenine when paired with 8-oxoG or guanine, it was hypothesized that nuclear levels of hMYH may be higher during S phase and immediately following DNA synthesis. Investigations were performed in normal diploid MRC5 cells to determine if there was a correlation between cell cycle stage and expression of hMYH. MRC5 cells were arrested by serum starvation and after initiation of cell proliferation the relative hMYH protein levels and localization were determined at various time points (0–30 h). Cell cycle progression of serum-stimulated proliferating cell cultures was determined by flow cytometry following standard procedures (28). Serum starvation of MRC5 cells for 120 h resulted in a 2N DNA content (G0/G1) in >97% of the cell population and did not change significantly in the first 6 h of serum stimulation (Fig. 3B).
Figure 3.

Changes in protein level and subcellular localization of hMYH during the cell cycle. Parallel cultures of serum-stimulated proliferating cells (MRC5) were fixed or lysed as indicated for immunostaining or western blot analyses. (A) Immunostaining of hMYH protein at indicated times following serum addition. (B) Percent of cells in S phase as a function of time after addition of complete medium to serum-arrested MRC5 cells determined by flow cytometry. (C) Western blot analyses of selected proteins that are implicated in the prevention of 8-oxoguanine-induced mutations and PCNA at various times following serum stimulation of arrested cells. (D) Quantitation of protein levels as a function of cell cycle stage. These data represent an average of two independent experiments. A significant increase in hMYH protein levels has been consistently observed in n > 6 experiments.
A low intensity hMYH-mediated fluorescence was observed in the nuclei of serum-arrested, G0/G1 MRC5 cells (Fig. 3A). As the cells progressed through the cell cycle nuclear staining of hMYH markedly increased. A similar increase in nuclear fluorescence was also observed for PCNA (data not shown). In contrast, only a slight increase in the fluorescence intensity of nuclear hMTH and hOGG1 was observed in the same preparations (data not shown). To confirm data obtained by immunochemical analyses, protein levels were investigated using western blot analyses. As shown in Figure 3C and D, the hMYH protein level increased in cells entering S phase. As compared to PCNA, the extent of the increase in hMYH was less, but occurred concomitantly with the increase in PCNA (Fig. 3C and D). hOGG1 and hMTH demonstrated no significant change in protein levels as a function of cell cycle, consistent with recent observations made by others (23,29,30).
To more specifically examine nuclear protein levels, nuclear lysates were prepared from cells following serum stimulation and analyzed for hMYH protein levels by western blot analyses. As shown in Figure 4, the nuclear forms of the protein increased ∼4-fold as the cells progressed through the G1/S phases of the cell cycle, reaching a maximum level in S phase.
Figure 4.

Nuclear hMYH protein levels increase during the cell cycle. (A) Western blot analysis of nuclear lysates of MRC5 cells at various times following serum stimulation of arrested cells. (B) Percent of cells in S phase as a function of time after addition of complete medium to serum-arrested MRC5 cells determined by flow cytometry.
Accumulation of hMYH in drug-induced S phase arrest
To further investigate the cell cycle-dependent expression of hMYH, we utilized human ovarian cell carcinoma cell lines, A2780 and A2780/100 cells, sensitive and resistant to radiation as well as DNA crosslinking agents (e.g. chlorambucil, melphalan and cisplatin) (31). We have previously shown that treatment of A2780 cells with chlorambucil resulted in a cell cycle arrest in S phase followed by apoptosis, while A2780/100 cells demonstrated a transitional G2/M arrest and survived drug treatment (32). In these experiments, A2780 and A2780/100 cells were synchronized using a double thymidine block (33) and treated with 100 µM chlorambucil for 1 h as previously described (32). Cells were harvested for cell cycle and western blot analyses at the indicated times (Fig. 5). hMYH levels increased dramatically in both cell lines and reached maximum levels coincident with S phase. A2780 cells arrested in S phase and hMYH protein levels were sustained and remained elevated, while in A2780/100 cells hMYH returned to a lower level as the cells entered the G1 phase of the cell cycle. Both A2780 and A2780/100 cells showed hMYH nuclear localization in the G1/S and S phases of the cell cycle, similar to normal diploid MRC5 cells (data not shown). Thus, these data offer independent evidence that hMYH protein levels are increased at the G1/S boundary and in S phase of the cell cycle.
Figure 5.

Accumulation of hMYH in the G1/S and S phases of the cell cycle in human ovarian carcinoma cells. Cell lines that are sensitive and relatively resistant to the cytotoxic effects of exposure to DNA crosslinking agents (A2780 and A2780/100, respectively) were synchronized by double thymidine block and treated with 100 µM chlorambucil. (A) Western blot analyses of hMYH levels following treatment of cells with chlorambucil. (B) Cell cycle stage distribution at time 0. (C) Cell cycle stage distribution at time 12 h following drug treatment. (D) Percent of cells in S phase as a function of time following chlorambucil treatment of cells determined by flow cytometry.
Nuclear hMYH is located at sites of DNA replication and co-localizes with PCNA
Given the many examples of PCNA interacting with various DNA repair or repair-related proteins and the conserved consensus PCNA-binding motif in hMYH, we examined if the nuclear forms of the protein were associated or co-localized with PCNA. At various times following serum stimulation of arrested cells, cells were fixed and subjected to separate or co-staining with hMYH and PCNA antibodies. Both PCNA- and hMYH-mediated fluorescence demonstrated bright intensely staining spots, ring- and rope-like structures and dark non-fluorescent compartments resembling nucleoli (Fig. 6). When hMYH [fluorescein isothiocianate (FITC), green fluorescence] and PCNA (rhodamine, red fluorescence) images were superimposed in the same preparations, this resulted in a yellow color, demonstrating co-localization of hMYH with PCNA (Fig. 6C). Co-localization of hMYH and PCNA was not observed in G0/G1 (0 time) cells, while the percentage of cells showing co-localization increased as a function of time (12, 18 and 21 h after serum stimulation), suggesting that these protein–protein interactions are regulated in concert with DNA synthesis (data not shown).
Figure 6.

Co-localization of hMYH, PCNA and BrdU in S phase MRC5 cells. (A) Co-localization of BrdU (anti-BrdU, green fluorescence) and hMYH (anti-MYH no. 32, red fluorescence) in nuclei of MRC5 cells. (B) Co-localization of BrdU (anti-BrdU, green fluorescence) and PCNA (anti-PCNA, red fluorescence). (C) Co-localization of PCNA (anti-PCNA, green fluorescence) and hMYH (anti hMYH no. 32, red fluorescence). These superimposed images were generated from photographs with double exposure. These images were contrast enhanced to emphasize nuclear staining. The bottom vertical panels represent superimposition of the two top panels of rhodamine and FITC fluorescence resulting in a yellow color, thus providing a direct observation of co-localization.
Due to the punctate nature of the hMYH nuclear staining pattern, we investigated if these intense sites of fluorescence were sites of DNA replication. Synchronized MRC5 cells were pulse labeled with 10 µM BrdU for 1 h at various times following serum stimulation. Discrete foci of replication were detectable within the nucleus by indirect immunofluorescence with anti-BrdU antibodies (34,35). Staining with anti-BrdU antibody was absent at 0 h (G0/G1 phase) and ∼60–80% of the nuclei stained for BrdU at 18–24 h (S phase). Superimposition of green (FITC/anti-BdrU antibody) and red (rhodamine/anti-hMYH antibody) images resulted in yellow fluorescence, demonstrating that hMYH is found at sites of DNA replication (Fig. 6A). PCNA has previously been shown to associate with DNA replication structures and was clearly co-localized with BrdU in our system (Fig. 6B).
DISCUSSION
Immediate post-replicative repair of DNA mispairs is critical for effective maintainence of the genome. Here evidence is provided that hMYH is localized to sites of DNA replication and that expression of this repair enzyme is dependent on the cell cycle. Thus, it appears that hMYH-initiated repair of adenine (or 2-hydroxyadenine) mispaired with 8-oxoguanine or guanine occurs in replication foci and the data are consistent with this process being mediated through interactions with PCNA. This pathway is analogous to that proposed for UNG2, the major human nuclear uracil DNA glycosylase (UDG) (22). UNG2 has been shown to exhibit increased protein levels in S phase and co-localizes with BrdU, PCNA and replication protein A (RPA) at replication foci. Direct interactions of UNG2 with PCNA and RPA have also been demonstrated, suggesting the formation of a BER complex in close association with DNA replication (22). A similar model has been suggested for hMYH-initiated BER in a recent report where immunoprecipitation experiments demonstrated that hMYH interacts not only with PCNA but also with RPA and APE (25). In addition, it has also recently been demonstrated that APE1 enhances the adenine glycosylase activity of mouse MYH, providing further evidence of protein–protein interactions among these BER enzymes (36). Our results are consistent with this model and provide the in situ co-localization evidence demonstrating that hMYH is indeed present in replication foci.
In addition, we have shown that nuclear levels of hMYH protein increased significantly as cells entered S phase, suggesting a cell cycle-dependent regulation of expression and/or subcellular targeting. Takoa et al. have shown that an epitope-tagged full-length hMYH in COS-7 cells localizes predominantly to the mitochondria and that an alternative transcript (type 2) is localized to the nucleus (12,15). Ohstubo et al. also identified various forms of the protein; nuclear (52/53 kDa) and mitochondrial (57 kDa) forms (14). The mitochondrial localization of p57 was specific to the inner membrane, similar to hOgg1-2a (37). This prompted a model of a BER complex sitting on the inner membrane of mitochondria (38). The nuclear and mitochondrial forms that we observed in these cells are consistent with hMYH type 2 being nuclear and the full-length type 1 being mitochondrial. Thus, the increased levels of nuclear hMYH may be due to increased expression of type 2 hMYH, as well as import into the nucleus. The exact mechanism for enhanced nuclear accumulation is currently under investigation. In these studies there was no evidence that the cytoplasmic or mitochondrial levels of hMYH decrease as nuclear levels increase. However, further investigations are necessary to examine whether this is a shuttling mechanism, as suggested by Tsai-Wu et al. (13), or differential regulation of expression of the type 2 transcript. Interestingly, nuclear MSH2 protein levels have been shown to increase rapidly by translocation from the cytoplasm and not via promoter activation or mRNA stabilization following exposure of cells to alkylating agents (39). In contrast, up-regulation of nuclear UNG2 protein appears to be an up-regulation of biosynthesis and not a translocation effect (19). Experiments are in progress to investigate protein and mRNA stability as well as promoter analyses.
We have shown that hMYH protein levels are enhanced and that the subcellular localization becomes predominantly nuclear as cells progress to the G1/S boundary. During S phase hMYH is specifically found at replication foci and in close association with PCNA for immediate post-replicative repair of adenine mispaired with guanine or 8-oxoG. Thus far, these data suggest that hMYH-initiated repair may be replication associated. It has not been determined if this repair is replication dependent. However, experiments demonstrating that 8-oxoG:A mispairs in a non-replicating shuttle vector system in mammalian cells were poorly repaired suggests that this pathway may be replication dependent (40). In conclusion, our data suggest that repair of A:G or 8-oxoG mispairs initiated by hMYH is in close proximity to DNA replication, poised for an efficient and rapid mutation prevention pathway.
Acknowledgments
ACKNOWLEDGEMENTS
We would like to thank Inez Petteway for technical support and Rosemary Martinez for clerical assistance on this manuscript. This work was supported by NIH grants RO1-AG17432 (to A.K.M.) and RO1-CA84461 (to I.B.) and the National Institute for Environmental Health Sciences Center at Galveston (P30 ES06676) Cell Biology Core Facility and Pilot Project funding (to A.K.M.).
References
- 1.Lindahl T. (2000) Suppression of spontaneous mutagenesis in human cells by DNA base excision-repair. Mutat. Res., 462, 129–135. [DOI] [PubMed] [Google Scholar]
- 2.Krokan H.E., Nilsen,H., Skorpen,F., Otterlei,M. and Slupphaug,G. (2000) Base excision repair of DNA in mammalian cells. FEBS Lett., 476, 73–77. [DOI] [PubMed] [Google Scholar]
- 3.Michaels M.L. and Miller,J.H. (1992) The GO system protects organisms from the mutagenic effect of the spontaneous lesion 8-hydroxyguanine (7,8-dihydro-8-oxoguanine). J. Bacteriol ., 174, 6321–6325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hayakawa H., Taketomi,A., Sakumi,K., Kuwano,M. and Sekiguchi,M. (1995) Generation and elimination of 8-oxo-7,8-dihydro-2′-deoxyguanosine 5′-triphosphate, a mutagenic substrate for DNA synthesis, in human cells. Biochemistry, 34, 89–95. [DOI] [PubMed] [Google Scholar]
- 5.Shibutani S., Takeshita,M. and Grollman,A.P. (1991) Insertion of specific bases during DNA synthesis past the oxidation-damaged base 8-oxodG. Nature, 349, 431–434. [DOI] [PubMed] [Google Scholar]
- 6.Einolf H.J. and Guengerich,F.P. (2001) Fidelity of nucleotide insertion at 8-oxo-7,8-dihydroguanine by mammalian DNA polymerase delta. J. Biol. Chem., 276, 3764–3771. [DOI] [PubMed] [Google Scholar]
- 7.Boiteux S. and Radicella,J.P. (2000) The human OGG1 gene: structure, functions, and its implication in the process of carcinogenesis. Arch. Biochem. Biophys ., 377, 1–8. [DOI] [PubMed] [Google Scholar]
- 8.Slupska M.M., Baikalov,C., Luther,W.M., Chiang,J.H., Wei,Y.F. and Miller,J.H. (1996) Cloning and sequencing a human homolog (hMYH) of the Escherichia coli mutY gene whose function is required for the repair of oxidative DNA damage. J. Bacteriol ., 178, 3885–3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hazra T.K., Izumi,T., Maidt,L., Floyd,R.A. and Mitra,S. (1998) The presence of two distinct 8-oxoguanine repair enzymes in human cells: their potential complementary roles in preventing mutation. Nucleic Acids Res., 26, 5116–5122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shinmura K., Yamaguchi,S., Saitoh,T., Takeuchi-Sasaki,M., Kim,S.R., Nohmi,T. and Yokota,J. (2000) Adenine excisional repair function of MYH protein on the adenine:8-hydroxyguanine base pair in double-stranded DNA. Nucleic Acids Res., 28, 4912–4918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Slupska M.M., Luther,W.M., Chiang,J.H., Yang,H. and Miller,J.H. (1999) Functional expression of hMYH, a human homolog of the Escherichia coli MutY protein. J. Bacteriol ., 181, 6210–6213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Takao M., Zhang,Q.M., Yonei,S. and Yasui,A. (1999) Differential subcellular localization of human MutY homolog (hMYH) and the functional activity of adenine:8-oxoguanine DNA glycosylase. Nucleic Acids Res., 27, 3638–3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tsai-Wu J.J., Su,H.T., Wu,Y.L., Hsu,S.M. and Wu,C.H. (2000) Nuclear localization of the human mutY homologue hMYH. J. Cell. Biochem., 77, 666–677. [PubMed] [Google Scholar]
- 14.Ohtsubo T., Nishioka,K., Imaiso,Y., Iwai,S., Shimokawa,H., Oda,H., Fujiwara,T. and Nakabeppu,Y. (2000) Identification of human MutY homolog (hMYH) as a repair enzyme for 2-hydroxyadenine in DNA and detection of multiple forms of hMYH located in nuclei and mitochondria. Nucleic Acids Res., 28, 1355–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Takao M., Aburatani,H., Kobayashi,K. and Yasui,A. (1998) Mitochondrial targeting of human DNA glycosylases for repair of oxidative DNA damage. Nucleic Acids Res., 26, 2917–2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Parker A., Gu,Y. and Lu,A.L. (2000) Purification and characterization of a mammalian homolog of Escherichia coli MutY mismatch repair protein from calf liver mitochondria. Nucleic Acids Res., 28, 3206–3215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu Y., Li,M., Lee,E.Y. and Maizels,N. (1999) Localization and dynamic relocalization of mammalian Rad52 during the cell cycle and in response to DNA damage. Curr. Biol., 9, 975–978. [DOI] [PubMed] [Google Scholar]
- 18.Luna L., Bjoras,M., Hoff,E., Rognes,T. and Seeberg,E. (2000) Cell-cycle regulation, intracellular sorting and induced overexpression of the human NTH1 DNA glycosylase involved in removal of formamidopyrimidine residues from DNA. Mutat. Res., 460, 95–104. [DOI] [PubMed] [Google Scholar]
- 19.Haug T., Skorpen,F., Aas,P.A., Malm,V., Skjelbred,C. and Krokan,H.E. (1998) Regulation of expression of nuclear and mitochondrial forms of human uracil-DNA glycosylase. Nucleic Acids Res., 26, 1449–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen F., Nastasi,A., Shen,Z., Brenneman,M., Crissman,H. and Chen,D.J. (1997) Cell cycle-dependent protein expression of mammalian homologs of yeast DNA double-strand break repair genes Rad51 and Rad52. Mutat. Res., 384, 205–211. [DOI] [PubMed] [Google Scholar]
- 21.Qiu J., Li,X., Frank,G. and Shen,B. (2001) Cell cycle-dependent and DNA damage-inducible nuclear localization of FEN-1 nuclease is consistent with its dual functions in DNA replication and repair. J. Biol. Chem., 276, 4901–4908. [DOI] [PubMed] [Google Scholar]
- 22.Otterlei M., Warbrick,E., Nagelhus,T.A., Haug,T., Slupphaug,G., Akbari,M., Aas,P.A., Steinsbekk,K., Bakke,O. and Krokan,H.E. (1999) Post-replicative base excision repair in replication foci. EMBO J., 18, 3834–3844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bouziane M., Miao,F., Bates,S.E., Somsouk,L., Sang,B., Denissenko,M. and O’Connor,T.R. (2000) Promoter structure and cell cycle dependent expression of the human methylpurine-DNA glycosylase gene. Mutat. Res., 461, 15–29. [DOI] [PubMed] [Google Scholar]
- 24.Matsumoto Y. (2001) Molecular mechanism of PCNA-dependent base excision repair. Prog. Nucleic Acid Res. Mol. Biol., 68, 129–138. [DOI] [PubMed] [Google Scholar]
- 25.Parker A., Gu,Y., Mahoney,W., Lee,S.H., Singh,K.K. and Lu,A.L. (2001) Human homolog of the MutY repair protein (hMYH) physically interacts with proteins involved in long-patch DNA base excision repair. J. Biol. Chem., 276, 5547–5555. [DOI] [PubMed] [Google Scholar]
- 26.Shapiro D.J., Sharp,P.A., Wahli,W.W. and Keller,M.J. (1988) A high-efficiency HeLa cell nuclear transcription extract. DNA, 7, 47–55. [DOI] [PubMed] [Google Scholar]
- 27.Tomkinson A.E. and Linn,S. (1986) Purification and properties of a single strand-specific endonuclease from mouse cell mitochondria. Nucleic Acids Res., 14, 9579–9593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bresnahan W.A., Boldogh,I., Thompson,E.A. and Albrecht,T. (1996) Human cytomegalovirus inhibits cellular DNA synthesis and arrests productively infected cells in late G1. Virology, 224, 150–160. [DOI] [PubMed] [Google Scholar]
- 29.Bialkowski K. and Kasprzak,K.S. (2000) Activity of the antimutagenic enzyme 8-oxo-2′-deoxyguanosine 5′-triphosphate pyrophosphohydrolase (8-oxo-dGTPase) in cultured chinese hamster ovary cells: effects of cell cycle, proliferation rate and population density. Free Radic. Biol. Med., 28, 337–344. [DOI] [PubMed] [Google Scholar]
- 30.Dhenaut A., Boiteux,S. and Radicella,J.P. (2000) Characterization of the hOGG1 promoter and its expression during the cell cycle. Mutat. Res., 461, 109–118. [DOI] [PubMed] [Google Scholar]
- 31.Horton J.K., Roy,G., Piper,J.T., Van Houten,B., Awasthi,Y.C., Mitra,S., Alaoui-Jamali,M.A., Boldogh,I. and Singhal,S.S. (1999) Characterization of a chlorambucil-resistant human ovarian carcinoma cell line overexpressing glutathione S-transferase mu. Biochem. Pharmacol., 58, 693–702. [DOI] [PubMed] [Google Scholar]
- 32.Roy G., Horton,J.K., Roy,R., Denning,T., Mitra,S. and Boldogh,I. (2000) Acquired alkylating drug resistance of a human ovarian carcinoma cell line is unaffected by altered levels of pro- and anti-apoptotic proteins. Oncogene, 19, 141–150. [DOI] [PubMed] [Google Scholar]
- 33.Chiu R.W. and Baril,E.F. (1975) Nuclear DNA polymerases and the HeLa cell cycle. J. Biol. Chem., 250, 7951–7957. [PubMed] [Google Scholar]
- 34.Hozak P., Jackson,D.A. and Cook,P.R. (1994) Replication factories and nuclear bodies: the ultrastructural characterization of replication sites during the cell cycle. J. Cell Sci., 107, 2191–2202. [DOI] [PubMed] [Google Scholar]
- 35.Nakayasu H. and Berezney,R. (1989) Mapping replicational sites in the eucaryotic cell nucleus. J. Cell Biol., 108, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang H., Clendenin,W.M., Wong,D., Demple,B., Slupska,M.M., Chiang,J.-H. and Miller,J. (2001) Enhanced activity of adenine-DNA glycosylase (Myh) by apurinic/apyrimidinic endonuclease (Ape1) in mammalian base excision repair of an A/GO mismatch. Nucleic Acids Res., 29, 743–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nishioka K., Ohtsubo,T., Oda,H., Fujiwara,T., Kang,D., Sugimachi,K. and Nakabeppu,Y. (1999) Expression and differential intracellular localization of two major forms of human 8-oxoguanine DNA glycosylase encoded by alternatively spliced OGG1 mRNAs. Mol. Biol. Cell, 10, 1637–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nakabeppu Y. (2001) Regulation of intracellular localization of human MTH1, Ogg1 and MYH proteins for repair of oxidative DNA damage. Prog. Nucleic Acid Res. Mol. Biol., 68, 75–94. [DOI] [PubMed] [Google Scholar]
- 39.Christmann M. and Kaina,B. (2000) Nuclear translocation of mismatch repair proteins MSH2 and MSH6 as a response of cells to alkylating agents. J. Biol. Chem., 275, 36256–36262. [DOI] [PubMed] [Google Scholar]
- 40.Le Page F., Guy,A., Cadet,J., Sarasin,A. and Gentil,A. (1998) Repair and mutagenic potency of 8-oxoG:A and 8-oxoG:C base pairs in mammalian cells. Nucleic Acids Res., 26, 1276–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]