

Abstract
N-terminal acetylation is an abundant modification influencing protein functions. Since ≈80% of mammalian cytosolic proteins are N-terminally acetylated, this potentially represents an untapped target for chemical control of their functions. Structural studies have revealed that, like lysine acetylation, N-terminal acetylation converts a positively charged amine into a hydrophobic handle that mediates protein interactions, suggesting it may be a druggable target. We report the development of chemical probes targeting the N-terminal acetylation-dependent interaction between an E2 conjugating enzyme (UBE2M, aka UBC12) and DCN1 (aka DCUN1D1), a subunit of a multiprotein E3 ligase for the ubiquitin-like protein NEDD8. The inhibitors are highly selective with respect to other protein acetyl amide binding sites, inhibit NEDD8 ligation in vitro and in cells, and suppress the anchorage-independent growth of a cell line harboring DCN1 amplification. Overall, the data demonstrate that N-terminal acetyl-dependent protein interactions are druggable targets, and provide insights into targeting multiprotein E2–E3 ligases.
Graphical abstract
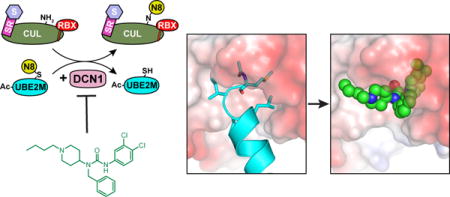
Introduction
Protein amino termini are hotbeds for modifications that influence the interactions and homeostasis of many proteins1. A widespread and conserved eukaryotic modification is N-terminal acetylation. Approximately 80% of mammalian cytosolic proteins are subject to either co- or post-translational modification by N-terminal acetyltransferases (NATs)2,3. N-terminal acetylation plays essential roles in cell proliferation, apoptosis, chromatin remodeling, protein trafficking, and other fundamental biological processes2,4–7. Mutations in some NAT enzymes have been linked to devastating human diseases8,9. At a molecular level, acetylation transforms the positively charged N-terminus into a hydrophobic handle by capping the amino group as an amide containing an additional methyl group3. N-terminal acetylation status can influence protein properties including folding, oligomerization, and intermolecular interactions3. Potentially therapeutically relevant roles for protein interactions regulated by N-terminal acetylation include assembly of an E2-E3 ubiquitin-like protein ligation complex, nucleosome binding by an epigenetic regulator, cytoskeletal organization, integrity of the Anaphase-Promoting complex, and E3 ligase-substrate interactions1–12.
Given the prevalence and importance of N-terminal acetylation, we sought to determine whether chemical disruption of a protein-protein interaction controlled by this modification could be useful for manipulating processes that it regulates. To our knowledge, there are no small molecules targeting binding pockets for acetylated N-termini. The recent development of small molecules targeting binding pockets for acetylated lysines suggests inhibiting interactions mediated by acetylated N-termini would be possible2,13–15. To address this, we focused on the interaction between the N-terminally acetylated E2 conjugating (UBE2M aka UBC12) and E3 ligase (DCN1, aka DCUN1D1, DCNL1, and SCCRO) enzymes for the ubiquitin-like protein NEDD8 (Fig. 1a). The molecular role of UBE2M’s acetylated N-terminus is structurally understood, with the acetyl group contributing around two orders-of-magnitude to the binding energy10.
Figure 1. Discovery of small molecule inhibitors targeting N-Acetyl-UBE2M interaction with DCN1.
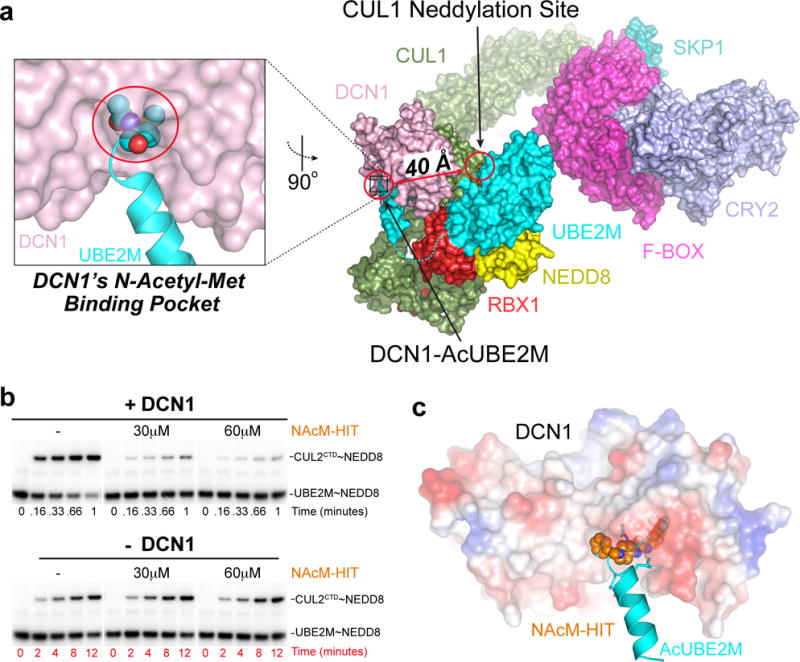
(a) Model of a neddylation complex, highlighting DCN1 (pink) interactions with acetylated N-terminus of UBE2M (cyan), including structures of CUL1 (green)-RBX1 (red) and SKP1 (pale cyan)-FBXL3 (magenta)-Substrate (CRY2, light blue)20. Although ≈40 Å from the CUL1 neddylation site, the DCN1-AcUBE2M interaction accelerates neddylation20.
(b) Pulse-chase assays monitoring effects of the indicated concentrations of NAcM-HIT on DCN1-dependent (top, timescale 0–1 min) or DCN1-independent (bottom, timescale 0–12 min) neddylation from AcUBE2M to CUL2CTD. The gel scans are representative of multiple biological replicates.
(c) Structure of DCN1 (surface colored by electrostatic potential) bound to NAcM-HIT (spheres, orange) aligned to DCN1 (omitted for clarity)-AcUBE2M (cyan) demonstrating NAcM-HIT binds to DCN1’s N-AcetylMet binding pocket.
The N-terminal acetylation-dependent interaction of UBE2M with DCN1 regulates assembly of a multiprotein complex catalyzing NEDD8 ligation to cullin proteins (Supplementary Information, Supplementary Fig. 1a). This process, termed “neddylation”, controls activities of the cullins in ubiquitin ligation cascades. During neddylation, the acetylated N-terminal methionine from UBE2M docks into a pocket in DCN1, which was named based on its loss of function in yeast and worms causing Defective Cullin Neddylation10,16–20. Within the multiprotein NEDD8 E3 ligase complex, the RING E3 RBX1 (which has multiple functions in neddylation and ubiquitination) binds both the E2 catalytic domain from UBE2M and the cullin substrate. DCN1 stabilizes the catalytic complex by binding the cullin’s substrate WHB domain and UBE2M’s acetylated N-terminus on a flexibly tethered helix (Fig. 1a)10,19–21. Mammals express five DCN paralogs with 30–80% sequence identity over their E3 “Potentiation of Neddylation” (PONY) domains, which combinatorially regulate neddylation of six homologous cullin proteins via distinctive but overlapping expression patterns and cellular locations10,16,19,22,23. Although downstream targets that are regulated by the human DCN1 pathway remain unknown, the DCN1 gene is amplified along the 3q26.3 region in most squamous cell carcinomas24. DCN1 amplification negatively correlates with cause-specific survival and high DCN1 protein levels have been associated with anchorage-independent growth in soft agar, suggesting that targeting DCN1 may be of clinical utility17,23–25.
Herein, we report discovery of potent and selective small molecule inhibitors of the N-acetyl UBE2M-DCN1 interaction. Overall, the data demonstrate that N-terminal acetylation is a druggable target, establish paradigms for selectively inhibiting N-terminal acetylation-dependent protein interactions, and provide routes for inhibiting a specific E2-E3 ubiquitin-like protein ligase complex.
Results
Antagonizing the N-acetyl-UBE2M–DCN1 complex
We developed a ligand competition assay based on prior mapping of the motifs mediating interactions between DCN1 (PONY domain alone) and UBE2M (acetylated amino terminal peptide)10,18,19 (Supplementary Fig. 1b). The TR-FRET signal between a biotinylated version of DCN1 (recognized by terbium-linked streptavidin) and a stapled peptide corresponding to N-terminally acetylated UBE2M (C-terminally labeled with AlexaFluor 488) was used to screen a library containing 601,194 unique chemicals at a fixed concentration of 30 μM for each test article (Supplementary Table 1, Supplementary Fig. 2). The primary screen gave an average final z’ of 0.56 and the fidelity of the assay for selecting true positives, determined by receiver operating characteristic (ROC) analysis, demonstrated good discriminatory power (area under the curve ~ 0.74) and indicated a cutoff of >45% activity would retain >80% of the true positives (Supplementary Fig. 2). 856 hits (0.15% hit rate) were tested for dose-dependent responses in the TR-FRET assay (Supplementary Dataset 1), yielding 182 validated hits with half-maximal inhibitory concentration (IC50) <15 μM. Chemical structure analysis by topology mapping and clustering26 revealed several chemotypes, including multiple scaffolds with promising structure-activity relationships (SARs) (Supplementary Fig. 3a). Scaffolds were prioritized based on dose-response potency, SAR range (≥10-fold), solubility (≥10 μM), permeability (≥200 × 10−6 cm/s), and low cytotoxicity to untransformed BJ fibroblasts. Scaffolds containing PAINS27 motifs or noncompliant with Lipinski’s rule-of-five28 were deprioritized.
Three structural clusters sharing a pharmacophore element consisting of a N-benzyl (or benzoyl) piperidine could be grouped into a single series of compounds represented by NAcM-HIT (1) (Supplementary Figs. 1c, 3b). The unifying feature among the three scaffolds is a piperidine ring that adds 3-dimensional character. Resynthesized NAcM-HIT inhibited DCN1 binding to the N-terminally acetylated UBE2M peptide (IC50 of 7 μM) (Supplementary Fig. 1c). We tested whether NAcM-HIT inhibits the neddylation reaction catalyzed by the larger multiprotein complex containing this E2-E3 subcomplex (Fig. 1a). To isolate the E2-E3-dependent step in NEDD8’s E1-E2-E3 cascade (Supplementary Fig. 1a), we utilized our pulse-chase assay10,20. First, the UBE2M~NEDD8 intermediate is generated in a “pulse” reaction catalyzed by the NEDD8 E1 in the presence of UBE2M, fluorescent NEDD8, and MgATP. After quenching this pulse reaction, neddylation is monitored by tracking the “chase” of fluorescent NEDD8 from UBE2M to the acceptor lysine on a CUL-RBX1 complex in the presence or absence of DCN110. NAcM-HIT blocked DCN1-dependent cullin neddylation with a IC50 of 4 μM in this assay (Fig. 1b, Supplementary Fig. 1c). Importantly, NAcM-HIT did not inhibit the much slower DCN1-independent NEDD8 ligation reaction, which does not depend on acetylation of UBE2M’s amino-terminus10 (Fig. 1b).
To aid compound optimization and determine mechanisms by which NAcM-HIT inhibits DCN1-UBE2M interactions and DCN1-dependent neddylation, we sought structural data. Initial crystal forms were plagued by the small molecule occupying crystal packing interfaces. To identify crystal forms suitable for structure-based inhibitor design, we tested 16 different versions of DCN1. Ultimately, a fusion of a mutant T4 lysozyme immediately N-terminal of DCN1’s PONY domain enabled obtaining crystals with DCN1’s N-acetyl-Met-binding pocket oriented toward solvent (Supplementary Fig. 4a, Supplementary Table 2). The 2.0 Å resolution co-crystal structure with NAcM-HIT demonstrated that the lysozyme fusion did not perturb DCN1’s fold (1.0 and 1.2 Å RMSD comparing to structures of DCN1 bound to acetylated peptide or full-length versions of UBE2M10,20) (Supplementary Fig. 4b). Importantly, NAcM-HIT occupies the targeted N-acetyl-methionine binding pocket in DCN1 (Fig. 1c).
Structure-based optimization of NAcM inhibitors
Our screening collection contained >300 compounds related to NAcM-HIT but with varying potency, which provided initial capture of SAR (Supplementary Dataset 2). The screening set SAR together with structural data for DCN1 binding to NAcM-HIT or N-terminally acetylated UBE2M10,19,20 revealed five sub-pockets within DCN1 that could be targeted to improve compound affinity (Fig. 2a). The Ile, N-acetyl, and Leu sub-pockets represent areas of the binding pocket occupied by those moieties from UBE2M, and the urea and hinge pockets represent areas populated by those substructures of NAcM-HIT (Fig. 2a). The SAR revealed the following trends 1) the solvent exposed Ile pocket tolerated a wide range of substituents; 2) the central piperidine core was critical to activity with alterations to its size or substitution pattern decreasing potency; 3) a hydrogen bond donor in the urea pocket was important, but the urea itself could be replaced with either an amide or a benzothiazole; 4) the hinge pocket only tolerated a narrow range of substituents, with even minor deviations from the meta-substituted 6-membered aromatic ring causing substantial decreases in activity.
Figure 2. Structure based optimization and development of a toolkit of small molecule probes inhibiting DCN’s interaction with UBE2M.
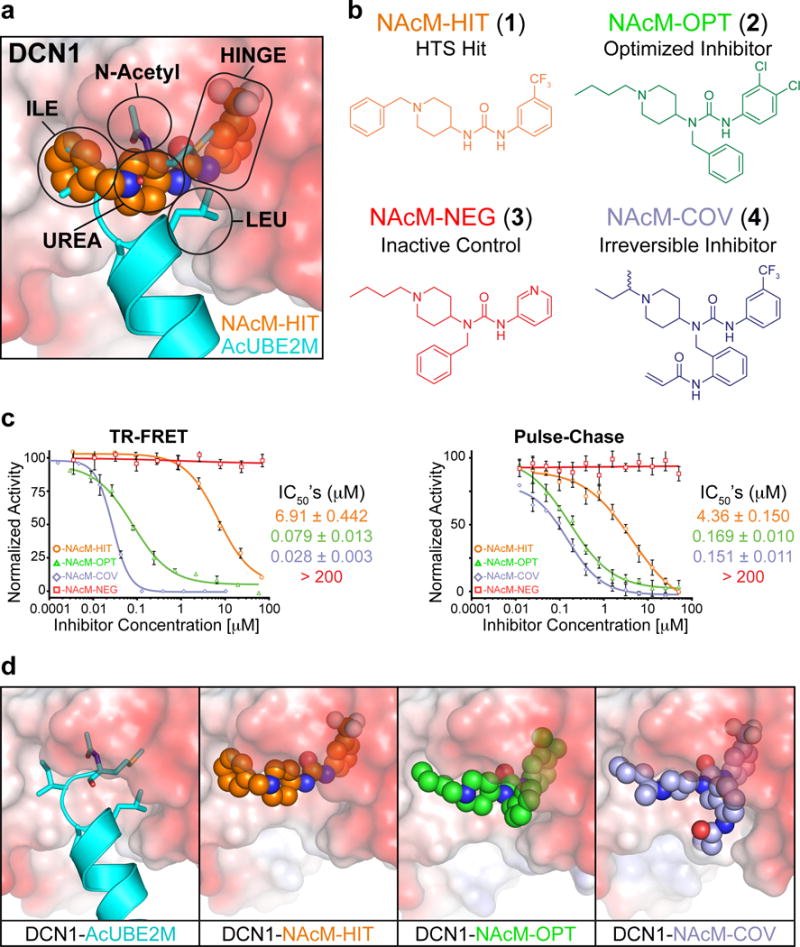
(a) Close-up of DCN1 UBE2M binding site with NAcM-HIT (orange) superimposed on N-terminally acetylated UBE2M (cyan, 3TDU.pdb). The sub-pockets targeted during structure-based compound optimization are highlighted.
(b) Chemical structures, nomenclature, and class of chemical probes targeting DCN’s N-Acetyl-Met binding pocket. NAcM-COV was synthesized and tested as a racemic mixture. Synthetic procedures and characterization data are in Supplementary Note.
(c) Inhibition of DCN1 binding to an N-terminally acetylated peptide from UBE2M (TR-FRET assay, left) or DCN1 activation of N-terminally acetylated UBE2M-dependent neddylation (pulse-chase enzyme assay, right). TR-FRET values represent averages of an experiment performed in triplicate and the pulse-chase values are averaged from three independent experiments.
(d) Comparison of co-crystal structures of DCN1 (surface electrostatic) bound to AcUBE2M (cyan, 3TDU.pdb), NAcM-HIT (orange), NAcM-OPT (green), and NAcM-COV (light blue).
We used iterative structure-based optimization to select moieties to fill the sub-pockets, paying particular attention to hydrogen bonding and electrostatic interactions. We generated more than 300 analogs and tested them in TR-FRET assays, and collected and refined X-ray crystallography data to the point of viewing FO-FC density showing more than 20 of these bound to DCN1. A key observation was that a moderately sized hydrophobic group fulfilling the Leu pocket significantly increased affinity. Overall, iterative examination of the TR-FRET-based dose-response curves, taken together with electron density maps, indicated four key drivers for potency: (1) the piperidine linker that orients the ends of the molecule into the hydrophobic Ile and hinge sub-pockets, (2) an H-bond between the urea aryl N-H and the backbone from DCN1’s Gln114 that anchors the inhibitor into the binding site, (3) tight steric packing of the hinge pocket around the substituted phenyl ring, and (4) hydrophobic interactions of the benzyl substituent within the Leu region. Ultimately, an optimized inhibitor, NAcM-OPT (2), was designed to exploit all four interactions. NAcM-OPT (IC50 of 80 nM) shows 100-fold enhanced potency relative to NAcM-HIT for inhibiting the interaction. The improved inhibition of the isolated E2-E3 interaction translated into 25-fold increased potency toward the neddylation reaction carried out by a fully assembled neddylation complex (Fig. 2c, Supplementary Fig. 5).
A 1.4 Å resolution co-crystal structure showed consolidation of numerous structural elements from DCN1 contributing to the high affinity interaction with NAcM-OPT (Fig. 2d, Supplementary Fig. 6a–d). First, side-chains from two internal aromatic residues, Phe109 and Phe117, are displaced relative to previous DCN1 structures establishing a significantly deeper hydrophobic pocket for the substituted phenyl ring to access. Second, several hydrophobic residues from DCN1 (Ile86, Phe89, Ala111, Phe117, Phe122, Phe164) coalesce around the phenyl ring to produce the tightly fitting hinge pocket. Third, the internal rearrangements of DCN1’s hinge pocket are seemingly necessary to place the urea aryl N-H for H-bonding with the backbone of Gln114 from DCN1, effectively positioning the compound (Supplementary Fig. 6c, 6d). The four-carbon alkyl linker anchors snugly into the Ile pocket through hydrophobic interactions with DCN1 Ala98, Leu184, and stacking with the aromatic ring of Tyr181. DCN1’s Ile83 and Ile86 contact the benzyl substituent in the Leu sub-pocket and stabilize the interaction.
The structural principles guided development of additional NAcM probes (Fig. 2b). First, we developed the inactive control molecule NAcM-NEG (3) by mimicking the electrostatic repulsion driven by the positive charge of unacetylated UBE2M’s amino terminus by replacing the hydrophobic di-chlorophenyl ring with a polar pyridine ring. (Supplementary Fig. 6e). As predicted, NAcM-NEG has no effect on the interaction between DCN1 and UBE2M as monitored by TR-FRET and no effect on neddylation as monitored by enzyme assays even at concentrations of 100 μM (Fig. 2c).
Second, we designed a covalent inhibitor targeting DCN1’s Cys115, located within 10 Å of the benzyl substituent (Supplementary Fig. 6f). Twelve analogs of NAcM-OPT, installing well-characterized cysteine-reactive electrophilic warheads including chloroacetamides and acrylamides29,30 onto the benzyl ring were profiled by TR-FRET (Supplementary Table 3). This revealed that warhead positioning was critical. Introduction of electrophiles onto ortho- or meta- positions of NAcM-OPT’s benzylic ring produced potent inhibitors (TR-FRET IC50 ≤250 nM; Supplementary Table 3: 4–7). Similar compounds including a methylene linker between the aromatic ring and the warhead were also potent inhibitors (Supplementary Table 3: 8–11). However, replacing the benzyl substituent with more flexible two or three carbon alkyl chains connecting the urea and the electrophile attenuated activity (Supplementary Table 3: 12–15). Assaying for covalent adduct formation after 24-hour incubation with DCN1 by LC/MS showed the ortho-N-acrylamide NAcM-COV (4) was most effective (Supplementary Fig. 7a). Therefore, we focused on NAcM-COV containing the acrylamide warhead, which can afford selectivity for cysteine over other endogenous nucleophiles and is well tolerated in cells29,30. Consistent with irreversible binding, apparent potency of NAcM-COV increased over time (Supplementary Fig. 8b). Although covalent inhibitors do not provide an equilibrium IC50, pre-incubating NAcM-COV with DCN1 overnight prior to the TR-FRET binding assay yields an apparent IC50 of ≤40 nM (Fig. 2c, Supplementary Fig. 8b). In our enzymatic assay, performing the pre-incubation overnight yields an IC50,app of 150 nM for blocking the neddylation reaction (Fig. 2c). The importance of the covalent warhead was confirmed with a matched control (NAcM-COVCTRL) 16 with a propionamide substitution incapable of covalent linkage to Cys115 replacing the electrophile (Supplementary Fig. 8a). Comparing properties of NAcM-COV and NAcM-COVCTRL revealed that the non-covalent inhibitor retains some activity, while the electrophile increases potency by >30-fold in the TR-FRET assay and 2.5-fold in the neddylation experiment (Supplementary Fig. 8b–e). Indeed, covalent linkage to the targeted Cys115 is observed in an X-ray co-structure of NAcM-COV bound to DCN1 (Fig. 2d, Supplementary Fig. 6f), and in mass spec analysis of DCN1 immunoprecipitates from NAcM-COV treated cells (Supplementary Fig. 8f, 8g). Thus, formation of the covalent linkage between NAcM-COV and Cys115 is a critical driver of biochemical potency.
Selectivity for N-acetyl-Met-driven interaction
Because acetyl dependent protein interactions are prevalent in cells, we broadly examined selectivity by testing if our compounds inhibit five classes of such interactions. First, we examined interactions utilizing acetylated lysines as drivers of binding including more than 50 histone acetylases, bromodomains, and histone deacetylases (Supplementary Fig. 9a)2,13. Screening at a fixed concentration of 10 μM and interrogating putative hits with dose-response studies to 100 μM revealed that our inhibitors do not significantly impact any of these interactions (Supplementary Fig. 9b; Supplemental Datasets 3, 4, and 5). This represents selectivity of >100-fold against tested acetyl-lysine binding sites.
To further interrogate selectivity, we tested activity toward NAT enzymes, which catalyze acetyl transfer from acetyl-CoA to cognate protein N-terminal sequences. UBE2M’s NAcMet-Ile N-terminus conforms to a substrate of NatC. Even at 25 μM, our NAcM compounds failed to inhibit recombinant trimeric human and yeast NatC enzymes, or NatA, NatE, or NatF from various organisms (Fig. 3a, 3b; Supplementary Figure 10)10,31–33.
Figure 3. Optimized NAcM inhibitors display exquisite selectivity for N-Acetyl pockets of DCN1 and DCN2.
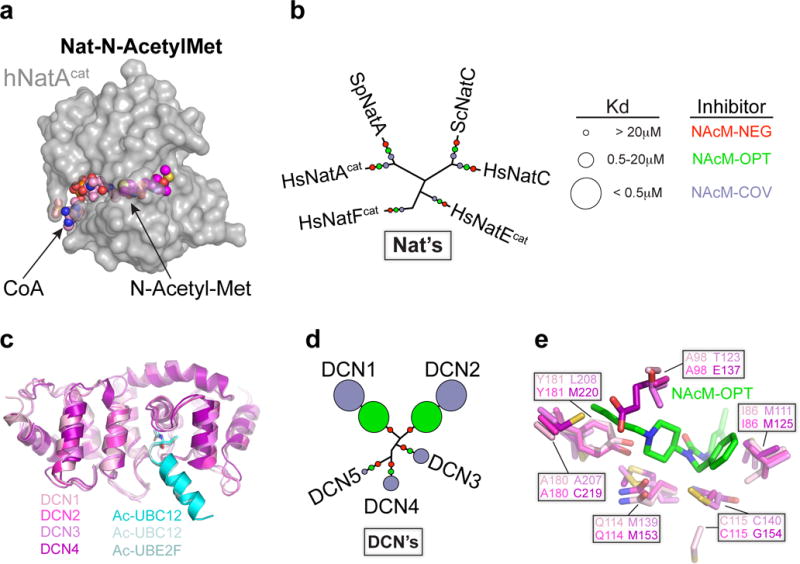
(a) Structure of complex containing N-Acetyl-Met peptide (purple), N-terminal acetyltransferase (hNaa50/NatA (gray), and CoA (pink) (4×5K.pdb).
(b) Effects of NAcM-NEG (red), NAcM-OPT (green), and NAcM-COV (light blue) on human (Hs), S. cerevisiae (Sc), or S. pombe (Sp) Nat enzymes. Kd values inferred from enzymatic assays (Supplementary Fig. 10) are plotted on similarity based dendrogram.
(c) Structural superimposition of DCN family members: DCN1 (100% identity, 0 RMSD, pink)-AcUBE2M (cyan, 3TDU.pdb), DCN2 (82%, 0.46, magenta)-AcUBE2M (pale cyan, 4GAO.pdb), DCN3 (36%, 1.35, violet)-AcUBE2F (teal, 4GBA.pdb), and DCN4 (34%, 1.57, purple).
(d) Effects of probes on DCN1 neddylation activity. Kd values inferred from pulse-chase assays (Supplementary Fig. 11) plotted as in (b).
(e) Subtle variations in targeted binding pocket determine NAcM selectivity profiles. Coloring as in (c) with residues potentially contributing to selectivity surrounding NAcM-OPT shown in stick representation.
Finally, to test selectivity amongst the most homologous systems, we examined activity against the five human DCN family members, which display high similarity in their PONY domains (Fig. 3c) and share overlapping in vitro activities of stimulating N-terminally acetylated UBE2M during cullin neddylation19,22. Using our enzymatic assay, examining neddylation in the presence of NAcM-OPT or NAcM-COV at 10 μM (>50-fold above the IC50 with DCN1), NAcM-OPT only inhibited DCN2, whose PONY domain is 82% identical to DCN1’s (100% identical in the N-Acetyl-Met binding pocket). NAcM-OPT showed no activity toward DCN3, DCN4, or DCN5 despite high structural similarity (Fig. 3c–e, Supplementary Fig 11). Side-chain differences explain selectivity of NAcM-OPT (Fig. 3e).
Partial activity of extremely high concentrations (50 μM) of NAcM-COV towards DCN3 and DCN4 is rationalized by conservation of the targeted Cys in DCN3, but was unexpected for DCN4, which has a glycine in this location (Fig. 3d, Supplementary Fig. 7b, 7c, 11). A 1.55 Å resolution crystal structure of the DCN4 PONY domain bound to its partner domain from CUL1 revealed Cys219 on the neighboring side of the pocket as having potential for reactivity with NAcM-COV (Supplementary Table 2, Supplementary Fig. 7b–d). Notably, the DCN5 sequence lacks cysteines corresponding to Cys115 in DCN1 or Cys219 in DCN4, potentially explaining lack of inhibition by NAcM-COV. We confirmed that the correlation between inhibition by NAcM-COV and the presence of a cysteine in the UBE2M-binding pocket involves covalent modification by performing TOF-based mass spectrometry after incubating 30 μM of the PONY domain from each DCN family member overnight with 60 μM NAcM-COV (Supplementary Fig. 7a, 7e–h). The predominant species of DCN1, DCN2, and DCN3 corresponded to adducts with NAcM-COV. Both modified and unmodified forms of DCN4 were detected, with no evidence of covalent linkage to DCN5. Replacement of Cys115 in DCN1 by either Ala or Gly residues prevented covalent adduct formation with NAcM-COV, thus confirming the essential role of Cys115.
Retrospective inspection of prior DCN1 structures shows Cys115’s side-chain located between N-acetyl Met1 and Leu4 from acetylated UBE2M (Supplementary Fig. 7k)10,20. Furthermore, DCN1’s massive stimulation of cullin neddylation was essentially eliminated upon mutating Cys115 to Ala, Gly, Ile, Leu, Thr, or Val (Supplementary Fig. 7l, 7m). Thus, while the capacity for the binding pocket to undergo structural rearrangement may be exploited for developing small molecules (Supplementary Fig. 6b–d), the structural requirement of Cys115 for catalytic activity provides a natural constraint that may prove useful for future covalent inhibitor development.
Taken together, our toolkit for probing DCN1 interactions with N-terminally acetylated UBE2M includes: a reversible inhibitor (NAcM-OPT), an irreversible inhibitor (NAcM-COV), and a structurally-matched but inactive control compound (NAcM-NEG; Fig. 2). These compounds are straightforward to synthesize in high yield and purity (Supplementary Note) 17-23, and are highly specific for DCN1 and DCN2 even across the largely structurally and functionally conserved DCN family (Fig. 3). Thus, it is possible to develop small molecule inhibitors selectively targeting an extraordinarily prevalent protein interaction motif.
NAcMs inhibit DCN1 binding to UBE2M in cells
We tested whether our small molecules inhibit DCN1-UBE2M interactions in cells using lentivirally expressed UBE2M with a C-terminal FLAG-HA tag at near-endogenous levels in 293T cells (Fig. 4a). C-terminal tagging of UBE2M in 293T cells was shown to allow its essentially complete N-terminal acetylation and affinity-purified (AP) UBE2M bound endogenous DCN110,34. Accordingly, the UBE2M-DCN1 interaction was observed after affinity purification from cells treated for 24 hours with DMSO or the inactive control compound NAcM-NEG (Fig. 4a). However, treatment with the reversible (NAcM-OPT) or irreversible (NAcM-COV) inhibitors blocked co-precipitation of DCN1 with UBE2M (Fig. 4a, 4b, Supplementary Dataset 6). Therefore, the inhibitors that disrupt interaction of UBE2M and DCN1 in reconstituted systems also disrupt complex formation in cells. Thus, one can inhibit formation of large cellular signaling complexes by disrupting binding of an N-acetylated amino terminus.
Figure 4. NAcM inhibitors are on target and disrupt AcUBE2M interaction network.
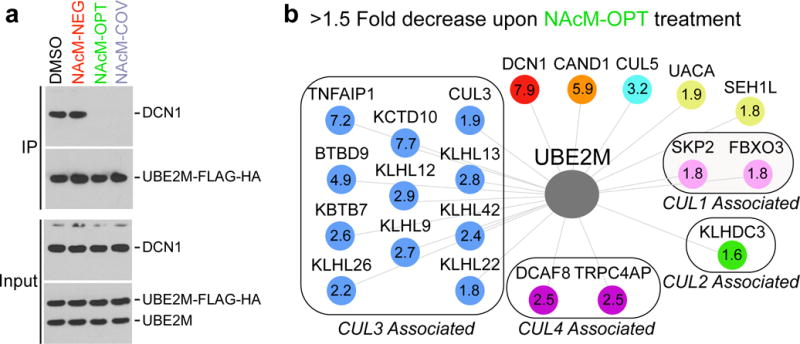
(a) NAcM-OPT and NAcM-COV, but not NAcM-NEG, disrupt co-immunoprecipitation of DCN1 with AcUBE2M. Experiments were performed with 293T cells expressing UBE2M with a C-terminal FLAG-HA tag at near-endogenous levels. Immunoblot of input and elutions from anti-FLAG immunoprecipitations were probed with the indicated antibodies (full gels shown in Supplementary Fig. 14)
(b) N-acetyl-UBE 2M interactions decreased by >1.5-fold with P <0.05 (calculated using the moderated t statistic (two-sided) with Benjamini–Hochberg false discovery rate adjustment; n = 3 independent experiments) by NAcM-OPT (DCN 1, red; CAND 1, orange) or by association with a particular CRL (CUL 1, pink; CUL 2, green; CUL 3, blue; CUL 4A, purple; CUL 5, cyan) (Supplementary Data Set 6). The fold decrease in binding is indicated for each protein.
Having shown that the inhibitors disrupted DCN1 binding to UBE2M, we examined effects on co-association with other proteins by AP-MS using a quantitative Tandem Mass Tagging (TMT)-based proteomic method34. Cells were treated with either negative controls (DMSO or NAcM-NEG) or active compounds (NAcM-OPT or NAcM-COV), affinity-purified complexes were processed for 10-plex TMT, and summed intensities for reporter ions were used to quantify components present within the complexes. Importantly, the interaction most greatly impacted by NAcM-OPT was UBE2M binding to DCN1, which was reduced ≈8-fold relative to controls (Fig. 4b, Supplementary Dataset 6). The overwhelming majority of other proteins whose interactions with UBE2M were substantially decreased were also part of the cullin-RING ligase (CRL) network, including 1.9- and 3.2-fold reduction of two cullins (CUL3 and CUL5, respectively), and several CRL substrate receptors. The 7.7-fold to 1.8-fold decrease in UBE2M association with a cohort of BTB proteins that serve as CUL3 substrate receptors was particularly striking (Fig. 4b), and suggests that in this cell line inhibiting UBE2M binding to DCN1 might especially impact CUL3. A drastic reduction in DCN1-binding to cullins was further implied by the 5.9-fold decrease in association of CAND1, as CAND1 was previously shown to associate with DCN1 in a cullin-dependent manner18,22. CAND1 marks inactive cullins that are neither neddylated nor bound to substrate receptors, raising the possibility that steady-state UBE2M recruitment to CAND1-bound cullins is DCN1-dependent, perhaps reflecting a mechanism for their rapid neddylation upon CAND1 displacement. We saw no significant changes in the level of acetylated UBE2M or any other acetylated peptide detected by our proteomics experiments (Supplemental Fig. 12, Supplemental Dataset 7).
NAcMs inhibit DCN1-dependent cellular neddylation
Although in lower eukaryotes that express only a single Dcn1 family member, eliminating Dcn1 or Ubc12 N-terminal acetylation leads to a 90 or 50% reduction in the steady-state level of neddylated cullins respectively10,16,21, in many mammalian cell lines, knocking down or knocking out DCN1 has relatively subtle effects on steady-state levels of cullin neddylation17,22,35,36. One possibility is that the other DCN-family members, or other regulators, compensate for loss of DCN119,22,37,38. Given the greater effects of deleting the singular yeast DCN116, and prior findings that high level expression of exogenous human DCN1 can modulate cullin neddylation and anchorage independent growth of human cells cultured on soft agar24, we sought to identify a cell line with high levels of endogenous DCN1 for testing effects of our inhibitors. Immunoblotting various human cell lines revealed elevated levels of DCN1 in non-small cell lung and tongue carcinoma cell lines HCC95 and CAL-33, consistent with amplification of the DCUN1D1 gene in most human squamous cell carcinomas (Fig. 5a).
Figure 5. NAcM inhibitors reduce CUL neddylation in cells and mimic effects of shRNA knockdown of DCN1.
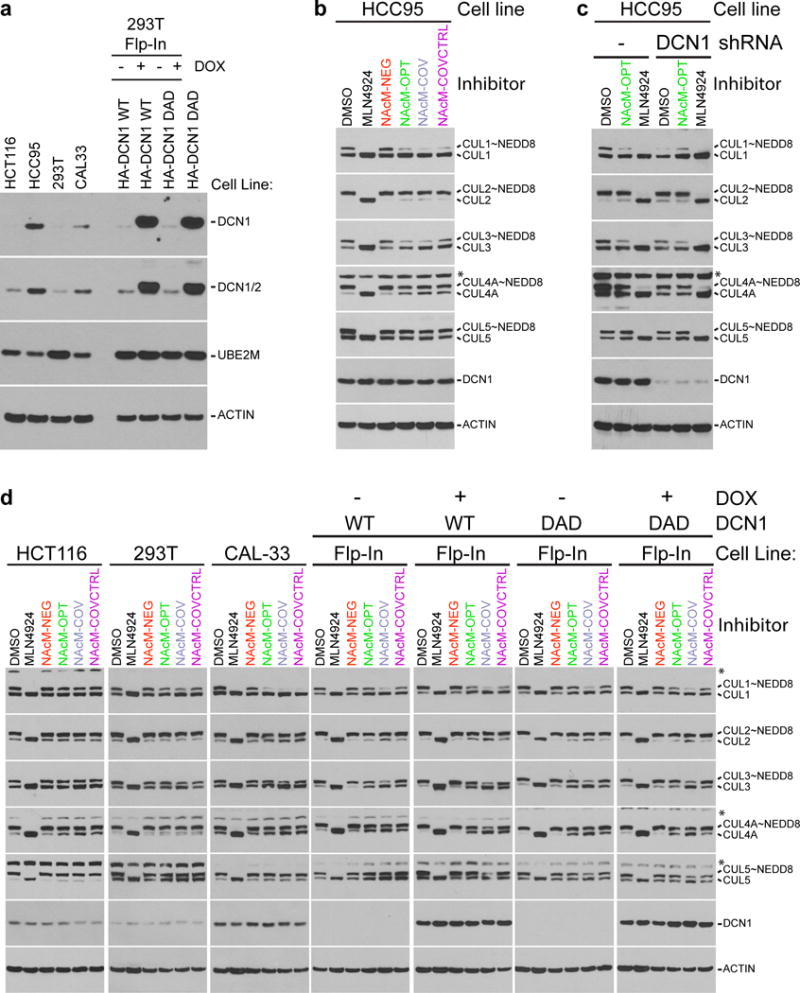
(a) HCC95 and CAL-33 have high levels of DCN1 as demonstrated by immunoblot of total cell extracts of indicated cell lines (full gels shown in Supplementary Fig. 15a).
(b) Effects of NAcM-NEG, NAcM-OPT, NAcM-COV, and NAcM-COVCTRL on cullin neddylation in HCC95 cells treated with DMSO, MLN4924 (single dose, 1 μM), or indicated compounds (10 μM, dosed at 0 and 24 hours). Cells were harvested at 48 hours, processed for immunoblotting, and probed with the indicated antibodies (full gels shown in Supplementary Fig. 15b).
(c) NAcM-OPT mimics shRNA knockdown of DCN1. Immunoblot of total cell extracts from HCC95, or a line stably expressing a DCN1 shRNA, treated and processed as in (b) (full gels shown in Supplementary Fig. 15c).
(d) Probe effects vary amongst cell lines. Immunoblot of total cell extracts from indicated cell lines, treated and processed as in (b). For all blots, * indicates non-specific bands in CUL1, CUL4A, and CUL5 immunoblots (full gels shown in Supplementary Fig. 15d–h).
Treatment of HCC95, the cell line expressing highest levels of DCN1, with NAcM-OPT or NAcM-COV reduced levels of neddylated cullins detected by immunoblotting to an extent comparable with that observed upon knocking down DCN1 through shRNA expression (Fig. 5b, 5c). Combining DCN1 knockdown and treatment with NAcM-OPT resulted in similar reductions of cullin neddylation as either treatment alone, consistent with the effects of NAcM-OPT depending on increased levels of DCN1 (Fig. 5b, 5c). As observed previously for other cell lines17,22,35,36, inhibiting DCN1 activity did not affect all cullins equally. In HCC95 cells, inhibiting DCN1 activity either by NAcM compounds or shRNA most affected the steady-state levels of neddylated CUL1 and CUL3, with lesser effects on CUL4A. Importantly, the inactive NAcM-NEG had no effect on cullin neddylation status. In concordance with our in vitro studies, NAcM-COV exhibited the most potent inhibition of cellular neddylation, while NAcM-COVCTRL exhibited relatively reduced potency, supporting our hypothesis that covalent linkage with Cys115 drives increased potency of NAcM-COV. Together, these results further support that the suite of NAcM compounds are on-target in cells.
To determine if sensitivity to the inhibitors correlates with DCN1 expression levels, we examined effects of NAcM compounds in multiple cell lines (Fig. 5a), two with DCN1 amplification (HCC95 and CAL-33), two that have not been reported to have amplification of the DCN1 gene (HCT116 and 293T), and Flp-IN 293T cell lines bearing doxycycline-inducible transgenes expressing either wild-type DCN1 or a mutant in the cullin-binding DAD patch21 that is defective in cullin binding (Fig. 5d). The effects of the NAcMs on steady state levels of cullin neddylation range from subtle effects on levels of neddylated CUL3 and CUL4A in HCT116 and CUL1 and CUL3 in the parental 293T line, to striking decreases in neddylated CUL1 and CUL3 in CAL-33 and HCC95. Levels of neddylated CUL2 and CUL5 were not obviously affected in any experiment. These differences suggest that DCN1 levels do correlate to some extent with sensitivity to the NAcMs but also that the DCN1 dependency of a particular cullin family member is cell-type specific. In support of this, induction of WT DCN1 in Flp-IN 293T cells, but not the cullin-binding defective DAD mutant, mildly sensitizes the levels of neddylated CUL4A to our inhibitors (Fig. 5d).
The effects of inhibiting DCN1-UBE2M interactions differ significantly from treating cells with the inhibitor of the NEDD8 E1 enzyme, MLN4924, which blocks upstream reactions, prevents forming the UBE2M~NEDD8 intermediate39, and eliminates all neddylation observed in cells (Fig. 5b–d). DCN1 inhibitors and MLN4924 show most pronounced cellular effects at concentrations above their respective biochemical IC95 (MLN4924: biochemical IC50 = 5 nM, cell based activity penetrant at 0.3–1 μM39; NAcMs: biochemical IC50 = 150–170 nm, cell based studies = 10 μM). Thus, our NAcM inhibitors of DCN1-UBE2M interactions disrupt function of the targeted multiprotein neddylation-catalyzing complex in cells.
Given the sensitivity of HCC95 cells to DCN1 inhibition, we sought to characterize cellular phenotypes induced by treatment with the suite of NAcM compounds. Although some roles for neddylated CRLs in non-degradative ubiquitination are emerging, neddylation is largely recognized for activating ubiquitin-dependent proteasomal degradation. Indeed, total blockade of neddylation by treatment with MLN4924 reduces total cellular ubiquitination by 10–20% leading to the stabilization of hundreds of CRL substrates39. Such a role has not been reported for DCN family members. We compared levels of several well-recognized proteasomal targets of cullin-RING ligases upon E1 or DCN1 inhibition by immunoblotting. Although control experiments with MLN4924 caused high-level accumulation of CRL substrates, there were no obvious effects on the levels of these substrates following either NAcM inhibition of DCN1 or its knockdown by shRNA (Fig. 6a). Likewise, in an unbiased proteome-wide search, inhibiting DCN1-dependent neddylation starkly contrasted with MLN4924 in not obviously increasing levels of any proteins measured by TMT-based mass spectrometry (Fig. 6b, Supplementary Dataset 8).
Figure 6. NAcM-OPT inhibits neddylation in and prevents anchorage-independent growth of a DCN1 amplified cell line without causing changes in protein homeostasis.
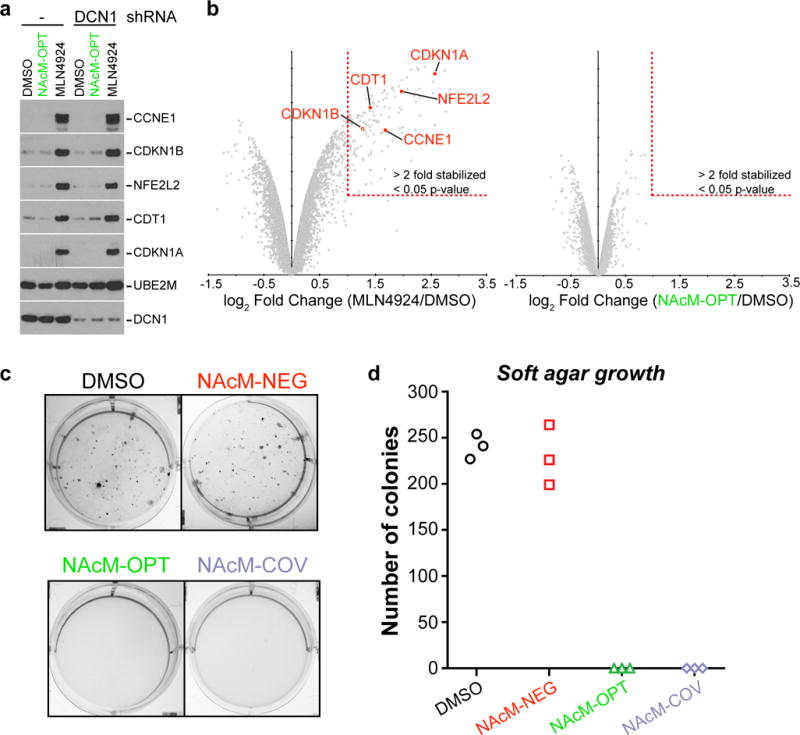
(a) Neither NAcM-OPT treatment nor DCN1 shRNA stabilizes cullin-RING ligase substrates in HCC95 cells. Cells treated and processed as in Fig. 5c using antibodies for the indicated proteins (full gels shown in Supplementary Fig. 16).
(b) NAcM-OPT does not globally change protein homeostasis in HCC95 cells. TMT-quantification of total proteome following treatment with DMSO, MLN4924 (left, 1 μM), or NAcM-OPT (right, 10 μM) for 24 hours. MLN4924 causes >2-fold stabilization of 136 proteins (p-value <0.05), whereas NAcM-OPT treatment fails to stabilize any protein (Supplementary Dataset 8). Substrates examined in (a) are highlighted in red.
(c) NAcM-OPT and NAcM-COV treatment blocks growth of HCC95 cells in soft agar. Representative data showing anchorage independent growth of HCC95 cells treated with NAcM-OPT (10 μM), NAcM-COV (10 μM), and NAcM-NEG (10 μM).
(d) Quantification of results from (c).
NAcM effects on DCN1-amplified cells
Human DCN1 was originally discovered due to its overexpression in tumor cells, with amplification conferring anchorage-independent growth24. Therefore, we examined effects of our DCN1/2-specific NAcM probes on growth of HCC95 cells in 3D culture. Indeed, at doses that are not growth inhibitory toward monolayer cultures (Supplementary Fig. 13), colony formation in soft agar was completely eliminated in the presence of active NAcMs, whereas treatment with the inactive control has no impact on colony formation (Fig. 6c, 6d). Thus, inhibiting cell proliferation in soft agar correlates with the functional blockade of DCN1 activity.
Discussion
We have shown that selective small molecule probes can be developed that competitively inhibit protein interactions mediated by N-terminal acetylation. Both the reversible (NAcM-OPT) and irreversible (NAcM-COV) inhibitors were exquisitely specific for blocking the targeted N-terminal acetylation-dependent interaction of UBE2M with DCN1.
Potency was achieved by both populating the pockets normally occupied by the N-terminal Met side-chain of UBE2M and engaging adjacent sites. Unexpected structural rearrangement of residues buried within the targeted pocket enabled our inhibitors to penetrate deeper into a remodeled groove that normally accommodates UBE2M’s Met side-chain. As this deeper groove was not observed in six prior structures of DCN family members complexed with N-terminally acetylated NEDD8 E2s10,19,20, our data underscore the value of empirical screening to harness cryptic protein conformations.
We fortuitously discovered that a cysteine in DCN1’s binding pocket is structurally crucial for binding N-terminally acetylated UBE2M. Although our intent was to validate targeting and increase potency, we note that covalent capture of structurally important cysteines has been proposed as an approach to drug targeting40. We anticipate that covalent targeting of non-mutable cysteines would also prove useful in mitigating potential resistance mutations.
The specific proteins targeted by inhibiting this N-terminal acetylation-dependent interaction are subunits of an ubiquitin-like protein ligase-conjugating enzyme complex. Despite great interest in targeting ubiquitin and ubiquitin-like protein cascades, discovering such inhibitors has been hindered by high homology of E2 and E3 catalytic domains. We demonstrate that exploiting non-homologous auxiliary E2-E3 interactions within such ligation complexes enables development of specific molecules that effectively inhibit ligase activity.
The effects of inhibiting this specific NEDD8 E2-E3 interaction vary significantly from those of MLN4924, which obliterates all neddylation by inhibiting the E1 enzyme initiating the NEDD8 cascade. While blocking binding to DCN1 clearly perturbs the UBE2M interactome, there are minimal effects on protein turnover. Perhaps the relatively lower level of neddylation seen with DCN1 inhibition in HCC95 and CAL-33 cells – particularly for CUL1 and CUL3 – is sufficient to sustain the CRL network and ubiquitination activity required for CRL substrate degradation37,38. It remains unknown when cellular neddylation of a particular cullin would involve DCN1 activity22.
Alternatively, DCN1 may specifically associate with a subset of CRLs that catalyze non-degradative ubiquitination. In this regard, it is intriguing that the NAcM inhibitors most greatly impaired neddylation of CUL3, as well as UBE2M interactions with CUL3-associated substrate receptors (Figs. 4, 5). Notably, proteasome-independent functions of neddylation are beginning to emerge from studies of CUL341. Challenges in finding non-degradative CRL targets may explain why specific downstream targets of DCN1-dependent cullin neddylation remain unknown. Our finding that inhibiting the DCN1-UBE2M interaction both decreases neddylation and anchorage-independent growth of HCC95 cells harboring DCUN1D1 gene amplification highlights the importance of identifying such substrates. In the long-term, developing DCN1 inhibitors may prove clinically relevant for cancers displaying amplification of the DCUN1D1 gene, in a setting requiring more nuanced regulation of neddylation than afforded by MLN4924 (Pevonedistat), or in combination with MLN4924 as an orthogonal means to inhibit the NEDD8 pathway.
Over the past 20 years, there has been widespread interest in discovering inhibitors of protein-protein interactions through targeting “hot spots” with small molecules that mimic the clusters of side-chains that drive the protein interactions, with several potential drugs in clinical trials. Our development of potent, selective inhibitors blocking UBE2M interactions with DCN1, without apparently influencing N-terminal acetylation status, demonstrates for the first time that these interactions are druggable. Notably, our NAcM inhibitors contrast from Nat enzyme inhibitors that would globally impact protein N-terminal acetylation31,32,42. Although specific interactions involving acetylated protein N-termini are only beginning to emerge, some potentially therapeutically relevant targets include ubiquitin E3 ligases, cytoskeletal assemblies, epigenetic regulators, and protein trafficking and quality control pathways1–7,11,12,43,44. Distinct hydrophobic N-termini may also prove useful targets for novel therapeutics against devastating pathogens, as formyl-Met at N-termini of bacterial proteins plays crucial roles in host-pathogen interactions45, and some proteins exported from malaria parasites into erythrocytes during infections undergo processing and post-translational N-terminal acetylation46. We predict that these or other pathways mediated by acetylation of protein N-termini or other non-histone sites can also be targeted selectively.
Online Methods
Constructs, Protein preparation, and Antibodies
Expression constructs were prepared by standard molecular biology techniques, with coding sequences entirely verified. Mutant versions were generated by QuickChange (Stratagene). DCN proteins used for in vitro assays correspond to the isolated PONY domain. For biotinylation of DCN1, an AviTag sequence (GSMSGLNDIFEAQKIEWHEGS) and flanking GlySer linkers between the TEV cleavage site and DCN1 PONY domain ORF were inserted into a pGEX based GST-TEV-DCN1 vector. Fusion between a mutant T4-lysozyme (C54T, C97A, A146T) and DCN1 was generated by overlap PCR and cloned as a His-TEV fusion in a pRSF-DUET-based vector. Where indicated, two additional mutations (D127A, R154A) were further incorporated into lysozyme in order to prevent inadvertent crystal packing contacts observed in initial crystals between some inhibitors and a lysozyme symmetry mate. For expression of the trimeric human NatC complex, Naa38 was cloned as a His-TEV fusion into pFastbac-HTB and Naa30 and Naa35 were cloned untagged into pFastbac1. hNaa10 was cloned as an N-terminal His-MBP-TEV fusion in a pRSFDUET based vector. hNaa50 was cloned with an N-terminal GST-TEV tag in a pGEX4T1 based vector. Vectors for expression and purification of hNaa60 and the S. pombe NatA complex were kind gifts from Ronen Marmorstein31,32.
The heterodimeric NEDD8 E1 complex (NAE1-UBA3), NEDD8, CUL1WHB, PONY domains of DCN1, DCN2, DCN3, DCN4, DCN5, yNatC, hNaa10, hNaa50, hNaa60, pombe NatA, and CUL2 (C-terminal domain, CTD)-RBX1 were expressed in E. coli and purified as described10,19,31,32,42. N-terminally acetylated UBE2M and full-length CUL3-RBX1 were expressed in insect cells as described10,47. Avi-Tag DCN1 was expressed as a GST-TEV fusion and purified as described previously for GST-TEV-DCN110. Lysozyme-DCN1 fusions were expressed with a N-terminal His-TEV tag in E. coli and purified by Ni-affinity chromatography. Eluted protein fractions from the Ni column were further purified by ion-exchange and gel filtration chromatography into 25 mM HEPES, 200 mM NaCl, 1 mM DTT, pH 7.5. The heterotrimeric human NatC complex was expressed in insect cells and purified by Ni-affinity chromatography, liberated by TEV cleavage on beads, and further purified by HiTrap Q ion-exchange and gel filtration chromatography.
Antibodies used in this study were: CUL1 (sc-17775), Actin (sc-1615), CDT1 (sc-365305), DCN1 (sc-81835), DCN1/2 (sc-398218), and p21 (sc-397) from Santa Cruz Biotechnology; CUL3 (A301-109A), Cul4A (A300-739A), and CCNE1 (A301-566A) from Bethyl Laboratories; CUL2 (ab166917), CUL5 (ab184177), and NFE2L2 (ab62352) from Abcam; p27 (610242) from BD Transduction Laboratories; DCN1 (clone 3D7) from Sigma. All commercial antibodies were used according to the manufacturers’ instructions. Antibodies against UBE2M and DCN1 have been described24,48.
Sample Sizes
The number of replicates used in each study represents the minimum number of samples required to obtain statistically reliable results, as based on our historical experience and power analysis. Briefly, the enclosed studies are largely exploratory studies, designed for the determination of gross changes in response (> 5-fold) at a power of 95% and a p-value of 5%, taking into account standard deviations observed in similar studies conducted in both the Guy and Schulman labs (100+ similar experiments over 15 years).
Preparation of Biotinylated-DCN1 and AcUBE2M1–12-AlexaFluor 488 for TR-FRET
Reaction conditions for biotinylation of DCN1 were initially established in small scale pilot experiments and the efficiency of Avi-DCN1 biotinylation was assessed by super shift of biotin-DCN1 in SDS-PAGE after the addition of a stoichiometric excess of avidin (Sigma Aldrich A9275). Biotinylation reactions contained 90 μM Avi-DCN1 and 4 μM BIRA in 10 mM Tris, 10 mM ATP, 10 mM magnesium acetate, 2 mM Biotin, pH 8.0 and were incubated at room temperature for 2 hours. Reaction mixtures were diluted four-fold into 25 mM Tris, 1 mM DTT, pH 8.0 and purified over a HiTrap Q ion-exchange column. Fractions containing biotin-DCN1 were pooled, concentrated, and further purified by size exclusion chromatography in 25 mM HEPES, 200 mM NaCl, 1 mM DTT, pH 7.5.
The hydrocarbon stapled Acetyl-UBE2M1–12 peptide corresponding to N-terminally acetylated residues 1–12 of human UBE2M with an additional C-terminal cysteine residue (sequence Acetyl-MIKLZ*SLKZ*QKKC, where Z* is 2,4′-pentenylalanine closed after synthesis to create the hydrocarbon staple), was synthesized and purified as previously described10. For labeling with Alexa Fluor 488 C5 maleimide (Invitrogen A10254), Acetyl-UBE2M peptide (7 mg) was dissolved in 5 mL of reaction buffer (50 mM HEPES pH 7.5, 200 mM NaCl, 10 mM TCEP) followed by a 15 minute incubation on ice to allow TCEP to completely reduce the C-terminally appended Cys residue. The peptide was added dropwise to 0.25 mL of a 26 mM solution of AlexaFluor dye in DMSO. The mixture was incubated at room temperature overnight, and cold acetone was added (~25 mL) until the solution became turbid. The mixture was spun at 4000 rpm for 3 minutes to remove unreacted dye. The precipitated orange residue was dissolved in water and purified by preparative HPLC (Waters 4000, in water:acetonitrile, 0.1% Formic acid, using a gradient from 5–50% acetonitrile, a flow rate or 15 mL/min, for a 27 minute run on a Gemini-NX 5u C18 110A 50×30.00mm column (Phenomenex). The purified sample was lyophilized, yielding ~5 mg of yellow powder with ~90% purity.
TR-FRET Assay
TR-FRET assays were carried out in black 384-well microtiter plates at a final volume of 20 μL per well. To screen library compounds, the assay cocktail was prepared as a mixture of 50 nM Biotin-DCN1, 20 nM Ac-UBE2M12-AlexaFluor488, 2.5 nM Tb-Streptavidin (ThermoFisher) in assay buffer (25 mM HEPES, 100 mM NaCl, 0.1% Triton X-100, 0.5 mM DTT, pH 7.5). The assay cocktail was incubated for 1 hour at room temperature and distributed using a WellMate instrument (Matrix). Compounds to be screened were added to assay plates from DMSO stock solutions by pin transfer using 50SS pins (V&P Scientific). The assay mixture was incubated for 1 hour at room temperature prior to measuring the TR-FRET signal with a PHERAstar FS plate reader (BMG Labtech) equipped with modules for excitation at 337 nm and emissions at 490 and 520 nm. The integration start was set to 100 μs and the integration time to 200 μs. The number of flashes was fixed at 100. The ratio of 520/490 was used as TR-FRET signal in calculations. Assay endpoints were normalized from 0% (DMSO only) to 100% inhibition (unlabeled competitor peptide) for hit selection and for curve fitting. Data curves in Figures 2c and Supplementary Figures 1c, 2b, 8b,c are fits to the average of n=3 independent experiments.
HTS campaign
Initial screening was performed at a single point concentration of 30 μM with a library of approximately 600,000 compounds. The HTS endeavor was implemented on a fully-automated system (HighRes Biosolutions) with an integrated robotic arm (Staübli). The protein and peptide master mixture was kept chilled at 4 °C, and dispensed into solid black 384-well assay plates (20 μL/well) using Matrix Wellmate bulk dispensers (ThermoFisher), followed by centrifugation using a V-Spin plate centrifuge (Agilent Technologies). Test articles and controls (stored as 10 mM solutions in DMSO donor plates) were transferred to the assay plates using a pin tool (V&P Scientific) equipped with FP1S50 pins resulting in final compound concentrations of 30 μM. The TR-FRET signal was acquired after the plates were incubated for 1 h at room temperature in dedicated incubators (Liconic Instruments). Dose-response curve fitting and chemical structure network graph analysis were performed as previously described49. Compound solubility, permeability, and inhibition of proliferation were performed as described previously50.
Enzyme assays in pulse-chase format monitoring DCN1-dependent cullin neddylation
Enzymatic neddylation of cullins was monitored using pulse-chase assays that isolate the reaction stimulated by DCN1 from upstream reactions in the E1-E2-E3 NEDD8 transfer cascade10,20. NEDD8 was labeled with Fluorescein-5-maleimide on an N-terminal Cys-containing tag as described20, and thioester-linked to N-terminally acetylated UBE2M in a “pulse” reaction incubating 10 μM UBE2M, 15 μM *NEDD8, and 400 nM APPBP1-UBA3 in 25 mM HEPES, 200 mM NaCl, 2.5 mM MgCl2, 1 mM ATP, pH 7.5 for 15 minutes at room temperature. After this reaction was quenched for 5 min on ice with 50 mM EDTA, NEDD8 was chased from AcUBE2M to a cullin, either CUL2 C-terminal domain (CTD) in complex with RBX1, or with full-length CUL3 in complex with RBX1. Our default assays generally utilize CUL2 as the neddylation substrate because (1) Cullin proteins generally express at low levels but our yields for recombinant CUL2 are ≈20 fold better which makes it most practical to perform experiments with CUL2. (2) DCN1 has a higher affinity for CUL2 (Kd ~500nM19) allowing for a higher percent saturation of CUL2 with DCN1 under the low protein concentration conditions of our chase assay. This is important for sensitizing the assays to detect DCN1-dependent neddylation as compared to reactions in the absence of DCN1, without having to use very high concentrations of DCN1. “Chase” reactions were performed by diluting the thioester-linked UBE2M~NEDD8 conjugate to 0.04 μM in 50 mM Tris, 50 mM NaCl, 50 mM EDTA, 0.5 mg/ml BSA, pH 6.8 on ice, reactions initiated by the addition of CUL-RBX complex alone or in a 1:1 mixture with the indicated DCN family member to a final concentration of 125 nM. Aliquots were removed at the indicated times and terminated with 2× SDS-PAGE sample buffer. Reaction products were separated on 4–12% NuPAGE gels (Invitrogen). Fluorescent gels were visualized by scanning on a Typhoon imager (GE).
Assaying N-terminal acetyltransferase activities
Synthetic peptide substrates NH2-MIKLFSLKQQKKEEESAGGTKGSSKK, NH2-EEEIAALRWGRPVGRRRRPVRVYP, NH2-SESSSKSRWGRPVGRRRRPVRVYP, NH2-MLGPEGGRWGRPVGRRRRPVRVYP, and NH2-MAPLDLDRWGRPVGRRRRPVRVYP were synthesized and HPLC purified to >98% purity by the Hartwell Center at St. Jude. N-terminal acetylation of peptide substrates was monitored utilizing a 5,5′-dithiobis-(2-nitrobenzoic acid (DTNB) based assay42. Purified Nat enzyme (100 nM S. pombe NatA, 200 nM yNatC, hNatC, hNaa10, hNaa50, or hNaa60) was mixed with 500 μM substrate peptide and 500 μM Acetyl-CoA (Sigma) in acetylation buffer (25 mM HEPES pH 7.5, 100 mM NaCl, 1 mM EDTA) in a final reaction volume of 50 μL. Reactions for yNatC, hNatC, pombe NatA, and hNaa10 were carried out at room temperature, while assays with hNaa50 and hNaa60 were at 37 °C. At the indicated times, the reactions were quenched by the addition of 100 μL 3.2 M guanidinium-HCl, 100 mM sodium phosphate dibasic, 10 mM EDTA, pH 6.8. 20 μL of a freshly made 10 mg/ml DTNB solution in 100 mM sodium phosphate dibasic, 10 mM EDTA, pH 6.8 was added to the quenched samples and incubated for 5 minutes at room temperature. Samples were further diluted to a final volume of 510 μL with 2.13 M guanidinium-HCl, 66.6 mM sodium phosphate dibasic, 6.66 mM EDTA, pH 6.8. The absorbance of the samples at 412 nM was recorded and the amount of product formed calculated using the extinction coefficient of 13,700 M−1 cm−1.
Crystallography
Crystals were grown by the hanging-drop vapor diffusion method. For lysozyme-DCN1-compound complex structures, mixtures were prepared by diluting lysozyme-DCN1 to 50 μM in 50 mM HEPES, 150 mM NaCl, pH 7.5. Compounds were subsequently added to a final concentration of 75 μM from concentrated DMSO stock solutions (1–2% DMSO final concentration) and incubated on ice for 1 hour to equilibrate binding. The mixture was then concentrated approximately 10–12-fold with an AmiconUltra concentrator prior to setting crystal screens. For the DCN4-CUL1 (WHB domain) complex structure, the purified proteins were mixed in stoichiometric levels, incubated on ice for 1 hour, and concentrated to a final complex concentration of approximately 700 μM.
For lysozyme-DCN1-NAcM complex structures we first crystallized apo-lysozyme-DCN1 at 4° C in 11–13% PEG3350, 0.2 M NH4Br resulting in large clumped crystals. Clusters of apo crystals were crushed and used as seed stocks for generating diffraction-quality lysozyme-DCN1-NAcM crystals by streak seeding into 6–9% PEG3350, 0.2 M NH4Br. Crystals were harvested in mother liquor supplemented with 25% MPD prior to flash-freezing in liquid nitrogen. Reflection data were collected at beamline 8.2.1 at the Advanced Light Source. The crystals belong to space group P21 with one lysozyme-DCN1-inhibitor complex in the asymmetric unit. Phases for all structures were obtained by molecular replacement using PHASER51 with the following search models: 1 copy each of (a) residues 6–158 from a prior structure of lysozyme (2LZM.pdb) and (b) residues 65–250 from a prior structure of DCN1 (3TDU.pdb). Following initial rounds of building and refinement, small molecules were fit into density with COOT52 utilizing compound restraint files generated with the PRODRG server53. In all cases manual building was performed in COOT and refinement was performed using Phenix54. Additional details of the refinement are provided in Supplementary Table 2. The final Ramachandran statistics are as follows: Lysozyme-DCN1-NAcM-HIT, 99% Favored, 0% Outliers; Lysozyme-DCN1-NAcM-OPT, 99% Favored, 0% Outliers; Lysozyme-DCN1-NAcM-COV, 98% Favored, 0% Outliers.
Crystals of DCN4 (PONY domain)-CUL1 (WHB domain) grew at 4° C in 25% PEG3350 0.2 M LiSO4 10 mM TCEP 0.1 M BTP pH 6.5 as sea urchin like needle clusters. Single, diffraction quality, crystals were obtained by streak seeding into 19% PEG3350 0.2 M LiSO4 10 mM TCEP 0.1 M BTP pH 6.3. Crystals were harvested from mother liquor and soaked in step gradients for 5 minutes, with sequential soaks in well solution supplemented with 12% and 24 % glycerol prior to flash freezing in liquid nitrogen. Reflection data were collected at the SERCAT 22-ID beamline at the Advanced Photon Source. The crystal belongs to space group C2 with one copy of the DCN4-CUL1 complex in the asymmetric unit. Phases were obtained by molecular replacement using PHASER51 using the following search models: 1 copy each of (a) residues 65–250 from a prior structure of DCN1 (3TDU.pdb) and (b) residues 705–770 from a prior structure of CUL1 (3TDI.pdb). The electron density surrounding the CUL1WHB domain was weak, presumably due to a lack of participation in crystal packing. Therefore we modeled and built this chain largely from a prior high resolution and high quality structure of CUL1WHB, 3TDU.pdb. Manual rebuilding was performed with COOT and refinement with Phenix52,54. Additional details of the refinement are provided in Supplementary Table 2. The final Ramachandran statistics were as follows: CUL1WHB:DCN4, 99% Favored, 0% Outliers.
Bromodomain Profiling
Compound profiling against a panel of 32 BRDs was performed by DiscoveRX Corp. at a single concentration of 10 μM. The amount of BRD captured on an immobilized ligand in the presence or absence of compound was measured using a quantitative real-time polymerase chain reaction (qPCR) method that detects the associated DNA label tagged to the BRD. Any target that demonstrated greater than or equal to 40% inhibition at 10 μM were subjected to dose-response profiling for Kd determination. An 11-point 3-fold serial dilution of each test compound was prepared in 100% DMSO at 1000× final test concentration (top concentration 120 μM) such that the final concentration of DMSO was 0.09% in the assay (Supplementary Dataset 4).
HDAC, SIRT, and HAT profiling
Compound profiling against a panel of 11 HDACs, 4 SIRTs, and 7 HATs was performed by Reaction Biology Corp. at a single concentration of 10 μM in triplicate. The results are reported as percent enzyme activity based on DMSO treated controls (Supplementary Dataset 3, 5).
Cell culture
HCC95 a squamous cell lung carcinoma line, BJ a normal human foreskin fibroblast cell line, HEK293T a embryonic kidney line, and HCT116 a human colon cancer cell line were purchased from the American Type Culture Collection (ATCC). CAL-33 a tongue squamous cell carcinoma line was purchased from the Leibniz Institute DSMZ-German Collection of Microorganisms and Cell Cultures. BJ cells were cultured according to recommendations. HCT116 cells were cultured in McCoy’s 5a medium modified with 10% fetal bovine serum and 2 mM L-glutamine. HCC95 cells were cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum and 2 mM L-glutamine. HEK293T and CAL-33 cells were cultured in DMEM (ThermoFisher) supplemented with 10% fetal bovine serum (Hyclone). Cell culture media was purchased from ATCC and fetal bovine serum from GE Healthcare Hyclone (Logan, Utah, USA). Cells were routinely tested for mycoplasma contamination using the MycoAlert Mycoplasma Detection Kit (Lonza, Walkersville, MD, USA).
Stable transfected human embryonic kidney (HEK) 293 Flp-In T-Rex cells (ThermoFisher) were cultured in DMEM supplemented with 10% fetal bovine serum, 2 mM L-glutamine, antibiotics (100 units/ml penicillin, 0.1 mg/ml streptomycin), and in the presence of the selection with 100 μg/ml hygromycin and 15 μg/ml blasticidin. HA-tagged DCNL1 expression vectors [pcDNA5-FRT/TO+HA-DCNL1 or pcDNA5-FRT/TO+HA-DCNL1(D211A, A235R, D241)] were stably transfected into HEK293 T-Rex using Invitrogen’s Flp-In T-Rex system according to the manufacturer’s instructions. Protein expression was induced for the times indicated using 1 μg/ml doxycycline.
Sequence validated, c-terminally FLAG-HA tagged UBE2M34, was transfected with TransIT-293 transfection reagent (Mirus Bio) in combination with viral helper constructs (VSVG, TAT1B, MGPM2, CMV-Rev1B). Viral particles were harvested and a stable cell line was created by infection of HEK293T cells and subsequent selection with puromycin (Life technologies).
SMARTvector lentiviral shRNA was obtained from Dharmacon (Source Clone ID:V3SVHS00_5390398; microRNA sequence TAGTCTGCGCTTACCTTAC). Lentivirus was transduced into HCC95 and a stable clone selected according to the manufactures recommendations.
Soft agar colony formation assays were conducted according to previously described protocols55. Colonies were visualized using a nitroblue tetrazolium chloride solution and imaged using a Nikon DSLR camera and light box. Colonies were defined as groupings of greater than 20 cells and counted manually. Shown are representative well images from individual experiments that were individually repeated at least three times.
Western Blot Analysis
Exponentially growing cells were plated in 6-well plates at 0.4 × 106 cells/well in 2 ml of media and incubated overnight at 37 °C in a humidified 5% CO2 incubator. 24 and 48 hrs after plating, the media was aspirated and replenished with 2 ml fresh media containing either 4 μL of DMSO or a 500× compound DMSO stock solution. The cells were harvested after 72 hrs via trypsinization, thoroughly washed with PBS, pelleted, flash frozen in liquid N2, and stored at −80 °C. Cell pellets were thawed on ice and lysed by harvested resuspension in 30–40 μL of lysis buffer [50 mM Tris, 150 mM NaCl, 0.5% NP-40, 0.1% SDS, 6.5 M Urea, 2 mM 1,10-orthophenanthroline, 1× Halt Protease and Phosphatase inhibitor cocktail (ThermoFisher), 0.25 kU Universal Nuclease (ThermoFisher), pH 7.5]. Cell suspensions were incubated on ice for 25 minutes with occasional mixing by pipetting up and down. Lysates were cleared by centrifugation at 13,000 rpm for 20 minutes and the supernatant collected. The protein concentration of total cell lysate was determined by BCA assay (Pierce) using BSA as a control. Cell lysates were diluted into 2× SDS-PAGE sample buffer such that 25 μg of total protein was loaded per well. Samples were heated at 95 °C for 2 minutes, briefly cleared by pulse centrifugation, separated on 4–12% NuPAGE gels (Invitrogen), and transferred to PVDF membranes (BIO-RAD) at 100 V for 90 minutes at 4 °C. Membranes were blocked for 1 hour in blocking buffer consisting of 1× TBS, 0.1% Tween-20, and 5% Blotting grade non-fat dry milk (BIO-RAD). Primary antibodies were prepared in blocking buffer and incubated with membranes overnight at 4 °C with rocking, followed by extensive washing in 1× TBS, 0.1% Tween-20. Secondary antibodies were prepared in blocking buffer according to the manufactures recommendations and incubated with membranes for 1 hour at room temperature. After extensive washing, membranes were developed with SuperSignal West Pico Chemiluminescent substrate (ThermoFisher) and developed by film exposure (HyBlot CL, Denville scientific). Shown are representative results from one experiment that was repeated independently three times.
Whole Proteome Profiling by Tandem Mass Tagging (TMT) and Mass Spectrometry
The experiment was performed with a previously published protocol with slight modification56,57. Briefly, proteins were extracted from cell pellets and digested with LysC (Wako) and trypsin (Promega). The resulting peptides were desalted by C18 cartridges (Harvard Apparatus) and chemically labeled with 10-plex TMT reagents (Thermo Fisher). The labeled samples were mixed equally, desalted and fractionated by offline basic pH reversed-phase liquid chromatography (RPLC, pH 8.0, XBridge C18 column, 4.6 mm × 25 cm, 3.5 μm particle size, Waters). The fractions were then analyzed by acidic pH RPLC-MS/MS analysis (75 μm × ~40 cm, 1.9 μm C18 resin from Dr. Maisch GmbH, Q-Exactive HF from Thermo Fisher).
Peptides/protein identification by MS/MS raw data was carried out by a newly developed tag-based hybrid search engine JUMP, which combines pattern matching and de novo sequencing to score putative peptides for high sensitivity58. The data was searched against the UniProt human database concatenated with a reversed decoy database to evaluate false discovery rate, and then filtered by mass accuracy and matching scores to reduce false discovery rate to ~1%. The quantitative analysis was processed by the JUMP software suite as described56 (Supplementary Dataset 8).
TMT Interaction Proteomics Data Analysis
Mass spectra were processed using a Sequest-based in-house software pipeline59. Searches were performed against all entries from the human UniProt database (March 11, 2014) including all protein sequences in reverse order. Precursor tolerance was set to 50 ppm, while product ion tolerance was set to 1.0 Da. TMT tags on lysines and N-termini (+229.163 Da) and carbamidomethylation of cysteine residues (+57.021 Da) were set as static modifications, while oxidation of methionine residues (+15.995 Da) was set as variable modification. Peptide- spectrum matches (PSMs) were adjusted to a 2% false discovery rate using linear discriminant analysis59. For each peptide, a total minimum signal to noise value of 100 was required60. Change in protein abundance was determined from reporter ion intensities averaged across all peptides for a given protein. Data analyses for TMT-IP were carried out using the R statistical package (Ver. 3.2.3). TMT channel intensities were normalized by bait peptide intensities, and then quantile normalized. The data were log-transformed then FDR adjusted p-values were determined using an empirical Bayes approach. Differential interactors were determined using a fold-change threshold of 1.5 and an FDR threshold of 0.05 (Supplementary Dataset 6).
IP-MS to Examine N-terminal Acetylation of UBE2M
UBE2M FLAG-HA cells were treated for 24 hours with 10 μM of NAcM-COV dissolved in DMSO or DMSO alone. Cells were lysed in 50 mM Tris pH 7.5, 150 mM NaCl, 0.5% NP40 with protease inhibitors (Roche) to generate whole cell lysates. Clarified lysates were immunoprecipitated with anti-FLAG magnetic beads (Sigma). Complexes were washed 4× with lysis buffer and 2× with PBS and eluted with FLAG peptide at room temperature. Eluted proteins were reduced using DTT and alkylated with iodoacetamide followed by TCA precipitation. TCA precipitated proteins were digested with Glu-C (ThermoFisher) then cleaned up on C-18 stage tips. Eluted peptides were resuspended in 5% formic acid and 5% acetonitrile. Data were collected using a Q Exactive mass spectrometer (ThermoFisher, San Jose, CA) coupled with a Famos autosampler (LC Packings) and an Accela600 liquid chromatography (LC) pump (ThermoFisher). Peptides were separated on a ~18 cm 100 μm inner diameter column. For each analysis, we loaded ~1 μg onto the column. Peptides were separated using a 70 min gradient of 5 to 29% acetonitrile in 0.125% formic acid with a flow rate of ~300 nL/min. For the MS1 scan resolution was set to 70,000 with an automatic gain control (AGC) target 1 × 105, and maximum injection time 250 ms. We selected the top twenty precursors for HCD MS2 analysis with the following parameters: resolution 17,500, AGC 1 × 105, maximum injection time 60 ms, isolation window 2 Th, normalized collision energy (NCE) 25, and centroid spectrum data type. In addition, unassigned and singly charged species were excluded from MS2 analysis and dynamic exclusion was set to automatic.
Data Availability Statement
Structural data have been deposited in the Protein DataBank (PDB) with coordinate accession numbers 5V83.pdb (DCN1-NAcM-HIT), 5V86.pdb (DCN1-NAcM-OPT), 5V88.pdb (DCN1:NAcM-COV), 5V89.pdb (DCN4PONY:CUL1WHB). All other data generated or analyzed during this study are included in this published article (and its supplementary information files) or are available from the corresponding author on reasonable request.
Supplementary Material
Acknowledgments
B.A.S., ALSAC, HHMI, and NIH R37GM069530, P30CA021765; J.T.H., NIH F32GM113310; J.W.H., NIH AG011085; J.A.P., NIH DK098285; J.P., NIH GM114260; SJCRH Proteomics Facility, NIH P30CA021765; American Lebanese Syrian Associated Charities. We acknowledge the High Throughput Biosciences Center, Medicinal Chemistry Center, Compound Management, and High Throughput Analytical Chemistry Centers in Chemical Biology and Therapeutics; Hartwell Center for use of their personnel and facilities. We thank J. Lee (Harvard Medical School) for assistance with TMT statistics, E.R. Watson (St. Jude Children’s Research Hospital) for purified hNatC enzyme, J. Earl (St. Jude Children’s Research Hospital) for photography, R. Marmorstein (Perelman School of Medicine, University of Pennsylvania) and T. Arnesen (University of Bergen) for NAT constructs, and staff at the BL8.2.1 and 22-ID beamlines at the Advanced Light Source and Advanced Photon Source.
Footnotes
Author contributions: D.C.S., J.T.H., B.S., J.W.H., B.A.S., and R.K.G. designed the research project and analyzed data; D.C.S., J.K.M., S.C.C., A.N.G., and J.M. contributed to assay development; D.C.S., V.O.S., and J.K.M. performed X-ray crystallography; J.T.H., D.B., and H.S.K. designed and performed compound optimization and syntheses; A.A.S. and T.C. contributed to the HTS campaign; D.C.S. performed in vitro neddylation experiments and western blotting; M.C. and D.Y.R. generated cell lines; S.-S.O. and M.C. generated the DCN1 shRNA line; I.R.K. and A.F.A. generated DCN1 Flp-In cell lines; Y.C. performed in vitro ADME-tox studies to aid in compound optimization; M.C., G.H., and J.L. performed cell-based experiments; D.Y.R., J.A.P., and J.R.C. performed cell-based AP-MS experiments; V.P., X.W., and J.P. performed TMT analysis on proteome samples; all authors contributed to specific parts of the manuscript, and D.C.S., J.T.H., B.A.S. and R.K.G. assume responsibility for the manuscript in its entirety.
Competing Financial Interest
The authors have filed a provisional application with the US Patent and Trademark Office on this work.
References
- 1.Varland S, Osberg C, Arnesen T. N-terminal modifications of cellular proteins: The enzymes involved, their substrate specificities and biological effects. Proteomics. 2015;15:2385–401. doi: 10.1002/pmic.201400619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Drazic A, Myklebust LM, Ree R, Arnesen T. The world of protein acetylation. Biochim Biophys Acta. 2016;1864:1372–401. doi: 10.1016/j.bbapap.2016.06.007. [DOI] [PubMed] [Google Scholar]
- 3.Aksnes H, Drazic A, Marie M, Arnesen T. First Things First: Vital Protein Marks by N-Terminal Acetyltransferases. Trends Biochem Sci. 2016;41:746–60. doi: 10.1016/j.tibs.2016.07.005. [DOI] [PubMed] [Google Scholar]
- 4.Hwang CS, Shemorry A, Varshavsky A. N-terminal acetylation of cellular proteins creates specific degradation signals. Science. 2010;327:973–7. doi: 10.1126/science.1183147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van Welsem T, et al. Synthetic lethal screens identify gene silencing processes in yeast and implicate the acetylated amino terminus of Sir3 in recognition of the nucleosome core. Mol Cell Biol. 2008;28:3861–72. doi: 10.1128/MCB.02050-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yi CH, et al. Metabolic regulation of protein N-alpha-acetylation by Bcl-xL promotes cell survival. Cell. 2011;146:607–20. doi: 10.1016/j.cell.2011.06.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Forte GM, Pool MR, Stirling CJ. N-terminal acetylation inhibits protein targeting to the endoplasmic reticulum. PLoS Biol. 2011;9:e1001073. doi: 10.1371/journal.pbio.1001073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Esmailpour T, et al. A splice donor mutation in NAA10 results in the dysregulation of the retinoic acid signalling pathway and causes Lenz microphthalmia syndrome. J Med Genet. 2014;51:185–96. doi: 10.1136/jmedgenet-2013-101660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rope AF, et al. Using VAAST to identify an X-linked disorder resulting in lethality in male infants due to N-terminal acetyltransferase deficiency. Am J Hum Genet. 2011;89:28–43. doi: 10.1016/j.ajhg.2011.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scott DC, Monda JK, Bennett EJ, Harper JW, Schulman BA. N-terminal acetylation acts as an avidity enhancer within an interconnected multiprotein complex. Science. 2011;334:674–8. doi: 10.1126/science.1209307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang Z, Kulkarni K, Hanrahan SJ, Thompson AJ, Barford D. The APC/C subunit Cdc16/Cut9 is a contiguous tetratricopeptide repeat superhelix with a homo-dimer interface similar to Cdc27. EMBO J. 2010;29:3733–44. doi: 10.1038/emboj.2010.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nguyen TV, et al. Glutamine Triggers Acetylation-Dependent Degradation of Glutamine Synthetase via the Thalidomide Receptor Cereblon. Mol Cell. 2016;61:809–20. doi: 10.1016/j.molcel.2016.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marmorstein R, Zhou MM. Writers readers of histone acetylation structure, mechanism, and inhibition. Cold Spring Harb Perspect Biol. 2014;6:a018762. doi: 10.1101/cshperspect.a018762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Filippakopoulos P, et al. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–73. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nicodeme E, et al. Suppression of inflammation by a synthetic histone mimic. Nature. 2010;468:1119–23. doi: 10.1038/nature09589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kurz T, et al. The conserved protein DCN-1/Dcn1p is required for cullin neddylation in C. elegans and S. cerevisiae. Nature. 2005;435:1257–61. doi: 10.1038/nature03662. [DOI] [PubMed] [Google Scholar]
- 17.Kim AY, et al. SCCRO (DCUN1D1) is an essential component of the E3 complex for neddylation. J Biol Chem. 2008;283:33211–20. doi: 10.1074/jbc.M804440200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scott DC, et al. A dual E3 mechanism for Rub1 ligation to Cdc53. Mol Cell. 2010;39:784–96. doi: 10.1016/j.molcel.2010.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Monda JK, et al. Structural conservation of distinctive N-terminal acetylation-dependent interactions across a family of mammalian NEDD8 ligation enzymes. Structure. 2013;21:42–53. doi: 10.1016/j.str.2012.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scott DC, et al. Structure of a RING E3 trapped in action reveals ligation mechanism for the ubiquitin-like protein NEDD8. Cell. 2014;157:1671–84. doi: 10.1016/j.cell.2014.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kurz T, et al. Dcn1 functions as a scaffold-type E3 ligase for cullin neddylation. Mol Cell. 2008;29:23–35. doi: 10.1016/j.molcel.2007.12.012. [DOI] [PubMed] [Google Scholar]
- 22.Keuss MJ, et al. Characterization of the mammalian family of DCN-type NEDD8 E3 ligases. J Cell Sci. 2016;129:1441–54. doi: 10.1242/jcs.181784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fu W, et al. Squamous Cell Carcinoma-related Oncogene (SCCRO) Family Members Regulate Cell Growth and Proliferation through Their Cooperative and Antagonistic Effects on Cullin Neddylation. J Biol Chem. 2016;291:6200–17. doi: 10.1074/jbc.M115.692756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sarkaria I, et al. Squamous cell carcinoma related oncogene/DCUN1D1 is highly conserved and activated by amplification in squamous cell carcinomas. Cancer Res. 2006;66:9437–44. doi: 10.1158/0008-5472.CAN-06-2074. [DOI] [PubMed] [Google Scholar]
- 25.Sarkaria IS, et al. SCCRO expression correlates with invasive progression in bronchioloalveolar carcinoma. Ann Thorac Surg. 2004;78:1734–41. doi: 10.1016/j.athoracsur.2004.05.056. [DOI] [PubMed] [Google Scholar]
- 26.Shelat AA, Guy RK. Scaffold composition and biological relevance of screening libraries. Nat Chem Biol. 2007;3:442–6. doi: 10.1038/nchembio0807-442. [DOI] [PubMed] [Google Scholar]
- 27.Baell JB, Holloway GA. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem. 2010;53:2719–40. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- 28.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46:3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 29.Finlay MR, et al. Discovery of a potent and selective EGFR inhibitor (AZD9291) of both sensitizing and T790M resistance mutations that spares the wild type form of the receptor. J Med Chem. 2014;57:8249–67. doi: 10.1021/jm500973a. [DOI] [PubMed] [Google Scholar]
- 30.Singh J, Petter RC, Baillie TA, Whitty A. The resurgence of covalent drugs. Nat Rev Drug Discov. 2011;10:307–17. doi: 10.1038/nrd3410. [DOI] [PubMed] [Google Scholar]
- 31.Liszczak G, et al. Molecular basis for N-terminal acetylation by the heterodimeric NatA complex. Nat Struct Mol Biol. 2013;20:1098–105. doi: 10.1038/nsmb.2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stove SI, et al. Crystal Structure of the Golgi-Associated Human Nalpha-Acetyltransferase 60 Reveals the Molecular Determinants for Substrate-Specific Acetylation. Structure. 2016;24:1044–56. doi: 10.1016/j.str.2016.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Van Damme P, et al. Proteome-derived peptide libraries allow detailed analysis of the substrate specificities of N(alpha)-acetyltransferases and point to hNaa10p as the post-translational actin N(alpha)-acetyltransferase. Mol Cell Proteomics. 2011;10 doi: 10.1074/mcp.M110.004580. M110 004580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huttlin EL, et al. The BioPlex Network: A Systematic Exploration of the Human Interactome. Cell. 2015;162:425–40. doi: 10.1016/j.cell.2015.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang G, Kaufman AJ, Ramanathan Y, Singh B. SCCRO (DCUN1D1) promotes nuclear translocation and assembly of the neddylation E3 complex. J Biol Chem. 2011;286:10297–304. doi: 10.1074/jbc.M110.203729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Meyer-Schaller N, et al. The human Dcn1-like protein DCNL3 promotes Cul3 neddylation at membranes. Proc Natl Acad Sci U S A. 2009;106:12365–70. doi: 10.1073/pnas.0812528106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lydeard JR, Schulman BA, Harper JW. Building and remodelling Cullin-RING E3 ubiquitin ligases. EMBO Rep. 2013;14:1050–61. doi: 10.1038/embor.2013.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Deshaies RJ, Emberley ED, Saha A. Control of cullin-ring ubiquitin ligase activity by nedd8. Subcell Biochem. 2010;54:41–56. doi: 10.1007/978-1-4419-6676-6_4. [DOI] [PubMed] [Google Scholar]
- 39.Soucy TA, et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature. 2009;458:732–6. doi: 10.1038/nature07884. [DOI] [PubMed] [Google Scholar]
- 40.Serafimova IM, et al. Reversible targeting of noncatalytic cysteines with chemically tuned electrophiles. Nat Chem Biol. 2012;8:471–6. doi: 10.1038/nchembio.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Genschik P, Sumara I, Lechner E. The emerging family of CULLIN3-RING ubiquitin ligases (CRL3s) cellular functions and disease implications. EMBO J. 2013;32:2307–20. doi: 10.1038/emboj.2013.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Foyn H, et al. Design, synthesis, and kinetic characterization of protein N-terminal acetyltransferase inhibitors. ACS Chem Biol. 2013;8:1121–7. doi: 10.1021/cb400136s. [DOI] [PubMed] [Google Scholar]
- 43.Shemorry A, Hwang CS, Varshavsky A. Control of protein quality and stoichiometries by N-terminal acetylation and the N-end rule pathway. Mol Cell. 2013;50:540–51. doi: 10.1016/j.molcel.2013.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Polevoda B, Cardillo TS, Doyle TC, Bedi GS, Sherman F. Nat3p and Mdm20p are required for function of yeast NatB Nalpha-terminal acetyltransferase and of actin and tropomyosin. J Biol Chem. 2003;278:30686–97. doi: 10.1074/jbc.M304690200. [DOI] [PubMed] [Google Scholar]
- 45.Bloes DA, Kretschmer D, Peschel A. Enemy attraction bacterial agonists for leukocyte chemotaxis receptors. Nat Rev Microbiol. 2015;13:95–104. doi: 10.1038/nrmicro3390. [DOI] [PubMed] [Google Scholar]
- 46.Boddey JA, Cowman AF. Plasmodium nesting remaking the erythrocyte from the inside out. Annu Rev Microbiol. 2013;67:243–69. doi: 10.1146/annurev-micro-092412-155730. [DOI] [PubMed] [Google Scholar]
Online Methods Only References
- 47.Scott DC, et al. Two Distinct Types of E3 Ligases Work in Unison to Regulate Substrate Ubiquitylation. Cell. 2016;166:1198–1214 e24. doi: 10.1016/j.cell.2016.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huang DT, et al. A unique E1–E2 interaction required for optimal conjugation of the ubiquitin-like protein NEDD8. Nat Struct Mol Biol. 2004;11:927–35. doi: 10.1038/nsmb826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guiguemde WA, et al. Chemical genetics of Plasmodium falciparum. Nature. 2010;465:311–5. doi: 10.1038/nature09099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Min J, et al. Optimization of a Novel Series of Ataxia-Telangiectasia Mutated Kinase Inhibitors as Potential Radiosensitizing Agents. J Med Chem. 2016;59:559–77. doi: 10.1021/acs.jmedchem.5b01092. [DOI] [PubMed] [Google Scholar]
- 51.McCoy AJ, et al. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Emsley P, Cowtan K. Coot model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–32. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 53.Schuttelkopf AW, van Aalten DM. PRODRG a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr D Biol Crystallogr. 2004;60:1355–63. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- 54.Adams PD, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–21. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Borowicz S, et al. The soft agar colony formation assay. J Vis Exp. 2014:e51998. doi: 10.3791/51998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Niu M, et al. Extensive Peptide Fractionation and y1 Ion-Based Interference Detection Method for Enabling Accurate Quantification by Isobaric Labeling and Mass Spectrometry. Anal Chem. 2017;89:2956–2963. doi: 10.1021/acs.analchem.6b04415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bai B, et al. Deep Profiling of Proteome and Phosphoproteome by Isobaric Labeling, Extensive Liquid Chromatography, and Mass Spectrometry. Methods Enzymol. 2017;585:377–395. doi: 10.1016/bs.mie.2016.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang X, et al. JUMP: a tag-based database search tool for peptide identification with high sensitivity and accuracy. Mol Cell Proteomics. 2014;13:3663–73. doi: 10.1074/mcp.O114.039586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huttlin EL, et al. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell. 2010;143:1174–89. doi: 10.1016/j.cell.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Paulo JA, O’Connell JD, Gygi SP. A Triple Knockout (TKO) Proteomics Standard for Diagnosing Ion Interference in Isobaric Labeling Experiments. J Am Soc Mass Spectrom. 2016;27:1620–5. doi: 10.1007/s13361-016-1434-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Structural data have been deposited in the Protein DataBank (PDB) with coordinate accession numbers 5V83.pdb (DCN1-NAcM-HIT), 5V86.pdb (DCN1-NAcM-OPT), 5V88.pdb (DCN1:NAcM-COV), 5V89.pdb (DCN4PONY:CUL1WHB). All other data generated or analyzed during this study are included in this published article (and its supplementary information files) or are available from the corresponding author on reasonable request.