

Abstract
Adenosine monophosphate (AMP)-activated protein kinase (AMPK), a highly conserved energy sensor, has a crucial role in cardiovascular, neurodegenerative and inflammatory diseases, as well as in cancer and metabolic disorders. Accumulating studies have demonstrated that AMPK activation enhances paracellular junctions, nutrient transporters, autophagy and apoptosis, and suppresses inflammation and carcinogenesis in the intestine, indicating an essential role of AMPK in intestinal health. AMPK inactivation is an aetiological factor in intestinal dysfunctions. This review summarizes the favourable outcomes of AMPK activation on intestinal health, and discusses AMPK as a potential therapeutic target for intestinal diseases.
Keywords: AMPK, absorption, barrier function, colorectal cancer, intestinal health, intestinal inflammation
1. Introduction
The intestine is the longest and largest vital epithelial organ. Its major functions are absorbing nutrients from food and establishing selectively permeable barriers against the external environment [1]. To execute these functions, the intestinal epithelium needs to form a barrier, which depends on a well-balanced cellular homeostasis, orchestrated by a delicate interaction and balance among differentiation, self-renewal, proliferation and the intestinal ecosystem [2]. Disruption of the balance in intestinal homeostasis causes enteritis and colitis, leading to malnutrition, diarrhoea, weight loss, constipation and fatigue [3,4]. Losses in intestinal homeostasis are associated with a broad range of pathological changes including metabolic disorders, inflammatory and autoimmune diseases, and cancers [5,6].
Adenosine monophosphate (AMP)-activated protein kinase (AMPK), a serine/threonine kinase, is evolutionarily conserved from yeast to mammals. As an energy sensor, AMPK is activated by upstream enzymes when the cellular ratio of AMP to adenosine triphosphate (ATP) is elevated due to nutrient deprivation [7]. After activation, AMPK phosphorylates downstream substrates to promote catabolism and impede anabolism, leading to ATP production and energy restoration [8,9]. AMPK activity can be altered owing to numerous physiological factors, such as hormones, cytokines and dietary nutrients, as well as pathological conditions such as obesity, chronic inflammation and type II diabetes [10]. Thus, AMPK activation can act as a therapeutic agent to treat various metabolic diseases [11,12]. Furthermore, the function of AMPK on the metabolism of liver and skeletal muscle has been well studied and documented [13,14]. AMPK also has a crucial role in regulating cellular development, such as adipogenesis [15], myogenesis [16] and osteogenesis [17].
Accumulating evidence supports the beneficial effects of AMPK on gut health, such as enhancing intestinal absorption [18], improving barrier function [19], suppressing colorectal carcinogenesis [20], and reducing intestinal inflammation [21] and metabolic-related disease [22]. Conversely, AMPK is inhibited under diabetic and obese conditions, which is correlated with impaired intestinal barrier function [23]. The inhibition of AMPK under pathological and physiological states has been comprehensively discussed in a previous review [10]. In this review, we summarize the regulatory role of AMPK in intestinal health and diseases, which links the important energy sensor to the maintenance of intestinal homeostasis.
2. AMPK and its regulators in the intestine
Intestinal mucosa contains the epithelial layer, the lamina propria, consisting mainly of connective tissue, and the muscularis mucosae layer, made of smooth muscle. AMPK is mainly located at the apical part of the villus epithelium in human adult jejunum [24]. AMPK is a heterotrimer, consisting of α catalytic subunits, and β and γ regulatory subunits. AMPK is activated by phosphorylation at Thr 172 of the α subunit by upstream kinases, such as liver kinase B1 (LKB1) and calmodulin-dependent protein kinase kinase (CaMKK) [25,26]. The binding of AMP to the γ subunit causes conformational changes which facilitate its phosphorylation by AMPK activators, such as LKB1 [27]. The α and β subunits each have two isoforms (α1 and α2, and β1 and β2), while the γ subunit has three isoforms (γ1, 2 and 3) [7]. The different heterotrimeric complexes display tissue specificity [24]. The AMPK α1/β2/γ1 complex is more abundant in differentiated intestinal epithelial cells [24]. Our recent study found that AMPK α1 deletion in intestinal epithelium suppresses intestinal differentiation in mouse jejunum with reduced mucosal height and villin content [19]. No change of epithelial architecture occurs in AMPKα2-deleted mice [28], which might be due to the predominance of the α1 subunit in intestinal epithelium. The layers of connective tissue and smooth muscle tend to be thinner [28], which is probably due to the expression of the α2 subunit in non-epithelial cells. In addition, α1 is expressed during the initial stages in myogenesis, while α2 becomes dominant in differentiated myogenic cells [29], illustrating the tissue-specific expression of AMPK α isoforms.
AMPK is linked to the beneficial effects of nutraceutical or pharmacological compounds in intestinal health (table 1). 5-Aminoimidazole-4-carboxamide ribonucleotide (AICAR) is commonly used as a pharmacological activator for AMPK. It triggers AMPK activation through conversion into ZMP (Z refers to imidazole), an AMP analogue mimicking the AMP effect [62]. As expected, AMPK is activated in Caco-2 cells in response to AICAR treatment [24]. It has been shown that AICAR promotes intestinal glucose transportation [63] and barrier function [19], and inhibits infiltration of inflammatory cells [33]. Another pharmacological compound, metformin, a dimethyl-biguanide, is a common anti-diabetic drug [64,65]. Metformin indirectly activates AMPK by inhibiting mitochondrial complex I in the respiratory chain to increase the AMP : ATP ratio [66]. In response to metformin, the phosphorylation of AMPK and its specific substrate acetyl-CoA carboxylase (ACC) increased 10-fold and 5-fold, respectively, in Caco-2 cells [24]. Metformin enhances intestinal glucose transportation [46] and inhibits inflammatory cytokines [67] and colitis [21]. Furthermore, microbial metabolite butyrate and other extracts from plants improve intestinal barrier function [38], suppress peptide transportation [42] and induce apoptosis in Caco-2 cells, associated with enhanced AMPK signalling. Though the mechanisms responsible for AMPK activation remain poorly defined, these plant-origin metabolites might inhibit mitochondrial function, including complex I in the respiratory chain and F1 ATP synthase, to increase the AMP : ATP ratio [63,68].
Table 1.
Compounds targeting AMPK pathways in the intestine.
| experimental setting | compounds | functions | references |
|---|---|---|---|
| HCT116; HT-29; LoVo cells | adiponectin | inhibits cyclin E and cell growth; promotes p21, p27, glucose utilization and fatty acid oxidation | [30,31] |
| mice jejunum | AICAR | inhibits SGLT1; facilitates glucose transportation by GLUT2 | [18] |
| Caco-2 cells | AICAR | inhibits PEPT1 | [32] |
| Caco-2 cells | AICAR | promotes ZO-1 assembly and E-cadherin; enhances barrier function; inhibits intestinal permeability | [19] |
| mice colon | AICAR | promotes goblet cells; inhibits infiltration of inflammatory cells; downregulates macrophages | [33] |
| Caco-2 cells; mice jejunum; human colonic mucosa | AICAR; metformin | inhibits chloride secretion | [34] |
| Caco-2 cells | alcohol | inhibits barrier function; disrupts cytoskeleton integrity | [35,36] |
| HCT116; SW480; LOVO cells; mice colon | berberine | increases mTOR activity and p53 phosphorylation | [37] |
| Caco-2 cells | butyrate | enhances barrier function; facilitates ZO-1/Occludin redistribution | [38] |
| T84 cells; mice colon | chitosan oligosaccharide | promotes tight junction assembly; inhibits NF-κB transcriptional activity; prevents the development of aberrant crypt foci | [34,39] |
| HT-29 cells | curcumin | induces COX-2 | [40] |
| HT-29 cells | combined 5-fluorouracil and genistein | induces COX-2 | [41] |
| Caco-2 cells | Compound C | promotes PEPT1 | [42] |
| HT-29 cells | EGCG | induces COX-2 | [20] |
| mice jejunum | leptin | promotes GLUT2 and GLUT5; decreases SGLT1 | [28] |
| mice jejunum and colon | high-fat diet | induces PPAR; triggers β-catenin activity; Increases intestinal tumorigenesis and villus length | [43,44] |
| pig jejunum and ileum | lipopolysaccharide | decreases oleic acid, glutamine and glucose in enterocytes | [45] |
| IL-10−/− mice colon | metformin | inhibits inflammatory cytokines and DSS-induced acute colitis | [21] |
| COLO205 cells | metformin | inhibits IL-8 expression and NF-κB transcriptional activity | [21] |
| rat small intestine | metformin | promotes GLUT5 expression | [46] |
| mice jejunum | metformin | facilitates localization of GLUT2 to apical membrane | [18] |
| HCT116 xenografts | metformin | inhibits tumour growth lacking P53 | [47] |
| rat caecum | metformin | increases short chain fatty acid-producing bacteria | [48,49] |
| rat duodenum | metformin | triggers GLP-1 from enteroendocrine L-cells; activates AMPK in hepatocytes in a non-autonomous manner | [50] |
| Caco-2 cells | MIYAIRI 588 | promotes ZO-1 | [51] |
| Pig jejunum | n-3 polyunsaturated fatty acids | promotes glucose uptake | [52,53] |
| db/db mice colon | pitavastatin | inhibits colonic preneoplastic lesions | [54,55] |
| mice colon | phenformin | inhibits chloride secretion | [54,56] |
| Caco-2 cells | propolis polyphenol | promotes tight junctions; enhances the barrier function | [56,57] |
| HT-29 cells | plumbagin | induces apoptosis via p53 | [57,58] |
| HT-29 cells | quercetin | induces apoptosis via p53 | [58,59] |
| HT-29 cells | selenium | induces COX-2 | [59,60] |
| Caco-2 cells | theaflavins | inhibits PEPT1 | [42,60] |
| HT-29 cells | 20(S)-ginsenoside Rg3 | induces apoptosis via p53 | [42,61] |
| pig jejunum and ileum | α-ketoglutarate | stimulates oxidation of energy substrates | [45,61] |
3. AMPK and intestinal absorption
The intestinal epithelium is composed of microvilli, villi and mucosal folds. To generate net influx, nutrients need to pass through the apical membrane of intestinal cells. Nutrients entering intestinal epithelial cells are either used by these cells or pass through the basolateral membrane of intestine cells into the circulatory system. The apical or basolateral transportation can be energy-dependent (active transport with a carrier) or independent (passive transport). The passive transportation depends on a concentration gradient. However, most of the sugars, amino acids, vitamins and minerals are transported by carriers or their respective transporters [69]. Thus, the functional regulation of intestinal transporters is critical for nutrient transportation.
Glucose is one of the most important nutrients in our body. Intestinal glucose absorption is mediated by glucose transporters, including glucose transporter 2 (GLUT2), glucose transporter 5 (GLUT5) and sodium–glucose transporter 1 (SGLT1) [70]. The temporal and quantitative regulation of glucose transporters governs glucose flux in and out of the intestine [71]. AMPK regulates glucose uptake not only in the heart [72], skeletal muscle [73], liver [74] and hippocampal neurons [75], but also in the gut. AMPKα2 knockout (KO) decreases protein levels of GLUT2 and GLUT5, while it increases protein levels of SGLT1 in the jejunum [28] (figure 1). Metformin increases GLUT5 expression [46], leading to translocation of GLUT2 to the apical membrane [18], which enhances glucose absorption in the gut [76,77]. Similarly, AICAR inhibits SGLT1 and promotes GLUT2 translocation in mice jejunal mucosa [18]. AMPK is upregulated in rats and pigs by feeding n-3 polyunsaturated fatty acids, which thereafter improve glucose uptake [52,53].
Figure 1.
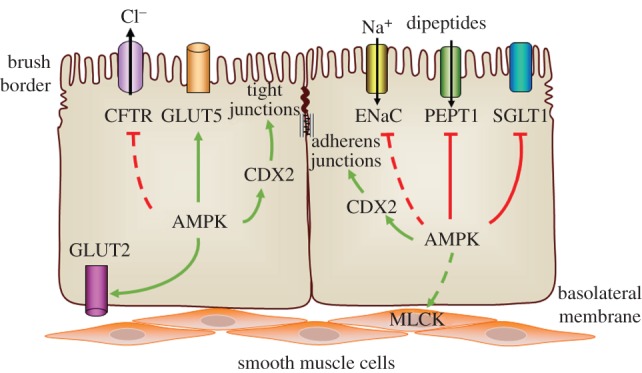
AMPK promotes intestinal absorption and barrier function. AMPK regulates glucose absorption via enhancing the function of glucose transporter (GLUT)2 and GLUT5, while inhibiting sodium–glucose transporter 1 (SGLT1). AMPK mediates ion absorption through possible inhibition of cystic fibrosis transmembrane regulator (CFTR) and epithelial Na+ channel (ENaC). Peptide transporter 1 (PEPT1) expression is attenuated by AMPK to reduce apical dipeptide uptake. In addition, AMPK may phosphorylate myosin light chain kinase (MLCK) to enhance vasodilatation and blood flow, further favouring intestinal absorption. Besides absorbing nutrients, the intestine also functions as a frontier barrier protecting the mucosal integrity. AMPK facilitates the establishment of paracellular junctions (tight junctions and adherens junctions) via caudal type homeobox 2 (CDX2), an intestinal transcription factor to upregulate intestinal differentiation. Green arrows indicate positive effects. Red lines indicate negative effects. Solid lines represent proven regulations, while dashed lines represent possible regulations.
Ion transporters in intestinal epithelium are critical in keeping ion homeostasis [78]. Ion imbalance leads to diarrhoea. AMPK plays an important role in maintaining this homeostasis. Loss of AMPKα1 enhances epithelial sodium (Na+) channel (ENaC) expression, which controls the reabsorption of Na+ [79] (figure 1). After blocking ubiquitin ligase or endocytosis, phenformin is unable to block ENaC activity, suggesting that ubiquitin ligase is crucial in mediating the effects of AMPK on ENaC ubiquitination [79]. By activating AMPK with AICAR and phenformin in lung epithelial cells, ENaC expression is downregulated, which may be derived from the adaptation to metabolic stress to limit energy dissipation [80]. Apart from ENaC, AMPK also inhibits chloride (Cl−) secretion [56], as indicated by the reduction of ion-transport proteins and the cystic fibrosis transmembrane regulator (CFTR) [81] (figure 1). The overstimulation of Cl− secretion by CFTR is the predominant aetiology for enterotoxigenic diarrhoea [82]. AICAR and metformin, which activate AMPK, can offset the increased Cl− efflux by cholera toxin in excised intestinal loops [34], thus preventing diarrhoea. These studies suggest strong clinical applications for AMPK as a potential pharmacological target for treating acute diarrhoeal disease.
Peptide transporters mediate amino acid absorption [83]. AICAR attenuates the expression of peptide transporter 1 (PEPT1) [32], while the AMPK inhibitor Compound C promotes peptide transportation [42] (figure 1). AICAR inhibits apical dipeptide uptake in Caco-2 cells on trans-well filters [32]. The negative correlation between peptide absorption and AMPK activation might be due to the energy-dependent process of peptide uptake, which is dependent on Na/K-ATPase [84]. Thus, peptide transportation is suppressed under nutrient deprivation, probably accompanied by AMPK-mediated signalling pathways.
Another explanation for absorptive dysfunction due to AMPK inhibition is associated with impaired peristaltic activity of visceral musculature. It has been shown that AMPKα mutation in Drosophila impairs the movement of food through the digestive tract, which results in smaller fat body cells, delayed metamorphosis and growth inhibition [85]. As mesenteric circulation is directly proportional to nutrient transportation out of the intestine [86], the regulation of capillary blood flow contributes to intestinal absorption. AMPK stimulates vasodilatation and blood flow by attenuating contraction of vascular smooth muscle [87], possibly due to phosphorylation of myosin light chain kinase (MLCK) [88] (figure 1).
4. AMPK and intestinal barrier function
Proper intestinal barrier function plays a critical role in our health. Besides absorbing nutrients and secreting fluid, the intestine also functions as a critical barrier maintaining mucosal integrity, which physically inhibits the penetration of harmful substances from the external environment [89]. Impaired barrier function increases intestinal permeability to cause a leaky gut, predisposing individuals to intestinal bowel disease [90], metabolic syndromes [91–94] and autoimmune disorders [95].
The major determinant of gut epithelial permeability is closure or opening of paracellular junctions between enterocyte intercellular spaces [89]. The gaps between adjacent cells are mechanically sealed by junctional complexes, including desmosomes, adherens junctions (AJs) and tight junctions (TJs) [96]. Tight junctions contribute to the selective paracellular permeability, while AJs are essential for TJ assembly [97]. Thus, intestinal barrier function depends on the performance of intestinal paracellular junctions, such as the establishment and reassembly of TJs, which is regulated by AMPK (figure 2).
Figure 2.
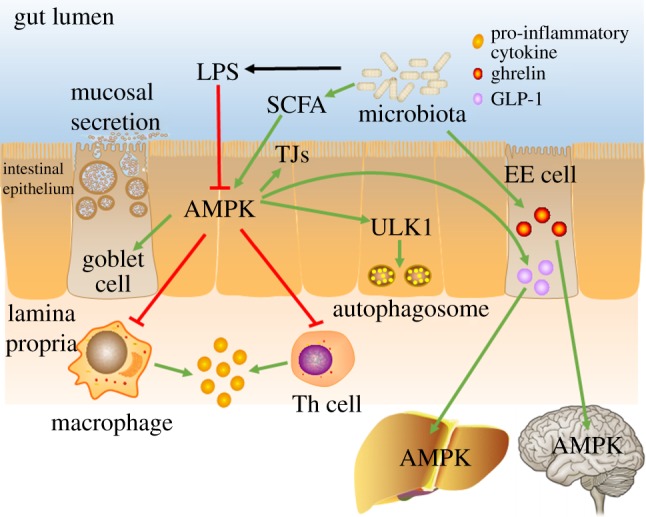
AMPK regulates intestinal inflammation and hormone secretion. AMPK suppresses intestinal inflammation through reducing pro-inflammatory cytokine production in macrophages, inhibiting the differentiation of T helper (Th) cells, promoting mucus secretion and enhancing autophagy, collaboratively. AMPK blocks the secretion of pro-inflammatory cytokines via inhibiting macrophage infiltration and differentiation of Th cells; AMPK triggers autophagy through activation of Unc-51-like autophagy activating kinase 1 (ULK1); AMPK increases goblet cells and associated mucus section, and enhances tight junctions (TJs) to strengthen intestinal barrier function. Gut microbiota and their metabolites such as short-chain fatty acids (SCFAs) regulate AMPK activation, exerting beneficial effects. Additionally, gut microbiota induces enteroendocrine (EE) cells to generate the gut hormone ghrelin, leading to AMPK activation in the hypothalamus to increase food intake. On the other hand, the microbiota upregulates glucagon like peptide 1 (GLP-1) production from EE cells, which augments AMPK phosphorylation in the liver, subsequently reducing hepatic glucose production. Green arrows indicate positive effects; red lines indicate negative effects.
In AMPK kinase-dead MDCK cells and Caco-2 cells, the formation of TJs (ZO-1) and establishment of transepithelial electric resistance (TEER) are delayed after calcium switch, while AICAR accelerates TJ reassembly [19,98]. Consistently, intestinal epithelium-specific deletion of AMPK α1 (AMPK VilCre) in mice enhanced intestinal permeability [19] (figure 1). Transmission electron microscopic observation further indicates that the ultrastructure of TJs is less compact in AMPK VilCre mice [19]. The paracellular junction is developed during intestinal differentiation [99], promoted by the intestinal transcription factor caudal type homeobox 2 (CDX2) [100]. AMPK promotes the expression of CDX2 and changes the CDX2 promoter-specific epigenetic modifications [19], providing a possible regulatory mechanism of AMPK on intestinal barrier function (figure 1). Interestingly, AMPK could be activated during TJ assembly via calcium switch, possibly due to the stimulated CaMKK (an AMPK activator) by an influx of calcium [98]. Furthermore, rapamycin rescues the delay of TJ assembly in AMPK kinase-dead cells, demonstrating that AMPK may, at least partially, mediate junction assembly via mammalian target of rapamycin (mTOR) signalling [98]. Additionally, AMPK profoundly promotes the formation of TJs in renal [101], mammary [102] and hepatic [103] epithelial cells.
Many environmental factors impact intestinal barrier function associated with alteration of AMPK. Chitosan oligosaccharide and polyphenol extracts from Propolis upregulates TJ assembly and enhances the barrier function associated with AMPK activation [39,57]. The consumption of alcohol, a potent metabolic stressor, diminishes cellular ATP production and increases intestinal permeability [35,36]. AMPK activators such as metformin and AICAR exert ameliorative effects on disrupted barrier function caused by bacterial [104] and viral pathogens [105], and pro-inflammatory cytokines [106]. Epithelial barriers are dysfunctional in AMPK VilCre mice [19], while metformin supplementation suppresses intestinal permeability, probably due to augmentation of epithelial differentiation [67].
Microbial metabolites, short-chain fatty acids (SCFAs), activate AMPK in colonocytes [38], associated with the enhanced TEER and redistribution of TJs [38]. Furthermore, butyrate protects against ethanol-induced intestinal barrier dysfunction, accompanied with AMPK activation [36]. Clostridium butyricum MIYAIRI 588, the butyrate-producing probiotic, enhances the activity of AMPK and strengthens TJs, resulting in mitigated gut permeability in non-alcoholic fatty liver disease [38,51]. Inhibition of AMPK either genetically or chemically abolishes the aforementioned therapeutic or prophylactic potential of SCFAs, demonstrating the modulatory role of AMPK in SCFAs’ enhanced barrier function [36].
5. AMPK and intestinal inflammation
The pathology of intestinal inflammation is mainly associated with immunological disorders [107]. Immune cells produce pro-inflammatory cytokines to defend against bacterial infections [108], triggering the activation of T cells and the recruitment of neutrophils [109]. Deficient regulatory T cells, excessive effector T cells and the overproduction of pro-inflammatory cytokines are all prone to inducing intestinal inflammation that exacerbates colitis [110,111]. Thus, the blocking of pro-inflammatory cytokines provides a therapeutic potential for inflammatory bowel disease (IBD), an autoimmune disease of the intestine [112].
Intestinal macrophages in the lamina propria are the primary source of pro-inflammatory cytokines [108]. The expression of inducible nitric oxide synthases (iNOSs) and tumour necrosis factor (TNF)α and phosphorylation of nuclear factor-kappa B (NF-κB) are inhibited in intestinal mucosal macrophages treated with AICAR [33]. In vitro, metformin suppresses TNFα-induced IL-8 expression and NF-κB inflammatory signalling [21], which facilitates T-cell differentiation [113]. AICAR suppresses the differentiation of Th1 and Th17 cells, possibly due to the downregulation of their transcriptional factors, T-bet and RORγt [33]. Lipopolysaccharides (LPSs), the major outer membrane component of Gram-negative bacteria, contribute to the inflammatory process of IBD [114]. LPS administration decreases phosphorylation of AMPK in pig jejunum and ileum (figure 2), which is ameliorated by dietary supplementation with α-ketoglutarate [45]. In addition, intraperitoneal injection with AICAR downregulates colonic macrophage activation in LPS-induced or 2,4,6-trinitrobenzenesulfonic acid (TNBS)-induced colitis [33]. Thus, AMPK may provide an intervening target to ameliorate LPS-induced gut damage.
The mucus layer produced by goblet cells provides an additional protective barrier to the gut epithelium. The defective formation of the mucus layer increases mouse susceptibility to colitis [115], and colitis reduces the size and number of goblet cells in human gut [116]. AICAR supplementation augments goblet cell differentiation and attenuates the infiltration of inflammatory cells in TNBS-induced acute colitis [33] (figure 2).
The immune defences and repair systems are activated to maintain tissue integrity upon pathogen infection. Autophagy enhances cell survival under stress conditions, and keeps the balance between immunity and inflammation. This may play a protective role against inflammatory diseases such as IBD [117]. Unc-51-like autophagy activating kinase 1 (ULK1) is the earliest trigger for autophagocytosis, which is phosphorylated and binds with mTORC1 when nutrients are sufficient [118]. When nutrients are deprived, ULK1 dissociates from mTORC1, subsequently leading to initiation of autophagy [118]. AMPK involves ULK1-engaged autophagy by directly phosphorylating ULK1 at Ser 317 and Ser 777 to initiate autophagy [119] (figure 2).
Inflammation is closely related to metabolism. To synthesize ATP, cells can undergo either glycolysis or aerobic oxidation. Inflammatory cells, such as macrophages and T helper (Th) cells, typically undergo glycolytic metabolism [120]; on the other hand, anti-inflammatory cells typically depend on oxidative metabolism through mitochondria [120]. AMPK activation creates a pseudo-starving state that promotes oxidative metabolism and inhibits inflammation [120]. Similarly, creatine can regulate metabolism by recycling ATP in cells. Mutation in the creatine biosynthesis enzyme increases mice colitis, while creatine supplementation ameliorates colitis, possibly related to ATP supply and AMPK activation [121]. Collectively, those studies suggest that AMPK regulates intestinal inflammation partially through altering cell metabolism. AMPK activation shifts pro-inflammatory to anti-inflammatory cytokine production in macrophages, facilitates the differentiation of Th cells, and promotes epithelial barrier function and epithelial autophagy.
Up to now, most studies on the effects of AMPK on inflammation have been short-term studies. Limited information also points to the long-term effects of AMPK in suppressing inflammation. In an epidemiologic study, metformin suppresses chronic inflammation as indicated by the ratio of neutrophils to lymphocytes in the blood sample of diabetic patients [122]. Considering the commonness of chronic intake of AMPK activators, such as phenformin and metformin in diabetic patients, the long-term effect of AMPK activation on inflammation needs to be further investigated.
6. AMPK and colorectal cancer
Intestinal epithelium is the most dynamic tissue in the body, as it is constantly being renewed. Perturbations of the balance among proliferation, differentiation and apoptosis could result in the predisposition to and initiation of colorectal cancer (CRC), the third most lethal cancer in the United States [123,124]. Unlike normal epithelial cells, cancer cells depend on glycolysis to provide energy, the so-called Warburg effect [125]. As a metabolic mediator, AMPK is an anti-apoptotic component when cells are injured from glucose deprivation [126], hyperglycaemia [127], ceramide production [128] and ischaemia [129]. In contrast, AMPK induces apoptosis in pancreatic cells [130], gastric cancer cells [131] and neuroblastoma cells [132]. Thus, the effects of AMPK on apoptosis are dependent on cell type and external stimuli.
AMPK activation in the intestine is inhibited in metabolic diseases. In rats with insulin resistance, the protein content of phospho-AMPK is markedly decreased in the jejunum [24]. A high-fat diet (HFD) inactivates AMPK, and triggers β-catenin activity due to the induction of the peroxisome proliferator-activated receptor, which thereafter increases intestinal tumorigenesis and villus length [43,44]. Pitavastatin has chemopreventive potential on obesity-related colorectal carcinogenesis, associated with AMPK activation [54]. The adiponectin secreted by adipose tissue links metabolic diseases to tumorigenesis, probably mediated through AMPK. The incidence of colonic polyps increases in colorectal cancer with adiponectin deficiency, concomitantly with the inactivation of AMPK [133]. Consistently, growth inhibition by adiponectin in HT-29 cells is attenuated due to AMPK deficiency, and promoted when metformin is presented [30].
Mechanistically, AMPK could inhibit colorectal carcinogenesis through upregulating p53, a tumour suppressor (figure 3). Quercetin [59], the major dietary flavonoid, 20(S)-ginsenoside Rg3 from Panax ginseng [61] and plumbagin from plants [58] cause apoptosis via an AMPK–p53 cascade. Oral administration of quercetin reduces tumour volume and activates AMPK in mice with HT-29 tumour xenografts [59]. Activated AMPK phosphorylates p53 at Ser 15, which is partially responsible for the inhibition of tumour growth [134]. Furthermore, berberine activates AMPK, decreases mTOR activity and phosphorylates p53 in HCT116 cells [37]. Similar effects of quercetin were also detected in lung [135], breast [136], leukaemia [137] and prostate cancer cells [138]. As a tumour suppressor, p53 is often mutated in cancer. Of note, metformin and AICAR selectively inhibit tumour growth in HCT116 xenografts lacking p53, but not in those with intact p53 [139]. This may suggest a potential anti-tumour effect of metformin in patients with p53-deficient tumours.
Figure 3.
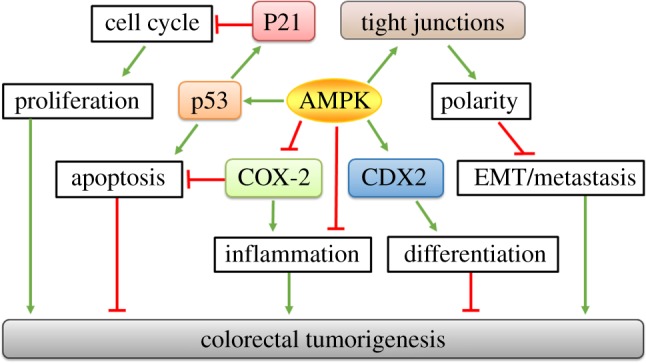
Suppressive mechanisms of AMPK in colorectal tumorigenesis. AMPK may suppress colorectal tumorigenesis through inhibiting proliferation, inflammation and metastasis as well as promoting apoptosis and differentiation collaboratively. AMPK induces apoptosis via p53 activation, which further upregulates p21 to impede cell cycling progression and cell proliferation; AMPK inhibits cancer progression by promoting caudal type homeobox 2 (CDX2) to induce differentiation, and by abating cyclooxygenase-2 (COX-2) to regulate apoptosis and inflammation; AMPK enhances the establishment of tight junctions to form epithelial polarity, which subsequently results in the amelioration of epithelial–mesenchymal transition (EMT) and colorectal metastasis. Green arrows indicate positive effects; red lines indicate negative effects.
AMPK could also inhibit colorectal cancer through inhibiting cyclooxygenase-2 (COX-2), an inflammatory enzyme (figure 3). Epigallocatechin gallate (EGCG), a polyphenol derived from green tea, stimulates AMPK in a dose-dependent manner [20]. The activated AMPK thereafter inhibits the production of COX-2 and prostaglandin E2 to induce apoptosis, while Compound C abolishes it [20]. Similarly, curcumin [40], selenium [60], and combined 5-fluorouracil and genistein [41] all demonstrate anti-tumorigenic effects via the AMPK–COX2 cascade.
In addition, AMPK exerts anti-tumour effects through arresting the cell cycle and inducing apoptosis (figure 3). Adiponectin stimulates AMPK, arrests cell cycle progression at the G1 phase, reduces cyclin E, and stimulates p21, p27, glucose utilization and fatty acid oxidation [31]. AMPK arrests the cell cycle to inhibit proliferation in many established cancer cells including prostate cancer PC-3 [140], hepatoma HepG2 [141], brain C6 glioma, astrocytoma U87MG, T-lymphoblast CEM and breast cancer MCF-7 [142] cells.
CDX2 overexpression inhibits the growth and proliferation of colorectal cancer cells [143,144]. CDX2 expression is dramatically decreased during the late stages of malignant colorectal cancer [145]. CDX2 is absent in 183 out of 621 colorectal cancers from patient specimens [146]. Its loss is positively correlated with tumour grade and stage [146]. AICAR treatment upregulates CDX2 expression in Caco-2 cells, while CDX2 expression is dramatically downregulated in AMPKα2 KO Caco-2 cells and AMPK VilCre KO mice [19] (figure 3). CDX2 deletion abolished the promoted effects of AMPK on intestinal differentiation markers [19]. CDX2 mutation upregulates colonic polyp number and increases the proliferation of colonic cells [147], whereas the re-expression of CDX2 inhibits cyclin D1 expression and cell proliferation in human intestinal epithelial crypt cells lacking Cdx2 [148]. As CDX2 is a transcription factor that facilitates intestinal differentiation, enhancing differentiation could be a promising strategy for anti-cancer therapy.
The loss of epithelial polarity leads to the damage of intestinal organization, which is probably the major step for neoplastic transformation [149], subsequently resulting in epithelial–mesenchymal transition and metastasis [150]. It has been shown that AMPKα mutation in Drosophila embryos leads to abnormal distribution of epithelial polarity markers [151]. The consequent loss of polarity along with over-proliferative aberration elicits tumorous growths [152,153]. Therefore, the strengthened tight junction by AMPK provides a possible method for inhibiting adenocarcinoma and tumorigenesis (figure 3).
Chronic inflammation dramatically increases the risk of tumorigenesis. The reactive nitrogen intermediates and reactive oxygen species associated with inflammation usually trigger genomic instability and induce genetic mutations [154]. The DNA damage in turn initiates colorectal carcinogenesis. Intestinal inflammation is strongly associated with colon cancer, which has been comprehensively discussed by Terzić and co-authors [155]. AMPK suppresses many aspects of intestinal inflammation, which is discussed in §5 ‘AMPK and intestinal inflammation’. AMPK VilCre KO mice demonstrate exacerbated dextran sodium sulfate (DSS)-induced colitis [19], while metformin administration reduces colitis in interleukin-10-deficient mice [67] as well as DSS-induced colitis in mice [21]. AMPK might inhibit intestinal tumorigenesis through mitigating intestinal inflammation (figure 3).
7. Gut microbiota regulates AMPK activity
Gut microbiota show a close relationship with intestinal health [156]. Metagenomic analysis shows that the populations of Firmicutes and Bacteroidetes are profoundly reduced in the gut microbiota from IBD patients [157]. Bifodobacteria, Lactobacillus and Bacteroides ameliorate IBD, while Helicobacter hepaticus exacerbates IBD [156], probably due to their difference in SCFA production. SCFAs activate AMPK in colonocytes; both venous infusion and oral administration of SCFAs to mice activate AMPK [158], which may explain the regulatory effect of gut microbiota on AMPK activity (figure 2). Oral administration of metformin or berberine increases the population of Allobaculum, Bacteroides, Blautia, Butyriciococcus and Phascolarctobacterium in gut microbiota, which promotes SCFA production [48].
Besides activation by low energy level, AMPK can also be regulated by intestinal hormones in a cell non-autonomous manner [159,160] (figure 2). Prebiotic treatment enhances the generation of gut hormones, glucagon-like peptide (GLP)-1 and GLP-2, due to an increase in enteroendocrine L-cells in the colon of obese mice [161] (figure 2). Likewise, metformin triggers L-cells in rat duodenum to secrete GLP-1 [50]. GLP-1 enhances AMP and subsequently activates AMPK in hepatocytes to reduce hepatic glucose production in a non-autonomous manner [162]. However, AMPK mutation in hepatocytes abolished the beneficial effects of the gut-derived peptide GLP-1 [162].
Constant activation of AMPK in the hypothalamus is able to increase food intake and body weight, while AMPK inactivation reduces appetite in rodents [163,164]. Intraperitoneal injection of ghrelin, the appetite-stimulating gastrointestinal hormone, upregulates rat food intake, associated with AMPK activation in presynaptic neurons [164,165] (figure 2). Consistently, AICAR injection into rat hypothalamus or cerebral ventricle enhances food intake [164], suggesting the integrative effect of AMPK in whole body metabolic regulation.
8. Conclusion
AMPK exerts protective effects on intestinal epithelial function through multiple mechanisms including improving intestinal absorption, enhancing barrier function, suppressing inflammation and preventing colorectal cancer. AMPK activation, either by pharmacological means or by nutraceuticals, might be a promising therapeutic strategy for treatment of intestinal disorders (figure 4).
Figure 4.
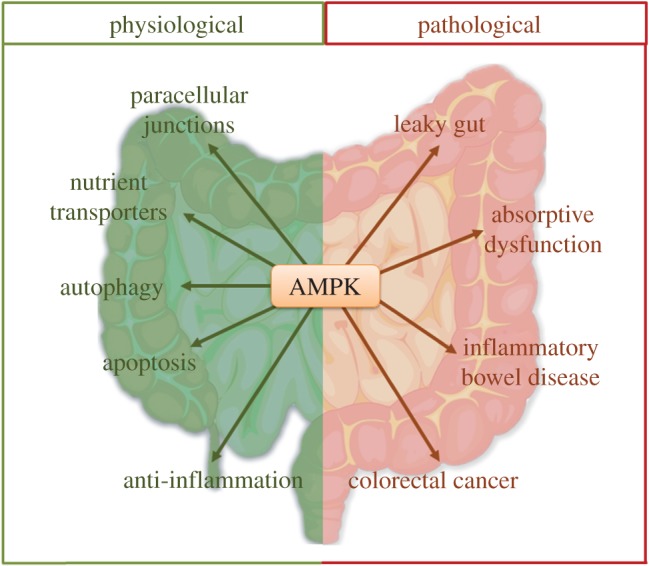
Implications and therapeutic perspectives of AMPK in the intestine. Under physiological conditions, AMPK activation strengthens paracellular junctions, enhances the function of nutrient transporters, promotes autophagy and apoptosis, and exerts anti-inflammatory effects. On the other hand, under pathological conditions, AMPK inactivation is implied in a number of intestinal diseases, such as leaky gut, absorptive dysfunction, inflammatory bowel disease and colorectal cancer.
Acknowledgements
We would like to thank Miss Sophie Corinne Trombetta and Ms. Jeanene Marie Deavila for their critical reading of the manuscript.
Data accessibility
This article has no additional data.
Competing interests
We declare we have no competing interests.
Funding
This work was partially supported by NIHR15HD073864 and Washington State University Agricultural Research Center Emerging Research Issues Internal Competitive grant no. (10A-3057-8640).
References
- 1.Stainier DY. 2005. No organ left behind: tales of gut development and evolution. Science 307, 1902–1904. (doi:10.1126/science.1108709) [DOI] [PubMed] [Google Scholar]
- 2.Moore KA, Lemischka IR. 2006. Stem cells and their niches. Science 311, 1880–1885. (doi:10.1126/science.1110542) [DOI] [PubMed] [Google Scholar]
- 3.Thompson WG. 1984. Gastrointestinal symptoms in the irritable bowel compared with peptic ulcer and inflammatory bowel disease. Gut 25, 1089–1092. (doi:10.1136/gut.25.10.1089) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Talley NJ, Stanghellini V, Heading R, Koch K, Malagelada J, Tytgat G. 1999. Functional gastroduodenal disorders. Gut 45(Suppl. 2), II37–II42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maloy KJ, Powrie F. 2011. Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature 474, 298–306. (doi:10.1038/nature10208) [DOI] [PubMed] [Google Scholar]
- 6.Rossi DJ, Jamieson CH, Weissman IL. 2008. Stems cells and the pathways to aging and cancer. Cell 132, 681–696. (doi:10.1016/j.cell.2008.01.036) [DOI] [PubMed] [Google Scholar]
- 7.Kahn BB, Alquier T, Carling D, Hardie DG. 2005. AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 1, 15–25. (doi:10.1016/j.cmet.2004.12.003) [DOI] [PubMed] [Google Scholar]
- 8.Hutber CA, Hardie D, Winder W. 1997. Electrical stimulation inactivates muscle acetyl-CoA carboxylase and increases AMP-activated protein kinase. Am. J. Physiol. Endocrinol. Metab. 272, E262–E266. [DOI] [PubMed] [Google Scholar]
- 9.Hardie DG. 2007. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat. Rev. Mol. Cell Biol. 8, 774–785. (doi:10.1038/nrm2249) [DOI] [PubMed] [Google Scholar]
- 10.Viollet B, Horman S, Leclerc J, Lantier L, Foretz M, Billaud M, Giri S, Andreelli F. 2010. AMPK inhibition in health and disease. Crit. Rev. Biochem. Mol. Biol. 45, 276–295. (doi:10.3109/10409238.2010.488215) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brusq J-M, Ancellin N, Grondin P, Guillard R, Martin S, Saintillan Y, Issandou M. 2006. Inhibition of lipid synthesis through activation of AMP kinase: an additional mechanism for the hypolipidemic effects of berberine. J. Lipid Res. 47, 1281–1288. (doi:10.1194/jlr.M600020-JLR200) [DOI] [PubMed] [Google Scholar]
- 12.Chen MB, McAinch AJ, Macaulay SL, Castelli LA, O'Brien PE, Dixon JB, Cameron-Smith D, Kemp BE, Steinberg GR. 2005. Impaired activation of AMP-kinase and fatty acid oxidation by globular adiponectin in cultured human skeletal muscle of obese type 2 diabetics. J. Clin. Endocrinol. Metab. 90, 3665–3672. (doi:10.1210/jc.2004-1980) [DOI] [PubMed] [Google Scholar]
- 13.Hardie DG, Sakamoto K. 2006. AMPK: a key sensor of fuel and energy status in skeletal muscle. Physiology 21, 48–60. (doi:10.1152/physiol.00044.2005) [DOI] [PubMed] [Google Scholar]
- 14.Ix JH, Sharma K. 2010. Mechanisms linking obesity, chronic kidney disease, and fatty liver disease: the roles of fetuin-A, adiponectin, and AMPK. J. Am. Soc. Nephrol. 21, 406–412. (doi:10.1681/ASN.2009080820) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hwang J-T, Park I-J, Shin J-I, Lee YK, Lee SK, Baik HW, Ha J, Pakr OJ. et al. 2005. Genistein, EGCG, and capsaicin inhibit adipocyte differentiation process via activating AMP-activated protein kinase . Biochem. Biophys. Res. Commun. 338, 694–699. (doi:10.1016/j.bbrc.2005.09.195) [DOI] [PubMed] [Google Scholar]
- 16.Al-Khalili L, Chibalin AV, Yu M, Sjödin B, Nylén C, Zierath JR, Krook A. 2004. MEF2 activation in differentiated primary human skeletal muscle cultures requires coordinated involvement of parallel pathways. Am. J. Physiol. Cell Physiol. 286, C1410–C1416. (doi:10.1152/ajpcell.00444.2003) [DOI] [PubMed] [Google Scholar]
- 17.Kim EK, Lim S, Park JM, Seo JK, Kim JH, Kim KT, Ryu SH, Suh P-G. 2012. Human mesenchymal stem cell differentiation to the osteogenic or adipogenic lineage is regulated by AMP-activated protein kinase. J. Cell. Physiol. 227, 1680–1687. (doi:10.1002/jcp.22892) [DOI] [PubMed] [Google Scholar]
- 18.Walker J, Jijon HB, Hugo D, Salehi P, Churchill T, Madsen KL. 2005. 5-Aminoimidazole-4-carboxamide riboside (AICAR) enhances GLUT2-dependent jejunal glucose transport: a possible role for AMPK. Biochem. J. 385, 485–491. (doi:10.1042/BJ20040694) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sun X, Yang Q, Rogers CJ, Du M, Zhu M-J. 2017. AMPK improves gut epithelial differentiation and barrier function via regulating Cdx2 expression. Cell Death Differ. 24, 819–831. (doi:10.1038/cdd.2017.14) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hwang J-T, Ha J, Park I-J, Lee S-K, Baik HW, Kim YM, Park O. 2007. Apoptotic effect of EGCG in HT-29 colon cancer cells via AMPK signal pathway. Cancer Lett. 247, 115–121. (doi:10.1016/j.canlet.2006.03.030) [DOI] [PubMed] [Google Scholar]
- 21.Koh SJ, Kim JM, Kim IK, Ko SH, Kim JS. 2014. Anti-inflammatory mechanism of metformin and its effects in intestinal inflammation and colitis-associated colon cancer. J. Gastroenterol. Hepatol. 29, 502–510. (doi:10.1111/jgh.12435) [DOI] [PubMed] [Google Scholar]
- 22.Zhang BB, Zhou G, Li C. 2009. AMPK: an emerging drug target for diabetes and the metabolic syndrome. Cell Metab. 9, 407–416. (doi:10.1016/j.cmet.2009.03.012) [DOI] [PubMed] [Google Scholar]
- 23.Meddings J. 2008. The significance of the gut barrier in disease. Gut 57, 438–440. (doi:10.1136/gut.2007.143172) [DOI] [PubMed] [Google Scholar]
- 24.Harmel E, et al. 2014. AMPK in the small intestine in normal and pathophysiological conditions. Endocrinology 155, 873–888. (doi:10.1210/en.2013-1750) [DOI] [PubMed] [Google Scholar]
- 25.Woods A, et al. 2003. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr. Biol. 13, 2004–2008. (doi:10.1016/j.cub.2003.10.031) [DOI] [PubMed] [Google Scholar]
- 26.Woods A, Dickerson K, Heath R, Hong S-P, Momcilovic M, Johnstone SR, Carlson M, Carling D. 2005. Ca2+/calmodulin-dependent protein kinase kinase-β acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab. 2, 21–33. (doi:10.1016/j.cmet.2005.06.005) [DOI] [PubMed] [Google Scholar]
- 27.Suter M, Riek U, Tuerk R, Schlattner U, Wallimann T, Neumann D. 2006. Dissecting the role of 5′-AMP for allosteric stimulation, activation, and deactivation of AMP-activated protein kinase. J. Biol. Chem. 281, 32 207–32 216. (doi:10.1074/jbc.M606357200) [DOI] [PubMed] [Google Scholar]
- 28.Sakar Y, Nazaret C, Lettéron P, Ait Omar A, Avenati M, Viollet B, Ducroc R, Bado A. 2009. Positive regulatory control loop between gut leptin and intestinal GLUT2/GLUT5 transporters links to hepatic metabolic functions in rodents. PLoS ONE 4, e7935 (doi:10.1371/journal.pone.0007935) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fu X, Zhao J-X, Zhu M-J, Foretz M, Viollet B, Dodson MV, Du M. 2013. AMP-activated protein kinase α1 but not α2 catalytic subunit potentiates myogenin expression and myogenesis. Mol. Cell. Biol. 33, 4517–4525. (doi:10.1128/MCB.01078-13) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zakikhani M, Dowling RJ, Sonenberg N, Pollak MN. 2008. The effects of adiponectin and metformin on prostate and colon neoplasia involve activation of AMP-activated protein kinase. Cancer Prev. Res. 1, 369–375. (doi:10.1158/1940-6207.CAPR-08-0081) [DOI] [PubMed] [Google Scholar]
- 31.Kim AY, et al. 2010. Adiponectin represses colon cancer cell proliferation via AdipoR1-and-R2-mediated AMPK activation. Mol. Endocrinol. 24, 1441–1452. (doi:10.1210/me.2009-0498) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pieri M, Christian HC, Wilkins RJ, Boyd C, Meredith D. 2010. The apical (hPepT1) and basolateral peptide transport systems of Caco-2 cells are regulated by AMP-activated protein kinase. Am. J. Physiol. Gastrointest. Liver Physiol. 299, G136–GG43. (doi:10.1152/ajpgi.00014.2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bai A, Ma AG, Yong M, Weiss CR, Ma Y, Guan Q, Bernstein CN, Perg Z. 2010. AMPK agonist downregulates innate and adaptive immune responses in TNBS-induced murine acute and relapsing colitis. Biochem. Pharmacol. 80, 1708–1717. (doi:10.1016/j.bcp.2010.08.009) [DOI] [PubMed] [Google Scholar]
- 34.Rogers AC, et al. 2013. Activation of AMPK inhibits cholera toxin stimulated chloride secretion in human and murine intestine. PLoS ONE 8, e69050 (doi:10.1371/journal.pone.0069050) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Manzo-Avalos S, Saavedra-Molina A. 2010. Cellular and mitochondrial effects of alcohol consumption. Int. J. Environ. Res. Public Health 7, 4281–4304. (doi:10.3390/ijerph7124281) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Elamin EE, Masclee AA, Dekker J, Pieters H-J, Jonkers DM. 2013. Short-chain fatty acids activate AMP-activated protein kinase and ameliorate ethanol-induced intestinal barrier dysfunction in Caco-2 cell monolayers. J. Nutr. 143, 1872–1881. (doi:10.3945/jn.113.179549) [DOI] [PubMed] [Google Scholar]
- 37.Li W, Hua B, Saud SM, Lin H, Hou W, Matter MS, Jia L, Colburn NH, Young MR. 2014. Berberine regulates AMP-activated protein kinase signaling pathways and inhibits colon tumorigenesis in mice. Mol. Carcinog. 54, 1096–1109. (doi:10.1002/mc.22179) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peng L, Li Z-R, Green RS, Holzman IR, Lin J. 2009. Butyrate enhances the intestinal barrier by facilitating tight junction assembly via activation of AMP-activated protein kinase in Caco-2 cell monolayers. J. Nutr. 139, 1619–1625. (doi:10.3945/jn.109.104638) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Muanprasat C, Wongkrasant P, Satitsri S, Moonwiriyakit A, Pongkorpsakol P, Mattaveewong T, Pichyangkura R, Chatsudthipong V. 2015. Activation of AMPK by chitosan oligosaccharide in intestinal epithelial cells: mechanism of action and potential applications in intestinal disorders. Biochem. Pharmacol. 96, 225–236. (doi:10.1016/j.bcp.2015.05.016) [DOI] [PubMed] [Google Scholar]
- 40.Lee YK, Park SY, Kim YM, Park OJ. 2009. Regulatory effect of the AMPK–COX-2 signaling pathway in curcumin-induced apoptosis in HT-29 colon cancer cells. Ann. N. Y. Acad. Sci. 1171, 489–494. (doi:10.1111/j.1749-6632.2009.04699.x) [DOI] [PubMed] [Google Scholar]
- 41.Hwang J-T, Ha J, Park OJ. 2005. Combination of 5-fluorouracil and genistein induces apoptosis synergistically in chemo-resistant cancer cells through the modulation of AMPK and COX-2 signaling pathways. Biochem. Biophys. Res. Commun. 332, 433–440. (doi:10.1016/j.bbrc.2005.04.143) [DOI] [PubMed] [Google Scholar]
- 42.Takeda J, Park H-Y, Kunitake Y, Yoshiura K, Matsui T. 2013. Theaflavins, dimeric catechins, inhibit peptide transport across Caco-2 cell monolayers via down-regulation of AMP-activated protein kinase-mediated peptide transporter PEPT1. Food Chem. 138, 2140–2145. (doi:10.1016/j.foodchem.2012.12.026) [DOI] [PubMed] [Google Scholar]
- 43.Mao J, Hu X, Xiao Y, Yang C, Ding Y, Hou N, Wang J, Cheng H, Zhang X. 2013. Overnutrition stimulates intestinal epithelium proliferation through β-catenin signaling in obese mice. Diabetes 62, 3736–3746. (doi:10.2337/db13-0035) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Beyaz S, et al. 2016. High-fat diet enhances stemness and tumorigenicity of intestinal progenitors. Nature 531, 53–58. (doi:10.1038/nature17173) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hou Y, et al. 2011. Effects of α-ketoglutarate on energy status in the intestinal mucosa of weaned piglets chronically challenged with lipopolysaccharide. Br. J. Nutr. 106, 357–363. (doi:10.1017/S0007114511000249) [DOI] [PubMed] [Google Scholar]
- 46.Lenzen S, Lortz S, Tiedge M. 1996. Effects of metformin on SGLT1, GLUT2, and GLUT5 hexose transporter gene expression in small intestine from rats. Biochem. Pharmacol. 51, 893–896. (doi:10.1016/0006-2952(95)02243-0) [DOI] [PubMed] [Google Scholar]
- 47.Wheaton WW, et al. 2014. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. Elife 3, e02242 (doi:10.7554/eLife.02242) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang X, Zhao Y, Xu J, Xue Z, Zhang M, Pang X, Zhang X, Zhao L. 2015. Modulation of gut microbiota by berberine and metformin during the treatment of high-fat diet-induced obesity in rats. Sci. Rep. 5, 14405 (doi:10.1038/srep14405) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shin N-R, Lee J-C, Lee H-Y, Kim M-S, Whon TW, Lee M-S, Bae J-W. 2013. An increase in the Akkermansia spp. population induced by metformin treatment improves glucose homeostasis in diet-induced obese mice. Gut 63, 727–735. (doi:10.1136/gutjnl-2012-303839) [DOI] [PubMed] [Google Scholar]
- 50.Duca FA, Côté CD, Rasmussen BA, Zadeh-Tahmasebi M, Rutter GA, Filippi BM, Lam TKT. 2015. Metformin activates a duodenal Ampk-dependent pathway to lower hepatic glucose production in rats. Nat. Med. 21, 506–511. (doi:10.1038/nm.3787) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Endo H, Niioka M, Kobayashi N, Tanaka M, Watanabe T. 2013. Butyrate-producing probiotics reduce nonalcoholic fatty liver disease progression in rats: new insight into the probiotics for the gut-liver axis. PLoS ONE 8, e633880 (doi:10.1371/journal.pone.0063388) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Suchankova G, Tekle M, Saha AK, Ruderman NB, Clarke SD, Gettys TW. 2005. Dietary polyunsaturated fatty acids enhance hepatic AMP-activated protein kinase activity in rats. Biochem. Biophys. Res. Commun. 326, 851–858. (doi:10.1016/j.bbrc.2004.11.114) [DOI] [PubMed] [Google Scholar]
- 53.Gabler NK, Radcliffe JS, Spencer JD, Webel DM, Spurlock ME. 2009. Feeding long-chain n-3 polyunsaturated fatty acids during gestation increases intestinal glucose absorption potentially via the acute activation of AMPK. J. Nutr. Biochem. 20, 17–25. (doi:10.1016/j.jnutbio.2007.11.009) [DOI] [PubMed] [Google Scholar]
- 54.Yasuda Y, et al. 2010. Pitavastatin inhibits azoxymethane-induced colonic preneoplastic lesions in C57BL/KsJ-db/db obese mice. Cancer Sci. 101, 1701–1707. (doi:10.1111/j.1349-7006.2010.01579.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang H, Zhao J-X, Hu N, Ren J, Du M, Zhu M-J. 2012. Side-stream smoking reduces intestinal inflammation and increases expression of tight junction proteins. World J. Gastroenterol. 18, 2180 (doi:10.3748/wjg.v18.i18.2180) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kongsuphol P, Hieke B, Ousingsawat J, Almaca J, Viollet B, Schreiber R, Kunzelmann K. 2009. Regulation of Cl− secretion by AMPK in vivo. Pflügers Arch. 457, 1071–1078. (doi:10.1007/s00424-008-0577-3) [DOI] [PubMed] [Google Scholar]
- 57.Wang K, Jin X, Chen Y, Song Z, Jiang X, Hu F, Conlon M, Topping D. 2016. Polyphenol-rich propolis extracts strengthen intestinal barrier function by activating AMPK and ERK signaling. Nutrients 8, 272 (doi:10.3390/nu8050272) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen M-B, Zhang Y, Wei M-X, Shen W, Wu X-Y, Yao C, Lu P-H. 2013. Activation of AMP-activated protein kinase (AMPK) mediates plumbagin-induced apoptosis and growth inhibition in cultured human colon cancer cells. Cell. Signal. 25, 1993–2002. (doi:10.1016/j.cellsig.2013.05.026) [DOI] [PubMed] [Google Scholar]
- 59.Kim H-J, et al. 2010. Apoptotic effect of quercetin on HT-29 colon cancer cells via the AMPK signaling pathway. J. Agric. Food Chem. 58, 8643–8650. (doi:10.1021/jf101510z) [DOI] [PubMed] [Google Scholar]
- 60.Hwang J-T, Kim YM, Surh Y-J, Baik HW, Lee S-K, Ha J, Park OJ. 2006. Selenium regulates cyclooxygenase-2 and extracellular signal-regulated kinase signaling pathways by activating AMP-activated protein kinase in colon cancer cells. Cancer Res. 66, 10 057–10 063. (doi:10.1158/0008-5472.CAN-06-1814) [DOI] [PubMed] [Google Scholar]
- 61.Yuan H-d, Quan H-Y, ZHanG Y, KiM SH. 2010. 20 (S)-Ginsenoside Rg3-induced apoptosis in HT-29 colon cancer cells is associated with AMPK signaling pathway. Apoptosis 2, 6. [DOI] [PubMed] [Google Scholar]
- 62.Corton JM, Gillespie JG, Hawley SA, Hardie DG. 1995. 5-Aminoimidazole-4-carboxamide ribonucleoside. Eur. J. Biochem. 229, 558–565. (doi:10.1111/j.1432-1033.1995.tb20498.x) [DOI] [PubMed] [Google Scholar]
- 63.Hawley SA, et al. 2010. Use of cells expressing γ subunit variants to identify diverse mechanisms of AMPK activation. Cell Metab. 11, 554–565. (doi:10.1016/j.cmet.2010.04.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mahmood K, Naeem M, Rahimnajjad NA. 2013. Metformin: the hidden chronicles of a magic drug. Eur. J. Intern. Med. 24, 20–26. (doi:10.1016/j.ejim.2012.10.011) [DOI] [PubMed] [Google Scholar]
- 65.Konopka AR, et al. 2016. Hyperglucagonemia mitigates the effect of metformin on glucose production in prediabetes. Cell Rep. 15, 1394–1400. (doi:10.1016/j.celrep.2016.04.024) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Doran E, Halestrap AP. 2000. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem. J. 348, 607–614. (doi:10.1042/bj3480343) [PMC free article] [PubMed] [Google Scholar]
- 67.Xue Y, Zhang H, Sun X, Zhu M-J. 2016. Metformin improves ileal epithelial barrier function in interleukin-10 deficient mice. PLoS ONE 11, e0168670 (doi:10.1371/journal.pone.0168670) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Huang S-L, Yu R-T, Gong J, Feng Y, Dai Y-L, Hu F, Hu Y-H, Tao Y-D, Leng Y. 2012. Arctigenin, a natural compound, activates AMP-activated protein kinase via inhibition of mitochondria complex I and ameliorates metabolic disorders in ob/ob mice. Diabetologia 55, 1469–1481. (doi:10.1007/s00125-011-2366-3) [DOI] [PubMed] [Google Scholar]
- 69.Ferraris RP, Diamond JM. 1989. Specific regulation of intestinal nutrient transporters by their dietary substrates. Annu. Rev. Physiol. 51, 125–141. (doi:10.1146/annurev.ph.51.030189.001013) [DOI] [PubMed] [Google Scholar]
- 70.Wood IS, Trayhurn P. 2003. Glucose transporters (GLUT and SGLT): expanded families of sugar transport proteins. Br. J. Nutr. 89, 3–9. (doi:10.1079/BJN2002763) [DOI] [PubMed] [Google Scholar]
- 71.Thorens B. 1996. Glucose transporters in the regulation of intestinal, renal, and liver glucose fluxes. Am. J. Physiol. Gastrointest. Liver Physiol. 270, G541–GG53. [DOI] [PubMed] [Google Scholar]
- 72.Russell RR, Bergeron R, Shulman GI, Young LH. 1999. Translocation of myocardial GLUT-4 and increased glucose uptake through activation of AMPK by AICAR. Am. J. Physiol. Heart Circ. Physiol. 277, H643–H6H9. [DOI] [PubMed] [Google Scholar]
- 73.Breen DM, Sanli T, Giacca A, Tsiani E. 2008. Stimulation of muscle cell glucose uptake by resveratrol through sirtuins and AMPK. Biochem. Biophys. Res. Commun. 374, 117–122. (doi:10.1016/j.bbrc.2008.06.104) [DOI] [PubMed] [Google Scholar]
- 74.Kawaguchi T, Osatomi K, Yamashita H, Kabashima T, Uyeda K. 2002. Mechanism for fatty acid ‘sparing’ effect on glucose-induced transcription. J. Biol. Chem. 277, 3829–3835. (doi:10.1074/jbc.M107895200) [DOI] [PubMed] [Google Scholar]
- 75.King TD, Song L, Jope RS. 2006. AMP-activated protein kinase (AMPK) activating agents cause dephosphorylation of Akt and glycogen synthase kinase-3. Biochem. Pharmacol. 71, 1637–1647. (doi:10.1016/j.bcp.2006.03.005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kellett G, Helliwell P. 2000. The diffusive component of intestinal glucose absorption is mediated by the glucose-induced recruitment of GLUT2 to the brush-border membrane. Biochem. J. 350, 155–162. (doi:10.1042/bj3500155) [PMC free article] [PubMed] [Google Scholar]
- 77.Au A, Gupta A, Schembri P, Cheeseman C. 2002. Rapid insertion of GLUT2 into the rat jejunal brush-border membrane promoted by glucagon-like peptide 2. Biochem. J. 367, 247–254. (doi:10.1042/bj20020393) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Field M. 2003. Intestinal ion transport and the pathophysiology of diarrhea. J. Clin. Invest. 111, 931 (doi:10.1172/JCI200318326) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Almaça J, Kongsuphol P, Hieke B, Ousingsawat J, Viollet B, Schreiber R, Amaral MD, Kunzelmanm K. 2009. AMPK controls epithelial Na+ channels through Nedd4-2 and causes an epithelial phenotype when mutated. Pflügers Arch. 458, 713–721. (doi:10.1007/s00424-009-0660-4) [DOI] [PubMed] [Google Scholar]
- 80.Woollhead AM, Scott JW, Hardie DG, Baines DL. 2005. Phenformin and 5-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside (AICAR) activation of AMP-activated protein kinase inhibits transepithelial Na+ transport across H441 lung cells. J. Physiol. 566, 781–792. (doi:10.1113/jphysiol.2005.088674) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hallows KR. 2005. Emerging role of AMP-activated protein kinase in coupling membrane transport to cellular metabolism. Curr. Opin. Nephrol. Hypertens. 14, 464–471. (doi:10.1097/01.mnh.0000174145.14798.64) [DOI] [PubMed] [Google Scholar]
- 82.Cottreau J, Tucker A, Crutchley R, Garey KW. 2012. Crofelemer for the treatment of secretory diarrhea. Expert Rev. Gastroenterol. Hepatol. 6, 17–23. (doi:10.1586/egh.11.87) [DOI] [PubMed] [Google Scholar]
- 83.Leibach FH, Ganapathy V. 1996. Peptide transporters in the intestine and the kidney. Annu. Rev. Nutr. 16, 99–119. (doi:10.1146/annurev.nu.16.070196.000531) [DOI] [PubMed] [Google Scholar]
- 84.Kennedy D, Leibach F, Ganapathy V, Thwaites D. 2002. Optimal absorptive transport of the dipeptide glycylsarcosine is dependent on functional Na+/H+ exchange activity. Pflügers Arch. 445, 139–146. (doi:10.1007/s00424-002-0910-1) [DOI] [PubMed] [Google Scholar]
- 85.Bland ML, Lee RJ, Magallanes JM, Foskett JK, Birnbaum MJ. 2010. AMPK supports growth in Drosophila by regulating muscle activity and nutrient uptake in the gut. Dev. Biol. 344, 293–303. (doi:10.1016/j.ydbio.2010.05.010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.McDaniel SS, et al. 2001. Anorexic effect of K+ channel blockade in mesenteric arterial smooth muscle and intestinal epithelial cells. J. Appl. Physiol. 91, 2322–2333. [DOI] [PubMed] [Google Scholar]
- 87.Goirand F, et al. 2007. Activation of AMP kinase α1 subunit induces aortic vasorelaxation in mice. J. Physiol. 581, 1163–1171. (doi:10.1113/jphysiol.2007.132589) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Horman S, et al. 2008. AMP-activated protein kinase phosphorylates and desensitizes smooth muscle myosin light chain kinase. J. Biol. Chem. 283, 18 505–18 512. (doi:10.1074/jbc.M802053200) [DOI] [PubMed] [Google Scholar]
- 89.Hollander D. 1999. Intestinal permeability, leaky gut, and intestinal disorders. Curr. Gastroenterol. Rep. 1, 410–416. (doi:10.1007/s11894-999-0023-5) [DOI] [PubMed] [Google Scholar]
- 90.Buhner S, et al. 2006. Genetic basis for increased intestinal permeability in families with Crohn's disease: role of CARD15 3020insC mutation? Gut 55, 342–347. (doi:10.1136/gut.2005.065557) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Miele L, et al. 2009. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology 49, 1877–1887. (doi:10.1002/hep.22848) [DOI] [PubMed] [Google Scholar]
- 92.Brun P, Castagliuolo I, Di Leo V, Buda A, Pinzani M, Palù G, Martines D. 2007. Increased intestinal permeability in obese mice: new evidence in the pathogenesis of nonalcoholic steatohepatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 292, G518–GG25. (doi:10.1152/ajpgi.00024.2006) [DOI] [PubMed] [Google Scholar]
- 93.Bosi E, Molteni L, Radaelli M, Folini L, Fermo I, Bazzigaluppi E, Piemonti L, Pastore MR, Paroni R. 2006. Increased intestinal permeability precedes clinical onset of type 1 diabetes. Diabetologia 49, 2824–2827. (doi:10.1007/s00125-006-0465-3) [DOI] [PubMed] [Google Scholar]
- 94.Teixeira TF, Collado MC, Ferreira CL, Bressan J, Maria do Carmo GP. 2012. Potential mechanisms for the emerging link between obesity and increased intestinal permeability. Nutr. Res. 32, 637–647. (doi:10.1016/j.nutres.2012.07.003) [DOI] [PubMed] [Google Scholar]
- 95.Jenkins R, Rooney P, Jones D, Bienenstock J, Goodacre R. 1987. Increased intestinal permeability in patients with rheumatoid arthritis: a side-effect of oral nonsteroidal anti-inflammatory drug therapy? Rheumatology 26, 103–107. (doi:10.1093/rheumatology/26.2.103) [DOI] [PubMed] [Google Scholar]
- 96.Farquhar MG, Palade GE. 1963. Junctional complexes in various epithelia. J. Cell Biol. 17, 375–412. (doi:10.1083/jcb.17.2.375) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Schneeberger EE, Lynch RD. 2004. The tight junction: a multifunctional complex. Am. J. Physiol. Cell Physiol. 286, C1213–C1228. (doi:10.1152/ajpcell.00558.2003) [DOI] [PubMed] [Google Scholar]
- 98.Zhang L, Li J, Young LH, Caplan MJ. 2006. AMP-activated protein kinase regulates the assembly of epithelial tight junctions. Proc. Natl Acad. Sci. USA 103, 17 272–17 277. (doi:10.1073/pnas.0608531103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ichikawa-Tomikawa N, Sugimoto K, Satohisa S, Nishiura K, Chiba H. 2011. Possible involvement of tight junctions, extracellular matrix and nuclear receptors in epithelial differentiation. Biomed Res. Int. 2011, 253048 (doi:10.1155/2011/253048) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Silberg DG, Swain GP, Suh ER, Traber PG. 2000. Cdx1 and cdx2 expression during intestinal development. Gastroenterology 119, 961–971. (doi:10.1053/gast.2000.18142) [DOI] [PubMed] [Google Scholar]
- 101.Hallows KR, Mount PF, Pastor-Soler NM, Power DA. 2010. Role of the energy sensor AMP-activated protein kinase in renal physiology and disease. Am. J. Physiol. Renal Physiol. 298, F1067–F1F77. (doi:10.1152/ajprenal.00005.2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yano T, Matsui T, Tamura A, Uji M, Tsukita S. 2013. The association of microtubules with tight junctions is promoted by cingulin phosphorylation by AMPK. J. Cell Biol. 203, 605–614. (doi:10.1083/jcb.201304194) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kojima T, et al. 2009. Tight junction proteins and signal transduction pathways in hepatocytes. Histol. Histopathol. 24, 1463–1472. (doi:10.14670/HH-24.1463) [DOI] [PubMed] [Google Scholar]
- 104.Amieva MR, Vogelmann R, Covacci A, Tompkins LS, Nelson WJ, Falkow S. 2003. Disruption of the epithelial apical-junctional complex by Helicobacter pylori CagA. Science 300, 1430–1434. (doi:10.1126/science.1081919) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Nava P, López S, Arias CF, Islas S, González-Mariscal L. 2004. The rotavirus surface protein VP8 modulates the gate and fence function of tight junctions in epithelial cells. J. Cell Sci. 117, 5509–5519. (doi:10.1242/jcs.01425) [DOI] [PubMed] [Google Scholar]
- 106.Utech M, Ivanov AI, Samarin SN, Bruewer M, Turner JR, Mrsny RJ, Parkos CA, Nusrat A. 2005. Mechanism of IFN-γ-induced endocytosis of tight junction proteins: myosin II-dependent vacuolarization of the apical plasma membrane. Mol. Biol. Cell 16, 5040–5052. (doi:10.1091/mbc.E05-03-0193) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Bouma G, Strober W. 2003. The immunological and genetic basis of inflammatory bowel disease. Nat. Rev. Immunol. 3, 521–533. (doi:10.1038/nri1132) [DOI] [PubMed] [Google Scholar]
- 108.Kamada N, et al. 2005. Abnormally differentiated subsets of intestinal macrophage play a key role in Th1-dominant chronic colitis through excess production of IL-12 and IL-23 in response to bacteria. J. Immunol. 175, 6900–6908. (doi:10.4049/jimmunol.175.10.6900) [DOI] [PubMed] [Google Scholar]
- 109.Sartor RB. 2006. Mechanisms of disease: pathogenesis of Crohn's disease and ulcerative colitis. Nat. Clin. Pract. Gastroenterol. Hepatol. 3, 390–407. (doi:10.1038/ncpgasthep0528) [DOI] [PubMed] [Google Scholar]
- 110.Xavier R, Podolsky D. 2007. Unravelling the pathogenesis of inflammatory bowel disease. Nature 448, 427–434. (doi:10.1038/nature06005) [DOI] [PubMed] [Google Scholar]
- 111.Nielsen O, Kirman I, Rüdiger N, Hendel J, Vainer B. 2003. Upregulation of interleukin-12 and -17 in active inflammatory bowel disease. Scand. J. Gastroenterol. 38, 180–185. (doi:10.1080/00365520310000672) [DOI] [PubMed] [Google Scholar]
- 112.Neurath MF. 2017. Current and emerging therapeutic targets for IBD. Nat. Rev. Gastroenterol. Hepatol. 14, 269–278. (doi:10.1038/nrgastro.2016.208) [DOI] [PubMed] [Google Scholar]
- 113.Gerondakis S, Fulford TS, Messina NL, Grumont RJ. 2014. NF-κB control of T cell development. Nat. Immunol. 15, 15–25. (doi:10.1038/ni.2785) [DOI] [PubMed] [Google Scholar]
- 114.Guo S, Al-Sadi R, Said HM, Ma TY. 2013. Lipopolysaccharide causes an increase in intestinal tight junction permeability in vitro and in vivo by inducing enterocyte membrane expression and localization of TLR-4 and CD14. Am. J. Pathol. 182, 375–387. (doi:10.1016/j.ajpath.2012.10.014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kim YS, Ho SB. 2010. Intestinal goblet cells and mucins in health and disease: recent insights and progress. Curr. Gastroenterol. Rep. 12, 319–330. (doi:10.1007/s11894-010-0131-2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Van Klinken BJ, Van der Wal JG, Einerhand A, Büller H, Dekker J. 1999. Sulphation and secretion of the predominant secretory human colonic mucin MUC2 in ulcerative colitis. Gut 44, 387–393. (doi:10.1136/gut.44.3.387) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Randall-Demllo S, Chieppa M, Eri R. 2013. Intestinal epithelium and autophagy: partners in gut homeostasis. Front. Immunol. 4, 301 (doi:10.3389/fimmu.2013.00301) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Yang Z, Klionsky DJ. 2010. Mammalian autophagy: core molecular machinery and signaling regulation. Curr. Opin. Cell Biol. 22, 124–131. (doi:10.1016/j.ceb.2009.11.014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kim J, Kundu M, Viollet B, Guan K-L. 2011. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 13, 132–141. (doi:10.1038/ncb2152) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.O'neill LA, Hardie DG. 2013. Metabolism of inflammation limited by AMPK and pseudo-starvation. Nature 493, 346–355. (doi:10.1038/nature11862) [DOI] [PubMed] [Google Scholar]
- 121.Turer E, McAlpine W, Wang K-w, Lu T, Li X, Tang M et al. 2017. Creatine maintains intestinal homeostasis and protects against colitis. Proc. Natl Acad. Sci. USA 114, E1273–E1281. (doi:10.1073/pnas.1621400114) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Cameron AR, et al. 2016. Anti-inflammatory effects of metformin irrespective of diabetes status. Circ. Res. 119, 652–665. (doi:10.1161/CIRCRESAHA.116.308445) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Siegel RL, Miller KD, Jemal A. 2015. Cancer statistics, 2016. CA Cancer J. Clin. 66, 7–30. (doi:10.3322/caac.21332) [DOI] [PubMed] [Google Scholar]
- 124.Hanahan D, Weinberg RA. 2000. The hallmarks of cancer. Cell 100, 57–70. (doi:10.1016/S0092-8674(00)81683-9) [DOI] [PubMed] [Google Scholar]
- 125.Seyfried TN, Flores RE, Poff AM, D'Agostino DP. 2014. Cancer as a metabolic disease: implications for novel therapeutics. Carcinogenesis 35, 515–527. (doi:10.1093/carcin/bgt480) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Culmsee C, Monnig J, Kemp BE, Mattson MP. 2001. AMP-activated protein kinase is highly expressed in neurons in the developing rat brain and promotes neuronal survival following glucose deprivation. J. Mol. Neurosci. 17, 45–58. (doi:10.1385/JMN:17:1:45) [DOI] [PubMed] [Google Scholar]
- 127.Ido Y, Carling D, Ruderman N. 2002. Hyperglycemia-induced apoptosis in human umbilical vein endothelial cells: inhibition by the AMP-activated protein kinase activation. Diabetes 51, 159–167. (doi:10.2337/diabetes.51.1.159) [DOI] [PubMed] [Google Scholar]
- 128.Peralta C, et al. 2001. Adenosine monophosphate–activated protein kinase mediates the protective effects of ischemic preconditioning on hepatic ischemia-reperfusion injury in the rat. Hepatology 34, 1164–1173. (doi:10.1053/jhep.2001.29197) [DOI] [PubMed] [Google Scholar]
- 129.Russell, et al. 2004. AMP-activated protein kinase mediates ischemic glucose uptake and prevents postischemic cardiac dysfunction, apoptosis, and injury. J. Clin. Invest. 114, 495 (doi:10.1172/JCI19297) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kefas BA, Cai Y, Kerckhofs K, Ling Z, Martens G, Heimberg H, Pipeleers D, Van de Casteele M. 2004. Metformin-induced stimulation of AMP-activated protein kinase in β-cells impairs their glucose responsiveness and can lead to apoptosis. Biochem. Pharmacol. 68, 409–416. (doi:10.1016/j.bcp.2004.04.003) [DOI] [PubMed] [Google Scholar]
- 131.Saitoh M, Nagai K, Nakagawa K, Yamamura T, Yamamoto S, Nishizaki T. 2004. Adenosine induces apoptosis in the human gastric cancer cells via an intrinsic pathway relevant to activation of AMP-activated protein kinase. Biochem. Pharmacol. 67, 2005–2011. (doi:10.1016/j.bcp.2004.01.020) [DOI] [PubMed] [Google Scholar]
- 132.Garcia-Gil M, Pesi R, Perna S, Allegrini S, Giannecchini M, Camici M, Tozzi MG. 2003. 5′-Aminoimidazole-4-carboxamide riboside induces apoptosis in human neuroblastoma cells. Neuroscience 117, 811–820. (doi:10.1016/S0306-4522(02)00836-9) [DOI] [PubMed] [Google Scholar]
- 133.Sugiyama M, et al. 2009. Adiponectin inhibits colorectal cancer cell growth through the AMPK/mTOR pathway. Int. J. Oncol. 34, 339–344. [PubMed] [Google Scholar]
- 134.Motoshima H, Goldstein BJ, Igata M, Araki E. 2006. AMPK and cell proliferation—AMPK as a therapeutic target for atherosclerosis and cancer. J. Physiol. 574, 63–71. (doi:10.1113/jphysiol.2006.108324) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Nguyen T, Tran E, Nguyen TH, Do P, Huynh T, Huynh H. 2004. The role of activated MEK-ERK pathway in quercetin-induced growth inhibition and apoptosis in A549 lung cancer cells. Carcinogenesis 25, 647–659. (doi:10.1093/carcin/bgh052) [DOI] [PubMed] [Google Scholar]
- 136.Singhal RL, Yeh YA, Prajda N, Olah E, Sledge G, Weber G. 1995. Quercetin down-regulates signal transduction in human breast carcinoma cells. Biochem. Biophys. Res. Commun. 208, 425–431. (doi:10.1006/bbrc.1995.1355) [DOI] [PubMed] [Google Scholar]
- 137.Shen SC, Chen YC, Hsu FL, Lee WR. 2003. Differential apoptosis-inducing effect of quercetin and its glycosides in human promyeloleukemic HL-60 cells by alternative activation of the caspase 3 cascade. J. Cell. Biochem. 89, 1044–1055. (doi:10.1002/jcb.10559) [DOI] [PubMed] [Google Scholar]
- 138.Lee H-J, Wang C-J, Kuo H-C, Chou F-P, Jean L-F, Tseng T-H. 2005. Induction apoptosis of luteolin in human hepatoma HepG2 cells involving mitochondria translocation of Bax/Bak and activation of JNK. Toxicol. Appl. Pharmacol. 203, 124–131. (doi:10.1016/j.taap.2004.08.004) [DOI] [PubMed] [Google Scholar]
- 139.Buzzai M, Jones RG, Amaravadi RK, Lum JJ, DeBerardinis RJ, Zhao F, Viollet B, Thompson CB. 2007. Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res. 67, 6745–6752. (doi:10.1158/0008-5472.CAN-06-4447) [DOI] [PubMed] [Google Scholar]
- 140.Xiang X, Saha AK, Wen R, Ruderman NB, Luo Z. 2004. AMP-activated protein kinase activators can inhibit the growth of prostate cancer cells by multiple mechanisms. Biochem. Biophys. Res. Commun. 321, 161–167. (doi:10.1016/j.bbrc.2004.06.133) [DOI] [PubMed] [Google Scholar]
- 141.Imamura K, Ogura T, Kishimoto A, Kaminishi M, Esumi H. 2001. Cell cycle regulation via p53 phosphorylation by a 5′-AMP activated protein kinase activator, 5-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside, in a human hepatocellular carcinoma cell line. Biochem. Biophys. Res. Commun. 287, 562–567. (doi:10.1006/bbrc.2001.5627) [DOI] [PubMed] [Google Scholar]
- 142.Rattan R, Giri S, Singh AK, Singh I. 2005. 5-Aminoimidazole-4-carboxamide-1-β-D-ribofuranoside inhibits cancer cell proliferation in vitro and in vivo via AMP-activated protein kinase. J. Biol. Chem. 280, 39 582–39 593. (doi:10.1074/jbc.M507443200) [DOI] [PubMed] [Google Scholar]
- 143.Mallo GV, Soubeyran P, Lissitzky JC, Andre F, Farnarier C, Marvaldi J, Dagorn J-C, Lovanna JL. 1998. Expression of the Cdx1 and Cdx2 homeotic genes leads to reduced malignancy in colon cancer-derived cells. J. Biol. Chem. 273, 14 030–14 036. (doi:10.1074/jbc.273.22.14030) [DOI] [PubMed] [Google Scholar]
- 144.Suh E, Traber PG. 1996. An intestine-specific homeobox gene regulates proliferation and differentiation. Mol. Cell. Biol. 16, 619–625. (doi:10.1128/MCB.16.2.619) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Ee HC, Erler T, Bhathal PS, Young GP, James RJ. 1995. Cdx-2 homeodomain protein expression in human and rat colorectal adenoma and carcinoma. Am. J. Pathol. 147, 586–592. [PMC free article] [PubMed] [Google Scholar]
- 146.Baba Y, et al. 2009. Relationship of CDX2 loss with molecular features and prognosis in colorectal cancer. Clin. Cancer Res. 15, 4665–4673. (doi:10.1158/1078-0432.CCR-09-0401) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Aoki K, Kakizaki F, Sakashita H, Manabe T, Aoki M, Taketo MM. 2011. Suppression of colonic polyposis by homeoprotein CDX2 through its nontranscriptional function that stabilizes p27Kip1. Cancer Res. 71, 593–602. (doi:10.1158/0008-5472.CAN-10-2842) [DOI] [PubMed] [Google Scholar]
- 148.Escaffit F, Paré F, Gauthier R, Rivard N, Boudreau F, Beaulieu J-F. 2006. Cdx2 modulates proliferation in normal human intestinal epithelial crypt cells. Biochem. Biophys. Res. Commun. 342, 66–72. (doi:10.1016/j.bbrc.2006.01.128) [DOI] [PubMed] [Google Scholar]
- 149.Sancho E, Batlle E, Clevers H. 2004. Signaling pathways in intestinal development and cancer. Annu. Rev. Cell Dev. Biol. 20, 695–723. (doi:10.1146/annurev.cellbio.20.010403.092805) [DOI] [PubMed] [Google Scholar]
- 150.Matter K, Aijaz S, Tsapara A, Balda MS. 2005. Mammalian tight junctions in the regulation of epithelial differentiation and proliferation. Curr. Opin. Cell Biol. 17, 453–458. (doi:10.1016/j.ceb.2005.08.003) [DOI] [PubMed] [Google Scholar]
- 151.Lee JH, et al. 2007. Energy-dependent regulation of cell structure by AMP-activated protein kinase. Nature 447, 1017–1020. (doi:10.1038/nature05828) [DOI] [PubMed] [Google Scholar]
- 152.Bilder D. 2004. Epithelial polarity and proliferation control: links from the Drosophila neoplastic tumor suppressors. Genes Dev. 18, 1909–1925. (doi:10.1101/gad.1211604) [DOI] [PubMed] [Google Scholar]
- 153.Mirouse V, Swick LL, Kazgan N, St Johnston D, Brenman JE. 2007. LKB1 and AMPK maintain epithelial cell polarity under energetic stress. J. Cell Biol. 177, 387–392. (doi:10.1083/jcb.200702053) [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 154.Grivennikov SI, Greten FR, Karin M. 2010. Immunity, inflammation, and cancer. Cell 140, 883–899. (doi:10.1016/j.cell.2010.01.025) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Terzić J, Grivennikov S, Karin E, Karin M. 2010. Inflammation and colon cancer. Gastroenterology 138, 2101–2114.e5. (doi:10.1053/j.gastro.2010.01.058) [DOI] [PubMed] [Google Scholar]
- 156.Round JL, Mazmanian SK. 2009. The gut microbiota shapes intestinal immune responses during health and disease. Nat. Rev. Immunol. 9, 313–323. (doi:10.1038/nri2515) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Frank DN, Amand ALS, Feldman RA, Boedeker EC, Harpaz N, Pace NR. 2007. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc. Natl Acad. Sci. USA 104, 13 780–13 785. (doi:10.1073/pnas.0706625104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Carvalho B, et al. 2012. Modulation of gut microbiota by antibiotics improves insulin signalling in high-fat fed mice. Diabetologia 55, 2823–2834. (doi:10.1007/s00125-012-2648-4) [DOI] [PubMed] [Google Scholar]
- 159.Minokoshi Y, Kim Y-B, Peroni OD, Fryer LG, Müller C, Carling D, Kahn BB. 2002. Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature 415, 339–343. (doi:10.1038/415339a) [DOI] [PubMed] [Google Scholar]
- 160.Yamauchi T, et al. 2002. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat. Med. 8, 1288–1295. (doi:10.1038/nm788) [DOI] [PubMed] [Google Scholar]
- 161.Everard A, et al. 2011. Responses of gut microbiota and glucose and lipid metabolism to prebiotics in genetic obese and diet-induced leptin-resistant mice. Diabetes 60, 2775–2786. (doi:10.2337/db11-0227) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Ben-Shlomo S, Zvibel I, Shnell M, Shlomai A, Chepurko E, Halpern Z, Barzilai N, Oren R, Fishmsn S. 2011. Glucagon-like peptide-1 reduces hepatic lipogenesis via activation of AMP-activated protein kinase. J. Hepatol. 54, 1214–1223. (doi:10.1016/j.jhep.2010.09.032) [DOI] [PubMed] [Google Scholar]
- 163.Minokoshi Y, et al. 2004. AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature 428, 569–574. (doi:10.1038/nature02440) [DOI] [PubMed] [Google Scholar]
- 164.Andersson U, Filipsson K, Abbott CR, Woods A, Smith K, Bloom SR, Carling D, Small CJ. 2004. AMP-activated protein kinase plays a role in the control of food intake. J. Biol. Chem. 279, 12 005–12 008. (doi:10.1074/jbc.C300557200) [DOI] [PubMed] [Google Scholar]
- 165.Hardie DG, Ross FA, Hawley SA. 2012. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 13, 251–262. (doi:10.1038/nrm3311) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This article has no additional data.