

Abstract
Compaction of the genome into the nuclear space is achieved by wrapping DNA around octameric assemblies of histone proteins to form nucleosomes, the fundamental repeating unit of chromatin. Aside from providing a means by which to fit larger genomes into the cell, chromatinization of DNA is a crucial means by which the cell regulates access to the genome. While the complex role that chromatin plays in gene transcription has been appreciated for a long time, it is now also apparent that crucial aspects of DNA replication are linked to the biology of chromatin. This review will focus on recent advances in our understanding of how the chromatin environment influences key aspects of DNA replication.
This article is part of the themed issue ‘Chromatin modifiers and remodellers in DNA repair and signalling'.
Keywords: DNA replication, nucleosome, chromatin
1. Introduction to replication origins
High-fidelity duplication of the genome is a basic requirement for organismal viability. Thus, understanding how the genome encodes the information for its own duplication is the preeminent question in the DNA replication field. The initial goal of early studies was to identify how replication is initiated. Seminal work in Escherichia coli revealed that DNA synthesis is initiated from a single point on the chromosome (origin) and proceeds bidirectionally until the two replication forks merge and terminate [1]. The E. coli chromosomal origin of replication (oriC) is specified by a single conserved sequence element that is subsequently bound by components of the bacterial replisome before initiating synthesis [2].
Contrasting with the situation in bacteria, eukaryotes use multiple origins that need to be fired at regular intervals to efficiently replicate larger chromosomes during each S-phase [3]. Origins in budding yeast are demarcated by conserved sequence elements called the autonomously replicating sequence (ARS) [4]. It is at these sites that the two essential, discrete steps of origin activation occur: origin licensing and origin firing. Licensing is mediated by the assembly of the pre-replication complex (pre-RC), which involves the binding of the hexameric origin recognition complex (ORC) to the origin, followed by the loading of the mini-chromosome maintenance (MCM) helicase complex onto DNA by ORC, along with Cdc6 and Cdt1. The loaded but inactive MCM helicase complex consists of a double hexamer of MCM2-7 proteins that encircle the double-stranded DNA [5,6]. In G1, any licensed origin has the potential to be fired in the subsequent S-phase, but origin firing is dependent upon the assembly of several additional factors, including CDC45 and GINS, which are regulated by two cell cycle–regulated protein kinases: Dbf4-dependent kinase (DDK) and cyclin-dependent kinase (CDK) [7]. After activation, the CDC45-MCM2-7-GINS (CMG) complex may begin translocation and unwinding of the parental genome, permitting DNA synthesis by the replicative DNA polymerases.
2. Chromatin and the initiation of DNA replication
The temporal separation of pre-RC assembly and activation represents a safeguard against over-replication, but the specific determinants that dictate which origins fire in any given S-phase are not completely understood. In yeast, the number of sequences that can specify origins far exceeds the actual number of origins used in S-phase [8]; and, in mammalian cells less than 10% of licensed origins are actually used in a given cell [9].
Many lines of evidence point to an important role for chromatin in the regulation of DNA replication. In the case of origin specification, recently developed in vitro replication systems from budding yeast have highlighted how chromatin helps specify sites of initiation [10,11]. Unlike the situation in vivo, pre-RC assembly and initiation on naked template DNA are not dependent upon specific initiator sequences [12,13]. However, chromatinization of the template dramatically influences the ability of ORC to stably bind to the DNA such that efficient binding and in vitro replication require a consensus sequence (ACS), which is a high-affinity ORC-binding site [10,11]. This finding ties in well with data from budding yeast where chromatin structure near replication origins appears to be tightly controlled: replication origins have a stereotypical nucleosome arrangement centred on a nucleosome-depleted region established by sequence-specific DNA-binding proteins as well as ORC [14]; indeed origin function is inhibited by encroachment of adjacent nucleosomes into the nucleosome-depleted region [15,16].
While budding yeast relies on DNA sequence elements for origin specification, the situation in metazoa is far more complex and less understood [17]. The first genome-wide replication-timing maps in Drosophila found a striking correlation between replication-timing and gene activity; early replicating regions of the genome coincided with a higher likelihood of gene activity on a genome-wide level [18]. Consistent with this, more recent work in Drosophila, Caenorhabditis elegans and mammalian cells has shown that sites of replication initiation are marked by the same histone modifications typically found at sites of active gene transcription [19–22]. In humans, Miotto et al. surveyed over 50 000 ORC-binding sites genome wide in order to distinguish chromatin features associated with selective ORC-binding and found the greatest predictor of ORC-binding patterns was accessible chromatin regions classified as DNase I hypersensitive sites (DHS) [19]. Based on this association with DHS, ORC-binding was also correlated with transcriptional activity and showed an enrichment of histone modifications typifying active chromatin, namely acetylation of histone H3 at lysine 27 (H3K27ac) and di-methylation of histone H3 at lysine 4 (H3K4me2). No other factors were found to be as predictive of ORC-binding, suggesting that selectivity of ORC localization in vivo largely results from opportunistic binding to accessible regions of DNA established by chromatin factors related to gene expression and not through a direct interaction with a DNA-binding factor or specific histone modification [19].
Nevertheless, there is considerable evidence that ORC makes specific interactions with histone modifications and, in particular, methylation states of histone H4 lysine 20 (H4K20me1/me2/me3) [23]. In mammals, H4K20 methylation depends on three known enzymes: PR-Set7 (Set8) is responsible for H4K20me1, Suv4-20H1 catalyses H4K20me2 and Suv4-20H2 catalyses H4K20me3. Of these, the most dynamic methylation state is H4K20me1, whose levels peak during M phase and steadily decline until reaching its lowest levels in S phase; these changes are mirrored by identical fluctuations in Set8 levels [23,24]. Initial reports in mammalian cells found severe replication defects associated with the stabilization or depletion of H4K20me1, suggesting a role for Set8 or H4K20me1 in regulating DNA replication [25]. The dynamic levels of Set8 (and thus, monomethylation on H4K20) are attributed to its degradation by the E3 ligase Cul4-Ddb1 through an interaction with PCNA, the homotrimeric ring that acts as a processivity factor for numerous replisome proteins [26]. Cells expressing a mutant form of Set8 that fails to interact with PCNA and cannot be degraded by Cul4-Ddb1, exhibit genome instability related to re-replication, indicating that the dynamic state of H4K20 methylation is important for regulating genome duplication [26]. The effect that disruption of H4K20 methylation state has on DNA replication was clarified by the Reinberg lab who showed that Suv4-20H1, which catalyses H4K20me2, facilitates the loading of ORC [27].
Supporting the role of H4K20 methylation in DNA replication is the finding that the ORC1 subunit possesses a bromo-adjacent homology domain (BAH), which is an evolutionary conserved chromatin recognition motif also found on the Sir3 chromatin silencing factor [28,29]. This BAH domain allows metazoan ORC1 to interact with methylated H4K20, with a significant preference for H4K20me2 over H4K20me1 and H4K20me3 [29]. Based on evidence that additional ORC subunits show lower binding to chromatin in ORC1-BAH mutant cells, the interaction with H4K20me2 probably helps stabilize the ORC complex association with chromatin [29]. Indeed, DNA replication in ORC1-BAH mutants is defective, with a large percentage of cells showing delayed S phase entry, a phenotype that is also seen in cells lacking the H4K20me2 methyltransferase, Suv4-20H1 [29,30].
Nonetheless, our understanding of ORC recruitment by H4K20 methylation remains incomplete. First, the profiling of ORC-binding sites and replication origins fails to show a strong enrichment for H4K20 methylation [19,20]; second, H4K20 methylation is associated with diverse biological outputs, including DNA repair, transcriptional silencing and the establishment of higher-order chromatin structure [23]. For example, H4K20 methylation states help to distinguish replicated from unreplicated chromatin during S phase [31], thus priming the HR repair machinery to bind newly replicated chromatin for downstream repair; this ancillary function of H4K20 methylation will be detailed in a later section. Given multitudinous roles in preserving genome stability, the delayed S phase entry in Set8 and Suv4-20 mutant cells could be related to their role in the DNA damage response or homologous recombination (HR) repair, rather than a deficiency in origin licensing and initiation. Indeed, using a combination of cytological, genetic and direct replication assays in Drosophila, the MacAlpine group identified that, while Set8 and H4K20me1 are important for cell-cycle progression, origin activation was not affected in the absence of Set8 or H4K20me1 [32]. In fact, greater than 50% of Drosophila mutants with alanine substitutions at H4K20 were viable, in stark contrast with the lethal phenotype of Set8 knockouts, indicating that the replication stress or DNA damage phenotypes reported in Set8 mutant cells could be related to other targets of Set8 methylation, including p53 and PCNA [32–34].
3. Replication in time and space
While nucleosome organization and histone modifications associated with transcription clearly influence where an origin can form, chromosomal context is important in defining when an origin fires during S phase. In budding yeast, simply by moving a given replication origin from a late-replicating region to an early replicating region of the chromosome, it is possible to advance the time of firing [35]. Furthermore, by targeting chromatin modifiers such as histone acetylases or deacetylases to specific sites on the chromosome, the origin efficiency, or probability of initiation, could be enhanced or suppressed [36,37]. Thus, particular origins are not pre-programmed to fire early or late during S-phase and the local chromatin environment could potentially permit or restrict an origin from firing.
The chromatin environment is intrinsically coupled to the organization of chromosomes within the nucleus [38]. Work from the Gilbert lab in vertebrate cells has characterized large chromosomal domains by their uniform and reproducible replication timing. These domains can be subdivided into two classes: constant timing regions (CTRs) typified by relatively synchronous and consistent firing of closely spaced origin clusters from cell to cell, and timing transition regions (TTRs) representing regions where replication-timing shifts from early to late [9,39,40]. Strikingly, the mapping of the CTRs overlaps with previously mapped topologically associating domains (TADs), which are chromosomal regions that permit specific and frequent interactions within a defined compartmentalized region of the chromosome and preclude interaction with neighbouring domains (figure 1) [38,39,41]. Thus, the programme by which the genome will be replicated appears to be determined by the structural organization of chromatin within the nucleus; and this structure is known to be established well before cells enter S phase [42]. Efforts to understand how the replication programme is determined will probably depend upon the challenging task of understanding the mechanisms by which chromosomes are organized within the nucleus; for example, in budding yeast, the forkhead proteins Fkh1/2 help early-firing of origins by promoting origin clustering in three-dimensional space [43]. In metazoa, the Rif1 protein potentially influences replication timing by mediating interactions between late-replicating regions and the nuclear lamina [44,45].
Figure 1.
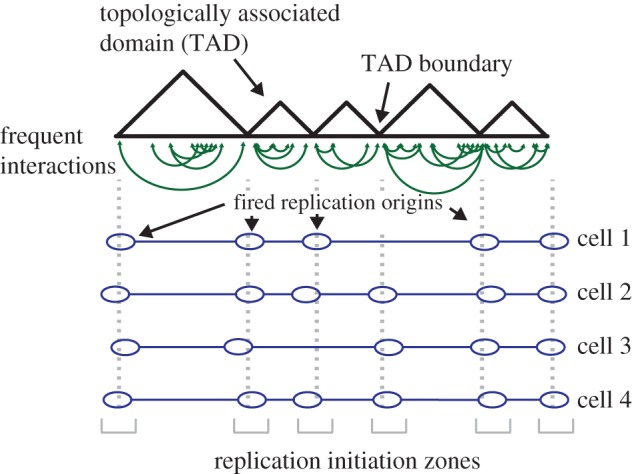
Topologically associated domain (TAD) structure and relation to replication origins. Representation of a region of a chromosome containing TADs. TADs contain elements of chromatin, such as gene promoters and enhancers that make frequent interactions with each other (depicted as green lines). TADs are separated by TAD boundaries; chromatin within one TAD seldom interacts with chromatin from another. Replication origins are enriched at TAD boundaries (marked as blue ovals), but the precise sites of initiation varies from cell to cell within a population; this gives rise to replication initiation zones, which are depicted at the bottom in grey.
The finding that nuclear organization delineates broad domains in the replication-timing programme is a significant development; however, further information is revealed by high-resolution mapping of origin location and efficiencies in human cells. The sequencing of Okazaki fragments (OF) allows the measurement of replication fork direction and represents a useful means by which to map the dynamics of replication at high resolution [20,46,47]. By sequencing OFs, the Hyrien group revealed that initiation in human cells is often confined to specific regions of chromosomes spanning tens of kilobases [20]. The precise sites of initiation within these zones vary from cell to cell within the population. Consistent with the proposed role of higher-order chromatin organization in replication origin function [40], OF mapping revealed that early-firing replication origins typically overlap TAD boundaries [20]. As would be expected from this correlation, the biology of TAD boundaries and replication origins is similar: both are broadly associated with active genes, but gene activity is not strictly required nor is it predictive [38,40].
How might origins and TAD boundaries be specified? The answer is far from clear, but a potential clue comes from the finding that many TADs are consistently demarcated through various cell types and developmental stages. These ‘constitutive' TADs are often bounded by so-called ‘housekeeping’ genes [41], whose consistent expression in cycling cells sets them apart from inducible genes with variable expression. Such an association makes logical sense: cells presumably must always express housekeeping genes when they are replicating, i.e. when origins are active. The connection between gene expression and DNA replication is further illustrated in a recent report investigating replication origin use in developing C. elegans embryos [21]. Here, similar to human cells, replication initiates from broad zones [20] but the midpoint of origins is demarcated by the histone modifications H3K27ac and H3K4me2 that are found at gene promoters and enhancers [48], and were recently found enriched at ORC-binding sites in human cells [19]. Essentially, all origins have these modifications and the vast majority of sites of modification are origins. Thus, the transcription and replication programmes are probably interlinked. How can coupling of the replication programme and transcription programme be executed with this arrangement? The answer lies in the finding that genes are non-randomly distributed across the genome: those next to replication origins are strongly biased for genes that are expressed during growth, which necessarily includes housekeeping genes. With this arrangement C. elegans embryonic cells can replicate within 20 min and simultaneously express genes necessary for growth [21].
In somatic cells, only a subset of replication initiation events occurs near actively transcribed genes. Indeed, perhaps the most interesting aspect of the high-resolution mapping of human replication origins is that vast regions of the genome that replicate late in S phase do not rely on initiation from specific zones [20]. Such late regions are broadly transcriptionally inactive and are enriched in heterochromatin. The initiation of late-replicating domains appears to be dictated by a cascade of replication origin firing events that are initiated from an early-firing region [20]. Licensed origins that are scattered through the late-replicating region are presumably induced to fire by replication forks encroaching from early-firing regions—perhaps by re-utilization of limiting replication factors. Indeed, factors required for replication initiation are known to be limiting and would permit a subset of origins to fire at the same time [49,50]. Thus, the local control of replication in late regions is, in part, reliant upon the timely replication of early regions. Collectively, these data support a model in which replication origins, sectored within their respective chromosomal domains, have varying probabilities of firing at the start of S phase and that the firing probability of origins increases as S phase progresses [51]. The origins that fire early and more uniformly, probably have a greater firing probability based on permissive chromosomal characteristics: proximity to housekeeping genes and TAD boundaries, DHS and accessible chromatin.
Why are genomes segregated into early- and late-replicating regions? One explanation would be that such temporal segregation in origin firing would allow cellular metabolism to provide consistent amounts of metabolites for efficient replication. Indeed, the growth of budding yeast strains that simultaneously fire early and late origins is partially restricted by dNTP levels [50]. Second, the segregation of replication into domains allows cells to more efficiently deal with replication stress. Problems encountered during early S phase can trigger the S phase checkpoint and suppress the initiation of new replication origins at distant sites [52]. Thus, when problems occur in one region of the chromosome, cells can ensure that issues are resolved before completing replication of the remainder of the genome [9]. Finally, the broad division of the genome into early- and late-replicating regions may provide a simple means to increase the robustness with which domains of histone modifications and chromatin states are reestablished following S phase. For example, in budding yeast, acetyl-CoA is intrinsically linked with growth and levels of acetyl-CoA and bulk histone acetylation peak at the beginning of S phase and then drop through S phase [53,54]. Thus, early replicating regions, which are typically transcriptionally active, may promote increased acetylation of the newly assembled chromatin, and thereby mark transcriptionally active regions for the next generation. Consistent with this hypothesis, microinjection experiments in human cells have shown that the assembly of transcriptionally competent chromatin is dependent upon the timing of the injection, with DNA injected early in S-phase being assembled into acetylated chromatin and expressed at higher levels [55,56]. Temporally separating the replication of active and repressed chromatin may therefore reinforce distinct chromatin types.
4. Replicating through chromatin: new views on the helicase
Once origins are fired a central issue in understanding how the replication fork proceeds through the genome is to unravel how the CMG helicase complex unwinds chromatinized DNA. New insights in this area have come from structural data that suggests the CMG helicase progresses through chromatin in the opposite orientation to what was previously thought [57]. The MCM2-7 replicative helicase consists of a hexameric ring, which, when combined with five accessory factors, comprise the 11-subunit CMG complex [58]. Each hexameric ring is composed of two tiers, comprising the C-terminal domain (CTD), which contains the ATP-binding sites and motor that drives translocation and DNA unwinding, and the N-terminal domain (NTD), respectively. When loaded onto DNA in G1, MCM2-7 double hexamers are oriented in a NTD to NTD manner, such that the motor CTD domains face away from one another [5,6]. Based on the orientation of the double hexamers in G1, it was believed that after activation, the CMG complexes simply migrated away from each other with the CTD on the leading end of 3′–5′ translocation [59]. By using cryo-EM to visualize CMG on forked DNA substrates, Georgescu et al. were able to capture the helicase in ‘translocation mode'; surprisingly, their structures revealed that the NTD is proximal to the fork and the CTD motor trails behind (figure 2). This finding is important for a number of reasons: first, because the leading strand polymerase (ɛ) associates with the CTD and polymerase α/primase, associates with the NTD [60], this orientation of CMG logically positions each polymerase for synthesis on their associated strands: polymerase ɛ can synthesize the leading strand as its template emerges from the CMG. Second, this orientation of CMG minimizes the amount of exposed single-stranded DNA on the lagging strand as the lagging-strand template, unwound at the front of the replication fork, can be primed by polymerase α/primase. Third, the model reveals an elegant quality control mechanism ensuring that each hexamer associates with the opposite strand of DNA before separating [57]. As CMGs are loaded onto double-stranded DNA prior to initiation and translocate on single-stranded DNA, the ‘NTD first' orientation implies that the hexameric rings must pass one another during initiation, which is only possible once both hexamers encircle single-stranded DNA. Finally, the new translocation orientation of CMG potentially reveals new modes for chromatin disassembly and parental histone recycling. The threading of the 3′ end of the DNA through the leading NTD positions the recently characterized MCM2 histone-binding domain at the very front of the CMG [61,62]. MCM2 chaperones H3:H4 tetramers both in vitro and in vivo by wrapping around the tetramer, much like nucleosomal DNA [61,62]. Therefore, the new model indicates that MCM2 could play a major role in disassembling parental nucleosomes in front of the replication fork (figure 3). The authors also speculate that the new position of the lagging-strand machinery at the leading edge of the CMG increases the likelihood that parental nucleosome deposition would preferentially occur on the lagging strand, opening up intriguing mechanisms for chromatin state inheritance and nucleosome assembly. However, with the exception of the remarkable, yet poorly understood inheritance of histone proteins in Drosophila male germline stem cells [63], there is little evidence of biased segregation of parental histones to the daughter genomes [64,65]. This may reflect the inherent asymmetry of the replication fork imparts little bias on histone segregation; or, that mechanisms have evolved—perhaps employing specific histone chaperones—to ensure the equal passage of parental histone to the daughter genomes.
Figure 2.
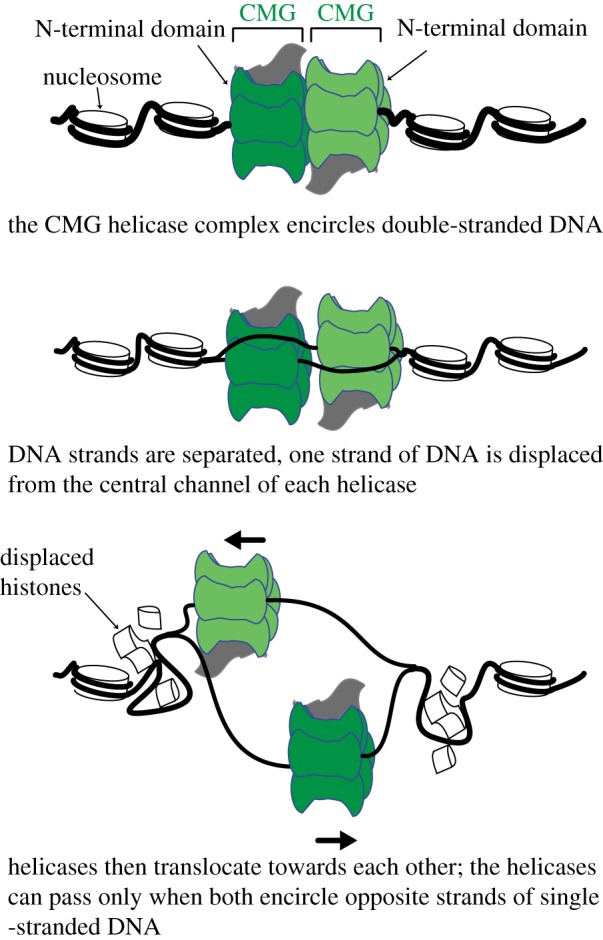
A model for helicase activation and separation. See main text for details.
Figure 3.

Dynamics at the replication fork. Representation of the replication fork progressing through chromatin. For simplicity, several proteins are omitted and only proteins discussed in the text are included. See text for details.
5. In vitro replication of chromatin template
Progression of the replication fork through chromatin is also stimulated by an array of other factors that probably alter the structure of chromatin. Here newly developed in vitro systems are beginning to reveal important clues. Using an in vitro replication system with purified components Kurat et al., were able to identify several proteins specifically associated with replicating chromatin [11]. Most prominent were histone chaperones (FACT, Nhp6, Asf1), chromatin remodelling complexes (INO80, Isw1), and histone acetyltransferase complexes (NuA4, SAGA). Importantly, efficient replication of chromatinized DNA required FACT, which is consistent with the noted role for FACT in promoting transcription from chromatinized templates and the general understanding of how FACT can disrupt the structural integrity of the nucleosome [66–68]. FACT associates with the replisome progression complex, [69] and interacts with multiple components at the replication fork, including DNA polymerase α [70] and the MCM2 N-terminal tail, where it forms a salt resistant complex with histones [71]. In the light of the recent findings regarding the orientation of translocating CMG [57] these results would place FACT at the leading edge of the helicase where it would presumably collaborate with the MCM2 tail to mediate the unwinding of parental histones (figure 3). Such a scenario is supported by the discovery that histones captured from FACT-MCM2 complexes lacked acetylation of histone H3 at lysine 56 [71]; which is a modification found on newly synthesized histones [72].
Kurat et al. also showed that the ATP-dependent chromatin remodelling complexes INO80 and ISW1a, and histone acetyltransferases, SAGA and NuA4, were all required in order to achieve rates of replication comparable with those measured in vivo [11]. Of these factors, Isw1 and INO80 have been implicated in various aspects of DNA replication: in budding yeast, Isw1 repositions nucleosomes on nascent DNA [73] and in human cells an ISW1-related complex (ACF1-ISWI) promotes efficient replication of heterochromatin [74]. INO80 has also been shown to interact with replication origins and the replication fork [75] and depletion of INO80 results in slowed replication fork progression [76]. Moreover, there is increasing evidence that INO80 is required for replication restart after fork stalling and can function in a pathway to evict RNA polymerase II from chromatin [77] upon replication stress [78]. However, with the exception of FACT, it remains to be determined whether the factors examined by Kurat et al. are specifically targeted to replication forks; and, if so, how they function in replication fork progression. Indeed, Devbhandari et al., who established a similar in vitro system achieved replication on chromatinized templates in the absence of FACT and many of the stimulatory factors described by Kurat et al. their reactions contained the histone chaperone NAP1 and Isw1, which were included to assemble nucleosomes on the template DNA. These factors presumably also stimulated replication through chromatin and nucleosome assembly on the nascent DNA. Thus, while many proteins appear capable of stimulating replication, it appears that no single factor is specifically required for replication through templates.
6. Chromatin regulates lagging-strand synthesis
The in vitro systems described by Devbhandari et al., and Kurat et al. also noted a profound alteration in lagging-strand synthesis when replication occurred on chromatin templates. The frequency at which the polymerase α/primase complex initiates each OF will influence the ultimate length of OFs produced by the replisome. On naked DNA templates, polymerase α/primase acts distributively: OFs become shorter—hence were more frequently initiated—with increasing amounts of polymerase α/primase [79]. When replication occurred through chromatin, Kurat et al., noted that the initiation frequency became much less sensitive to the concentration of polymerase α/primase—indicating that polymerase α/primase may act processively in the context of chromatin [11]. It is unclear how chromatin should influence the frequency of OF initiation: one mechanism would be that chromatin somehow helps sequester polymerase α/primase to the replisome; but another interesting possibility is that MCM helicase progression may slow each time a nucleosome is encountered. If the rate-limiting step in replication fork progression is assumed to be the unwinding of nucleosomal DNA ahead of the replication fork, then initiation of OFs by polymerase α/primase may occur as the fork slows from one nucleosome to the next. Thus, the initiation site and the frequency of initiation events (hence the ultimate length of the OF) could, at least in part, be dictated by nucleosome structure and how many nucleosomes the fork moves through each cycle.
Aside from the frequency of initiation, the lengths of OFs are also dictated by a processing reaction in which polymerase δ simultaneously extends the 3′ end of a nascent OF and triggers the degradation of the 5′ end of the preceding OF [80]. Repeated cycles of extension and DNA cleavage produce a nick that migrates away from the replication fork and can be sealed by DNA ligase I [80]. This reaction—known as strand displacement synthesis—relies upon structure specific nucleases such as Fen1 to degrade the RNA or DNA displaced by polymerase δ (figure 3). Devbhandari et al. incorporated the basic components for OF processing, including the flap endonuclease Fen1 and DNA ligase I in their in vitro system [10]. Interestingly, they noted that while the replication of naked plasmid DNA occurred robustly, much of the synthesized DNA was greater than unit length—meaning that unconstrained synthesis was occurring (most probably by polymerase δ) and only a fraction of the lagging-strand products were small and competent for ligation [10]. Replication of chromatinized templates dramatically suppressed the extent of DNA polymerization, resulting in short lagging-strand products that were readily ligated. Since the DNA replication reactions were conducted with an excess of core histones, Nap1 and Isw1 (which are potent nucleosome assembly factors [81]) the suppression of polymerase δ is most readily explained by the reassembly of nucleosomes on the lagging strand, which presumably prevent extensive strand displacement synthesis by polymerase δ [10]. These data support earlier results from budding yeast which showed that nucleosome assembly on daughter strands is required for optimal processing of OFs in vivo [47].
The requirement for nucleosome assembly to constrain DNA synthesis on the lagging strand potentially provides a means to ensure the removal of error prone DNA synthesized by DNA polymerase α while preventing excessive strand displacement synthesis by polymerase δ [47]. In addition, inhibition of polymerase δ by newly assembled nucleosomes may allow components of the lagging-strand machinery to be recycled from one OF to the next; and, in doing so, allow the fidelity of nucleosome assembly to be communicated to the replication fork. In this scenario, defects in nucleosome assembly on the lagging strand result in extensive strand displacement synthesis by polymerase δ. Thus, in the absence of timely nucleosome assembly, the synthesis of each OF would be slowed, which may ultimately slow the replication fork—allowing the rate of nucleosome assembly to be coupled with the rate of DNA synthesis. Studies in mammalian cells indicate that replication fork progression through chromatin requires efficient delivery of newly synthesized histones [82] and replication forks are slowed when the nucleosome assembly is impaired [83]. With this reasoning, it is worth considering whether some of the stimulatory effects on DNA synthesis of histone chaperones and chromatin remodelling enzymes seen in vitro replication systems may be attributed to the promotion of nucleosome assembly and efficient lagging-strand synthesis.
7. Priming for recombination during DNA replication
Even in the absence of exogenous DNA damage, faithful completion of DNA replication relies upon the HR pathway to protect stalled replication forks or to restart collapsed forks [84]. DNA replication produces sister chromosomes, but because chromosomes are replicated at different times during S phase, an interesting question is how do cells know when they have a sister with which to repair? Recent data suggests that events occurring at the replication fork help the HR machinery discriminate between replicating and non-replicating chromatin [31]. TONSL-MMS22 L is a heterodimeric complex capable of interacting with histones as well as the histone chaperones Asf1 and MCM2 via the TONSL ankyrin repeat domain (ARD) [85,86]. Structural characterization shows that the binding of soluble histones H3-H4 by TONSL bridges the connection to ASF1 and MCM2, creating a co-chaperone complex prior to deposition of histones during replication coupled chromatin assembly [31]. The co-chaperone complex is dependent upon a number of interactions on the histone H4 tail, including lysine 20 (H4K20), the methylation of which is associated with replication and repair processes that maintain genome integrity [23]. Importantly, H4K20 methylation (H4K20me) abolishes TONSL binding to nucleosomes, suggesting that TONSL-MMS22 L specifically recognizes unmodified histones at H4K20. Since newly synthesized histones deposited in S phase are unmethylated at K20 [72], TONSL-MMS22 L is thus likely to bind to replication forks and nascent chromatin. However, TONSL-MMS22 L recruitment to chromatin occurs within a narrow temporal window as H4K20 is methylated by the histone methyltransferase SET8 [23] in late S phase. Based on this timely recruitment of TONSL-MMS22L and its known role as a mediator of HR, the recognition of nascent chromatin by the complex probably promotes expedient HR repair at compromised replication forks [31,85,86]. Indeed, a subsequent report provided direct evidence for the recruitment of TONSL-MMS22 L to collapsed replication forks and its promotion of Rad51-dependent recombination [87]. Presumably, recruitment of TONSL-MMS22 L to replication forks, allows efficient interaction with RPA, which coats stretches of single-stranded DNA formed during collapse of replication forks [87]. While it is known that the TONSL-MMS22 L heterodimer is required for RAD51 foci formation upon DNA damage [85,86], it is also required to recruit RAD51 to stalled replication forks [87]. An intact TONSL-MMS22 L heterodimer forms a tight interaction with two molecules of RAD51 and was shown to facilitate strand exchange by reducing the affinity of RAD51 for double-stranded DNA, a function similar to the tumour suppressor BRCA2 [87–89]. These findings illustrate an interesting new paradigm, in which replication coupled chromatin assembly provides an opportunity to prime newly replicated daughter genomes with repair complexes in the event of replication stress or DNA damage. In the light of these findings, it would be of interest to identify whether complexes similar to TONSL-MMS22 L are able to recognize chromatin specific transitions as a signal to load the DNA repair components best suited to fix the damage within the specific cell-cycle phase.
8. Conclusion
The advances in biochemical characterization of the replisome and its components have reinforced our understanding of how integrated the passage of the replication fork is with chromatin dynamics. Indeed, while in vitro systems have shown that DNA can be efficiently replicated in the absence of chromatin, it is clear that the presence of nucleosomes on the template DNA can constrain sites of initiation and the processivity of the DNA polymerases. But rather than a simple impediment, chromatin should more realistically be viewed as a modulator that can fine tune many aspects of DNA replication. The new in vitro systems offer tantalizing insights into how replication occurs on chromatin, yet they remain incomplete. Most notably, neither report examined components of the replication-associated chromatin assembly system that couples nucleosome assembly, mediated in part through the histone chaperone CAF-1, to the replication fork through PCNA. Once this pathway is included, it may be possible to faithfully recapitulate nucleosome assembly on nascent DNA in vitro. But a considerable challenge with the biochemical systems will be to achieve a stoichiometry of the components, which faithfully recapitulates the situation in vivo. Thus, more quantitative assays that interrogate in vivo replication will be needed to supplement the in vitro systems.
From the demonstration that ORC binds to accessible DNaseI hypersensitive sites, to the association of replication timing with higher-order chromatin folding, it is apparent that DNA replication is profoundly influenced by chromatin organization. Given that the same chromatin features are implicated in gene transcription and DNA replication, some immediate challenges will be to disentangle causal relationships between the processes at play. This may prove challenging as perturbation of one process will probably affect the other; nevertheless the realization that chromatin structures typically associated with gene transcription are also used in DNA replication may provide a new perspective from which we may better understand how and why such chromatin structures are established and maintained. While ORC-binding represents the critical first step in origin licensing in G1, it is yet unknown how ORC is targeted to chromatin or whether chromatin structural changes precede ORC-binding and MCM loading. Most certainly, a deeper understanding of how chromatin is organized within the nucleus and the factors responsible for such organization will prove valuable to many aspects of genome research.
Acknowledgements
We thank D. Remus, X. Zhao and members of the Whitehouse lab at MSKCC for helpful discussions and comments on this manuscript.
Data accessibility
This article has no additional data.
Authors' contributions
The manuscript was written and edited by J.M.B. and I.W.
Competing interests
We declare we have no competing interests.
Funding
Work in the Whitehouse lab is supported by the NIH (R01 GM102253, P30CA008748) and the American Cancer Society (128073-RSG-15-041-01-DMC).
References
- 1.Jacob F, Brenner S, Cuzin F (eds). 1963. On the regulation of DNA replication in bacteria. Cold Spring Harbor symposia on quantitative biology. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press. [Google Scholar]
- 2.O'Donnell M, Langston L, Stillman B. 2013. Principles and concepts of DNA replication in bacteria, Archaea, and Eukarya. Cold Spring Harb. Perspect. Biol. 5, a010108 ( 10.1101/cshperspect.a010108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hyrien O. 2015. Peaks cloaked in the mist: the landscape of mammalian replication origins. J. Cell Biol. 208, 147–160. ( 10.1083/jcb.201407004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huberman JA, Spotila LD, Nawotka KA, el-Assouli SM, Davis LR. 1987. The in vivo replication origin of the yeast 2 microns plasmid. Cell 51, 473–481. ( 10.1016/0092-8674(87)90643-X) [DOI] [PubMed] [Google Scholar]
- 5.Remus D, Beuron F, Tolun G, Griffith JD, Morris EP, Diffley JF. 2009. Concerted loading of Mcm2-7 double hexamers around DNA during DNA replication origin licensing. Cell 139, 719–730. ( 10.1016/j.cell.2009.10.015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Evrin C, Clarke P, Zech J, Lurz R, Sun J, Uhle S, Li H, Stillman B, Speck C. 2016. A double-hexameric MCM2-7 complex is loaded onto origin DNA during licensing of eukaryotic DNA replication. Proc. Natl Acad. Sci. USA 106, 20 240–20 245. ( 10.1073/pnas.0911500106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bell SP, Labib K. 2009. Chromosome duplication in Saccharomyces cerevisiae. Genetics 203, 1027–1067. ( 10.1534/genetics.115.186452) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kaykov A, Nurse P. 2015. The spatial and temporal organization of origin firing during the S-phase of fission yeast. Genome Res. 25, 391–401. ( 10.1101/gr.180372.114) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rivera-Mulia JC, Gilbert DM. 2016. Replicating large genomes: divide and conquer. Mol. Cell 62, 756–765. ( 10.1016/j.molcel.2016.05.007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Devbhandari S, Jiang J, Kumar C, Whitehouse I, Remus D. 2017. Chromatin constrains the initiation and elongation of DNA replication. Mol. Cell 65, 131–141. ( 10.1016/j.molcel.2016.10.035) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kurat CF, Yeeles JT, Patel H, Early A, Diffley JF. 2017. Chromatin controls DNA replication origin selection, lagging-strand synthesis, and replication fork rates. Mol. Cell 65, 117–130. ( 10.1016/j.molcel.2016.11.016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gros J, Devbhandari S, Remus D. 2014. Origin plasticity during budding yeast DNA replication in vitro. EMBO J. 33, 621–636. ( 10.1002/embj.201387278) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.On KF, Beuron F, Frith D, Snijders AP, Morris EP, Diffley JF. 2014. Prereplicative complexes assembled in vitro support origin-dependent and independent DNA replication. EMBO J. 33, 605–620. ( 10.1002/embj.201387369) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eaton ML, Galani K, Kang S, Bell SP, MacAlpine DM. 2010. Conserved nucleosome positioning defines replication origins. Genes Dev. 24, 748–753. ( 10.1101/gad.1913210) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Simpson RT. 1990. Nucleosome positioning can affect the function of a cis-acting DNA element in vivo. Nature 343, 387–389. ( 10.1038/343387a0) [DOI] [PubMed] [Google Scholar]
- 16.Lipford JR, Bell SP. 2001. Nucleosomes positioned by ORC facilitate the initiation of DNA replication. Mol. Cell 7, 21–30. ( 10.1016/S1097-2765(01)00151-4) [DOI] [PubMed] [Google Scholar]
- 17.Prioleau MN, MacAlpine DM.. 2016. DNA replication origins-where do we begin? Genes Dev. 30, 1683–1697. ( 10.1101/gad.285114.116) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schubeler D, Scalzo D, Kooperberg C, van Steensel B, Delrow J, Groudine M. 2002. Genome-wide DNA replication profile for Drosophila melanogaster: a link between transcription and replication timing. Nat. Genet. 32, 438–442. ( 10.1038/ng1005) [DOI] [PubMed] [Google Scholar]
- 19.Miotto B, Ji Z, Struhl K. 2016. Selectivity of ORC binding sites and the relation to replication timing, fragile sites, and deletions in cancers. Proc. Natl Acad. Sci. USA 113, E4810– E4819. ( 10.1073/pnas.1609060113) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Petryk N, Kahli M, d'Aubenton-Carafa Y, Jaszczyszyn Y, Shen Y, Silvain M, Thermes C, Chen C-L, Hyrien O. 2016. Replication landscape of the human genome. Nat. Commun. 7, 10208 ( 10.1038/ncomms10208) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pourkarimi E, Bellush JM, Whitehouse I. 2016. Spatiotemporal coupling and decoupling of gene transcription with DNA replication origins during embryogenesis in C. elegans. Elife 5, e21728 ( 10.7554/eLife.21728) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rodríguez-Martínez M, Pinzon N, Ghommidh C, Beyne E, Seitz H, Cayrou C, Méchali M. 2017. The gastrula transition reorganizes replication-origin selection in Caenorhabditis elegans. Nat. Struct. Mol. Biol. 24, 290–299. ( 10.1038/nsmb.3363) [DOI] [PubMed] [Google Scholar]
- 23.Jorgensen S, Schotta G, Sorensen CS. 2013. Histone H4 lysine 20 methylation: key player in epigenetic regulation of genomic integrity. Nucleic Acids Res. 41, 2797–2806. ( 10.1093/nar/gkt012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nishioka K, et al. 2002. PR-Set7 is a nucleosome-specific methyltransferase that modifies lysine 20 of histone H4 and is associated with silent chromatin. Mol. Cell 9, 1201–1213. ( 10.1016/S1097-2765(02)00548-8) [DOI] [PubMed] [Google Scholar]
- 25.Tardat M, Murr R, Herceg Z, Sardet C, Julien E. 2007. PR-Set7-dependent lysine methylation ensures genome replication and stability through S phase. J. Cell Biol. 179, 1413–1426. ( 10.1083/jcb.200706179) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tardat M, Brustel J, Kirsh O, Lefevbre C, Callanan M, Sardet C, Julien E. 2010. The histone H4 Lys 20 methyltransferase PR-Set7 regulates replication origins in mammalian cells. Nat. Cell Biol. 12, 1086–1093. ( 10.1038/ncb2113) [DOI] [PubMed] [Google Scholar]
- 27.Beck DB, Burton A, Oda H, Ziegler-Birling C, Torres-Padilla ME, Reinberg D. 2012. The role of PR-Set7 in replication licensing depends on Suv4-20 h. Genes Dev. 26, 2580–2589. ( 10.1101/gad.195636.112) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Armache KJ, Garlick JD, Canzio D, Narlikar GJ, Kingston RE. 2011. Structural basis of silencing: Sir3 BAH domain in complex with a nucleosome at 3.0 A resolution. Science 334, 977–982. ( 10.1126/science.1210915) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kuo AJ, Song J, Cheung P, Ishibe-Murakami S, Yamazoe S, Chen JK, Patel DJ, Gozani O. 2012. The BAH domain of ORC1 links H4K20me2 to DNA replication licensing and Meier-Gorlin syndrome. Nature 484, 115–119. ( 10.1038/nature10956) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schotta G, Lachner M, Sarma K, Ebert A, Sengupta R, Reuter G, Reinberg D, Jenuwein T. 2004. A silencing pathway to induce H3-K9 and H4-K20 trimethylation at constitutive heterochromatin. Genes Dev. 18, 1251–1262. ( 10.1101/gad.300704) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Saredi G, et al. 2016. H4K20me0 marks post-replicative chromatin and recruits the TONSL-MMS22 L DNA repair complex. Nature 534, 714–718. ( 10.1038/nature18312) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li Y, Armstrong RL, Duronio RJ, MacAlpine DM. 2016. Methylation of histone H4 lysine 20 by PR-Set7 ensures the integrity of late replicating sequence domains in Drosophila. Nucleic Acids Res. 44, 7204–7218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shi X, Kachirskaia I, Yamaguchi H, West LE, Wen H, Wang EW, Dutta S, Appella E, Gozani O. 2007. Modulation of p53 function by SET8-mediated methylation at lysine 382. Mol. Cell 27, 636–646. ( 10.1016/j.molcel.2007.07.012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Takawa M, et al. 2012. Histone lysine methyltransferase SETD8 promotes carcinogenesis by deregulating PCNA expression. Cancer Res. 72, 3217–3227. ( 10.1158/0008-5472.CAN-11-3701) [DOI] [PubMed] [Google Scholar]
- 35.Ferguson BM, Fangman WL. 1992. A position effect on the time of replication origin activation in yeast. Cell 68, 333–339. ( 10.1016/0092-8674(92)90474-Q) [DOI] [PubMed] [Google Scholar]
- 36.Goren A, Tabib A, Hecht M, Cedar H. 2008. DNA replication timing of the human beta-globin domain is controlled by histone modification at the origin. Genes Dev. 22, 1319–1324. ( 10.1101/gad.468308) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vogelauer M, Rubbi L, Lucas I, Brewer BJ, Grunstein M. 2002. Histone acetylation regulates the time of replication origin firing. Mol. Cell 10, 1223–1233. ( 10.1016/S1097-2765(02)00702-5) [DOI] [PubMed] [Google Scholar]
- 38.Sexton T, Cavalli G. 2015. The role of chromosome domains in shaping the functional genome. Cell 160, 1049–1059. ( 10.1016/j.cell.2015.02.040) [DOI] [PubMed] [Google Scholar]
- 39.Pope BD, et al. 2014. Topologically associating domains are stable units of replication-timing regulation. Nature 515, 402–405. ( 10.1038/nature13986) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rivera-Mulia JC, Gilbert DM. 2016. Replication timing and transcriptional control: beyond cause and effect-part III. Curr. Opin Cell Biol. 40, 168–178. ( 10.1016/j.ceb.2016.03.022) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B. 2012. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376–380. ( 10.1038/nature11082) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dileep V, Ay F, Sima J, Vera DL, Noble WS, Gilbert DM. 2015. Topologically associating domains and their long-range contacts are established during early G1 coincident with the establishment of the replication-timing program. Genome Res. 25, 1104–1113. ( 10.1101/gr.183699.114) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Knott SR, Peace JM, Ostrow AZ, Gan Y, Rex AE, Viggiani CJ, Tavaré S, Aparicio OM. 2012. Forkhead transcription factors establish origin timing and long-range clustering in S. cerevisiae . Cell 148, 99–111. ( 10.1016/j.cell.2011.12.012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cornacchia D, et al. 2012. Mouse Rif1 is a key regulator of the replication-timing programme in mammalian cells. EMBO J. 31, 3678–3690. ( 10.1038/emboj.2012.214) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Foti R, et al. 2016. Nuclear architecture organized by Rif1 underpins the replication-timing program. Mol. Cell 61, 260–273. ( 10.1016/j.molcel.2015.12.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McGuffee SR, Smith DJ, Whitehouse I. 2013. Quantitative, genome-wide analysis of eukaryotic replication initiation and termination. Mol. Cell 50, 123–135. ( 10.1016/j.molcel.2013.03.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Smith DJ, Whitehouse I. 2012. Intrinsic coupling of lagging-strand synthesis to chromatin assembly. Nature 483, 434–438. ( 10.1038/nature10895) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ho JW, et al. 2014. Comparative analysis of metazoan chromatin organization. Nature 512, 449–452. ( 10.1038/nature13415) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Collart C, Allen GE, Bradshaw CR, Smith JC, Zegerman P. 2013. Titration of four replication factors is essential for the Xenopus laevis midblastula transition. Science 341, 893–896. ( 10.1126/science.1241530) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mantiero D, Mackenzie A, Donaldson A, Zegerman P. 2011. Limiting replication initiation factors execute the temporal programme of origin firing in budding yeast. EMBO J. 30, 4805–4814. ( 10.1038/emboj.2011.404) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rhind N, Gilbert DM. 2013. DNA replication timing. Cold Spring Harb. Perspect Biol. 5, a010132 ( 10.1101/cshperspect.a010132) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Blow JJ, Ge XQ. 2009. A model for DNA replication showing how dormant origins safeguard against replication fork failure. EMBO Rep. 10, 406–412. ( 10.1038/embor.2009.5) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cai L, Tu BP. 2011. Acetyl-CoA drives the transcriptional growth program in yeast. Cell Cycle 10, 3045–3046. ( 10.4161/cc.10.18.17000) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tu BP, Mohler RE, Liu JC, Dombek KM, Young ET, Synovec RE, McKnight SL. 2009. Cyclic changes in metabolic state during the life of a yeast cell. Proc. Natl Acad. Sci. USA 104, 16 886–16 891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lande-Diner L, Zhang J, Cedar H. 2007. Shifts in replication timing actively affect histone acetylation during nucleosome reassembly. Mol. Cell 34, 767–774. ( 10.1016/j.molcel.2009.05.027) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang J, Xu F, Hashimshony T, Keshet I, Cedar H. 2002. Establishment of transcriptional competence in early and late S phase. Nature 420, 198–202. ( 10.1038/nature01150) [DOI] [PubMed] [Google Scholar]
- 57.Georgescu R, Yuan Z, Bai L, de Luna Almeida Santos R, Sun J, Zhang D, Yurieva O, Li H, O'Donnell ME. 2013. Structure of eukaryotic CMG helicase at a replication fork and implications to replisome architecture and origin initiation. Proc. Natl Acad. Sci. USA 114, E697–E706. ( 10.1073/pnas.1620500114) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bell SD, Botchan MR. 2017. The minichromosome maintenance replicative helicase. Cold Spring Harb. Perspect Biol. 5, a012807 ( 10.1101/cshperspect.a012807) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Remus D, Diffley JF. 2009. Eukaryotic DNA replication control: lock and load, then fire. Curr. Opin Cell Biol. 21, 771–777. ( 10.1016/j.ceb.2009.08.002) [DOI] [PubMed] [Google Scholar]
- 60.Sun J, Shi Y, Georgescu RE, Yuan Z, Chait BT, Li H, O'Donnell ME. 2015. The architecture of a eukaryotic replisome. Nat. Struct. Mol. Biol. 22, 976–982. ( 10.1038/nsmb.3113) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Huang H, Stromme CB, Saredi G, Hödl M, Strandsby A, González-Aguilera C, Chen S, Groth A, Patel DJ. 2015. A unique binding mode enables MCM2 to chaperone histones H3-H4 at replication forks. Nat. Struct. Mol. Biol. 22, 618–626. ( 10.1038/nsmb.3055) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Richet N, et al. 2015. Structural insight into how the human helicase subunit MCM2 may act as a histone chaperone together with ASF1 at the replication fork. Nucleic Acids Res. 43, 1905–1917. ( 10.1093/nar/gkv021) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tran V, Lim C, Xie J, Chen X. 2012. Asymmetric division of Drosophila male germline stem cell shows asymmetric histone distribution. Science 338, 679–682. ( 10.1126/science.1226028) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Annunziato AT. 2005. Split decision: what happens to nucleosomes during DNA replication? J. Biol. Chem. 280, 12 065–12 068. ( 10.1074/jbc.R400039200) [DOI] [PubMed] [Google Scholar]
- 65.Ramachandran S, Henikoff S. 2015. Replicating nucleosomes. Sci. Adv. 1, e1500587 ( 10.1126/sciadv.1500587) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hsieh FK, Kulaeva OI, Patel SS, Dyer PN, Luger K, Reinberg D, Studitsky VM. 2013. Histone chaperone FACT action during transcription through chromatin by RNA polymerase II. Proc. Natl Acad. Sci. USA 110, 7654–7659. ( 10.1073/pnas.1222198110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Xin H, Takahata S, Blanksma M, McCullough L, Stillman DJ, Formosa T. 2009. yFACT induces global accessibility of nucleosomal DNA without H2A-H2B displacement. Mol. Cell 35, 365–376. ( 10.1016/j.molcel.2009.06.024) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tsunaka Y, Fujiwara Y, Oyama T, Hirose S, Morikawa K. 2016. Integrated molecular mechanism directing nucleosome reorganization by human FACT. Genes Dev. 30, 673–686. ( 10.1101/gad.274183.115) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gambus A, Jones RC, Sanchez-Diaz A, Kanemaki M, van Deursen F, Edmondson RD, Labib K. 2006. GINS maintains association of Cdc45 with MCM in replisome progression complexes at eukaryotic DNA replication forks. Nat. Cell Biol. 8, 358–366. ( 10.1038/ncb1382) [DOI] [PubMed] [Google Scholar]
- 70.Wittmeyer J, Formosa T. 1997. The Saccharomyces cerevisiae DNA polymerase alpha catalytic subunit interacts with Cdc68/Spt16 and with Pob3, a protein similar to an HMG1-like protein. Mol. Cell. Biol. 17, 4178–4190. ( 10.1128/MCB.17.7.4178) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Foltman M, Evrin C, De Piccoli G, Jones RC, Edmondson RD, Katou Y, Nakato R, Shirahige K, Labib K. 2013. Eukaryotic replisome components cooperate to process histones during chromosome replication. Cell Rep. 3, 892–904. ( 10.1016/j.celrep.2013.02.028) [DOI] [PubMed] [Google Scholar]
- 72.Hammond CM, Stromme CB, Huang H, Patel DJ, Groth A. 2017. Histone chaperone networks shaping chromatin function. Nat. Rev. Mol. Cell Biol. 18, 141–158. ( 10.1038/nrm.2016.159) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yadav T, Whitehouse I. 2016. Replication-coupled nucleosome assembly and positioning by ATP-dependent chromatin-remodeling enzymes. Cell Rep. 15, 715–723. ( 10.1016/j.celrep.2016.03.059) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Collins N, Poot RA, Kukimoto I, García-Jiménez C, Dellaire G, Varga-Weisz PD. 2002. An ACF1-ISWI chromatin-remodeling complex is required for DNA replication through heterochromatin. Nat. Genet. 32, 627–632. ( 10.1038/ng1046) [DOI] [PubMed] [Google Scholar]
- 75.Shimada K, Oma Y, Schleker T, Kugou K, Ohta K, Harata M, Gasser SM. 2008. Ino80 chromatin remodeling complex promotes recovery of stalled replication forks. Curr. Biol. 18, 566–575. ( 10.1016/j.cub.2008.03.049) [DOI] [PubMed] [Google Scholar]
- 76.Lee HS, Lee SA, Hur SK, Seo JW, Kwon J. 2014. Stabilization and targeting of INO80 to replication forks by BAP1 during normal DNA synthesis. Nat. Commun. 5, 5128 ( 10.1038/ncomms6128) [DOI] [PubMed] [Google Scholar]
- 77.Lafon A, Taranum S, Pietrocola F, Dingli F, Loew D, Brahma S, Bartholomew B, Papamichos-Chronakis M. 2015. INO80 chromatin remodeler facilitates release of RNA polymerase II from chromatin for ubiquitin-mediated proteasomal degradation. Mol. Cell 60, 784–796. ( 10.1016/j.molcel.2015.10.028) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Poli J, et al. 2016. Mec1, INO80, and the PAF1 complex cooperate to limit transcription replication conflicts through RNAPII removal during replication stress. Genes Dev. 30, 337–354. ( 10.1101/gad.273813.115) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yeeles JT, Janska A, Early A, Diffley JF. 2017. How the Eukaryotic replisome achieves rapid and efficient DNA replication. Mol. Cell 65, 105–116. ( 10.1016/j.molcel.2016.11.017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jin YH, Ayyagari R, Resnick MA, Gordenin DA, Burgers PM. 2003. Okazaki fragment maturation in yeast. II. Cooperation between the polymerase and 3'-5'-exonuclease activities of Pol delta in the creation of a ligatable nick. J. Biol. Chem. 278, 1626–1633. ( 10.1074/jbc.M209803200) [DOI] [PubMed] [Google Scholar]
- 81.Vary JC Jr, Fazzio TG, Tsukiyama T. 2004. Assembly of yeast chromatin using ISWI complexes. Methods Enzymol. 375, 88–102. ( 10.1016/S0076-6879(03)75006-X) [DOI] [PubMed] [Google Scholar]
- 82.Groth A, Ray-Gallet D, Quivy JP, Lukas J, Bartek J, Almouzni G. 2005. Human Asf1 regulates the flow of S phase histones during replicational stress. Mol. Cell 17, 301–311. ( 10.1016/j.molcel.2004.12.018) [DOI] [PubMed] [Google Scholar]
- 83.Mejlvang J, et al. 2014. New histone supply regulates replication fork speed and PCNA unloading. J. Cell Biol. 204, 29–43. ( 10.1083/jcb.201305017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.San Filippo J, Sung P, Klein H. 2008. Mechanism of eukaryotic homologous recombination. Annu. Rev. Biochem. 77, 229–257. ( 10.1146/annurev.biochem.77.061306.125255) [DOI] [PubMed] [Google Scholar]
- 85.O'Donnell L, et al. 2010. The MMS22 L-TONSL complex mediates recovery from replication stress and homologous recombination. Mol. Cell 40, 619–631. ( 10.1016/j.molcel.2010.10.024) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Duro E, et al. 2010. Identification of the MMS22 L-TONSL complex that promotes homologous recombination. Mol. Cell 40, 632–644. ( 10.1016/j.molcel.2010.10.023) [DOI] [PubMed] [Google Scholar]
- 87.Piwko W. 2010. The MMS22 L-TONSL heterodimer directly promotes RAD51-dependent recombination upon replication stress. EMBO J. 35, 2584–2601. ( 10.15252/embj.201593132) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Jensen RB, Carreira A, Kowalczykowski SC. 2016. Purified human BRCA2 stimulates RAD51-mediated recombination. Nature 467, 678–683. ( 10.1038/nature09399) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Liu J, Doty T, Gibson B, Heyer WD. 2010. Human BRCA2 protein promotes RAD51 filament formation on RPA-covered single-stranded DNA. Nat. Struct. Mol. Biol. 17, 1260–1262. ( 10.1038/nsmb.1904) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This article has no additional data.