

Abstract
Deforestation has detrimental consequences on biodiversity, affecting species interactions at multiple scales. The associations among vertebrates, pathogens and their commensal/symbiotic microbial communities (i.e. microbiomes) have important downstream effects for biodiversity conservation, yet we know little about how deforestation contributes to changes in host microbial diversity and pathogen abundance. Here, we tested the effects of landcover, forest connectivity and infection by the chytrid fungus Batrachochytrium dendrobatidis (Bd) on amphibian skin bacterial diversity along deforestation gradients in Brazilian landscapes. If disturbance to natural habitat alters skin microbiomes as it does in vertebrate host communities, then we would expect higher host bacterial diversity in natural forest habitats. Bd infection loads are also often higher in these closed-canopy forests, which may in turn impact skin-associated bacterial communities. We found that forest corridors shaped composition of host skin microbiomes; high forest connectivity predicted greater similarity of skin bacterial communities among host populations. In addition, we found that host skin bacterial diversity and Bd loads increased towards natural vegetation. Because symbiotic bacteria can potentially buffer hosts from Bd infection, we also evaluated the bi-directional microbiome-Bd link but failed to find a significant effect of skin bacterial diversity reducing Bd infections. Although weak, we found support for Bd increasing bacterial diversity and/or for core bacteria dominance reducing Bd loads. Our research incorporates a critical element in the study of host microbiomes by linking environmental heterogeneity of landscapes to the host–pathogen–microbiome triangle.
Keywords: microbial diversity, deforestation, biodiversity, chytrid, spatial connectivity
1. Introduction
Symbiotic microbial communities can protect their hosts from diseases by directly inhibiting pathogens or increasing host immune capacity [1]. At the same time, invading pathogens can alter the structure of these microbiomes [2,3], potentially reducing host fitness and their ability to fight recurrent or novel infections [4]. These complex multi-species interactions are likely influenced by environmental conditions [5]. Although multiple studies have described links between microbial species composition and wildlife diseases [6–8], landscape-level studies focused on the host–pathogen–microbiome responses to environmental change are few and sorely needed to understand and manage the accelerated declines of animal populations.
Loss of natural vegetation is one of the largest human-induced threats to biodiversity [9]. Habitat disturbances lead to simplified trophic networks and lower functional redundancy in natural ecosystems [10], which often facilitate biological invasions [11] and result in further detrimental effects on homogenized communities [12]. For instance, depauperate animal communities often show higher risk of disease due to mechanisms such as shifts in interactions among host species [13] or environmental stress affecting both hosts and microbes [14,15]. Nonetheless, it is unclear to what degree interactions among hosts, diversity of commensal microbes, and pathogens change with deforestation. Whether deforestation alters these interactions, and to what extent those changes lead to further biodiversity loss, is a challenge for modern conservation biology.
Amphibians are among the most threatened vertebrates due to the dual impacts of anthropogenic habitat modification [16,17] and the emerging fungal pathogen Batrachochytrium dendrobatidis (Bd) [17–19] that has caused population declines of hundreds of frog species globally [17]. Contrary to most wildlife pathogens, Bd disproportionately impacts hosts in pristine forests [19], creating a challenge for amphibian conservation. Bd is a waterborne pathogen that infects the epidermal tissue of amphibians and often disrupts their ability to osmoregulate [20]. Because amphibian skin microbes and Bd may occupy the same microenvironment (epidermal tissue), the amphibian skin microbiome could play a vital role in host defences against Bd [21,22]. Recent studies have reported strong inhibitory effects of particular bacterial species on Bd, both in culture [23–25] and in vivo [6,7]. Bacterial diversity was also linked to Bd suppression in experimentally assembled biofilm communities [26]. By disrupting the amphibian skin, Bd may also change host microbial communities. High Bd infection loads may reduce the number of bacterial OTUs in amphibian skin [2,3], though hosts infected with Bd could increase microbial diversity by recruiting beneficial bacteria [5]. Given the importance of the bi-directional link between amphibian skin microbiome and Bd, there is an urgent need to understand how environmental change impacts the interaction between commensal and pathogenic host microorganisms.
Here, we tested the impacts of deforestation, forest connectivity and Bd infection loads on amphibian skin bacterial diversity. We expect that habitat fragmentation will lower overall host skin bacterial diversity and alter bacterial composition if microbiomes respond to reduced host-immune capacity or are negatively affected by homogeneous vegetation and the harsher abiotic conditions of disturbed habitats. In contrast, Bd infection loads tend to be higher in amphibians from natural forests [19,27], which may either decrease host bacterial diversity through disrupting amphibian skin or increase diversity by leading hosts to recruit beneficial bacteria in those habitats [2,3,5].
To evaluate mechanisms leading to changes in symbiotic microbial diversity, we screened 20 populations of a habitat generalist treefrog (Dendropsophus minutus) along two deforestation gradients in Brazil's Atlantic forest. We determined Bd infection loads on each individual frog and quantified skin bacterial diversity using 16S rRNA amplicon sequencing. Because biodiversity can buffer pathogens in natural systems [13], we also investigated the potential effect of host skin bacterial diversity on Bd. Microbiome-induced resistance and pathogen-induced disturbance in amphibian hosts are not mutually exclusive mechanisms that could explain changes in host bacterial diversity [2]. Therefore, we compared the fit of competing structural equation models testing the effects of (i) skin bacterial diversity on Bd loads and (ii) Bd loads on skin bacterial diversity. Combined, our results provide a critical element to the study of microbiomes by considering environmental heterogeneity and its consequences for host–pathogen–microbiome interactions.
2. Methods
(a). Field sampling
We sampled 10 populations of the golden lesser treefrog (Dendropsophus minutus, Hylidae) in a landscape of Serra do Mar coastal forest (hereafter Serra do Mar), State of São Paulo (23°20′ S, 45°12′ W), and 10 populations in a landscape of the Araucaria Moist forest (Araucaria forest), State of Rio Grande do Sul (29°24′ S, 50°24′ W), Brazil [19]. Our focal landscapes show contrasting deforestation rates: Serra do Mar landscape harbours large remnants of continuous natural forest (51.63% remaining natural vegetation), whereas Araucaria forest shows high levels of habitat loss and fragmentation (19.23% remaining natural vegetation). Our focal amphibian host species breeds in ponds during most of the year and, although no records of population declines have been reported for this species, Bd has sublethal impacts on D. minutus in the laboratory [28]. We captured 187 individuals (average of 9.3 ± 3.65 s.d. frogs per pond), rinsed each frog in the field with distilled water, and used sterile swabs to sample host epidermis. To avoid DNA degradation during field collection we preserved swabs in sterile vials containing 2.5 ml of 200 proof ethanol for later Bd quantification and bacterial sequencing.
(b). Spatial data
We quantified per cent deforestation for each sampling location based on high-resolution satellite images from 2010 (SPOT, 2-m resolution). In each landscape, the selected study sites (ponds) represented a gradient of natural vegetation cover. We calculated the percentage of deforestation within a diameter of 300 m surrounding each sampling site using ArcGIS 9.3.1 [29]. To avoid effects of elevation and macroclimate, we chose landscapes with low climatic and topographic variability (mean elevation ± s.d. of sampling sites for Serra do Mar = 929.9 ± 71.4 m; for Araucaria forest = 918.0 ± 10.2 m). For the habitat connectivity analyses among sampling locations we used the national land cover database for Brazil (1 : 50 000 scale shapefiles; [30]) and used a binary classification of natural and non-natural land cover. To avoid biases due to seasonality, we sampled ponds continuously over 30 days in Serra do Mar, and 41 days in Araucaria forest in the summer of 2009/2010.
(c). qPCR and bacterial sequencing
We extracted DNA from skin swabs using 50 µl PrepMan Ultra® and screened samples for presence and infection intensity of Bd using Taqman qPCR assays [31] with a Bd standard curve ranging from 0.1 to 1000 zoospore genome equivalents. To build standard curves we used Bd isolate CLFT023; a genotype from the global panzootic lineage isolated from a Hylodes frog approximately 100 km from our Serra do Mar landscape [32].
To quantify bacterial diversity, we followed the Earth Microbiome Project protocol (v. 4.13 [33]) using DNA extracted from skin swabs. We PCR-amplified the V4 region of the 16S rRNA gene using universal primers 515F and 806R. We ran reactions in triplicate and included a negative water control for each barcoded primer. We included blank reactions with water instead of DNA in each plate. Amplicons were visualized in 2% agarose gel, pooled, and quantified using QuantiFluor (Promega, USA). Equimolar quantities from each sample (100 ng) were combined to generate the amplicon library, and then cleaned using Agencourt AMPure XP beads (Beckman Coulter, USA). We sequenced the pooled library using Illumina MiSeq (2× 250 bp) at the Biotechnology Research Center (BRC) Genomics Facility at Cornell University; only forward reads were used for the analyses.
We used Quantitative Insights into Microbial Ecology v. 1.9.0 (QIIME) to split libraries and assign sequences into operational taxonomic units (OTUs) [34] with default quality filtering parameters. We assigned OTUs using the open reference protocol in QIIME, which clusters sequences against the Greengenes database from May 2013, using uclust [35,36]; our final dataset was limited to bacterial sequences only (no mitochondria or chloroplast sequences). We excluded samples with less than 10 K reads and OTUs with fewer than 866 reads in coverage, based on the 0.005% OTU threshold suggested by Bokulich et al. [37]. Although ethanol-based sample preservation can have mixed effects on prokaryotic DNA quality [38], our coverage was exceptionally high. We normalized the OTU table using cumulative sum scaling [39]. After all filtering steps, we detected a total of 638 bacterial OTUs and 132 core OTUs (present in 90% of our samples). In addition, we used a database of bacterial sequences with known functional activity against Bd [22] to identify putatively inhibitory OTUs. We filtered this database to only include sequences with known Bd-inhibitory function, and then matched these OTUs to our OTU table at 97% sequence similarity. We deposited the full database in Dryad (http://dx.doi.org/10.5061/dryad.812rc) [40].
(d). Statistical analyses
To determine the role of spatial connectivity on similarity of amphibian skin bacteria among our sampling locations, we used two connectivity indices: Euclidean distance (minimum straight-line distance between each pair of sampling populations) and a spatial metric of forest connectivity. We calculated habitat connectivity (natural forest habitat) among pairwise populations by employing our land cover raster as resistance grid using CIRCUITSCAPE v. 3.3 [41]. We coded low resistance pixels (raster value = 1) for natural forest cover and high resistance pixels (raster value = 10) for non-natural vegetation cover. We employed a cell connection scheme linking each node to four neighbours. Spatial connectivity analyses weighed all possible paths between pairs of sampling ponds and produced both pairwise resistance matrices and summary connectivity rasters. To quantify microbiome similarity, we measured pairwise beta diversity using both unweighted and weighted UniFrac distance matrices after collapsing samples by site in QIIME. We used simple and partial Mantel tests to correlate forest connectivity and Euclidean distance matrices with OTU similarity (unweighted and weighted UniFrac distance values) among pairwise sampling populations using PASSAGE v. 2.0 [42].
We detected indicator OTUs by testing for differences in OTU occurrence between hosts sampled in ponds with low (less than 45%) and high (more than 45%) natural forest cover for each landscape. To do this, we applied function multipatt implemented in R package indicspecies [43], selected OTUs with high association values (i.e. IndVal.g > 0.70, [24]), and visualized patterns with a heatmap using R package phyloseq [44].
We used QIIME to compute the following measures of alpha-diversity of skin bacterial communities: Faith's phylogenetic diversity of OTUs, OTU richness and Shannon diversity. We compared Faith's phylogenetic diversity of OTUs among hosts sampled across deforestation gradients (continuous variable ranging from 0% to 100% natural vegetation) using generalized linear mixed models (GLMMs), including landscape, the interaction between per cent natural vegetation and landscape as fixed effects, and sampling pond as random effect. We performed Pearson correlations (r) between OTU's relative abundance and both Faith's phylogenetic diversity of OTUs and Bd infection loads (log) across samples. We then reported Pearson's r average (±95% CI) of 90% core OTUs and tested whether Pearson's r was associated with OTU rank abundance using linear regressions (standard least squares), independently, for Faith's phylogenetic diversity of OTUs and Bd infection loads. We repeated these analyses using solely OTUs with known antifungal properties. To assess the potential role of Bd recruiting beneficial bacteria we used linear regressions with Bd loads (log) as the explanatory variable and the number of OTUs with known anti-Bd properties [22] as the response variable; we repeated this analysis including both Bd loads and Faith's phylogenetic diversity as fixed effects. To test the potential effect of beneficial bacteria suppressing Bd loads we used linear regressions with the relative abundance of anti-Bd OTUs as the explanatory variable and Bd loads (log) as the response variable.
We used principal coordinate analysis (PCoA) to consolidate information of bacterial community composition for amphibian hosts using unweighted and weighted UniFrac distance matrices. We then performed PermANOVAs to test for differences in skin bacterial composition between Bd+ and Bd− hosts from the best-sampled pond from each landscape.
We applied path analyses with maximum Wishart likelihood (500 iterations) to test for the relative strength of direct and indirect associations among infection loads, per cent natural vegetation (continuous variable), and host skin bacterial diversity. We built two competing models, and in both models we allowed natural vegetation to influence Bd infection loads, skin bacterial diversity (Faith's phylogenetic diversity) and bacterial community composition (PCoA Axis 1; unweighted UniFrac distances). In the first model, we only allowed bacterial diversity and composition to predict Bd infection loads. In contrast, in the second model, we only allowed Bd infection loads to predict bacterial diversity and composition. We compared the fit of these two contrasting models using the root mean square error of approximation (RMSEA), and inferred the direction of the relationship between Bd and skin bacterial diversity. In both models we included latent variables (u) independently explaining Faith's phylogenetic diversity of OTUs, PCoA Axis 1 and Bd infection loads. We controlled for sampling pond by adding its random effect to all biotic variables in both models.
3. Results
(a). Spatial connectivity and skin microbiome similarity among host populations
Reconstruction of land cover resistance in both study landscapes showed lower forest connectivity among ponds in Araucaria forest (figure 1a) than in Serra do Mar (figure 1b). We found that forest connectivity was positively correlated with bacterial community similarity among amphibian populations from Araucaria forest when Euclidean distance was held constant (unweighed UniFrac distances: r = 0.522, p = 0.018; figure 1a). In this highly disturbed landscape, Euclidean distance alone had no effect on bacterial community similarity (r = −0.029, p = 0.436). Forest connectivity was also positively correlated with bacterial community similarity in Serra do Mar (r = 0.537, p = 0.019; figure 1b), while Euclidean distance showed a negative non-significant trend (r = −0.293, p = 0.071). In addition, forest connectivity was a positive predictor of bacterial community similarity when Euclidean distance was held constant in a partial Mantel test (r = 0.475, p = 0.048; figure 1b). We performed additional partial Mantel tests using weighted UniFrac distance matrices and found non-significant associations between forest connectivity and skin bacterial community similarity.
Figure 1.

Pairwise forest connectivity and microbiome similarity (unweighed UniFrac distances—uud) of amphibian skin bacteria (OTUs) among 10 sampling populations of Dendropsophus minutus in (a) Araucaria forest and (b) Serra do Mar landscapes. Maximum Euclidean distance between sampling populations for Serra do Mar (18.88 km) and Araucaria forest (21.33 km). (Online version in colour.)
(b). Indicators of habitat quality using amphibian skin bacteria
Amphibian skin microbial communities were dominated by phylum Proteobacteria (more than 70%), and we found similar proportions of the most abundant classes in both Serra do Mar and Araucaria forest landscapes (electronic supplementary material, figure S1). Multilevel pattern analyses identified 16 OTUs associated with habitat type in Serra do Mar and only two OTUs in Araucaria forest (Agrobacterium sp. and Rhabdochlamydia sp.). In Serra do Mar, all indicator OTUs were significantly associated with areas of high natural forest cover (electronic supplementary material, figure S2), further indicating an ordered loss of amphibian skin bacterial diversity with increasing deforestation.
(c). Skin bacterial diversity and Bd across gradients of habitat quality
Faith's phylogenetic diversity of OTUs increased towards areas of natural forest cover (t = 2.88, β = 0.026, p = 0.010; figure 2, electronic supplementary material, table S1); both richness and Shannon diversity of OTUs showed a similar non-significant trend of higher diversity in hosts from natural habitats (electronic supplementary material, table S1). Besides showing higher bacterial diversity, individual hosts from areas of high natural vegetation also had average higher Bd infection loads (t = 2.79, β = 0.009, p = 0.014; figure 2, electronic supplementary material, table S1).
Figure 2.

Skin bacterial diversity (Faith's phylogenetic diversity of OTUs) in individual frogs from areas of low (less than 45%) and high (more than 45%) natural forest cover. Bd infection load data are overlaid in a size/colour-coded gradient. Box plot depicts median (horizontal line) and the first and third quartiles; vertical lines delimit the maximum and minimum values, except for one outlier. (Online version in colour.)
The relative abundance of D. minutus' core microbiome (90% core OTUs; N = 132) was, on average, negatively correlated with both Faith's phylogenetic diversity of OTUs (β = −0.361, 95% CI = (−0.413, −0.309); figure 3a) and Bd infection loads (β = −0.116, 95% CI = (−0.139, −0.093); figure 3b); this pattern was consistent when considering only core OTUs with known antifungal properties (Faith's phylogenetic diversity: (β = −0.517, 95% CI = (−0.683, −0.351); figure 3a); Bd infection loads: (β = −0.181, 95% CI = (−0.249, −0.113); figure 2a)). OTU relative abundance positively predicted Faith's phylogenetic diversity (β = 0.001, t = 20.66, p < 0.0001) and Bd infection loads (β = 0.0003, t = 13.11, p < 0.0001); this pattern was also consistent when considering only core OTUs with known antifungal properties (Faith's phylogenetic diversity: (β = 0.001, t = 5.70, p < 0.0001); Bd infection loads: (β = 0.0005, t = 4.76, p < 0.0001)). Higher Bd loads marginally predicted an increase in the number of anti-Bd OTUs per host (β = 0.567, t = 1.93, p = 0.055), though this trend disappeared when controlling for Faith's phylogenetic diversity of OTUs (β = 0.229, t = 0.85, p = 0.394). Furthermore, the relative abundance of anti-Bd OTUs did not predict Bd infection loads (β = −8.957, t = −1.38, p = 0.168).
Figure 3.
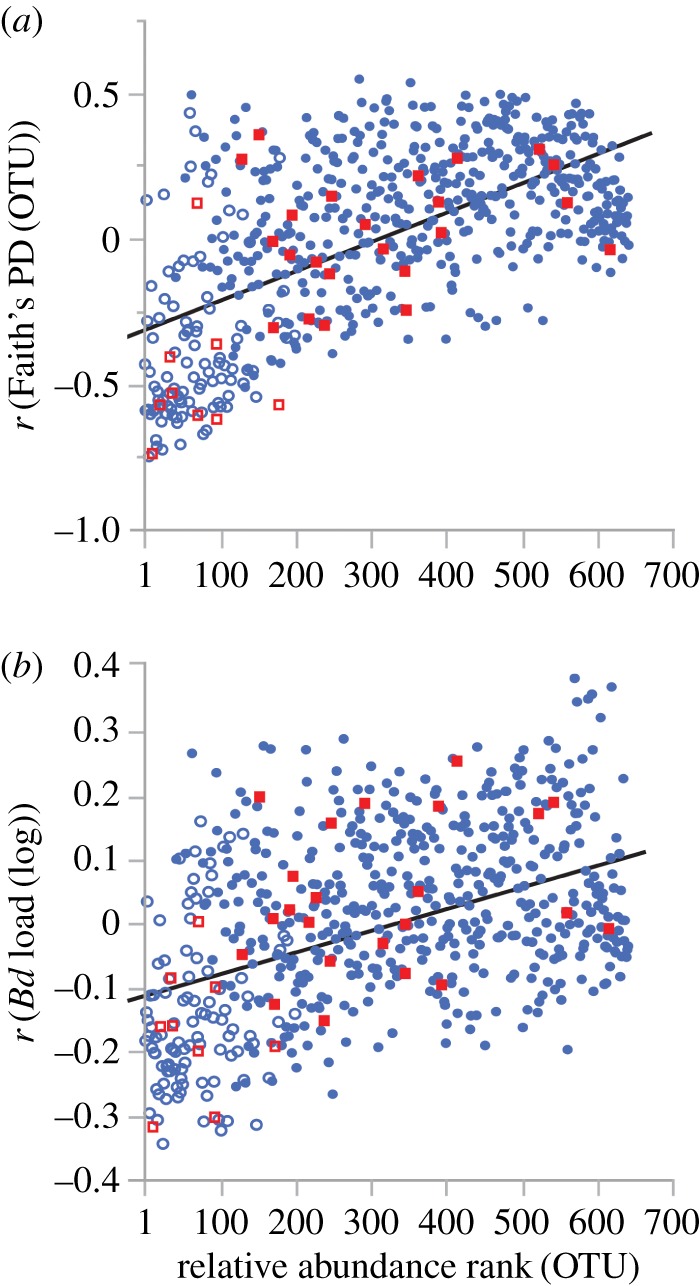
Relative abundance rank of each OTU correlated with Faith's phylogenetic diversity of OTUs (a) and Bd infection loads (b) across individual hosts. Ninety per cent core OTUs are shown in hollow symbols. OTUs with potential anti-Bd properties are shown in red squares. (Online version in colour.)
Our PCoA ordinations (unweighted UniFrac distances) showed that skin bacterial community composition significantly differed between Bd+ and Bd− individuals from the best-sampled pond in Araucaria forest (PermANOVA: F1,16 = 3.75, R2 = 0.20, p = 0.002; electronic supplementary material, figure S3a), with a similar trend in Serra do Mar (F1,10 = 1.53, R2 = 0.15, p = 0.090; electronic supplementary material, figure S3b). We repeated these two PermANOVAs using weighted UniFrac distance matrices and results remained consistent. Land cover was also a good predictor of PCoA axes I and II when entered as continuous variables in GLMMs (electronic supplementary material, table S1).
(d). Direct and indirect effects of natural vegetation on host skin microbiomes and Bd loads
Our most parsimonious path model indicated that higher natural vegetation was linked to a significant increase in Bd infection loads, which was in turn associated to an increase in skin bacterial diversity (Faith's phylogenetic diversity: RMSEA = 0.122; figure 4a). Conversely, our second model allowing causal effects of both bacterial diversity and composition on Bd loads did not show any significant association among biotic variables (RMSEA = 0.138; figure 4b). In both models we found a strong direct effect of natural vegetation increasing skin bacterial diversity and altering composition; we detected a weaker association between skin bacterial diversity and Bd.
Figure 4.
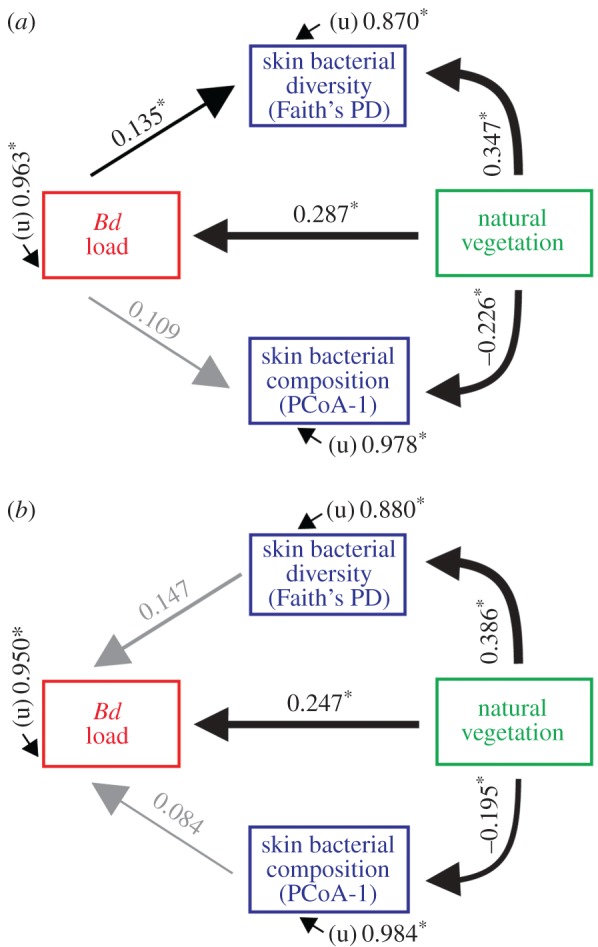
Path analyses showing direct and indirect effects of natural vegetation on both amphibian skin microbiome and Bd. Model (a) shows the relative strengths of natural vegetation (%) on skin bacterial diversity (Faith's phylogenetic diversity), skin bacterial composition (PCoA axis I; unweighted UniFrac distances), and Bd loads (log–G.E.), including the effect of Bd loads on both skin bacterial diversity and composition. Model (b) includes the effects of skin bacterial diversity and composition on Bd loads. Numbers are standardized path coefficients (*p < 0.05). Latent (unmeasured) variables represented by ‘u’. The thickness of the arrows represents the relative strength of each relationship. Direct effect probabilities: effect of skin bacterial diversity on Bd loads (p = 0.041); Bd loads on skin bacterial diversity (p = 0.062). Non-significant paths are shown in grey. (Online version in colour.)
4. Discussion
We showed that spatial connectivity among natural habitats leads to increased similarity of amphibian skin bacterial communities and that bacterial diversity is higher in frogs from natural than disturbed habitats. These results indicate that corridors of natural vegetation maintain movement of amphibian-associated bacteria through their hosts and/or the environment. Our data also showed that the observed higher skin microbial diversity in continuous natural habitats could be linked to higher Bd loads in closed-canopy forests. Taken together, our results confirm our predictions that habitat quality and connectivity may impact the interaction between Bd and host skin microbiomes, underscoring the complexity of host–pathogen dynamics in fragmented landscapes. Combined, our findings highlight the importance of environmental change and the spatial context on the host–pathogen–microbiome triangle.
Deforestation often leads to fragmentation and reduced habitat connectivity [45], both of which are linked to genetic erosion and biodiversity loss at multiple scales [46]. The distributions of hosts and associated microbes are expected to respond to habitat change at different spatial scales [47,48]. Nonetheless, the effects of habitat change may cascade down from hosts to their skin microbiomes due to shifts in host's habitat use and immune capacity. Our focal host species likely uses a much more diverse array of microenvironments (e.g. arboreal, aquatic, terrestrial) in natural (complex) than in disturbed (homogeneous) habitats, which may translate in exposure to a broader suite of microbial taxa from natural habitats that is able to colonize amphibian skin. Additionally, canopy removal creates harsher microclimates that may exclude several groups of bacteria the same way it reduces Bd [19]. Thus, it is plausible that more bacterial OTUs are present in forests than in disturbed and homogenized environments due to specific abiotic conditions, and this alone could in turn result in the observed higher diversity of skin bacteria in hosts from natural habitats.
In agreement with the above prediction, we found that increased forest connectivity was linked to high skin bacterial similarity among pairs of host populations when analysing with unweighted distance matrices. Habitat connectivity was a much better predictor of similarity between skin bacterial communities than Euclidean distance, indicating that habitat quality is key for bacterial community assembly and/or filtering. Nonetheless, we observed a non-significant effect of forest connectivity on bacterial community similarity when analysing our weighted matrix data. These findings indicate that forest connectivity could determine whether a species of bacteria can persist or not (unweighted data), though the impact of habitat quality on OTU relative abundance (weighted data) may be more diffuse. For instance, our analyses revealed 16 skin bacterial OTUs that were always found in amphibians from continuous natural forests, contrasting with zero OTUs unique to neighbouring areas of disturbed vegetation. These results highlight that habitat loss and fragmentation may reduce similarity and diversity of host microbiomes through filtering out several host skin bacteria.
A second possibility is that host genetics may directly impact selection for skin bacteria, and thus microbiome structure might mirror host population genetics from natural and disturbed habitats. Although we do not have direct measures of genetic diversity for our focal populations, we think that strong genetic differentiation by distance/resistance at this sampling scale is unlikely [49]. Our host species is found in equally high abundances in both natural and disturbed environments and does not appear to be limited in gene flow by barriers in these landscapes. Nonetheless, a study of fine-scale population genetic diversity and microbial diversity would be fruitful. Combined, our results show that declines in alpha diversity and pairwise similarity of amphibian skin bacteria are parallel to what has been reported for vertebrate communities in fragmented habitats [45,50], indicating a consistency across spatial scales perceived by vertebrate hosts and their associated microorganisms.
Mounting evidence supports the pattern that biodiversity loss increases disease risk [13]. However, we are just beginning to focus on whether the diversity of host microbial communities might afford a protective role against pathogen infections [3,26,51–54]. Diversity is expected, on average, to give rise to ecosystem stability [10–12]. Analogously, diversity of commensal microbial species may be positively correlated with host health [51,52]. Although OTU richness may suppress Bd growth in biofilm communities [26], previous experimental studies using live amphibians so far failed to find a clear effect of bacterial OTU richness on Bd infection loads [2,53,55]. Furthermore, a field-based study focusing on a congener host from Panama found no association between the number of bacterial OTUs and Bd [56]. While variance partitioning in our path analyses did not show a significant effect of bacterial diversity on Bd infection loads, it pointed to a weak but significant effect of Bd increasing amphibian skin bacterial diversity (figure 4). Furthermore, our PermANOVAs indicate that Bd is a strong predictor of bacterial composition independent of land cover. Therefore, the impact of Bd on skin bacteria might be stronger than the effect of bacterial diversity buffering Bd infections in our focal host species. These results, however, should be interpreted with caution due to the correlative nature of our data and also because our path models indicated that land cover had a stronger impact than Bd on host skin bacterial diversity.
Bd-infected hosts may increase host microbial diversity by recruiting protective bacteria with antifungal properties [3,5]. We found a positive non-significant trend between number of anti-Bd OTUs and Bd infection loads, indicating that frogs highly infected with Bd may recruit anti-Bd bacteria. However, our results showed that this association was confounded by OTU diversity because diverse microbiomes are expected to have a larger number of anti-Bd OTUs than depauperate microbiomes. The relative abundance of these anti-Bd OTUs was also not a good predictor of Bd loads. Furthermore, the abundances of core and non-core OTUs showed similar correlations with Bd loads using either the full dataset or just the anti-Bd OTUs. Although our results may indicate that common and abundant bacteria may reduce Bd loads, and/or that Bd may recruit rare beneficial bacteria, the OTU rank abundance was also tightly correlated with bacterial diversity across our samples. This pattern may be due to the expected (and observed) inverse correlation between OTU diversity and dominance, and not necessarily due to Bd recruiting skin bacteria.
Alternatively, Bd infection often leads to immunosuppression in the host [57,58], which could promote colonization and/or proliferation of opportunistic and pathogenic bacteria, leading to changes in skin bacterial diversity in amphibians. Contrary to our findings, recent studies found lower skin bacterial richness on amphibian hosts highly infected with Bd at the same time that bacterial dominance increased with infection loads [2,3,5,55]. Over time, a destabilized microbial community could well be dominated by strong competitors, which could possibly lead to a sharp drop in skin bacterial diversity in advanced stages of chytridiomycosis. Our contrasting findings are likely a result of higher tolerance of D. minutus to the local Bd genotype(s) compared to species that have declined due to Bd in the wild. The Antillean frog Eleutherodactylus coqui is more susceptible to local Bd isolates than our focal host D. minutus after being experimentally infected in the laboratory [28,59,60], indicating that opportunistic bacteria in E. coqui may become dominant, and lead to a reduction of skin bacterial diversity when infected with Bd. A recent study demonstrated that our focal host species showed thickening of the skin when infected with a locally endemic Bd strain [28], which could create a complex skin environment for bacterial colonization. Furthermore, E. coqui experimentally infected with Bd reduced OTU richness immediately after skin shedding [5]. These findings suggest that non-clinical chytridiomycosis may lead to a thicker and more complex skin environment where multiple groups of bacteria could thrive, whereas high infection loads of clinical chytridiomycosis may increase rates of skin shedding [61] with likely negative effects on symbiotic skin microbes.
In conclusion, our data demonstrate that host microbial diversity and composition can respond to anthropogenic habitat modification at the landscape scale. Our data also showed that the interaction between deforestation and pathogen infections could impact host microbial diversity. Given the correlative nature of the bi-directional link between host skin microbiomes and Bd, manipulative studies will be key to effectively test whether and under what circumstances amphibian skin bacterial diversity translates to host defences against pathogens [5,26,51,52]. These studies will also contribute to the body of literature testing whether or not biodiversity decline leads to ecological instability at multiple scales [10]. We are just beginning to comprehend the complexity of the host–pathogen–microbiome triangle. Understanding multi-level community interactions on a spatial context will be critical to efficiently tackle environmental health in the wake of accelerated anthropogenic habitat change.
Supplementary Material
Acknowledgements
We thank F. Adams, N. Alves, D. Becker and K.G.D. Kochhann for field support, and D. T. Correa for providing feedback on the manuscript. We thank D. Rodriguez for support during the OTU picking step.
Ethics
Research permits were provided by Instituto Chico Mendes–Instituto Brasileiro do Meio Ambiente e dos Recursos Naturais Renováveis/Brazil (Permits 20621-1 and 21.125-1), Instituto Florestal do Estado de São Paulo (Permit 260108-009.662), and the Cornell University Institutional Animal Care and Use Committee.
Data accessibility
Sequence data are available from NCBI Sequence Read Archive (SUB2686692). Both the mapping file and final dataset are available from Dryad: http://dx.doi.org/10.5061/dryad.812rc [40].
Authors' contributions
C.G.B. designed research, carried out fieldwork, performed molecular and statistical analyses, and wrote the paper with important contributions from A.V.L., C.F.B.H. and K.R.Z. C.F.B.H. carried out fieldwork. A.V.L. carried out molecular lab work and participated in data analysis. All authors gave final approval for publication.
Competing interests
We declare we have no competing interests.
Funding
Our work was funded by grants from the American Philosophical Society, Sigma Xi, Andrew Mellon Foundation, Atkinson Center for a Sustainable Future, Department of Ecology and Evolutionary Biology at Cornell University, Conselho Nacional de Desenvolvimento Científico e Tecnológico - CNPq (no. 312895/2014-3, and no. 302518/2013-4), National Science Foundation (DEB-1209382, DEB-1120249, and DEB-1310036), and Fundação de Amparo à Pesquisa do Estado de São Paulo - FAPESP (no. 2013/50741-7 and no. 2014/50342-8).
References
- 1.Harris RN, et al. 2009. Skin microbes on frogs prevent morbidity and mortality caused by a lethal skin fungus. ISME J. 3, 818–824. ( 10.1038/ismej.2009.27) [DOI] [PubMed] [Google Scholar]
- 2.Jani AJ, Briggs CJ. 2014. The pathogen Batrachochytrium dendrobatidis disturbs the frog skin microbiome during a natural epidemic and experimental infection. Proc. Natl Acad. Sci. USA 111, E5049–E5058. ( 10.1073/pnas.1412752111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rebollar EA, Hughey MC, Medina D, Harris RN, Ibanez R, Belden LK. 2016. Skin bacterial diversity of Panamanian frogs is associated with host susceptibility and presence of Batrachochytrium dendrobatidis. ISME J. 10, 1682–1695. ( 10.1038/ismej.2015.234) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Glasl B, Herndl GJ, Frade PR. 2016. The microbiome of coral surface mucus has a key role in mediating holobiont health and survival upon disturbance. ISME J. 10, 2280–2292. ( 10.1038/ismej.2016.9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Longo AV, Zamudio KR. 2017. Environmental fluctuations and host skin bacteria shift survival advantage between frogs and their fungal pathogen. ISME J. 11, 349–361. ( 10.1038/ismej.2016.138) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Becker MH, Harris RN. 2010. Cutaneous bacteria of the redback salamander prevent morbidity associated with a lethal disease. PLoS ONE 5, e10957 ( 10.1371/journal.pone.0010957) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Muletz C, Myers JM, Domangue RJ, Herrick JD, Harris RN. 2012. Soil bioaugmentation with amphibian cutaneous bacteria protects amphibian host from infection by Batrachochytrium dendrobatidis. Biol. Conserv. 152, 119–126. ( 10.1016/j.biocon.2012.03.022) [DOI] [Google Scholar]
- 8.Hoyt JR, et al. 2015. Bacteria isolated from bats inhibit the growth of Pseudogymnoascus destructans, the causative agent of white-nose syndrome. PLoS ONE 10, e0121329 ( 10.1371/journal.pone.0121329) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pereira HM, et al. 2010. Scenarios for global biodiversity in the 21st century. Science 330, 1496–1501. ( 10.1126/science.1196624) [DOI] [PubMed] [Google Scholar]
- 10.McCann KS. 2000. The diversity–stability debate. Nature 405, 228–233. ( 10.1038/35012234) [DOI] [PubMed] [Google Scholar]
- 11.Elton CS. 1958. Ecology of invasions by animals and plants. London, UK: Chapman & Hall. [Google Scholar]
- 12.MacArthur RH. 1955. Fluctuations of animal populations and a measure of community stability. Ecology 36, 533–536. ( 10.2307/1929601) [DOI] [Google Scholar]
- 13.Ostfeld RS, Keesing F. 2012. Effects of host diversity on infectious disease. Annu. Rev. Ecol. Evol. Syst. 43, 157–182. ( 10.1146/annurev-ecolsys-102710-145022) [DOI] [Google Scholar]
- 14.Cushman SA. 2006. Effects of habitat loss and fragmentation on amphibians: a review and prospectus. Biol. Conserv. 128, 231–240. ( 10.1016/j.biocon.2005.09.031) [DOI] [Google Scholar]
- 15.Yuan X, Knelman JE, Gasarch E, Wang DL, Nemergut DR, Seastedt TR. 2016. Plant community and soil chemistry responses to long-term nitrogen inputs drive changes in alpine bacterial communities. Ecology 97, 1543–1554. ( 10.1890/15-1160.1) [DOI] [PubMed] [Google Scholar]
- 16.Becker CG, Fonseca CR, Haddad CFB, Batista RF, Prado PI. 2007. Habitat split and the global decline of amphibians. Science 318, 1775–1777. ( 10.1126/science.1149374) [DOI] [PubMed] [Google Scholar]
- 17.IUCN, Conservation International, NatureServe. 2016. Global amphibian assessment. See www.amphibians.org/publications/threatened-amphibians-of-the-world/ (accessed 15 March 2016).
- 18.Lips KR, Diffendorfer J, Mendelson JR, Sears MW. 2008. Riding the wave: reconciling the roles of disease and climate change in amphibian declines. PLoS Biol. 6, e72 ( 10.1371/journal.pbio.0060072) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Becker CG, Zamudio KR. 2011. Tropical amphibian populations experience higher disease risk in natural habitats. Proc. Natl Acad. Sci. USA 108, 9893–9898. ( 10.1073/pnas.1014497108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Voyles J, et al. 2009. Pathogenesis of chytridiomycosis, a cause of catastrophic amphibian declines. Science 326, 582–585. ( 10.1126/science.1176765) [DOI] [PubMed] [Google Scholar]
- 21.Bletz MC, Loudon AH, Becker MH, Bell S, Woodhams DC, Minbiole KPC, Harris RN, Gaillard J-M. 2013. Mitigating amphibian chytridiomycosis with bioaugmentation: characteristics of effective probiotics and strategies for their selection and use. Ecol. Lett. 16, 807–820. ( 10.1111/ele.12099) [DOI] [PubMed] [Google Scholar]
- 22.Woodhams DC, et al. 2015. Antifungal isolates database of amphibian skin-associated bacteria and function against emerging fungal pathogens. Ecology 96, 595 ( 10.1890/14-1837.1) [DOI] [Google Scholar]
- 23.Flechas SV, Sarmiento C, Cárdenas ME, Medina EM, Restrepo S, Amézquita A. 2012. Surviving chytridiomycosis: differential anti-Batrachochytrium dendrobatidis activity in bacterial isolates from three lowland species of Atelopus . PLoS ONE 7, e44832 ( 10.1371/journal.pone.0044832) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Becker MH, et al. 2015. Phylogenetic distribution of symbiotic bacteria from Panamanian amphibians that inhibit growth of the lethal fungal pathogen Batrachochytrium dendrobatidis. Mol. Ecol. 24, 1628–1641. ( 10.1111/mec.13135) [DOI] [PubMed] [Google Scholar]
- 25.Muletz-Wolz CR, et al. 2017. Antifungal bacteria on woodland salamander skin exhibit high taxonomic diversity and geographic variability. Appl. Environ. Microbiol. 83, e00186-17 ( 10.1128/AEM.00186-17) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Piovia-Scott J, Rejmanek D, Woodhams DC, Worth SJ, Kenny H, McKenzie V, Lawler SP, Foley JE. 2017. Greater species richness of bacterial skin symbionts better suppresses the amphibian fungal pathogen Batrachochytrium dendrobatidis. Microb. Ecol. 74, 217–226. ( 10.1007/s00248-016-0916-4) [DOI] [PubMed] [Google Scholar]
- 27.Becker CG, Rodriguez D, Longo-Berrios AV, Talaba A, Zamudio KR. 2012. Disease risk in temperate amphibian populations is higher at closed-canopy sites. PLoS ONE 7, e48205 ( 10.1371/journal.pone.0048205) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bovo RP, Andrade DV, Toledo LF, Longo AV, Rodriguez D, Haddad CFB, Zamudio KR, Becker CG. 2016. Physiological responses of Brazilian amphibians to an enzootic infection of the chytrid fungus Batrachochytrium dendrobatidis. Dis. Aquat. Organ. 117, 245–252. ( 10.3354/dao02940) [DOI] [PubMed] [Google Scholar]
- 29.ESRI. 2014. Arcview 10.2.1 (Redlands, CA).
- 30.Fundação SOS Mata Atlântica, INPE. 2015. Atlas dos remanescentes florestais da Mata Atlântica: Período 2013–2014. See http://mapas.sosma.org.br/ (accessed 22 March 2016).
- 31.Boyle DG, Boyle DB, Olsen V, Morgan JAT, Hyatt AD. 2004. Rapid quantitative detection of chytridiomycosis (Batrachochytrium dendrobatidis) in amphibian samples using real-time Taqman PCR assay. Dis. Aquat. Organ. 60, 141–148. ( 10.3354/dao060141) [DOI] [PubMed] [Google Scholar]
- 32.Schloegel LM, et al. 2012. Novel, panzootic and hybrid genotypes of amphibian chytridiomycosis associated with the bullfrog trade. Mol. Ecol. 21, 5162–5177. ( 10.1111/j.1365-294X.2012.05710.x) [DOI] [PubMed] [Google Scholar]
- 33.Caporaso JG, et al. 2012. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 6, 1621–1624. ( 10.1038/ismej.2012.8) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Caporaso JG, et al. 2010. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. ( 10.1038/nmeth.f.303) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.DeSantis TZ, et al. 2006. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 72, 5069–5072. ( 10.1128/AEM.03006-05) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Edgar RC. 2010. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. ( 10.1093/bioinformatics/btq461) [DOI] [PubMed] [Google Scholar]
- 37.Bokulich NA, Subramanian S, Faith JJ, Gevers D, Gordon JI, Knight R, Mills DA, Caporaso JG. 2012. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 10, 57–59. ( 10.1038/nmeth.2276) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thakuria D, Schmidt O, Liliensiek A-K, Egan D, Doohan FM. 2009. Field preservation and DNA extraction methods for intestinal microbial diversity analysis in earthworms. J. Microbiol. Methods 76, 226–233. ( 10.1016/j.mimet.2008.10.015) [DOI] [PubMed] [Google Scholar]
- 39.Paulson JN, Stine OC, Bravo HC, Pop M. 2013. Differential abundance analysis for microbial marker-gene surveys. Nat. Methods 10, 1200–1202. ( 10.1038/nmeth.2658) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Becker CG, Longo AV, Haddad CFB, Zamudio KR. 2017. Data from: Land cover and forest connectivity alter the interactions among host, pathogen, and skin microbiome. Dryad Digital Repository. ( 10.5061/dryad.812rc) [DOI] [PMC free article] [PubMed]
- 41.McRae BH, Shah VB. 2009. Circuitscape user's guide. Santa Barbara, CA: University of California; See http://www.circuitscape.org. [Google Scholar]
- 42.Rosenberg MS, Anderson CD. 2011. Passage: pattern analysis, spatial statistics, and geographic exegesis. V. 2. Methods Ecol. Evol. 2, 229–232. ( 10.1111/j.2041-210X.2010.00081.x) [DOI] [Google Scholar]
- 43.De Cáceres M, Legendre P. 2009. Associations between species and groups of sites: indices and statistical inference. Ecology 90, 3566–3574. ( 10.1890/08-1823.1) [DOI] [PubMed] [Google Scholar]
- 44.McMurdie PJ, Holmes EC. 2013. Phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 8, e61217 ( 10.1371/journal.pone.0061217) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fahrig L. 2003. Effects of habitat fragmentation on biodiversity. Annu. Rev. Ecol. Evol. Syst. 34, 487–515. ( 10.1146/annurev.ecolsys.34.011802.132419) [DOI] [Google Scholar]
- 46.Pimm SL, Russell GJ, Gittleman JL, Brooks TM. 1995. The future of biodiversity. Science 269, 347–350. ( 10.1126/science.269.5222.347) [DOI] [PubMed] [Google Scholar]
- 47.Horner-Devine MC, Lage M, Hughes JB, Bohannan BJM. 2004. A taxa–area relationship for bacteria. Nature 432, 750–753. ( 10.1038/nature03073) [DOI] [PubMed] [Google Scholar]
- 48.Fierer N, Jackson R. 2006. The diversity and biogeography of soil bacterial communities. Proc. Natl Acad. Sci. USA 103, 626–631. ( 10.1073/pnas.0507535103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dixo M, Metzger JP, Morgante JS, Zamudio KR. 2009. Habitat fragmentation reduces genetic diversity and connectivity among toad populations in the Brazilian Atlantic Coastal Forest. Biol. Conserv. 142, 1560–1569. ( 10.1016/j.biocon.2008.11.016) [DOI] [Google Scholar]
- 50.Chisholm C, Lindo Z, Gonzalez A. 2011. Metacommunity diversity depends on connectivity and patch arrangement in heterogeneous habitat networks. Ecography 34, 415–424. ( 10.1111/j.1600-0587.2010.06588.x) [DOI] [Google Scholar]
- 51.Dillon RJ, Vennard CT, Buckling A, Charnley AK. 2005. Diversity of locust gut bacteria protects against pathogen invasion. Ecol. Lett. 8, 1291–1298. ( 10.1111/j.1461-0248.2005.00828.x) [DOI] [Google Scholar]
- 52.Van Elsas JD, Chiurazzi M, Mallon CA, Elhottova D, Kristufek V, Salles JF. 2012. Microbial diversity determines the invasion of soil by a bacterial pathogen. Proc. Natl. Acad. Sci. USA 109, 1159–1164. ( 10.1073/pnas.1109326109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Walke JB, et al. 2015. Community structure and function of amphibian skin microbes: an experiment with bullfrogs exposed to a chytrid fungus. PLoS ONE 10, e0139848 ( 10.1371/journal.pone.0139848) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Loudon AH, et al. 2016. Vertebrate hosts as islands: dynamics of selection, immigration, loss, persistence and potential function of bacteria on salamander skin. Front. Microbiol. 7, 333 ( 10.3389/fmicb.2016.00333) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Longo AV, Savage AE, Hewson I, Zamudio KR. 2015. Seasonal and ontogenetic variation of skin microbial communities and relationships to natural disease dynamics in declining amphibians. R. Soc. Open Sci. 2, 140377 ( 10.1098/rsos.140377) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Belden LK, et al. 2015. Panamanian frog species host unique skin bacterial communities. Front. Microbiol. 6, 1171 ( 10.3389/fmicb.2015.01171) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Carey C, Pessier AP, Peace AD. 2003. Pathogens, infectious disease and immune defenses. In Amphibian conservation (ed. Semlitch RD.), pp. 191–225. Washington, DC: Smithsonian Press. [Google Scholar]
- 58.Fites JS, et al. 2013. The invasive chytrid fungus of amphibians paralyzes lymphocyte responses. Science 9, 342–366. ( 10.1126/science.1243316) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Longo AV, Burrowes PA, Joglar RL. 2010. Seasonality of Batrachochytrium dendrobatidis infection in direct-developing frogs suggests a mechanism for persistence. Dis. Aquat. Organ. 92, 253–260. ( 10.3354/dao02054) [DOI] [PubMed] [Google Scholar]
- 60.Becker CG, Rodriguez D, Toledo LF, Longo AV, Lambertini C, Correa DT, Leite DS, Haddad CFB, Zamudio KR. 2014. Partitioning the net effect of host diversity on an emerging amphibian pathogen. Proc. R. Soc. B 281, 20141796 ( 10.1098/rspb.2014.1796) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ohmer MEB, Cramp RL, White CR, Franklin CE. 2015. Skin sloughing rate increases with chytrid fungus infection load in a susceptible amphibian. Funct. Ecol. 29, 674–682. ( 10.1111/1365-2435.12370) [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- Becker CG, Longo AV, Haddad CFB, Zamudio KR. 2017. Data from: Land cover and forest connectivity alter the interactions among host, pathogen, and skin microbiome. Dryad Digital Repository. ( 10.5061/dryad.812rc) [DOI] [PMC free article] [PubMed]
Supplementary Materials
Data Availability Statement
Sequence data are available from NCBI Sequence Read Archive (SUB2686692). Both the mapping file and final dataset are available from Dryad: http://dx.doi.org/10.5061/dryad.812rc [40].