

Abstract
Box H/ACA small nucleolar ribonucleoprotein particles (H/ACA snoRNPs) play key roles in the synthesis of eukaryotic ribosomes. How box H/ACA snoRNPs are assembled remains unknown. Here we show that yeast Nhp2p, a core component of these particles, directly binds RNA. In vitro, Nhp2p interacts with high affinity with RNAs containing irregular stem–loop structures but shows weak affinity for poly(A), poly(C) or for double-stranded RNAs. The central region of Nhp2p is believed to function as an RNA-binding domain, since it is related to motifs found in various RNA-binding proteins. Removal of two amino acids that shortens a putative β-strand element within Nhp2p central domain impairs the ability of the protein to interact with H/ACA snoRNAs in cell extracts. In vivo, this deletion prevents cell viability and leads to a strong defect in the accumulation of H/ACA snoRNAs and Gar1p. These data suggest that proper direct binding of Nhp2p to H/ACA snoRNAs is required for the assembly of H/ACA snoRNPs and hence for the stability of some of their components. In addition, we show that converting a highly conserved glycine residue (G59) within Nhp2p central domain to glutamate significantly reduces cell growth at 30 and 37°C. Remarkably, this modification affects the steady-state levels of H/ACA snoRNAs and the strength of Nhp2p association with these RNAs to varying degrees, depending on the nature of the H/ACA snoRNA. Finally, we show that the modified Nhp2p protein whose interaction with H/ACA snoRNAs is impaired cannot accumulate in the nucleolus, suggesting that only the assembled H/ACA snoRNP particles can be efficiently retained in the nucleolus.
INTRODUCTION
Small nuclear ribonucleoprotein particles (snRNPs) are involved in at least four major processes: the synthesis of ribosomes, the processing of pre-tRNAs and pre-mRNAs and the replication of telomeres. To understand the mode of assembly and action of these particles, it is essential that we (i) identify all their protein and RNA constituents and (ii) gain a detailed knowledge of how these interact with each other. The structure and mode of assembly of snRNPs involved in pre-mRNA splicing are becoming quite well understood (1). Far less information is available for the snRNPs taking part in ribosome synthesis [C/D- and H/ACA-type small nucleolar RNPs (snoRNPs) and RNase MRP], pre-tRNA processing (RNase P) or telomere replication (telomerase). However, knowledge of the structure and assembly path of the Sm protein complex of splicing snRNPs is relevant to the study of the mode of assembly of Saccharomyces cerevisiae telomerase, since it has recently been shown that its RNA component contains a binding site for Sm proteins (2). Similarly, study of the structure and mode of synthesis of H/ACA-type snoRNPs will be useful for mammalian telomerase research given that the telomerase RNA component in vertebrates possesses a domain related to H/ACA snoRNAs (3,4).
H/ACA snoRNPs are required for the post-transcriptional conversion of uridines to pseudouridines in pre-ribosomal RNA (pre-rRNA) (5,6) and the removal of some spacer regions of the pre-rRNA (7). Both processes require direct binding of H/ACA snoRNPs to the pre-rRNA by base-pairing interactions between the latter and the RNA component (H/ACA snoRNA) of the particles. H/ACA snoRNAs are the constituents of H/ACA snoRNPs that have been studied the most so far. Dozens have been identified in yeast and human (8–10) and they all share a common two-dimensional structure. This structure is characterized by the succession of two irregular stem–loops containing an internal bulge, called a ‘pseudouridylation pocket’. The stem–loops are separated by a single-stranded hinge region containing the conserved H box (consensus 5′-ANANNA-3′) and followed by a single-stranded tail containing the ACA or AUA triplet situated 3 nt from the 3′-end of the snoRNA (8,9). Selection of a pre-rRNA uridine residue destined for conversion into pseudouridine occurs by base pairing of the pseudouridylation pocket of the cognate snoRNA guide on each side of the uridine (5). The integrity of the stems, the H and ACA box is essential for both the stability and function of H/ACA snoRNAs (8,9,11). In addition, mutating the H or ACA box or disrupting the stems prevents nucleolar accumulation of mutant RNAs injected in Xenopus oocytes (12). Some, if not all, of these RNA elements probably constitute protein-binding sites.
Yeast H/ACA snoRNPs contain at least four essential proteins, Cbf5p (13–16), Gar1p (8,9,17), Nhp2p (16,18,19) and Nop10p (19). The Drosophila, rat and human orthologs of Cbf5p, respectively called Nop60B (20,21), Nap57p (22,23) and Dyskerin (24), as well the human orthologs of Gar1p, hGar1p (25), Nhp2p, hNhp2p (26) and Nop10p, hNop10p (19,26) have been characterized. In addition, putative archaebacterial orthologs of Cbf5p, Gar1p and Nop10p have been found, raising the possibility that RNP particles related to eukaryotic H/ACA snoRNPs exist in archaebacteria (26,27). Cbf5p, Nhp2p and Nop10p are required for the accumulation of H/ACA snoRNPs in yeast (15,16,19). Cbf5p contains a conserved domain found in pseudouridine synthases (28). A point mutation within this domain, while preserving the integrity of H/ACA snoRNPs, abolishes pre-rRNA pseudouridylation in vivo (29). Thus Cbf5p most probably provides the catalytic activity responsible for the snoRNP-directed uridine to pseudouridine isomerization reaction. The functions of the other protein components of H/ACA snoRNPs remain to be established or clarified. The absence of Gar1p inhibits interactions between H/ACA snoRNPs and the pre-rRNA (30). Under in vitro conditions, Gar1p has the ability to bind to the snR10 and snR30 H/ACA snoRNAs (31) but the Gar1p binding site(s) on the snoRNAs have not been identified and the in vivo relevance of this RNA–protein interaction is not obvious. Koonin et al. (32) and Vilardell and Warner (33) proposed that the Nhp2p protein also directly binds to RNA. Indeed, the central part of Nhp2p is clearly homologous to domains found in proteins believed or shown to directly interact with RNA, such as ribosomal proteins and RNP components (16,19,32–35). One of these ribosomal proteins, L30 from yeast (formerly known as L32), is quite remarkable in that it binds to its own pre-mRNA in the nucleus to inhibit its splicing, to its mature mRNA in the cytoplasm to inhibit its translation and to 25S rRNA (33,36–41).
In this work, we show that Nhp2p is indeed an RNA-binding protein. Alterations of the central domain of the protein proposed to interact with RNA impair cell growth and affect the accumulation of H/ACA snoRNAs.
MATERIALS AND METHODS
Strains, media and plasmids
The standard haploid and diploid yeast strains used were JG540 (Mata, ade2, ade3, leu2, lys2.1, trp1.1, tyr7.1, ura3, can1) and JG540 2n (Mata/Matα, ade2/ade2, ade3/ade3, leu2/leu2, lys2.1/lys2.1, trp1.1/trp1.1, tyr7.1/tyr7.1, ura3/ura3, can1/can1). Saccharomyces cerevisiae strains were grown in YNB medium [0.17% yeast nitrogen base, 0.5% (NH4)2SO4] supplemented with 2% glucose or 2% galactose and the required amino acids.
A chromosomal nhp2::LEU2 haploid strain was obtained as follows. A LEU2 gene cassette flanked by the NsiI and HindIII restriction sites was PCR amplified from plasmid Yep13 using oligonucleotides 5′-CCCCCATGCATCATTATTTTTTTCCTCAACATAACG-3′ and 5′-GGGGGAAGCTTGTGTTTTTTATTTGTTGTATTTTTTTTTTTTTAGAG-3′. This LEU2 cassette digested by NsiI and HindIII was used to replace in plasmid pHA100 (19) a NsiI/HindIII fragment containing the NHP2 gene, creating plasmid pFH80. A genomic DNA fragment encompassing the disrupted nhp2::LEU2 allele was released from pFH80 by Bsu36I/NheI digestion. This fragment was used to transform the diploid strain JG540 2n to produce a NHP2/nhp2::LEU2 heterozygous diploid strain. Correct integration at the NHP2 locus was checked by Southern analysis (data not shown). The NHP2/nhp2::LEU2 diploid strain was transformed with plasmid pJPG250, a centromeric plasmid containing the URA3 marker and NHP2ZZ genes (19). Sporulation of the NHP2/nhp2::LEU2 heterozygous diploid strain containing pJPG250 was induced, dissection of tetrads was performed and a haploid strain, termed YO342, resulting from the germination of a nhp2::LEU2 spore carrying the pJPG250 plasmid was selected.
To obtain nhp2::LEU2 haploid strains expressing the ZZ-tagged proteins Nhp2pZZ, Nhp2V56KpZZ, Nhp2G59EpZZ, Nhp2R68ApZZ or Nhp2D80ApZZ, strain YO342 was transformed with centromeric plasmids pNHP2ZZ, pnhp2V56KZZ, pnhp2G59EZZ, pnhp2R68AZZ or pnhp2D80AZZ, respectively (see below for construction of plasmids). The loss of plasmid pJPG250 was then selected for by streaking the resulting transformed strains on YNB proline medium supplemented with the required amino acids and containing 0.6 g/l fluoroorotic acid (42). Haploid strains nhp2::LEU2/pNHP2ZZ, nhp2::LEU2/pnhp2V56KZZ, nhp2::LEU2/pnhp2G59EZZ, nhp2::LEU2/pnhp2R68AZZ, nhp2::LEU2/pnhp2D80AZZ obtained in this way were termed YO346, YO359, YO352, YO357 and YO358, respectively.
Plasmids pNHP2ZZ, pnhp2V56KZZ, pnhp2G59EZZ, pnhp2R68AZZ and pnhp2D80AZZ were obtained as follows. A yeast expression vector containing the ZZ-tag gene was constructed first. A BamHI/BclI ZZ-gene cassette was PCR amplified using pUN100Nt (a gift from Prof. T. Bergès, Université de Poitiers, France) and oligonucleotides PAN2 (5′-CCCGGATCCTTCAGCATGCCTTGCGCAACAG-3′) and PAC#4 (5′-CCCCCCCTGATCATTATTTCGGCGCCTGAGCATCATT-3′). This cassette was inserted in the BamHI site of plasmid pJPG53 (43), a centromeric vector containing the TRP1 gene marker and the promoter and terminator sequences of the GAR1 gene, separated by a unique BamHI restriction enzyme site. In the resulting plasmid, pHA113, the BamHI site remains unique and is located 5′ to the ZZ-gene. An NHP2 gene cassette was then obtained by PCR amplification using oligonucleotides Nhp2-5′-BamHI (5′-CCCCCGGATCCAATGGGTAAAGACAACAAGG-3′) and Nhp2-3′-BamHI (5′-CCCCCGGATCCCATAAAGCTTGAACTTCTTTGAC-3′). nhp2 gene cassettes containing mutations resulting in the V56K, G59E, R68A and D80A substitutions were obtained in two PCR steps. First, partial cassettes containing the mutated sites at their 3′-end were amplified using oligonucleotides Nhp2-5′-BamHI and either Nhp2-V56K (5′-GACAACTTCCTTGACACCTCTTTTCTTATTCTTGGCCTTGGAAGC-3′), Nhp2-G59E (5′-TGACAACTTCCTTGACTTCTCTTTTAACATTCTTGGCCTT-3′), Nhp2-R68A (5′-CTAAACCTTTTTCACCCTTTGCTAAGGCCTTGACAACTTC-3′) or Nhp2-D80A (5′-GGAAATAACATCAGCTGGAGAAATGGCACCGGCGATGACGAC-3′). The fragments obtained in the first amplification step were used in the second amplification round combined with oligonucleotide Nhp2-3′-BamHI. The NHP2, nhp2V56K, nhp2G59E, nhp2R68A and nhp2D80A cassettes obtained were digested by BamHI and inserted in the BamHI site of pHA113, creating plasmids pNHP2ZZ, pnhp2V56KZZ, pnhp2G59EZZ, pnhp2R68AZZ and pnhp2D80AZZ, respectively.
The strains used for the depletion experiment (see Fig. 4) resulted from transformation of strain GAL::nhp2 [YO253, (19)] with plasmids pNHP2ZZ (see above), pnhp2ΔV76I77ZZ or the plasmid pMCGZZ3 (19), a centromeric vector containing the TRP1 marker gene and devoid of any NHP2 sequence. Plasmid pnhp2ΔV76I77ZZ was obtained as follows. An nhp2ΔV76I77 gene cassette flanked by BamHI restriction sites was produced in two PCR steps. First, a partial cassette containing the mutated site at the 3′-end was amplified using oligonucleotides Nhp2-5′-BamHI and Nhp2-Δβ (5′-AATAACATCAGCTGGAGAAATGTCACCGGCGACTAAACCTTTTTCACCCTTTCTTAAGGC-3′). The resulting PCR fragment was used in a second PCR round combined with oligonucleotide Nhp2-3′-BamHI. The PCR fragment obtained was digested with BamHI and inserted in the BamHI site of plasmid pHA113, creating plasmid pnhp2Δβ. A fragment from pnhp2Δβ containing the GAR1 promoter, the nhp2ΔV76I77ZZ gene and the GAR1 terminator was released by an EcoRI/PstI double digestion and inserted in pFL45S (44) digested by the same enzymes, creating plasmid pnhp2ΔV76I77ZZ.
Figure 4.
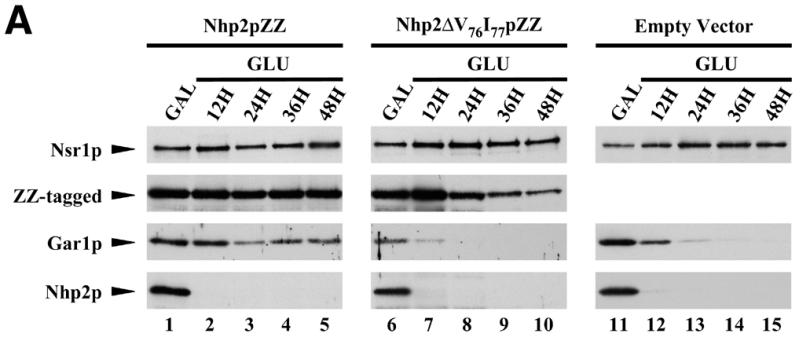
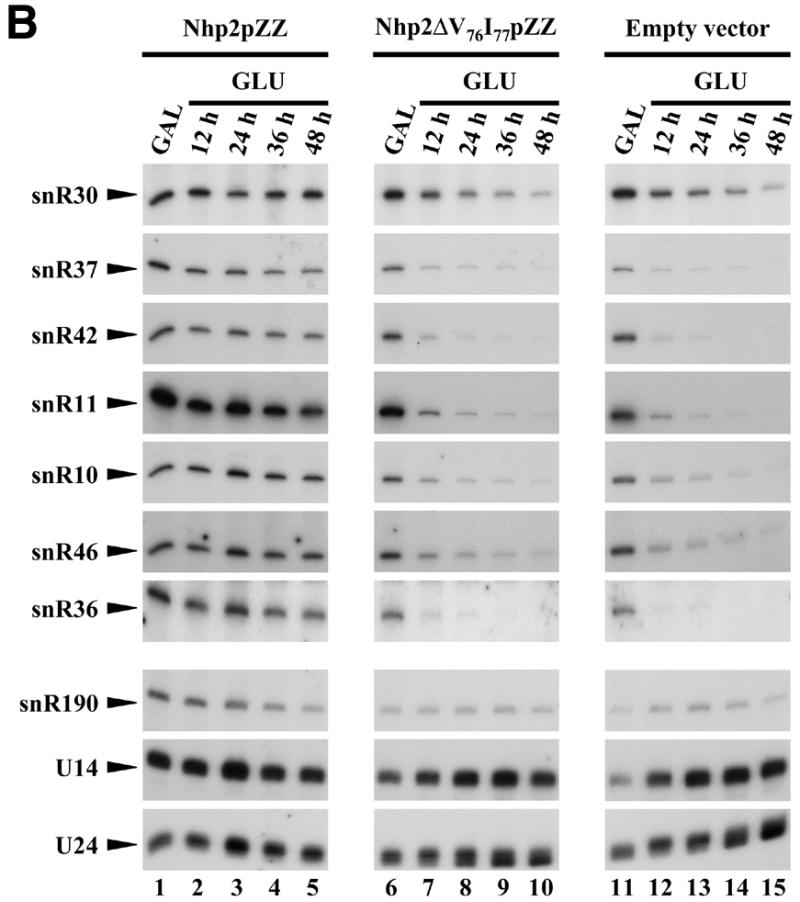
Inhibition of the accumulation of Gar1p and H/ACA snoRNAs due to the ΔV76I77 deletion within Nhp2p. Strains GAL::nhp2/pNHP2ZZ (Nhp2pZZ, lanes 1–5), GAL::nhp2/pnhp2ΔV76I77ZZ (Nhp2ΔV76I77pZZ, lanes 6–10) and GAL::nhp2/empty vector (empty vector, lanes 11–15) were grown in galactose-containing medium (lanes 1, 6 and 11), then were shifted to glucose-containing medium for 12 (lanes 2, 7 and 12), 24 (lanes 3, 8 and 13), 36 (lanes 4, 9 and 14) or 48 h (lanes 5, 10 and 15). Culture aliquots were collected and total proteins or total RNAs were extracted for western (A) or northern (B) analysis. (A) Proteins were separated by SDS–PAGE and transferred to cellulose membranes. Wild-type Nhp2p and ZZ-tagged proteins, Gar1p and Nsr1p proteins were detected by use of anti-Nhp2p, anti-Gar1p, anti-Nsr1p sera, respectively. (B) Extracted RNAs were separated on denaturing 6% polyacrylamide gels and transferred to nylon membranes. snoRNAs were detected by hybridization with specific oligonucleotide probes.
To purify Nhp2pZZ used in band retardation assays, strain GAL::nhp2ZZ (19) grown to mid-exponential phase in YNB medium supplemented with 2% galactose was used. To purify the Nhp2ΔV76I77pZZ and Nhp2G59EpZZ proteins, plasmids directing their expression from the GAL/CYC1 hybrid promoter were constructed. This was achieved in two steps. First, a high copy yeast expression vector containing a ZZ-gene cassette was produced as follows. The ZZ gene was PCR amplified as described above. The resulting PCR fragment was digested by BamHI and BclI and inserted in the BamHI site of pYEDP60.2 (45), creating plasmid pHA114. In pHA114, the BamHI site remains unique. nhp2ΔV76I77 and nhp2G59E gene cassettes were PCR amplified using plasmids pnhp2Δβ and pnhp2G59EZZ, respectively, and oligonucleotides Nhp2-5′-BamHI and Nhp2-3′-BamHI. These nhp2ΔV76I77 and nhp2G59E gene cassettes were digested with BamHI and inserted in the BamHI site of pHA114, creating plasmids pHA133 and pFH99, respectively. The Nhp2ΔV76I77pZZ and Nhp2G59EpZZ proteins were purified from strain JG540 transformed with pHA133 or strain nhp2::LEU2/pnhp2G59EZZ (YO352, see above) transformed with pFH99, respectively, grown to mid-exponential phase in YNB medium supplemented with 2% galactose.
Escherichia coli DH5α strain [F′, endA1, hsdr17 (rk– mk+), supE44, thi-1, recA1, gyrA (Nalr), relA1, Δ(lacIZYA-argF)U169, deoR, (φ80dlacΔ(lacZ)M15)] grown in LB (1% bacto-tryptone, 0.5% bacto-yeast extract, 1% NaCl) liquid or solid media was used for all cloning procedures. Escherichia coli strain BL21(DE3)pLysS [F–, ompT, hsdSB(rB–mB–), gal, dcm (DE3), pLysS] was used for expression of histidine-tagged Nhp2p. It was grown in LB or M9 (0.1% NH4Cl, 0.3% KH2PO4, 0.6% Na2HPO47H2O, 0.4% glucose, 0.05% NaCl, 1 mM MgSO4) media supplemented with chloramphenicol (34 µg/ml).
With the aim of purifying recombinant histidine-tagged Nhp2p from E.coli, the NHP2 open reading frame was PCR amplified using oligonucleotides NHP2PET5′ (5′-GGGGCATATGGGTAAAGACAACAAGGAAC-3′) and NHP2PET3′ (5′-GGGGGATCCTCATAAAGCTTGAACTTCTTTGAC-3′), the resulting PCR fragment was digested by BamHI and NdeI and inserted in the pET15b vector (Novagen) cut by the same enzymes, creating plasmid pET15b-NHP2. This plasmid was transformed into E.coli BL21(DE3)pLysS strain. The resulting transformed strain was propagated in M9 medium supplemented with ampicillin (50 µg/ml) and chloramphenicol (34 µg/ml). To induce the expression of histidine-tagged Nhp2p, IPTG (1 mM final concentration) was added to a culture at OD600 0.6 and incubation was continued for 4 h at 37°C.
Plasmids allowing the in vitro transcription of snR36 and derivatives, which were used in band retardation experiments, were constructed as follows. Open reading frames encoding full-length snR36 or portions of it, either the 5′ stem–loop + insertion element + H box, the 5′ stem–loop + insertion element, the 3′ stem–loop + AUA box or the 3′ stem–loop were PCR amplified using psnR36 plasmid (5) and the following combinations of oligonucleotides: snR36-5′-EcoRI (5′-CCCCCGAATTCTTGCCCTGTGCCTCGCTCGG-3′) + snR36-3′-PmlI.HindIII (5′-CCCCCAAGCTTCACGTGATATGAGACGTTCTAATT-3′) (snR36 full-length); snR36-5′-EcoRI + snR36#2B-PmlI.HindIII (5′-CCCCCAAGCTTCACGTGAATTGTTTTAGCCCGTTGATC-3′) (5′ stem–loop + insertion element + H box); snR36-5′-EcoRI + snR36#2A-PmlI.HindIII (5′-CCCCCAAGCTTCACGTGGCCCGTTGATCAAAAAAATAA-3′) (5′ stem–loop + insertion element); snR36#3-EcoRI (5′-CCCCCGAATTCAGACTTCTTTGAGTACGAGG-3′) + snR36-3′-PmlI.HindIII (3′ stem–loop + AUA box); snR36#3-EcoRI + snR36#5-PmlI.HindIII (5′-CCCCCAAGCTTCACGTGAGACGTTCTAATTACAATACG-3′) (3′ stem–loop). The resulting PCR fragments were digested by EcoRI and HindIII and inserted in the pGEM1 vector (Promega) cut by the same enzymes, producing plasmids pHA106, pHA108, pHA107, pHA110 and pHA109, respectively. A plasmid allowing the in vitro transcription of snR78 was constructed as follows. The SNR78 open reading frame was amplified by PCR using oligonucleotides snR78-EcoRI (5′-CCCCCGAATTCTCCCTTGATGACCAAAATAAATTTTTAC-3′) and snR78-HindIII (5′-CCCCCAAGCTTCACGTGAAAAACCTCAGAAATAAGAATAAACG-3′). The resulting PCR fragment was digested by EcoRI and HindIII and inserted into the pGEM1 vector (Promega) cut by the same enzymes, producing pHA135. To transcribe yeast U24 in vitro, plasmid pyU24 was constructed as follows. The open reading frame of yU24 was amplified by PCR using the oligonucleotides yU24-EcoRI (5′-CCCGAATTCCAAATGATGTAATAACATA-3′) and yU24-HindIII (5′-CCCAAGCTTTCAGAGATCTTGGTGATA-3′). The resulting PCR fragment was digested by BamHI and HindIII and inserted in vector pT7/T3z.19 digested by the same enzymes, creating plasmid pyU24.
Protein purification from yeast
Cell pellets (nature of the strains used is indicated in the previous paragraph) corresponding to 2 l of culture at mid-exponential phase were resuspended in 10 ml ice-cold lysis buffer (50 mM Tris–HCl, pH 8.0, 100 mM NaCl, 5 mM EDTA, 10% glycerol) supplemented with a complete protease inhibitor cocktail (Boehringer Mannheim). Cells were broken in a ‘one shot cell disrupter’ (Constant Systems) set at 1.7 kbar. Extracts were centrifuged at 4°C for 20 min at 10 000 g. Supernatants, to which Triton X-100 was added to reach a final concentration of 1%, were loaded on 0.5 ml IgG–sepharose columns previously packed and equilibrated according to the manufacturer’s recommendations (Amersham Pharmacia Biotech). The columns were washed first with 100 ml ice-cold lysis buffer supplemented with Triton X-100, then with 5 ml ice-cold 0.5 M MgCl2, 20 mM Tris–HCl, pH 8.0, 0.2% Triton X-100, finally with 5 ml ice-cold 2.2 M MgCl2, 20 mM Tris–HCl, pH 8.0, 0.2% Triton X-100. Bound ZZ-tagged proteins were eluted using 5 ml ice-cold 4.5 M MgCl2, 20 mM Tris–HCl, pH 8.0, 0.2% Triton X-100. Presence of the proteins in the eluted fractions was checked by SDS–PAGE, followed by western blotting or Coomassie blue staining of gels. Protein-containing fractions were dialyzed against a 30 mM Tris–HCl, pH 7.4, 150 mM KCl, 2 mM MgCl2 buffer.
Production of anti-Nhp2p antibodies
Recombinant histine-tagged Nhp2p was purified from strain BL21(DE3)pLysS transformed with pET15b-NHP2 (treated as described above) by affinity chromatography using Ni-NTA agarose (Qiagen) and heparin Sepharose (Amersham Pharmacia Biotech) columns, as instructed by the suppliers. The purified protein was used to immunize a rabbit (performed by Eurogentec S.A.). The anti-Nhp2p polyclonal serum obtained after 3 months reacted specifically at 5000-fold dilution with Nhp2p from total yeast cellular extracts.
Western analysis
Proteins from total extracts were separated on 12 or 15% polyacrylamide gels and transferred to hybond-C extra membranes (Amersham Pharmacia Biotech). Gar1p and Nsr1p were detected as described in (46). Nhp2p was detected by use of the anti-Nhp2p serum (see above) diluted 5000-fold.
RNA extractions, northern hybridizations and immunoprecipitations
These were performed as described by Henras et al. (19). Sequences of anti-sense oligonucleotides used to detect snR10, snR11, snR36, snR42 and snR46 have been reported in (19). The sequence of the oligonucleotide used to detect snR37 is the following: 5′-GATAGTATTAACCACTACTG-3′.
Band retardation assays
32P-labeled in vitro transcribed snoRNA (10 fmol), resuspended in a 30 mM Tris–HCl, pH 7.4, 150 mM KCl, 2 mM MgCl2 buffer, was heat denatured at 70°C, 10 min, followed by slow cooling to room temperature. When required, unlabeled competitor RNAs, previously heat denatured and cooled in the same way, were then added to the labeled RNAs. The RNAs were subsequently mixed with the required amounts of purified protein, previously resuspended in the appropriate buffer to obtain a final binding buffer containing 30 mM Tris–HCl, pH 7.4, 150 mM KCl, 2 mM MgCl2, 0.1% Triton X-100, 1 mM DTT, 20% glycerol, 100 ng/µl poly(A) or poly(C), 100 ng/µl BSA. The 20 µl binding reaction was incubated 12 min at room temperature and was then loaded on a 10–12% polyacrylamide, 50 mM Tris, 50 mM glycine gel. Electrophoresis was carried out at 20 mA, 4°C, using a 50 mM Tris, 50 mM glycine running buffer.
In vitro transcription of RNAs was performed with T3, T7 or SP6 RNA polymerases (Promega) following standard procedures. Full-length snR36, snR36 5′ stem–loop + insertion element + H box, snR36 5′ stem–loop + insertion element, snR36 3′ stem–loop + AUA box, snR36 3′ stem–loop or full-length snR78 were in vitro transcribed using T7 RNA polymerase and plasmids pHA106, pHA108, pHA107, pHA110, pHA109 or pHA135, respectively (see above), cut by BbrPI. yU24 was in vitro transcribed using T3 RNA polymerase and plasmid pyU24 cut by HindIII. To produce double-stranded RNAs, each strand of the pGEM1 (Promega) polylinker was in vitro transcribed using T7 or SP6 RNA polymerase and pGEM1 templates digested by HindIII or EcoRI, respectively. Poly(A), poly(C), poly(G) and poly(U) RNAs were purchased from Sigma.
Immunoelectron microscopy
Detection of ZZ-tagged proteins by immunoelectron microscopy was performed as described by Henras et al. (19).
RESULTS
Nhp2p binds directly to the 5′ and 3′ halves of snR36 H/ACA snoRNA in vitro
To establish whether Nhp2p can directly bind RNA, we decided to carry out band retardation experiments with purified Nhp2p. Protein purification was performed by affinity chromatography using extracts from a S.cerevisiae strain expressing Nhp2p tagged at its C-terminus with two IgG-binding domains (ZZ domains) derived from Staphylococcus aureus protein A. We have previously shown that ZZ-tagged Nhp2p (Nhp2pZZ) is fully functional (19). We optimized the protocols for yeast cell lysate production and elution from the IgG–Sepharose affinity column to ensure that we would not purify other H/ACA snoRNP components along with Nhp2pZZ (for details see Materials and Methods). The resulting protein preparation seems free of contaminating proteins (Fig. 1, lane 1).
Figure 1.
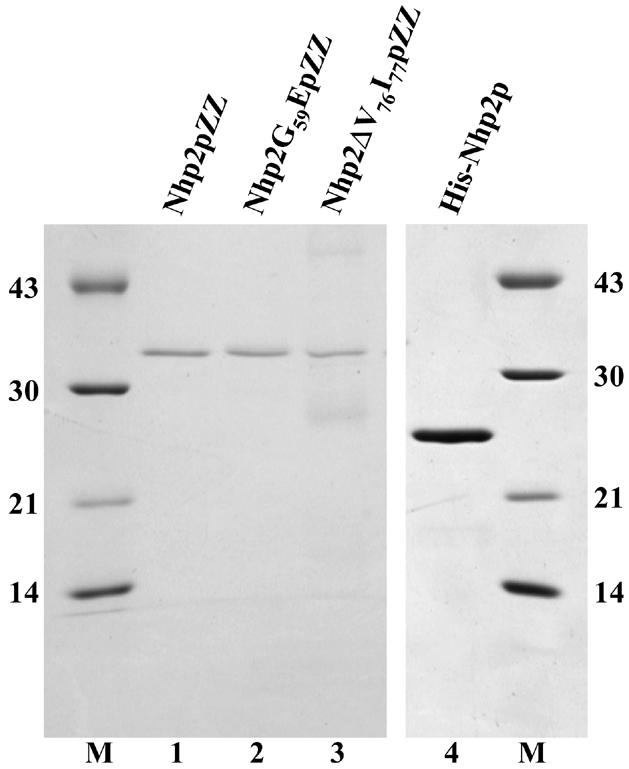
Purified proteins used in band retardation assays. Purified proteins were submitted to SDS–PAGE and revealed by Coomassie blue staining. Lane 1, Nhp2pZZ; lane 2, Nhp2G59EpZZ; lane 3, Nhp2ΔV76I77pZZ; lane 4, His-Nhp2p. Molecular weights (kDa) of markers (M) indicated on the left and right.
The ability of purified Nhp2pZZ to directly interact with an H/ACA snoRNA was tested by band retardation assays using labeled in vitro transcribed snR36. As shown in Figure 2A, several retarded complexes are obtained. These data show that Nhp2pZZ can bind RNA directly and suggest that snR36 contains several binding sites for this protein. More than half of the labeled snR36 RNA is already shifted at a protein concentration of 4 nM (Fig. 2A, lane 2). Thus Nhp2pZZ displays a high affinity for this RNA.
Figure 2.
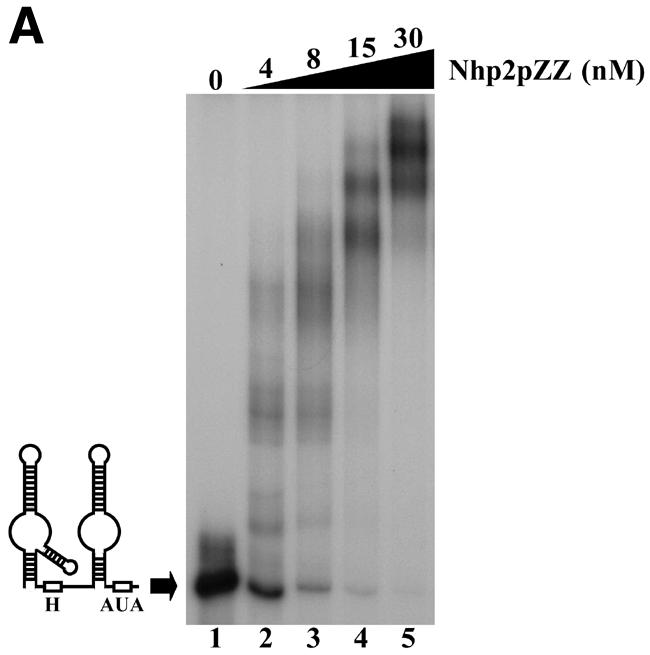


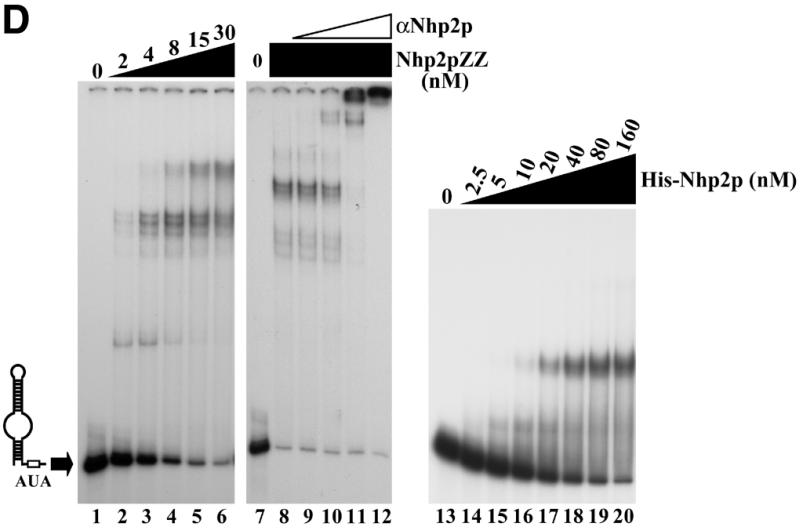
Nhp2p is an RNA-binding protein. Band retardation assays were performed with 10 fmol of labeled full-length snR36 (A), snR36 5′ stem–loop + insertion element + H box (B), snR36 5′ stem–loop + insertion element (C), snR36 3′ stem–loop + AUA box (D), snR36 3′ stem–loop (E). Nhp2pZZ purified from yeast or His-Nhp2p purified from E.coli were used at the concentrations indicated. Protein was omitted in reactions loaded in lanes 1 (A–E), lane 7 (D), lanes 9 (B, C and E) and lane 13 (D). One microliter of anti-Nhp2p polyclonal serum diluted 104-, 103-, 102- and 10-fold was added to binding reactions loaded in lanes 9–12 of (D), respectively. All reactions contained 100 ng/µl poly(A). Complexes were resolved by electrophoresis on native 10 or 12% polyacrylamide gels.
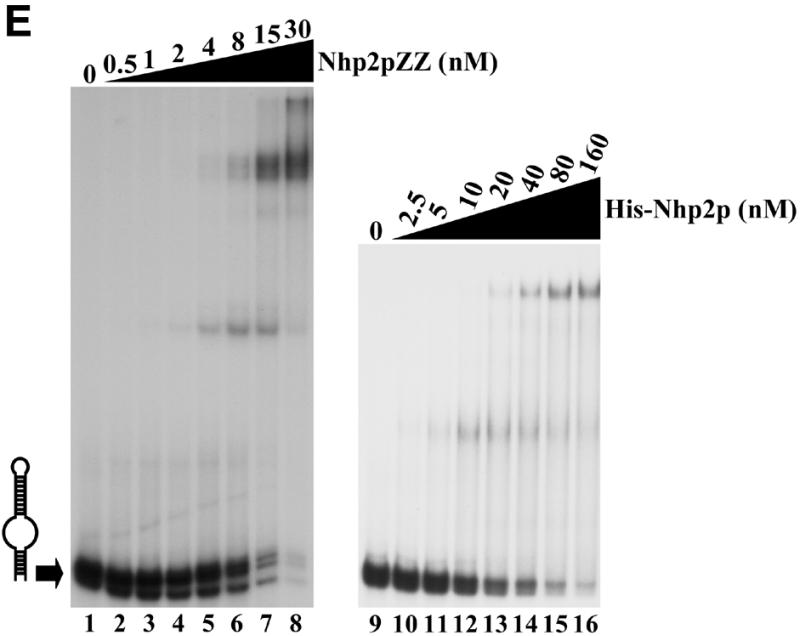
To locate binding sites for Nhp2pZZ within the snR36 molecule, band retardation assays were performed with snR36 sub-domains (for a cartoon of snR36, see Fig. 2A): the 5′ stem–loop, insertion element and H box (snR36 structure differs slightly from the canonical one of H/ACA snoRNAs in that it contains an additional stem–loop inserted at the base of the 5′ stem–internal loop); the 5′ stem–loop and insertion element; the 3′ stem–loop and AUA box (in snR36, the canonical ACA box is replaced by the variant AUA); the 3′ stem–loop. As shown in Figure 2B–E, Nhp2pZZ can bind to all these elements with high affinity. Addition of an anti-Nhp2p serum to binding reactions containing Nhp2pZZ and labeled snR36 3′ stem–loop + AUA box lowered the mobility of all complexes (Fig. 2D, lanes 9–12), indicating that they contain Nhp2pZZ. To rule out the possibility that the ZZ tag mediates RNA binding, band retardation assays using snR36 sub-domains were repeated with histidine-tagged Nhp2p (His-Nhp2p) purified from E.coli. Clearly, His-Nhp2p also binds strongly to the four snR36 sub-fragments (Fig. 2B, C and E, lanes 10–16 and D, lanes 14–20).
These data demonstrate that Nhp2p can bind in vitro to RNAs lacking the single-stranded H or AUA boxes. They show that a single irregular stem–loop with an internal bulge is efficiently bound by Nhp2p. Indeed, the formation of several retarded complexes with the 3′ stem–loop (Fig. 2E) suggests that this structure can be contacted by several Nhp2p molecules.
Nhp2p binds C/D-type snoRNAs in vitro but displays little affinity for perfect double-stranded RNAs or for poly(A) or poly(C) RNAs
The high number of retarded complexes obtained in band retardation experiments suggests that Nhp2p can interact with significant affinity with a broad range of RNA sequences/structures, at least under our in vitro assay conditions (Fig. 2). To determine what RNA characteristics favor interaction with Nhp2p in vitro, competition experiments were performed. Band retardation assays were carried out by first mixing labeled snR36 3′ stem–loop + AUA box at a final concentration of 0.5 nM with various unlabeled competitor RNAs at increasing concentrations. Purified Nhp2pZZ was then added at a final concentration of 30 nM. At such protein concentration, 90% of the probe is shifted in the presence of 100 ng/µl unlabeled poly(A) RNA as sole competitor. Addition of 150-fold molar excess of unlabeled snR36 3′ stem–loop + AUA box reduces the proportion of the shifted probe to 50% (Fig. 3A, lane 3). Essentially the same results are obtained when unlabeled C/D-type U24 or snR78 snoRNAs, whose sizes are similar to that of snR36 3′ stem–loop + AUA box, are added (Fig. 3A, lanes 5–8). Interestingly, the central part of yU24 (47) likely folds into a stem–loop containing an internal bulge consisting of parts of the internal D′ and C′ boxes. This structure, predicted by the RNA folding software available on the mfold version 3.0 server of Zuker and Turner (http://www.ib c.wustl.edu/~zuker/rna/; 48), ensures that the C′ and D′ boxes are maintained in close proximity to each other (49). In the case of snR78 (50), it is probable that the terminal C and D boxes are brought together by the formation of two short stems, creating a stem–internal bulge–stem structure closing the snR78 molecule.
Figure 3.
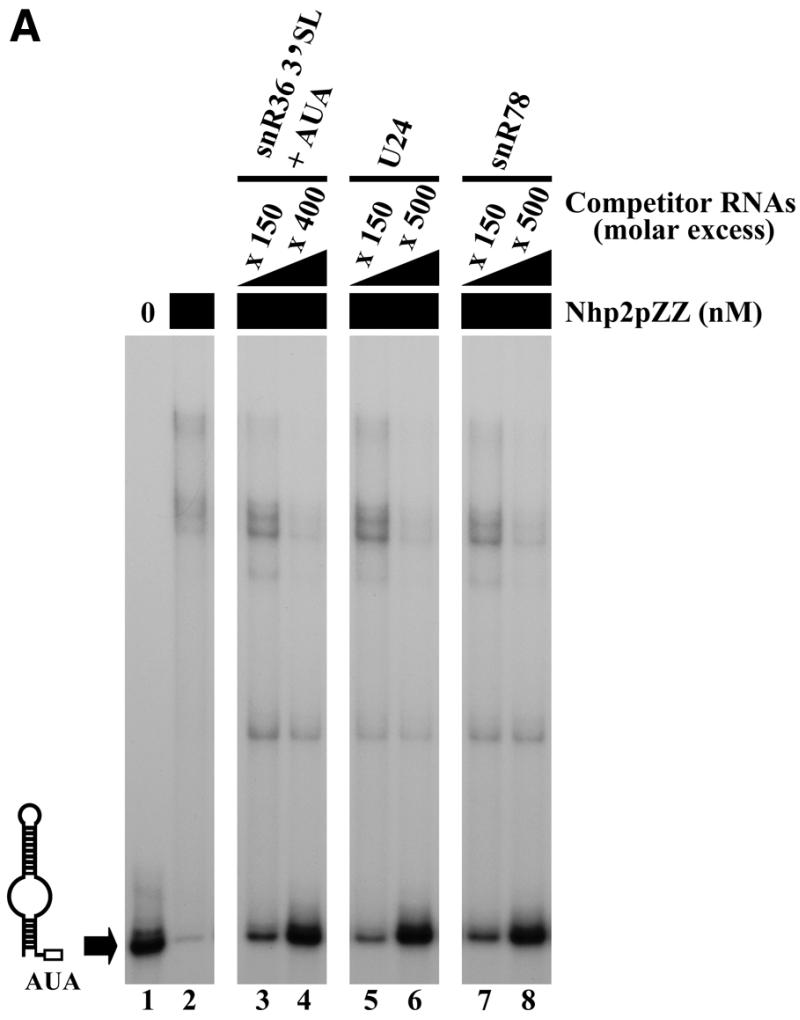
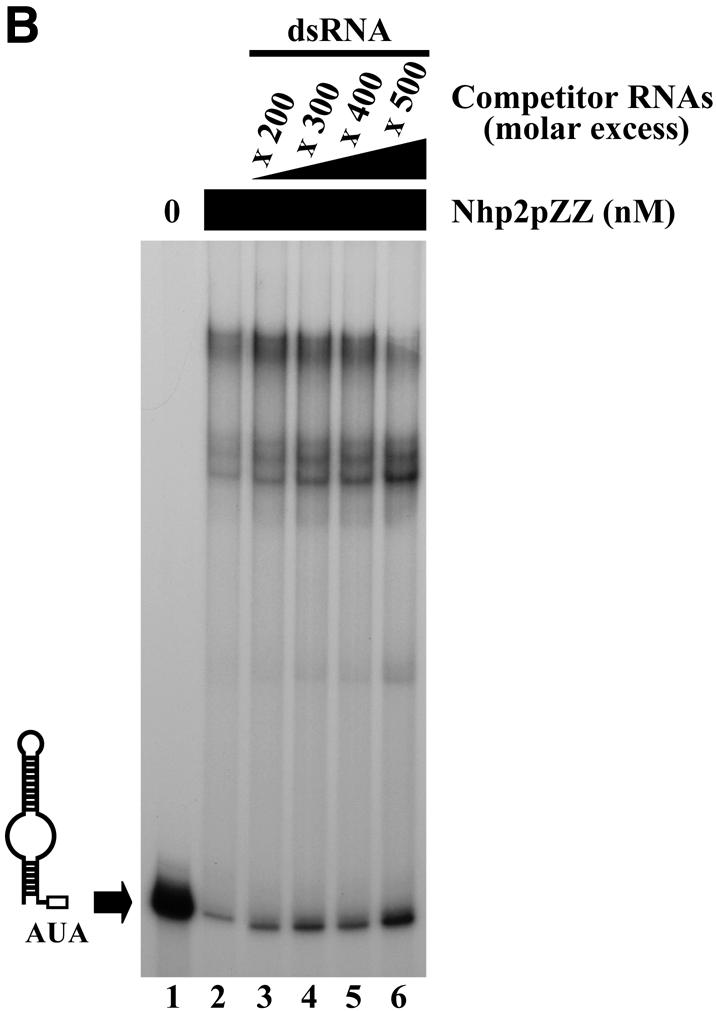
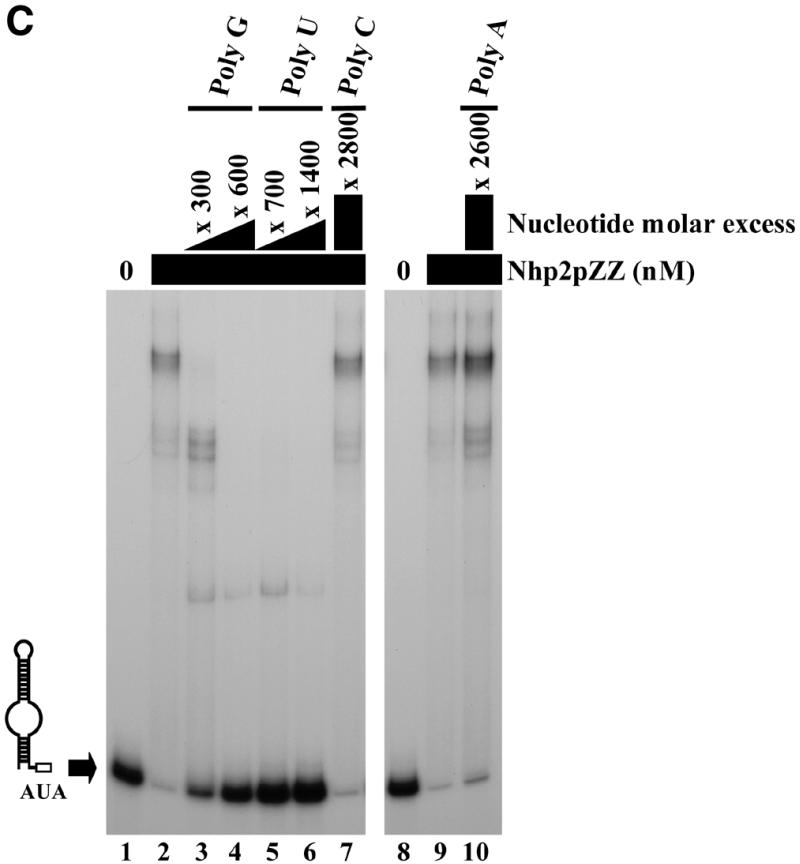
Competition experiments. Band retardation assays were performed with 10 fmol of labeled snR36 3′ stem–loop + AUA box and 100 ng/µl poly(A), except for the retardation assays of lanes 8–10 in (C), for which 100 ng/µl poly(C) were used. All reactions contained Nhp2pZZ at a concentration of 30 nM, except for those loaded in lanes 1 (A–C), and lane 8 (C), which lacked protein. Complexes were resolved by electrophoresis on native 12% polyacrylamide gels. (A) Nhp2pZZ interacts with C/D-type snoRNAs in vitro. The following competitor RNAs were added to binding reactions loaded in lanes 3–8: lane 3, snR36 3′ stem–loop + AUA box, 150× molar excess; lane 4, snR36 3′ stem–loop + AUA box, 400× molar excess; lane 5, U24, 150× molar excess; lane 6, U24, 500× molar excess; lane 7, snR78, 150× molar excess; lane 8, snR78, 500× molar excess. (B) Nhp2pZZ binds poorly to double-stranded RNAs. Double-stranded competitor RNAs, obtained by transcribing each strand of the pGEM1 polylinker followed by strand annealing, were added to the reactions loaded in lanes 3–6 at increasing concentrations: lane 3, 200× molar excess; lane 4, 300× molar excess; lane 5, 400× molar excess; lane 6, 500× molar excess. (C) Nhp2pZZ binds poorly to poly(A) and poly(C). The following competitor RNAs were added to binding reactions loaded in lanes 3–7 and 10: lane 3, poly(G), 300× molar excess of nucleotides; lane 4, poly(G), 600× molar excess of nucleotides; lane 5, poly(U), 700× molar excess of nucleotides; lane 6, poly(U), 1400× molar excess of nucleotides; lane 7, poly(C), 2800× molar excess of nucleotides; lane 10, poly(A), 2600× molar excess of nucleotides.
To investigate the possibility that Nhp2p can bind any double-stranded RNA, irrespective of its nucleotide composition, double-stranded RNA molecules were used as competitors in band retardation experiments. These molecules were obtained by transcribing each strand of the pGEM1 polylinker separately, followed by strand annealing. At the concentrations used, these 53-nt double-stranded RNA molecules proved very weak competitors (Fig. 3B, lanes 3–6). We next tested the ability of Nhp2pZZ to interact with poly(A), poly(C), poly(G) and poly(U) RNAs. Competition experiments with poly(G), poly(U) or poly(C) were performed in the presence of 100 ng/µl poly(A), those with poly(A) in the presence of 100 ng/µl poly(C). Poly(G) or poly(U) RNAs inhibit the formation of retarded complexes but somewhat less efficiently than unlabeled snR36 3′ stem–loop + AUA box (Fig. 3C, lanes 3–6). Typically, a 600-fold molar excess of nucleotides in the case of poly(G) achieves the same level of competition as a 400-fold molar excess of unlabeled snR36 3′ stem–loop + AUA box. In contrast, poly(A) or poly(C) RNAs proved to be extremely inefficient competitors (Fig. 3C, lanes 7 and 10).
We conclude that Nhp2pZZ can bind in vitro with reasonable affinity to RNA molecules displaying diverse sequences and structures. It does, however, discriminate between RNA types. Among the molecules we tested, those most efficiently bound contain irregular stem–loops. Double-stranded molecules are bound very weakly. In addition, Nhp2pZZ seems to have very low affinity for poly(A) or poly(C).
Integrity of the conserved central domain of Nhp2p is crucial for normal cell growth
The central region of Nhp2p, encompassing amino acids 50–82, is related to domains found in several proteins suspected or shown to directly interact with various RNAs (16,19,32–35,39). It has been postulated that these domains constitute RNA-binding motifs of the same type (32,33). When these domains are aligned, it becomes apparent that several positions are occupied by amino acids displaying similar physico-chemical properties. One glycine residue (at position 59 within Nhp2p primary sequence) is invariant (32). Moreover, conserved elements of secondary structure can be identified within these domains (32,33). The size of these elements may be critical. Indeed, removal of a single isoleucine residue within a conserved hydrophobic β-strand of ribosomal protein L30 prevents its interaction with its specific RNA-binding site in vitro and in vivo (33).
We wanted to determine the in vivo phenotypes resulting from the alteration of the highly conserved hydrophobic β-strand (containing the hydrophobic residues V75, V76 and I77) and of the invariant glycine residue at position 59 within the putative RNA-binding domain of Nhp2p. Moreover, we decided to compare these phenotypes with those resulting from substitutions of amino acids within the central domain of Nhp2p that are less conserved than G59. The valine residue at position 56, the arginine 68 and the aspartate 80 were selected for modification. Unlike the invariant glycine, these amino acids are not conserved in all putative RNA-binding motifs of the ‘L30-type’ but their positions are always occupied by amino acids of similar physico-chemical properties (32). We constructed a set of plasmids directing expression of modified versions of ZZ-tagged Nhp2p, either lacking residues V76 and I77 within the proposed hydrophobic β-strand (Nhp2ΔV76I77pZZ) or containing K instead of V at position 56 (Nhp2V56KpZZ), E instead of G at position 59 (Nhp2G59EpZZ), A instead of R at position 68 (Nhp2R68ApZZ) or A instead of D at position 80 (Nhp2D80ApZZ). These plasmids were respectively termed pnhp2ΔV76I77ZZ, pnhp2V56KZZ, pnhp2G59EZZ, pnhp2R68AZZ or pnhp2D80AZZ. A yeast plasmid directing expression of ZZ-tagged wild-type Nhp2p was also produced (termed pNHP2ZZ).
These plasmids were transformed in a haploid yeast strain whose NHP2 chromosomal copy is controlled by the GAL1-10 promoter [strain GAL::nhp2 (19)]. In this strain, chromosomal NHP2 transcription is induced by galactose and repressed by glucose. The GAL::nhp2 strain is therefore unable to grow on glucose-containing media. Transformants of the GAL::nhp2 strain were grown on a medium supplemented with galactose, then streaked on glucose-containing plates. Only those transformants containing plasmid pnhp2ΔV76I77ZZ or a control plasmid lacking the NHP2 gene were unable to grow on plates containing glucose (data not shown). We conclude that the ΔV76I77 deletion is lethal.
To detect whether substitutions of Nhp2p residues V56 to K, G59 to E, R68 to A and D80 to A induce subtle growth defects, the centromeric plasmids directing expression of the corresponding ZZ-tagged altered versions of Nhp2p or the wild-type protein were introduced by plasmid shuffling in a haploid nhp2::LEU2 null strain (for details, see Materials and Methods). All strains grew as the wild-type control, except the strain containing plasmid pnhp2G59EZZ, which displayed significant growth impairment at 30 and 37°C. In selective medium at 30°C, the latter strain grows with a doubling-time of 160 min compared to 120 min for the wild-type control (data not shown). The growth defect does not result from lower levels of Nhp2G59EpZZ compared to those of the wild-type protein, as demonstrated by western analysis (data not shown).
The ΔV76I77 deletion within Nhp2p central domain impairs the accumulation of H/ACA snoRNAs and Gar1p in vivo and inhibits the association of Nhp2p with H/ACA snoRNAs
Nhp2p is required for normal accumulation of H/ACA snoRNAs and the Gar1p protein (19). We tested therefore whether the lethality due to the ΔV76I77 deletion correlates with impaired accumulation of H/ACA snoRNAs and Gar1p. This was done by shifting the GAL::nhp2/pnhp2ΔV76I77ZZ strain (see previous paragraph) from a galactose-containing medium to one containing glucose to repress expression of the wild-type protein. GAL::nhp2 transformants containing either pNHP2ZZ or the vector lacking insert were treated similarly to serve as controls. Total proteins or RNAs were extracted from culture aliquots collected before the carbon-source shift or after 12, 24, 36 or 48 h of growth in glucose-containing medium. Western analysis shows that after 12 or 24 h of growth in a glucose-containing medium, at which times the endogenous wild-type Nhp2p protein has become almost undetectable (Fig. 4A, compare lanes 1, 6, 11 with lanes 2, 7 and 12), levels of Nhp2ΔV76I77pZZ are comparable to those of the tagged wild-type protein (Fig. 4A, compare lanes 1–3 with 6–8). Only at later time points after the carbon-source shift do Nhp2ΔV76I77pZZ levels start to decrease. In the strain expressing Nhp2ΔV76I77pZZ, Gar1p protein levels have already significantly declined after 12 h following the carbon-source shift, and after 24 h these levels are strongly reduced (Fig. 4A, lanes 7 and 8). These results are strictly identical to those obtained with the GAL::nhp2 strain transformed with an empty vector (Fig. 4A, lanes 12 and 13). Furthermore, northern analysis reveals that in the GAL::nhp2 strain expressing Nhp2ΔV76I77pZZ after 24 h of growth on glucose-containing medium, steady-state levels of most H/ACA snoRNAs are drastically reduced (Fig. 4B, compare lanes 6 and 8) and mature 18S rRNA levels are diminished (data not shown), exactly to the same extent as in the GAL::nhp2 strain transformed with the empty vector. In contrast, levels of C/D-type snoRNAs are not reduced (Fig. 4B). We conclude that removal of amino acids V76 and I77 within Nhp2p strongly inhibits the accumulation of H/ACA snoRNPs.
The above described phenotypes could result from impaired association of Nhp2ΔV76I77pZZ with H/ACA snoRNAs. To test this, immunoprecipitation experiments were carried out. These had to be performed using a strain expressing both the ZZ-tagged mutant and the wild-type Nhp2p protein since, in absence of the latter, levels of H/ACA snoRNPs are strongly reduced (see above). Such an approach is validated by the finding that in a strain expressing both ZZ-tagged and endogenous wild-type Nhp2p, Nhp2pZZ is efficiently incorporated into H/ACA snoRNPs (Fig. 5, lanes 1 and 2, and data not shown). When immunoprecipitations were carried out using IgG–Sepharose and a total cell extract obtained from wild-type strain JG540 expressing Nhp2ΔV76I77pZZ, none of the H/ACA snoRNAs tested could be detected in the pellet (Fig. 5, lanes 3 and 4). These results strongly suggest that the interaction of Nhp2ΔV76I77pZZ with H/ACA snoRNAs is severely inhibited in vivo.
Figure 5.
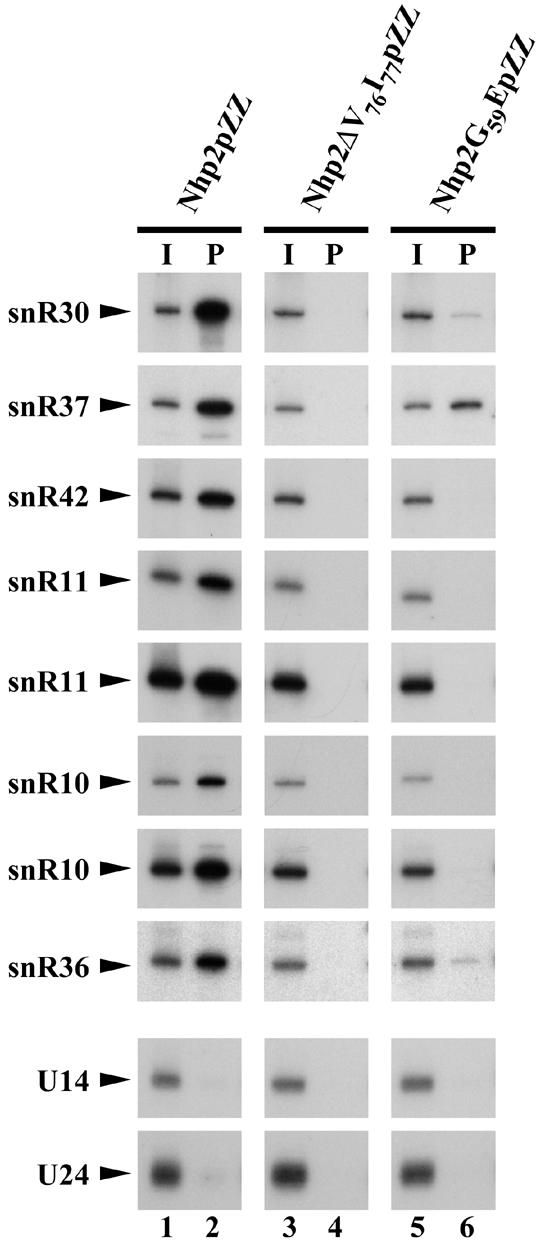
Effects of the ΔV76I77 deletion or the G59E substitution on the interaction of Nhp2p with H/ACA snoRNAs in cell extracts. Immunoprecipitation experiments were carried out using IgG–Sepharose and extracts from strains obtained by transforming JG540 with plasmids directing expression of either Nhp2pZZ (lanes 1 and 2), Nhp2ΔV76I77pZZ (lanes 3 and 4) or Nhp2G59EpZZ (lanes 5 and 6). RNAs extracted from one-fifteenth of the input extracts (I, lanes 1, 3 and 5) or from the pellets following immunoprecipitation (P, lanes 2, 4 and 6) were separated on a denaturing 6% polyacrylamide gel and transferred to a nylon membrane. Various H/ACA and C/D snoRNAs were detected by hybridization with specific oligonucleotide probes.
Deletion ΔV76I77 inhibits the specific accumulation of Nhp2p within the nucleolus
We assessed by immunoelectron microscopy the effect on Nhp2p sub-cellular localization of the ΔV76I77 deletion, which impairs the association of Nhp2p with H/ACA snoRNAs in cell extracts and H/ACA snoRNP accumulation in vivo. ZZ-tagged wild-type or mutant proteins expressed in a wild-type strain, in order to preserve intact nuclear domains, were detected by use of anti-protein A antibodies and colloidal gold-conjugated protein A. Most ZZ-tagged wild-type Nhp2p proteins are found concentrated in the nucleolus, although some can also be detected in the nucleoplasm (Fig. 6) (19). Nhp2ΔV76I77pZZ is efficiently imported into the nucleus but unlike the wild-type protein, it is found evenly distributed throughout the nucleus (Fig. 6). These results strongly suggest that Nhp2p can accumulate in the nucleolus only when it is associated with H/ACA snoRNAs.
Figure 6.

The ΔV76I77 deletion prevents the specific accumulation of Nhp2p within the nucleolus. Shown are immunolocalizations of Nhp2pZZ (WT) or Nhp2ΔV76I77pZZ (ΔV76I77) proteins expressed in wild-type strain JG540. ZZ-tagged proteins were detected by treatment with anti-protein A antibodies followed by incubation with colloidal gold-conjugated protein A. No, nucleolus; Nu, nucleoplasm.
Conversion of G59 to E within the putative RNA-binding domain of Nhp2p affects the steady-state levels of H/ACA snoRNAs to different extents
We tested next by northern analysis whether the growth defect resulting from the conversion of glycine 59 to glutamate correlates with an alteration of the steady-state levels of H/ACA snoRNAs. Surprisingly, the steady-state levels of these snoRNAs are not all affected in the same way by the G59E conversion (Fig. 7, lane 3). In the strain expressing Nhp2G59EpZZ, levels of some H/ACA snoRNAs are severely decreased (by ∼85% for snR10 and snR11), decreased (by ∼50% for snR42), increased (by 40–50% for snR30 or snR37) or only mildly affected (e.g. snR36), compared to the wild-type strain. Levels of H/ACA snoRNAs in the other mutant strains differ somewhat from wild-type levels but to a far lesser extent (Fig. 7, lanes 2, 4 and 5).
Figure 7.
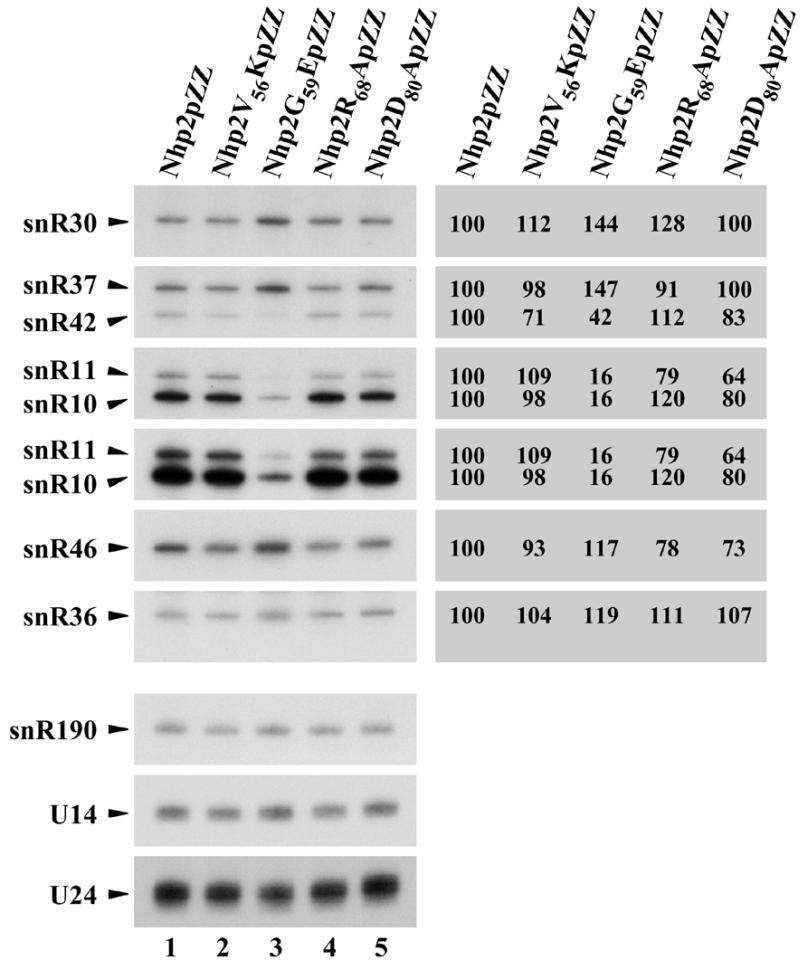
Northern analysis of snoRNA levels in nhp2::LEU2 strains expressing Nhp2V56KpZZ, Nhp2G59EpZZ, Nhp2R68ApZZ or Nhp2D80ApZZ. Total RNAs were extracted from strains nhp2::LEU2/pNHP2ZZ (Nhp2pZZ, lane 1), nhp2::LEU2/pnhp2V56KZZ (Nhp2V56KpZZ, lane 2), nhp2::LEU2/pnhp2G59EZZ (Nhp2G59EpZZ, lane 3), nhp2::LEU2/pnhp2R68AZZ (Nhp2R68ApZZ, lane 4) and nhp2::LEU2/pnhp2D80AZZ (Nhp2D80ApZZ, lane 5), separated on a denaturing 6% polyacrylamide gel and transferred to a nylon membrane. Various H/ACA-type and C/D-type snoRNAs were detected by hybridization with specific oligonucleotide probes. Phosphorimager scans of the northern blot were used to quantify H/ACA snoRNA levels. These are given as percentage level in wild-type cells (to the right of the northern blot). Values were normalized using the levels of the C/D-type snR190 and U14 snoRNAs as internal standards. Values oscillate on average by ±5%.
The variations in the way steady-state levels of H/ACA snoRNAs are changed by the G59E conversion could be, at least partly, explained by the fact that this substitution modifies the strength of Nhp2p association to different H/ACA snoRNAs to varying degrees. To test this idea, immunoprecipitation experiments were carried out with IgG–Sepharose under stringent conditions using extracts from strains expressing both ZZ-tagged protein (wild-type or modified) and endogenous wild-type Nhp2p. It is obvious that the G59E substitution significantly reduces the efficiency with which all tested H/ACA snoRNAs are co-precipitated with the tagged protein (Fig. 5, lanes 5 and 6). Moreover, the extent of the decrease of co-immunoprecipitation efficiency depends on the tested H/ACA snoRNA. The most efficiently co-precipitated snoRNA is snR37, whose levels are increased the most by the G59E conversion. snR30, whose steady-state levels are also increased by the G59E conversion, is co-precipitated to some extent, although clearly far less efficiently than by the tagged wild-type protein. snR10, snR11 and snR42 snoRNAs, whose levels are significantly reduced by the G59E conversion, can hardly be detected in the pellet fraction obtained after immunoprecipitation of Nhp2G59EpZZ. Thus, the above data show that the G59E substitution weakens, at least in cell extracts, the interaction of Nhp2p with all tested H/ACA snoRNAs, but to different degrees.
The ΔV76I77 deletion and the G59E substitution within Nhp2p central domain modify the pattern of complexes obtained in band retardation assays
In order to test the effects of the ΔV76I77 deletion and the G59E substitution within Nhp2p central domain on the interactions of the protein with RNA in vitro, band retardation assays were performed with Nhp2ΔV76I77pZZ and Nhp2G59EpZZ proteins purified from yeast (see Materials and Methods) (Fig. 1, lanes 2 and 3). As shown in Figure 8 (lanes 8–12) Nhp2ΔV76I77pZZ is still able to interact with snR36 3′ stem + AUA box in vitro. However, the pattern of complexes obtained with this modified protein bears no resemblance to the one produced by wild-type Nhp2pZZ. The small reduction in molecular weight of Nhp2p due to the removal of just two amino acids is unlikely to cause such major differences. The G59E conversion does not significantly reduce the overall affinity of the protein for snR36 3′ stem + AUA box (Fig. 8, lanes 14–18). The pattern of retarded bands obtained using Nhp2G59EpZZ does not differ profoundly from the wild-type profile, except that the complex of highest mobility (Fig. 8, asterisk) is absent. Among the H/ACA snoRNAs tested, snR10 and snR11 display the strongest decrease in steady-state levels in the strain expressing Nhp2G59EpZZ (see above). Thus band retardation assays were also performed with the Nhp2G59EpZZ protein and in vitro transcribed snR10 5′ and 3′ halves (data not shown). Surprisingly, however, the modifications induced by the G59E conversion on the pattern of complexes obtained with snR10 5′ and 3′ halves do not differ substantially from those observed in the case of snR36 3′ stem + AUA box (i.e. the complex of highest mobility disappears and some of the complexes are shifted slightly upwards).
Figure 8.
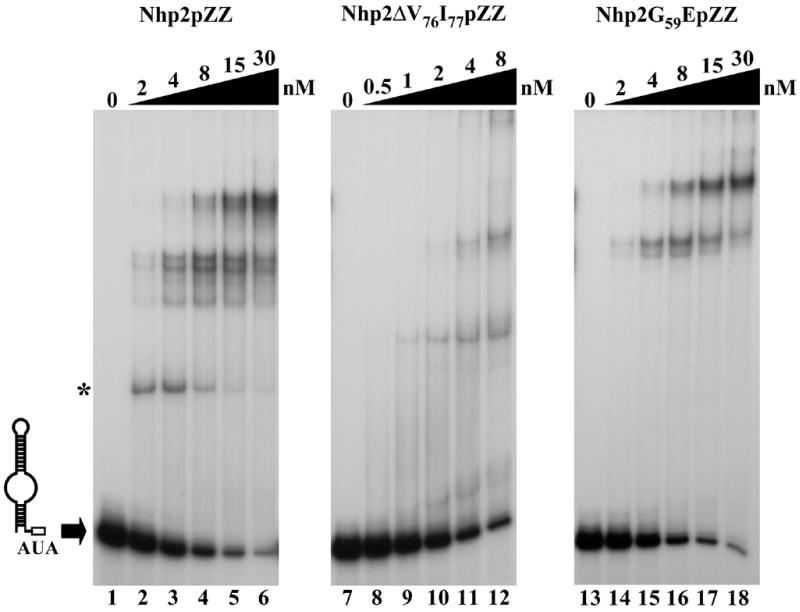
The ΔV76I77 deletion or the G59E substitution within Nhp2p central domain alters the pattern of complexes obtained in band retardation assays. Band retardation experiments were performed using 10 fmol of labeled snR36 3′ stem + AUA box and Nhp2pZZ (lanes 2–6), Nhp2ΔV76I77pZZ (lanes 8–12) or Nhp2G59EpZZ (lanes 14–18) added at the indicated concentrations. In reactions loaded in lanes 1, 7 and 13, protein was omitted. Complexes were resolved by electrophoresis on native 12% polyacrylamide gels.
These data suggest that shortening the putative hydrophobic β-strand significantly alters the way Nhp2p interacts with RNA in vitro while the effects of the G59E conversion are less dramatic.
DISCUSSION
While the protein composition of the core of H/ACA snoRNPs has been determined, the precise functions of most of its constituents, the assembly pathway and the final structure of the particles remain unknown. We are currently trying to clarify these issues. We focused our attention first on the Nhp2p protein, which is essential for proper accumulation of H/ACA snoRNPs (19) and was proposed to contain an RNA-binding domain of the type found in yeast ribosomal protein L30 (16,19,32,33). These properties prompted us to test whether Nhp2p can bind H/ACA snoRNAs directly. Band retardation assays performed with a purified Nhp2p protein show unambiguously that this is the case. Interestingly, recent data by Dragon et al. (25) strongly suggest that the human counterpart of Nhp2p does also bind directly to H/ACA snoRNAs. These authors showed that four proteins from HeLa cell extracts can be specifically UV-cross-linked to the U17 H/ACA snoRNA. One of them, a protein of 23 kDa apparent molecular weight and the likely counterpart of yeast Nhp2p, is by far the most efficiently cross-linked protein, lending strong support to the notion that it is in direct contact with RNA.
In vitro, yeast Nhp2p interacts with good affinity with a broad range of RNAs. Moreover, the intricate pattern of complexes obtained in band retardation assays performed with snR36 5′ or 3′ halves suggests that Nhp2p can bind in vitro to several different sites within H/ACA snoRNAs. This behavior contrasts with that displayed by ribosomal protein L30, which exhibits a very tight specificity for a well characterized binding site found on its own pre- and mRNA (33,39,51–53). The human 15.5 kDa [U4/U6.U5] tri-snRNP protein contains an RNA-binding domain of the L30-type and it too binds with high specificity to a site found on U4 snRNA (34). Both L30 and the 15.5 kDa protein specifically interact with an asymmetric internal loop closed by two stems of different lengths. The integrity of the stems as well as the particular nature of several nucleotides within the loop are crucial for high affinity binding (33,34,39,51–53). The human 15.5 kDa tri-snRNP protein not only interacts with U4 snRNA but also with all tested C/D-box snoRNAs and, remarkably, the specific binding site on these snoRNAs is constituted by the box C/D motif that can be folded into a stem–internal loop–stem structure (35). Thus, within the family of proteins containing the L30-type RNA-binding domain, proteins exhibiting very tight RNA-binding specificity (L30, 15.5 kDa protein) and at least one protein that is capable of efficient binding to a broad range of RNAs in vitro (Nhp2p) are to be found. This is not unprecedented: it has already been well documented for the family of proteins sharing the RNA recognition motif (RRM) domain (54,55).
Yeast Nhp2p does not, however, interact with all types of RNA with equal efficiency. In particular, its interaction with single-stranded poly(A) or poly(C) RNAs is extremely poor. This is interesting because poly(A) is in effect a succession of canonical H boxes. Thus, if Nhp2p does bind to the H box of H/ACA snoRNAs in vivo, the box is unlikely to suffice for tight binding. In fact, our data show that neither the H nor the AUA box are necessary for Nhp2p interaction with RNA in vitro.
Clearly, Nhp2p interacts in vitro with RNA molecules, in particular C/D-type snoRNAs, with which it is not associated in a stable manner in vivo. In cells, Nhp2p interaction with these RNAs may be prevented by preferential binding of C/D-type snoRNP proteins (23,35,56–63). In addition, Nhp2p interaction with other protein component(s) of H/ACA snoRNPs may increase its specificity for H/ACA snoRNAs in vivo. Determining the RNA site(s) bound by Nhp2p in the context of the assembled H/ACA snoRNPs will require reconstitution studies with the remaining protein components of these particles.
In order to determine the in vivo consequences of altering the central domain of Nhp2p proposed to mediate interactions with RNA, we undertook a mutational analysis of the protein. To start with, hydrophobic residues V76 and I77 within a putative β-strand of Nhp2p central domain were removed. Such alteration was chosen because it mimics a deletion within the corresponding β-strand of ribosomal protein L30 that inhibits binding of the latter protein to its pre-mRNA but does not prevent its incorporation into ribosomes (33). These data demonstrate that slightly shortening the β-strand does not drastically alter protein folding. In support of the notion that removal of amino acids V76 and I77 of Nhp2p does not induce major structural rearrangements of the protein, we observe that Nhp2ΔV76I77pZZ is efficiently targeted to the nucleus. In vivo, the modified protein cannot support normal accumulation of H/ACA snoRNPs: in cells expressing Nhp2ΔV76I77pZZ under conditions of wild-type Nhp2p depletion, levels of Gar1p and H/ACA snoRNAs are drastically reduced. Moreover, the extent of the reduction is identical in the similarly treated control cells depleted of wild-type Nhp2p and lacking the modified protein. Prolonged culturing of cells under conditions of wild-type Nhp2p depletion eventually leads to a decrease in Nhp2ΔV76I77pZZ levels. However, this may be a consequence but cannot be the cause of the Gar1p and H/ACA snoRNA accumulation defects since these are detected well before the amounts of Nhp2ΔV76I77pZZ start declining. Results of the immunoprecipitation experiments strongly suggest that the phenotypes of the ΔV76I77 deletion are a direct consequence of the inability of the modified Nhp2p protein to participate in productive H/ACA snoRNP assembly. While this could be caused by protein–protein interaction defects, the band retardation data favor the idea that it is, at least partly, due to impaired direct interaction of Nhp2ΔV76I77pZZ with H/ACA snoRNAs in vivo. Indeed, removal of amino acids V76 and I77 seems to drastically alter the way Nhp2p interacts with RNA in vitro, since the profile of retarded bands obtained in retardation assays using Nhp2ΔV76I77pZZ is totally different from the one produced by the wild-type protein. This is somewhat reminiscent of effects reported for DNA-binding proteins that induce DNA bending. For such proteins, modifying the position of their binding site relative to the ends of a given DNA fragment substantially alters the mobility of retarded complexes obtained in band retardation assays (64). We believe that the above-described data lend support to the view that proper and efficient direct binding of Nhp2p to H/ACA snoRNAs is important for H/ACA snoRNP assembly and hence for the stability of some of their constituents.
Koonin et al. (32) noted that only one amino acid, a glycine (at position 59 within the primary sequence of Nhp2p), is strictly conserved among all known putative RNA-binding domains of the L30-type. This observation strongly suggests that the presence of this conserved glycine is needed for optimum binding of these domains to RNA. Indeed, in the solution structure of L30 bound to its specific RNA, solved by NMR spectroscopy, as well as in the crystal structure of the 15.5 kDa protein bound to a U4 snRNA fragment, the invariant glycine is positioned at the center of the RNA binding surface of the protein and in close proximity to RNA (65,66). In the case of L30 and the 15.5 kDa protein at least, the glycine residue is probably favored for sterical reasons, as a more bulky amino acid might hinder the protein from making close contact with the RNA (65). Since the functional importance of the invariant glycine residue in Nhp2p had never been tested experimentally, we analyzed the effects of converting residue G59 to E within Nhp2p. This modification does not alter the steady-state levels of the protein. Moreover, we confirmed by immunoelectron microscopy that Nhp2G59EpZZ is efficiently imported into the nucleus (data not shown). In vivo, the G59 to E conversion leads to substantial growth defects at 30 and 37°C, the growth impairment at lower temperatures being far milder. Consistent with the fact that residues V56, R68 and D80 within the putative RNA-binding domain of Nhp2p are less conserved than G59, conversion of V56 to K, R68 to A or D80 to A has little effect on cell growth. The conversion of G59 to E also modifies the steady-state levels of H/ACA snoRNAs. Unexpectedly, the effects vary depending on the H/ACA snoRNA considered, levels of some snoRNAs being severely decreased (by 85% for snR10 and snR11), little affected (e.g. snR36) or actually substantially increased (e.g. snR37). One explanation accounting for these results is that the G59E mutation affects the affinity of Nhp2p for different H/ACA snoRNAs to different extents. In an attempt to test this, Nhp2G59EpZZ proteins were precipitated from cell extracts containing also wild-type Nhp2p devoid of tag and the levels of different H/ACA snoRNAs brought down were assessed. As it turns out, co-immunoprecipitation efficiencies of all H/ACA snoRNAs are reduced by the G59E conversion but to varying degrees. We observe that snR37, whose levels are increased most by the G59E conversion is co-precipitated best with Nhp2G59EpZZ. Conversely, those snoRNAs whose levels are lowered most, snR10 and snR11, are hardly co-precipitated at all with this modified protein. Why the levels of some H/ACA snoRNAs should actually be increased in the strain expressing Nhp2G59EpZZ is not immediately obvious. This may happen if the activity of the machinery involved in the maturation/turnover of H/ACA snoRNAs is limiting. For example, increased turnover of snR10 (one the most abundant H/ACA snoRNAs) due to defective interaction with Nhp2G59EpZZ may increase the stability of those H/ACA snoRNAs with which the mutant protein interacts, comparatively, in a more stable fashion.
The in vivo effects of the G59E conversion are likely to be chiefly consequences of altered binding of Nhp2p to snoRNAs as such. Indeed, results of band retardation assays suggest that interaction of Nhp2G59EpZZ with RNA is altered in vitro. Moreover, while this work was in progress, Nottrott et al. (34) reported that converting the conserved G to K in the human 15.5 kDa [U4/U6.U5] tri-snRNP protein abolished its specific binding to the 5′ stem–loop of U4.
Little is known thus far regarding the stage at which H/ACA snoRNP proteins associate with H/ACA (pre)-snoRNAs and where within the nucleus the assembly process takes place. It is also unclear whether H/ACA snoRNP proteins can accumulate in the nucleolus independently from H/ACA snoRNAs. The protein domains required for nucleolar accumulation have been analyzed in the case of Dyskerin (67), yeast Gar1p and human Gar1p (hGar1p) (26,43). The central cores of Gar1p and hGar1p accumulate within nucleoli (26,43). Removal of the 20 C-terminal amino acids of hGar1p core region prevents accumulation within nucleoli and coiled bodies (26). Interestingly, this truncated version of hGar1p fails to interact with the H/ACA snoRNA U17 as judged from immunoprecipitation assays (26). Removal of amino acids V76 and I77 within the Nhp2p protein has very similar effects. Nhp2ΔV76I77pZZ is not excluded from the nucleolus as such but found evenly distributed throughout the nucleus. This observation is consistent with the idea that nucleolar accumulation is above all the consequence of a specific retention within the nucleolus. It also strongly suggests that Nhp2p cannot accumulate in the nucleolus when it is not integrated into H/ACA snoRNPs.
Acknowledgments
ACKNOWLEDGEMENTS
We are particularly grateful to Prof. P. Bouvet (Ecole Normale Supérieure, Lyon, France) for repeated advice and discussions. We also wish to thank him and Dr T. Kiss (LBME, Toulouse, France) for critical reading of the manuscript and Drs E. Izaurralde and B. Séraphin (EMBL, Heidelberg, Germany) for helpful discussions. The gifts of monoclonal anti-Nsr1p antibody from Prof. J. Woolford (Carnegie Mellon University) and of plasmid pUN100Nt from Prof. T. Bergès (Université de Poitiers, France) are gratefully acknowledged. We are thankful to members of the Ferrer laboratory for help and numerous discussions. We thank Y. de Préval for synthesis of oligonucleotides and D. Villa for art work. A.H. and C.D. are supported by grants from la Ligue Nationale contre le Cancer and the Ministère de l’Education Nationale, de l’Enseignement Supérieur et de la Recherche, respectively. This work was financed by la Ligue Nationale contre le Cancer, the CNRS and Université Paul Sabatier, Toulouse, France.
References
- 1.Kambach C., Walke,S. and Nagai,K. (1999) Structure and assembly of the spliceosomal small nuclear ribonucleoprotein particles. Curr. Opin. Struct. Biol., 9, 222–230. [DOI] [PubMed] [Google Scholar]
- 2.Seto A.G., Zaug,A.J., Sobel,S.G., Wolin,S.L. and Cech,T.R. (1999) Saccharomyces cerevisiae telomerase is an Sm small nuclear ribonucleoprotein particle. Nature, 401, 177–180. [DOI] [PubMed] [Google Scholar]
- 3.Mitchell J.R., Cheng,J. and Collins,K. (1999) A box H/ACA small nucleolar RNA-like domain at the human telomerase RNA 3′ end. Mol. Cell. Biol., 19, 567–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen J.L., Blasco,M.A. and Greider,C.W. (2000) Secondary structure of vertebrate telomerase RNA. Cell, 100, 503–514. [DOI] [PubMed] [Google Scholar]
- 5.Ganot P., Bortolin,M.-L. and Kiss,T. (1997) Site-specific pseudouridine formation in preribosomal RNA is guided by small nucleolar RNAs. Cell, 89, 799–809. [DOI] [PubMed] [Google Scholar]
- 6.Ni J., Tien,A.L. and Fournier,M.J. (1997) Small nucleolar RNAs direct site-specific synthesis of pseudouridine in ribosomal RNA. Cell, 89, 565–573. [DOI] [PubMed] [Google Scholar]
- 7.Tollervey D. and Kiss,T. (1997) Function and synthesis of small nucleolar RNAs. Curr. Opin. Cell Biol., 9, 337–342. [DOI] [PubMed] [Google Scholar]
- 8.Balakin A.G., Smith,L. and Fournier,M.J. (1996) The RNA world of the nucleolus: two major families of small RNAs defined by different box elements with related functions. Cell, 86, 823–834. [DOI] [PubMed] [Google Scholar]
- 9.Ganot P., Caizergues-Ferrer,M. and Kiss,T. (1997) The family of box ACA small nucleolar RNAs is defined by an evolutionarily conserved secondary structure and ubiquitous sequence elements essential for RNA accumulation. Genes Dev., 11, 941–956. [DOI] [PubMed] [Google Scholar]
- 10.Samarsky D.A. and Fournier,M.J. (1999) A comprehensive database for the small nucleolar RNAs from Saccharomyces cerevisiae. Nucleic Acids Res., 27, 161–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bortolin M.-L., Ganot,P. and Kiss,T. (1999) Elements essential for accumulation and function of small nucleolar RNAs directing site-specific pseudouridylation of ribosomal RNAs. EMBO J., 18, 457–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Narayanan A., Lukowiak,A., Jady,B.E., Dragon,F., Kiss,T., Terns,R.M. and Terns,M.P. (1999) Nucleolar localization signals of box H/ACA small nucleolar RNAs. EMBO J., 18, 5120–5130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jiang W., Middleton,K., Yoon,H.J., Fouquet,C. and Carbon,J. (1993) An essential yeast protein, CBF5p, binds in vitro to centromeres and microtubules. Mol. Cell. Biol., 13, 4884–4893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cadwell C., Yoon,H.J., Zebarjadian,Y. and Carbon,J. (1997) The yeast nucleolar protein Cbf5p is involved in rRNA biosynthesis and interacts genetically with the RNA polymerase I transcription factor RRN3. Mol. Cell. Biol., 17, 6175–6183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lafontaine D.L., Bousquet-Antonelli,C., Henry,Y., Caizergues-Ferrer,M. and Tollervey,D. (1998) The box H + ACA snoRNAs carry Cbf5p, the putative rRNA pseudouridine synthase. Genes Dev., 12, 527–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Watkins N.J., Gottschalk,A., Neubauer,G., Kastner,B., Fabrizio,P., Mann,M. and Luhrmann,R. (1998) Cbf5p, a potential pseudouridine synthase, and Nhp2p, a putative RNA-binding protein, are present together with Gar1p in all H BOX/ACA-motif snoRNPs and constitute a common bipartite structure. RNA, 4, 1549–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Girard J.-P., Lehtonen,H., Caizergues-Ferrer,M., Amalric,F., Tollervey,D. and Lapeyre,B. (1992) GAR1 is an essential small nucleolar RNP protein required for pre-rRNA processing in yeast. EMBO J., 11, 673–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kolodrubetz D. and Burgum,A. (1991) Sequence and genetic analysis of NHP2: a moderately abundant high mobility group-like nuclear protein with an essential function in Saccharomyces cerevisiae. Yeast, 7, 79–90. [DOI] [PubMed] [Google Scholar]
- 19.Henras A., Henry,Y., Bousquet-Antonelli,C., Noaillac-Depeyre,J., Gélugne,J.-P. and Caizergues-Ferrer,M. (1998) Nhp2p and Nop10p are essential for the function of H/ACA snoRNPs. EMBO J., 17, 7078–7090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Phillips B., Billin,A.N., Cadwell,C., Buchholz,R., Erickson,C., Merriam,J.R., Carbon,J. and Poole,S.J. (1998) The Nop60B gene of Drosophila encodes an essential nucleolar protein that functions in yeast. Mol. Gen. Genet., 260, 20–29. [DOI] [PubMed] [Google Scholar]
- 21.Giordano E., Peluso,I., Senger,S. and Furia,M. (1999) minifly, a Drosophila gene required for ribosome biogenesis. J. Cell Biol., 144, 1123–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meier U.T. and Blobel,G. (1994) NAP57, a mammalian nucleolar protein with a putative homolog in yeast and bacteria. J. Cell Biol., 127, 1505–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang Y., Isaac,C., Wang,C., Dragon,F., Pogacic,V. and Meier,U.T. (2000) Conserved composition of mammalian box H/ACA and box C/D small nucleolar ribonucleoprotein particles and their interaction with the common factor Nopp140. Mol. Biol. Cell, 11, 567–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heiss N.S., Knight,S.W., Vulliamy,T.J., Klauck,S.M., Wiemann,S., Mason,P.J., Poustka,A. and Dokal,I. (1998) X-linked dyskeratosis congenita is caused by mutations in a highly conserved gene with putative nucleolar functions. Nature Genet., 19, 32–38. [DOI] [PubMed] [Google Scholar]
- 25.Dragon F., Pogacic,V. and Filipowicz,W. (2000) In vitro assembly of human H/ACA small nucleolar RNPs reveals unique features of U17 and telomerase RNAs. Mol. Cell. Biol., 20, 3037–3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pogacic V., Dragon,F. and Filipowicz,W. (2000) Human H/ACA small nucleolar RNPs and telomerase share evolutionarily conserved proteins NHP2 and NOP10. Mol. Cell. Biol., 20, 9028–9040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Watanabe Y. and Gray,M.W. (2000) Evolutionary appearance of genes encoding proteins associated with box H/ACA snoRNAs: cbf5p in Euglena gracilis, an early diverging eukaryote, and candidate Gar1p and Nop10p homologs in archaebacteria. Nucleic Acids Res., 28, 2342–2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koonin E.V. (1996) Pseudouridine synthases: four families of enzymes containing a putative uridine-binding motif also conserved in dUTPases and dCTP deaminases. Nucleic Acids Res., 24, 2411–2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zebarjadian Y., King,T., Fournier,M.J., Clarke,L. and Carbon,J. (1999) Point mutations in yeast CBF5 can abolish in vivo pseudouridylation of rRNA. Mol. Cell. Biol., 19, 7461–7472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bousquet-Antonelli C., Henry,Y., Gélugne J.-P., Caizergues-Ferrer,M. and Kiss,T. (1997) A small nucleolar RNP protein is required for pseudouridylation of eukaryotic ribosomal RNAs. EMBO J., 16, 4770–4776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bagni C. and Lapeyre,B. (1998) Gar1p binds to the small nucleolar RNAs snR10 and snR30 in vitro through a nontypical RNA binding element. J. Biol. Chem., 273, 10868–10873. [DOI] [PubMed] [Google Scholar]
- 32.Koonin E.V., Bork,P. and Sander,C. (1994) A novel RNA-binding motif in omnipotent suppressors of translation termination, ribosomal proteins and a ribosome modification enzyme? Nucleic Acids Res., 22, 2166–2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vilardell J. and Warner,J.R. (1997) Ribosomal protein L32 of Saccharomyces cerevisiae influences both the splicing of its own transcript and the processing of rRNA. Mol. Cell. Biol., 17, 1959–1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nottrott S., Hartmuth,K., Fabrizio,P., Urlaub,H., Vidovic,I., Ficner,R. and Luhrmann,R. (1999) Functional interaction of a novel 15.5kD S[U4/U6.U5] tri-snRNP protein with the 5′ stem–loop of U4 snRNA. EMBO J., 18, 6119–6133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Watkins N.J., Ségault,V., Charpentier,B., Nottrott,S., Fabrizio,P., Bachi,A., Wilm,M., Rosbash,M., Branlant,C. and Luhrmann,R. (2000) A common core RNP structure shared between the small nucleoar box C/D RNPs and the spliceosomal U4 snRNP. Cell, 103, 457–466. [DOI] [PubMed] [Google Scholar]
- 36.Dabeva M.D., Post-Beittenmiller,M.A. and Warner,J.R. (1986) Autogenous regulation of splicing of the transcript of a yeast ribosomal protein gene. Proc. Natl Acad. Sci. USA, 83, 5854–5857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Eng F.J. and Warner,J.R. (1991) Structural basis for the regulation of splicing of a yeast messenger RNA. Cell, 65, 797–804. [DOI] [PubMed] [Google Scholar]
- 38.Dabeva M.D. and Warner,J.R. (1993) Ribosomal protein L32 of Saccharomyces cerevisiae regulates both splicing and translation of its own transcript. J. Biol. Chem., 268, 19669–19674. [PubMed] [Google Scholar]
- 39.Vilardell J. and Warner,J.R. (1994) Regulation of splicing at an intermediate step in the formation of the spliceosome. Genes Dev., 8, 211–220. [DOI] [PubMed] [Google Scholar]
- 40.Li B., Vilardell,J. and Warner,J.R. (1996) An RNA structure involved in feedback regulation of splicing and of translation is critical for biological fitness. Proc. Natl Acad. Sci. USA, 93, 1596–1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vilardell J., Yu,S.J. and Warner,J.R. (2000) Multiple functions of an evolutionarily conserved RNA binding domain. Mol. Cell, 5, 761–766. [DOI] [PubMed] [Google Scholar]
- 42.McCusker J.H. and Davis,R.W. (1991) The use of proline as a nitrogen source causes hypersensitivity to, and allows more economical use of 5FOA in Saccharomyces cerevisiae. Yeast, 7, 607–608. [DOI] [PubMed] [Google Scholar]
- 43.Girard J.-P., Bagni,C., Caizergues-Ferrer,M., Amalric,F. and Lapeyre,B. (1994) Identification of a segment of the small nucleolar ribonucleoprotein-associated protein GAR1 that is sufficient for nucleolar accumulation. J. Biol. Chem., 269, 18499–18506. [PubMed] [Google Scholar]
- 44.Bonneaud N., Ozier-Kalogeropoulos,O., Li,G.Y., Labouesse,M., Minvielle-Sebastia,L. and Lacroute,F. (1991) A family of low and high copy replicative, integrative and single-stranded S. cerevisiae/E. coli shuttle vectors. Yeast, 7, 609–615. [DOI] [PubMed] [Google Scholar]
- 45.Urban P., Cullin,C. and Pompon,D. (1990) Maximizing the expression of mammalian cytochrome P-450 monooxygenase activities in yeast cells. Biochimie, 72, 463–472. [DOI] [PubMed] [Google Scholar]
- 46.Bousquet-Antonelli C., Vanrobays,E., Gélugne,J.-P., Caizergues-Ferrer,M. and Henry,Y. (2000) Rrp8p is a yeast nucleolar protein functionally linked to Gar1p and involved in pre-rRNA cleavage at site A2. RNA, 6, 826–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Qu L.-H., Henry,Y., Nicoloso,M., Michot,B., Azum,M.-C., Renalier,M.-H., Caizergues-Ferrer,M. and Bachellerie,J.-P. (1995) U24, a novel intron-encoded small nucleolar RNA with two 12 nt long, phylogenetically conserved complementarities to 28S rRNA. Nucleic Acids Res., 23, 2669–2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mathews D.H., Sabina,J., Zuker,M. and Turner,D.H. (1999) Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J. Mol. Biol., 288, 911–940. [DOI] [PubMed] [Google Scholar]
- 49.Kiss-Laszlo Z., Henry,Y. and Kiss,T. (1998) Sequence and structural elements of methylation guide snoRNAs essential for site-specific ribose methylation of pre-rRNA. EMBO J., 17, 797–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Qu L.-H., Henras,A., Lu,Y.-J., Zhou,H., Zhou,W.-X., Zhu,Y.-Q., Zhao,J., Henry,Y., Caizergues-Ferrer,M. and Bachellerie,J.-P. (1999) Seven novel methylation guide small nucleolar RNAs are processed from a common polycistronic transcript by Rat1p and RNase III in yeast. Mol. Cell. Biol., 19, 1144–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li H., Dalal,S., Kohler,J., Vilardell,J. and White,S.A. (1995) Characterization of the pre-mRNA binding site for yeast ribosomal protein L32: the importance of a purine-rich internal loop. J. Mol. Biol., 250, 447–459. [DOI] [PubMed] [Google Scholar]
- 52.White S.A. and Li,H. (1996) Yeast ribosomal protein L32 recognizes an RNA G:U juxtaposition. RNA, 2, 226–234. [PMC free article] [PubMed] [Google Scholar]
- 53.Li H. and White,S.A. (1997) RNA apatamers for yeast ribosomal protein L32 have a conserved purine-rich internal loop. RNA, 3, 245–254. [PMC free article] [PubMed] [Google Scholar]
- 54.Dreyfuss G., Matunis,M.J., Pinol-Roma,S. and Burd,C.G. (1993) hnRNP proteins and the biogenesis of mRNA. Annu. Rev. Biochem., 62, 289–321. [DOI] [PubMed] [Google Scholar]
- 55.Mattaj I.W. (1993) RNA recognition: a family matter? Cell, 73, 837–840. [DOI] [PubMed] [Google Scholar]
- 56.Gautier T., Bergès,T., Tollervey,D. and Hurt,E. (1997) Nucleolar KKE/D repeat proteins Nop56p and Nop58p interact with Nop1p and are required for ribosome biogenesis. Mol. Cell. Biol., 17, 7088–7098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Caffarelli E., Losito,M., Giorgi,C., Fatica,A. and Bozzoni,I. (1998) In vivo identification of nuclear factors interacting with the conserved elements of box C/D small nucleolar RNAs. Mol. Cell. Biol., 18, 1023–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Watkins N.J., Newman,D.R., Kuhn,J.F. and Maxwell,E.S. (1998) In vitro assembly of the mouse U14 snoRNP core complex and identification of a 65-kDa box C/D-binding protein. RNA, 4, 582–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wu P., Brockenbrough,J.S., Metcalfe,A.C., Chen,S. and Aris,J.P. (1998) Nop5p is a small nucleolar ribonucleoprotein component required for pre-18 S rRNA processing in yeast. J. Biol. Chem., 273, 16453–16463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lafontaine D.L. and Tollervey,D. (1999) Nop58p is a common component of the box C+D snoRNPs that is required for snoRNA stability. RNA, 5, 455–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fatica A., Galardi,S., Altieri,F. and Bozzoni,I. (2000) Fibrillarin binds directly and specifically to U16 box C/D snoRNA. RNA, 6, 88–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lafontaine D.L. and Tollervey,D. (2000) Synthesis and assembly of the box C+D small nucleolar RNPs. Mol. Cell. Biol., 20, 2650–2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Newman D.R., Kuhn,J.F., Shanab,G.M. and Maxwell,E.S. (2000) Box C/D snoRNA-associated proteins: two pairs of evolutionarily ancient proteins and possible links to replication and transcription. RNA, 6, 861–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lane D., Prentki,P. and Chandler,M. (1992) Use of gel retardation to analyze protein–nucleic acid interactions. Microbiol. Rev., 56, 509–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mao H., White,S.A. and Williamson,J.R. (1999) A novel loop–loop recognition motif in the yeast ribosomal protein L30 autoregulatory RNA complex. Nature Struct. Biol., 6, 1139–1147. [DOI] [PubMed] [Google Scholar]
- 66.Vidovic I., Nottrott,S., Hartmuth,K., Luhrmann,R. and Ficner,R. (2000) Crystal structure of the spliceosomal 15.5kD protein bound to a U4 snRNA fragment. Mol. Cell, 6, 1331–1342. [DOI] [PubMed] [Google Scholar]
- 67.Heiss N.S., Girod,A., Salowsky,R., Wiemann,S., Pepperkok,R. and Poustka,A. (1999) Dyskerin localizes to the nucleolus and its mislocalization is unlikely to play a role in the pathogenesis of dyskeratosis congenita. Hum. Mol. Genet., 8, 2515–2524. [DOI] [PubMed] [Google Scholar]