

Abstract
DNA methylation patterns are frequently deregulated in t(8;21) acute myeloid leukaemia (AML), but little is known of the mechanisms by which specific gene sets become aberrantly methylated. Here, we found that the promoter DNA methylation signature of t(8;21)+ AML blasts differs from that of t(8;21)− AMLs. This study demonstrated that a novel hypermethylated zinc finger‐containing protein, THAP10, is a target gene and can be epigenetically suppressed by AML1‐ETO at the transcriptional level in t(8;21) AML. Our findings also show that THAP10 is a bona fide target of miR‐383 that can be epigenetically activated by the AML1‐ETO recruiting co‐activator p300. In this study, we demonstrated that epigenetic suppression of THAP10 is the mechanistic link between AML1‐ETO fusion proteins and tyrosine kinase cascades. In addition, we showed that THAP10 is a nuclear protein that inhibits myeloid proliferation and promotes differentiation both in vitro and in vivo. Altogether, our results revealed an unexpected and important epigenetic mini‐circuit of AML1‐ETO/THAP10/miR‐383 in t(8;21) AML, in which epigenetic suppression of THAP10 predicts a poor clinical outcome and represents a novel therapeutic target.
Keywords: AML1‐ETO, epigenetics, miR‐383, t(8;21) AML, THAP10
Subject Categories: Cancer; Chromatin, Epigenetics, Genomics & Functional Genomics; Haematology
Introduction
t(8;21) acute myeloid leukaemia (AML) is a highly heterogeneous disease from a biological and clinical standpoint (Hsiao et al, 2015). This remains a significant barrier to the development of accurate clinical classification, risk stratification and targeted therapy for this disease. Epigenetic control of gene expression has been suggested to play a pivotal role in the biological behaviour of cells (Wouters & Delwel, 2016). Disruption of normal DNA methylation distribution is a hallmark of cancer and has critical roles in the initiation, progression and maintenance of malignant phenotypes (Figueroa et al, 2010). Several tumour suppressor genes [e.g. miR‐193a (Li et al, 2013b) and miR‐223 (Fazi et al, 2007a; Pulikkan et al, 2010)] have been shown to be abnormally methylated in t(8;21) AML patients. Moreover, aberrant distribution of promoter DNA methylation occurring in specific and distinct patterns was identified as a universal feature in t(8;21) AML (Figueroa et al, 2010). However, the mechanisms underlying these aberrant methyl cytosine patterns have not been defined.
The thanatos‐associated proteins (THAPs), a novel family of cellular factors, are characterized by the presence of an evolutionarily conserved protein motif (Cayrol et al, 2007). Overall, there are 12 distinct human proteins that contain the THAP domain (THAP 0–11; Zhu et al, 2009). Among them, THAP1 has been identified as a nuclear protein associated with promyelocytic leukaemia nuclear bodies (PML NBs; Cayrol et al, 2007). THAP7 has both histone‐binding and putative DNA‐binding motifs (Macfarlan et al, 2005). THAP9 was shown to be an active DNA transposase and still retains the catalytic activity to mobilize P transposable elements across species (Majumdar et al, 2013). Most recently, the mouse homolog of THAP11, Ronin, was identified as an essential factor underlying embryogenesis and embryonic stem cell pluripotency (Dejosez et al, 2008). In contrast, Zhu et al (2009) reported that THAP11 functions as a negative regulator of cell growth in human hepatoma cells through transcriptional repression of the proto‐oncogene MYC. These studies, together with data obtained in C. elegans, indicated that THAPs are zinc‐dependent, sequence‐specific DNA‐binding factors involved in cell proliferation, apoptosis, the cell cycle, chromatin modification and transcriptional regulation. However, the transcriptional regulatory properties of most human THAPs and their roles in pathological processes remain largely unknown.
Given the function of AML1‐ETO as a transcriptional repressor (DeKelver et al, 2013), we investigated whether AML1‐ETO was associated with aberrant epigenetic programming in t(8;21) AML. Here, we report an unexpected and functional epigenetic mini‐circuit of AML1‐ETO/THAP10/miR‐383 in t(8;21) AML, which might represent a novel therapeutic target as well as a biomarker for predicting clinical outcome of patients with AML.
Results
t(8;21) AML displays a distinct signature of aberrant DNA methylation
First, we used a DNA methylation microarray for 450 k CpG sites to profile genome‐wide DNA methylation in AML blasts of patients with or without t(8;21) (Appendix Table S1) and normal bone marrow (NBM) CD34+ cells. The heatmap displayed a unique epigenetic signature with global hypermethylation for t(8;21)+ AML, which differed from either the t(8;21)− AML or NBM samples (Fig 1A). Unsupervised hierarchical clustering analysis revealed a clear segregation of t(8;21)+ AML from t(8;21)− AML blasts or normal CD34+ cells (Appendix Fig S1), despite the heterogeneity of AML blasts. It also showed that t(8;21)+ cases were further divided into two sub‐clusters (A and B) with distinguishable methylation profiles (Appendix Fig S1). Interestingly, sub‐cluster A contained 5 of 7 t(8;21)+ AML cases carrying Y chromosome deletion (Appendix Table S1).
Figure 1. AML1‐ETO+ AML displays a unique genome‐wide methylation profile compared to AML1‐ETO− AML and normal bone marrow CD34+ cells.
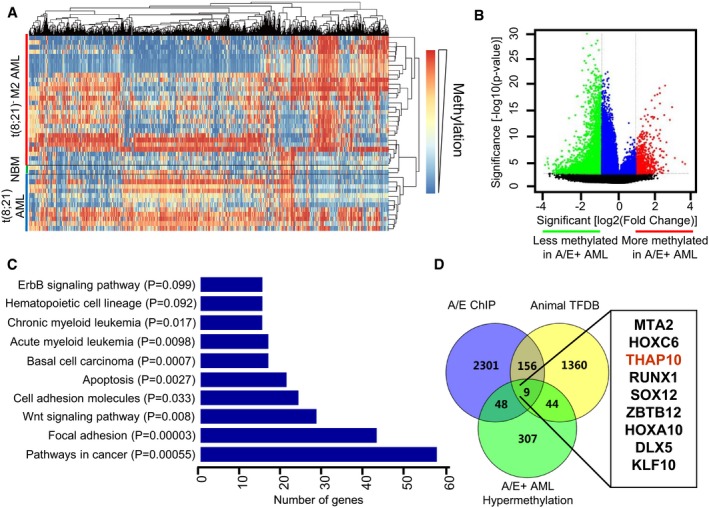
- Overview of two‐way (genes against samples) hierarchical clustering of AML1‐ETO+ (n = 12), AML1‐ETO− (n = 28) M2 AML blasts and normal bone marrow cells (NBM, n = 2) using the genes that vary the most among samples. Representative heatmap is shown to identify the aberrant DNA methylation signatures of specific genes in AML, compared to normal CD34+ haematopoietic cells obtained from healthy donors. In this heatmap, each column represents one gene, and each row represents one AML patient or healthy donor. Red bar, t(8;21)− M2 AML. Blue bar, t(8;21)+ AML. Green bar, normal bone marrow (NBM).
- Volcano plot illustrating the methylation difference between AML1‐ETO+ (n = 12) and AML1‐ETO− (n = 28) AML samples, with the corresponding moderated t‐test P‐values. Red, probe sets that were significantly hypermethylated (P < 0.001, methylation difference > 1.5); green, probe sets that were significantly hypomethylated (P < 0.001, methylation difference < −1.5); blue, probe sets that did not have an absolute methylation difference > 1.5.
- Functional enrichment analysis of differential DNA methylation genes (DMGs), based on the pathway enrichment analysis of hypergeometric distribution.
- Venn diagrams for systematic survey of the publicly available database, by combining with 408 differentially methylated genes identified in the present study (see panel B), to identify the 9‐gene set in t(8;21) AML.
Analysis of the differentially methylated probes within 10 kb from each gene transcriptional start site (TSS) identified a total of 888 differentially methylated probe sets (corresponding to 408 genes) in t(8;21)+ AML [Fig 1B, P < 0.001 and methylation log ratio difference > 1.5 vs. t(8;21)− AML]. In t(8;21)+ AML, functional pathway analysis indicated that 57 and 17 hypermethylated genes were grouped into the categories of cancer and AML, respectively (Fig 1C). Among these genes (Appendix Table S2), many are involved in tumour suppression [e.g. RB1 (Dyson, 2016)], apoptosis [e.g. BCL2L1 (Liu et al, 2015)], the cell cycle [e.g. CCND1 (Mende et al, 2015), CCND2 (Khanjyan et al, 2013)] or protein kinases [e.g. KIT (Wichmann et al, 2015), GSK3‐β (Orme et al, 2016)]. By combined with 408 differentially methylated genes (Fig 1B), a systematic survey of the publicly available databases (Gardini et al, 2008; Zhang et al, 2015) identified nine transcriptional factor genes targeted by AML1‐ETO that were differentially hypermethylated in t(8;21) AML (Fig 1D). The same approach was then used to analyse the data from TCGA methylome (Ley et al, 2013), which identified 13 genes differentially hypermethylated in t(8;21) AML (Fig EV1). Of note, 4 of these 13 genes matched those identified in our distinct signature of t(8;21) AML (Fig 1D).
Figure EV1. TCGA methylome database reanalysis showed that AML1‐ETO + AML displays a similar unique genome‐wide methylation signature compared to our data.
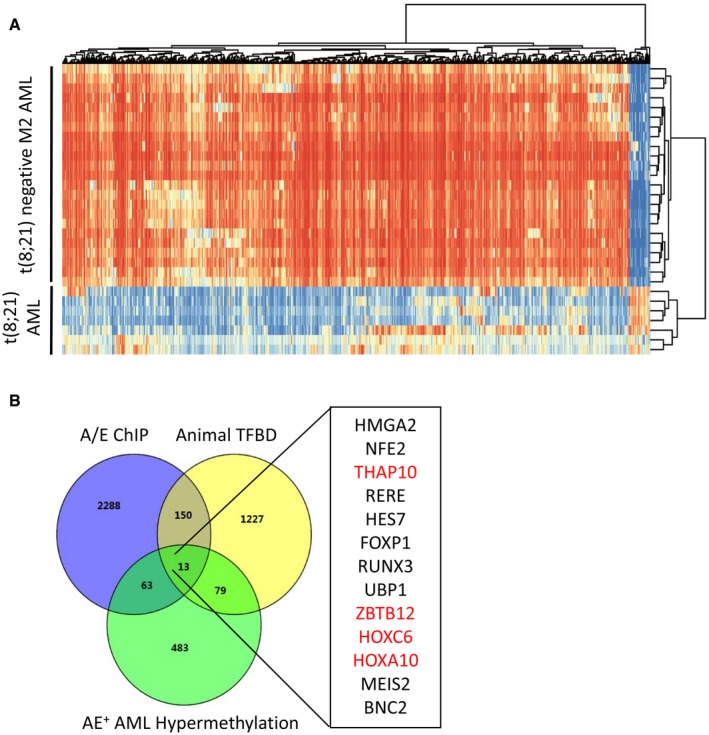
- Overview of two‐way (genes against samples) hierarchical clustering of AML1‐ETO+ (n = 7) and AML1‐ETO− (n = 37) M2 AML blasts using the genes that vary the most among samples. Representative heatmap is shown to identify the aberrant DNA methylation signatures of specific genes in AML. Each column of the heatmap represents one gene, and each row represents one AML patient.
- Venn diagrams for systematic survey of the publicly available database to identify the 13 genes in t(8;21) AML. Four out of 13 genes overlapped with our distinct signature in t(8;21) AML and are shown in red, including THAP10.
Source data are available online for this figure.
A strong hypermethylated signature of t(8;21) AML suggests a therapeutic benefit from hypomethylating agents (e.g. 5‐azacytidine/5‐Aza). Notably, 5‐Aza treatment significantly increased apoptosis of AML1‐ETO+ AML cells (Appendix Fig S2), compared to their AML1‐ETO− counterparts. Together, these findings indicate a distinct signature of aberrant DNA methylation in t(8;21) AML, consistent with the hypothesis that AML1‐ETO alters epigenetic patterning to drive leukaemogenesis via deregulation of specific complementary gene sets, which may be susceptible to hypomethylating agents.
THAP10 is epigenetically suppressed in t(8;21) leukaemia cells
Differential methylated region (DMR) analysis was then performed to explore the genes aberrantly methylated in t(8;21) AML. The top 10 genes with higher methylation levels in t(8;21)+ than t(8;21)− AML and NBM are shown in Appendix Table S3. Notably, both sets of these top 10 genes and nine genes described above (Fig 1D) included THAP10, while the other nine genes listed in Appendix Table S3 were not found in the latter gene set. Moreover, methylation levels of all 14 probes in the THAP10 promoter region were higher in AML1‐ETO+ than AML1‐ETO− blasts or NBM cells (Appendix Fig S3), supporting THAP10 as a novel target gene epigenetically regulated by AML1‐ETO.
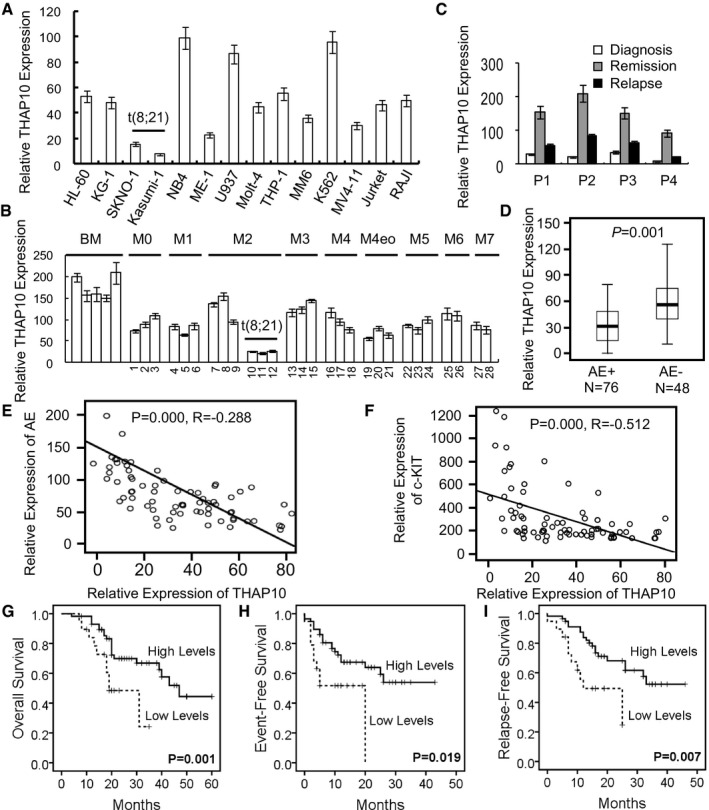
THAP10 mRNA levels were then determined to validate the functional role of its promoter hypermethylation. Significantly, core‐binding factor [CBF, including t(8;21)] AML cell lines displayed lower THAP10 mRNA levels than non‐CBF cells (Fig 2A). In a cohort of AML patients (Appendix Table S4), THAP10 mRNA levels in AML blasts were lower than NBM cells, while the lowest level of THAP10 was observed in t(8;21)+ AML (Fig 2B). Virtually identical results were obtained from the analysis of data from 200 AML samples in TCGA database (Fig EV2; Ley et al, 2013). Interestingly, THAP10 mRNA levels in bone marrow mononuclear cells of AML1‐ETO+ AML patients who achieved complete remission after induction chemotherapy were relatively higher than the same patients at relapse (Fig 2C). In an additional cohort of 124 AML/M2 patients (Appendix Table S5), THAP10 mRNA levels were 0.9‐fold lower in AML1‐ETO+ than AML1‐ETO− cases (Fig 2D).
Figure 2. THAP10 is down‐regulated in t(8;21) AML and correlates with adverse clinical outcome.
-
A, BRelative qRT–PCR quantification of THAP10 mRNA levels in the indicated leukaemia cell lines (A) and mononuclear cells (MNC) isolated from leukaemia patients (B) (n = 28).
-
CSequential analyses of THAP10 mRNA levels in mononuclear cells isolated from bone marrow samples of four individual leukaemia patients at different stages of disease, including newly diagnosed, remission and relapse.
-
DComparison of THAP10 mRNA levels in two categories of AML M2 subtype patients (total n = 124): AML1‐ETO+ (n = 76) and AML1‐ETO− (n = 48) t(8;21) AML, P = 0.001. Data are represented as a boxplot, which shows the median value of mRNA levels (line in the middle of the box), the first and the third quartiles (lower and upper limits of each box, respectively). Wiskers display the highest and lowest data values.
-
E, FCorrelations in gene expression between THAP10 and AML1‐ETO (R = −0.288, P = 0.000, E) or c‐KIT (R = −0.512, P = 0.000, F), the two‐sided paired Pearson test was used for the significance of association.
-
G–ICorrelations of THAP10 expression (high, n = 56; low, n = 20) with overall survival (P = 0.001, G), event‐free survival (P = 0.046, H) and relapse‐free survival (P = 0.039, I). The cut‐off value (dash line) is 2 × 101 of THAP10 level, by which high vs. low THAP10 mRNA expression is defined, the log‐rank test was used for the survival analysis.
Figure EV2. THAP10 mRNA levels in AML subtypes from TCGA databases.
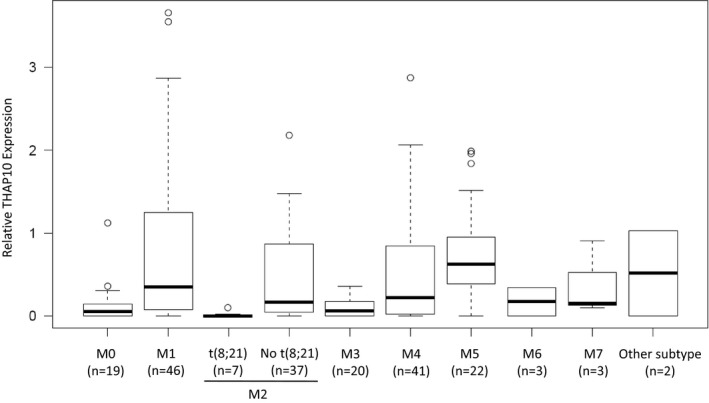
Boxplot analysis showed that THAP10 mRNA levels were the lowest in t(8;21)+ AML. AML subtypes were classified according to the FAB classification. Data are represented as a boxplot, which shows the median value of mRNA levels (line in the middle of the box), the first and third quartiles (lower and upper limits of each box, respectively). THAP10 expression is derived and normalized from RPKM (Reads Per Kilobase per Million mapped reads) of RNA‐Seq data for 200 AML samples. Whiskers display the highest and lowest data values.
As AML1‐ETO and c‐KIT gene abnormalities are associated with poor outcome of patients with t(8;21) AML (Jiao et al, 2009; Qin et al, 2014), we examined the relationships among expression of THAP10, AML1‐ETO and c‐KIT in newly diagnosed t(8;21) AML patients (Appendix Table S5). Notably, THAP10 mRNA level was inversely correlated with those of AML1‐ETO (Fig 2E) and c‐KIT (Fig 2F). Then, 76 patients with t(8;21) AML (Appendix Table S5) were grouped into quartiles according to THAP10 levels and divided into THAP10‐high (n = 56) and THAP10‐low patients (n = 20; Appendix Fig S4). The results revealed that THAP10‐high patients had significantly longer overall (Fig 2G), event‐free (Fig 2H) or relapse‐free (Fig 2I) survival than THAP10‐low patients. Together, these findings indicate that THAP10 gene silencing is associated with unfavourable outcomes of t(8;21) AML patients.
AML1‐ETO epigenetically suppresses THAP10 by binding to AML1‐binding sites
AML1‐ETO retains the binding capacity to AML1‐binding sites at gene promoters, and it can also recruit additional transcriptional cofactors, mostly corepressors (Li et al, 2013b). Notably, AML1‐ETO knock‐down resulted in sharp increases (~15‐fold compared to controls) in THAP10 mRNA and protein levels in AML1‐ETO+ SKNO‐1 (Fig 3A). In contrast, ectopically expressing zinc‐inducible HA‐tagged AML1‐ETO significantly reduced THAP10 levels in U937 cells (Fig EV3A). However, despite the significant difference in mRNA levels of THAP10, there was only a moderate difference in THAP10 protein levels between AML1‐ETO− HL60 and AML1‐ETO+ Kasumi‐1 cells (Fig 3A). This inconsistency suggests that other mechanism(s) may also contribute to the protein level of THAP10. Nevertheless, these results indicate that AML1‐ETO inhibits THAP10 expression.
Figure 3. AML1‐ETO epigenetically suppresses THAP10 via direct binding to its promoter region.
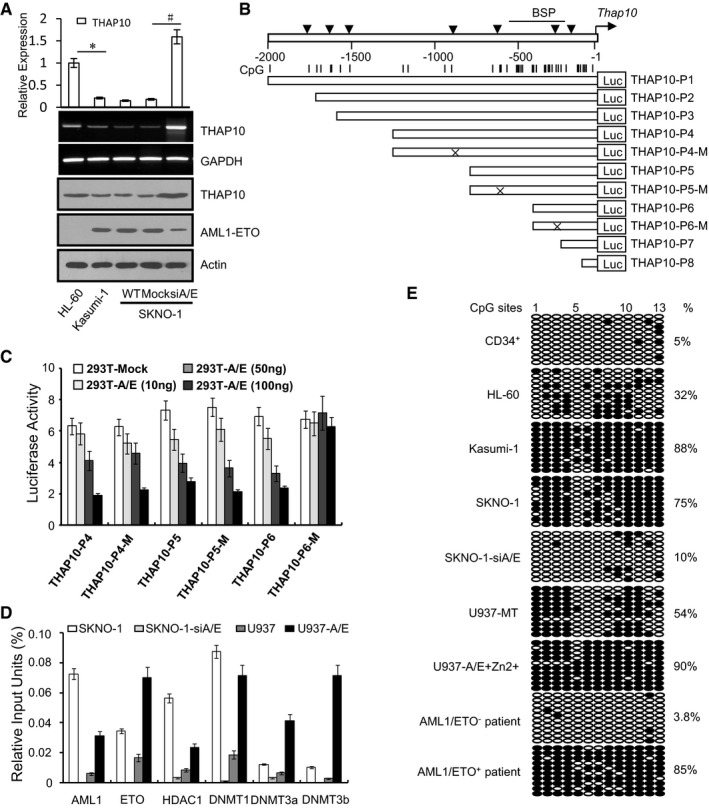
- Upper panel: Relative quantification of THAP10 mRNA levels in the indicated leukaemia cell lines. The results represent the mean of three independent evaluations ± SD (*P = 0.001, # P = 0.0007, Student's t‐test was used for the comparisons). Middle and lower panels: mRNA and protein levels of THAP10 and AML1‐ETO, respectively.
- Upper panel: Schematic diagrams of the AML1‐binding sites and the CpG islands along the THAP10 genes. Numbers indicate the nucleotides relative to pre‐THAP10 (−1 nt). Vertical arrowheads indicate the AML1‐binding sites; vertical lines indicate CpG dinucleotides; horizontal bar illustrates the regions analysed by bisulphite sequencing. Lower panels: A series of constructs containing different AML1‐binding sites and their mutants.
- Luciferase reporter activities of human 293T cells transiently co‐transfected for 48 h with luciferase reporters containing the wild‐type sequence of the THAP10 promoter or its mutant counterparts, together with increasing amounts of pcDNA3.0 vectors containing AML1‐ETO or mock cDNA (293T‐Mock). Triplicate values are defined as mean ± SD.
- Chromatin immunoprecipitation (ChIP) using the indicated antibodies or IgG, after which qRT–PCR was performed to evaluate the specificity of protein binding. Triplicate values are defined as mean ± SD.
- Genomic bisulphite sequencing to detect the methylation status of the DNA sequences surrounding the AML1‐binding site (−280 nt) in the pre‐THAP10 gene upstream region in CD34+ haematopoietic progenitors isolated from peripheral blood (PB) of healthy donors, the indicated leukaemia cell lines and primary leukaemic blasts. Each row of circles represents the sequence of an individual clone. Solid and empty circles represent methylated and unmethylated CpG dinucleotides, respectively.
Figure EV3. Epigenetically down‐regulated THAP10 can be reactivated by DNMT inhibitors and HDAC inhibitors.
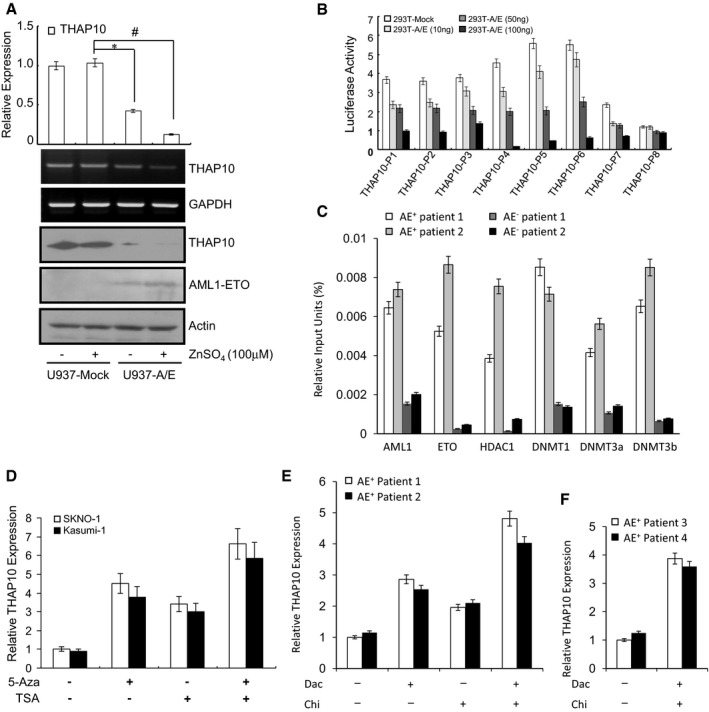
- Upper panel: Relative quantification of THAP10 levels in U937 (Mock and A/E‐HA) cells. The results represent the average of three independent evaluations ± SD (*P = 0.038, # P = 0.008, two‐sided Student's t‐test was used for the comparisons). Middle panels: RNA levels of THAP10 analysed by agarose electrophoresis. GAPDH was used as a loading control. Lower panels: Protein levels of THAP10 and AML1‐ETO monitored by immunoblot analysis. β‐actin was used as a loading control. Treatment for 16 h with 100 μM Zn2+ was employed to induce the expression of AML1‐ETO.
- Human 293T cells were transiently co‐transfected for 48 h with luciferase reporter containing wild‐type sequences of the THAP10 promoter regions, together with increasing amounts (10, 50 and 100 ng) of pcDNA3.0 with AML1‐ETO cDNA or without (293T‐Mock). Data represent the mean of three independent evaluations ± SD for luciferase activity relative to the empty vector (pGL3‐LUC) control. pRL‐TK was used as an internal control.
- Chromatin immunoprecipitation (ChIP) using the indicated antibodies or IgG in AML blasts, after which qRT–PCR was performed to evaluate the specificity of protein binding. Triplicate values are defined as mean ± SD.
- SKNO‐1 and Kasumi‐1 cells were treated with the DNMT inhibitor 5‐azacytidine (5‐Aza, 2.5 μM) +/− the HDAC inhibitor TSA (3.0 μM) for 40 h, after which RT–PCR was performed to detect the mRNA levels of THAP10. Data represent the normalized values (mean ± SD) of relative THAP10 expression from three independent experiments. Normalized THAP10 level in SKNO‐1 cells was arbitrarily set to 1.
- AML‐ETO+ patient blasts were treated with the DNMT inhibitor decitabine (Dac, 2.5 μM) +/− the HDAC inhibitor chidamide (Chi, 3.0 μM) for 40 h, after which RT–PCR was performed to detect the mRNA levels of THAP10. Data represent the normalized values (mean ± SD) of relative THAP10 expression from three independent experiments. Normalized THAP10 level in SKNO‐1 cells was arbitrarily set to 1.
- In primary blasts from clinical trial (NCT02886559) patients, qRT–PCR detected the THAP10 levels right after completion of the first therapeutic program. Normalized THAP10 level in patient blasts was arbitrarily set to 1. Triplicate values are defined as mean ± SD.
A bioinformatics search (http://www.cbrc.jp/research/db/TFSEARCH.html) at the 5′ end of the predicted “core promoter” sequence revealed seven putative AML1‐binding sites surrounded by CpG islands in the THAP10 upstream region (−2,000 nt to +1 nt relative to TSS; Fig 3B, upper panel). Accordingly, luciferase reporters containing a series of wild‐type (P1–P8) or mutated (P4‐M to P6‐M) sequences of the THAP10 promoter region (Fig 3B, lower panels) were designed, and 293T cells were co‐transfected with each reporter together with increasing amounts of AML1‐ETO or empty vectors. Notably, ectopic AML1‐ETO expression resulted in a dose‐dependent decrease in luciferase activity of THAP10‐P1‐6 or THAP10‐P4‐M and THAP10‐P5‐M but not THAP10‐P6‐M and THAP10‐P8, which lack an AML1‐binding site (Figs 3C and EV3B), suggesting that AML1‐ETO binds to the proximal AML1‐binding sites of the THAP10 promoter to suppress THAP10. Moreover, ChIP assays revealed that shRNA knock‐down of AML1‐ETO in SKNO‐1 cells resulted in a sharp decrease in binding of AML1‐ETO, HDAC1, DNMT1, DNMT3a and DNMT3b to the region around the AML1‐binding sites of the THAP10 promoter compared to control SKNO‐1 cells (Fig 3D). In contrast, ectopic overexpression of AML1‐ETO in U937 cells led to a marked increase in binding of these proteins compared to control U937 cells (Fig 3D). Similar results were obtained in primary AML1‐ETO+ vs. AML1‐ETO− AML blasts (Fig EV3C).
Further, bisulphite sequencing assays revealed a higher frequency of methylated CpG dinucleotides encompassing THAP10 in AML1‐ETO+ SKNO‐1 (75%) and U937‐A/E cells (90%) than U937 (54%) and HL‐60 (32%) or normal CD34+ cells (5%) (Fig 3E). In contrast, AML1‐ETO knock‐down in SKNO‐1 cells dramatically diminished THAP10 CpG methylation (Fig 3E). The frequency of methylated CpG dinucleotides encompassing endogenous THAP10 was higher in AML1‐ETO+ (85%) than AML1‐ETO− (3.8%) AML blasts (Fig 3E). Interestingly, while treatment with 5‐Aza increased THAP10 expression by ~4.0‐fold in AML1‐ETO+ cells displaying strong hypermethylation of THAP10 (Fig 3E), co‐treatment with 5‐Aza and the HDAC inhibitor TSA further increased its expression (Fig EV3D). These findings are likely due to increasing acetylation (activation) of the THAP10 promoter via inhibition of the co‐repressor HDAC1, which was recruited to the AML1‐binding sites of THAP10 (Fig 3D) by HDAC inhibitors (e.g. TSA). Treatment with another hypomethylating agent (decitabine) +/− the clinically relevant HDAC inhibitor chidamide also increased THAP10 expression ex vivo in primary blasts (Fig EV3E) as well as in vivo in patients enrolled in an ongoing clinical trial (NCT02886559; Fig EV3F). Together, these findings suggest that AML1‐ETO in an abnormal chromatin remodelling complex with other cofactors (e.g. DNMTs) mediates hypermethylation of CpG islands around AML1‐binding sites on the THAP10 promoter, while probably increasing histone deacetylation (e.g. by HDAC1) on the THAP10 promoter as well, which might represent a novel mechanism responsible for suppression of THAP10 expression in t(8;21) AML.
THAP10 is a bona fide target of miR‐383 that is up‐regulated in AML1‐ETO+ leukaemia cells
In addition to the mechanism described above in which AML1‐ETO directly suppresses the THAP10 gene by methylating its promoter, we then examined whether other mechanism(s) were also involved in suppression of THAP10 expression in AML‐ETO+ AML. MicroRNAs (miRNAs) play an important role in the lineage differentiation of haematopoietic cells by regulating expression of oncogenes or tumour suppressors (Vian et al, 2014). Deregulation of miRNA expression has been shown to be involved in multistep carcinogenesis and has rapidly emerged as a novel therapeutic target (Li et al, 2013b; Dorrance et al, 2015; Tarighat et al, 2016). Computational analysis for miRNA target prediction using the TargetScan algorithm (http://www.targetscan.org) revealed THAP10 as a putative target of miR‐383 (Fig 4A). To validate the functional role of miR‐383 in regulation of THAP10 expression, 293T cells were co‐transfected with THAP10 3′‐UTR luciferase reporters and pre‐miR‐383. As shown in Fig EV4A, expression of miR‐383 resulted in a ~50% reduction in luciferase activity of wild‐type THAP10 3′‐UTR but not in miR‐383‐binding site‐mutated THAP10 3′‐UTR (Fig 4A). Furthermore, administration of synthetic miR‐383 also reduced endogenous THAP10 expression at the mRNA level in HL‐60 and NB4 cells (Fig EV4B) and the protein level (Fig 4B). Taken together, these results indicate that THAP10 is a bona fide target of miR‐383.
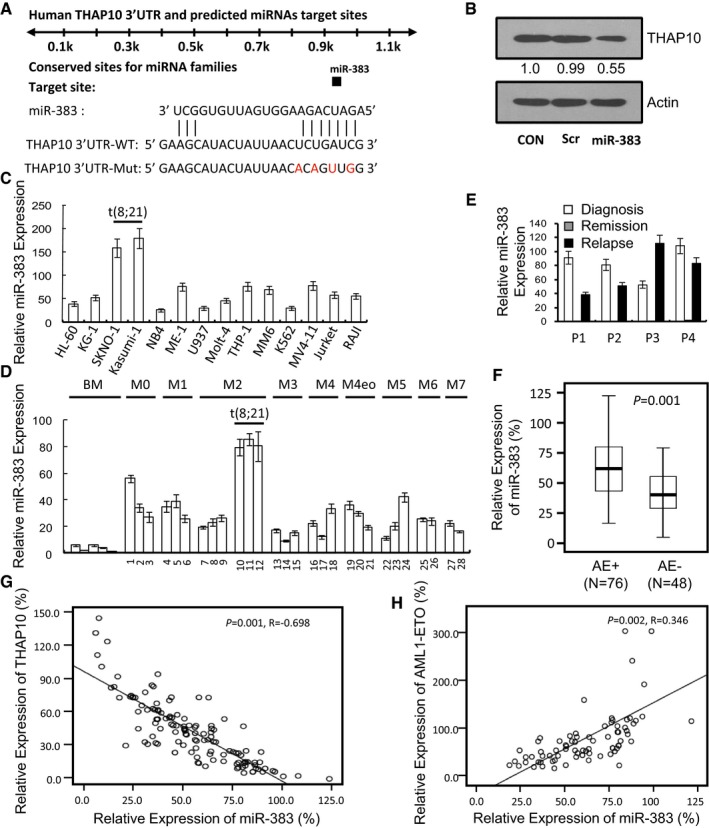
Figure 4. miR‐383 negatively correlates with THAP10 levels but positively correlates with AML1‐ETO .
-
AThe sequences indicating the putative interaction site between the miR‐383 seed region (nucleotides 2–8) and the 3′‐UTRs of THAP10 wild‐type (WT) or mutated derivatives.
-
BWestern blot analysis monitoring the effect of synthetic miR‐383 on THAP10 protein levels.
-
C, DRelative qRT–PCR quantification of THAP10 mRNA levels in the indicated leukaemia cell lines (C) and mononuclear cells (MNC) isolated from leukaemia patients (D) (n = 28). Triplicate values are defined as mean ± SD.
-
ESequential analyses of miR‐383 levels in mononuclear cells isolated from bone marrow samples of four individual leukaemia patients at the newly diagnosed, remission and relapse stages, respectively. Triplicate values are defined as mean ± SD.
-
FRelative qRT–PCR quantification of THAP10 mRNA levels in two groups of AML M2 subtype patients (total n = 124): AML1‐ETO+ and AML1‐ETO− t(8;21) AML (P = 0.001). Data are represented as a boxplot, which shows the median value of mRNA levels (line in the middle of the box), the first and third quartiles (lower and upper limits of each box, respectively). Whiskers display the highest and lowest data values.
-
G, HCorrelation of miR‐383 expression with THAP10 (R = −0.689, P = 0.001, G) or AML1‐ETO levels (R = 0.346, P = 0.002, H). Two‐sided paired Pearson test was used for the significance of association.
Figure EV4. miR‐383 levels in indicated cell lines and the localization of THAP10.
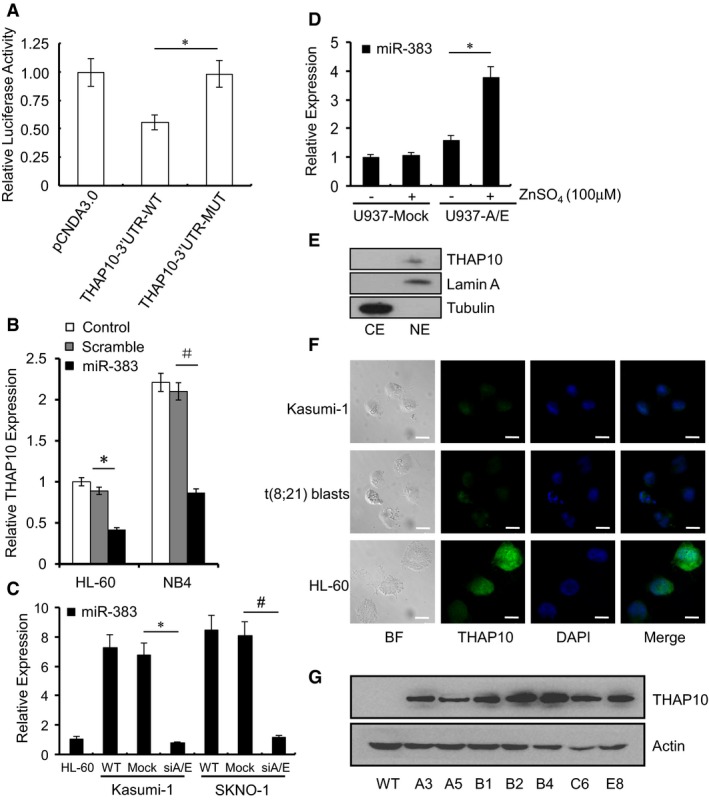
- HEK293T cells were co‐transfected for 48 h with pCDNA3.0 control and miR‐383 expressing plasmids, together with a modified luciferase pGL‐3 control vector containing THAP10 3′‐UTRs or a predicted miR‐383 binding site mutant as indicated, after which luciferase activity assays were performed. Data represent the normalized values (mean ± SD) of relative luciferase activity from three independent experiments (*P = 0.009, two‐sided Student's t‐test was used for the comparison). Normalized luciferase activity in the absence of the miR‐383 expression vector was set to 1.
- Relative quantification of THAP10 levels in HL‐60 and NB4 leukaemic cells after transfection with synthetic miR‐383 or scramble for 48 h. The results represent the mean of three independent evaluations ± SD (*P = 0.0001, # P = 0.0001, two‐sided Student's t‐test was used for the comparisons).
- Relative quantification of miR‐383 expression in the indicated cells. The results represent the mean of three independent evaluations ± SD (*P = 0.001, # P = 0.0015, two‐sided Student's t‐test was used for the comparisons).
- Relative quantification of miR‐383 levels in U937 (mock and A/E‐HA) cells. The results represent the mean of three independent evaluations ± SD (*P = 0.004, two‐sided Student's t‐test was used for the comparison).
- Endogenous expression of THAP10 in nuclei. Cytoplasmic (CE) and nuclear extracts (NE) were isolated from Kasumi‐1 cells transduced with lentivirus vector expressing THAP10, after which Western blot analysis was performed using polyclonal anti‐THAP10 antibody. Tubulin and lamin A were used as loading controls for the cytoplasm and nuclei, respectively.
- Immunofluorescence microscopy shows the endogenous THAP10 localization in Kasumi‐1, HL‐60 and t(8;21) AML blasts. BF, bright field. Scale bar, 10 μm.
- Ectopic expression of THAP10 in Kasumi‐1 cells. After transduction with lentivirus vector expressing THAP10, multiple stable clones indicated with capital letters and numbers were selected after incubation with puromycin for 30 days. Western blot analysis using anti‐THAP10 antibody was performed to screen and monitor the protein levels of THAP10. β‐actin was used as a loading control. WT, wild type.
Source data are available online for this figure.
qRT–PCR analysis revealed higher miR‐383 levels in t(8;21)+ than t(8;21)− AML cell lines (Fig 4C) and blasts (Fig 4D) in a cohort of AML patients (Appendix Table S4). Interestingly, miR‐383 expression was decreased in bone marrow mononuclear cells of AML1‐ETO+ patients who achieved complete remission following induction chemotherapy, compared to the same patients at relapse (Fig 4E). These findings were further validated by results showing that miR‐383 levels were twofold higher in AML1‐ETO+ than AML1‐ETO− AML blasts (Fig 4F) in an additional cohort of 124 AML cases (Appendix Table S5).
Furthermore, AML1‐ETO knock‐down decreased miR‐383 expression by ~sevenfold in AML1‐ETO+ cells, compared to controls (Fig EV4C). In contrast, ectopic expression of AML1‐ETO in U937 cells increased miR‐383 levels by 2.4‐fold (Fig EV4D). Consistently, qRT–PCR analysis revealed an inverse correlation between miR‐383 and THAP10 expression (Fig 4G), but a positive correlation between miR‐383 and AML1‐ETO expression (Fig 4H), in a cohort of newly diagnosed t(8;21) AML patients (Appendix Table S5). Together, these findings suggest that miR‐383 expression correlates negatively with THAP10 but positively with AML1‐ETO in t(8;21) AML.
AML1‐ETO transcriptionally induces expression of miR‐383
The NHR1 domain of AML1‐ETO provides a docking site for p300 binding, allowing it to co‐localize at the regulatory regions of many AML1‐ETO target genes (Wang et al, 2011). A bioinformatics search (http://www.cbrc.jp/research/db/TFSEARCH.html) of the miR‐383 at the 5′ end of the predicted “core promoter” sequence revealed three putative AML1‐binding sites and one p300‐binding site (Fig 5A, upper panel). Accordingly, luciferase reporters containing a series of wild‐type (MIR‐P1 to MIR‐P4) or mutated (MIR‐P1‐M to MIR‐P3‐M) sequences of the miR‐383 promoter region (Fig 5A, lower panels) were designed, and 293T cells were co‐transfected with each reporter together with increasing amounts of AML1‐ETO or empty vectors. Ectopic AML1‐ETO expression resulted in a dose‐dependent increase in luciferase activity of MIR‐P1, MIR‐P1‐M and MIR‐P2, but not MIR‐P2‐M, MIR‐P3 and MIR‐P4 (Fig 5B), suggesting that the AML1‐binding sites in the promoter region (−676 nt relative to +1 nt) of miR‐383, which is close to the p300‐binding site (−669 nt relative to +1), might be involved in AML1‐ETO‐mediated expression of miR‐383.
Figure 5. AML1‐ETO epigenetically activates expression of miR‐383 .
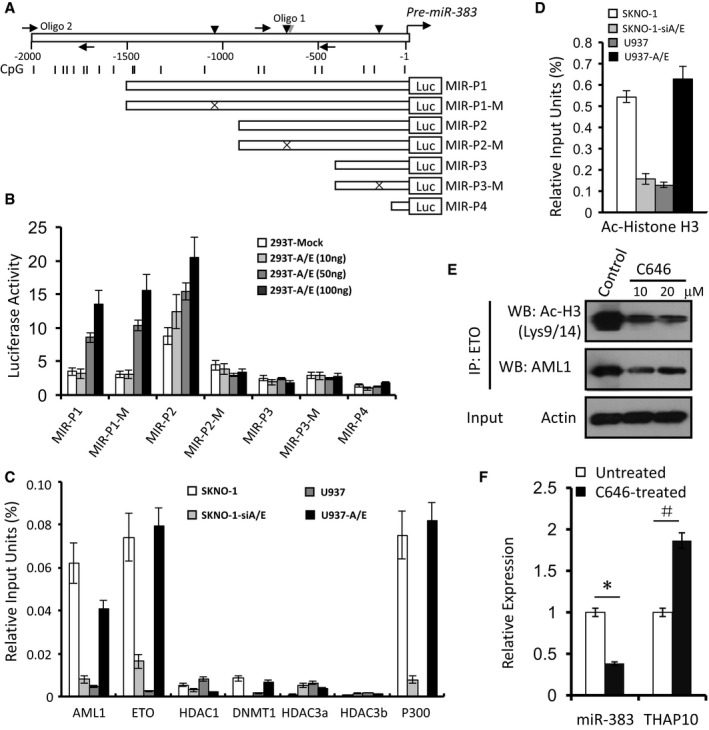
- Upper panel: Schematic diagrams of the AML1‐ and p300‐binding sites and the CpG islands along the pre‐miR‐383 genes. Numbers indicate the nucleotides relative to pre‐miR‐383 (+1 nt). Vertical arrowheads indicate AML1 (dark) and p300 (grey) binding sites; horizontal arrows indicate the location of the primers used for the ChIP assays; vertical lines indicate CpG dinucleotides. Lower panels: A series of constructs containing different AML1‐binding sites and their mutants.
- Luciferase reporter activities of human 293T cells transiently co‐transfected for 48 h with luciferase reporter constructs containing the wild‐type sequence of the miR‐383 regulatory regions or its mutant counterparts, together with increasing amounts of pcDNA3.0 containing AML1‐ETO or mock cDNA (293T‐Mock). Triplicate values are defined as mean ± SD.
- ChIP using the indicated antibodies or IgG, after which qRT–PCR was performed to evaluate the specificity of protein binding. Triplicate values are defined as mean ± SD.
- ChIP using Ac‐histone H3 antibody or IgG, after which qRT–PCR was performed for amplification of the region in miR‐383 containing the predicted AML1‐binding site in the upstream region of miR‐383 with the predicted p300‐binding site, to evaluate the specificity of protein binding. Triplicate values are defined as mean ± SD.
- Kasumi‐1 cells were treated with C646 (10 or 20 μM) for 24 h, after which acetylation of histone H3 was analysed by immunoprecipitation (IP) and Western blot.
- qRT–PCR quantification of miR‐383 and THAP10 mRNA levels in the Kasumi‐1 cells treated with or without C646 (10 μM) for 24 h. The results represent the mean of three independent evaluations ± SD (*P = 0.0001, # P = 0.002). Student's t‐test was used for the comparisons.
ChIP analysis revealed the presence of AML1‐ETO and p300, rather than HDAC1, DNMT1, DNMT3a and DNMT3b, in the miR‐383 promoter region surrounding the AML1‐binding sites (Fig 5C). Furthermore, ChIP analysis using antibody against acetylated histone H3 showed that acetylation levels within the miR‐383 promoter region around the AML1‐ETO‐ and p300‐binding sites were higher in AML1‐ETO+ than AML1‐ETO− cells (Fig 5D). Given the role of the interaction between AML1‐ETO and p300 in regulation of histone acetylation (Wang et al, 2011), the chemical p300 inhibitor C646 was then used to determine the function of p300 in acetylation of the miR‐383 promoter region around the AML1‐ETO‐ and p300‐binding sites. Indeed, C646 treatment substantially decreased acetylation of histone H3 (lysine 9/14) as well as AML1‐ETO in Kasumi‐1 cells (Fig 5E), while it increased THAP10 levels (Fig 5F). Together, these findings indicate that AML‐ETO recruits p300 to the AML1‐binding sites in the pre‐miR‐383 regulatory region and therefore transcriptionally activates miR‐383 expression, which in turn suppresses THAP10 expression, suggesting an alternative indirect mechanism for AML‐ETO to negatively regulate THAP10 expression in t(8;21) AML.
THAP10 is a nuclear protein that inhibits proliferation but promotes differentiation of t(8;21) AML cells
In Drosophila, several THAP proteins are putative transcription factors (Sabogal et al, 2010), indicating that human THAP10 may also regulate transcription. Thus, we first examined whether THAP10 was localized in the nucleus, where transcriptional factors function to regulate gene expression. To this end, NIH3T3 cells were transfected with EGFP‐fused THAP10 cDNA to monitor subcellular localization of THAP10 protein. In contrast to GFP, which was distributed throughout the cell, GFP‐THAP10 was found only in the nucleus (Fig 6A). Western blot analysis further validated nuclear localization of endogenous THAP10 protein in Kasumi‐1 cells transduced with lentivirus vector expressing THAP10 (Fig EV4E). Moreover, immunofluorescence staining for THAP10 also revealed that the endogenous THAP10 protein predominantly localizes in the nuclei of HL‐60 cells and, to a lesser extent, in Kasumi‐1 cells and t(8;21) AML blasts (Fig EV4F). Together, these findings suggest that the THAP10 gene encodes a nuclear protein.
Figure 6. THAP10 localizes in the nuclei and inhibits proliferation but promotes differentiation of t(8;21) AML cells.
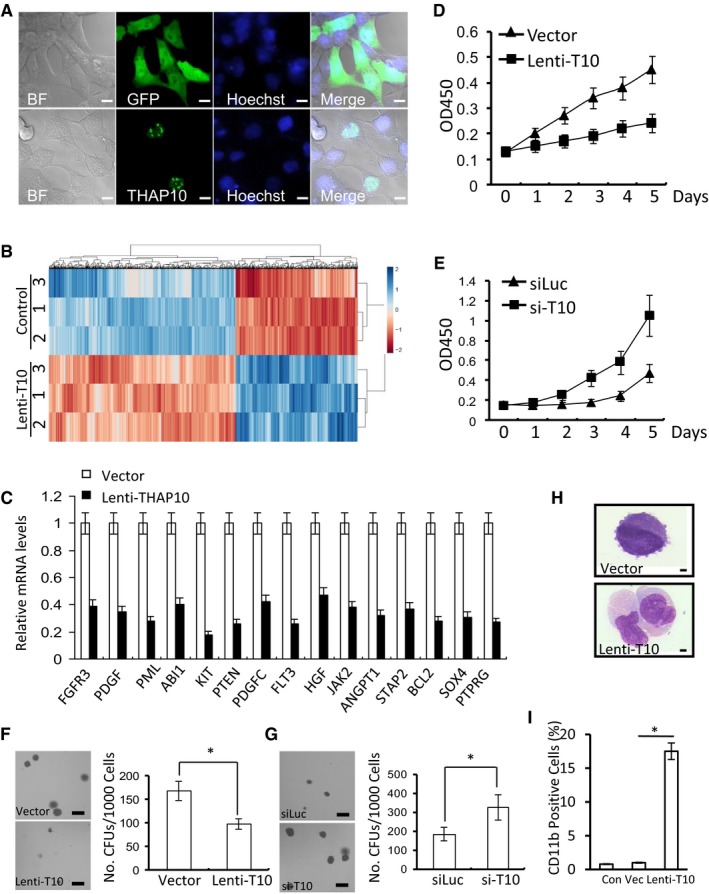
-
ANuclear localization of THAP10 in NIH3T3 cells. Scale bar, 10 μm.
-
BRepresentative heatmap of the aberrant gene profile of Kasumi‐1 cells transduced with or without THAP10 using lentivirus. Each column indicates a gene, and 1–3 indicate three independent pools of cells.
-
CFold changes in the mRNA levels for 15 selected genes measured by RT–PCR.
-
DGrowth curve of Kasumi‐1 cells transduced with THAP10 (Lenti‐T10) or lentiviral empty vector (Vector).
-
EGrowth curve of HL‐60 cells transfected with synthetic si‐THAP10 (si‐T10) or scramble (siLuc) sequences.
-
F, GAnalysis of CFUs in Kasumi‐1 (F, *P = 0.026) and HL‐60 cells (G, *P = 0.018) using a methylcellulose culture system. Scale bar, 1 mm. Two‐sided Student's t‐test was used for the comparisons.
-
HMorphological analysis of Kasumi‐1 cells at 4 days after transduction with empty vector or Lenti‐THAP10 (T10). Scale bar, 10 μm.
-
IFlow cytometric analysis of CD11b expression in Kasumi‐1 cells at 144 h after transduction with Lenti‐THAP10 (T10) or control vector (Vec). Wild‐type Kasumi‐1 cells as control (Con) (*P = 0.005). Two‐sided Student's t‐test was used for the comparisons.
To examine the functional role of THAP10 in leukaemogenesis of t(8;21) AML, microarray analysis of global gene expression was performed to identify the genes and pathways that correlate with THAP10 (Fig 6B) in Kasumi‐1 cells stably expressing THAP10 (Fig EV4G). Strikingly, classification based on the gene ontology (GO) annotation showed that in THAP10‐expressing Kasumi‐1 cells, the significantly down‐regulated genes were related to either receptor tyrosine kinase signalling pathways or cell differentiation (Appendix Table S6), suggesting the selectivity of THAP10 in inhibition of gene expression. These results were further validated by qPCR using specific primers for 15 distinct genes that were selected either from the transmembrane receptor protein tyrosine kinase signalling pathway or from the haematopoietic progenitor cell differentiation pathway. All these 15 genes were down‐regulated by at least twofold in THAP10‐expressing Kasumi‐1 cells compared to controls (Fig 6C). For detection of the total tyrosine phosphorylation, Western blot analyses showed a diminution of approximately 50% in p‐Tyr levels in THAP10‐expressing Kasumi‐1 cells compared with mock‐transduced cells (Appendix Fig S5). Thus, these findings suggest that THAP10 inhibits cell proliferation by suppressing the genes associated with the enzyme‐linked receptor signalling cascade, which may promote cell differentiation.
The role of THAP10 in myeloid cell proliferation/differentiation was then examined. Ectopic THAP10 expression significantly reduced proliferation of Kasumi‐1 cells (Fig 6D). Conversely, shRNA knock‐down of THAP10 (Fig EV5A) markedly promoted cell proliferation (Fig 6E) in HL‐60 cells that displayed a relatively high basal level of THAP10 (Fig 3A). Moreover, ectopic THAP10 expression led to significant reductions in both number (~45%) and size of CFUs in Kasumi‐1 cells (Fig 6F), while THAP10 shRNA knock‐down increased the number (~75%) and size of CFUs in HL‐60 cells (Fig 6G). Finally, ectopic THAP10 expression also induced myelomonocytic differentiation of SKNO‐1 cells, as shown by morphological changes indicating maturation towards granulocytes (e.g. chromatin condensation, decreased nuclear/cytoplasmic ratio and appearance of primary granules; Fig 6H), as well as an increase (1–17.5%) in CD11b+ differentiated cells (Fig 6I). Taken together, these findings indicated that the nuclear protein THAP10 inhibits proliferation but promotes differentiation of t(8;21) AML cells.
Figure EV5. Levels of THAP10 and miR‐383 in cell lines or tumours and the mouse model.
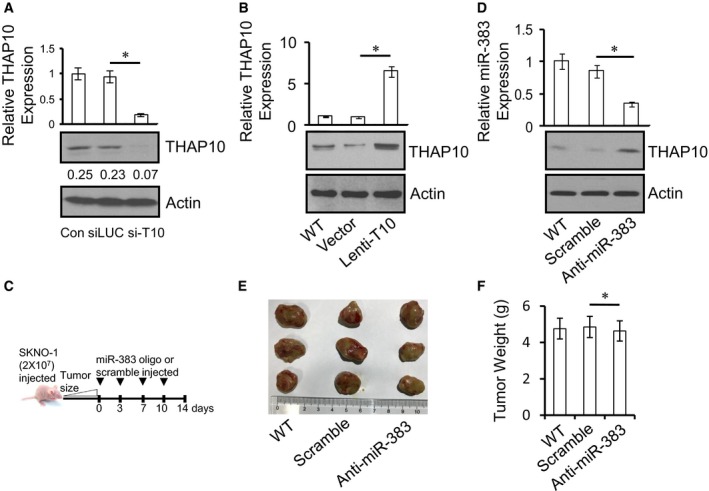
- Upper panel: Relative quantification of THAP10 mRNA levels in HL‐60 cells (wild type, siLUC and si‐T10). The results represent the mean of three independent evaluations ± SD (*P = 0.002, two‐sided Student's t‐test was used for the comparison). Lower panels: Immunoblot analysis of THAP10 protein levels using an anti‐THAP10 antibody. β‐actin was used for a loading control. Values indicate the ratios between THAP10 and actin.
- Expression of THAP10 in mouse Kasumi‐1 cell xenograft models (wild type, lenti‐empty vector and lenti‐THAP10). Upper panel: Relative quantification of THAP10 mRNA levels. The results represent the mean of three independent evaluations ± SD (*P = 0.0013, two‐sided Student's t‐test was used for the comparison). Lower panels: Protein levels of THAP10 by immunoblot analysis for THAP10 with an anti‐THAP10 antibody in xenografts indicated. β‐actin was used as a loading control.
- Diagram illustrating the workflow of the mouse study for testing the function of miR‐383.
- Effects of miR‐383 on THAP10 expression in mouse Kasumi‐1 cell xenograft models (wild type, scramble and anti‐miR‐383). Upper panel: Relative quantification of miR‐383 levels. The results represent the mean of three independent evaluations ± SD (*P = 0.0029, two‐sided Student's t‐test was used for the comparison). Lower panels: Protein levels of THAP10 monitored by immunoblot analysis. β‐actin was used as a loading control.
- Representative photographs of mouse xenografts in the scramble and synthetic anti‐miR‐383 treated AML1‐ETO− HL‐60 cells, captured at the end of the experiment (day 14).
- Triplicate values are defined as mean ± SD of tumour weight at the end of the experiment (WT, wild type; *P = 0.700, two‐sided Student's t‐test was used for the comparison).
Source data are available online for this figure.
The AML1‐ETO/miR‐383/THAP10 axis plays a functional role in leukaemogenesis of t(8;21) AML in vivo
Finally, the anti‐leukaemic function of THAP10 was examined in vivo. Kasumi‐1 cells ectopically expressing THAP10 were engrafted subcutaneously in the flank of immunocompromised BALB/c nude mice. Tumours formed from THAP10‐expressing cells were significantly smaller than the empty‐vector control (P = 0.0016) or parental counterparts (P = 0.0015) at day 28 post‐cell inoculation (Fig 7A–C), with an average tumour weight of 1.80 g for THAP10‐expressing cells vs. 5.25 g for empty‐vector controls (P = 0.0009, Fig 7D). Ectopic THAP10 expression markedly increased THAP10 mRNA (Fig EV5B, upper panel) and protein levels (Fig EV5B, lower panels) in xenografts.
Figure 7. THAP10 expression or miR‐383 inhibition suppresses tumour growth of t(8;21) AML cells in vivo .
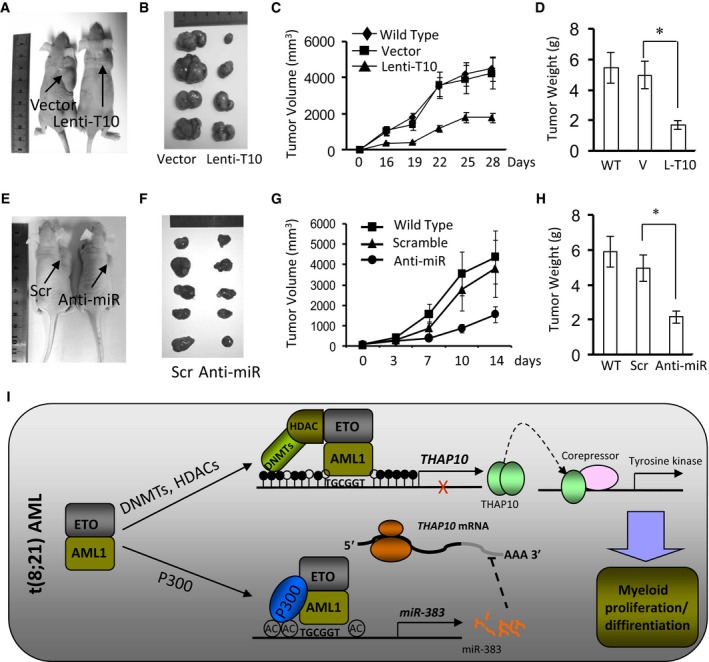
- Representative photographs of mice in the lentivirus empty vector and Lenti‐THAP10 (T10) groups captured at the end of the experiment (day 28).
- Tumour growth in mice engrafted with Kasumi‐1 cells ectopically expressing THAP10.
- Tumour volumes measured at the indicated days during the experiment (n = 6 for each group of wild type, empty vector and Lenti‐T10). Values are defined as mean ± SD.
- The mean ± SD of tumour weight at day 28 for the indicated groups (WT, wild type; V, empty vector; L‐T10, Lenti‐THAP10; n = 6 for each group; *P = 0.006). Two‐sided Student's t‐test was used for the comparisons.
- Representative photographs of mice in the scramble and synthetic anti‐miR‐383 treated groups, captured at the end of the experiment (day 14).
- Tumour growth in mice engrafted with SKNO‐1 cells ectopically expressing anti‐miR‐383.
- Tumour volumes measured at the indicated days during the experiment (wild type, n = 5; scramble, n = 6; synthetic anti‐miR‐383, n = 6). Values are defined as mean ± SD.
- The mean ± SD of tumour weight at the end of the experiment (WT, wild type, n = 5; Scr, scramble, n = 6; Anti‐miR, anti‐miR‐383, n = 6; *P = 0.009). Two‐sided Student's t‐test was used for the comparisons.
- A schematic model for the epigenetic suppression of THAP10 by AML1‐ETO directly or indirectly via miR‐383 and the interplays between AML1‐ETO and the co‐repressors DNMTs and HDACs or the co‐activator p300. Epigenetic suppression of THAP10 represents a mechanistic link between AML1‐ETO fusion protein and the tyrosine kinase cascades by targeting their downstream genes, which mediate inhibition of myeloid proliferation and promotion of cell differentiation in t(8;21) AML.
Similar experiments were performed to examine the effect of miR‐383 on tumour growth. t(8;21)+ SKNO‐1 cells were inoculated subcutaneously into the flank of BALB/c nude mice as described above. When tumour size reached 50 mm3, synthetic anti‐miR‐383 or scramble oligonucleotides were intratumourally injected as illustrated in Fig EV5C. On day 14 post‐first treatment, treatment with anti‐miR‐383 significantly reduced tumour size (P = 0.0025 vs. scramble oligonucleotide; P = 0.0018 vs. untreated control; Fig 7E–G), with an average tumour weight of 2.10 g for the anti‐miR‐383 vs. 5.15 g for the scramble oligonucleotide groups (P = 0.001, Fig 7H). Finally, anti‐miR‐383 treatment markedly diminished miR‐383 expression (Fig EV5D, upper panel), while it increased THAP10 protein levels (Fig EV5D, lower panels) in tumour tissue. To determine whether the in vivo activity of anti‐miR‐383 is selective against AML‐ETO+ AML, t(8;21)− HL‐60 cells were inoculated subcutaneously and intratumourally injected with anti‐miR‐383 or scramble oligonucleotides as described above. As shown in Fig EV5E and F, there was no significant difference in either tumour size or weight between anti‐miR‐383 and scramble oligonucleotide (4.85 g vs. 4.63 g, P = 0.700). Collectively, these findings strongly indicate that THAP10 expression, which is negatively regulated by miR‐383, functionally inhibits leukaemogenesis of t(8;21) AML.
Discussion
Aberrant DNA methylation and/or deacetylation are major factors in epigenetic gene silencing. Indeed, disruption of DNA methylation patterns is frequently present in aberrant development and leukaemogenesis (Oakes et al, 2016). The present study unveiled a novel epigenetic mini‐circuitry of AML1‐ETO/THAP10/miR‐383 in t(8;21) AML, which plays a functional role in leukaemogenesis of this poor‐prognostic AML subtype. The oncogenic transcription factor AML1‐ETO, a fusion protein generated by t(8;21) translocation, can recruit DNMTs to and subsequently hypermethylate the promoters of its target genes (Fazi et al, 2007a; Li et al, 2013b). In this study, we showed that t(8;21) AML blasts displayed a specific methylation signature, distinct from those of t(8;21)− AML and normal CD34+ cells, which was functionally associated with AML1‐ETO.
We identified THAP10 as a target gene of AML1‐ETO. The oncogenic effect of AML1‐ETO is primarily linked to its ability to form oligomeric complexes with an increased affinity for HDACs and DNMTs, which render AML1‐ETO a potent transcriptional repressor of AML1‐target genes. In this context, it has been reported that RARβ2, a retinoic acid (RA)‐regulated tumour suppressor gene, is silenced by aberrant DNA methylation in a variety of human malignancies, including t(8;21) AML (Fazi et al, 2007b). Here, we showed that AML1‐ETO directly bound to and hypermethylated the promoter region of THAP10, resulting in epigenetic suppression of this gene. THAP10 and LRRC49 (leucine rich repeat containing 49), both located on chromosome 15q23 in close proximity, were simultaneously suppressed due to hypermethylation of the bidirectional promoter region in breast cancer (De Souza Santos et al, 2008). The present findings indicate that THAP10 acts as a tumour suppressor in t(8;21) AML, suggesting that AML1‐ETO promotes leukaemogenesis via epigenetic suppression of THAP10. Therefore, THAP10 expression may predict a favourable clinical outcome of patients with t(8;21) AML. The relevance of this finding in AMLs is underlined by the fact that aberrant heterochromatic gene silencing can represent an alternative mechanism to gene mutation or deletion for the transcriptional repression of tumour suppressor genes (Baylin & Ohm, 2006; Mack et al, 2015). However, we could not rule out the possibility that other AML1‐ETO‐targeted genes (e.g. those depicted in Fig 1D) might also be involved in leukaemogenesis driven by this oncogenic fusion protein.
In addition to direct epigenetic suppression of the THAP10 gene via methylation of its promoter by AML1‐ETO, a question then arose whether this represents the sole mechanism for AML1‐ETO to inhibit THAP10 expression or whether other mechanism(s) could also be involved in suppression of THAP10 expression in AML‐ETO+ AML. We further demonstrated that THAP10 is also a bona fide target of miR‐383, a miRNA associated with several types of solid tumour (Lian et al, 2010; He et al, 2013; Li et al, 2013a; Riaz et al, 2013; Zhao et al, 2014). Interestingly, AML1‐ETO bound to the promoter of miR‐383 and activated its expression, while miR‐383 in turn suppressed expression of THAP10. Therefore, these findings indicate that AML1‐ETO inhibits expression of the tumour suppressor THAP10 directly via epigenetic suppression of the THAP10 promoter and indirectly through transcriptional activation of miR‐383 in t(8;21) AML (Fig 7I). This hypothesis is supported by findings indicating that the differentiation response to increased THAP10 can be restored in t(8;21) cell lines and blasts by (i) changing the methylation status at regulatory sites on the AML1‐ETO target gene THAP10 by AML1‐ETO knock‐down via RNAi or pharmacologic treatment with the demethylating agent 5‐azacytidine and (ii) impairing the interaction between AML1‐ETO and the transcriptional co‐activator p300 using the chemical inhibitor C646 or treatment with synthetic anti‐miR‐383.
Recent evidence suggests that alterations in transcription factors and tyrosine kinases represent two classes of the most frequently detected genetic events in human leukaemias. However, whether a causal relationship exists between the two genetic events remains largely unknown. The large‐scale gene expression profiling in the present study showed that THAP10 acts as a transcriptional repressor to selectively modulate the enzyme‐linked receptor tyrosine kinase signalling cascade and cell differentiation in t(8;21) AML. Thus, epigenetic suppression of THAP10 may link to both the first hit (e.g. AML1‐ETO) and the second hit (e.g. tyrosine kinase signalling cascade). Thus, we hypothesize that “loss of function” of the transcriptional repressor THAP10 may result in activation of a key tyrosine kinase pathway, which then promotes cell proliferation but blocks cell differentiation of myeloid progenitors. Indeed, ectopic expression of THAP10 resulted in a clear increase in differentiated cells with specific morphologic features and the CD11b marker. However, the mechanism(s) underlying THAP10‐mediated cell differentiation remains to be defined. Interestingly, there were significant inverse correlations between THAP10 and AML1‐ETO or tyrosine kinase C‐KIT mRNA levels, implying that high levels of THAP10 may indicate a good clinical outcome of patients with t(8;21) AML. Therefore, these findings strongly suggest that THAP10 is a tumour suppressor candidate as well as a potential biomarker for leukaemogenesis of t(8;21) AML.
In summary, this study identified AML1‐ETO/THAP10/miR‐383 as a novel epigenetic mini‐circuitry in t(8;21) AML, thereby providing new insight into the mechanism of action of AML1‐ETO in driving leukaemogenesis. These findings also suggest that THAP10 and miR‐383 are novel biomarkers and therapeutic targets in this AML subtype. Furthermore, this study presents new evidence for a functional link between the AML1‐ETO protein, cell proliferation and differentiation, and the abnormal tyrosine kinase signalling pathway in human t(8;21) AML. Our study also provided new insight into the molecular processes required for myeloid differentiation and identified possible new targets for the development of novel therapeutic approaches to leukaemia. Future studies should aim to (i) determine the roles of other human THAP proteins in cell proliferation, (ii) define the composition of the endogenous THAP10 protein complexes and (iii) characterize the potential links between THAP10 and its targets in regulating specific promoters involved in tyrosine kinase signalling pathways.
Materials and Methods
Cell lines and patient samples
HL‐60, NB4, ME‐1, Molt‐4, THP‐1, MM6, K5652, MV4‐11, Jurkat, RAJI, U937, U937‐A/E‐HA, Kasumi‐1, SKNO‐1, SKNO‐1‐siA/E and KG1 cell lines were maintained in RPMI 1640 medium supplemented with 50 μg/ml streptomycin, 50 IU penicillin and 10% FCS. SKNO‐1 cells were infected with the pRRLcPPT.hPGK, the lentiviral vector encoding the previously described siAGF1 oligonucleotides (Appendix Table S7) against the AML1‐ETO mRNA fusion site (siA/E‐RNAs) for silencing of AML1‐ETO (Fazi et al, 2007a; Martinez Soria et al, 2009; Li et al, 2013b). The U937‐A/E‐HA clone was obtained by transfection via electroporation of U937 wild‐type (WT) cells with an HA‐tagged AML1‐ETO cDNA subcloned into a vector carrying the Zn2+‐inducible mouse MT‐1 promoter, as previously reported (Fazi et al, 2007b). Human embryonic kidney (HEK) 293T cells were cultured in DMEM medium supplemented with 50 μg/ml streptomycin, 50 IU penicillin and 10% FCS. Treatments with 5‐azacytidine (2.5 μM, Sigma‐Aldrich), decitabine (2.5 μM, Xi′an Janssen Pharmaceutical Ltd.) and chidamide (3.0 μM, Shenzhen Chipscreen Biosciences Ltd.) were performed for 40 h.
This study was approved by the Human Subject Ethics Committee in Chinese PLA General Hospital and conducted in accordance with the Declaration of Helsinki. Informed consent was obtained from each subject. Mononuclear cells from bone marrow samples of newly diagnosed, untreated AML patients and healthy donors were isolated using Ficoll‐Hypaque (Sigma‐Aldrich, St Louis, MO) gradient centrifugation. Patient characteristics are summarized in Appendix Tables S1, S4 and S5.
RNA and protein extraction and analysis
Total RNA was extracted from cells using the TriPure isolation system (Roche). The relative quantity of THAP10 mRNA was measured using 200 ng of total RNA by qRT–PCR with qRT‐PCR Detection Kit (Ambion, Applied Biosystems, Milan) and the ABI PRISM 7500 Sequence Detection System (Applied Biosystems). Gene expression was determined by the comparative C T method using ABL levels for normalization as recommended in the manufacturer's instructions. Whole cell proteins were extracted using RIPA lysis buffer. Nuclear and cytoplasmic proteins were separated using the NE‐PER Nuclear and Cytoplasmic Extraction Kit (Thermo Scientific, Waltham, MA) as per the manufacturer's instructions.
Immunofluorescence and confocal microscopy
Analysis for nuclear localization of endogenous THAP10 was performed in transfected cells [NIH3T3 cells transfected with pEGFP or pTHAP10‐EGFP; cells were grown on poly‐D‐lysine‐coated cover slips (BD Biosciences, San Jose, CA, USA) 24 h after transfection] or Kasumi‐1, HL‐60 and t(8;21) AML blasts. Briefly, cells were fixed for 20 min with 4% cold paraformaldehyde and then permeabilized for 5 min with 0.05% Triton X‐100. Cells were incubated with 10 μl of Hoechst33342 (100 mg/l, Thermo Fisher Scientific) for 15 min for transfected cells. For Kasumi‐1, HL‐60 and t(8;21) AML blasts, permeabilized cells were then blocked with PBS‐BSA (PBS with 1% bovine serum albumin) and incubated with THAP10 primary antibodies (2 μg/ml, NBP1‐86226, Novus Biologicals, USA) overnight at 4°. Cells were then washed three times in PBA‐BSA and incubated in the dark for 1 h with Alexa Fluor 488‐conjugated secondary antibodies (1/500, #4412, Cell Signalling Technology, USA) diluted in PBA‐BSA. After three PBS washes, nuclei were counterstained with DAPI (0.2 μg/ml, #4083, Cell Signalling Technology, USA). Following extensive washing in PBS, samples were air‐dried and mounted in Rubber Cement (Union Rubber, Inc., Trenton, NJ, USA). Immunofluorescence images were captured using confocal microscopy (LSM 880, ZEISS, Germany). Green, GFP‐THAP10 or endogenous THAP10; blue, nucleus.
DNA methylation microarray for 450 k CpG sites
Microarray‐based DNA methylation analysis with 450 k arrays was used to profile genome‐wide DNA methylation in primary cells, using an Infinium Human Methylation 450 BeadChip with an Infinium HD methylation assay kit and HiScanSQ (Illumina), which assesses the methylation status at more than 485,000 individual CpG sites, encompassing 99% of reference sequence genes and 96% of CpG islands.
Total DNA was extracted using Wizard a genomic DNA purification kit (Promega Corp., WI, USA). DNA quality was determined with a Nano Drop 2000c spectrophotometer (Thermo Fisher Scientific Inc., Wilmington, DE), and 1 μg of DNA was bisulphite converted using a EZ DNA methylation gold kit (Zymo Research Corp., Irvine, CA) according to the manufacturer's protocol. Then, 500 ng of the bisulphate‐converted DNA was assayed with an Infinium Human Methylation 450 BeadChip using the Infinium HD methylation assay kit (Illumina), which assesses the methylation status at more than 485,000 individual CpG sites encompassing 99% of reference sequence genes and 96% of CpG islands. Each DNA sample first underwent an overnight isothermal whole‐genome amplification step. Amplified DNA was then fragmented, precipitated and resuspended. Samples were hybridized to BeadChips overnight at 48°C in an Illumina hybridization oven. Using an automated protocol on the TecanEvo robot (Tecan Group Ltd., Mannedorf, Switzerland), hybridized arrays were processed through a single‐base extension reaction on the probe sequence using DNP‐ or biotin‐labelled nucleotides, with subsequent immunostaining. The BeadChips were then coated, dried and imaged on an Illumina HiScanSQ. Image data were extracted using the Genome Studio version 2010.3 methylation module (Illumina). Beta values were calculated at each locus (β = intensity of methylated allele/intensity of unmethylated allele + intensity of methylated allele + 100), followed by analysis with the R package to normalize the data. The X and Y chromosome methylation data were excluded from the results. Quality control inclusion valuation depended on hybridization detection P < 0.05 (Accession number: GSE80508). Differential methylation loci were shown in a volcano plot: red points (P < 0.05, FC ≥ 2), and green points (P < 0.05, FC ≤ −2) indicate hypermethylation and hypomethylation loci, respectively. Furthermore, a heatmap was generated to demonstrate the indentified genes with abnormal expression/methylation profiles. DAVID online tools were used to implement the enrichment and pathway analysis. Venn diagrams were used to reveal the genes with hypermethylation profiles: These genes are both transcription factor and target of AML1‐ETO. The hypermethylation list was derived from the differential analysis of our 450K BeadChips, while transcription factors and targets of AML1‐ETO were from animal TFDB database (Zhang et al, 2015) and Gardini et al (2008), respectively. All statistical analyses were carried out with R 3.2.3.
Cell transfection
For siRNA‐mediated gene knock‐down, 1 × 106 Kasumi‐1 cells or SKNO‐1 cells were seeded into 6‐well plate for overnight incubation before transfection. miRNAs and siRNAs used in the study were synthesized by GenePharma (Shanghai, China). The sequences of siRNA or miRNA are provided in Appendix Table S7. Transfection with siRNAs and miRNAs was performed using HiPerFect (Qiagen, Hilden, Germany) according to the manufacturer's protocol. In brief, for each well, 24 μl HiPerFect and 12 μl siRNA or 20 μM miRNA were added to 400 μl serum‐free RPMI‐1640. The mixture was added to cells with 400 μl complete medium and incubated for 6 h, and then, 1.6 ml of complete medium was replenished. The final concentration of miRNA or siRNA was 100 nM. Total RNA and protein were prepared 48 h after transfection and subjected to qRT–PCR or Western blot analysis. Transfection efficiency was determined using FAM‐labelled control RNA by a FACScalibur flow cytometer (Becton Dickinson, Franklin Lakes, NJ, USA). This reagent yielded at least 90% transfection efficiency.
For gene overexpression, 2 × 105 293 T cells were seeded into a 6‐well plate for overnight incubation before transfection. Cells were transfected with a total of 0.5 μg expression plasmids or the corresponding empty vectors using Lipofectamine™ 2000 reagent (Life Technologies) as per the manufacturer's instruction. For co‐transfection experiments, the total amount of DNA or siRNA was kept the same between the individual gene and co‐transfection groups by adding equal amounts of empty‐vector or negative control.
THAP10‐expressing lentivirus preparation and transduction
Lentiviruses expressing EGFP and THAP10 were purchased from HanBio (www.hanbio.net). For lentivirus constructs, the CDS of THAP10 was inserted into hU6‐MCS‐PGK‐EGFP lentiviral vectors (Hanbio, Shanghai, China). The recombinant lentiviruses were produced by co‐transfection of 293T cells with the plasmids PSPAX2 and PMD2G with LipoFiter™ (Hanbio, Shanghai, China). Lentivirus‐containing supernatants were harvested 48 h after transduction and filtered through 0.22‐μm cellulose acetate filters (Millipore, USA). Recombinant lentiviruses were then concentrated by ultracentrifugation (2 h at 50,000 × g). To establish stable cell lines, Kasumi‐1 cells were transduced with lentiviral vector with or without THAP10 at an MOI of approximately 100 in the presence of 5 μg/ml polybrene. Approximately 48 h after transduction, the medium was changed, and puromycin was added for selection of stable transduced cells. Puromycin‐resistant colonies were selected for 3 weeks and then expanded. Gene modification via lentiviral delivery was confirmed by Western blot analysis.
Chromatin immunoprecipitation (ChIP)
Three biological replicate ChIP‐seq experiments were conducted for the specific detection of AML1‐bound genomic regions on THAP10 and miR‐383 promoters according to standard procedures with several modifications. Briefly, cross‐linked chromatin from approximately 5–10 × 107 SKNO‐1, SKNO‐1‐siA/E, U937 and U937‐A/E cells was prepared and fragmented to an average size of approximately 200 bp by 30–40 cycles of sonication (30 s each) in 15 ml tubes using the Bioruptor UCD‐200 sonicator (Diagenode). For immunoprecipitation, AML1 (sc‐8563, Santa Cruz Biotechnology), ETO (sc‐9737, Santa Cruz Biotechnology), HDAC1 (ab7028, Abcam), DNMT1 (ab13537, Abcam), DNMT3a (ab13888, Abcam), DNMT3b (ab13604, Abcam) and p300 (ab14984, Abcam) antibodies were added to 12 ml of diluted, fragmented chromatin. Nonimmunized rabbit serum served as a control. ChIP using the normal mouse IgG (Abcam) antibody was performed on naked and sonicated DNA extracted from the same cell samples and assayed using the EZ‐ChIP™ Chromatin Immunoprecipitation kit (Millipore) as per the manufacturer's instructions. Genomic THAP10 and pre‐miR‐383 upstream regions close to the putative AML1‐binding site were amplified. Primer sequences are shown in Appendix Table S7. GAPDH served as a control for nonspecific precipitated sequences.
Immunoprecipitation and Western blot analysis
Kasumi‐1 cells were treated with or without C646 (10 and 20 μM) for 24 h, after which cells were lysed in RIPA buffer containing 1 mM DTT, 1 μg/ml DNase I and a proteinase inhibitor cocktail (Roche) and then incubated on ice for 30 min. Anti‐ETO agarose (Santa Cruz Biotechnology) was used for immunoprecipitation assays. For Western blot analysis, protein samples were separated by electrophoresis on denaturing 10% SDS–PAGE gels and blotted to PVDF membranes (Millipore). Immunoblotting was performed using monoclonal antibodies, including anti‐AML1 (MABD126, Calbiochem), anti‐p‐Tyr (sc‐51688, Santa Cruz Biotechnology) and anti‐Ac‐histone H3 (Lys9/14, sc‐8655, Cell Signalling Technology). Anti‐β‐actin (sc‐47778, Santa Cruz Biotechnology) was used as loading control. The proteins were visualized by the ECL method (Amersham Biosciences).
Transactivation assays
DNA fragments were amplified by PCR from human genomic DNA. Primer sequences are shown in Appendix Table S7. All fragments were inserted in pGL3‐LUC reporter vectors (Promega). Various mutants were generated using a QuikChange Lightning Site‐Directed Mutagenesis kit as per the manufacturer's instructions (QuikChange, Agilent Technologies). All constructs were verified by DNA sequencing. 2 × 105 HEK293T cells were plated in 24‐well plates and transiently co‐transfected by SuperFect (Qiagen) with 10, 50 or 100 ng of pcDNA3 vectors with or without AML1‐ETO cDNA, together with 400 ng of the LUC reporter constructs as described above. Co‐transfection with a pRL‐TK Renilla luciferase reporter vector (Promega) was used as an internal control. Cells were harvested at 48 h post‐transfection and analysed using the Dual Luciferase Assay (Promega) as per the manufacturer's instructions.
Bisulphite sequencing assay
Bisulphite sequencing assays were performed to examine the methylation status of the CpG dinucleotides within the promoter region of the THAP10 gene, using EpiTect Bisulfite kit (Qiagen) as per the manufacturer's instructions. The assay was performed using 1 μg of bisulphite‐treated genomic DNA from the indicated cell lines and blasts from untreated patients. After bisulphite conversion performed with EpiTect Bisulfite kit (Qiagen) as previously described (Yu et al, 2005; Li et al, 2013b), the fragments of interest were amplified. Primer sequences are shown in Appendix Table S7. PCR products were gel‐purified and cloned into the pGEM®‐T vector systems (Promega). Individual bacterial colonies were used for PCR using vector‐specific primers, and PCR products were sequenced for the analyses of DNA methylation.
Luciferase assays for reverse screening the miRNAs regulating THAP10
To screen the miRNAs that directly regulate expression of THAP10, the 3′‐UTR or its mutant was cloned into a modified pGL‐3 control vector downstream of the coding sequence of luciferase. The transfection mixtures contained 100 ng of firefly luciferase reporter and 400 ng of plasmids expressing miR‐383 primary transcripts, derived from a constructed miRNA expression library. Transfection with pRL‐TK (Promega) was used as an internal control. The firefly luciferase activities were analysed using the Dual Luciferase Assay (Promega) at 48 h after transfection.
Microarray analysis and functional enrichment of differentially expressed gene (DEGs)
Total RNA was isolated from Kasumi‐1 cells after transduction with lentivirus expressing THAP10 or control using an RNeasy Mini kit (QIAGEN) according to the protocol. RNA quality, concentration and integrity were assessed by an Agilent 2100 Bioanalyzer. Sample labelling, microarray hybridization and washing were performed as per the manufacturer's instructions. Briefly, total RNA was transcribed to double‐stranded cDNA, synthesized into cRNA and labelled with cyanine 3‐CTP. The labelled cRNAs were then hybridized onto the microarray. After washing, the arrays were scanned by the Agilent Scanner G2505C (Agilent Technologies). Gene expression was analysed with Affymetrix U133 plus2.0 Human Gene Expression Arrays (Affymetrix Technologies). Data collection and analysis were performed according to the protocols by the Gene Biotechnology Corporation (Shanghai, China). DEGs of three independent experiments were used for biological process enrichment with Gorilla, an online gene ontology analyser. In addition, DEGs were used for pathway enrichment using DAVID functional annotation bioinformatics microarray.
Cell differentiation, apoptosis and colony‐forming analysis
Cell differentiation was evaluated by morphological examination of Wright‐Giemsa‐stained cytospins using light microscopy. Cells were also stained with anti‐CD11b (BD Biosciences PharMingen) and then subjected to flow cytometric analysis (a minimum of 50,000 events were collected for each sample) by FACScan (Becton Dickinson) using CellQuest software (Becton Dickinson) for data acquisition and analysis. For cell proliferation assays, cells post‐transfection were seeded into 96‐well plates (5,000 cells/well), and cell proliferation was then assessed using the CCK‐8 kit (Dojindo, Japan) as per the manufacturer's instructions. Briefly, 10 μl of CCK‐8 solution was added to the cell culture and incubated for an additional 4 h. Absorbance was determined at 450 nm wavelength. For apoptosis analysis, cells were stained with Annexin V and 7AAD (BD Biosciences) and analysed by a FACSCalibur flow cytometer (Becton Dickinson). Colony formation assays were performed using the methylcellulose H4230 culture system (Stem Cell Technologies) as per the manufacturer's instructions. After incubation for 10 days at 37°C and 5% CO2 in a humidified atmosphere, colonies of > 50 cells with morphological haematopoietic characteristics were counted.
Animal studies
BALB/c nude female mice (5 weeks old and weight > 15 g) were utilized. All mice used in this study were bred and maintained in a pathogen‐free environment. Splenectomy was performed at least 10 days before inoculation. After the mice recovered from the incisions, general irradiation at a dose of 400 cGy was performed at 24 h before tumour cell inoculation. The mice were then randomly divided into two groups. Xenograft models were established by subcutaneous injection into the right flank with 2 × 107 Kasumi‐1 cells (n = 4) or Kasumi‐1‐THAP10 cells (n = 5). The mice were sacrificed on day 28, and tumour weights were measured. A xenograft model was also established by subcutaneous injection with 2 × 107 SKNO‐1 cells into the right flank of nude mice. Tumour size was measured daily until it reached 50 mm3, after which 5 μg of synthetic miR‐383 or scramble diluted in Lipofectamine solution (Invitrogen) with a total volume of 100 μl was injected directly into tumours on days 1, 3, 7 and 10. Tumours were measured on the day of the first treatment and 4 days after the last treatment when the animals were sacrificed. Necropsies were performed, and tumours were weighed. Tumour volumes were calculated using the equation V (mm3) = A×B2/2, where A is the longest diameter and B is the perpendicular diameter.
Statistical analysis
SPSS 13.0 software was used to analyse the data. Wilcoxon signed‐rank test was performed to determine the differences in THAP10 and miR‐383 expression in clinical samples. Seventy‐six patients were grouped into quartiles according to THAP10 expression levels and divided into high (THAP10‐H, n = 56) and low (THAP10‐L, n = 20) groups based on the trend observed in clinical outcome after performing a Cox regression analysis of event‐free survival (EFS) and overall survival (OS) with THAP10 quartile grouping as the independent variable. Multivariate survival analysis was carried out using a Cox regression model. Student's t‐test was used to determine the statistical significance of experimental variables, and P‐values < 0.05 were considered statistically significant.
Author contributions
YL, QN, JS and YC performed experiments and analysed the data; MJ constructed plasmids and detected luciferase; LG, WH and YJ provided AML patient samples and clinical data; SH and AL performed ChIP experiments; ZH, DL and LW performed statistical analysis; CN, YD and MQZ commented on the paper; YL wrote the paper; YL and LY designed the research; and LY supervised the work.
Conflict of interest
The authors declare that they have no conflict of interest.
The paper explained.
Problem
Aberrant distribution of promoter DNA methylation occurring in specific and distinct patterns has been shown to be a universal feature in t(8;21) AML patients. However, the mechanisms that mediate these aberrant methyl cytosine patterns have not been defined.
Results
We conducted a large‐scale DNA methylation profiling study and found strong evidence that AML1‐ETO contributes to aberrant promoter DNA methylation patterning in t(8;21) AML. We found that the promoter DNA methylation signature of t(8;21) AML blasts differs from those of normal CD34+ bone marrow cells and non‐t(8;21) AMLs. This signature contains 408 differentially methylated genes, many of which are involved in cancer pathways and myeloid leukaemia. Then, database systematic survey and differential methylated regions (DMR) analysis were performed, which demonstrated that a novel hypermethylated zinc finger‐containing protein, THAP10, is a target gene and can be epigenetically suppressed by AML1‐ETO at the transcriptional level in t(8;21) AML, and suppression of THAP10 predicts a poor clinical outcome. Our findings also showed that THAP10 is a bona fide target of miR‐383 that can be epigenetically activated by AML1‐ETO. In this study, we demonstrated that epigenetic suppression of THAP10 is the mechanistic link between AML1‐ETO fusion proteins and tyrosine kinase cascades. In addition, we showed that THAP10 is a nuclear protein that inhibits myeloid proliferation and promotes differentiation both in vitro and in vivo.
Clinical impact
Our results identified AML1‐ETO/THAP10/miR‐383 as a novel epigenetic mini‐circuitry in t(8;21) AML, thereby providing a new insight into the mechanism of AML1‐ETO in driving leukaemogenesis. It also offers an opportunity to develop THAP10 and miR‐383 as novel biomarkers and therapeutic targets in this AML subtype. Finally, genome‐wide DNA methylation profiling is a powerful tool for characterization and clinical stratification of AML and warrants attention in clinical practice.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grants 81370635, 81570137, 81270611, 81470010 and 81670162) and the Beijing Natural Science Foundation (Grant 7151009). We thank Professor Qishou Xu for experimental support, and Ms. Li Cheng for technical assistance.
EMBO Mol Med (2017) 9: 933–949
References
- Baylin SB, Ohm JE (2006) Epigenetic gene silencing in cancer – a mechanism for early oncogenic pathway addiction? Nat Rev Cancer 6: 107–116 [DOI] [PubMed] [Google Scholar]
- Cayrol C, Lacroix C, Mathe C, Ecochard V, Ceribelli M, Loreau E, Lazar V, Dessen P, Mantovani R, Aguilar L et al (2007) The THAP‐zinc finger protein THAP1 regulates endothelial cell proliferation through modulation of pRB/E2F cell‐cycle target genes. Blood 109: 584–594 [DOI] [PubMed] [Google Scholar]
- De Souza Santos E, De Bessa SA, Netto MM, Nagai MA (2008) Silencing of LRRC49 and THAP10 genes by bidirectional promoter hypermethylation is a frequent event in breast cancer. Int J Oncol 33: 25–31 [PubMed] [Google Scholar]
- Dejosez M, Krumenacker JS, Zitur LJ, Passeri M, Chu LF, Songyang Z, Thomson JA, Zwaka TP (2008) Ronin is essential for embryogenesis and the pluripotency of mouse embryonic stem cells. Cell 133: 1162–1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeKelver RC, Yan M, Ahn EY, Shia WJ, Speck NA, Zhang DE (2013) Attenuation of AML1‐ETO cellular dysregulation correlates with increased leukemogenic potential. Blood 121: 3714–3717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorrance AM, Neviani P, Ferenchak GJ, Huang X, Nicolet D, Maharry KS, Ozer HG, Hoellarbauer P, Khalife J, Hill EB et al (2015) Targeting leukemia stem cells in vivo with antagomiR‐126 nanoparticles in acute myeloid leukemia. Leukemia 29: 2143–2153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyson NJ (2016) RB1: a prototype tumor suppressor and an enigma. Genes Dev 30: 1492–1502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fazi F, Racanicchi S, Zardo G, Starnes LM, Mancini M, Travaglini L, Diverio D, Ammatuna E, Cimino G, Lo‐Coco F et al (2007a) Epigenetic silencing of the myelopoiesis regulator microRNA‐223 by the AML1/ETO oncoprotein. Cancer Cell 12: 457–466 [DOI] [PubMed] [Google Scholar]
- Fazi F, Zardo G, Gelmetti V, Travaglini L, Ciolfi A, Di Croce L, Rosa A, Bozzoni I, Grignani F, Lo‐Coco F et al (2007b) Heterochromatic gene repression of the retinoic acid pathway in acute myeloid leukemia. Blood 109: 4432–4440 [DOI] [PubMed] [Google Scholar]
- Figueroa ME, Lugthart S, Li Y, Erpelinck‐Verschueren C, Deng X, Christos PJ, Schifano E, Booth J, van Putten W, Skrabanek L et al (2010) DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer Cell 17: 13–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardini A, Cesaroni M, Luzi L, Okumura AJ, Biggs JR, Minardi SP, Venturini E, Zhang DE, Pelicci PG, Alcalay M (2008) AML1/ETO oncoprotein is directed to AML1 binding regions and co‐localizes with AML1 and HEB on its targets. PLoS Genet 4: e1000275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Z, Cen D, Luo X, Li D, Li P, Liang L, Meng Z (2013) Downregulation of miR‐383 promotes glioma cell invasion by targeting insulin‐like growth factor 1 receptor. Med Oncol 30: 557 [DOI] [PubMed] [Google Scholar]
- Hsiao HH, Liu YC, Wang HC, Tsai YF, Wu CH, Cho SF, Hsu JF, Huang CT, Hsiao SY, Lee CP et al (2015) Additional chromosomal abnormalities in core‐binding factor acute myeloid leukemia. Genet Mol Res 14: 17028–17033 [DOI] [PubMed] [Google Scholar]
- Jiao B, Wu CF, Liang Y, Chen HM, Xiong SM, Chen B, Shi JY, Wang YY, Wang JH, Chen Y et al (2009) AML1‐ETO9a is correlated with C‐KIT overexpression/mutations and indicates poor disease outcome in t(8;21) acute myeloid leukemia‐M2. Leukemia 23: 1598–1604 [DOI] [PubMed] [Google Scholar]
- Khanjyan MV, Yang J, Kayali R, Caldwell T, Bertoni C (2013) A high‐content, high‐throughput siRNA screen identifies cyclin D2 as a potent regulator of muscle progenitor cell fusion and a target to enhance muscle regeneration. Hum Mol Genet 22: 3283–3295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley TJ, Miller C, Ding L, Raphael BJ, Mungal AJ, Robertson AG, Hoadley K, Triche TJ, Laird PW, Baty JD et al (2013) Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med 368: 2059–2074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li KK, Pang JC, Lau KM, Zhou L, Mao Y, Wang Y, Poon WS, Ng HK (2013a) MiR‐383 is downregulated in medulloblastoma and targets peroxiredoxin 3 (PRDX3). Brain Pathol 23: 413–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Gao L, Luo X, Wang L, Gao X, Wang W, Sun J, Dou L, Li J, Xu C et al (2013b) Epigenetic silencing of microRNA‐193a contributes to leukemogenesis in t(8;21) acute myeloid leukemia by activating the PTEN/PI3K signal pathway. Blood 121: 499–509 [DOI] [PubMed] [Google Scholar]
- Lian J, Tian H, Liu L, Zhang XS, Li WQ, Deng YM, Yao GD, Yin MM, Sun F (2010) Downregulation of microRNA‐383 is associated with male infertility and promotes testicular embryonal carcinoma cell proliferation by targeting IRF1. Cell Death Dis 1: e94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Clegg HV, Leslie PL, Di J, Tollini LA, He Y, Kim TH, Jin A, Graves LM, Zheng J et al (2015) CHCHD2 inhibits apoptosis by interacting with Bcl‐x L to regulate Bax activation. Cell Death Differ 22: 1035–1046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macfarlan T, Kutney S, Altman B, Montross R, Yu J, Chakravarti D (2005) Human THAP7 is a chromatin‐associated, histone tail‐binding protein that represses transcription via recruitment of HDAC3 and nuclear hormone receptor corepressor. J Biol Chem 280: 7346–7358 [DOI] [PubMed] [Google Scholar]
- Mack SC, Rich JN, Scacheri PC (2015) “PEAR‐ing” genomic and epigenomic analyses for cancer gene discovery. Cancer Discov 5: 1018–1020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumdar S, Singh A, Rio DC (2013) The human THAP9 gene encodes an active P‐element DNA transposase. Science 339: 446–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez Soria N, Tussiwand R, Ziegler P, Manz MG, Heidenreich O (2009) Transient depletion of RUNX1/RUNX1T1 by RNA interference delays tumour formation in vivo . Leukemia 23: 188–190 [DOI] [PubMed] [Google Scholar]
- Mende N, Kuchen EE, Lesche M, Grinenko T, Kokkaliaris KD, Hanenberg H, Lindemann D, Dahl A, Platz A, Hofer T et al (2015) CCND1‐CDK4‐mediated cell cycle progression provides a competitive advantage for human hematopoietic stem cells in vivo . J Exp Med 212: 1171–1183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakes CC, Seifert M, Assenov Y, Gu L, Przekopowitz M, Ruppert AS, Wang Q, Imbusch CD, Serva A, Koser SD et al (2016) DNA methylation dynamics during B cell maturation underlie a continuum of disease phenotypes in chronic lymphocytic leukemia. Nat Genet 48: 253–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orme MH, Liccardi G, Moderau N, Feltham R, Wicky‐John S, Tenev T, Aram L, Wilson R, Bianchi K, Morris O et al (2016) The unconventional myosin CRINKLED and its mammalian orthologue MYO7A regulate caspases in their signalling roles. Nat Commun 7: 10972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulikkan JA, Dengler V, Peramangalam PS, Peer Zada AA, Muller‐Tidow C, Bohlander SK, Tenen DG, Behre G (2010) Cell‐cycle regulator E2F1 and microRNA‐223 comprise an autoregulatory negative feedback loop in acute myeloid leukemia. Blood 115: 1768–1778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin YZ, Zhu HH, Jiang Q, Jiang H, Zhang LP, Xu LP, Wang Y, Liu YR, Lai YY, Shi HX et al (2014) Prevalence and prognostic significance of c‐KIT mutations in core binding factor acute myeloid leukemia: a comprehensive large‐scale study from a single Chinese center. Leuk Res 38: 1435–1440 [DOI] [PubMed] [Google Scholar]
- Riaz M, van Jaarsveld MT, Hollestelle A, Prager‐van der Smissen WJ, Heine AA, Boersma AW, Liu J, Helmijr J, Ozturk B, Smid M et al (2013) miRNA expression profiling of 51 human breast cancer cell lines reveals subtype and driver mutation‐specific miRNAs. Breast Cancer Res 15: R33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabogal A, Lyubimov AY, Corn JE, Berger JM, Rio DC (2010) THAP proteins target specific DNA sites through bipartite recognition of adjacent major and minor grooves. Nat Struct Mol Biol 17: 117–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarighat SS, Santhanam R, Frankhouser D, Radomska HS, Lai H, Anghelina M, Wang H, Huang X, Alinari L, Walker A et al (2016) The dual epigenetic role of PRMT5 in acute myeloid leukemia: gene activation and repression via histone arginine methylation. Leukemia 30: 789–799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vian L, Di Carlo M, Pelosi E, Fazi F, Santoro S, Cerio AM, Boe A, Rotilio V, Billi M, Racanicchi S et al (2014) Transcriptional fine‐tuning of microRNA‐223 levels directs lineage choice of human hematopoietic progenitors. Cell Death Differ 21: 290–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Gural A, Sun XJ, Zhao X, Perna F, Huang G, Hatlen MA, Vu L, Liu F, Xu H et al (2011) The leukemogenicity of AML1‐ETO is dependent on site‐specific lysine acetylation. Science 333: 765–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wichmann C, Quagliano‐Lo Coco I, Yildiz O, Chen‐Wichmann L, Weber H, Syzonenko T, Doring C, Brendel C, Ponnusamy K, Kinner A et al (2015) Activating c‐KIT mutations confer oncogenic cooperativity and rescue RUNX1/ETO‐induced DNA damage and apoptosis in human primary CD34 + hematopoietic progenitors. Leukemia 29: 279–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wouters BJ, Delwel R (2016) Epigenetics and approaches to targeted epigenetic therapy in acute myeloid leukemia. Blood 127: 42–52 [DOI] [PubMed] [Google Scholar]
- Yu L, Liu C, Vandeusen J, Becknell B, Dai Z, Wu YZ, Raval A, Liu TH, Ding W, Mao C et al (2005) Global assessment of promoter methylation in a mouse model of cancer identifies ID4 as a putative tumor‐suppressor gene in human leukemia. Nat Genet 37: 265–274 [DOI] [PubMed] [Google Scholar]
- Zhang HM, Liu T, Liu CJ, Song S, Zhang X, Liu W2, Jia H, Xue Y, Guo AY (2015) AnimalTFDB 2.0: a resource for expression, prediction and functional study of animal transcription factors. Nucleic Acids Res 43: D76–D81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Gu H, Chang J, Wu J, Wang D, Chen S, Yang X, Qian B (2014) MicroRNA‐383 regulates the apoptosis of tumor cells through targeting Gadd45 g. PLoS ONE 9: e110472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu CY, Li CY, Li Y, Zhan YQ, Li YH, Xu CW, Xu WX, Sun HB, Yang XM (2009) Cell growth suppression by thanatos‐associated protein 11(THAP11) is mediated by transcriptional downregulation of c‐Myc. Cell Death Differ 16: 395–405 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5