

Abstract
Key points
Ionotropic glutamate receptor (iGluR) subunits are N‐glycosylated at 4–12 sites, and Golgi processing produces mature receptors that contain high‐mannose, hybrid and complex oligosaccharides. N‐glycosylation is crucial for receptor biogenesis, influences receptor trafficking and provides a binding site for carbohydrate binding proteins.
Glycan moieties are large, polar and occasionally charged, and they are attached at sites along iGluRs that position them for involvement in the structural changes underlying gating.
Altering glycan content on kainate receptors (KARs), a subfamily of iGluRs, changes functional properties of the receptor, such as desensitization, recovery from desensitization and deactivation.
We report the first observation that the charged trisaccharide HNK‐1 is conjugated to native KARs, and we find that it substantially alters recombinant KAR functional properties.
Our results show that the molecular composition of N‐glycans can influence KAR biophysical properties, revealing a potential mechanism for fine‐tuning the function of these receptors.
Abstract
Ionotropic glutamate receptors (iGluRs) are tetrameric proteins with between four and 12 consensus sites for N‐glycosylation on each subunit, which potentially allows for a high degree of structural diversity conferred by this post‐translational modification. N‐glycosylation is required for proper folding of iGluRs in mammalian cells, although the impact of oligosaccharides on the function of successfully folded receptors is less clear. Glycan moieties are large, polar, occasionally charged and mediate many protein–protein interactions throughout the nervous system. Additionally, they are attached at sites along iGluR subunits that position them for involvement in the structural changes underlying gating. In the present study, we show that altering glycan content on kainate receptors (KARs) changes the functional properties of the receptors in a manner dependent on the identity of both the modified sugars and the subunit composition of the receptor to which they are attached. We also report that native KARs carry the complex capping oligosaccharide human natural killer‐1. Glycosylation patterns probably differ between cell types, across development or with pathologies, and thus our findings reveal a potential mechanism for context‐specific fine‐tuning of KAR function through diversity in glycan structure.
Keywords: deactivation, desensitization, HNK‐1
Key points
Ionotropic glutamate receptor (iGluR) subunits are N‐glycosylated at 4–12 sites, and Golgi processing produces mature receptors that contain high‐mannose, hybrid and complex oligosaccharides. N‐glycosylation is crucial for receptor biogenesis, influences receptor trafficking and provides a binding site for carbohydrate binding proteins.
Glycan moieties are large, polar and occasionally charged, and they are attached at sites along iGluRs that position them for involvement in the structural changes underlying gating.
Altering glycan content on kainate receptors (KARs), a subfamily of iGluRs, changes functional properties of the receptor, such as desensitization, recovery from desensitization and deactivation.
We report the first observation that the charged trisaccharide HNK‐1 is conjugated to native KARs, and we find that it substantially alters recombinant KAR functional properties.
Our results show that the molecular composition of N‐glycans can influence KAR biophysical properties, revealing a potential mechanism for fine‐tuning the function of these receptors.
Abbreviations
- AMPAR
AMPA receptor
- ATD
amino‐terminal domain
- CI
confidence interval
- CNS
central nervous system
- eGFP
enhanced green fluorescent protein
- GlcAT‐P
glucuronyltransferase P
- GlcNAc
N‐acetylglucosamine
- HNK‐1
human natural killer‐1
- HNK‐1ST
HNK‐1 sulphotransferase
- iGluR
ionotropic glutamate receptor
- KAR
kainate receptor
- LBD
ligand binding domain
- MW
molecular weight
- NMDAR
NMDA receptor
- PNGase F
protein N‐glycosidase F
- Popen
open probability
- PSA
polysialic acid
- ST‐3
α‐2,3‐sialyltransferase
- ST‐6
α‐2,6‐sialyltransferase
Introduction
Post‐translational modifications of ionotropic glutamate receptor (iGluR) subunits have the potential to diversify channel function and impact intracellular trafficking (Traynelis et al. 2010). Fundamental steps in the biogenesis of mammalian iGluRs, such as efficient protein folding and egress from the endoplasmic reticulum, require N‐glycosylation, which consists of the conjugation and processing of oligosaccharides attached to asparagines in the extracellular domains of subunit proteins (Everts et al. 1999). For tetrameric iGluRs, the potential structural diversity conferred by variable oligosaccharide content far exceeds any other form of post‐translational modification. Individual iGluR subunit proteins have between four and 12 N‐glycosylation consensus motifs, and oligosaccharides can constitute ∼10% or more of the mass of the mature receptors (Rogers et al. 1991; Roche et al. 1994). Discrete sites of N‐glycosylation in neuronal AMPA, kainate and NMDA receptors were identified in a proteomic analysis of glycans and glycopeptides (Parker et al. 2013). These sites of oligosaccharide conjugation included asparagines within the first extracellular linker domain connecting the amino terminal and ligand binding domains in the receptor subunits, which positions oligosaccharide chains on native receptors adjacent to key structural components involved in channel gating. The precise glycan composition of oligosaccharide chains conjugated to iGluRs is not clear, although functional surface receptors carry both high‐mannose and complex glycans (Hanus et al. 2016; Kaniakova et al. 2016). In addition, the sulfated trisaccharide human natural killer‐1 (HNK‐1) epitope is added to the AMPA receptor subunit GluA2, where it mediates interactions with N‐cadherin necessary for the induction of plasticity in hippocampal CA1 pyramidal neurons (Yamamoto et al. 2002; Morita et al. 2009).
The proximity of substantial, highly polar and potentially charged sugar chains to a critical functional domain in iGluRs suggested that oligosaccharides might directly affect receptor gating. This hypothesis has been tested in a number of ways over the last two decades, although no clear consensus has emerged. For example, tunicamycin inhibition of N‐glycosylation in Xenopus oocytes diversely altered (but did not preclude) expression of functional AMPA and kainate receptors and had little effect on agonist EC50 values; however, a possible effect of glycans on desensitization properties was inferred from changes in relative glutamate and kainate current amplitudes (Everts et al. 1997). On the other hand, elimination by mutagenesis of two N‐glycosylation sites in the initial S1 segment of the ligand‐binding domain (LBD) of GluA4 AMPA receptors had no measurable effect on receptor function or ligand binding affinity (Pasternack et al. 2003). Radioligand binding assays have been similarly equivocal; for example, rat brain AMPA receptors exhibit two discrete [3H]AMPA binding affinities (Hall et al. 1992), which was ascribed to differentially glycosylated subunit isoforms (Standley et al. 1998).
In the present study, we took a different tack to test the hypothesis that glycan chemical content impacts iGluR function. Rather than precluding all N‐glycosylation with tunicamycin or mutagenesis, we pharmacologically inhibited key oligosaccharide processing enzymes in the Golgi or over‐expressed enzymes responsible for transferring capping sugars to complex oligosaccharides. We found that these manipulations altered iGluR gating to differing degree, with the addition of HNK‐1 epitopes having the largest effect. Our results suggest that alterations in N‐glycan identity produce subtle differences in recombinant kainate receptor (KAR) functional properties in a manner that depends on both the receptor subunit composition and the identity of the sugars attached to the protein.
Materials and methods
Ethical approval
All animals used in these studies were treated in accordance with protocols approved by Northwestern University's Institutional Animal Care and Use Committee, which were consistent with standards of care established by the Guide for the Care and Use of Animals (8th edition). Animals were fed standard chow and had ad libitum access to food and water. Male and female mice over 3 weeks old were used for all of the experiments. Wild‐type animals are C57Bl/6 from Charles River (Wilmington, MA, USA). Five‐KAR subunit knockout mice were provided Dr Anis Contractor (Xu et al. 2017). HNK‐1ST knockout mice (B6;129‐Chst10tm1Mifu/Mmjax) were obtained from Jackson Laboratories (Bar Harbor, ME, USA) (RRID:MMRRC_037129‐JAX). All mice were killed by decapitation following anaesthesia with inhaled isoflurane and all steps were taken to minimize pain and suffering. We understand the ethical principles under which the Journal of Physiology operates and our work complies with the animal ethics checklist outlined in Grundy (2015).
Materials
DNAs used in these studies were provided to us by Dr Derek Bowie (McGill University, Montreal, QC, Canada; rat GluK2∆NG5,6,7 cDNA), Dr Sakari Kellokumpu (University of Oulu, Oulu, Finland; eGFP‐ST3 and eGFP‐ST6 cDNAs), Dr Shogo Oka (Kyoto University, Kyoto, Japan; pIRES‐GlcAT‐P‐HNK‐1ST cDNA) and Dr Susumu Tomita (Yale University School of Medicine, New Haven, CY, USA; Neto2 cDNA). In GluK2ΔNG5,6,7, an S/T to A mutation is present in the consensus N‐glycosylation sequences (N‐X‐S/T, where X ≠ P) at T414, T425 and S432 (Fay & Bowie, 2006). The GluK1 and GluK2 cDNAs used in these experiments expressed unedited (glutamine‐containing) receptors. Swainsonine (product number S9263) and kifunensine (product number K1140) were purchased from Sigma‐Aldrich (St Louis, MO, USA).
Cell culture and transfection
Human embryonic kidney cells expressing T‐antigen, clone 17 (HEK293T/17) from the American Type Culture Collection (Manassas, VA, USA) were cultured in Dulbecco's modified essential medium (Corning Cellgro, Manassas, VA, USA) supplemented with 10% heat‐inactivated fetal bovine serum (Gemini Bio‐Products, West Sacramento, CA, USA), 100 μg ml−1 penicillin and 100 μg ml−1 streptomycin (Corning Cellgro), at 37°C with 5% CO2. Transfections were performed in accordance with the manufacturer's instructions using a ratio of 1 μg of cDNA to 3 μl of Mirius Bio Trans‐IT reagent (Mirius Bio Corporation, Madison, WI, USA). An enhanced green fluorescent protein (eGFP) was co‐transfected to identify receptor‐expressing cells. Swainsonine (20 μm) and kifunensine (5 μm) treatments were added to the culture medium at least 4 h prior to transfection.
Electrophysiology
Whole‐cell or outside‐out patch recordings were made from transfected HEK293T/17 cells held at –70 mV as described previously (Vivithanaporn et al. 2007). Currents were elicited by rapid application of glutamate to receptor‐expressing cells using a piezoceramic system or a Siskiyou MXPZT‐300 solution switcher (Siskiyou Corporation, Grants Pass, OR, USA). Rise times (10–90%) were < 2.0 ms from whole‐cell recordings and < 1.0 ms from outside‐out patch recordings. Weighted desensitization rates were calculated from bi‐exponential fits of 1 s glutamate applications in Clampfit, version 10 (Molecular Devices, Sunnyvale, CA, USA). We note that the very rapid desensitization of GluK2/GluK5 receptors approaches the effective temporal limit of solution exchange in whole‐cell recordings, and thus it is possible that we might have been unable to resolve faster desensitization time courses induced by experimental manipulations to cells expressing this particular kainate receptor. Recovery rates were calculated with single exponential association fits in Prism, version 5 (GraphPad Software, La Jolla, CA, USA). Glutamate applications of 1–2 ms for deactivation were confirmed by recording the junction potential after each experiment. External solution contained (in mm): 150 NaCl, 2.8 KCl, 1.8 CaCl2, 1.0 MgCl2, 10 glucose and 10 Hepes, adjusted to pH 7.3. Intracellular solution contained (in mm): 110 CsF, 30 CsCl, 4 NaCl, 0.5 CaCl2, 10 Hepes and 5 EGTA, adjusted to pH 7.3.
Western blotting
Recombinant proteins were expressed in HEK293T/17 for 72 h prior to washing with ice‐cold Dulbecco's PBS and lysing in lysis buffer containing (in mm) 150 NaCl, 25 Tris buffer, 1 EDTA, 1 NaF, 1 Na3VO4, 1 phenylmethane sulphonyl fluoride, Protease Inhibitor Cocktail (product number P2714; Sigma‐Aldrich) and 1% Triton X‐100, adjusted to pH 7.4. Samples were solubilized for 1 h at 4°C and cleared of cell debris by centrifugation. For immunoprecipitations, tissue was isolated and homogenized in lysis buffer, and then solubilized and cleared of cell debris. KAR subunits were precipitated from 1 mg of total protein using 2 μl of antibody (see below) and Pierce Protein A/G Agarose Beads (product number 20421; Thermo Scientific, Waltham, MA, USA) overnight at 4°C. Proteins were separated on a denaturing 8% polyacrylamide gel and transferred to a polyvinylidene fluoride membrane. Proteins were precipitated or detected using a rabbit anti‐myc antibody (product number 06‐549 RRID:AB_310165; EMD Millipore, Darmstadt, Germany), a rabbit anti‐GluR6/7 (GluK2/3) antibody (product number 04‐921 RRID:AB_1587072; EMD Millipore) or a mouse anti‐HNK‐1 epitope antibody (product number 1C10 RRID:AB_10570406, deposited in the University of Iowa Developmental Studies Hybridoma Bank by W. M. Halfter, Iowa City, IA, USA). Antibody binding was imaged on an Odyssey CLx Imager (LI‐COR Biosciences, Lincoln, NE, USA) or using Immobilon Western Chemilluminescent HRP Substrate (product number WBKLS0500; EMD Millipore) and film. Protein molecular weight was determined using Image Studio Software (LI‐COR Biosciences).
Statistical analysis
Comparisons between two conditions were made by unpaired t test. Comparisons between three or more groups were made with a one‐way ANOVA followed by Dunnett's multiple comparison. Equivalent results were obtained using either parametric or non‐parametric tests, and the statistical results are reported from parametric tests. The time courses of recovery from desensitization were fit with a one‐phase association exponential function. Values are reported as the mean ± SEM. Statistical tests were performed in Prism, version 5 (GraphPad Software).
Results
Biochemical manipulation of glycan content
We tested the hypothesis that restricting glycan processing can alter KAR functional properties by expressing recombinant receptors in the presence of enzyme inhibitors of α‐mannosidases, the cis‐Golgi enzymes responsible for trimming branches from immature, mannose‐containing oligosaccharide branches. Receptor‐transfected HEK293T/17 cells were treated with either kifunensine, an α‐mannosidase I inhibitor, or swainsonine, an α‐mannosidase II inhibitor. These pharmacological tools restrict processing to immature or hybrid glycan structures, respectively (Fig. 1 A). Treatment with the two inhibitors incrementally reduced the molecular weight (MW) of recombinant myc‐tagged GluK2a protein, as detected with anti‐myc antibody (Fig. 1 B). Swainsonine reduced myc‐GluK2a monomers from 136 ± 2 kDa to 128 ± 1 kDa; similarly, kifunensine decreased the myc‐GluK2a MW to 127 ± 1 kDa (repeated measures ANOVA, P = 0.0002; n = 3 experiments) (Fig. 1 B). Thus, pharmacological inhibition of two Golgi‐resident processing enzymes reduces the mass of oligosaccharide chains on GluK2a KAR subunits.
Figure 1. α‐mannosidase inhibition reduces myc‐GluK2a molecular weight and alters myc‐GluK2a‐containing receptor desensitization.
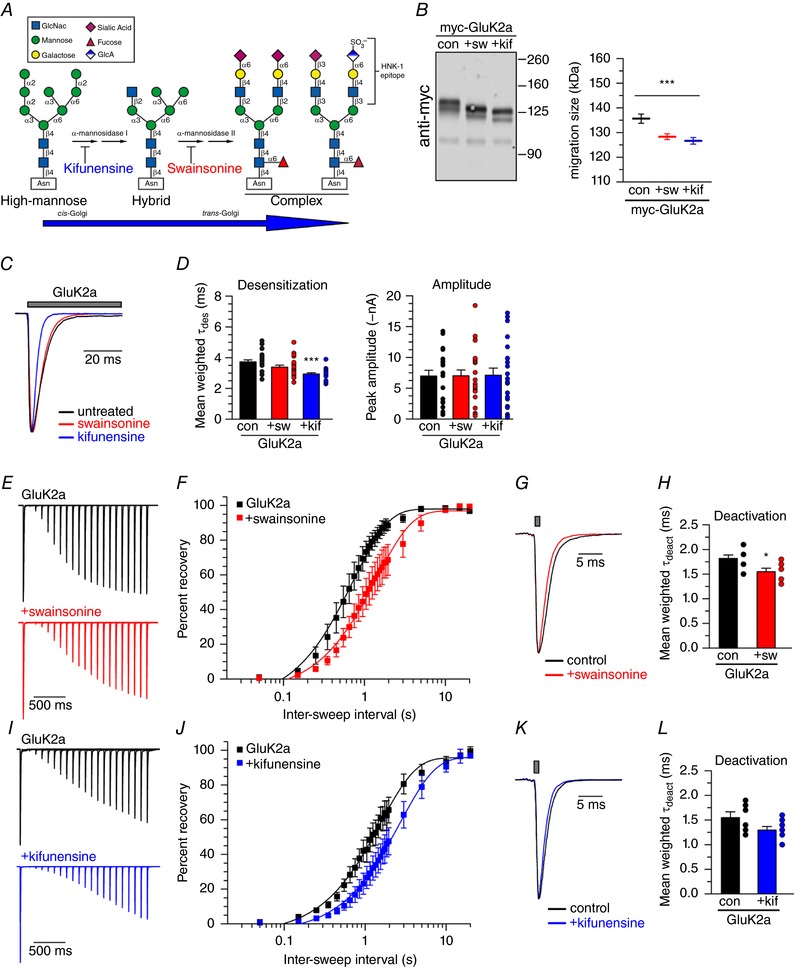
A, kifunensine inhibits α‐mannosidase I and swainsonine inhibits α‐mannosidase II, blocking hybrid and complex oligosaccharide formation, respectively. B, myc‐GluK2a was expressed in untreated control, swainsonine‐treated or kifunensine‐treated HEK293T/17 cells, detected by immunoblotting with an anti‐myc antibody, and myc‐GluK2a MW was measured from gel migration. C, representative current traces from untreated control, swainsonine‐treated and kifunensine‐treated myc‐GluK2a‐expressing HEK293T/17 cells. Grey bar indicates glutamate (10 mm) application. Amplitudes are scaled. D, quantification of glutamate‐evoked desensitization and current amplitude from untreated and treated myc‐GluK2a‐expressing HEK293T/17 cells. E, representative current traces in two‐pulse glutamate (10 mm) recovery experiments recorded from untreated and swainsonine‐treated myc‐GluK2a‐expressing HEK293T/17 cells. Intervals between glutamate exposures range from 50 ms to 2 s in the traces shown. Amplitudes of the first glutamate application are scaled. F, quantification of mean glutamate recovery for myc‐GluK2a‐expressing cells with and without swainsonine. Amplitude of the second glutamate application in a two‐pulse experiment is reported as a normalized percentage of the first glutamate application. Results were fitted with a single component exponential equation and the best‐fit τrec values differ significantly (P < 0.0001). G, representative myc‐GluK2a deactivation current traces. Grey bar indicates glutamate (10 mm) application. Amplitudes are scaled. H, quantification of glutamate‐evoked deactivation from myc‐GluK2a‐containing outside‐out patches pulled from untreated and swainsonine‐treated HEK293T/17 cells. I, representative current traces in recovery experiments recorded from untreated and kifunensine‐treated myc‐GluK2a‐expressing HEK293T/17 cells, as in (E). J, quantification of mean glutamate recovery for myc‐GluK2a‐expressing cells with and without kifunensine, as in (F). Results were fitted with a single component exponential equation and the best‐fit τrec values differ significantly (P < 0.0001). K, representative myc‐GluK2a deactivation current traces. Grey bar indicates glutamate (10 mm) application. Amplitudes are scaled. L, quantification of glutamate‐evoked deactivation from myc‐GluK2a‐containing outside‐out patches pulled from untreated and kifunensine‐treated HEK293T/17 cells. con, control; sw, swainsonine treatment; kif, kifunensine treatment. Error bars indicate the SEM. Statistical significance is indicated: *** P < 0.001. [Color figure can be viewed at wileyonlinelibrary.com]
Functional consequences of restricting glycan processing
The biochemical data show that we are able to manipulate the oligosaccharide moieties attached to KAR subunits. We next determined whether these manipulations altered the functional properties of GluK2a‐containing KARs in patch clamp recordings from transfected HEK293T/17 cells that had been treated with normal media, swainsonine or kifunensine. Glutamate (10 mm) was applied to the cells for 100 ms to evoke whole‐cell currents; representative traces are shown in Fig. 1 C. We found that currents elicited from cells incubated with the α‐mannosidase I inhibitor kifunensine exhibited more rapid desensitization (control: τdes = 3.7 ± 0.1 ms, n = 22; kif: 2.9 ± 0.1 ms, n = 22; P < 0.0001, Dunnett's multiple comparison with control P < 0.001), whereas currents from swainsonine‐treated GluK2a KARs were not different from the control (3.4 ± 0.1 ms; n = 23) (Fig. 1 D). Whole‐cell current amplitudes were not altered by either α‐mannosidase inhibitor, with peak amplitudes of 7.0 ± 0.9 nA in control cells, 7.0 ± 1.0 nA in swainsonine‐treated cells and 7.1 ± 1.1 nA in kifunensine‐treated cells (P = 0.99) (Fig. 1 D), suggesting that the pharmacological treatments did not grossly alter biogenesis and functional expression of GluK2a KARs. Because kifunensine treatment speeded up desensitization, we tested whether α‐mannosidase I inhibition altered other functional properties of GluK2a KARs.
We tested whether the pharmacological treatments altered two other kinetic parameters: recovery from desensitization and deactivation. To evaluate recovery from desensitization, recovery curves were generated by applying glutamate twice at varying intervals ranging from 50 ms to 20 s (Fig. 1 E). Swainsonine‐treated GluK2a‐containing receptors recovered from glutamate‐evoked desensitization with a tau of 1.4 s [95% confidence interval (CI) = 1.2–1.7], which was two‐fold slower than the control recovery of 0.7 s (95% CI = 0.6–0.9) (one‐phase association exponential equation, parameters of best fit lines differ with P < 0.0001) (Fig. 1 F). We also tested whether restricting glycan processing altered deactivation of GluK2a‐containing receptors (Fig. 1 G). Brief (1 ms) application of glutamate to outside‐out patches from swainsonine‐treated GluK2a KARs evoked currents that deactivated with a τ of 1.6 ± 0.07 ms (n = 8), which is faster than the 1.8 ± 0.07 ms (n = 7) deactivation measured from untreated patches (unpaired t test P = 0.023) (Fig. 1 H). Kifunensine‐treated GluK2a‐containing receptors recovered from glutamate‐evoked desensitization with a tau of 2.7 s (95% CI = 2.3–3.3), which again was slower than the interleaved control recoveries of 1.5 s (95% CI = 1.4–1.8) (parameters of best fit lines differ with P < 0.0001) (Fig. 1 I, J). We note that fitting of the two sets of data for recovery from glutamate desensitization produced somewhat different time constants (Fig. 1 F and J). These data were collected several years apart and suggest that the variable expression of other factors in the heterologous cells influences recovery time course; for the purposes of the present study, however, analysis of the effects of swainsonine and kifunensine were performed contemporaneously with their interleaved control recordings. Finally, kifunensine‐treated GluK2a KARs deactivated with a τ of 1.3 ± 0.07 ms (n = 8), which was not different from the 1.6 ± 0.1 ms (n = 6) deactivation time course in untreated patches (P = 0.075) (Fig. 1 K and L). These data show that restricting glycan processing on GluK2a‐containing KARs alters receptor desensitization kinetics, suggesting that sugar composition can influence the complex structural rearrangements underlying this functional property.
We next attempted to determine the gating properties of GluK2a KARs lacking all N‐glycans by treating live cells with protein N‐glycosidase F (PNGase F). PNGase F cleaves glycans at the linkage between asparagine side chain and the inner‐most N‐acetylglucosamine (GlcNAc), removing entire glycan chains from the protein structure. However, we found that the MW of GluK2a protein was only slightly reduced following treatment of live GluK2a‐transfected cells with PNGase F for 2 h (Fig. 2 A). Longer incubations of up to 24 h also were ineffective at catalysing cleavage (data not shown). By contrast, PNGase F treatment effectively removed oligosaccharides from the denatured subunit protein (Fig. 2 A). These data suggest that the PNGase F substrate site is inaccessible in the native receptor, perhaps as a result of steric hindrance by oligosaccharide chains, by the receptor itself or by a combination of both structural components. PNGase F also did not alter glutamate‐evoked current amplitudes or desensitization measured in recordings from GluK2a‐expressing cells (Fig. 2 B and C) (untreated: 7.5 ± 0.9 nA, τdes = 4.3 ± 0.1 ms, n = 23; PNGase F: 8.9 ± 1.0 nA, τdes = 4.1 ± 0.2 ms, n = 20). Thus, we were unable to test the effect of complete enzymatic removal of glycans on KAR functional properties.
Figure 2. Glycan chains attached between the ATD and LBD are critical modulators of KAR desensitization.
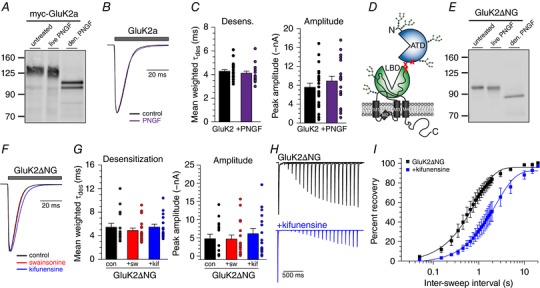
A, western blot showing myc‐GluK2a MW after PNGase F treatment. Myc‐GluK2a‐expressing cells were incubated in buffer (untreated) or buffer containing PNGase F (live PNGF) for 2 h prior to lysing. Protein from untreated cell lysates was denatured, digested with PNGase F (den. PNGF) and run as a positive control for PNGase F‐mediated glycan removal. B, representative traces of currents evoked from untreated (control) or live PNGase F‐incubated (PNGF) HEK293T/17 cells expressing myc‐GluK2a. Grey bar indicates glutamate (10 mm) application. C, quantification of whole‐cell glutamate‐evoked desensitization and amplitude from untreated and PNGase F‐incubated cells expressing myc‐GluK2a. D, cartoon depiction of the mutated glycosylation sites in a GluK2ΔNG5,6,7 subunit. E, western blot showing GluK2ΔNG5,6,7 MW after PNGase F treatment, as in (A). F, representative current traces from untreated control, swainsonine‐treated and kifunensine‐treated GluK2ΔNG5,6,7‐expressing HEK293T/17 cells. Grey bar indicates glutamate (10 mm) application. Amplitudes are scaled. G, quantification of glutamate‐evoked desensitization and amplitude from untreated and treated GluK2ΔNG5,6,7‐expressing HEK293T/17 cells. H, representative current traces in two‐pulse glutamate (10 mm) recovery experiments recorded from untreated and kifunesine‐treated GluK2ΔNG5,6,7‐expressing HEK293T/17 cells. Intervals between glutamate exposures range from 50 ms to 2 s in the traces shown. Amplitudes from the first glutamate application are scaled. I, quantification of mean glutamate recovery for GluK2ΔNG5,6,7‐expressing cells with and without kifunensine treatment. Amplitude of the second glutamate application in a two‐pulse experiment is reported as a normalized percentage of the first glutamate application. Results were fitted with a single component exponential equation and the best‐fit τrec values differ significantly (P < 0.0001). con, control; sw, swainsonine treatment; kif, kifunensine treatment. Error bars indicate the SEM. Statistical significance is indicated: *** P < 0.001. [Color figure can be viewed at wileyonlinelibrary.com]
In the GluK2a subunit, plant and vertebrate lectins that allosterically modulate receptor function predominantly bind to oligosaccharides conjugated to one or more of three asparagines at the interface between the amino‐terminal domain (ATD) and LBD (glycan sites 5, 6 and 7) (Fig. 2 D) (Fay & Bowie, 2006; Copits et al. 2014). We hypothesized that these oligosaccharide chains also acted as the key mediators of the glycan‐dependent functional changes observed in the preceding experiments. To test this idea, we analysed the biochemical and functional properties of a mutant receptor, GluK2∆NG5,6,7, in which these N‐glycosylation consensus sites were eliminated by mutating requisite serine or threonine residues to alanines, thus preventing glycan transfer to the asparagine (for positions of mutations, see Materials and methods). Biochemical analysis of the mutant protein demonstrated that ∼10 kDa of oligosaccharide was conjugated to the mutant receptor, as measured from the difference in MW between the untreated and PNGase F digested GluK2∆NG5,6,7 protein (96 ± 0.9 kDa vs. 85 ± 0.9 kDa, respectively) (Fig. 2 E, lanes 1 and 3). This is significantly less than the ∼40 kDa of oligosaccharide added to wild‐type GluK2 (137 ± 2.0 kDa for untreated vs. 98 ± 0.7 kDa PNGase‐treated denatured GluK2a) (Fig. 2 A). Thus, > 70% of the sugar content on GluK2a protein is conjugated to at most three of nine potential glycosylation sites, and oligosaccharides attached to the remaining sites also exhibited almost complete resistance to PNGase F‐mediated cleavage under non‐denaturing conditions.
Homomeric GluK2∆NG5,6,7 receptors desensitized more slowly than wild‐type myc‐GluK2a receptors on average (unpaired t test, P = 0.0056). Additionally, mutant receptors desensitized with the same time course regardless of enzyme inhibitor treatment (Fig. 2 F), in contrast to wild‐type myc‐GluK2a receptors (untreated: τdes = 5.4 ± 0.7 ms, n = 15; swainsonine: τdes = 4.9 ± 0.4 ms, n = 19; kifunensine τdes = 5.5 ± 0.5 ms, n = 16; P = 0.66) (Fig. 2 G). Restricting glycan processing did not alter peak current amplitudes from GluK2∆NG5,6,7 receptors (Fig. 2 G). These data suggest that glycans conjugated to one or more of these discrete sites between the ATD and LBD influence GluK2a KAR receptor gating. Surprisingly, kifunensine treatment significantly slowed recovery from glutamate‐evoked desensitization of GluK2∆NG5,6,7 receptors despite its lack of an effect on the rate of entry into desensitization (Fig. 2 H). Untreated GluK2∆NG5,6,7 receptor currents recovered from desensitization with a τ of 0.9 s (95% CI = 0.7–1.0; n = 3), which was faster than untreated wild‐type receptors (parameters of best fit lines differ P < 0.0001). Treatment with kifunensine slowed recovery of these mutant receptors to a τ of 2.4 s (95% CI = 1.9–3.0; n = 6) (parameters of best fit lines differ P < 0.0001) (Fig. 2 I), which was similar to the rate at which wild‐type kifunensine‐treated receptors recovered from desensitization. These data suggest that the glycans with the greatest impact on macroscopic desensitization and those that most strongly affect recovery from desensitization are located at non‐overlapping or only partially overlapping sites along the GluK2a receptor subunit.
We next tested whether oligosaccharide composition influenced desensitization rates of other KARs. Homomeric GluK1‐2a KARs (Fig. 3 A) expressed in untreated cells desensitized with a mean weighted τdes of 13.4 ± 1.9 ms (n = 19), which was significantly slower than the 8.3 ± 0.9 ms τdes measured from swainsonine‐treated cells (n = 15, P < 0.05 vs. control) and 8.2 ± 1.1 ms from kifunensine‐treated cells (n = 18, P < 0.05 vs. control) (P = 0.018) (Fig. 3 B), showing that restricted glycan processing similarly affects homomeric GluK1‐2a and GluK2a‐containing receptors. However, inhibition of α‐mannosidase II speeded up GluK1‐2a KAR desensitization but did not affect GluK2a KARs, which might be a result of GluK1‐2a containing one fewer N‐glycosylation site or differential interactions between glycans and distinct receptors. Neither swainsonine, nor kifunensine treatment altered mean peak current amplitudes (P = 0.72) (Fig. 3 B). By contrast, heteromeric GluK2a/GluK5 KARs were not measurably affected by restricted glycan processing (Fig. 3 C). Glutamate‐evoked currents from control cells desensitized at a rate of 1.8 ± 0.1 ms (n = 17) and this was not significantly altered by either swainsonine (2.1 ± 0.2 ms, n = 17) or by kifunensine (2.4 ± 0.1 ms, n = 17) (P = 0.26) (Fig. 3 D). We note, however, that limitations in the speed of solution exchange in whole‐cell recording makes it possible that we were unable to resolve small effects of the enzyme inhibitors on these particular receptors, which desensitize more rapidly than others that we examined. Peak current amplitudes for GluK2a/K5‐containing receptors were also unaltered by restricted glycan processing (P = 0.92) (Fig. 3 D).
Figure 3. α‐mannosidase inhibition speeds up the desensitization of other KAR combinations.
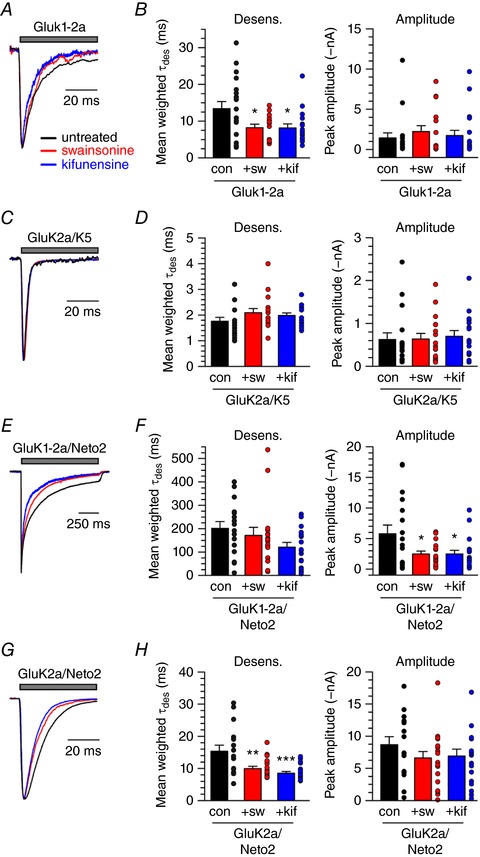
A, representative current traces from untreated control, swainsonine‐treated and kifunensine‐treated HEK293T/17 cells expressing myc‐GluK1‐2a. Grey bar indicates glutamate (10 mm) application. Amplitudes are scaled. B, quantification of glutamate‐evoked desensitization and amplitude from untreated and treated HEK293T/17 cells expressing myc‐GluK1‐2a. C and D, as in (A) and (B). Cells express myc‐GluK2a and myc‐GluK5. E and F, as in (A) and (B). Cells express myc‐GluK1‐2a and Neto2. G and H, as in (A) and (B). Cells express myc‐GluK2a and Neto2. Error bars indicate the SEM. con, control; sw, swainsonine treatment; kif, kifunensine treatment. Statistical significance is indicated: * P < 0.05, ** P < 0.01, *** P < 0.001. [Color figure can be viewed at wileyonlinelibrary.com]
We next tested whether glycan composition affected functional properties of KARs containing auxiliary subunits and again observed differential effects in GluK1 and GluK2 containing receptors. We found that assembly of GluK1‐2a with its auxiliary subunit Neto2 eliminated any functional effect of restricting oligosaccharide processing (Fig. 3 E). GluK1‐2a/Neto2 desensitization was highly variable, as expected, and currents decayed with a τdes of 202 ± 29 ms (n = 16) in control cells. Swainsonine and kifunensine treatment resulted in τdes of 172 ± 35 ms (n = 16) and 121 ± 21 ms (n = 18), respectively, which were not different from untreated cells (P = 0.13) (Fig. 3 F). Current amplitudes were slightly smaller following α‐mannosidase inhibition (control: 5.8 ± 1.4 nA; swainsonine: 2.5 ± 0.5 nA; kifunensine: 2.5 ± 0.6 nA; P = 0.020) (Fig. 3 F). By contrast, glycan processing did affect the desensitization rate of GluK2a‐containing receptors that were co‐assembled with Neto2 (Fig. 3 G). Currents mediated by GluK2a/Neto2 KARs desensitized with a τdes of 15.3 ± 1.9 ms in untreated cells (n = 16). Desensitization was faster for currents evoked from cells treated with both swainsonine (10.0 ± 0.7 ms n = 19) and kifunensine (8.5 ± 0.6 ms, n = 19; P = 0.0003) (Fig. 3 H). The amplitude of currents from GluK2a/Neto2 KARs were not different in untreated cells compared to α‐mannosidase inhibition (P = 0.38; Fig. 3 H).
Functional consequences of incorporating the negatively‐charged glycan HNK‐1
The sulphated trisaccharide HNK‐1 is incorporated into select, high MW central nervous system (CNS) glycoproteins (Schwarting et al. 1987), as well neuronal GluA2 AMPA receptor subunits, where it increases receptor stability in the plasma membrane (Morita et al. 2009). It contributes a negative charge to the distal end of oligosaccharide antennae and could, in principle, interact with determinants in receptor proteins to modulate function. We tested whether this trisaccharide was incorporated into KARs and, if so, how HNK‐1 might shape receptor gating in recordings from recombinant KARs in HEK293T/17 cells co‐expressing the enzymes necessary for generation of the HNK‐1 epitope found in the brain, glucuronyltransferase‐P (GlcAT‐P) and the HNK‐1 sulphotransferase (HNK‐1ST).
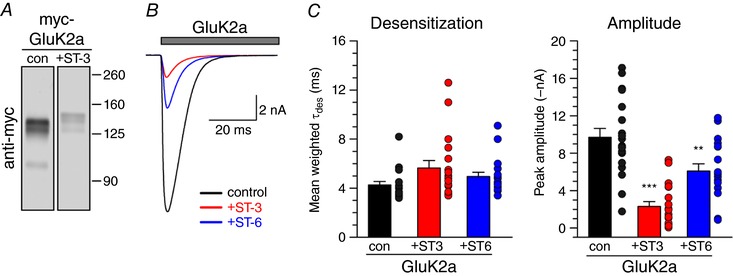
The enzymes were expressed together with myc‐GluK1‐2a, myc‐GluK2a, GluK3 or myc‐GluK5, and the receptor proteins were immunoprecipitated with either anti‐myc or anti‐GluK2/3 antibody before immunoblotting with an anti‐HNK‐1 antibody (Fig. 4 A). We found that myc‐GluK2a and GluK3a subunits exhibited HNK‐1 immunoreactivity, whereas myc‐GluK1‐2a and myc‐GluK5 subunits did not. In Fig. 4 A, a representative western blot from these experiments shows the anti‐HNK‐1 staining (Fig. 4 A, top), as well control staining following re‐probing the membranes for anti‐myc (Fig. 4 A, centre) and anti‐GluK2/3 (Fig. 4 A, bottom). HNK‐1 conjugation of KARs occurs in vivo as well; we observed immunoreactivity for the trisaccharide on neuronal GluK2/3 subunits immunoprecipitated with anti‐GluK2/3 antibody from the hippocampus dissected from wild‐type mice but not from tissue lacking all five KAR subunits (KAR−/−) (Fig. 4 B) or lacking the HNK‐1ST (HNK−/−) (Fig. 4 C). This is the first detection of the HNK‐1 epitope on native KARs and it highlights the diversity of oligosaccharides on these receptors. Moreover, GluK2/3 KARs represent one of relatively few CNS proteins carrying this glycan epitope that are not components of the extracellular matrix.
Figure 4. KAR subunits are substrates for HNK‐1 conjugation.
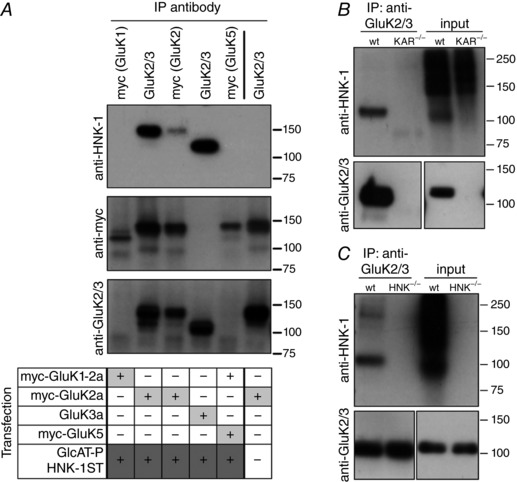
A, immunoprecipitation of the HNK‐1 epitope with KAR subunits. Recombinant KAR subunits were co‐expressed in HEK293T/17 cells with or without GlcAT‐P and HNK‐1ST, as denoted by the table of transfection combinations. KAR subunits were immunoprecipitated from cell lysates with an antibody against myc or against the GluK2 and GluK3 subunits. HNK‐1 was detected by immunoblotting for the HNK‐1 epitope; enrichment of KAR subunits was confirmed by immunoblotting for myc or GluK2/3. B, western blot showing that KAR subunits in the hippocampus contain the HNK‐1 epitope. KARs were precipitated from hippocampal homogenates using an antibody against GluK2 and GluK3. HNK‐1 is detected by immunoblotting in immunoprecipitates from wild‐type mice but not from mice lacking all five KAR subunits (KAR−/−). Input lanes (10 μg) show that HNK‐1 is present on other proteins in KAR−/− tissue. Membranes were stripped and re‐probed for GluK2/3 to confirm KAR subunit enrichment. C, HNK‐1 was detected on hippocampal KAR subunits following immunoprecipitation with anti‐GluK2/3 from wild‐type mice but not from mice lacking the HNK‐1ST (HNK−/−). Membranes were stripped and re‐probed for GluK2/3 to confirm KAR subunit enrichment.
Because HNK‐1 modification of KARs occurs both in vitro and in vivo, we tested how this conjugation might alter the functional properties of myc‐GluK2a KARs (Fig. 5 A). Co‐expression of GluK2a with HNK‐1 synthesizing enzymes resulted in glutamate‐evoked currents that desensitized with a more than two‐fold slower time course, from a mean τdes of 3.5 ± 0.3 ms (n = 9) in control recordings to 7.7 ± 0.6 ms (n = 19) with HNK‐1 (P = 0.0001) (Fig. 5 B). Mean peak amplitudes were unaffected. HNK‐1 conjugation slowed deactivation of the receptors in outside‐out patch recordings from a control τ of 1.6 ± 0.03 ms (n = 4) to 2.7 ± 0.3 ms (n = 5; P = 0.0094) (Fig. 5 C and D). Finally, recovery from desensitization was more rapid (control: 4.0 s, 95% CI = 3.6–4.6, n = 4; HNK‐1: 1.0 s, 95% CI = 0.9–1.1, n = 7; P < 0.0001) (Fig. 5 E and F).
Figure 5. HNK‐1 conjugation alters desensitization and deactivation of GluK2a‐containing receptors.
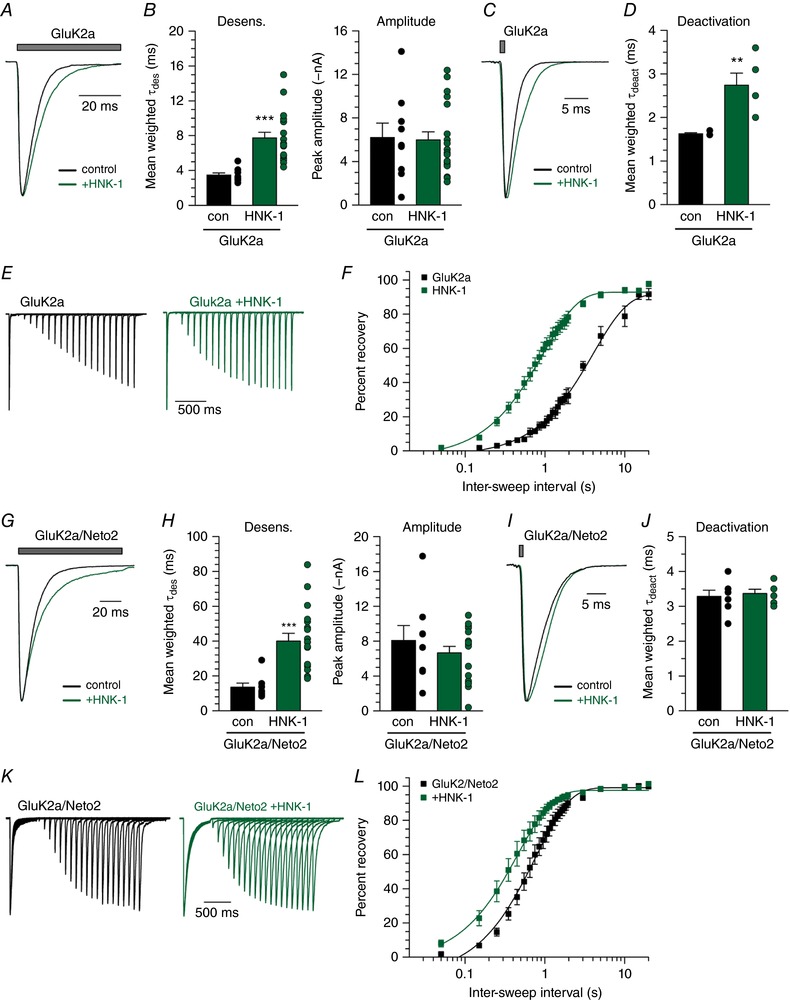
A, representative current traces from control HEK293T/17 cells expressing only myc‐GluK2a and from cells where myc‐GluK2a was co‐expressed with the HNK‐1‐conjugating transferases GlcAT‐P and HNK‐1ST. Grey bar indicates glutamate (10 mm) application. Amplitudes are scaled. B, quantification of glutamate‐evoked desensitization and amplitude from myc‐GluK2a alone and HNK‐1 co‐expressing HEK293T/17 cells. C, representative GluK2a deactivation current traces with and without HNK‐1. Grey bar indicates glutamate (10 mm) application. Amplitudes are scaled. D, quantification of glutamate‐evoked deactivation from myc‐GluK2a‐containing outside‐out patches pulled from control and HNK‐1 co‐transfected HEK293T/17 cells. E, representative current traces in two‐pulse glutamate (10 mm) recovery experiments recorded from myc‐GluK2a expressing HEK293T/17 cells transfected either alone or with HNK‐1‐conjugating transferases. Intervals between glutamate exposures range from 50 ms to 2 s in the traces shown. Amplitudes from the first glutamate application are scaled. F, quantification of mean glutamate recovery for myc‐GluK2a‐expressing cells with and without HNK‐1 conjugation. Amplitude of the second glutamate application in a two‐pulse experiment is reported as a normalized percentage of the first glutamate application. Results were fitted with a single component exponential equation and the best‐fit τrec values differ significantly (P < 0.0001). G, representative current traces from control HEK293T/17 cells expressing myc‐GluK2a and Neto2 alone or with HNK‐1 transferases. Grey bar indicates glutamate (10 mm) application. Amplitudes are scaled. H, quantification of glutamate‐evoked desensitization and amplitude from myc‐GluK2a/Neto2 alone and HNK‐1 co‐expressing HEK293T/17 cells. I, representative GluK2a/Neto2 deactivation current traces with and without HNK‐1. Grey bar indicates glutamate (10 mm) application. Amplitudes are scaled. J, quantification of glutamate‐evoked deactivation from myc‐GluK2a/Neto2‐containing outside‐out patches pulled from control and HNK‐1 co‐transfected HEK293T/17 cells. K, representative current traces in two‐pulse glutamate (10 mm) recovery experiments recorded from myc‐GluK2a/Neto2 expressing HEK293T/17 cells transfected either alone or with HNK‐1‐conjugating transferases. As in (E). L, quantification of mean glutamate recovery for myc‐GluK2a/Neto2‐expressing cells with and without HNK‐1 conjugation, as in (F). Results were fitted with a single component exponential equation and the best‐fit τrec values differ significantly (P < 0.0001). con, control transfection of receptor alone; HNK‐1, co‐transfection with GlucAT‐P and HNK‐1ST. Error bars indicate the SEM. Statistical significance is indicated: ** P < 0.01, *** P < 0.001. [Color figure can be viewed at wileyonlinelibrary.com]
Functional properties of Neto2‐containing KARs also were altered by HNK‐1 incorporation. The desensitization rate of myc‐GluK2a/Neto2 KARs was three‐fold slower in cells co‐transfected with the HNK‐1 transferases, increasing the τdes from 13.5 ± 2.4 ms (n = 8) to 40.0 ± 4.5 ms (n = 18, P = 0.0009) in the presence of HNK‐1 (Fig. 5 G and H). GluK2a peak current amplitudes with Neto2 were not affected in HNK‐1‐expressing cells (P = 0.37) (Fig. 5 H). HNK‐1 conjugation did not change GluK2a/Neto2 deactivation from 3.3 ± 0.2 (n = 7) to 3.4 ± 0.1 ms (n = 6; P = 0.72) (Fig. 5 I and J), although it speeded up GluK2a/Neto2 recovery from desensitization (control: 0.8 s, 95% CI = 0.7–0.9, n = 6; HNK‐1: 0.5 s, 95% CI = 0.4–0.5, n = 6; P < 0.0001) (Fig. 5 K and L). These data therefore demonstrate that the sulphated HNK‐1 oligosaccharide modifies a number of receptor functional parameters, including deactivation.
We tested whether HNK‐1 modulation of receptor gating was dependent on the trio of glycan conjugation sites in the linker between the ATD and LBD that are eliminated in the GluK2∆NG5,6,7 receptor mutant (Fig. 6 A). The mean weighted tau of desensitization measured from currents evoked from cells expressing GluK2∆NG5,6,7 alone was 6.9 ± 1.1 ms (n = 7). Surprisingly, HNK‐1 conjugation caused glutamate‐evoked currents to desensitize faster (4.0 ± 0.2 ms, n = 12; P = 0.0034) (Fig. 6 B) and to recover from desensitization slower (control τrec of 1.2 s, 95% CI = 1.0–1.5, n = 5; HNK‐1 τrec of 2.3 s, 95% CI = 2.0–2.8, n = 6; P < 0.0001) (Fig. 6 C and D), which is opposite the effect that HNK‐1 addition had on wild‐type receptors. Indeed, HNK‐1 modification of GluK2∆NG5,6,7 ‘normalized’ desensitization properties to time courses that were indistinguishable from wild‐type receptors lacking HNK‐1 (unpaired t test P = 0.14). We conclude from these data that HNK‐1 modification of GluK2a subunits can occur at two or more sites of glycosylation to effect opposing changes on receptor kinetics.
Figure 6. HNK‐1 conjugation differentially modulates desensitization of KARs lacking glycan attachment sites.
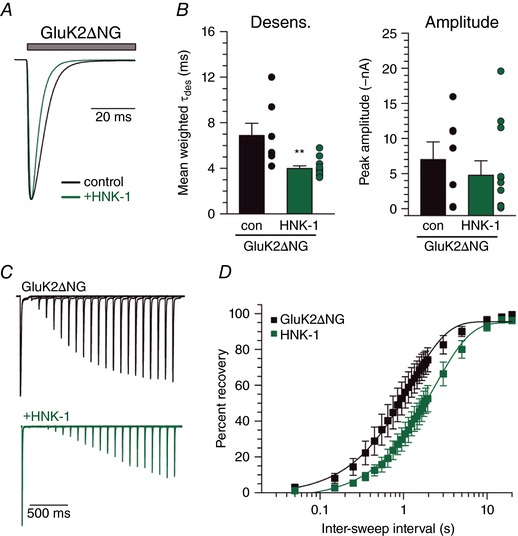
A, representative current traces from HEK293T/17 cells expressing GluK2ΔNG5,6,7 alone or with HNK‐1 transferases. Grey bar indicates glutamate (10 mm) application. Amplitudes are scaled. B, quantification of glutamate‐evoked desensitization and amplitude from GluK2ΔNG5,6,7 alone and HNK‐1 co‐expressing HEK293T/17 cells. C, representative current traces in two‐pulse glutamate (10 mm) recovery experiments recorded from GluK2ΔNG5,6,7 expressing HEK293T/17 cells. Intervals between glutamate exposures range from 50 ms to 2 s in the traces shown. Amplitudes from the first glutamate application are scaled. D, quantification of mean glutamate recovery for GluK2ΔNG5,6,7‐expressing cells with and without HNK‐1 transferase expression. Amplitude of the second glutamate application in a two‐pulse experiment is reported as a normalized percentage of the first glutamate application. Results were fitted with a single component exponential equation and the best‐fit τrec values differ significantly (P < 0.0001). con, control transfection of receptor alone; HNK‐1, co‐transfection with GlucAT‐P and HNK‐1ST. Error bars indicate the SEM. Statistical significance is indicated: ** P < 0.01, *** P < 0.001. [Color figure can be viewed at wileyonlinelibrary.com]
The marked effect that HNK‐1 conjugation had on GluK2a currents prompted us to test whether HNK‐1 had equivalent modulatory actions on receptors composed of another KAR subunit, GluK3a. These receptors exhibit a very low sensitivity to glutamate (Schiffer et al. 1997), which arises in part because partially occupied receptors desensitize rapidly (Perrais et al. 2009). We hypothesized that, if HNK‐1 addition to GluK3a slowed desensitization of currents evoked from KARs containing this subunit, as is the case for homomeric GluK2a KARs, we should observe an increase in peak current amplitudes in addition to the change in desensitization kinetics. To test this, we evoked whole‐cell currents with 30 mm glutamate from cells expressing GluK3a with and without HNK‐1‐generating transferases (Fig. 7 A). Similar to GluK2a KARs, the desensitization rate of GluK3a‐containing receptors was slowed, from a τdes of 6.0 ± 0.2 ms in control cells (n = 23) to 9.7 ± 0.3 ms with the addition of HNK‐1 (n = 20, P < 0.0001) (Fig. 7 B). Moreover, mean peak current amplitudes increased by three‐fold at this concentration of glutamate, from a control of 1.2 ± 0.1 nA to 3.9 ± 0.5 nA in cells expressing GluK3a with HNK‐1 (P < 0.0001) (Fig. 7 B). HNK‐1 made the rate of recovery of GluK3 KARs around three‐fold faster (control: τrec of 1.4 s, 95% CI = 1.2–1.5, n = 4; HNK‐1: τrec of 0.6 s, 95% CI = 0.5–0.7, n = 4; P < 0.0001) (Fig. 7 C and D). If the increase in amplitude we observed was a result of slowed desensitization of partially occupied receptors, we would also expect to observe a leftward shift in the apparent glutamate affinity when HNK‐1 was conjugated to receptors. Indeed, concentration–response relationships revealed that HNK‐1 conjugation shifted the macroscopic EC50 of GluK3a KARs from a control of 12.3mm (95% CI = 7.1–21.4, d.f. = 21) to 5.6mm (95% CI = 3.5–9.0, d.f. = 17; P = 0.0001) (Fig. 7 E and F). Finally, we tested whether HNK‐1 addition slowed deactivation of homomeric GluK3a KARs (Fig. 7 G). A 1 ms application of 30mm glutamate evoked currents with mean weighted τdes of 0.7 ± 0.04 ms (n = 5), which did not detectably change with HNK‐1 conjugation (τdes 0.8 ± 0.09 ms, n = 6; P = 0.31) (Fig. 7 H). These data show that HNK‐1 also modifies GluK3a KARs in similar ways as GluK2a receptors, with the exception that deactivation did not appear to be slowed. It is possible, however, that the extremely rapid deactivation of GluK3a receptors might have precluded detection of a modest slowing of this aspect of channel function.
Figure 7. HNK‐1 conjugation alters desensitization and apparent glutamate affinity of GluK3a‐containing KARs.
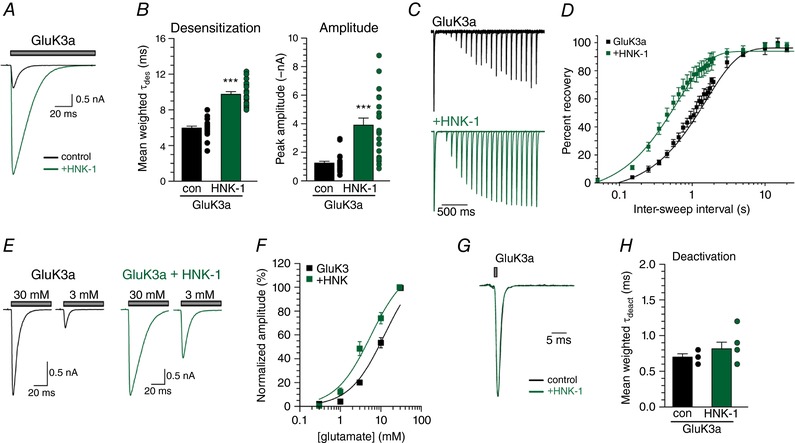
A, representative current traces from control HEK293T/17 cells expressing GluK3a alone or with the HNK‐1 transferases GlcAT‐P and HNK‐1ST. Grey bar indicates glutamate (30 mm) application. Amplitudes are scaled. B, quantification of glutamate‐evoked desensitization and amplitude from GluK3a alone and HNK‐1 co‐expressing HEK293T/17 cells. C, representative current traces in two‐pulse glutamate (30 mm) recovery experiments recorded from GluK3a‐expressing HEK293T/17 cells. Intervals between glutamate exposures range from 50 ms to 2 s in the traces shown. Amplitudes from the first glutamate application are scaled. D, quantification of mean glutamate recovery for GluK3a‐expressing cells with and without HNK‐1 conjugation. Amplitude of the second glutamate application in a two‐pulse experiment is reported as a normalized percentage of the first glutamate application. Results were fitted with a single component exponential equation, and the best‐fit τrec values differ significantly (P < 0.0001). E, representative traces of currents evoked with 3 mm or 30 mm glutamate application from HEK293T/17 cells expressing GluK3a, either alone or with HNK‐1 transferases. F, current were evoked from GluK3a‐expressing HEK293T/17 cells with 0.3 mm, 1mm, 3 mm and 10 mm glutamate, and were normalized to the amplitude of currents evoked with 30 mm glutamate. These measurements were log‐transformed, the log‐transformation was fit with a single component exponential, and the best‐fit EC50 values differ significantly (P = 0.0001). G, representative GluK3a deactivation current traces. Grey bar indicates glutamate (30mm) application. Amplitudes are scaled. H, quantification of glutamate‐evoked deactivation from GluK3a‐containing outside‐out patches pulled from control and HNK‐1 expressing HEK293T/17 cells. con, control transfection of receptor alone; HNK‐1, co‐transfection with GlucAT‐P and HNK‐1ST. Error bars indicate the SEM. Statistical significance is indicated: *** P < 0.001. [Color figure can be viewed at wileyonlinelibrary.com]
The effects of HNK‐1 do not generalize to all negatively‐charged glycans
To determine whether another negatively‐charged glycan common in the CNS (Zamze et al. 1998), sialic acid, had similar effects on KAR function, we co‐expressed GFP‐tagged sialyltransferases with myc‐GluK2a receptors. Co‐expression of myc‐GluK2a with α‐2,3‐sialyltransferase (ST‐3) increased the MW of the protein by 7 ± 1 kDa (one‐sample t test P = 0.0099, n = 3) at the same time as clearly reducing the total protein expression (Fig. 8 A). The basis for the observed reduction in equilibrium protein expression is unclear but could include enhanced degradation or slowed biogenesis induced by the addition of sialic acid to GluK2a‐containing receptors. Neither ST‐3, nor α‐2,6‐sialyltransferase (ST‐6) altered desensitization of glutamate‐evoked currents (Fig. 8 B and C, P = 0.096). In agreement with reduced protein expression shown in Fig. 8 A, peak current amplitudes were lower from ST‐3 expressing cells (2.3 ± 0.5 nA, n = 18) and ST‐6 expressing cells (6.1 ± 0.8 nA, n = 18) compared to control recordings (9.7 ± 1.0 nA, n = 18) (P < 0.0001) (Fig. 8 C). These data show that the effect of HNK‐1 on slowing GluK2a KAR currents is not replicated by adding a different charged capping sugar, suggesting that there is specificity in the contribution that HNK‐1 makes to receptor function.
Figure 8. Sialyltransferase overexpression increases GluK2 molecular weight but does not alter desensitization.
A, western blot showing MW of myc‐GluK2a with and without sialyltransferase overexpression. Myc‐GluK2a was expressed alone or with ST‐3 in HEK293T/17 cells, myc‐GluK2a was detected by immunoblotting with an anti‐myc antibody. B, representative current traces from control HEK293T/17 cells expressing only myc‐GluK2a (con) and from cells where myc‐GluK2a was co‐expressed with either ST‐3 or ST‐6. Grey bar indicates glutamate (10 mm) application. Amplitudes are scaled. C, quantification of glutamate‐evoked current desensitization and amplitude from myc‐GluK2a alone and ST‐3 or ST‐6 co‐expressing HEK293T/17 cells. Error bars indicate the SEM. Statistical significance is indicated: ** P < 0.01, *** P < 0.001. [Color figure can be viewed at wileyonlinelibrary.com]
Discussion
The data obtained in the present study provide the first evidence that the functional properties of KARs depend on the identity and complexity of the N‐glycosylation moieties attached to these receptors, and they suggest a nuanced relationship between glycan identity, receptor composition and receptor function. This finding suggests that differential expression of oligosaccharide‐processing machinery in distinct cell types, brain regions or at distinct developmental stages could fine‐tune KAR function, and represents another layer of complexity in the molecular control of these receptors.
Indirect evidence has suggested previously that oligosaccharide content might modulate iGluR function. For example, exogenously applied polysialic acid (PSA) increased the open probability (P open) of AMPA receptors (AMPARs) purified from rat brain and reconstituted in lipid bilayers, raising the possibility that sialylated glycans in the extracellular matrix could similarly alter P open. Furthermore, PSA application increased the current density through AMPARs from young but not adult animals (Vaithianathan et al. 2004), which might arise from developmental differences in iGluR subunit composition. AMPAR ligand affinity is also altered by Golgi processing; as receptors move through the secretory system, [3H]AMPA binding affinity in hippocampal sections is reduced, suggesting that increased oligosaccharide complexity reduces the AMPA affinity of the receptor (Standley et al. 1998). However, for AMPARs and KARs expressed in oocytes, tunicamycin treatment did not alter the EC50 of either glutamate or kainate, and only mutation of NG8 changed GluK2 EC50 (Everts et al. 1997; Everts et al. 1999). Precluding N‐glycosylation with tunicamycin treatment or consensus site mutations increased KAR current amplitudes in Xenopus oocytes, whereas, in mammalian cells, desensitization was not different between receptors lacking individual consensus glycosylation sites (Everts et al. 1997; Everts et al. 1999). We found that mutation of three key consensus sites did alter GluK2a desensitization and recovery, and that promoting HNK‐1 conjugation to the remaining glycans normalized these properties. These observations show that the identity and location of glycans along KARs are critical components of their contribution to receptor function.
We do not yet know how glycans produce the functional changes that we observed, but, given the range of effects, it is most probable that the glycans are interacting with one or more determinants in receptor ligand‐binding domains to alter the stability of different gating conformations. The attachment locations, relatively large size and diversity of chemical constituents of N‐linked oligosaccharide chains allow the possibility that they could mediate intra‐domain, inter‐domain and potentially even intra‐subunit interactions.
In silico modelling supports an intra‐domain interaction between core oligosaccharides attached to the NMDA receptor (NMDAR) subunit GluN1 and elements of the GluN1 LBD. The N‐glycosylation site at N440 is located in the upper lobe of the GluN1 LBD and was predicted to stabilize a closed LBD conformation (Sinitskiy et al. 2017) and mutation of this consensus site for glycosylation reduced NMDAR glycine affinity in functional studies. GluN1‐N440 is analogous to the GluK2‐N430 glycosylation site, which is one of three sites mutated in GluK2ΔNG. Sinitskiy et al. (2017) predicted that polar interactions between the mannose constituents of immature glycans and a hydrophilic region of the lower LBD lobe occur only when the LBD is in a closed conformation, and these interactions reciprocally increase the likelihood of a closed LBD state. Almost all hydroxyl groups on the glycan mannose constituents interacted with the target residues on GluN1 in this model, with oligosaccharide flexibility highlighted by the lack of a single preferred structure and binding mode. This region of the LBD is partially conserved in GluK2 and similar interactions probably occur in KARs because we found that glycan chains at GluK2‐N430 and adjacent sites were required for kifunensine treatment to speed desensitization and for HNK‐1 conjugation to slow desensitization. The negative charges in HNK‐1 conferred by the sulphate and glucuronic acid constituents potentially contribute distinct interactions with receptor subunits. For example, in the GluN1 model, the oligomannosidic chain at GluN1‐N440 primarily interacts with the negatively‐charged E712, E716 and D723, as well as Q719, which project away from the lip of the ligand‐binding pocket along helix H in D2. In GluK2, the analogous amino acids K719, E723, T730 and Q726, respectively, present a less negative surface and thus represent potential sites of interaction for the HNK‐1 glycan. Moreover, the core oligosaccharide modelled in the study by Sinitskiy et al. (2017), Man5GlcNAc2, was both uncharged and predicted to be significantly smaller in mass relative to HNK‐1‐containing complex oligosaccharides, and thus HNK‐1 could possibly interact with a variety of different interaction partners on the LBD.
The relatively large structure of complex oligosaccharides suggests that conjugation at key sites instead could mediate inter‐domain interactions that alter the stability of the desensitized state of the receptors. N‐glycosylation sites 3, 5, 6 and 7 in GluK2 are positioned around a ‘sandwich’ interface between the ATD and LBD of the A/C subunits of desensitized KARs (Meyerson et al. 2016). Furthermore, a glycan at GluK2‐N430 (NG7), and potentially at adjacent sites, would be positioned to mediate cross‐subunit interactions near the D1 LBD dimer interface, which critically influences KAR desensitization (Weston et al. 2006; Wong et al. 2006; Nayeem et al. 2009). The opposite changes in desensitization kinetics that we observed with HNK‐1 conjugation to wild‐type and GluK2ΔNG KARs suggest that HNK‐1–receptor interactions in distinct functional domains differentially influence receptor gating, and modelling studies such as those performed with NMDAR subunits would be a useful approach for understanding these interactions (Sinitskiy et al. 2017).
Differential glycosylation of iGluRs or their auxiliary proteins might also modify their protein–protein interactions. Plant lectins impact both AMPAR and KAR desensitization by binding to carbohydrate substrates (Everts et al. 1997), an effect that, for KARs, depends on the sugar affinity of the lectin (Thalhammer et al. 2002). Mammalian lectins also alter AMPAR and KAR desensitization in a manner that depends on both receptor subunit identity and their affinity for the sugars attached to receptors, and we showed that galectin‐1 slows desensitization of KAR currents evoked from sensory neurons (Copits et al. 2014). Additionally, GluA2 is a substrate for HNK‐1 in the hippocampus and this modification stabilizes GluA2‐containing receptors at the cell surface via HNK‐1‐mediated interactions with N‐cadherin during hippocampal long‐term potentiation (Morita et al. 2009). Hybrid and complex glycans are critical for surface retention of GluA2‐containing receptors and increasing the oligomannosidic content promotes receptor internalization (Hanus et al. 2016). Our observation that KAR subunits are substrates for HNK‐1 invites the question of how this trisaccharide modulates receptor function in the brain. Perhaps HNK‐1 alters the strength of KAR–protein interactions, such as the trans‐synaptic interaction with C1q proteins that determines KAR retention at mossy fibre–CA3 synapses (Matsuda et al. 2016; Straub et al. 2016). The structural pattern of oligosaccharides on native iGluRs and the functional implication of changes to glycan content are outstanding questions that are of critical importance for understanding receptor function in neuronal circuits.
Determining the glycan content of native iGluRs is no easy task, considering that each subunit has between four and 12 N‐glycosylation consensus sequences and each site could be differentially modified by the oligosaccharide‐processing enzymes of a given cell. AMPARs and NMDARs not only have high oligomannosidic content, but also are conjugated to hybrid and complex sugars, which probably contribute important elements to receptor function (Hanus et al. 2016; Kaniakova et al. 2016). A glycoproteomics scan in the rat brain confirmed GluK2, GluK3 and GluK5 to be N‐glycosylated at multiple sites, including GluK2 sites 5, 6 and 7 (Parker et al. 2013), which we found to be critical determinants of oligosaccharide modulation of receptor properties. These studies confirm that multiple oligosaccharide structures are present on native iGluRs and at functionally relevant locations, although the question of which sugars occupy what space along the receptor remains unanswered.
iGluRs are not the only receptor–channels whose function is affected by N‐glycosylation. Constitutive activation can be manipulated in TRPC3 and TRPC6 by the addition or removal of a glycosylation site (Dietrich et al. 2003). Temperature sensitivity of TRPM8 is altered when glycosylation is precluded, both in sensory neurons and recombinant systems (Pertusa et al. 2012). TRPV5 is stabilized on the membrane of kidney cells following glycan cleavage by the lectin klotho and the subsequent binding of galectin‐1 to the newly‐exposed sugars (Chang et al. 2005; Cha et al. 2008). For GABAA receptors, the ß2‐subunits require their two most C‐terminal glycosylation sites for proper assembly, and removal of any of their three consensus sequences reduces the channel P open (Lo et al. 2010). How these findings inform native TRPC, TRPM and GABAA receptor function remains to be clarified, although it is clear that the attached oligosaccharides can modulate diverse functional properties of ligand‐gated ion channels.
Glycans also contribute to voltage‐gated channel properties. Charged sialic acid moieties set the voltage dependence of Kv4.3 gating in cardiomyocytes (Ufret‐Vincenty et al. 2001b). Myocytes from mice with genetic ablation of muscle LIM (Lin11, Isl‐1 and Mec‐3 domain) protein exhibit depolarized sodium channel gating and slowed inactivation, which are properties that can be reproduced in wild‐type myocytes by desialylation with neuraminidase (Ufret‐Vincenty et al. 2001a). Additionally, developmental control of sialylation affects sodium channel properties in both cardiac and neural tissues. Sodium channel sialylation is initially low in ventricular myocytes and increases over development, whereas sialylation is high at all ages in atrial myocytes. As a result, sodium channel gating threshold differs regionally in neonatal but not adult myocytes (Stocker & Bennett, 2006). In sensory neurons, a transient post‐natal increase in Nav1.9 sialylation hyperpolarizes the voltage of inactivation (Tyrrell et al. 2001). These experiments highlight that glycosylation‐mediated differences in channel function occur between cell types and across development, and this is almost certain to be true for native iGluRs that might be affected by oligosaccharide composition. As discussed above, there is evidence that association or attachment to PSA differentially impacts AMPARs from neonatal and adult animals. The possibility that N‐linked glycans could be a cellular tool for developmental and cell‐type specific regulation of iGluR function is exciting; much work remains to be carried out both to reveal the regional and developmental patterns of glycosylation on iGluRs and to determine what precise functional properties are altered by different oligosaccharide fingerprints.
In the present study, we provide evidence that the oligosaccharides attached to KARs impact receptor functional properties beyond protein folding, forward trafficking and lectin binding. Our data suggest that oligosaccharides on KARs are important components of the structural changes that occur upon agonist binding. The study of KAR glycosylation in a reduced, recombinant system is an important step towards achieving a better understanding of the full spectrum of elements contributing to iGluR function in neurons. How these findings inform the function of endogenous receptors is an important and exciting open question.
Additional information
Competing interests
The authors declare that they have no competing financial interests.
Author contributions
CGV, BAC and GTS conceived and designed the work. CGV, BAC, JRS and YFG acquired, analysed and interpreted the data. CGV and GTS drafted the manuscript and all authors revised it critically. All authors approved the final version of the manuscript and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was supported by grant from the National Institute of Neurological Disorders and Stroke to G.T.S. (R01NS071952).
References
- Cha SK, Ortega B, Kurosu H, Rosenblatt KP, Kuro OM & Huang CL (2008). Removal of sialic acid involving klotho causes cell‐surface retention of TRPV5 channel via binding to galectin‐1. Proc Natl Acad Sci USA 105, 9805–9810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Q, Hoefs S, van der Kemp AW, Topala CN, Bindels RJ & Hoenderop JG (2005). The beta‐glucuronidase klotho hydrolyzes and activates the TRPV5 channel. Science 310, 490–493. [DOI] [PubMed] [Google Scholar]
- Copits BA, Vernon CG, Sakai R & Swanson GT (2014). Modulation of ionotropic glutamate receptor function by vertebrate galectins. J Physiol 592, 2079–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich A, Mederos Y Schnitzler M, Emmel J, Kalwa H, Hofmann T & Gudermann T (2003). N‐linked protein glycosylation is a major determinant for basal TRPC3 and TRPC6 channel activity. J Biol Chem 278, 47842–47852. [DOI] [PubMed] [Google Scholar]
- Everts I, Petroski R, Kizelsztein P, Teichberg VI, Heinemann SF & Hollmann M (1999). Lectin‐induced inhibition of desensitization of the kainate receptor GluR6 depends on the activation state and can be mediated by a single native or ectopic N‐linked carbohydrate side chain. J Neurosci 19, 916–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everts I, Villmann C & Hollmann M (1997). N‐Glycosylation is not a prerequisite for glutamate receptor function but Is essential for lectin modulation. Mol Pharmacol 52, 861–873. [DOI] [PubMed] [Google Scholar]
- Fay AM & Bowie D (2006). Concanavalin‐A reports agonist‐induced conformational changes in the intact GluR6 kainate receptor. J Physiol 572, 201–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundy D (2015). Principles and standards for reporting animal experiments in the journal of physiology and experimental physiology. J Physiol 593, 2547–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall RA, Kessler M & Lynch G (1992). Evidence that high‐ and low‐affinity DL‐alpha‐amino‐3‐hydroxy‐5‐methylisoxazole‐4‐propionic acid (AMPA) binding sites reflect membrane‐dependent states of a single receptor. J Neurochem 59, 1997–2004. [DOI] [PubMed] [Google Scholar]
- Hanus C, Geptin H, Tushev G, Garg S, Alvarez‐Castelao B, Sambandan S, Kochen L, Hafner AS, Langer JD & Schuman EM (2016). Unconventional secretory processing diversifies neuronal ion channel properties. eLife 5, e20609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaniakova M, Lichnerova K, Skrenkova K, Vyklicky L & Horak M (2016). Biochemical and electrophysiological characterization of N‐glycans on NMDA receptor subunits. J Neurochem 138, 546–556. [DOI] [PubMed] [Google Scholar]
- Lo WY, Lagrange AH, Hernandez CC, Harrison R, Dell A, Haslam SM, Sheehan JH & Macdonald RL (2010). Glycosylation of {beta}2 subunits regulates GABAA receptor biogenesis and channel gating. J Biol Chem 285, 31348–31361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda K, Budisantoso T, Mitakidis N, Sugaya Y, Miura E, Kakegawa W, Yamasaki M, Konno K, Uchigashima M, Abe M, Watanabe I, Kano M, Watanabe M, Sakimura K, Aricescu AR & Yuzaki M (2016). Transsynaptic modulation of kainate receptor functions by C1q‐like proteins. Neuron 90, 752–767. [DOI] [PubMed] [Google Scholar]
- Meyerson JR, Chittori S, Merk A, Rao P, Han TH, Serpe M, Mayer ML & Subramaniam S (2016). Structural basis of kainate subtype glutamate receptor desensitization. Nature 537, 567–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita I, Kakuda S, Takeuchi Y, Itoh S, Kawasaki N, Kizuka Y, Kawasaki T & Oka S (2009). HNK‐1 glyco‐epitope regulates the stability of the glutamate receptor subunit GluR2 on the neuronal cell surface. J Biol Chem 284, 30209–30217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayeem N, Zhang Y, Schweppe DK, Madden DR & Green T (2009). A nondesensitizing kainate receptor point mutant. Mol Pharmacol 76, 534–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker BL, Thaysen‐Andersen M, Solis N, Scott NE, Larsen MR, Graham ME, Packer NH & Cordwell SJ. (2013). Site‐specific glycan‐peptide analysis for determination of N‐glycoproteome heterogeneity. J Proteome Res 12, 5791–5800. [DOI] [PubMed] [Google Scholar]
- Pasternack A, Coleman SK, Fethiere J, Madden DR, LeCaer JP, Rossier J, Pasternack M & Keinanen K (2003). Characterization of the functional role of the N‐glycans in the AMPA receptor ligand‐binding domain. J Neurochem 84, 1184–1192. [DOI] [PubMed] [Google Scholar]
- Perrais D, Coussen F & Mulle C (2009). Atypical functional properties of GluK3‐containing kainate receptors. J Neurosci 29, 15499–15510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertusa M, Madrid R, Morenilla‐Palao C, Belmonte C & Viana F (2012). N‐glycosylation of TRPM8 ion channels modulates temperature sensitivity of cold thermoreceptor neurons. J Biol Chem 287, 18218–18229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche KW, Raymond LA, Blackstone C & Huganir RL (1994). Transmembrane topology of the glutamate receptor subunit GluR6. J Biol Chem 269, 11679–11682. [PubMed] [Google Scholar]
- Rogers SW, Hughes TE, Hollmann M, Gasic GP, Deneris ES & Heinemann S (1991). The characterization and localization of the glutamate receptor subunit GluR1 in the rat brain. J Neurosci 11, 2713–2724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiffer HH, Swanson GT & Heinemann SF (1997). Rat GluR7 and a carboxy‐terminal splice variant, GluR7b, are functional kainate receptor subunits with a low sensitivity to glutamate. Neuron 19, 1141–1146. [DOI] [PubMed] [Google Scholar]
- Schwarting GA, Jungalwala FB, Chou DK, Boyer AM & Yamamoto M (1987). Sulfated glucuronic acid‐containing glycoconjugates are temporally and spatially regulated antigens in the developing mammalian nervous system. Dev Biol 120, 65–76. [DOI] [PubMed] [Google Scholar]
- Sinitskiy AV, Stanley NH, Hackos DH, Hanson JE, Sellers BD & Pande VS (2017). Computationally Discovered Potentiating Role of Glycans on NMDA Receptors. Sci Rep 7:44578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Standley S, Tocco G, Wagle N & Baudry M (1998). High‐ and low‐affinity alpha‐[3H]amino‐3‐hydroxy‐5‐methylisoxazole‐4‐propionic acid ([3H]AMPA) bindin g sites represent immature and mature forms of AMPA receptors and are composed of differentially glycosylated subunits. J Neurochem 70, 2434–2445. [DOI] [PubMed] [Google Scholar]
- Stocker PJ & Bennett ES (2006). Differential sialylation modulates voltage‐gated Na+ channel gating throughout the developing myocardium. J Gen Physiol 127, 253–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub C, Noam Y, Nomura T, Yamasaki M, Yan D, Fernandes HB, Zhang P, Howe JR, Watanabe M, Contractor A & Tomita S (2016). Distinct subunit domains govern synaptic stability and specificity of the kainate receptor. Cell Reports 16, 531–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thalhammer A, Everts I & Hollmann M (2002). Inhibition by lectins of glutamate receptor desensitization is determined by the lectin's sugar specificity at kainate but not AMPA receptors. Mol Cell Neurosci 21, 521–533. [DOI] [PubMed] [Google Scholar]
- Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, Hansen KB, Yuan H, Myers SJ & Dingledine R (2010). Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev 62, 405–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyrrell L, Renganathan M, Dib‐Hajj SD & Waxman SG (2001). Glycosylation alters steady‐state inactivation of sodium channel Nav1.9/NaN in dorsal root ganglion neurons and is developmentally regulated. J Neurosci 21, 9629–9637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ufret‐Vincenty CA, Baro DJ, Lederer WJ, Rockman HA, Quinones LE & Santana LF (2001a). Role of sodium channel deglycosylation in the genesis of cardiac arrhythmias in heart failure. J Biol Chem 276, 28197–28203. [DOI] [PubMed] [Google Scholar]
- Ufret‐Vincenty CA, Baro DJ & Santana LF (2001b). Differential contribution of sialic acid to the function of repolarizing K(+) currents in ventricular myocytes. Am J Physiol Cell Physiol 281, C464–C474. [DOI] [PubMed] [Google Scholar]
- Vaithianathan T, Matthias K, Bahr B, Schachner M, Suppiramaniam V, Dityatev A & Steinhauser C (2004). Neural cell adhesion molecule‐associated polysialic acid potentiates alpha‐amino‐3‐hydroxy‐5‐methylisoxazole‐4‐propionic acid receptor currents. J Biol Chem 279, 47975–47984. [DOI] [PubMed] [Google Scholar]
- Vivithanaporn P, Lash LL, Marszalec W & Swanson GT (2007). Critical roles for the M3‐S2 transduction linker domain in kainate receptor assembly and postassembly trafficking. J Neurosci 27, 10423–10433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weston MC, Gertler C, Mayer ML & Rosenmund C (2006). Interdomain interactions in AMPA and kainate receptors regulate affinity for glutamate. J Neurosci 26, 7650–7658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong AY, Fay AM & Bowie D (2006). External ions are coactivators of kainate receptors. J Neurosci 26, 5750–5755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Marshall JJ, Fernandes HB, Nomura T, Copits BA, Procissi D, Mori S, Wang L, Zhu Y, Swanson GT & Contractor A (2017). Complete disruption of the kainate receptor gene family results in corticostriatal dysfunction in mice. Cell Reports 18, 1848–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto S, Oka S, Inoue M, Shimuta M, Manabe T, Takahashi H, Miyamoto M, Asano M, Sakagami J, Sudo K, Iwakura Y, Ono K & Kawasaki T (2002). Mice deficient in nervous system‐specific carbohydrate epitope HNK‐1 exhibit impaired synaptic plasticity and spatial learning. J Biol Chem 277, 27227–27231. [DOI] [PubMed] [Google Scholar]
- Zamze S, Harvey DJ, Chen YJ, Guile GR, Dwek RA & Wing DR (1998). Sialylated N‐glycans in adult rat brain tissue – a widespread distribution of disialylated antennae in complex and hybrid structures. Eur J Biochem 258, 243–270. [DOI] [PubMed] [Google Scholar]