

Abstract
Key points
The ascending brainstem transmitter acetylcholine depolarizes thalamocortical relay neurons while it induces hyperpolarization in local circuit inhibitory interneurons.
Sustained K+ currents are modulated in thalamic neurons to control their activity modes; for the interneurons the molecular nature of the underlying ion channels is as yet unknown.
Activation of TASK‐1 K+ channels results in hyperpolarization of interneurons and suppression of their action potential firing.
The modulation cascade involves a non‐receptor tyrosine kinase, c‐Src.
The present study identifies a novel pathway for the activation of TASK‐1 channels in CNS neurons that resembles cholinergic signalling and TASK‐1 current modulation during hypoxia in smooth muscle cells.
Abstract
The dorsal part of the lateral geniculate nucleus (dLGN) is the main thalamic site for state‐dependent transmission of visual information. Non‐retinal inputs from the ascending arousal system and inhibition provided by γ‐aminobutyric acid (GABA)ergic local circuit interneurons (INs) control neuronal activity within the dLGN. In particular, acetylcholine (ACh) depolarizes thalamocortical relay neurons by inhibiting two‐pore domain potassium (K2P) channels. Conversely, ACh also hyperpolarizes INs via an as‐yet‐unknown mechanism. By using whole cell patch‐clamp recordings in brain slices and appropriate pharmacological tools we here report that stimulation of type 2 muscarinic ACh receptors induces IN hyperpolarization by recruiting the G‐protein βγ subunit (Gβγ), class‐1A phosphatidylinositol‐4,5‐bisphosphate 3‐kinase, and cellular and sarcoma (c‐Src) tyrosine kinase, leading to activation of two‐pore domain weakly inwardly rectifying K+ channel (TWIK)‐related acid‐sensitive K+ (TASK)‐1 channels. The latter was confirmed by the use of TASK‐1‐deficient mice. Furthermore inhibition of phospholipase Cβ as well as an increase in the intracellular level of phosphatidylinositol‐3,4,5‐trisphosphate facilitated the muscarinic effect. Our results have uncovered a previously unknown role of c‐Src tyrosine kinase in regulating IN function in the brain and identified a novel mechanism by which TASK‐1 channels are activated in neurons.
Keywords: interneuron, K2P channels, muscarinic modulation, thalamus, tyrosine kinase
Key points
The ascending brainstem transmitter acetylcholine depolarizes thalamocortical relay neurons while it induces hyperpolarization in local circuit inhibitory interneurons.
Sustained K+ currents are modulated in thalamic neurons to control their activity modes; for the interneurons the molecular nature of the underlying ion channels is as yet unknown.
Activation of TASK‐1 K+ channels results in hyperpolarization of interneurons and suppression of their action potential firing.
The modulation cascade involves a non‐receptor tyrosine kinase, c‐Src.
The present study identifies a novel pathway for the activation of TASK‐1 channels in CNS neurons that resembles cholinergic signalling and TASK‐1 current modulation during hypoxia in smooth muscle cells.
Abbreviations
- ACh
acetylcholine
- AChR
acetylcholine receptor
- c‐Src
cellular and sarcoma
- DAG
diacylglycerol
- DAQ B1
demethylasterriquinone B1
- dLGN
dorsal lateral geniculate nucleus
- EGFP
enhanced green fluorescent protein
- GABA
γ‐aminobutyric acid
- Gαi/o
G‐protein α subunit of the subtype i/o
- Gαq
G‐protein α subunit of the subtype q
- Gβγ
G‐protein βγ subunit
- GAD67
glutamate decarboxylase 67
- GPCR
G‐protein‐coupled receptor
- GRK2
G‐protein‐coupled receptor kinase 2
- IKL
K+ leak current
- IN
local circuit interneuron
- IRS‐1‐Y608P
insulin receptor substrate‐1 Tyr608 peptide
- ISO
standing outward current
- K2P channel
two pore domain potassium channel
- MAChR
muscarinic ACh receptor
- M2AChR
muscarinic ACh receptor type 2
- OxoM
oxotremorine M
- PI3K
class‐1A phosphatidylinositol‐4,5‐bisphosphate 3‐kinase
- PIP2
phosphatidylinositol‐4,5‐bisphosphate
- PIP3
phosphatidylinositol‐3,4,5‐trisphosphate
- PLCβ
phospholipase Cβ
- RMP
resting membrane potential
- SFK
Src family of kinases
- SH2
Src homology 2
- TASK
TWIK‐related acid‐sensitive K+
- TREK
TWIK‐related K+
- TRPC
transient receptor potential canonical channel
- TWIK
two‐pore domain weakly inwardly rectifying K+
- TC
thalamocortical relay
- TK
tyrosine kinase
Introduction
The thalamus is strategically placed to control the flow of signals from the sensory organs to the cerebral cortex in a state‐dependent manner (Steriade et al. 1997). Processing of information by thalamocortical networks is dynamically modulated by several neurotransmitters. Within the dorsal lateral geniculate nucleus (dLGN), γ‐aminobutyric acid (GABA)ergic local circuit interneurons (INs) that inhibit thalamocortical relay (TC) cells, the principal projection neurons of the thalamus, are key players for shaping the output of this circuitry (Gabbott & Bacon, 1994). In particular, INs play a critical role in feed‐forward inhibition, thereby dynamically regulating the transfer of information from primary sensory afferents to TC neurons, shaping receptive field properties in the visual thalamus and configuring thalamic network oscillations (Blitz & Regehr, 2005; Lorincz et al. 2009; Antal et al. 2010; Wang et al. 2011; Crandall & Cox, 2013). Besides inhibition via conventional axodendritic synapses (F1 terminals), INs exert a powerful inhibitory control over the excitability of TC neurons by releasing GABA from vesicle‐containing dendrodendritic F2 terminals organized in complex triadic synaptic arrangements (Sherman & Guillery, 2006). The output of F2 terminals is strongly controlled by acetylcholine (ACh) released from cholinergic fibres of the ascending arousal system that are closely associated with the triad. In addition, it has been established that INs express muscarinic ACh receptor type 2 (M2AChR; Plummer et al. 1999). Stimulation of these receptors activates a K+ leak current (I KL), hyperpolarizes INs, decreases spontaneous inhibitory inputs onto TC neurons, and regulates feed‐forward inhibition in a stimulus intensity‐dependent manner (McCormick & Pape, 1988; Pape & McCormick, 1995; Cox & Sherman, 2000; Zhu & Heggelund, 2001; Antal et al. 2010). However, the molecular nature of I KL and the intracellular signalling cascade in INs have not yet been assessed.
The negative value of the resting membrane potential (RMP) in neurons critically depends on the sustained activity of members of the K2P channel family (Enyedi & Czirjak, 2010). In TC neurons, two‐pore domain weakly inwardly rectifying K+ (TWIK)‐related acid‐sensitive K+ (TASK) and TWIK‐related K+ (TREK) channels represent the central elements for determination of RMP. They both play a role in controlling the state‐dependent switch in activity modes (tonic firing and bursting activity), and thus influence sensory information processing as well as the generation of natural sleep oscillations and pathophysiological rhythms (Bista et al. 2015a). Our previous studies demonstrated that membrane depolarization induced by muscarinic agonists in TC neurons depends on the inhibition of TASK and TREK channels after activation of M1/M3 acetylcholine receptor (AChRs) coupled to G‐protein α subunit of the subtype q (Gαq protein) and phospholipase Cβ (PLCβ), which is then followed by phosphatidylinositol‐4,5‐bisphosphate (PIP2; an activator of TREK channels) breakdown and diacylglycerol (DAG; an inhibitor of TASK channels) production (Meuth et al. 2003, 2006; Broicher et al. 2008; Bista et al. 2012, 2015b; Wilke et al. 2014). Based on these findings, we hypothesized that K2P channels are also central for muscarinic signalling of INs (Bista et al. 2015a). Previous studies on signal transduction pathways that regulate smooth muscle function suggested that cellular and sarcoma (c‐Src) tyrosine kinase (TK) might be involved in the modulation of TASK‐1 channels (Gerthoffer, 2005; Nagaraj et al. 2013; MacKay & Knock, 2015). In PC12 cells, it was shown that phosphorylation of the tyrosine residue 370 of rat TASK‐1 channels (corresponding to Y368 in mouse and Y353 in human TASK‐1) was associated with activation of c‐Src TK by class‐1A phosphatidylinositol‐4,5‐bisphosphate 3‐kinase (PI3K) and transient co‐localization of c‐Src with the channel (Matsuoka & Inoue, 2015). In addition, PI3Ks play important roles in most cell types and affect multiple biological functions (Vanhaesebroeck et al. 2010; Dbouk & Backer, 2013; Gross & Bassell, 2014). The enzymatic activity of PI3Ks in phosphorylation of membrane‐associated phosphoinositides is mediated by a group of catalytic subunits (class IA: p110α/PIK3CA, p110β/PIK3CB, p110δ/PIK3CD; class IB: p110γ/PIK3CG) that are associated with one of seven regulatory subunits (p85α, p55α, p50α, p85β, p55γ, p101, p87). Some regulatory subunits have protein–protein interaction domains including Src homology 2 (SH2) domains. In particular, class IA but not class IB PI3Ks bind the SH2 domains of p85 type regulatory subunits. While in non‐neuronal cells PI3K catalytic subunits may have partially redundant functions, there is increasing evidence that their roles are more confined to distinct receptor‐dependent pathways in neurons. The analysis of their role in neuronal signalling as well as dysfunction has been identified as an emerging field in neuroscience (Gross & Bassell, 2014).
In this study, we performed whole cell patch clamp recordings in INs and used the muscarinic agonist oxotremorine M (OxoM) to activate MAChRs while monitoring the standing outward current (I SO). This is to a large degree carried through K2P channels in different neuronal cell types (Millar et al. 2000; Meuth et al. 2003; Deng et al. 2007; Dobler et al. 2007). To improve targeting of INs in brain slices containing the dLGN, we used previously established transgenic mice expressing enhanced green fluorescent protein (EGFP) in GABAergic neurons (glutamate decarboxylase 67 (GAD67)–enhanced green fluorescent protein (EGFP) mice) (Leist et al. 2016). Our findings point to a novel neuronal signalling pathway that leads to activation of K2P channels via M2AChR, PI3K and c‐Src TK activity in INs.
Methods
Ethical approval
The authors understand the ethical principles under which The Journal operates and our work complies with the animal ethics checklist (Grundy, 2015). All animal care and experimental work has been approved by local authorities (LANUV NRW; approval ID: 84‐02.05.50.15.026) and was performed in accordance with Directive 2010/63/EU of the European Parliament and of the Council of 22 September 2010 on the protection of animals used for scientific purposes. A total number of 84 animals were used and all efforts were made to minimize the animals’ pain and suffering. Experiments were performed on tissue from GAD67‐EGFP mice (Leist et al. 2016) of both sexes ranging in age from postnatal day (P) 14 to P24. To visualize INs in TASK‐1‐deficient mice, we crossbred GAD67‐EGFP with TASK‐1−/− mice (Mulkey et al. 2007; Meuth et al. 2009). Mice were bred and kept in institutional facilities in a 12 h light–dark cycle with food and water available ad libitum. For removal of the brain, mice were deeply anaesthetized using isoflurane (2‐chloro‐2‐(difluoromethoxy)‐1,1,1‐trifluoro‐ethane; 4% in O2), decapitated and brain tissue was used for electrophysiological analyses in vitro.
Acute slice preparation procedure
After decapitation and removal of the skull, a block of brain tissue containing the thalamus was removed from the cranial vault and submerged in ice‐cold aerated (O2) saline containing (in mm): sucrose, 200; Pipes, 20; KCl, 2.5; NaH2PO4, 1.25; MgSO4, 10; CaCl2, 0.5; dextrose, 10; pH 7.35, with NaOH. Thalamic slices of 270–300 μm thickness were prepared as coronal sections on a vibrating blade microtome (Leica Biosystems, Nussloch, Germany) by advancing the blade from the frontal to the caudal side of the brain. Finally, slices were transferred into a holding chamber and initially kept submerged at 30°C in a solution containing (in mm): NaCl, 125; KCl, 2.5; NaH2PO4, 1.25; NaHCO3, 24; MgSO4, 2; CaCl2, 2; dextrose, 10; pH adjusted to 7.35; equilibrated with 95% O2–5% CO2 (v/v) gas mixture. After 30 min the slices were cooled down to room temperature.
Whole‐cell patch clamp recordings
Brain slices were transferred to a recording chamber and continuously perfused (1–1.5 ml min−1) at 33–35°C (In‐Line Solution Heater, Harvard Apparatus, March, Germany) with aerated (O2) artificial cerebrospinal fluid (ACSF) containing (in mm): NaCl, 120; KCl, 2.5; NaH2PO4, 1.25; Hepes, 30; MgSO4, 2; CaCl2, 2; dextrose, 10; pH 7.25, adjusted with HCl (osmolality 305 mosmol kg−1). Slices were viewed using infrared video microscopy (DIC‐IR) and fluorescence imaging. Patch pipettes were pulled from borosilicate glass (GC150T‐10; Clark Electromedical Instruments, Pangbourne, UK) and had a resistance of 2.5–4.5 MΩ when filled with intracellular solution containing (in mm): potassium gluconate, 95; K3‐citrate, 20; NaCl, 10; Hepes, 10; MgCl2, 1; CaCl2, 0.5; BAPTA, 3; Mg‐ATP, 3; Na2‐GTP, 0.5. The internal solution was set to a pH of 7.25 with KOH and an osmolality of 295 mosmol kg−1. The glass electrode was subsequently connected to an EPC‐10 amplifier (HEKA Elektronik, Lambrecht, Germany) via a chlorinated silver wire. Tight seals were obtained on the soma of labelled dLGN interneurons. When seal resistance was > 1.5 GΩ, the pipette capacitance was compensated and a fluorescence image of the pipette touching the membrane patch was made for documentation. After obtaining the whole‐cell configuration, recordings were filtered and digitized using Pulse software (HEKA Elektronik). Series resistance at the start of experiments was 6–14 MΩ and compensation of > 40% was routinely applied. Series resistance was monitored and recordings were terminated whenever a significant increase (> 25%) occurred. Input resistance of INs ranged from 100 to 500 MΩ. A liquid junction potential of 8 mV was corrected offline for all recordings. RMP was measured when the access resistance had stabilized and perfusion of the intracellular milieu was expected to be complete (≥ 5 min after establishing the whole cell configuration). Only cells displaying overshooting action potentials (APs) and a negative RMP of up to −50 mV were included for analysis. Finally, only recordings of cells in which the EGFP signal was dialysed and depleted by the pipette solution were taken into consideration for the present study.
Determination of the intrinsic electrophysiological properties of INs
Current clamp recordings were used to analyse passive and active membrane properties. Analysis was performed according to established procedures (Leist et al. 2016). Electrophysiological parameters were measured from responses to step current injections of 800 ms duration that were applied from the RMP. Injected currents varied between −120 and +130 pA (increments of 50 pA). Membrane input resistance (R in) was deduced from the slope of the current–voltage (I–V) relationship obtained from the current injections to −20 and +30 pA. Membrane time constants (τm) were obtained by fitting single or double exponentials (FitMaster, HEKA Elektronik) to negative voltage deflections induced by hyperpolarizing current injections of −20 pA. The membrane capacitance (C m) was calculated using the equation: C m = τm/R in. The voltage sag (V sag) of the membrane potential was measured for potentials reaching a maximal negative value of about −95 mV and was calculated as the relative change between the maximal (V max; typically reached within 200 ms) and steady state voltage deflection (V ss; at the end of hyperpolarizing current injections) using the equation: ((V max − V ss)/V max) × 100%.
Drug application
Pharmacological substances were applied in the absence or presence of the muscarinic agonist OxoM while monitoring the amplitude of I SO. The phosphatidylinositol‐3,4,5‐trisphosphate (PIP3) analogue (1‐(1,2‐dihexadecanoylphosphatidyl)inositol‐3,4,5‐trisphosphate tetra‐sodium salt), the Src family of kinases (SFK) activator EPQpYEEIPIYL phosphopeptide and the insulin receptor substrate‐1 (IRS‐1) (Tyr608) peptide (IRS‐1‐Y608P) were dissolved in the intracellular recording solution and directly applied via the patch pipette. The tips of pipettes containing peptides or PIP3 were filled with drug‐free intracellular solution for obtaining the whole‐cell configuration. For some cell‐permeant pharmacological substances which act on intracellular targets, brain slices were pre‐incubated for at least 60 min (including gallein, LY294002, U73122, PP2, genistein, KBsrc4). K2P channel antagonists (including A293, A1899, PK‐THPP, norfluoxetine and spadin), muscarinic antagonists (including 1,1‐dimethyl‐4‐diphenylacetoxypiperidinium iodide (4‐DAMP), pirenzipine, AF‐DX 116 and AF‐DX 384) and PI3K activators (including demethylasterriquinone B1 (DAQ B1) and β‐oestradiol) were applied with the extracellular solution. In the case where substances were dissolved in dimethylsulfoxide (DMSO), the final concentration did not exceed 2‰. Drug suppliers were as follows: Sigma‐Aldrich Chemie GmbH (Taufkirchen, Germany) for Src kinase family activator; Santa Cruz Biotechnology Inc. (Dallas, TX, USA) for PIP3 analogue, IRS‐1‐Y608P and β‐oestradiol; and Sanofi‐Aventis Deutschland GmbH (Frankfurt, Germany) for A293 and A1899. All other drugs were purchased from Tocris Bioscience Ltd (Bristol, UK).
Statistics
If not mentioned otherwise all results are presented as the mean ± SEM. A two‐sample test for data variance was used to indicate differences in variance. Depending on variance homology (normally distributed) or significant alternations in data variance, Student's paired/unpaired t test (P t) or the non‐parametric Mann–Whitney test (P MW) was used to test for statistical significance (OriginPro 8 software, Additive GmbH, Friedrichsdorf, Germany). For multiple comparisons, one‐way ANOVA (P ANOVA) testing (OriginPro 8 software) was used followed by Tukey's test for indicating significance levels. Levels of significance were indicated as * (P < 0.05), ** (P < 0.01) and *** (P < 0.001). n represents the total number of cells (usually only one cell was analysed per brain slice; slices from ≥ 3 different animals were used per experimental condition.
Results
Muscarinic stimulation of INs is associated with membrane hyperpolarization and an increase in I SO
A total of 234 GAD67–EGFP‐labelled dLGN INs were analysed using the whole‐cell patch clamp technique (Fig. 1 A). Under the present experimental conditions, INs revealed an RMP of −63.5 ± 0.4 mV (n = 234) when recorded in current clamp mode. Bath application of OxoM (10 μm) induced a significant hyperpolarization of the RMP by 7.8 ± 1.2 mV (paired P t = 0.003; n = 5; Fig. 1 B). The generation of an AP was triggered by a short depolarizing current step (15 ms, +120 pA) under control conditions that was prevented after OxoM application (Fig. 1 C). Some INs (8%) revealed spontaneous AP generation in the form of tonic firing (n = 21; Fig. 1 D) or bursting (n = 6; Fig. 1 E). Bath application of the muscarinic agonist OxoM induced hyperpolarization of membrane potential and was sufficient to abolish spontaneous AP firing (Fig. 1 D and E). These findings indicate that membrane hyperpolarization induced by muscarinic signalling prevents AP generation in INs.
Figure 1. Muscarinic receptor stimulation causes hyperpolarisation of the RMP that is sufficient to abolish AP generation in dLGN INs.
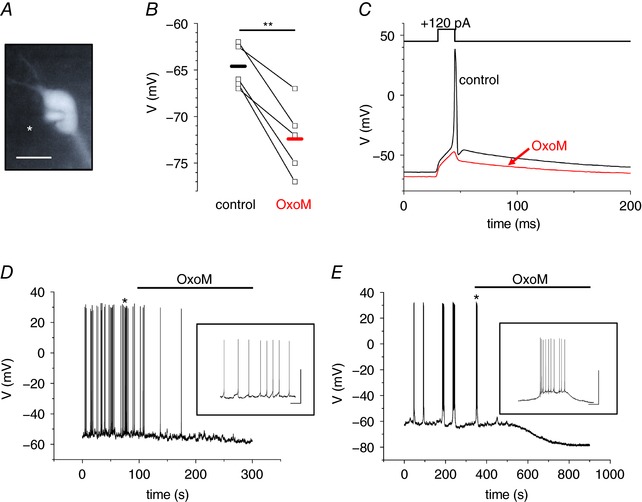
A, fluorescence image of a dLGN IN obtained from a brain slice of an EGFP‐GAD67 mouse. Note the patch pipette on the left side of the cell (marked with asterisk in image). The scale bar represents 10 μm. B, scatter diagram of the RMP under control conditions and during the application of OxoM. Points connected by lines indicate individual cell changes. The horizontal bars indicate the mean RMP under the two recording conditions (black, control; red, OxoM). C, current‐clamp recording of an IN in the absence (black trace) and presence (red trace) of OxoM (10 μm). OxoM mediates membrane hyperpolarization, which is sufficient to abolish the induction of an AP during a short lasting (15 ms) depolarizing current step (+120 pA). D and E, whole cell voltage recordings of spontaneously active INs revealing tonic firing (D) and rhythmic bursting (E). Muscarinic receptor stimulation by OxoM (10 μm; as indicated by the horizontal line) leads to membrane hyperpolarization and silencing of INs. Insets show the activity patterns at higher temporal resolution. Scale bars represent 50 mV and 2 s, respectively.
Next, INs were recorded under voltage clamp conditions. Cells were held at a potential of −28 mV to induce I SO, a current that is carried by persistently open ion channels like K2P channels (Fig. 2 A). Current measurements started 8–15 min after establishing the whole cell configuration when I SO amplitudes revealed a constant level (Fig. 2 B). In a total of 132 INs, the steady state level of I SO revealed an amplitude of 244.0 ± 9.9 pA (n = 132). For current amplitude vs. time histograms (Fig. 2 B), I SO amplitudes were determined every 25 s at a 200 ms epoch just before a voltage ramp protocol was initiated. Voltage ramps (duration 800 ms, rate of hyperpolarization of 7 ms mV−1) were used (Bista et al. 2015b) to adjust the membrane potential from −28 to −138 mV (Fig. 2 A, inset). Bath application of OxoM (10 μm) increased I SO by 13.4 ± 3.5% (n = 10; Fig. 2 B; the maximal effect was determined by taking time periods of 4 min before washing in and out of the drug into account). The OxoM effect was reversible (Fig. 2 B) and repeatable within the same IN (data not shown). Without any further experimental manipulation, I SO amplitudes were stable over a time period of at least 40 min (the mean normalized I SO at 40 min was 99.5 ± 1.5% compared to the level at the beginning; n = 7; compare Fig. 4 C). The I–V relationship of the OxoM‐sensitive current was obtained by graphical subtraction of control currents from those recorded in the presence of OxoM (i.e. OxoM – control; Fig. 2 C). The resulting graph revealed a reversal potential of the OxoM‐sensitive current at −109.1 ± 1.6 mV (n = 10, Fig. 3 C), which was close to the expected K+ equilibrium potential (E K) of −104 mV under the present experimental conditions. We noticed that subtracted I–V relationships did not always reveal ideal Goldman–Hodgkin–Katz rectification. Multiple modifying influences may have contributed to the appearance of the native macroscopic current measured here (Renigunta et al. 2015).
Figure 2. MAChR stimulation increases the standing outward current (I SO) in INs.
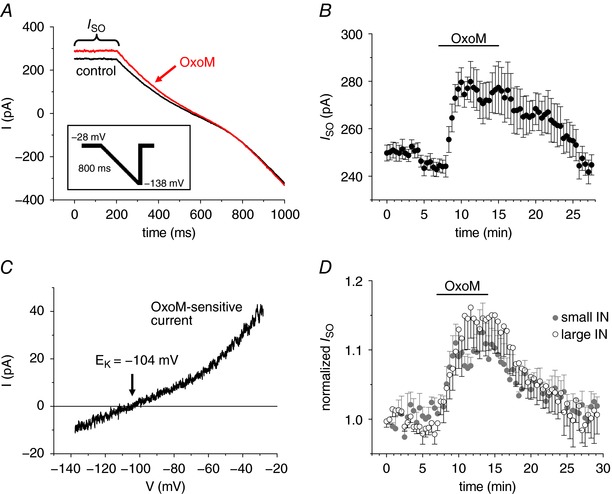
A, representative current traces induced by hyperpolarizing voltage ramps (see inset) under control conditions (black trace) and in the presence of OxoM (10 μm; red trace). The inset shows the applied voltage clamp protocol. B, mean values of I SO vs. time plot indicating a reversible increase in I SO (n = 10). C, exemplar current–voltage (I–V) relationship of the OxoM‐sensitive current that was obtained by subtracting control current traces from traces during drug application and assigning each point in time to the corresponding membrane potential. D, normalized mean values of I SO vs. time plots in dLGN INs categorized as small (grey circles, n = 7) or large (open circles, n = 7) as previously described (Leist et al. 2016).
Figure 4. I SO modulation via M2/4AChR requires non‐receptor c‐Src tyrosine kinase activity.
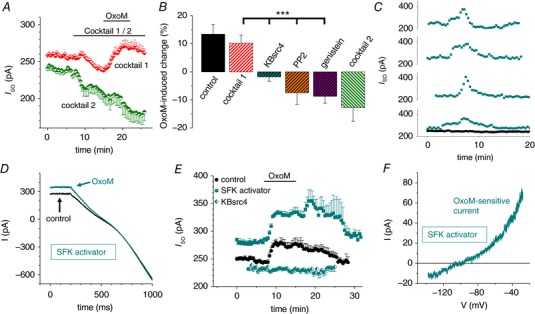
A, mean I SO vs. time plot illustrating the OxoM‐mediated current modulation in the presence of M2/4AChR‐ (cocktail 2, green symbols, n = 8) and M1/3AChR‐specific blockers (cocktail 1, red symbols, n = 10). Note that M2/4AChR stimulation is required for the OxoM‐mediated increase in I SO. B, summary bar graph of the maximal I SO change during the application of OxoM alone (black control bar) and in combination with different muscarinic receptor antagonist cocktails as indicated. Inhibitors of non‐receptor SFKs were applied in the presence of cocktail 1 (i.e. blocking M1/3AChR; shown by hatched bars) as indicated. C, I SO vs. time plots illustrating a considerably current increase, which appears spontaneously in some INs following intracellular application of the SFK activator (turquoise squares; t = 0 corresponds to a time point of about 5 min after establishing the whole cell configuration). Drug free control measurements of the I SO (black squares, n = 7) were stable (mean: 0.995 ± 0.015) over time. D, representative current traces induced by hyperpolarizing voltage ramps in the presence of the SFK activator (control, black trace) and following additional application of OxoM (turquoise trace). E, mean I SO vs. time plot with the SFK activator peptide applied via the recording pipette (turquoise squares, n = 10). I SO modulation during OxoM application was markedly increased (P MW = 0.037) compared to control (black circles, as in Fig. 2 B). No current modulation was found in the presence of the selective c‐Src TK inhibitor KBsrc4 (half turquoise symbols). F, exemplar I–V relationship of the OxoM‐sensitive current in the presence of the SFK activator.
Figure 3. TASK‐1 channel blockers reduce the OxoM‐mediated increase in I SO .
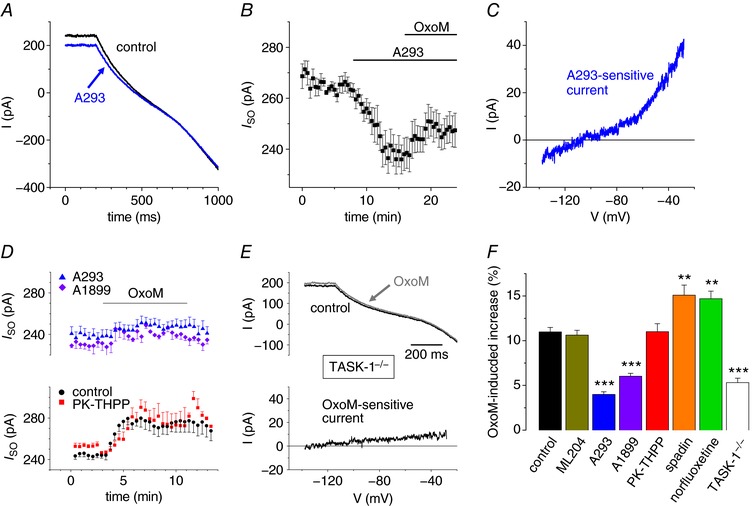
A, representative current traces induced by hyperpolarizing voltage ramps under control conditions (black trace) and in the presence of A293 (5 μm; blue trace). B, mean I SO vs. time plot illustrating current reduction during application of A293 followed by a small current increase when OxoM was applied (n = 8). C, a representative I–V relationship of the A293‐sensitive current. D, I SO vs. time plots of the OxoM‐mediated modulation of I SO in the presence of different TASK channel blockers. Note that the TASK‐3 channel blocker PK‐THPP (red squares, n = 8) does not significantly alter the OxoM response seen under control conditions (black circles; as in Fig. 2 B), whereas the TASK‐1 channel blockers A1899 (purple diamonds, n = 6) and A293 (blue triangles, as in Fig. 3 B) reduced the OxoM effect. E, representative current traces (upper panel) induced by hyperpolarizing voltage ramps (control conditions, black trace; OxoM, 10 μm, grey trace) and the OxoM‐sensitive current component (lower panel) recorded in TASK‐1‐deficient mice. F, summary bar graph illustrating the mean I SO change during the application of OxoM under control conditions (black bar), TASK‐1‐deficient mice (white bar) and in the presence of different ion channel blockers as indicated. Black stars indicate significance levels obtained by the use of ANOVA testing with respective controls.
Previously, we categorized INs based on their cell size as small and large neurons (Leist et al. 2016). When small (72.7 ± 2.5 pF, n = 65) and large (136.5 ± 3.5 pF, n = 67) INs were analysed, the current density of I SO was not different between the two types of IN (small IN: I SO = 2.56 ± 0.12 pA pF−1, n = 65; large IN: I SO = 2.28 ± 0.11 pA pF−1, n = 67; P MW = 0.08). To ensure that the two subtypes of INs in the mouse dLGN showed no differences concerning the OxoM‐mediated modulation of I SO, we further investigated an additional set of cells. INs that were identified as large in size revealed significantly higher C m values (129.1 ± 11.5 pF; P t = 0.002; n = 7) and significantly smaller V sag amplitudes (3.9 ± 0.8%; P t = 0.002; n = 7) compared to small INs (C m: 62.5 ± 5.5 pF; V sag: 28.4 ± 5.3%; n = 7). In contrast, averaged I SO current density in large INs (1.86 ± 0.26 pA pF−1; n = 7) was not significantly different when compared to small INs (2.38 ± 0.21 pA pF−1; n = 7; P MW = 0.35). Moreover, the maximal increase in I SO due to stimulation of muscarinic receptors was not significantly different in large (14.5 ± 4.2%; n = 7) and small (12.5 ± 4.2%; n = 7; Pt = 0.74; Fig. 2 D) INs. Under current clamp conditions, the two types of IN exhibited both spontaneous tonic firing and bursting activity (small IN: 3 bursting, 18 tonic firing; large IN: 3 bursting, 3 tonic firing). Data from small and large INs were, therefore, pooled in subsequent experiments.
These findings indicate that muscarinic receptor stimulation activates a K+ outward current with the typical characteristics of K2P channels.
TASK‐1 channels are modulated by MAChR stimulation
In order to identify the effector channels activated by muscarinic stimulation, selective K2P channel blockers were used to determine functional expression of TASK and TREK channels. As a first step, the selective TASK channel blocker A293 was tested (Putzke et al. 2007). Bath application of A293 (5 μm) decreased I SO by 11.1 ± 2.2% (n = 8, Fig. 3 A; maximal effect was determined in a time period of 4 min before and after wash‐in of the drug). In addition, the mean I SO increase following OxoM application was significantly smaller in the presence of A293 (4.0 ± 0.3%; Fig. 3 B and F) compared to the control effect (P MW < 0.001). Moreover, the I–V of the A293‐sensitive current was obtained by graphical subtraction of currents obtained in the presence of the blocker from control currents (i.e. control – A293) and revealed outward rectification and a negative reversal potential of −110.5 ± 2.0 mV (n = 9; Fig. 3 C). Furthermore, in the continuous presence of OxoM, application of A293 completely reversed the OxoM‐induced increase in I SO amplitude (data not shown). We next used PK‐THPP and A1899 as these two compounds show opposite selectivity for TASK‐1 (IC50 = 300 nm for PK‐THPP; IC50 = 35 nm for A1899) in comparison to TASK‐3 (IC50 = 35 nm for PK‐THPP; IC50 = 350 nm for A1899) channels (Streit et al. 2011; Coburn et al. 2012). In the presence of PK‐THPP (300 nm), the mean I SO increase (11.0 ± 0.9%; n = 8) was not significantly different compared to the control effect (Fig. 3 D and F). In contrast, bath application of OxoM in the presence of A1899 (200 nm) resulted in a significantly smaller mean I SO increase (6.0 ± 0.3%; n = 6) compared to OxoM control application as well as the combination of OxoM with PK‐THPP (P ANOVA < 0.001; Fig. 3 D and F). The use of TASK‐1‐deficient mice (Mulkey et al. 2007; Meuth et al. 2009) further confirmed the importance of TASK‐1 channels for muscarinic modulation in INs (Fig. 3 E). In the absence of TASK‐1 channels, the OxoM‐induced current increase was significantly (P ANOVA < 0.001) reduced to 5.3 ± 0.5% (n = 8) and revealed a rather linear I–V relationship that approached zero at the most hyperpolarized potentials (Fig. 3 E and F)
Next, we applied the active metabolite of fluoxetine known as norfluoxetine to INs, which has been shown to block TREK‐1 and TREK‐2 currents in the low micromolar concentration range (Kennard et al. 2005). Bath application of norfluoxetine (20 μm) decreased I SO by 10.8 ± 1.0% (n = 7). Additional application of OxoM increased I SO mean values by 14.8 ± 0.9% (n = 7), which was significantly (P MW = 0.007) stronger compared to the mean effect of OxoM under control conditions (11.0 ± 0.5%; n = 10; Fig. 3 F). Moreover, INs showed sensitivity for the selective TREK‐1 channel blocker spadin (Moha Ou Maati et al. 2012). The mean I SO decrease induced by spadin (1 μm) application was 14.7 ± 1.7% (n = 4; P MW = 0.002). It should be noted that spadin blocks homomeric TREK‐1 and heteromeric TREK‐1/TREK‐2 channels but not homomeric TREK‐2 channels (Moha Ou Maati et al. 2012). Application of OxoM in the presence of spadin increased I SO mean values by 15.1 ± 1.1%, which was significantly stronger compared to the OxoM control effect (P MW = 0.009; Fig. 3 F).
The transient receptor potential canonical channel 4 (TRPC4) is involved in the control of GABA release in INs (Munsch et al. 2003), and is activated by M2AChR (Jeon et al. 2012). Therefore, we applied 10 μm of the selective TRPC4 channel blocker ML204 (Miller et al. 2011), which, however, resulted only in a 3.1 ± 1.0% (n = 7) reduction in I SO amplitude. In the presence of ML204, the mean I SO change during OxoM application (10.6 ± 0.5%; P MW = 0.41) was not different compared to the control effect of OxoM (Fig. 3 D).
These findings indicate that homomeric TASK‐1 (or possibly heteromeric TASK‐1/TASK‐3) channels but not homomeric TASK‐3, TREK‐1‐, TREK‐2‐ or TRPC4‐containing channels form the molecular basis of OxoM‐mediated I SO increase.
The increase in I SO via M2/M4AChRs requires c‐Src TK activity
We next addressed the question which muscarinic receptor subtypes were involved in the OxoM‐mediated activation of TASK‐1 channels by using specific antagonists. Thus, we applied a combination of M2/M4AChR antagonists (10 μm of each AF‐DX 166 and AF‐DX 384, termed here cocktail 2) and observed a reduction in I SO by 15.7 ± 3.7% (n = 11; Fig. 4 A) pointing to some basal receptor activity. Under these conditions, additional application of OxoM further reduced I SO by 12.8 ± 4.9% (n = 8; Fig. 4 A and B). On the other hand, when a cocktail containing the M1/M3AChR antagonists pirenzipine and 4‐DAMP (10 μm each, termed here cocktail 1) was applied, an initial reduction in I SO by 5.1 ± 1.7% (n = 20) was followed by an I SO increase of 10.2 ± 1.6% (n = 11) when OxoM was added to the external solution (Fig. 4 A and B). The latter was not significantly different compared to the OxoM effect under control conditions, indicating that M2/M4AChRs were relevant for the positive modulation of I SO. In smooth muscle cells M2AChR‐dependent stimulation of c‐Src TK was described, and c‐Src TK auto‐phosphorylation was found to be associated with the phosphorylation of TASK‐1 channels and increased current activity (Nagaraj et al. 2013). To test whether similar mechanisms exist in CNS INs, the following experiments were conducted in the presence of cocktail 1 (blocking M1/M3AChR). When slices were pre‐incubated (100 μm, > 1 h) with the TK inhibitor genistein (Akiyama et al. 1987), the application of OxoM revealed a reduction of I SO by 8.7 ± 2.5% (n = 9; Fig. 4 B) that was significantly different (P ANOVA < 0.001) compared to the change in I SO induced by OxoM under control conditions and in the presence of cocktail 1. A similar reduction of I SO by OxoM (7.5 ± 4.2%; n = 8) was observed following pre‐incubation (1 μm, > 1 h) with the unspecific SFK inhibitor PP2 (Hanke et al. 1996). Moreover, pre‐treatment of slices with 0.1 μm of the selective c‐Src TK inhibitor KBsrc4 (Brandvold et al. 2012) completely abolished any I SO modulation mediated by OxoM (I SO reduction: 1.7 ± 1.6%; n = 7; Fig. 4 B; P ANOVA < 0.001 compared to cocktail 1). As an additional indicator for the involvement of c‐Src TK, we applied the SFK activator peptide (EPQpYEEIPIYL phosphopeptide) via the patch pipette (Eck et al. 1993). While the SFK activator peptide diffused into the cell interior a strong transient increase in I SO was observed in 4 (53.2 ± 9.6%; n = 4; maximal increase calculated for these INs) out of 10 cells, which was never seen under control conditions within the same time period (0.5 ± 2.0% change in amplitude within 20 min; n = 7; Fig. 4 C). In addition, application of OxoM in the presence of the SFK activator peptide significantly enhanced the increase in I SO (19.1 ± 1.5%; n = 10; P MW = 0.037; Fig. 4 D and E) compared to the control effect of OxoM. Note that in the presence of the SFK activator peptide, a transient additional increase in I SO occurred when OxoM was washed out (26.5 ± 7.4%; P MW = 0.014 compared to control wash‐out 7.4 ± 2.7%). The OxoM‐activated current in the presence of SKF activator revealed outward rectification and reversal close to E K (Fig. 4 F).
These findings indicate the involvement of c‐Src TK in the increase in I SO amplitude following M2/M4AChR stimulation.
Coupling of M2/M4AChRs to Gβγ and PI3K is involved in the modulation of I SO in INs
M2AChRs in smooth muscle cells have been shown to preferentially couple to G‐protein α subunit of the subtype i/o (Gαi/o), PI3K and c‐Src TK to modulate ion channel activity (Gerthoffer, 2005). Furthermore, rapid deactivation of Gαi/o is mediated by c‐Src TK via a G‐protein βγ subunit (Gβγ)‐ and PI3K‐dependent pathway (Huang et al. 2014). Following stimulation of Gαi/o type G‐protein‐coupled receptors (GPCRs), SFK members are known to form protein complexes with PI3Ks via the N‐terminal SH2 (nSH2) domain of the p85 subunit (Nozu et al. 2000; Gentili et al. 2002). Using the SMALI prediction for SH2 domains (Li & Li, 2017), we found that mouse c‐Src TK (Uniprot ID: P05480) has at least one site (including Y144) for binding the nSH2 domain of the p85β (score = 1.09) or p85α (score = 1.06) regulatory subunit of PI3K (data not shown). To assess the possible involvement of these signalling proteins, the following measurements were obtained in the presence of cocktail 1 (blocking M1/M3AChR). First, we pre‐incubated (> 1 h) dLGN INs with gallein (10 μm), an antagonist for Gβγ (Lehmann et al. 2008), which significantly reduced the OxoM‐induced increase of I SO (2.6 ± 2.3%; n = 5; P ANOVA < 0.01) compared to the effects recorded in the presence of cocktail 1 (Fig. 5 A and C). Next, we tested inhibitors and activators of PI3K activity. Preincubation (> 1 h) with LY294002 (5 μm), which inhibits PI3K activity (Vlahos et al. 1994), strongly reduced the M2/M4AChR‐mediated increase in I SO (2.3 ± 1.8%, n = 8; Fig. 5 C). This effect was significantly different compared to the OxoM effect in the presence of cocktail 1 (P ANOVA < 0.01). PI3K activators were applied with the extracellular medium (DAQ B1, β‐estradiol) or via the patch pipette (IRS‐1‐Y608P). Wash‐in of DAQ B1 (10 μm), an insulin mimetic compound (Weber et al. 2000), increased I SO by about 10% (Fig. 5 B). Additional application of OxoM resulted in a strong further increase (P ANOVA < 0.01, with respect to the OxoM effect in cocktail 1) in I SO (23.5 ± 1.8%, n = 8; Fig. 5 C). The OxoM‐activated current was characterized by outward rectification and reversal close to E K (Fig. 5 A). The use of IRS‐1‐Y608P (10 μm), which activates PI3K by association with the SH2 domains of the p85 subunit (Shoelson et al. 1992; Noh et al. 2013), confirmed the effect of insulin receptor activation. With IRS‐1‐Y608P included in the internal solution, the OxoM effect was strongly increased (28.1 ± 2.1%, n = 7; P ANOVA < 0.01, with respect to the OxoM effect in cocktail 1) and the I–V relationship of the OxoM‐sensitive current was typical for outwardly rectifying K+ channels (Fig. 5 A and C). Activation of PI3K by interaction with the regulatory p85 subunit is a non‐genomic mechanism of the steroid hormone estradiol (Simoncini et al. 2000). Therefore we tested the effect of β‐estradiol on muscarinic activation of I SO. In the presence of β‐estradiol, the OxoM effect on I SO was significantly increased (25.4 ± 2.3, n = 6; P ANOVA < 0.01, with respect to the OxoM effect in cocktail 1; Fig. 5 C) with the sensitive component revealing outward rectification and reversal at E K (Fig. 5 A).
Figure 5. Characterization of the signalling pathway underlying the increase of I SO in dLGN INs.
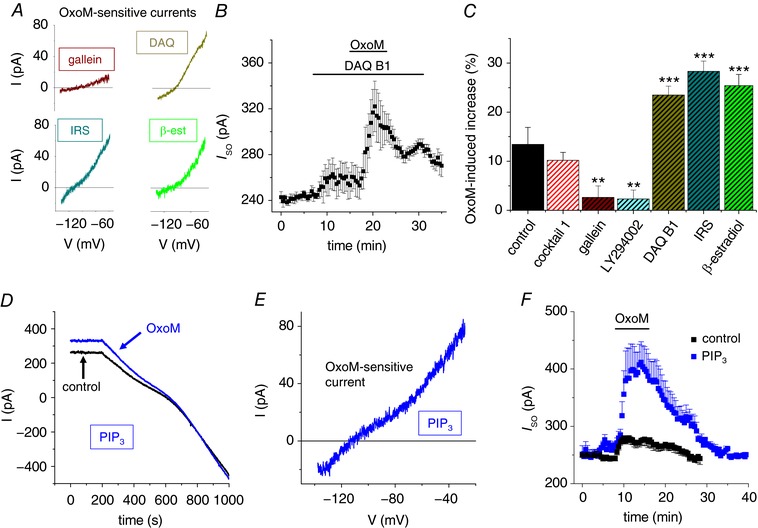
A, I–V relationships of the OxoM‐sensitive currents in the presence of different drugs (as indicated; DAQ, DAQ B1; IRS, IRS‐1‐Y608P; β‐est, β‐estradiol). B, mean I SO vs. time plot illustrating the strongly enhanced I SO increase in the presence of DAQ B1 (n = 8). C, summary bar graph illustrating the maximal I SO change during the application of OxoM. Hatched bars indicate that M1/3AChR antagonists were present during the application of OxoM. D, examplar traces of voltage clamp recordings in the absence (black trace) and in the presence of OxoM (blue trace) when 50 μm PIP3 was added to the pipette solution. E, exemplar I–V relationship of the OxoM‐sensitive current with PIP3 added to the pipette solution. F, mean I SO vs. time plot illustrating the strongly enhanced and prolonged I SO increase in experiments in which PIP3 was added to the pipette solution (blue symbols, n = 7) in comparison to the control effect (black symbols, as in Fig. 2 B). Note that muscarinic receptor stimulation is still required to cause a detectable change in I SO.
Next, we assessed the role of the product of catalytic PI3K activity, namely PIP3. Application of OxoM in the presence of intracellular PIP3 (50 μm) revealed a strongly enhanced I SO increase of 62.8 ± 13.7% (n = 7; P MW = 0.001; Fig. 5 D and F) compared to control conditions. It is important to note that PIP3 diffusion into the cell did not alter I SO amplitudes per se. Muscarinic stimulation was necessary to modulate I SO in a fully reversible manner (Fig. 5 F). Moreover, the OxoM‐sensitive current in the presence of PIP3 was characterized by outward rectification and reversal close to E K (Fig. 5 E). To further assess the role of phospholipids, slices were pre‐incubated (10 μm, > 1 h) with the PLCβ antagonist U73122 (Bleasdale et al. 1990; Bista et al. 2012). Under these conditions, OxoM induced a strong increase in I SO (32.4 ± 9.9%; n = 7; data not shown). Moreover, OxoM‐mediated I SO modulation following U73122 treatment was completely reversed by A293 application (data not shown).
These findings indicate that the interaction of the p85 SH2 domain with c‐Src is essential for I SO modulation following M2/M4AChR stimulation. Furthermore, PIP3 signalling increases the muscarinic effect, possibly facilitating the SH2‐dependent protein interaction.
Discussion
The findings presented here indicate that stimulation of M2/M4AChRs mediates the up‐regulation of TASK‐1 channels in dLGN INs, eventually causing hyperpolarization and silencing of neuronal firing. The modulation pathway (Fig. 6) resembles muscarinic signalling that is observed in smooth muscle cells (Gerthoffer, 2005). Therefore, we have identified a novel and unique pathway that activates K2P channels under physiological conditions in central neurons.
Figure 6. Proposed signalling pathway for the modulation of TASK‐1 channels in dLGN IN.
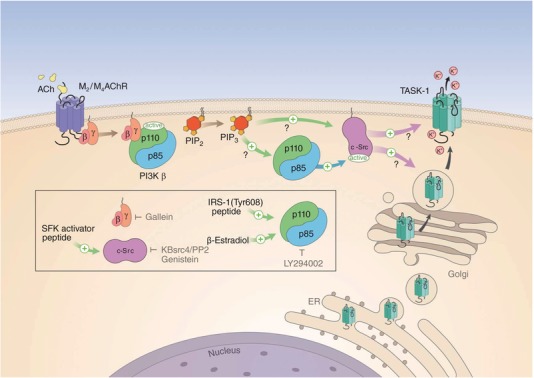
Binding of ACh to Gi/o‐protein‐coupled M2/M4AChR (the α subunit is not shown) leads to the activation of PI3Kβ via binding of the G‐protein βγ subunit. While the inhibition of the catalytic p110 subunit by the regulatory p85 subunit is released, the SH2 domain‐dependent interaction of p85 with c‐Src leads to activation of TK activity. c‐Src activation increases current through TASK‐1 (or TASK‐1,3) channels by direct interaction (as indicated by the short time scale of minutes of the Oxo effect) with the channel protein. Facilitation of trafficking and integration of channels into the membrane (possible via the adapter proteins p11 and 14‐3‐3) is a potential second mechanism. The product of p110 enzymatic activity, the phospholipid PIP3, potentially facilitates the interaction between the components of the receptor cascade, which may be arranged in close spatial proximity. The box indicates the sites of action of enzyme activators (green arrows) and inhibitors (depicted by grey ┬ symbols) used in the present study.
Components of the signalling pathway: MAChR, Gβγ, PI3K, c‐Src TK and TASK‐1
Based on the effects of the subtype‐specific blockers pirenzepine (M1AChR), AF‐DX 116 (M2AChR), 4‐DAMP (M3AChR) and AF‐DX 384 (M2/M4AChR), we conclude that M2/M4AChRs, but not M1/M3AChRs, are involved in the activation of I SO in INs (Hammer et al. 1980, 1986; Michel et al. 1989; Miller et al. 1991). Moreover, the blocking effect of gallein confirmed the involvement of G‐protein Gβγ subunits (Lehmann et al. 2008; Ukhanov et al. 2011).
Downstream of Gβγ‐dependent activation, an interaction exists between PI3K class IA and c‐Src TK that is established following GPCR stimulation through formation of a complex via the nSH2 domain of the p85 subunit (Chan et al. 1990; von Willebrand et al. 1998; Yu et al. 1998; Nozu et al. 2000; Cuevas et al. 2001; Chan et al. 2002; Gentili et al. 2002). In particular, the nSH2 domain of p85 is known to mediate an inhibitory influence via the helical domain of the catalytic p110 subunit (Miled et al. 2007; Mandelker et al. 2009), which can be disrupted by the binding of tyrosine‐phosphorylated proteins or peptides (Yu et al. 1998; Burke et al. 2011; Zhang et al. 2011). To probe the involvement of p85‐related SH2 domains in the modulation of TASK‐1 channels in dLGN INs, we used the PI3K antagonist LY294002 and the PI3K activators DAQ B1 (Salituro et al. 2001; Webster et al. 2003), IRS‐1‐Y608P (Shoelson et al. 1992; Ito et al. 1997) and β‐estradiol (Simoncini et al. 2000; Marin et al. 2005). Indeed, the modulation of I SO was blocked by LY294002 application and enhanced by the three PI3K activators. Activation of the insulin receptor by DAQ B1 results in tyrosine phosphorylation of IRS‐1, which associates with the p85 subunit of PI3K followed by activation of the enzyme (Salituro et al. 2001). In a similar way IRS‐1‐Y608P applied via the patch pipette bound to the p85 subunit via SH2 domain recognition (Backer et al. 1992; Shoelson et al. 1992). Also the activated oestrogen receptor signals via the p85 subunit (Simoncini et al. 2000), thereby pointing to the central role of this regulatory subunit for the modulation cascade investigated here. Besides this direct interaction, p85 is also capable of activating c‐Src indirectly (Walker et al. 2007; Boroughs et al. 2014). Furthermore, our results indicate that downstream of Gβγ proteins, c‐Src activity is regulated by PI3Kβ but not by PI3K class IB enzymes.
The involvement of c‐Src TK was further confirmed through use of the TK inhibitor genistein (Akiyama et al. 1987), the SFK inhibitor PP2 (Hanke et al. 1996), and the specific c‐Src kinase antagonist KBsrc4 (Brandvold et al. 2012), all of which abolished the I SO increase mediated by OxoM. It should be noted, however, that genistein may modulate ion channel function via inhibition of TK enzymatic activity (Yu et al. 2004; Missan et al. 2006) and/or direct blocking (Paillart et al. 1997; Belevych et al. 2002; Altomare et al. 2006; Gierten et al. 2008; Zhao et al. 2008). Furthermore, we found a positive effect of the SFK activator peptide (EPQpYEEIPIYL phosphopeptide) on the OxoM‐induced increase in I SO. The transient increase in I SO seen in some cells during diffusion of the SFK activator peptide into the cell indicates that receptor stimulation of the signalling cascade is necessary for sustained activation. Here the production of PIP3 may play a role (see below). From our data it is not clear whether direct phosphorylation of the channel protein and/or additional interacting proteins like the adapter protein 14‐3‐3 contributes to TASK‐1 modulation by TK. While mutations at the single TK phosphorylation site (Y323) within the TASK‐1 protein were not found to be involved in genistein modulation of the channel (Gierten et al. 2008), several members of the TASK channel family reveal 14‐3‐3‐dependent surface expression which is sensitive to the phosphorylation/dephosphorylation status of the trafficking control region of the channels (Kilisch et al. 2015, 2016; Fernandez‐Orth et al. 2016). However the fast time course of minutes of the OxoM‐induced response may point to increased channel opening in the present study.
The I SO current component modulated by muscarinic stimulation revealed hallmarks of a current through TASK‐1 channels. The I–V relationship of the OxoM‐sensitive current was characterized by outward rectification and the reversal potential was found to be close to the K+ equilibrium potential. Moreover, bath application of the selective TASK‐1 (A293, A1899) but not TASK‐3 (PK‐THPP) channel blockers decreased I SO (Putzke et al. 2007; Streit et al. 2011; Coburn et al. 2012; Chokshi et al. 2015). Importantly, the OxoM‐mediated I SO modulation was significantly smaller in TASK‐1‐deficient mice. However, since pharmacological block and genetic knock‐out of TASK‐1 did not completely block the OxoM effect, a contribution of heteromeric TASK‐1/TASK‐3 (Berg et al. 2004; Kang et al. 2004; Aller et al. 2005; Kim et al. 2009; Enyedi & Czirjak, 2010; Turner & Buckler, 2013; Rinné et al. 2015) channels or other outward rectifying K+ channels cannot be excluded.
The present results therefore point to a scenario (Fig. 6) in which c‐Src TK acts downstream of Gβγ dimers (Igishi & Gutkind, 1998) with direct activation of PI3K by Gβγ (Vanhaesebroeck et al. 2010; Dbouk & Backer, 2013) increasing the activity of c‐Src and modulating additional downstream elements (Schwindinger & Robishaw, 2001). The interaction of p85 containing PI3Ks with c‐Src kinase is essential for the increase in I SO that is carried by the modulation of monomeric TASK‐1 or heteromeric TASK‐1/3 channels.
Muscarinic control of K2P channels in the thalamus
The findings that TASK channels are reciprocally modulated by Gαi/o‐coupled M2/M4AChRs and Gαq/11‐coupled M1/M3AChR pathways that activate and inhibit them, respectively, point to a possible crosstalk between signalling pathways (Gerthoffer, 2005; Bista et al. 2015a). Indeed strong evidence for a partially cooperative signalling between M2AChR and M3AChR as well as between Gαi and Gαq on the levels of second messengers and effector kinases, especially PI3K, has been presented (Blaukat et al. 2000; Hornigold et al. 2003; Arrighi et al. 2013). While the reduced I SO amplitude after M2/4AChR inhibition may be straightforwardly explained by effects due to ambient ACh in the slice and/or basal receptor activity, the current reduction in response to M1/3AChR inhibition may be based on the convergence of pathways on PI3K. The finding that block of M1/3AChR in slices pre‐incubated with LY294002 had no effect on I SO may point to this scenario (data not shown). Also differences in the dynamics of signalling pathways when basally active or stimulated by synthetic agonists may be of relevance here (Geppetti et al. 2015). The effect of ambient ACh and/or basal MAChR activity has been observed in dLGN TC neurons before (Broicher et al. 2008).
Furthermore, while blocking M2/M4AChRs, OxoM induced a decrease in I SO, which in the present study was in agreement with the inhibition of TASK channels by DAG due to M1/M3AChR stimulation (Wilke et al. 2014; Bista et al. 2015b). However, blocking c‐Src TK activity (i.e. blocking the activation of TASK channels) by application of genistein and PP2 was associated with a decrease in I SO during OxoM treatment. Although M1/M3AChRs were blocked during these experiments, several additional factors may have contributed to this observation. (1) The blocking of M1/M3AChRs may have been incomplete; (2) Gαq/11‐coupled M5AChRs that are present in the dLGN may have been activated (Wei et al. 1994; Broicher et al. 2008); (3) GPCRs and SFKs show multi‐layered forms of crosstalk including direct association of SFKs with GPCRs or receptor‐associated proteins (Luttrell & Luttrell, 2004). Therefore c‐Src TK activity may have influenced critical components of MAChR pathways. For instance, G‐protein‐coupled receptor kinase 2 (GRK2) phosphorylation by c‐Src TK is known to increase its activity following M2AChR stimulation (Sarnago et al. 1999; Mahavadi et al. 2007) and the interaction between Gαq and GRK2 is associated with the inhibition of M3AChRs as well as Gαq‐dependent PLCβ activity (Huang et al. 2007; Luo et al. 2008; Wolters et al. 2015). The latter will reduce and increase DAG and PIP2 levels, respectively, resulting in facilitation of currents through TASK and TREK channels (Wilke et al. 2014; Bista et al. 2015b). Indeed, we found that pre‐incubation with the PLCβ inhibitor U73122 significantly increased the modulation of I SO by OxoM, thereby resembling the effect of PIP3 application. It is not clear, however, whether the activation of PI3K can significantly reduce the availability of PIP2 to serve as a substrate for PLCβ to form DAG and whether increased availability of PIP2 can stimulate PIP3 production. Nevertheless it is interesting to note that M1AChR receptor‐induced intracellular DAG transients have been shown to increase DAG levels by about 50% with respect to baseline (Wilke et al. 2014).
The role of PIP3 in the modulation of TASK channels
Intracellular application of PIP3 via the patch pipette strongly increased the OxoM effect in dLGN INs. Once synthesized, PIP3 signals via direct PIP3‐binding effector proteins (Kriplani et al. 2015). In addition PIP3 may act via stabilizing and recruiting protein–protein interactions: (1) PIP3 is known to play a role in recruiting SH2‐containing signalling proteins including PI3K and c‐Src TK to the plasma membrane (Chellaiah et al. 2001). Therefore, PIP3 interacts with the cSH2, but not the nSH2 domain of the p85 subunit (Ching et al. 2001). Specific phosphopeptides such as pYEEI compete for the binding of PIP3 to the SH2 domain of c‐Src TK (Rameh et al. 1995). Thus, PIP3 could facilitate the assembly of a c‐Src/PI3K‐containing complex by promoting SH2 binding to c‐Src‐phosphorylated sites. For instance, the p85 subunit was found to bind with 100‐fold greater affinity to c‐Src‐phosphorylated epidermal growth factor receptors (EGFRs) in comparison to autophosphorylated EGFRs (Stover et al. 1995). (2) Both lipid kinase and protein kinase activity of PI3K are required for the transactivation of EGFRs by β2‐adrenergic receptors, prototypical GPCRs (Watson et al. 2016). Mechanistically, the generation of PIP3 stabilizes the GPCR complex while phosphorylation of c‐Src by PI3K activates its TK activity. Since MAChRs also signal as part of large multiprotein complexes (Nelson et al. 2007; Huster et al. 2010), the role of PIP3 may be to stabilize protein interactions in the activated signalling cascade (Fig. 6).
TK‐ and PI3K‐dependent signalling in the thalamus
c‐Src TK has been identified as a crucial element for setting a negative resting membrane potential and a low resting pulmonary vascular tone by activating TASK‐1 channels in smooth muscle cells (Gerthoffer, 2005; Nagaraj et al. 2013; MacKay & Knock, 2015). Therefore, the modulation of TASK‐1 channels in INs, exemplifies a smooth muscle‐like signal transduction pathway in the brain. Also, vascular L‐type Ca2+ channels are up‐regulated following M2AChR stimulation by a signalling pathway involving Gβγ, PI3Kβ and c‐Src TK (Macrez et al. 2001; Oldenburg et al. 2003; Callaghan et al. 2004). Since L‐type Ca2+ channels are expressed in dLGN INs, a similar modulation may be expected in these cells (Munsch et al. 1997; Budde et al. 1998). Indeed, activation of M2AChR led to long‐lasting, nimodipine‐sensitive plateau potentials when INs were challenged by stimulation of the optic tract (Antal et al. 2010). Future studies have to assess the involvement of c‐Src TK in this modulation. Different PI3K subtypes are characterized by neuron‐specific signalling and function (Gross & Bassell, 2014). Here, we add activation of TASK‐1 channels via PI3K and c‐Src TK to this growing list indicating that there are currently emerging roles of TK signalling in modulating thalamocortical circuits.
Functional significance of muscarinic modulation in INs
Feed‐forward inhibition exerted by INs plays critical roles in the dynamic regulation of the precision of firing and the refinement of receptive field properties of TC neurons and thus the information transfer of primary sensory afferents to the cortex (Blitz & Regehr, 2005; Antal et al. 2010; Crandall & Cox, 2013). Phasic inhibition from INs leads to effective temporal framing of the relay mode output of TC neurons in the α‐frequency range, which is essential for visual perception (Lorincz et al. 2009). Besides phasic inhibition, synaptic GABA release in the dLGN generates a GABAA receptor‐mediated tonic inhibition that hyperpolarizes TC neurons, thereby favouring thalamic burst firing and synchronized slow rhythmic activity in the thalamocortical network and modulating burst precision (Cope et al. 2005; Bright et al. 2007).
Here, we show that muscarinic receptor stimulation causes hyperpolarization of INs, due to the increased activity of TASK‐1 channels. In the absence of intense optic tract stimulation, activation of MAChRs leads to the cessation of spontaneous or induced AP firing of INs (McCormick & Pape, 1988; Pape & McCormick, 1995; Antal et al. 2010). Since bursting from hyperpolarized potentials is less pronounced in dLGN INs, and oscillatory activity occurs at more depolarized potentials compared to TC neurons (McCormick & Pape, 1988; Pape et al. 1994; Zhu et al. 1999; Broicher et al. 2007; Halnes et al. 2011; Seabrook et al. 2013; Leist et al. 2016), a reduced GABAergic inhibition of TC neurons may be expected following muscarinic stimulation. Indeed, GABA release and inhibitory connections in the thalamus are suppressed by the activation of M2AChRs (Cox & Sherman, 2000; Castro‐Alamancos, 2002; Antal et al. 2010), whereas ACh activation of M1/M3AChR increases the ability of TC neurons to excite cortical cells through increased tonic firing (Bista et al. 2015a). This is important since the desynchronized cortical state during active behaviour in mice is driven by increased thalamic AP firing rates (Poulet et al. 2012).
Our findings point to an intriguing scenario where INs in the rodent dLGN possess distinctive modulatory pathways for K2P channels in comparison with those of TC neurons. While TASK‐1/3 channels are inhibited via activation of M1/M3AChR in TC neurons, TASK‐1 channels are increased via M2AChR in INs. Together, the direct excitation of TC neurons and inhibition of INs allow the ascending cholinergic brainstem system to exert a powerful facilitatory influence on the transfer of visual information to the cerebral cortex (McCormick & Pape, 1988). Cholinergic signalling within the thalamocortical system is known to enhance the attentional modulation of neuronal activity (Herrero et al. 2008). Therefore, it is of interest to note that similar to the circuit modulation in the thalamus, ACh acts via M2/M4AChR on fast‐spiking GABAergic interneurons in the cortex and decreases the probability of GABA release (Kruglikov & Rudy, 2008). Thereby, ACh reduces the amount of feed‐forward inhibition of pyramidal neurons (Gabernet et al. 2005; Higley & Contreras, 2006), while ACh depolarizes pyramidal neurons via activation of M1AChR (McCormick & Prince, 1985, 1986; Kruglikov & Rudy, 2008). Thus, ACh release in the thalamocortical system results in the enhancement of neuronal activity during wakefulness. These findings point towards the idea that mechanisms facilitating the informational relay are conserved within different parts of the thalamocortical system.
Additional information
Competing interests
The authors declare no competing financial interests.
Author contributions
M.L., S.R., M.D. and A.A. designed and performed the experiments and analysed the data. T.B., N.D. and S.G.M designed and supervised the project and reviewed all experiments. H.C.P. provided important scientific input. M.L. and T.B. wrote the manuscript. All authors edited and agreed on the final version of the manuscript. All authors agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed. This work was done in partial fulfilment of the Ph.D. work of M.L.
Funding
This work was supported by DFG (Cells‐in‐Motion Cluster of Excellence; CRC128‐B06; INST2105/27‐1; BU1019/15‐1) and IZKF Münster (Bud3/001/16).
Acknowledgements
The authors thank Elke Naß for excellent technical assistance and Heike Blum for helping to design Fig. 6.
References
- Akiyama T, Ishida J, Nakagawa S, Ogawara H, Watanabe S, Itoh N, Shibuya M & Fukami Y (1987). Genistein, a specific inhibitor of tyrosine‐specific protein kinases. J Biol Chem 262, 5592–5595. [PubMed] [Google Scholar]
- Aller MI, Veale EL, Linden AM, Sandu C, Schwaninger M, Evans LJ, Korpi ER, Mathie A, Wisden W & Brickley SG (2005). Modifying the subunit composition of TASK channels alters the modulation of a leak conductance in cerebellar neurons. J Neurosci 25, 11455–11467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altomare C, Tognati A, Bescond J, Ferroni A & Baruscotti M (2006). Direct inhibition of the pacemaker (I f) current in rabbit sinoatrial node cells by genistein. Br J Pharmacol 147, 36–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antal M, Acuna‐Goycolea C, Pressler RT, Blitz DM & Regehr WG (2010). Cholinergic activation of M2 receptors leads to context‐dependent modulation of feedforward inhibition in the visual thalamus. PLoS Biol 8, e1000348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrighi N, Bodei S, Zani D, Michel MC, Simeone C, Cosciani Cunico S, Spano P & Sigala S (2013). Different muscarinic receptor subtypes modulate proliferation of primary human detrusor smooth muscle cells via Akt/PI3K and MAP kinases. Pharmacol Res 74, 1–6. [DOI] [PubMed] [Google Scholar]
- Backer JM, Myers MG Jr, Shoelson SE, Chin DJ, Sun XJ, Miralpeix M, Hu P, Margolis B, Skolnik EY, Schlessinger J & White MF (1992). Phosphatidylinositol 3′‐kinase is activated by association with IRS‐1 during insulin stimulation. EMBO J 11, 3469–3479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belevych AE, Warrier S & Harvey RD (2002). Genistein inhibits cardiac L‐type Ca2+ channel activity by a tyrosine kinase‐independent mechanism. Mol Pharmacol 62, 554–565. [DOI] [PubMed] [Google Scholar]
- Berg AP, Talley EM, Manger JP & Bayliss DA (2004). Motoneurons express heteromeric TWIK‐related acid‐sensitive K+ (TASK) channels containing TASK‐1 (KCNK3) and TASK‐3 (KCNK9) subunits. J Neurosci 24, 6693–6702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bista P, Cerina M, Ehling P, Leist M, Pape HC, Meuth SG & Budde T (2015a). The role of two‐pore‐domain background K+ (K2P) channels in the thalamus. Pflügers Arch 467, 895–905. [DOI] [PubMed] [Google Scholar]
- Bista P, Meuth SG, Kanyshkova T, Cerina M, Pawlowski M, Ehling P, Landgraf P, Borsotto M, Heurteaux C, Pape HC, Baukrowitz T & Budde T (2012). Identification of the muscarinic pathway underlying cessation of sleep‐related burst activity in rat thalamocortical relay neurons. Pflügers Arch 463, 89–102. [DOI] [PubMed] [Google Scholar]
- Bista P, Pawlowski M, Cerina M, Ehling P, Leist M, Meuth P, Aissaoui A, Borsotto M, Heurteaux C, Decher N, Pape HC, Oliver D, Meuth SG & Budde T (2015b). Differential phospholipase C‐dependent modulation of TASK and TREK two‐pore domain K+ channels in rat thalamocortical relay neurons. J Physiol 593, 127–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaukat A, Barac A, Cross MJ, Offermanns S & Dikic I (2000). G protein‐coupled receptor‐mediated mitogen‐activated protein kinase activation through cooperation of Gαq and Gαi signals. Mol Cell Biol 20, 6837–6848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleasdale JE, Thakur NR, Gremban RS, Bundy GL, Fitzpatrick FA, Smith RJ & Bunting S (1990). Selective inhibition of receptor‐coupled phospholipase C‐dependent processes in human platelets and polymorphonuclear neutrophils. J Pharmacol Exp Ther 255, 756–768. [PubMed] [Google Scholar]
- Blitz DM & Regehr WG (2005). Timing and specificity of feed‐forward inhibition within the LGN. Neuron 45, 917–928. [DOI] [PubMed] [Google Scholar]
- Boroughs LK, Antonyak MA & Cerione RA (2014). A novel mechanism by which tissue transglutaminase activates signaling events that promote cell survival. J Biol Chem 289, 10115–10125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandvold KR, Steffey ME, Fox CC & Soellner MB (2012). Development of a highly selective c‐Src kinase inhibitor. ACS Chem Biol 7, 1393–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bright DP, Aller MI & Brickley SG (2007). Synaptic release generates a tonic GABAA receptor‐mediated conductance that modulates burst precision in thalamic relay neurons. J Neurosci 27, 2560–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broicher T, Kanyshkova T, Landgraf P, Rankovic V, Meuth P, Meuth SG, Pape HC & Budde T (2007). Specific expression of low‐voltage‐activated calcium channel isoforms and splice variants in thalamic local circuit interneurons. Mol Cell Neurosci 36, 132–145. [DOI] [PubMed] [Google Scholar]
- Broicher T, Wettschureck N, Munsch T, Coulon P, Meuth SG, Kanyshkova T, Seidenbecher T, Offermanns S, Pape HC & Budde T (2008). Muscarinic ACh receptor‐mediated control of thalamic activity via Gq/G11‐family G‐proteins. Pflügers Arch 456, 1049–1060. [DOI] [PubMed] [Google Scholar]
- Budde T, Munsch T & Pape H‐C (1998). Distribution of L‐type calcium channels in rat thalamic neurons. Eur J Neurosci 10, 586–597. [DOI] [PubMed] [Google Scholar]
- Burke JE, Vadas O, Berndt A, Finegan T, Perisic O & Williams RL (2011). Dynamics of the phosphoinositide 3‐kinase p110δ interaction with p85α and membranes reveals aspects of regulation distinct from p110α. Structure 19, 1127–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callaghan B, Koh SD & Keef KD (2004). Muscarinic M2 receptor stimulation of Cav1.2b requires phosphatidylinositol 3‐kinase, protein kinase C, and c‐Src. Circ Res 94, 626–633. [DOI] [PubMed] [Google Scholar]
- Castro‐Alamancos MA (2002). Different temporal processing of sensory inputs in the rat thalamus during quiescent and information processing states in vivo . J Physiol 539, 567–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan TO, Rodeck U, Chan AM, Kimmelman AC, Rittenhouse SE, Panayotou G & Tsichlis PN (2002). Small GTPases and tyrosine kinases coregulate a molecular switch in the phosphoinositide 3‐kinase regulatory subunit. Cancer Cell 1, 181–191. [DOI] [PubMed] [Google Scholar]
- Chan TO, Tanaka A, Bjorge JD & Fujita DJ (1990). Association of type I phosphatidylinositol kinase activity with mutationally activated forms of human pp60c‐src. Mol Cell Biol 10, 3280–3283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chellaiah MA, Biswas RS, Yuen D, Alvarez UM & Hruska KA (2001). Phosphatidylinositol 3,4,5‐trisphosphate directs association of Src homology 2‐containing signaling proteins with gelsolin. J Biol Chem 276, 47434–47444. [DOI] [PubMed] [Google Scholar]
- Ching TT, Lin HP, Yang CC, Oliveira M, Lu PJ & Chen CS (2001). Specific binding of the C‐terminal Src homology 2 domain of the p85α subunit of phosphoinositide 3‐kinase to phosphatidylinositol 3,4,5‐trisphosphate. Localization and engineering of the phosphoinositide‐binding motif. J Biol Chem 276, 43932–43938. [DOI] [PubMed] [Google Scholar]
- Chokshi RH, Larsen AT, Bhayana B & Cotten JF (2015). Breathing stimulant compounds inhibit TASK‐3 potassium channel function likely by binding at a common site in the channel pore. Mol Pharmacol 88, 926–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coburn CA, Luo Y, Cui M, Wang J, Soll R, Dong J, Hu B, Lyon MA, Santarelli VP, Kraus RL, Gregan Y, Wang Y, Fox SV, Binns J, Doran SM, Reiss DR, Tannenbaum PL, Gotter AL, Meinke PT & Renger JJ (2012). Discovery of a pharmacologically active antagonist of the two‐pore‐domain potassium channel K2P9.1 (TASK‐3). ChemMedChem 7, 123–133. [DOI] [PubMed] [Google Scholar]
- Cope DW, Hughes SW & Crunelli V (2005). GABAA receptor‐mediated tonic inhibition in thalamic neurons. J Neurosci 25, 11553–11563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox CL & Sherman SM (2000). Control of dendritic outputs of inhibitory interneurons in the lateral geniculate nucleus. Neuron 27, 597–610. [DOI] [PubMed] [Google Scholar]
- Crandall SR & Cox CL (2013). Thalamic microcircuits: presynaptic dendrites form two feedforward inhibitory pathways in thalamus. J Neurophysiol 110, 470–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuevas BD, Lu Y, Mao M, Zhang J, LaPushin R, Siminovitch K & Mills GB (2001). Tyrosine phosphorylation of p85 relieves its inhibitory activity on phosphatidylinositol 3‐kinase. J Biol Chem 276, 27455–27461. [DOI] [PubMed] [Google Scholar]
- Dbouk HA & Backer JM (2013). Novel approaches to inhibitor design for the p110β phosphoinositide 3‐kinase. Trends Pharmacol Sci 34, 149–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng PY, Poudel SK, Rojanathammanee L, Porter JE & Lei S (2007). Serotonin inhibits neuronal excitability by activating two‐pore domain K+ channels in the entorhinal cortex. Mol Pharmacol 72, 208–218. [DOI] [PubMed] [Google Scholar]
- Dobler T, Springauf A, Tovornik S, Weber M, Schmitt A, Sedlmeier R, Wischmeyer E & Döring F (2007). TRESK two‐pore‐domain K+ channels constitute a significant component of background potassium currents in murine dorsal root ganglion neurones. J Physiol 585, 867–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eck MJ, Shoelson SE & Harrison SC (1993). Recognition of a high‐affinity phosphotyrosyl peptide by the Src homology‐2 domain of p56lck. Nature 362, 87–91. [DOI] [PubMed] [Google Scholar]
- Enyedi P & Czirjak G (2010). Molecular background of leak K+ currents: two‐pore domain potassium channels. Physiol Rev 90, 559–605. [DOI] [PubMed] [Google Scholar]
- Fernandez‐Orth J, Ehling P, Ruck T, Pankratz S, Hofmann MS, Landgraf P, Dieterich DC, Smalla KH, Kahne T, Seebohm G, Budde T, Wiendl H, Bittner S & Meuth SG (2016). 14‐3‐3 proteins regulate K2P5.1 surface expression on T lymphocytes. Traffic 18, 29–43. [DOI] [PubMed] [Google Scholar]
- Gabbott PL & Bacon SJ (1994). An oriented framework of neuronal processes in the ventral lateral geniculate nucleus of the rat demonstrated by NADPH diaphorase histochemistry and GABA immunocytochemistry. Neuroscience 60, 417–440. [DOI] [PubMed] [Google Scholar]
- Gabernet L, Jadhav SP, Feldman DE, Carandini M & Scanziani M (2005). Somatosensory integration controlled by dynamic thalamocortical feed‐forward inhibition. Neuron 48, 315–327. [DOI] [PubMed] [Google Scholar]
- Gentili C, Morelli S & Russo De Boland A (2002). Involvement of PI3‐kinase and its association with c‐Src in PTH‐stimulated rat enterocytes. J Cell Biochem 86, 773–783. [DOI] [PubMed] [Google Scholar]
- Geppetti P, Veldhuis NA, Lieu T & Bunnett NW (2015). G protein‐coupled receptors: dynamic machines for signaling pain and itch. Neuron 88, 635–649. [DOI] [PubMed] [Google Scholar]
- Gerthoffer WT (2005). Signal‐transduction pathways that regulate visceral smooth muscle function. III. Coupling of muscarinic receptors to signaling kinases and effector proteins in gastrointestinal smooth muscles. Am J Physiol Gastrointest Liver Physiol 288, G849–G853. [DOI] [PubMed] [Google Scholar]
- Gierten J, Ficker E, Bloehs R, Schlomer K, Kathofer S, Scholz E, Zitron E, Kiesecker C, Bauer A, Becker R, Katus HA, Karle CA & Thomas D (2008). Regulation of two‐pore‐domain (K2P) potassium leak channels by the tyrosine kinase inhibitor genistein. Br J Pharmacol 154, 1680–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross C & Bassell GJ (2014). Neuron‐specific regulation of class I PI3K catalytic subunits and their dysfunction in brain disorders. Front Mol Neurosci 7, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundy D (2015). Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology . J Physiol 593, 2547–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halnes G, Augustinaite S, Heggelund P, Einevoll GT & Migliore M (2011). A multi‐compartment model for interneurons in the dorsal lateral geniculate nucleus. PLoS Comput Biol 7, e1002160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer R, Berrie CP, Birdsall NJ, Burgen AS & Hulme EC (1980). Pirenzepine distinguishes between different subclasses of muscarinic receptors. Nature 283, 90–92. [DOI] [PubMed] [Google Scholar]
- Hammer R, Giraldo E, Schiavi GB, Monferini E & Ladinsky H (1986). Binding profile of a novel cardioselective muscarine receptor antagonist, AF‐DX 116, to membranes of peripheral tissues and brain in the rat. Life Sci 38, 1653–1662. [DOI] [PubMed] [Google Scholar]
- Hanke JH, Gardner JP, Dow RL, Changelian PS, Brissette WH, Weringer EJ, Pollok BA & Connelly PA (1996). Discovery of a novel, potent, and Src family‐selective tyrosine kinase inhibitor. Study of Lck‐ and FynT‐dependent T cell activation. J Biol Chem 271, 695–701. [DOI] [PubMed] [Google Scholar]
- Herrero JL, Roberts MJ, Delicato LS, Gieselmann MA, Dayan P & Thiele A (2008). Acetylcholine contributes through muscarinic receptors to attentional modulation in V1. Nature 454, 1110–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higley MJ & Contreras D (2006). Balanced excitation and inhibition determine spike timing during frequency adaptation. J Neurosci 26, 448–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornigold DC, Mistry R, Raymond PD, Blank JL & Challiss RA (2003). Evidence for cross‐talk between M2 and M3 muscarinic acetylcholine receptors in the regulation of second messenger and extracellular signal‐regulated kinase signalling pathways in Chinese hamster ovary cells. Br J Pharmacol 138, 1340–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Nalli AD, Mahavadi S, Kumar DP & Murthy KS (2014). Inhibition of Gαi activity by Gβγ is mediated by PI 3‐kinase‐γ‐ and cSrc‐dependent tyrosine phosphorylation of Gαi and recruitment of RGS12. Am J Physiol Gastrointest Liver Physiol 306, G802–G810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Zhou H, Mahavadi S, Sriwai W & Murthy KS (2007). Inhibition of Gaq‐dependent PLC‐β1 activity by PKG and PKA is mediated by phosphorylation of RGS4 and GRK2. Am J Physiol Cell Physiol 292, C200–C208. [DOI] [PubMed] [Google Scholar]
- Huster M, Frei E, Hofmann F & Wegener JW (2010). A complex of CaV1.2/PKC is involved in muscarinic signaling in smooth muscle. FASEB J 24, 2651–2659. [DOI] [PubMed] [Google Scholar]
- Igishi T & Gutkind JS (1998). Tyrosine kinases of the Src family participate in signaling to MAP kinase from both Gq and Gi‐coupled receptors. Biochem Biophys Res Commun 244, 5–10. [DOI] [PubMed] [Google Scholar]
- Ito Y, Ariga M, Takahashi S, Takenaka A, Hidaka T & Noguchi T (1997). Changes in tyrosine phosphorylation of insulin receptor and insulin receptor substrate‐1 (IRS‐1) and association of p85 of phosphatidylinositol 3‐kinase with IRS‐1 after feeding in rat liver in vivo. J Endocrinol 154, 267–273. [DOI] [PubMed] [Google Scholar]
- Jeon JP, Hong C, Park EJ, Jeon JH, Cho NH, Kim IG, Choe H, Muallem S, Kim HJ & So I (2012). Selective Gαi subunits as novel direct activators of transient receptor potential canonical (TRPC)4 and TRPC5 channels. J Biol Chem 287, 17029–17039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang D, Han J, Talley EM, Bayliss DA & Kim D (2004). Functional expression of TASK‐1/TASK‐3 heteromers in cerebellar granule cells. J Physiol 554, 64–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennard LE, Chumbley JR, Ranatunga KM, Armstrong SJ, Veale EL & Mathie A (2005). Inhibition of the human two‐pore domain potassium channel, TREK‐1, by fluoxetine and its metabolite norfluoxetine. Br J Pharmacol 144, 821–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilisch M, Lytovchenko O, Arakel EC, Bertinetti D & Schwappach B (2016). A dual phosphorylation switch controls 14‐3‐3‐dependent cell surface expression of TASK‐1. J Cell Sci 129, 831–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilisch M, Lytovchenko O, Schwappach B, Renigunta V & Daut J (2015). The role of protein‐protein interactions in the intracellular traffic of the potassium channels TASK‐1 and TASK‐3. Pflügers Arch 467, 1105–1120. [DOI] [PubMed] [Google Scholar]
- Kim D, Cavanaugh EJ, Kim I & Carroll JL (2009). Heteromeric TASK‐1/TASK‐3 is the major oxygen‐sensitive background K+ channel in rat carotid body glomus cells. J Physiol 587, 2963–2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriplani N, Hermida MA, Brown ER & Leslie NR (2015). Class I PI 3‐kinases: Function and evolution. Adv Biol Regul 59, 53–64. [DOI] [PubMed] [Google Scholar]
- Kruglikov I & Rudy B (2008). Perisomatic GABA release and thalamocortical integration onto neocortical excitatory cells are regulated by neuromodulators. Neuron 58, 911–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann DM, Seneviratne AM & Smrcka AV (2008). Small molecule disruption of G protein βγ subunit signaling inhibits neutrophil chemotaxis and inflammation. Mol Pharmacol 73, 410–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leist M, Datunashvili M, Kanyshkova T, Zobeiri M, Aissaoui A, Cerina M, Romanelli MN, Pape HC & Budde T (2016). Two types of interneurons in the mouse lateral geniculate nucleus are characterized by different h‐current density. Sci Rep 6, 24904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li SS & Li L (2017). SH2 ligand prediction‐guidance for in‐silico screening. Methods Mol Biol 1555, 77–81. [DOI] [PubMed] [Google Scholar]
- Lorincz ML, Kekesi KA, Juhasz G, Crunelli V & Hughes SW (2009). Temporal framing of thalamic relay‐mode firing by phasic inhibition during the alpha rhythm. Neuron 63, 683–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Busillo JM & Benovic JL (2008). M3 muscarinic acetylcholine receptor‐mediated signaling is regulated by distinct mechanisms. Mol Pharmacol 74, 338–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luttrell DK & Luttrell LM (2004). Not so strange bedfellows: G‐protein‐coupled receptors and Src family kinases. Oncogene 23, 7969–7978. [DOI] [PubMed] [Google Scholar]
- MacKay CE & Knock GA (2015). Control of vascular smooth muscle function by Src‐family kinases and reactive oxygen species in health and disease. J Physiol 593, 3815–3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macrez N, Mironneau C, Carricaburu V, Quignard JF, Babich A, Czupalla C, Nurnberg B & Mironneau J (2001). Phosphoinositide 3‐kinase isoforms selectively couple receptors to vascular L‐type Ca2+ channels. Circ Res 89, 692–699. [DOI] [PubMed] [Google Scholar]
- Mahavadi S, Huang J, Sriwai W, Rao KR & Murthy KS (2007). Cross‐regulation of VPAC2 receptor internalization by m2 receptors via c‐Src‐mediated phosphorylation of GRK2. Regul Pept 139, 109–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandelker D, Gabelli SB, Schmidt‐Kittler O, Zhu J, Cheong I, Huang CH, Kinzler KW, Vogelstein B & Amzel LM (2009). A frequent kinase domain mutation that changes the interaction between PI3Kα and the membrane. Proc Natl Acad Sci USA 106, 16996–17001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin R, Guerra B, Alonso R, Ramirez CM & Diaz M (2005). Estrogen activates classical and alternative mechanisms to orchestrate neuroprotection. Curr Neurovasc Res 2, 287–301. [DOI] [PubMed] [Google Scholar]
- Matsuoka H & Inoue M (2015). Src mediates endocytosis of TWIK‐related acid‐sensitive K+ 1 channels in PC12 cells in response to nerve growth factor. Am J Physiol Cell Physiol 309, C251–C263. [DOI] [PubMed] [Google Scholar]
- McCormick DA & Pape H‐C (1988). Acetylcholine inhibits identified interneurons in the cat lateral geniculate nucleus. Nature 334, 246–248. [DOI] [PubMed] [Google Scholar]
- McCormick DA & Prince DA (1985). Two types of muscarinic response to acetylcholine in mammalian cortical neurons. Proc Natl Acad Sci USA 82, 6344–6348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick DA & Prince DA (1986). Mechanisms of action of acetylcholine in the guinea‐pig cerebral cortex in vitro . J Physiol 375, 169–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meuth SG, Aller MI, Munsch T, Schuhmacher T, Seidenbecher T, Kleinschnitz C, Pape HC, Wiendl H, Wisden W & Budde T (2006). The contribution of TASK‐1‐containing channels to the function of dorsal lateral geniculate thalamocortical relay neurons. Mol Pharmacol 69, 1468–1476. [DOI] [PubMed] [Google Scholar]
- Meuth SG, Budde T, Kanyshkova T, Broicher T, Munsch T & Pape H‐C (2003). Contribution of TWIK‐related acid‐sensitive K+ channel 1 (TASK1) and TASK3 channels to the control of activity modes in thalamocortical neurons. J Neurosci 23, 6460–6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meuth SG, Kleinschnitz C, Broicher T, Austinat M, Braeuninger S, Bittner S, Fischer S, Bayliss DA, Budde T, Stoll G & Wiendl H (2009). The neuroprotective impact of the leak potassium channel TASK1 on stroke development in mice. Neurobiol Dis 33, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel AD, Stefanich E & Whiting RL (1989). Direct labeling of rat M3‐muscarinic receptors by [3H]4DAMP. Eur J Pharmacol 166, 459–466. [DOI] [PubMed] [Google Scholar]
- Miled N, Yan Y, Hon WC, Perisic O, Zvelebil M, Inbar Y, Schneidman‐Duhovny D, Wolfson HJ, Backer JM & Williams RL (2007). Mechanism of two classes of cancer mutations in the phosphoinositide 3‐kinase catalytic subunit. Science 317, 239–242. [DOI] [PubMed] [Google Scholar]
- Millar JA, Barratt L, Southan AP, Page KM, Fyffe RE, Robertson B & Mathie A (2000). A functional role for the two‐pore domain potassium channel TASK‐1 in cerebellar granule neurons. Proc Natl Acad Sci USA 97, 3614–3618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JH, Gibson VA & McKinney M (1991). Binding of [3H]AF‐DX 384 to cloned and native muscarinic receptors. J Pharmacol Exp Ther 259, 601–607. [PubMed] [Google Scholar]
- Miller M, Shi J, Zhu Y, Kustov M, Tian JB, Stevens A, Wu M, Xu J, Long S, Yang P, Zholos AV, Salovich JM, Weaver CD, Hopkins CR, Lindsley CW, McManus O, Li M & Zhu MX (2011). Identification of ML204, a novel potent antagonist that selectively modulates native TRPC4/C5 ion channels. J Biol Chem 286, 33436–33446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Missan S, Linsdell P & McDonald TF (2006). Tyrosine kinase and phosphatase regulation of slow delayed‐rectifier K+ current in guinea‐pig ventricular myocytes. J Physiol 573, 469–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moha Ou Maati H, Veyssiere J, Labbal F, Coppola T, Gandin C, Widmann C, Mazella J, Heurteaux C & Borsotto M (2012). Spadin as a new antidepressant: Absence of TREK‐1‐related side effects. Neuropharmacology 62, 278–288. [DOI] [PubMed] [Google Scholar]
- Mulkey DK, Talley EM, Stornetta RL, Siegel AR, West GH, Chen X, Sen N, Mistry AM, Guyenet PG & Bayliss DA (2007). TASK channels determine pH sensitivity in select respiratory neurons but do not contribute to central respiratory chemosensitivity. J Neurosci 27, 14049–14058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munsch T, Budde T & Pape H‐C (1997). Voltage‐activated intracellular calcium transients in thalamic relay cells and interneurons. Neuroreport 8, 2411–2418. [DOI] [PubMed] [Google Scholar]
- Munsch T, Freichel M, Flockerzi V & Pape HC (2003). Contribution of transient receptor potential channels to the control of GABA release from dendrites. Proc Natl Acad Sci USA 100, 16065–16070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaraj C, Tang B, Balint Z, Wygrecka M, Hrzenjak A, Kwapiszewska G, Stacher E, Lindenmann J, Weir EK, Olschewski H & Olschewski A (2013). Src tyrosine kinase is crucial for potassium channel function in human pulmonary arteries. Eur Respir J 41, 85–95. [DOI] [PubMed] [Google Scholar]
- Nelson CD, Perry SJ, Regier DS, Prescott SM, Topham MK & Lefkowitz RJ (2007). Targeting of diacylglycerol degradation to M1 muscarinic receptors by β‐arrestins. Science 315, 663–666. [DOI] [PubMed] [Google Scholar]
- Noh MY, Kim YS, Lee KY, Lee YJ, Kim SH, Yu HJ & Koh SH (2013). The early activation of PI3K strongly enhances the resistance of cortical neurons to hypoxic injury via the activation of downstream targets of the PI3K pathway and the normalization of the levels of PARP activity, ATP, and NAD+ . Mol Neurobiol 47, 757–769. [DOI] [PubMed] [Google Scholar]
- Nozu F, Owyang C & Tsunoda Y (2000). Involvement of phosphoinositide 3‐kinase and its association with pp60src in cholecystokinin‐stimulated pancreatic acinar cells. Eur J Cell Biol 79, 803–809. [DOI] [PubMed] [Google Scholar]
- Oldenburg O, Critz SD, Cohen MV & Downey JM (2003). Acetylcholine‐induced production of reactive oxygen species in adult rabbit ventricular myocytes is dependent on phosphatidylinositol 3‐ and Src‐kinase activation and mitochondrial KATP channel opening. J Mol Cell Cardiol 35, 653–660. [DOI] [PubMed] [Google Scholar]
- Paillart C, Carlier E, Guedin D, Dargent B & Couraud F (1997). Direct block of voltage‐sensitive sodium channels by genistein, a tyrosine kinase inhibitor. J Pharmacol Exp Ther 280, 521–526. [PubMed] [Google Scholar]
- Pape H‐C, Budde T, Mager R & Kisvarday Z (1994). Prevention of Ca2+‐mediated action potentials in GABAergic local circuit neurons of the thalamus by a transient K+ current. J Physiol 478, 403–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pape HC & McCormick DA (1995). Electrophysiological and pharmacological properties of interneurons in the cat dorsal lateral geniculate nucleus. Neuroscience 68, 1105–1125. [DOI] [PubMed] [Google Scholar]
- Plummer KL, Manning KA, Levey AI, Rees HD & Uhlrich DJ (1999). Muscarinic receptor subtypes in the lateral geniculate nucleus: a light and electron microscopic analysis. J Comp Neurol 404, 408–425. [DOI] [PubMed] [Google Scholar]
- Poulet JF, Fernandez LM, Crochet S & Petersen CC (2012). Thalamic control of cortical states. Nat Neurosci 15, 370–372. [DOI] [PubMed] [Google Scholar]
- Putzke C, Wemhöner K, Sachse FB, Rinne S, Schlichthörl G, Li XT, Jae L, Eckhardt I, Wischmeyer E, Wulf H, Preisig‐Müller R, Daut J & Decher N (2007). The acid‐sensitive potassium channel TASK‐1 in rat cardiac muscle. Cardiovasc Res 75, 59–68. [DOI] [PubMed] [Google Scholar]
- Rameh LE, Chen CS & Cantley LC (1995). Phosphatidylinositol (3,4,5)P3 interacts with SH2 domains and modulates PI 3‐kinase association with tyrosine‐phosphorylated proteins. Cell 83, 821–830. [DOI] [PubMed] [Google Scholar]
- Renigunta V, Schlichthörl G & Daut J (2015). Much more than a leak: structure and function of K2P‐channels. Pflügers Arch 467, 867–894. [DOI] [PubMed] [Google Scholar]
- Rinné S, Kiper AK, Schlichthörl G, Dittmann S, Netter MF, Limberg SH, Silbernagel N, Zuzarte M, Moosdorf R, Wulf H, Schulze‐Bahr E, Rolfes C & Decher N (2015). TASK‐1 and TASK‐3 may form heterodimers in human atrial cardiomyocytes. J Mol Cell Cardiol 81, 71–80. [DOI] [PubMed] [Google Scholar]
- Salituro GM, Pelaez F & Zhang BB (2001). Discovery of a small molecule insulin receptor activator. Recent Prog Horm Res 56, 107–126. [DOI] [PubMed] [Google Scholar]
- Sarnago S, Elorza A & Mayor F Jr (1999). Agonist‐dependent phosphorylation of the G protein‐coupled receptor kinase 2 (GRK2) by Src tyrosine kinase. J Biol Chem 274, 34411–34416. [DOI] [PubMed] [Google Scholar]
- Schwindinger WF & Robishaw JD (2001). Heterotrimeric G‐protein βγ‐dimers in growth and differentiation. Oncogene 20, 1653–1660. [DOI] [PubMed] [Google Scholar]
- Seabrook TA, Krahe TE, Govindaiah G & Guido W (2013). Interneurons in the mouse visual thalamus maintain a high degree of retinal convergence throughout postnatal development. Neural Dev 8, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman SM & Guillery RW (2006). Exploring the Thalamus and its Role in Cortical Function. MIT Press, Cambridge, MA. [Google Scholar]
- Shoelson SE, Chatterjee S, Chaudhuri M & White MF (1992). YMXM motifs of IRS‐1 define substrate specificity of the insulin receptor kinase. Proc Natl Acad Sci USA 89, 2027–2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simoncini T, Hafezi‐Moghadam A, Brazil DP, Ley K, Chin WW & Liao JK (2000). Interaction of oestrogen receptor with the regulatory subunit of phosphatidylinositol‐3‐OH kinase. Nature 407, 538–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steriade M, Jones EG & McCormick DA (1997). Thalamus. Elsevier, Amsterdam. [Google Scholar]
- Stover DR, Becker M, Liebetanz J & Lydon NB (1995). Src phosphorylation of the epidermal growth factor receptor at novel sites mediates receptor interaction with Src and P85α. J Biol Chem 270, 15591–15597. [DOI] [PubMed] [Google Scholar]
- Streit AK, Netter MF, Kempf F, Walecki M, Rinne S, Bollepalli MK, Preisig‐Müller R, Renigunta V, Daut J, Baukrowitz T, Sansom MS, Stansfeld PJ & Decher N (2011). A specific two‐pore domain potassium channel blocker defines the structure of the TASK‐1 open pore. J Biol Chem 286, 13977–13984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner PJ & Buckler KJ (2013). Oxygen and mitochondrial inhibitors modulate both monomeric and heteromeric TASK‐1 and TASK‐3 channels in mouse carotid body type‐1 cells. J Physiol 591, 5977–5998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ukhanov K, Brunert D, Corey EA & Ache BW (2011). Phosphoinositide 3‐kinase‐dependent antagonism in mammalian olfactory receptor neurons. J Neurosci 31, 273–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhaesebroeck B, Guillermet‐Guibert J, Graupera M & Bilanges B (2010). The emerging mechanisms of isoform‐specific PI3K signalling. Nat Rev Mol Cell Biol 11, 329–341. [DOI] [PubMed] [Google Scholar]
- Vlahos CJ, Matter WF, Hui KY & Brown RF (1994). A specific inhibitor of phosphatidylinositol 3‐kinase, 2‐(4‐morpholinyl)‐8‐phenyl‐4H‐1‐benzopyran‐4‐one (LY294002). J Biol Chem 269, 5241–5248. [PubMed] [Google Scholar]
- von Willebrand M, Williams S, Saxena M, Gilman J, Tailor P, Jascur T, Amarante‐Mendes GP, Green DR & Mustelin T (1998). Modification of phosphatidylinositol 3‐kinase SH2 domain binding properties by Abl‐ or Lck‐mediated tyrosine phosphorylation at Tyr‐688. J Biol Chem 273, 3994–4000. [DOI] [PubMed] [Google Scholar]
- Walker VG, Ammer A, Cao Z, Clump AC, Jiang BH, Kelley LC, Weed SA, Zot H & Flynn DC (2007). PI3K activation is required for PMA‐directed activation of cSrc by AFAP‐110. Am J Physiol Cell Physiol 293, C119–C132. [DOI] [PubMed] [Google Scholar]
- Wang X, Vaingankar V, Soto Sanchez C, Sommer FT & Hirsch JA (2011). Thalamic interneurons and relay cells use complementary synaptic mechanisms for visual processing. Nat Neurosci 14, 224–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson LJ, Alexander KM, Mohan ML, Bowman AL, Mangmool S, Xiao K, Naga Prasad SV & Rockman HA (2016). Phosphorylation of Src by phosphoinositide 3‐kinase regulates beta‐adrenergic receptor‐mediated EGFR transactivation. Cell Signal 28, 1580–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber MA, Lidor A, Arora S, Salituro GM, Zhang BB & Sidawy AN (2000). A novel insulin mimetic without a proliferative effect on vascular smooth muscle cells. J Vasc Surg 32, 1118–1126. [DOI] [PubMed] [Google Scholar]
- Webster NJ, Park K & Pirrung MC (2003). Signaling effects of demethylasterriquinone B1, a selective insulin receptor modulator. Chembiochem 4, 379–385. [DOI] [PubMed] [Google Scholar]
- Wei J, Walton EA, Milici A & Buccafusco JJ (1994). m1‐m5 muscarinic receptor distribution in rat CNS by RT‐PCR and HPLC. J Neurochem 63, 815–821. [DOI] [PubMed] [Google Scholar]
- Wilke BU, Lindner M, Greifenberg L, Albus A, Kronimus Y, Bunemann M, Leitner MG & Oliver D (2014). Diacylglycerol mediates regulation of TASK potassium channels by Gq‐coupled receptors. Nat Commun 5, 5540. [DOI] [PubMed] [Google Scholar]
- Wolters V, Krasel C, Brockmann J & Bunemann M (2015). Influence of Gαq on the dynamics of M3‐acetylcholine receptor‐G‐protein‐coupled receptor kinase 2 interaction. Mol Pharmacol 87, 9–17. [DOI] [PubMed] [Google Scholar]
- Yu HG, Lu Z, Pan Z & Cohen IS (2004). Tyrosine kinase inhibition differentially regulates heterologously expressed HCN channels. Pflugers Arch 447, 392–400. [DOI] [PubMed] [Google Scholar]
- Yu J, Wjasow C & Backer JM (1998). Regulation of the p85/p110α phosphatidylinositol 3′‐kinase. Distinct roles for the N‐terminal and C‐terminal SH2 domains. J Biol Chem 273, 30199–30203. [DOI] [PubMed] [Google Scholar]
- Zhang X, Vadas O, Perisic O, Anderson KE, Clark J, Hawkins PT, Stephens LR & Williams RL (2011). Structure of lipid kinase p110β/p85β elucidates an unusual SH2‐domain‐mediated inhibitory mechanism. Mol Cell 41, 567–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Liu B, Zhang G, Jia Z, Jia Q, Geng X & Zhang H (2008). Molecular basis for genistein‐induced inhibition of Kir2.3 currents. Pflugers Arch 456, 413–423. [DOI] [PubMed] [Google Scholar]
- Zhu J & Heggelund P (2001). Muscarinic regulation of dendritic and axonal outputs of rat thalamic interneurons: a new cellular mechanism for uncoupling distal dendrites. J Neurosci 21, 1148–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu JJ, Uhlrich DJ & Lytton WW (1999). Burst firing in identified rat geniculate interneurons. Neuroscience 91, 1445–1460. [DOI] [PubMed] [Google Scholar]