

Abstract
Key points
Ivermectin (IVM) is a widely used antiparasitic drug in humans and pets which activates glutamate‐gated Cl− channel in parasites.
It is known that IVM binds to the transmembrane domains (TMs) of several ligand‐gated channels, such as Cys‐loop receptors and P2X receptors.
We found that the G‐protein‐gated inwardly rectifying K+ (GIRK) channel, especially GIRK2, is activated by IVM directly in a Gβγ‐independent manner, but the activation is dependent on phosphatidylinositol‐4,5‐biphosphate (PIP2).
We identified a critical amino acid residue of GIRK2 for activation by IVM, Ile82, located in the slide helix between the TM1 and the N‐terminal cytoplasmic tail domain (CTD).
The results demonstrate that the TM–CTD interface in GIRK channel, rather than the TMs, governs IVM‐mediated activation and provide us with novel insights on the mode of action of IVM in ion channels.
Abstract
Ivermectin (IVM) is a widely used antiparasitic drug in humans and pets which activates glutamate‐gated Cl− channel in parasites. It is also known that IVM binds to the transmembrane domains (TMs) of several ligand‐gated channels, such as Cys‐loop receptors and P2X receptors. In this study, we found that the G‐protein‐gated inwardly rectifying K+ (GIRK) channel is activated by IVM directly. Electrophysiological recordings in Xenopus oocytes revealed that IVM activates GIRK channel in a phosphatidylinositol‐4,5‐biphosphate (PIP2)‐dependent manner, and that the IVM‐mediated GIRK activation is independent of Gβγ subunits. We found that IVM activates GIRK2 more efficiently than GIRK4. In cultured hippocampal neurons, we also observed that IVM activates native GIRK current. Chimeric and mutagenesis analyses identified an amino acid residue unique to GIRK2 among the GIRK family, Ile82, located in the slide helix between the TM1 and the N‐terminal cytoplasmic tail domain (CTD), which is critical for the activation. The results demonstrate that the TM–CTD interface in GIRK channels, rather than the TMs, governs IVM‐mediated activation. These findings provide us with novel insights on the mode of action of IVM in ion channels that could lead to identification of new pharmacophores which activate the GIRK channel.
Keywords: GIRK channel, ivermectin, PIP2
Key points
Ivermectin (IVM) is a widely used antiparasitic drug in humans and pets which activates glutamate‐gated Cl− channel in parasites.
It is known that IVM binds to the transmembrane domains (TMs) of several ligand‐gated channels, such as Cys‐loop receptors and P2X receptors.
We found that the G‐protein‐gated inwardly rectifying K+ (GIRK) channel, especially GIRK2, is activated by IVM directly in a Gβγ‐independent manner, but the activation is dependent on phosphatidylinositol‐4,5‐biphosphate (PIP2).
We identified a critical amino acid residue of GIRK2 for activation by IVM, Ile82, located in the slide helix between the TM1 and the N‐terminal cytoplasmic tail domain (CTD).
The results demonstrate that the TM–CTD interface in GIRK channel, rather than the TMs, governs IVM‐mediated activation and provide us with novel insights on the mode of action of IVM in ion channels.
Abbreviations
- ABM
abamectin
- Ci‐VSP
Ciona intestinalis voltage sensing phosphatase
- CTD
cytoplasmic tail domain
- DOM
doramectin
- EPM
eprinomectin
- GIRK
G‐protein‐gated inwardly rectifying K+
- GPCR
G‐protein‐coupled receptor
- IVM
ivermectin
- PIP2
phosphatidylinositol‐4,5‐biphosphate
- PIP3
phosphatidylinositol‐3,4,5‐trisphosphate
- PTX
pertussis toxin
- TM
transmembrane domain
- TPN‐Q
tertiapin‐Q
- TTX
tetrodotoxin
Introduction
Ivermectin (IVM) is a well‐known and commonly used antiparasitic drug which was discovered and modified by Drs Omura and Campbell (Campbell et al. 1983; Ikeda et al. 1987). IVM kills parasite by increasing the activity of glutamate‐gated Cl− channels at nanomolar concentrations and this results in the hyperpolarization of the nerve and muscle cells of parasite (Cully et al. 1996; Martin, 1996; Burkhart, 2000). IVM also targets several Cys‐loop receptors of ligand‐gated channels, including the glycine receptor, GABAAR, α7 nicotinic acetylcholine receptor and histamine receptor (Sigel & Baur, 1987; Krause et al. 1998; Adelsberger et al. 2000; Shan et al. 2001; Zheng et al. 2002). The P2X4 receptor and farnesoid X receptor are also activated by IVM (Silberberg et al. 2007; Jin et al. 2013). Previous studies demonstrated that IVM modulates channel function by inserting deeply into subunit interfaces on the transmembrane domains (TMs) of these Cys‐loop receptors and the P2X4 receptor, and that the IVM‐binding site is near the extracellular membrane surface (Silberberg et al. 2007; Jelinkova et al. 2008; Lynagh & Lynch, 2012).
In the course of screening of a small‐molecule library towards the identification of modulators of a Gi/o‐coupled orphan G‐protein‐coupled receptor (GPCR) by monitoring G‐protein‐gated inwardly rectifying K+ (GIRK) current, we happened to observe that IVM activates GIRK channels even in the absence of the orphan GPCR. The GIRK family has four members: Kir3.1 (GIRK1), Kir3.2 (GIRK2), Kir3.3 (GIRK3) and Kir3.4 (GIRK4). Most of the GIRK channels are heterotetramers in native tissues, especially Kir3.1–Kir 3.2 in the brain and Kir3.1–Kir 3.4 in the heart, and these channels control the excitability of neurons and the rate of heart beat, respectively (Noma & Trautwein, 1978; Kubo et al. 1993; Lesage et al. 1994; Krapivinsky et al. 1995; Liao et al. 1996; Inanobe et al. 1999; Hibino et al. 2010). GIRK channels are directly activated by Gβγ subunits when Gi/o‐coupled GPCR on the membrane of the same cell is activated by agonists, which results in the dissociation of Gβγ from Gα (Logothetis et al. 1987; Ito et al. 1992; Clapham & Neer, 1993; Reuveny et al. 1994). The channel activity is also known to be directly modulated by phosphatidylinositol 4,5‐bisphosphate (PIP2) and intracellular Na+ (Huang et al. 1998; Sui et al. 1998; Ho & Murrell‐Lagnado, 1999), and activated by some chemicals, including alcohols, dithiothreitol, narigin and ML297 (GIRK1 only) (Kobayashi et al. 1999; Lewohl et al. 1999; Zeidner et al. 2001; Aryal et al. 2009; Yow et al. 2011; Kaufmann et al. 2013).
Since the mechanism of IVM‐mediated GIRK activation is unclear, we aimed to clarify the structural background and action mode of IVM on the GIRK channel function. In the present study, we demonstrate that IVM activates Kir3.1–Kir3.2 (including native GIRK in hippocampal neurons) strongly and Kir3.1–Kir3.4 weakly, in a PIP2‐dependent manner, and we reveal a unique mode of action of IVM on GIRK channels unlike its mode of action on Cys‐loop and P2X4 receptors. We observed that the slide helix between TM1 and the N‐terminal cytoplasmic tail domain (CTD), rather than the TMs of the GIRK channel, acts as a primarily structural determinant for IVM‐mediated activation. Our results present strong evidence indicating that IVM possesses higher efficacy of activation of Kir3.2 than Kir3.4, and that a unique amino acid residue located in the slide helix at the TM–CTD interfaces of Kir3.2, Ile82, plays a critical role for activation by IVM.
Methods
Ethical approval
All experiments were approved by the Animal Care Committee of the National Institutes of Natural Sciences (an umbrella institution of National Institute for Physiological Sciences, Japan), and were performed in accordance with its guidelines. The animal experiments also comply with the policy and regulations of The Journal of Physiology (Grundy, 2015).
cDNA and molecular biology
The cRNA of rat Kir3.1, mouse Kir3.2, mouse Kir3.3, rat Kir3.4, mouse Kir2.1, porcine M2R and Ci‐VSP were used in this study. The Ci‐VSP cDNA was kindly provided by Dr Yasushi Okamura (Osaka University, Osaka, Japan). Mutations in Kir3.2 and Kir3.4 were introduced using the PfuUltra II Fusion HS DNA Polymerase kit (Agilent Technologies, Santa Clara, CA, USA) and verified by DNA sequencing. Complementary RNAs were transcribed from the linearized cDNA by mMessage mMachine kit (Ambion).
Oocyte preparations
Adult female Xenopus laevis (90–120 g) were purchased from Hamamatsu Seibutsu Kyouzai (Hamamatsu, Japan) and housed in the institutional aquatic facility. The total number of frogs for obtaining oocytes in the present study was about 30. Ten to fifteen frogs at a time were maintained in a water tank with sufficient space (L900 × W660 × H500 mm) and normal feeding at 18°C on a light–dark cycle of 12 h light and 12 h dark. Frogs were anaesthetized by 0.15% tricaine for 30 min through skin absorption in a separated water box. Oocytes were then surgically removed from the frogs by making a small incision on the abdomen on ice. The depth of anaesthesia was monitored by the disappearance of withdrawal reflexes. After surgery, frogs recovered from anaesthesia and were then returned to another water tank. Post‐operational frogs did not show signs of distress and recuperated for at least 3 months before the next surgery. After emptying of oocytes, anaesthetized frogs were killed by double pithing. Oocytes were treated with collagenase (2 mg ml–1, Sigma‐Aldrich) for 3–4 h to remove the follicular membrane before injection with 50 nl of cRNA. Injected oocytes were incubated at 17°C in frog Ringer solution containing: 88 mm NaCl, 1 mm KCl, 2.4 mm NaHCO3, 0.3 mm Ca(NO3)2, 0.41 mm CaCl2, 0.82 mm MgSO4 and 15 mm Hepes (pH 7.6) with penicillin–streptomycin solution (0.1%, Sigma‐Aldrich). Oocytes for single‐channel recording were incubated in a hypertonic solution (a double concentration of frog Ringer solution) for 10 min to manually remove the vitelline membrane by forceps. Currents were recorded 2–4 days after the cRNA injection. Oocytes used in some experiments were pretreated with pertussis toxin (PTX) by injection of 3.25 ng PTX 12 h before electrophysiological recording.
Electrophysiological recording of oocytes
Membrane currents were recorded by two‐electrode voltage clamp using an OC‐725C amplifier (Warner Instruments) at room temperature (22–25°C). The bath solution contained: 96 mm KCl, 3 mm MgCl2 and 5 mm Hepes (pH 7.5 with KOH). In some experiments, ND96 was used as bath solution, which contained: 96 mm NaCl, 2 mm KCl, 1 mm MgCl2, 1.8 mm CaCl2 and 5 mm Hepes (pH 7.5 with NaOH). The resistance of glass electrodes was 0.2–0.5 MΩ when filled with the pipette solution containing 3 m potassium acetate and 10 mm KCl. Data acquisition was performed by a digital converter (Digidata 1440, Molecular Devices) and pCLAMP 10.5 software (Molecular Devices). The dose‐dependent response of IVM was measured by sequential application of IVM to the bath solution using an electronic adjustable pipette C5000 (Gilson; 1 ml s−1) and washed out using a perfusion system (1 ml 15 s−1) for inlet and suction pipettes with negative pressure at the outlet. The EC50 and dose–response curve were calculated by fitting data to the Hill equation with SigmaPlot 13 (Hulinks): y = min + (max – min)/(1 + (x/EC50) ^ (–Hill slope)), where x refers to the IVM concentration, y refers to the normalized response (I IVM/I Basal; see detail in Fig. 1 and Results) at a given concentration, ‘max’ refers to the maximum response, ‘min’ refers to the minimum response, EC50 is the concentration of agonist generating half‐maximal response and ‘Hill slope’ is the slope.
Figure 1. Characterization of IVM‐induced GIRK current in Xenopus oocytes.
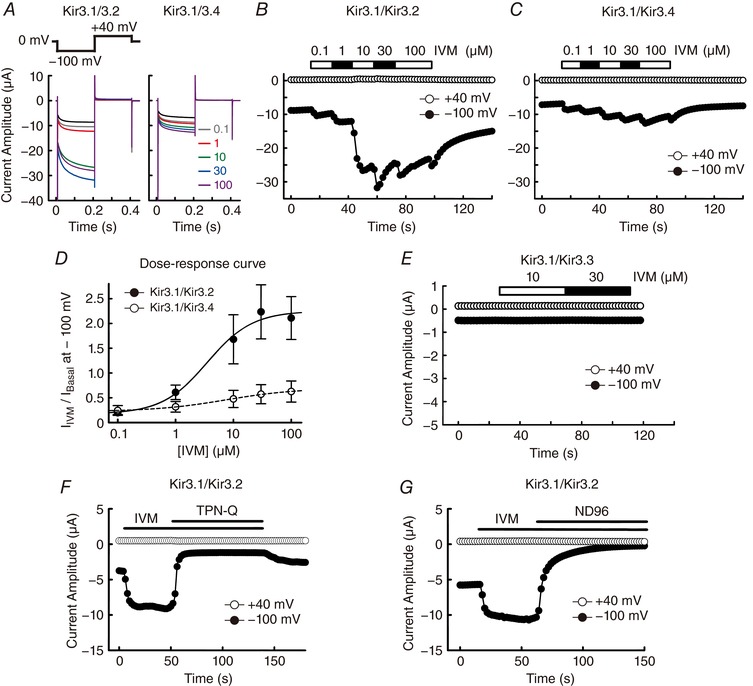
A, currents were recorded in the presence of IVM (0.1–100 μm, coloured traces) using the protocol shown above. The current traces in the oocytes expressing Kir3.1–Kir3.2 (left) and Kir3.1–Kir3.4 (right) are shown. Traces in black indicate the basal current before the application of IVM. B and C, time courses of IVM‐induced current at –100 mV (filled circles) and +40 mV (open circles) in Kir3.1–Kir3.2 (B) and Kir3.1–Kir3.4 (C). D, dose–response curves of the Kir3.1–Kir3.2 (filled circles) and Kir3.1–Kir3.4 groups (open circles). I Basal indicates the current before the application of IVM; I IVM indicates the maximum current in the presence of IVM at a given concentration at –100 mV with the basal current level subtracted. I Basal was normalized to 1. Data are means ± SD, for n = 6 cells. E, time courses of IVM‐induced current at –100 mV (filled circles) and +40 mV (open circles) in an oocyte expressing Kir3.1–Kir3.3. F, 10 μm IVM‐induced current was inhibited by application of 0.1 μm tertiapin‐Q (TPN‐Q) in oocytes expressing Kir3.1–Kir3.2. G, 10 μm IVM‐induced current was eliminated by replacing the extracellular high K+ (96 mm) solution with ND96 solution (96 mm Na+, 2 mm K+) in oocytes expressing Kir3.1–Kir3.2.
Single channel currents were recorded using the cell‐attached patch clamp configuration (Axopatch 200B, Molecular Devices) at room temperature, and the current traces were low‐pass filtered at 2 kHz. The bath and pipette solution contained: 96 mm KCl, 3 mm MgCl2 and 5 mm Hepes (pH 7.4 with KOH). The resistance of the glass electrodes was 2–5 MΩ when filled with the pipette solution. Data acquisition was performed using a digital converter (Digidata 1440) and pCLAMP 10.3 software (Molecular Devices). IVM was added to the bath solution and perfused into a recording chamber. The channel open probability was calculated as NP open. The mean NP open was calculated as (T o)/(T o + T c), where T o refers to total open time, T c refers to total closed time and N refers to the number of channels in the patch.
Primary culture of rat hippocampal neurons
Primary cultures of rat hippocampal neurons were prepared basically as described previously (Fukata et al. 2013). Pregnant Wistar/ST rats were purchased from Japan SLC, Inc. (Hamamatsu, Japan) and used on the day the rats were received. A pregnant rat with embryonic rats (embryonic days 19) was killed by CO2 inhalation (two rats in total) and then embryos (10 embryos per pregnant rat) were removed and decapitated. Hippocampi were dissected from embryonic rat brain and placed in a 10 cm dish on ice with a Ca2+‐ and Mg2+‐free Hanks‐buffered saline (CMF‐HBSS) containing: Hanks’ Balanced Salt solution (Sigma‐Aldrich), 10 mm glucose, 10 mm Hepes (pH 7.3 with NaOH). To dissociate hippocampal neurons, hippocampi were treated with 10 units ml−1 papain (Worthington Biochemical) for 10 min at 37°C. Dissociated neurons were plated onto 12 mm poly‐l‐lysine (Sigma‐Aldrich)‐coated glass coverslips in 24‐well plates (3 × 104 cells well−1) with a plating medium containing: neurobasal medium (ThermoFisher Scientific), 10% FBS and 10 mm Hepes (pH 7.3 with NaOH). Neurons were incubated at 37°C and 5% CO2 for 3 h, and then the medium was replaced by a medium containing: neurobasal medium, B‐27 supplement (ThermoFisher Scientific), 2 mm GlutaMax supplement‐I (ThermoFisher Scientific) and 10 mm Hepes (pH 7.3 with NaOH). Half of the medium was removed and replaced with fresh medium every 7 days. After 13–16 days in culture, neurons were used for electrophysiological recordings.
Electrophysiological recording of primary cultured rat hippocampal neurons
Whole‐cell currents of hippocampal neurons were recorded using the patch‐clamp technique (Axopatch 200B, Molecular Devices) at room temperature, and the current traces were low‐pass filtered at 2 kHz. Coverslips with cultured neurons were submerged in a chamber with a low K+ bath solution containing: 140 mm NaCl, 4 mm KCl, 0.3 mm MgCl2, 1 mm CaCl2, 5.5 mm d‐glucose and 5 mm Hepes (pH 7.4 with NaOH). The resistance of the glass electrodes was 2–5 MΩ when filled with a pipette solution containing: 130 mm KCl, 5 mm Na2ATP, 4 mm MgCl2, 0.1 mm CaCl2, 3 mm EGTA, 0.2 mm GTP and 10 mm Hepes (pH 7.4 with KOH). IVM‐induced currents were recorded in a Ca2+‐free high K+ bath solution containing: 120 mm NaCl, 25 mm KCl, 3 mm MgCl2, 5.5 mm d‐glucose and 5 mm Hepes (pH 7.4 with NaOH), supplemented with 1 μm tetrodotoxin (TTX, Wako, Osaka, Japan) and 10 μm ZD7288 (Abcam, a blocker of hyperpolarization‐activated cation current (I h)). Data acquisition was performed using a digital converter (Digidata 1322) and pCLAMP 9.2 software (Molecular Devices). IVM and a GIRK channel blocker, tertiapin‐Q (TPN‐Q, Abcam), were applied to the Ca2+‐free high K+ bath solution and perfused to the recording chamber using VC‐8 valve controller (Warner Instruments) and SF‐77B perfusion fast‐step (Warner Instruments).
Chemicals
In the initial screening to search for modulator(s) of an orphan Gi/o‐coupled GPCR, a small‐molecule library in the laboratory of one of the authors (M. U.) was used. Other chemicals were purchased from Sigma‐Aldrich. IVM was dissolved in DMSO by sonication; abamectin (ABM), doramectin (DOM) and eprinomectin (EPM) were dissolved in ethanol. Application of chemicals in experiments were diluted to a final solvent concentration <0.1% in the bath solution, which elicited a negligible effect on GIRK channel.
Data analysis and statistics
Data were analysed by Clampfit 10.5 (Molecular Devices) and SigmaPlot 13 (Hulinks). All data are shown as means ± SD from n cells. Statistical differences were evaluated using one‐way ANOVA and Dunnett's or Dunn's multiple comparison tests, comparing every group with the control group. A Student's paired t test was used to evaluate the statistical difference between the IVM‐induced current before and after application of TPN‐Q in neurons. Values of P < 0.05 were judged to be statistically significant.
Results
IVM activates GIRK current in Xenopus oocytes
Through heterologous expression of GIRK heteromeric complexes, Kir3.1–Kir3.2 or Kir3.1–Kir3.4, in Xenopus oocytes, we observed electrophysiologically that application of IVM (0.1–100 μm) induced an increase in inward current at –100 mV (Fig. 1 A–C). The EC50 of IVM for the Kir3.1–Kir3.2 heterotetramer was 3.5 ± 2.0 μm (n = 6); that for the Kir3.1–Kir3.4 heterotetramer was 7.5 ± 1.9 μm (n = 6) (Fig. 1 D). The dose–response relationship was calculated from the ratio of the IVM‐induced current (I IVM, the basal current was subtracted) to the basal current amplitude before the application of IVM (I Basal) (Fig. 1 A–D). In the presence of a saturated concentration of IVM (100 μm), the ratio of I IVM to I Basal (I IVM/I Basal) of Kir3.1–Kir3.2 was 2.11 ± 0.43 (n = 6), whereas that of Kir3.1–Kir3.4 was 0.63 ± 0.21 (n = 6). The results show that IVM possesses a higher efficacy in the activation of Kir3.1–Kir3.2 than that of Kir3.1–Kir3.4. We also examined the effect of IVM on the Kir3.1–Kir3.3 heterotetramer and observed a negligible current in the presence or absence of IVM (Fig. 1 E). This is consistent with previous studies showing that expression of Kir3.3 reduces the activity of Kir3.3–Kir3.x heterotetramers (Kofuji et al. 1995; Lesage et al. 1995).
IVM‐induced current was blocked by TPN‐Q, a specific inhibitor of GIRK channel (Fig. 1 F). When we removed extracellular K+ ions by replacing high K+ solution (96 mm) with high Na+ solution (96 mm), ion flux was eliminated at both –100 mV and 40 mV (Fig. 1 G). These results confirm that IVM activates GIRK channels and does not change the ion selectivity.
Avermectins activate Kir3.1–Kir3.2 current in Xenopus oocytes
IVM and its analogues are derived from avermectin, which was isolated from Streptomyces avermitilis. These 16‐membered macrocyclic lactone derivatives, known as avermectins, have potent antiparasitic property and possess similar structures to IVM (Fig. 2 A). Here we examined the effects of these avermectins on GIRK current in the same cell, using abamectin (ABM), doramectin (DOM), eprinomectin (EPM) and IVM. At 10 μm these avermectins activated GIRK current in oocytes expressing Kir3.1–Kir3.2, and ABM showed a similarly high efficacy to IVM (Fig. 2 B and C). ABM, DOM and EPM possess low water solubility and they are only partially dissolved in DMSO; therefore in our experiments these drugs were fully dissolved in ethanol and diluted to a final solvent concentration <0.1% in the bath solution. Since ethanol is known to activate GIRK channels directly (Kobayashi et al. 1999), we compared the effects of vehicle (0.1% ethanol) with ABM (10 μm) on GIRK activation in the same cell. We observed that application of vehicle induced a small increase in current amplitude and ABM‐induced GIRK current was significantly larger than that of vehicle (Fig. 2 D and E). These results demonstrate that avermectins act as GIRK activators.
Figure 2. Activation of Kir3.1–Kir3.2 current by avermectins in Xenopus oocytes.
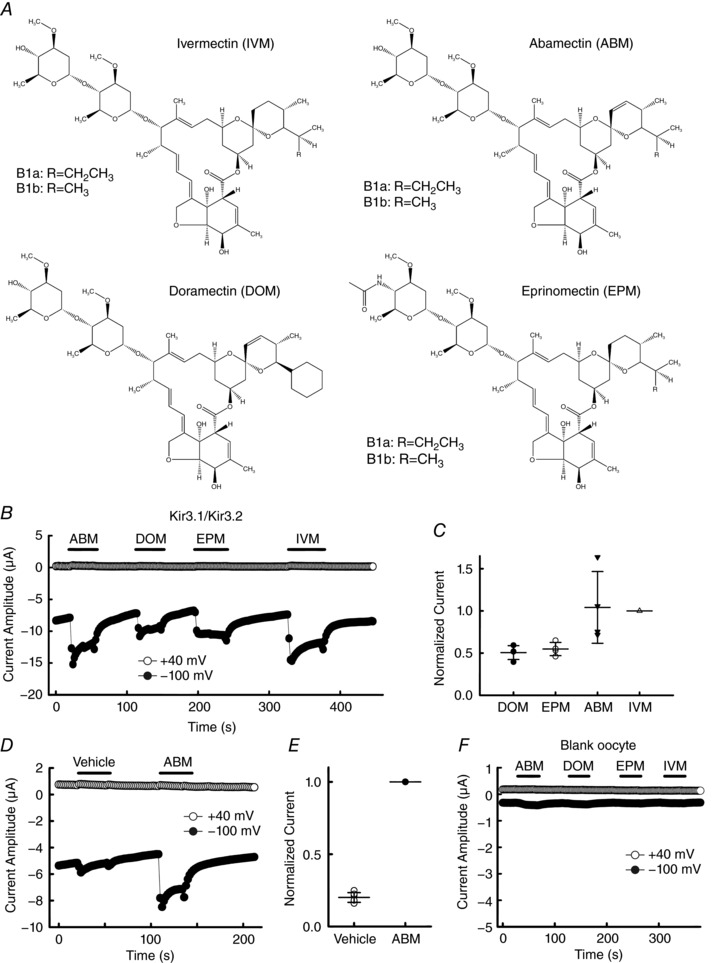
A, chemical structure of IVM analogues (avermectins). B, time courses of IVM analogue‐induced current at –100 mV (filled circles) and +40 mV (open circles) in oocytes expressing Kir3.1–Kir3.2. C, normalized current induced by application of 10 μm of each IVM analogue to the same cell. Current induced by 10 μm IVM was normalized to 1. Each symbol shows raw data (from n = 4 cells) and some of the symbols overlap with each other. Each horizontal bar shows the mean of the group with error bars (SD). D, effects of 10 μm abamectin (ABM) and the vehicle (0.1% ethanol) for ABM, doramectin (DOM) and eprinomectin (EPM) on GIRK current. E, normalized current induced by application of the vehicle (0.1% ethanol, open symbols) and ABM (filled symbol). Current induced by 10 μm ABM was normalized to 1. Data are means ± SD, n = 6 cells. F, current induced by 10 μm of each IVM analogue in ‘blank’ oocytes.
A previous study suggested that ABM formed hydrophobic pores in an artificial bilayer lipid membrane in a Teflon cell (Fomkina et al. 2001). Here we examined the effects of 10 μm ABM, DOM, EPM and IVM in ‘blank’ oocytes with no injection of cRNA. We observed that only ABM showed a negligible increase in current amplitude and the others showed almost no change in current amplitude (Fig. 2 F). Since the current amplitude of ABM‐induced current in oocytes expressing Kir3.1–Kir3.2 was clearly larger than that of blank oocytes, we can exclude the contribution of an ion flux through the ABM‐formed pores in the plasma membrane of oocytes.
IVM activates native GIRK current in rat hippocampal neurons
Since we observed that IVM strongly activates Kir3.1–Kir3.2 using an in vitro expression system as shown in Fig. 1, we next examined the effects of IVM on native GIRK current in neurons. Kir3.1–Kir3.2 heterotetramers are widely distributed in the brain, especially hippocampus, cerebral cortex and cerebellum (Liao et al. 1996). Here we used cultured neurons of rat hippocampus for the measurement of native GIRK current. Whole‐cell currents of hippocampal neurons were recorded by voltage clamp at a holding potential of –80 mV with a voltage ramp from –100 mV to 40 mV for 0.7 s, repeated every 2 s. A Ca2+‐free high K+ (25 mm) bath solution supplemented with TTX and ZD7288 was used to block the current through Na+ channels, Ca2+‐activated channels and hyperpolarization‐activated cation current (I h). Current–voltage (I–V) relationships of the basal neuronal current before (black traces) and after application of IVM (10 μm) without (red traces) or with TPN‐Q (3 μm, blue traces) are shown in Fig. 3 A and C, and the average of the reversal potential was –34.8 ± 7.8 mV (n = 7). Application of TPN‐Q alone did not show a significant change in current amplitude of basal neuronal current. Application of 10 μm IVM increased the amplitude of the inward current (Fig. 3 A–D). The IVM‐induced current in hippocampal neurons (hippocampal I IVM) was partially inhibited by 3 μm TPN‐Q: TPN‐Q inhibited 71.5% of hippocampal I IVM in Fig. 3 A–B and 35.8% in Fig. 3 C–D at –100 mV. The average of the hippocampal I IVM was 188.2 ± 122.3 pA (n = 7, shown in red) in the presence of IVM alone and 105.4 ± 115.2 pA (n = 7, shown in blue) in the presence of IVM and TPN‐Q (Fig. 3 E). The presence of TPN‐Q significantly decreased the current amplitude of hippocampal I IVM (normalized ΔI was 0.49 ± 0.27 in the presence of IVM and TPN‐Q, each n = 7, P = 0.003 in Fig. 3 F), suggesting that IVM activates native GIRK current in hippocampal neurons.
Figure 3. IVM‐induced GIRK current in rat hippocampal neurons.
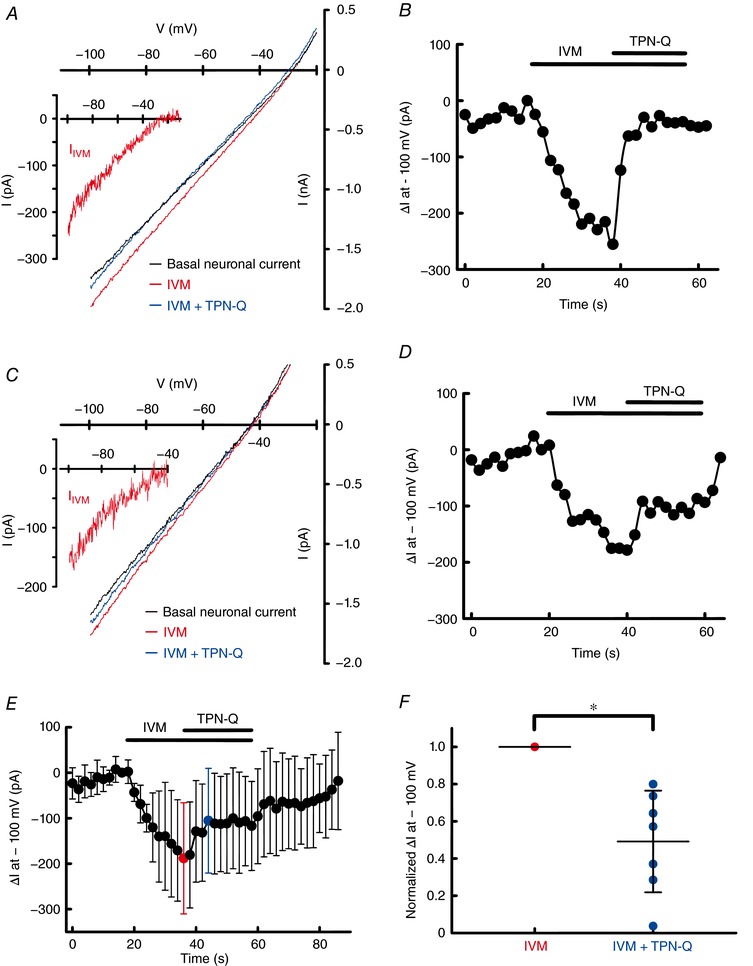
A and C, whole‐cell currents of hippocampal neurons were recorded in the absence (black traces) and presence of IVM (10 μm) without (red traces) or with TPN‐Q (3 μm, blue traces) in a Ca2+‐free high K+ (25 mm) bath solution at a holding potential of –80 mV with a voltage ramp from –100 mV to +40 mV for 0.7 s, repeated every 2 s. The inset I–V relationship of IVM‐induced current in hippocampal neurons (hippocampal I IVM) was obtained by subtracting the basal neuronal current from the current recorded in the presence of IVM. B and D, time courses of change in current amplitude (ΔI, the basal neuronal current was subtracted) in the absence and presence of IVM and TPN‐Q at –100 mV. E, time courses of averaged ΔI at –100 mV (n = 7 cells). F, normalized ΔI induced by IVM in the absence (red symbols) and presence of TPN‐Q (blue symbols). Current induced by IVM was normalized to 1. Each symbol shows raw data (from n = 7 cells) and some of the symbols overlap with each other. Each horizontal bar shows the mean of the group with error bars (SD). *Statistically significant difference (P < 0.05).
The partial block of the hippocampal I IVM by TPN‐Q (Fig. 3 A–F) may be due to an insufficient block of GIRK current by TPN‐Q in the patched single neuron. Another possibility is that IVM activates not only GIRK channels in hippocampal neurons, but also an inward current via an unknown ion channel which is insensitive to TPN‐Q.
IVM‐mediated Kir3.1–Kir3.2 activation is independent of Gβγ in Xenopus oocytes
Since the GIRK channel is known to be directly activated by Gβγ subunits via the stimulation of GPCRs, here we examined the possibility that the effect of IVM is on the endogenous GPCR linked to the GIRK channel, using application of pertussis toxin (PTX), which uncouples Gi/o from GPCRs and thereby prevents the activation of GIRK channels due to increased Gβγ. In the oocytes co‐expressing Kir3.1–Kir3.2 with M2 muscarinic receptor (M2R), pretreatment with PTX inhibited the M2R‐mediated activation of GIRK current in the presence of the M2R agonist ACh (Fig. 4 A). However, pretreatment with PTX did not inhibit the IVM‐induced GIRK current, suggesting that IVM‐induced Kir3.1–Kir3.2 activation is independent of Gi/o. To further confirm this finding, we examined the effect of IVM on a Gβγ‐insensitive mutant of Kir3.2. Leu344 in the βL–βM loop of the cytoplasmic domain of Kir3.2 is critical for Gβγ binding (Whorton & MacKinnon, 2011) and a mutation of Leu344 to Glu (L344E) prevents Gβγ‐mediated activation of Kir3.2 (Finley et al. 2004). In the oocytes co‐expressing Kir3.2 L344E with M2R, we observed that L344E showed no response to the application of 1 μm ACh but it was sensitive to IVM (10 μm) (Fig. 4 B), showing that IVM‐induced Kir3.2 activation is independent of Gβγ. These results demonstrate that IVM directly activates GIRK channel without activating endogenous GPCRs or G‐proteins in oocytes.
Figure 4. IVM‐induced Kir3.1–Kir3.2 current is independent of Gβγ .
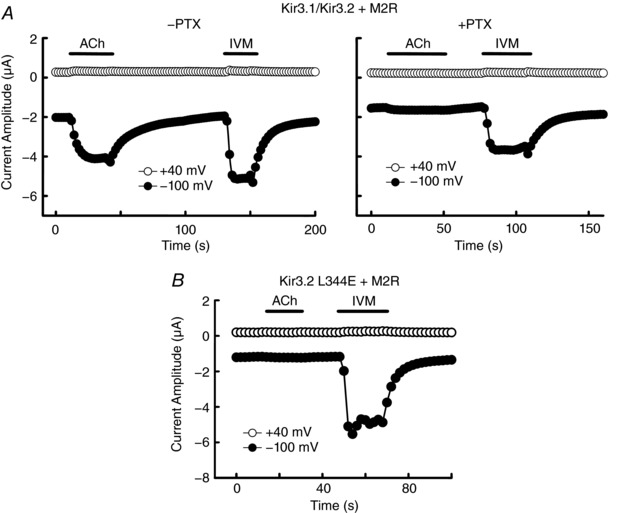
A, currents were recorded in the presence of 1 μm ACh or 10 μm IVM at –100 mV (filled circles) and +40 mV (open circles) in the oocytes expressing Kir3.1–Kir3.2 and M2R without (left) or with (right) pretreatment by pertussis toxin (PTX). B, currents were recorded in the presence of 1 μm ACh or 10 μm IVM in the oocytes expressing the Kir3.2 L344E mutant and M2R.
Single channel activity of Kir3.1–Kir3.2 in the presence of IVM in Xenopus oocytes
We also analysed the single channel behaviour of Kir3.1–Kir3.2 in the presence of IVM using cell‐attached patch recording in oocytes. Single channel activity was recorded at a holding potential of –100 mV which showed transitions between the closed and open state in the absence (left) and presence (right) of 10 μm IVM (Fig. 5 A). Since there was no exogenous Gβγ in the oocytes expressing Kir3.1–Kir3.2 alone, the single channel activity showed a few flickering openings between the long closures before applying IVM. After application of IVM to the bath solution, burst activities with longer open durations were observed. The open probability (NP open) of Kir3.1–Kir3.2 was significantly increased by the application of IVM (NP open in the presence of IVM was 0.04 ± 0.02, n = 6, P = 0.008) (Fig. 5 B). The results suggest that the binding of IVM shifts the equilibrium between states to the open state of GIRK channel.
Figure 5. Effects of IVM on the single channel activity of Kir3.1–Kir3.2.
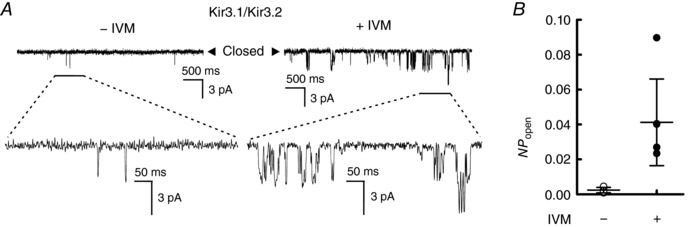
A, single channel activity of Kir3.1–Kir3.2 was recorded at a holding potential of –100 mV before (left) and after (right) the application of 10 μm IVM in the same oocyte under the cell‐attached configuration. B, open probability (NP open) of Kir3.1–Kir3.2 in the absence (open symbols) and presence (filled symbols) of 10 μm IVM. The NP open was measured from continues single channel recording for 5 s. Data show the raw data of NP open (circles) and the means ± SD from the recording with a duration of 30 s before and after the application of IVM in the same cell. Some symbols overlap with each other.
IVM‐mediated Kir3.1–Kir3.2 activation is dependent on the presence of PIP2 in Xenopus oocytes
Since GIRK channel is modulated by PIP2, here we co‐expressed Kir3.1–Kir3.2 with Ciona intestinalis voltage sensing phosphatase (Ci‐VSP) (Murata et al. 2005) in oocytes to control endogenous PIP2 levels. At highly depolarized potentials, the enzyme activity of Ci‐VSP increases, resulting in the dephosphorylation of phosphatidylinositol‐3,4,5‐trisphosphate (PIP3) and PIP2 (Sakata et al. 2011). We analysed the effect of the depolarization‐dependent phosphatase activity of Ci‐VSP on the co‐expressed Kir3.1–Kir3.2 using various interval potentials (0, 10, 20, 40, 60, 80 mV) between each hyperpolarized test pulse (–100 mV) (Fig. 6 A and B). The effect of depolarization‐mediated activation of Ci‐VSP reached a saturated level in terms of the reduction of GIRK current when the interval potential was higher than 60 mV, and the current recovered after changing the interval potential from a depolarized potential (80 mV) to resting potential (0 mV) (Fig. 6 B and C). We also confirmed the Ci‐VSP is indeed functional in our heterologous expression system by analysing the effect of depolarization‐mediated activation of Ci‐VSP on the channel activity of Kir2.1 (Fig. 6 D), which is known to possess a higher affinity for PIP2 than Kir3.2 (Murata & Okamura, 2007). IVM‐induced GIRK current was almost eliminated by depolarization (60 mV)‐mediated reduction in PIP2 levels in the presence of Ci‐VSP and not in the absence of Ci‐VSP (Fig. 6 E and F). Depolarization of the interval potential from 0 mV to 60 mV reduced 81.5 ± 5.1% (n = 5) of I IVM by Ci‐VSP. A high concentration of wortmannin (a phosphoinositide 3‐kinase and phosphoinositide 4‐kinase inhibitor), which is commonly used to reduce PIP2 production, reduced the response to IVM and prevented the recovery of GIRK current after changing the interval potential from a depolarized potential (60 mV) to resting potential (0 mV) (Fig. 6 G). These results show that IVM‐mediated Kir3.1–Kir3.2 activation is dependent on PIP2.
Figure 6. Effects of PIP2 level modulation by Ci‐VSP on IVM‐induced Kir3.1–Kir3.2 current.
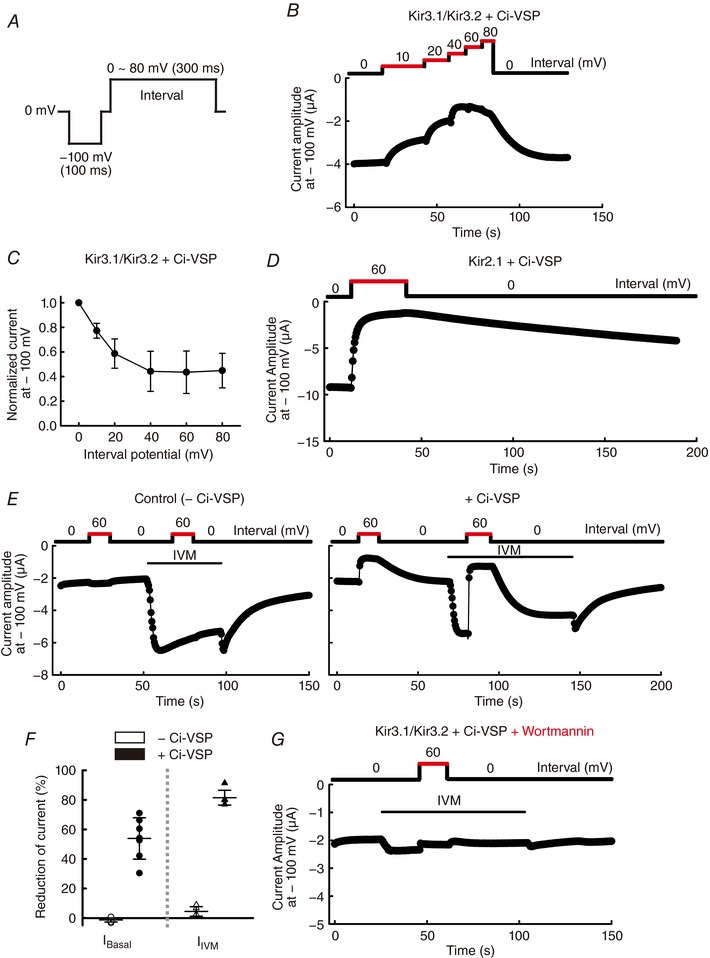
A, recording protocol for measurement of the effects of the voltage‐dependent phosphatase activity of Ci‐VSP on GIRK current in oocytes expressing Kir3.1–Kir3.2 with Ci‐VSP. Recording pulse was repeated every 0.5 s. B, time course of basal GIRK current at –100 mV with different interval potentials (0–80 mV) shown above was recorded in the same cell. C, normalized current plotted against the interval potential. Current with 0 mV of interval potential was normalized to 1 (n = 6 cells). D, time course of Kir2.1 current at –100 mV with 0 mV or 60 mV of interval potential shown above, in oocytes expressing Kir2.1 and Ci‐VSP. E, current cells at –100 mV with 0 mV or 60 mV of interval potential in oocytes without (left) or with (right) co‐expression of Ci‐VSP triggered by application of 10 μm IVM. F, reduction of current amplitude from the level with 0 mV interval potential (as 100%) to that with 60 mV interval potential in oocytes without Ci‐VSP (open symbols) and with Ci‐VSP (filled symbols). Data show the raw data and the means ± SD, n = 6 cells for I Basal (circles); n = 5 for I IVM (triangles). Some symbols overlap with each other. G, pretreatment by 30 μm wortmannin for 1 h before recording prevented the recovery of GIRK current upon altering the interval potential from 60 mV to 0 mV in the presence of 10 μm IVM in oocytes expressing Kir3.1–Kir3.2 and Ci‐VSP.
Identification of the structural determinants of Kir3.2 for IVM‐mediated activation
Since Kir3.2 channels showed an especially remarkable response to IVM among GIRK subtypes (Fig. 1), we speculated that some unique amino acid residue(s) in Kir3.2 channel may play important roles in IVM action (Fig. 7 A). Previous reports showed that IVM binds to the TMs of Cys‐loop receptors and P2X4 receptor (Silberberg et al. 2007; Lynagh & Lynch, 2012), and therefore our foremost speculation was that the TMs of Kir3.2 also determine the IVM‐mediated activation of GIRK channel. To examine this hypothesis, we constructed chimeras between Kir3.2 and Kir3.4 by replacing various regions of Kir3.2 with those of Kir3.4 (Fig. 7 A and B). These chimeric channels were functional when co‐expressed with Kir3.1 in oocytes and showed responses to the application of 10 μm IVM (Fig. 7 C). Most of the chimeric channels showed similar I IVM/I Basal to wild‐type (WT) Kir3.1–Kir3.2 (1.44 ± 0.43, n = 9), but only the chimera which possessed the N‐terminal cytoplasmic segment of Kir3.4 showed a significant decrease (0.42 ± 0.13, n = 6, P = 0.008) and this was close to the I IVM/I Basa of Kir3.1–Kir3.4 (0.37 ± 0.08, n = 10). This suggests that the IVM effect is most likely modulated by the N‐terminus region. The TMs, pore‐forming region and C‐terminal region of GIRK channel are not the primarily structural determinants for the IVM effect. Therefore, we next focused on the N‐terminal region and constructed mutants by replacing the unique amino acid residues of Kir3.2 with those of Kir3.4 (Fig. 7 A and D). Among these mutants, only the mutants containing the Ile82 to Leu (I82L) mutation showed a significant decrease in I IVM/I Basal in comparison to Kir3.2 WT (1.51 ± 0.46 for WT and 0.31 ± 0.04 for T80S and I82L, each n = 6, P = 0.002 in Fig. 7 E; 1.56 ± 0.34 for WT and 0.49 ± 0.10 for I82L, each n = 6–8, P < 0.001 in Fig. 7 F), showing that Ile82 of Kir3.2 is critical for the IVM‐mediated GIRK activation. We also examined the mutant of a critical cysteine for dithiothreitol‐dependent GIRK activation, C65A, and observed that Cys65 is not necessary for IVM‐induced current increase (Fig. 7 E). In Kir3.4, mutation of Leu77 (a residue located at the same position as Ile82 in Kir3.2) to Ile (L77I) increased the response to IVM (I IVM/I Basal was 0.65 ± 0.23 for Kir3.4 WT; 1.53 ± 0.27 for L77I; 1.67 ± 0.46 for S75T and L77I, each n = 6–8 in Fig. 7 F), suggesting that the L77I mutation of Kir3.4 rescues the sensitivity to IVM compared with Kir3.4 WT. These results show that the Ile82 in Kir3.2 is indispensable for the IVM effect.
Figure 7. Identification of structural determinants of Kir3.2 for IVM‐mediated activation.
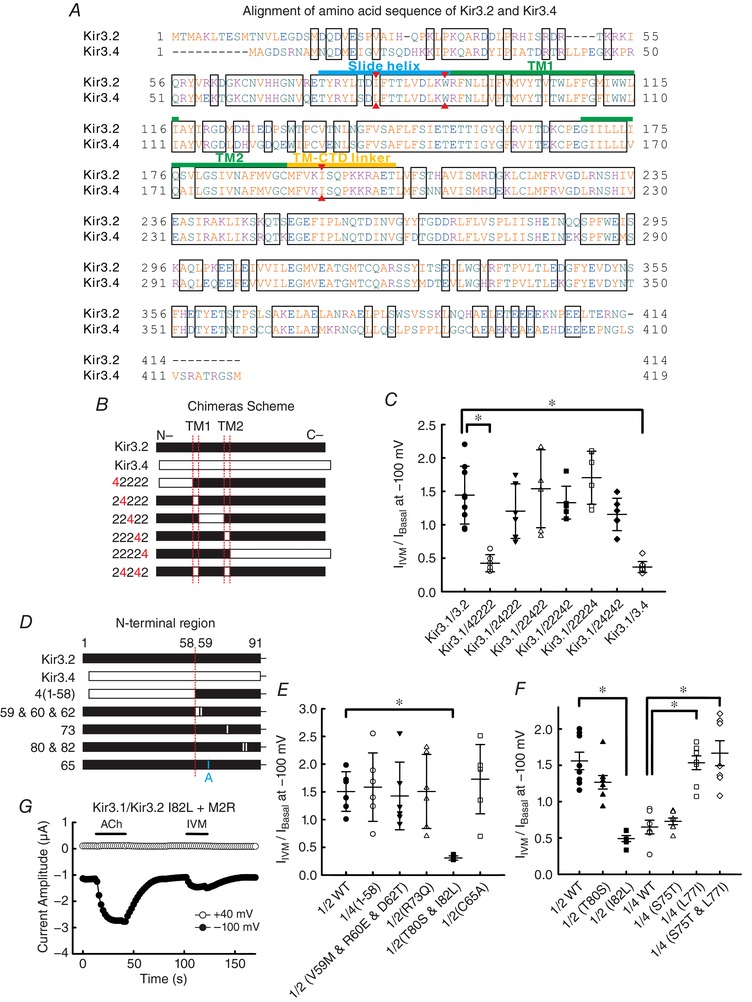
A, alignment of amino acid sequence of Kir3.2 and Kir3.4. Red triangles indicate the locations of I82, W91 and I195 in Kir3.2 and their corresponding positions in Kir3.4. B, schematic representations of Kir3.2, Kir3.4 and chimeras. Segments from Kir3.2 are shown in filled bars and segments from Kir3.4 are shown in open bars. C, the ratio of I Basal to I IVM (I Basal/I IVM) was recorded with the protocol shown in Fig. 1 A in oocytes co‐expressing Kir3.1 with Kir3.2 WT, Kir3.4 WT or each chimera shown in B (n = 9 cells for Kir3.1–Kir3.2 WT; n = 10 for Kir3.1–Kir3.4 WT; n = 6 for others). D, schematic presentations of the N‐terminal region of Kir3.2, Kir3.4, chimeras and mutants. E, the I Basal/I IVM of each channel subunit shown in D co‐expressed with Kir3.1 (n = 6 cells for each). F, the I Basal/I IVM of Kir3.2 and Kir3.4 mutants with co‐expressed with Kir3.1 (n = 6 cells for Kir3.1–Kir3.2 I82L and Kir3.1–Kir3.4 WT; n = 7 for Kir3.1–Kir3.4 S75T and L77I; n = 8 for others). All data show the raw data and the means ± SD. *Statistically significant difference (P < 0.05). G, currents were recorded in the presence of 1 μm ACh or 10 μm IVM in the oocytes expressing Kir3.1–Kir3.2 I82L and M2R.
There is a possibility that the weak IVM effect of Kir3.2 I82L is due to a high basal activity of the channel. To examine this possibility, we analysed the GPCR‐induced activation of this mutant in oocytes co‐expressing Kir3.1–Kir3.2 I82L and M2R (Fig. 7 G). We observed that the 1 μm ACh‐induced current (I ACh) was much larger than the 10 μm IVM‐induced current. The I ACh/I Basal was 1.42 and the I IVM/I Basal was 0.37, suggesting that Kir3.1–Kir3.2 I82L is still gated by Gβγ/GPCR. The normal Gβγ‐gated property of Kir3.2 I82L is also consistent with that of Kir3.4 WT, in which a corresponding Leu is also located. The result shows that the weak IVM effect of Kir3.1–Kir3.2 I82L is not due to a high basal channel activity.
Roles of the branched side‐chain of Ile82 of Kir3.2 in IVM‐mediated activation
In the N‐terminal region of Kir3.2, Ile82 locates in the slide helix between the TM1 and N‐terminal CTDs (Figs 7 A and 8 A) (Whorton & MacKinnon, 2011), and the amino acid residue at the corresponding position in Kir3.1, Kir3.3 and Kir3.4 is Leu. Ile82 of Kir3.2 is a branched side‐chain amino acid with a methyl group towards the inner transmembrane helix (TM2), and this methyl group is lacking in the Leu77 of Kir3.4 (Fig. 8 B and C). Therefore, we speculated that this methyl group on the branched side‐chain is critical for IVM‐mediated GIRK activation. To verify this hypothesis, we mutated Ile82 to Val (I82V), a branched side‐chain amino acid with an inner methyl group, and compared the effect of IVM on I82V with other mutants at position 82, including mutations of Ile82 to Leu (I82L), Ala (I82A) and Trp (I82W) (Fig. 8 D). Co‐expression of Kir3.1 with Kir3.2 I82V showed significantly larger I IVM/I Basal than those of I82L, I82A and I82W (1.29 ± 0.13 for I82V; 0.49 ± 0.10 for I82L; 0.36 ± 0.06 for I82A; 0.17 ± 0.05 for I82W, each n = 6, P < 0.001 for I82L, I82A and I82W when compared with I82V). These results support our speculation that the branched side‐chain amino acid with a methyl group towards TM2 contributes to the IVM‐dependent activation and that the large side‐chain of Trp facing towards the outer side of the channel may disturb the rearrangement of TM‐CTD interface upon IVM binding.
Figure 8. Effects of single point mutants of Kir3.2 I82 on IVM‐induced current.
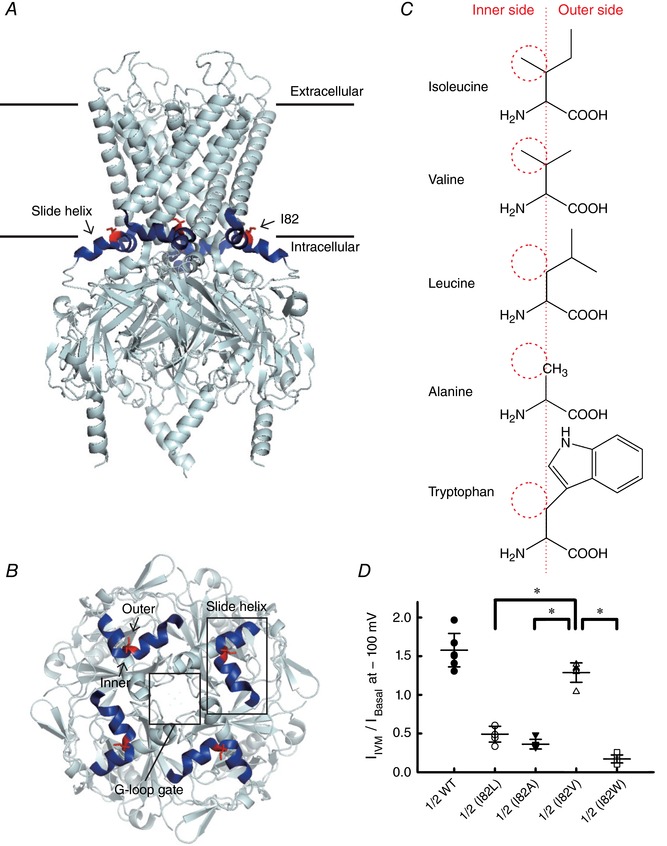
A, the crystal structure of Kir3.2 (Protein Data Bank, PDB ID 3SYA) (Whorton & MacKinnon, 2011). The location of the slide helix between the TM1 and the N‐terminal CTD is shown in blue and I82 is shown in red. B, top view of the slide helix, I82 and the G‐loop gate of Kir3.2. C, structure of amino acid used in the single point mutation experiment shown in D. Red dashed circles indicate the presence and absence of the methyl group towards the inner TM helix (TM2). D, the I Basal/I IVM of single point mutants of Kir3.2 I82 co‐expressed with Kir3.1. Data show the raw data and the means ± SD, n = 7 cells for Kir3.1–Kir3.2 WT; n = 6 for others. *Statistically significant difference (P < 0.05).
Roles of other amino acid residues surrounding Ile82 of Kir3.2
We further examined several mutants of amino acid residues surrounding Ile82 of Kir3.2 based on the Kir3.2 crystal structure (Whorton & MacKinnon, 2011) (Fig. 9 A). Mutations of Phe83 to Ala (F83A) and Phe186 to Trp (F186W) induced very little basal current which hindered the investigation of their roles in IVM effect. Mutations of Trp91 to Ala (W91A) and Ile195 to Ala (I195A) reduced the effects of IVM on GIRK current (I IVM/I Basal was 0.61 ± 0.08 for W91A; 0.26 ± 0.06 for I195A, each n = 6–7) and mutations of Leu86 to Ala (L86A) and Phe186 to Ala (F186A) showed no significant difference in I IVM/I Basal from that of WT (Fig. 9 B). The results show that Ile82 and Trp91 located in the slide helix and Ile195 located in the TM–CTD linker contribute to the IVM‐mediated GIRK activation, presumably by forming part of the IVM‐binding pocket in the TM–CTD interface (Fig. 9 C).
Figure 9. Effects of single point mutants of other amino acid residues surrounding Kir3.2 I82 on IVM‐induced current.
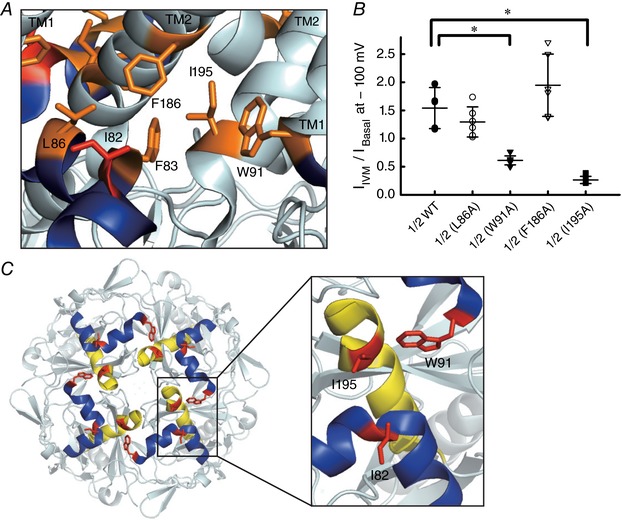
A, the expanded view of the crystal structure of Kir3.2 surrounding I82 (PDB ID 3SYA). The location of single point mutants surrounding I82 (red) in this study are shown in orange. The slide helix is shown in blue. B, the I Basal/I IVM of single point mutants of Kir3.2 co‐expressed with Kir3.1. Data show the raw data and the means ± SD, n = 7 cells for Kir3.1–Kir3.2 WT and Kir3.1–Kir3.2 W91A; n = 6 for others. *Significant difference (P < 0.05). C, top view of the slide helix (blue) and TM–CTD linker (yellow), and the locations of I82, W91 and I195 (red) in Kir3.2.
Discussion
The novel findings in the present study are as follows: (1) IVM and its analogues act as GIRK activators in a PIP2‐dependent manner and the IVM‐mediated GIRK activation is independent of Gβγ; (2) IVM activates Kir3.2 more efficiently than Kir3.4; (3) IVM activates native GIRK current in hippocampal neurons; (4) a unique amino acid residue among GIRK family, Ile82, located in the slide helix between the TM1 and the N‐terminal CTD of Kir3.2, is critical for the activation; (5) the methyl group of the branched side‐chain of Ile82 towards TM2 acts as a switch for IVM‐dependent activation; (6) Ile82 and Trp91 located in the slide helix and Ile195 located in the TM–CTD linker contribute to the IVM‐dependent activation. The results demonstrate that the TM–CTD interface of Kir3.2 channels, rather than the TMs, acts as the structural determinant for IVM‐mediated GIRK activation.
Insights into the roles of GIRK channel in the medication for parasitic infection
In the present study, we demonstrate that IVM and other avermectins activate GIRK channel (Kir3.1–Kir3.2 strongly and Kir3.1–Kir3.4 weakly) in a PIP2‐dependent manner, and the IVM‐mediated GIRK activation is independent of Gβγ (Figs 1, 2, 3, 4, 5, 6). A recent study also showed the effect of IVM on the activation of the Kir3.2 homotetramer in HEK293 cells (Su et al. 2016), but no detailed information was reported. We observed that the EC50 of IVM for the activation of GIRK channel (Fig. 1 D) is similar to the ligand‐gated channels of IVM targets in mammalian cells, such as the glycine receptor and GABAAR (1–10 μm) (Robertson, 1989; Schonrock & Bormann, 1993; Shan et al. 2001), and this is much higher than the nanomolar concentration which activates the glutamate‐gated Cl− channel in nematodes (Martin & Pennington, 1989; McCavera et al. 2009). However, adverse effects of IVM and other avermectins have been reported in several cases under medication for parasitic infection in humans and various animal species (Barragry, 1987; Lovell, 1990; Campbell, 1991). Clinical trials in humans reported adverse effects of IVM in patients, including itching, oedema, dizziness and the Mazzotti reaction (Campbell, 1991). Overdoses of IVM in some animal species that are hypersensitive to IVM, especially dogs, resulted in toxic symptoms, including listlessness, blindness, ataxia, bradycardia, depression, mydriasis, tremors, blindness, coma, and death in some cases (Barragry, 1987; Hopper et al. 2002). A summarized report showed that the maximum plasma concentration of IVM in several animal species after drug administration was about 3–150 μm (calculated from ng ml−1), which depends on the species, route of administration, commercial formulation, body weight, physiological status and so on (Gonzalez Canga et al. 2009). This range of maximum plasma concentrations of IVM could be sufficient to influence GIRK channel activity in various organs. Since Kir3.1–Kir3.2 and Kir3.1–Kir3.4 heterotetramers are well known to regulate the excitability of neurons and the rate of heart beat, respectively (Noma & Trautwein, 1978; Krapivinsky et al. 1995; Liao et al. 1996; Inanobe et al. 1999; Hibino et al. 2010), IVM‐mediated activation of GIRK channel could contribute to some of the adverse effects, including the disorders of the central nervous system and bradycardia.
A unique action mode of IVM in GIRK channel
We showed that the Ile82 of Kir3.2 at the slide helix between the TM1 and the N‐terminus CTD is the primary structural determinant for IVM‐mediated GIRK activation, and the methyl group of the branched side‐chain of Ile82 towards TM2 acts as a switch for IVM action (Figs 7, 8, 9). Here we considered two hypotheses to explain how the slide helix serves for IVM‐dependent activation: (1) IVM binds to the slide helix and reinforces the hydrophobic core around the TM–CTD interface by forming novel hydrophobic interactions between IVM and Ile82/Trp91/Ile195, and thereby stabilizes the open state of channel (Meng et al. 2016); (2) IVM binds to another place and causes global conformational rearrangement of the channel, and this disturbs the hydrophobic interaction around the TM–CTD interface by forming novel hydrophobic interactions between the Ile82/Trp91/Ile195 and TM2 residues, thereby stabilizing the open state of channel (Meng et al. 2016). Based on the results of chimera and point mutant experiments (Fig. 7), it is most likely that, except for the unique Ile82 in Kir3.2, IVM does not bind to the amino acid residues which differ between Kir3.2 and Kir3.4. Therefore, we speculated that the Ile82 or the amino acid residues close to the TM–CTD interface could be the most likely potential binding site. Presumably Ile82 and Trp91 in the slide helix and Ile195 in the TM–CTD linker form part of the binding pocket for IVM (Fig. 9). Since Trp91 and Ile195 are conserved in Kir3.2 and Kir3.4, the presence of these two residues could contribute to the weak response to IVM in Kir3.4, which lacks the critical Ile in the slide helix (Figs 1 A–D and 7 A). The presence of the methyl group in the branched side‐chain of Ile towards TM2 in Kir3.2 WT or the Kir3.4 L77I mutant could shift the equilibrium from the closed to the open state upon IVM binding to the pocket (Figs 7 F and 8). However, we cannot exclude the possibility that IVM may bind to other positions which are conserved in the GIRK family.
In Cys‐loop receptors and the P2X4 receptor, as shown through X‐ray structure, mutagenesis and molecular modelling, IVM binds to the interface between two adjacent subunits in the TMs near the extracellular membrane surface (Silberberg et al. 2007; Jelinkova et al. 2008; Hibbs & Gouaux, 2011; Lynagh & Lynch, 2012; Zemkova et al. 2014; Du et al. 2015). A Gly residue on the M3 helix of the Cys‐loop receptor contributes to the high affinity for IVM binding, and some residues in the M1, M2 and M3 helices of TMs interact with IVM by forming an H‐bond (Lynagh & Lynch, 2010, 2012; Hibbs & Gouaux, 2011; Zemkova et al. 2014; Du et al. 2015). The binding of two molecules of IVM is sufficient to activate Cys‐loop receptors (Shan et al. 2001; Grudzinska et al. 2005; Lynagh & Lynch, 2010, 2012), and this is shared with the action mode of IVM in GIRK channels in which two of the structural determinant (Ile82) in the Kir3.1–Kir3.2 heterotetramers are sufficient to activate the channel.
Roles of PIP2 in IVM‐mediated GIRK activation
There are two functional gates in the GIRK channel, the inner helix gate and the G‐loop gate, and they open and close rapidly during G‐protein stimulation in the presence of PIP2 (Nishida et al. 2007; Whorton & MacKinnon, 2011). G‐protein binds to the βL–βM loop of the CTD and only opens the G‐loop gate; PIP2 binds near the interface between TMs and CTDs but PIP2 alone is insufficient to open each gate in the GIRK channel (Finley et al. 2004; Hansen et al. 2011; Whorton & MacKinnon, 2011). Since we observed that IVM could not activate GIRK channels in the absence of PIP2 (Fig. 6), we considered two possible reasons: (1) the binding of PIP2 in GIRK channel causes a rotation of the slide helix (Whorton & MacKinnon, 2011), allowing the approach of IVM to the Ile82. Thus, in the absence of PIP2, it is hard for IVM to be inserted into the TM–CTD interface; (2) the binding of IVM to GIRK opens only the G‐loop gate in the absence of PIP2, and both gates can be opened in the presence of PIP2. Some studies have demonstrated that Lys64, Lys90, Arg92, Lys194, Lys199 and Lys200 in Kir3.2 contribute to PIP2 binding (Hansen et al. 2011; Whorton & MacKinnon, 2011). Since the critical residues for IVM‐mediated GIRK activation, Trp91 and Ile195, are located just next to the PIP2‐coordinating residues Lys90, Arg92 and Lys194, it is most likely that the association of PIP2 with the GIRK channel induces stabilization of the TM–CTD interface, providing a suitable distance between the slide helix and the TM–CTD linker to receive IVM (Fig. 9 C).
In summary, our results have demonstrated the critical role of PIP2 in the modulation of IVM‐mediated GIRK activation and revealed the unique mode of action of IVM in the GIRK channel. The TM–CTD interface, rather than the TMs, acts as the structural determinant for IVM‐mediated GIRK activation. The critical amino acid residue, Ile82 in the slide helix of Kir3.2, contributes to the high efficacy of the IVM response. This study provides novel insights into the mode of action of IVM in ion channels, and may also provide information that leads to identification of new pharmacophores which activate the GIRK channel.
Additional information
Competing interests
The authors declare no competing financial interests.
Author contributions
Oocyte preparations and all of the electrophysiological experiments were performed in the laboratory of Y.K. in the Division of Biophysics and Neurobiology, National Institute for Physiological Sciences (NIPS); hippocampal cultures were developed in the laboratory of Dr M. Fukata in the Division of Membrane Physiology, NIPS. I‐S.C. performed all experiments on Xenopus oocytes and data analyses; M.T. performed electrophysiological recordings from neurons and data analyses; Y.F. established primary cultures of rat hippocampal neurons; M.U. contributed to the initiation of research and provided the small‐molecule library; I‐S.C. and Y.K. designed the study and wrote the paper. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This study was supported by the Grant‐in‐Aid for Young Scientists (B) 16K18999 (to I‐S.C.), the Scientific Research (B) 26293044 (to Y.K.) and the Scientific Research (S) 26220206 (to M.U.) from the Japan Society for the Promotion of Science.
Acknowledgments
We thank Dr Y. Okamura for providing us with the Ci‐VSP cDNA, and Dr Y. Yoshimura for discussion and valuable advice on the patch clamp recording from neurons. We also thank Ms C. Naito for technical assistance, and all members in Kubo laboratory for discussion. iCeMS is supported by World Premier International Research Centre Initiative (WPI), MEXT, Japan.
References
- Adelsberger H, Lepier A & Dudel J (2000). Activation of rat recombinant α1β2γ2S GABAA receptor by the insecticide ivermectin. Eur J Pharmacol 394, 163–170. [DOI] [PubMed] [Google Scholar]
- Aryal P, Dvir H, Choe S & Slesinger PA (2009). A discrete alcohol pocket involved in GIRK channel activation. Nat Neurosci 12, 988–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barragry TB (1987). A review of the pharmacology and clinical uses of ivermectin. Can Vet J 28, 512–517. [PMC free article] [PubMed] [Google Scholar]
- Burkhart CN (2000). Ivermectin: an assessment of its pharmacology, microbiology and safety. Vet Hum Toxicol 42, 30–35. [PubMed] [Google Scholar]
- Campbell WC (1991). Ivermectin as an antiparasitic agent for use in humans. Annu Rev Microbiol 45, 445–474. [DOI] [PubMed] [Google Scholar]
- Campbell WC, Fisher MH, Stapley EO, Albers‐Schonberg G & Jacob TA (1983). Ivermectin: A potent new antiparasitic agent. Science 221, 823–828. [DOI] [PubMed] [Google Scholar]
- Clapham DE & Neer EJ (1993). New roles for G‐protein βγ‐dimers in transmembrane signalling. Nature 365, 403–406. [DOI] [PubMed] [Google Scholar]
- Cully DF, Paress PS, Liu KK, Schaeffer JM & Arena JP (1996). Identification of a Drosophila melanogaster glutamate‐gated chloride channel sensitive to the antiparasitic agent avermectin. J Biol Chem 271, 20187–20191. [DOI] [PubMed] [Google Scholar]
- Du J, Lu W, Wu S, Cheng Y & Gouaux E (2015). Glycine receptor mechanism elucidated by electron cryo‐microscopy. Nature 526, 224–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finley M, Arrabit C, Fowler C, Suen KF & Slesinger PA (2004). βL–βM loop in the C‐terminal domain of G protein‐activated inwardly rectifying K+ channels is important for Gβγ subunit activation. J Physiol 555, 643–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fomkina MG, Khashaev Z, Drinyaev VA & Chailakhyan LM (2001). Channel‐forming properties of the neuropeptide preparation abamectin in bilayer lipid membranes. Dokl Biochem Biophys 378, 221–224. [DOI] [PubMed] [Google Scholar]
- Fukata Y, Dimitrov A, Boncompain G, Vielemeyer O, Perez F & Fukata M (2013). Local palmitoylation cycles define activity‐regulated postsynaptic subdomains. J Cell Biol 202, 145–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez Canga A, Sahagun Prieto AM, Jose Diez Liebana M, Martinez NF, Vega MS & Vieitez JJ (2009). The pharmacokinetics and metabolism of ivermectin in domestic animal species. Vet J 179, 25–37. [DOI] [PubMed] [Google Scholar]
- Grudzinska J, Schemm R, Haeger S, Nicke A, Schmalzing G, Betz H & Laube B (2005). The β subunit determines the ligand binding properties of synaptic glycine receptors. Neuron 45, 727–739. [DOI] [PubMed] [Google Scholar]
- Grundy D (2015). Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology . J Physiol 593, 2547–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen SB, Tao X & MacKinnon R (2011). Structural basis of PIP2 activation of the classical inward rectifier K+ channel Kir2.2. Nature 477, 495–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibbs RE & Gouaux E (2011). Principles of activation and permeation in an anion‐selective Cys‐loop receptor. Nature 474, 54–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibino H, Inanobe A, Furutani K, Murakami S, Findlay I & Kurachi Y (2010). Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol Rev 90, 291–366. [DOI] [PubMed] [Google Scholar]
- Ho IH & Murrell‐Lagnado RD (1999). Molecular determinants for sodium‐dependent activation of G protein‐gated K+ channels. J Biol Chem 274, 8639–8648. [DOI] [PubMed] [Google Scholar]
- Hopper K, Aldrich J & Haskins SC (2002). Ivermectin toxicity in 17 collies. J Vet Intern Med 16, 89–94. [DOI] [PubMed] [Google Scholar]
- Huang CL, Feng S & Hilgemann DW (1998). Direct activation of inward rectifier potassium channels by PIP2 and its stabilization by Gβγ . Nature 391, 803–806. [DOI] [PubMed] [Google Scholar]
- Ikeda H, Kotaki H & Omura S (1987). Genetic studies of avermectin biosynthesis in Streptomyces avermitilis . J Bacteriol 169, 5615–5621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inanobe A, Yoshimoto Y, Horio Y, Morishige KI, Hibino H, Matsumoto S, Tokunaga Y, Maeda T, Hata Y, Takai Y & Kurachi Y (1999). Characterization of G‐protein‐gated K+ channels composed of Kir3.2 subunits in dopaminergic neurons of the substantia nigra. J Neurosci 19, 1006–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito H, Tung RT, Sugimoto T, Kobayashi I, Takahashi K, Katada T, Ui M & Kurachi Y (1992). On the mechanism of G protein βγ subunit activation of the muscarinic K+ channel in guinea pig atrial cell membrane. Comparison with the ATP‐sensitive K+ channel. J Gen Physiol 99, 961–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jelinkova I, Vavra V, Jindrichova M, Obsil T, Zemkova HW, Zemkova H & Stojilkovic SS (2008). Identification of P2X4 receptor transmembrane residues contributing to channel gating and interaction with ivermectin. Pflugers Archiv 456, 939–950. [DOI] [PubMed] [Google Scholar]
- Jin L, Feng X, Rong H, Pan Z, Inaba Y, Qiu L, Zheng W, Lin S, Wang R, Wang Z, Wang S, Liu H, Li S, Xie W & Li Y (2013). The antiparasitic drug ivermectin is a novel FXR ligand that regulates metabolism. Nat Commun 4, 1937. [DOI] [PubMed] [Google Scholar]
- Kaufmann K, Romaine I, Days E, Pascual C, Malik A, Yang L, Zou B, Du Y, Sliwoski G, Morrison RD, Denton J, Niswender CM, Daniels JS, Sulikowski GA, Xie XS, Lindsley CW & Weaver CD (2013). ML297 (VU0456810), the first potent and selective activator of the GIRK potassium channel, displays antiepileptic properties in mice. ACS Chem Neurosci 4, 1278–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Ikeda K, Kojima H, Niki H, Yano R, Yoshioka T & Kumanishi T (1999). Ethanol opens G‐protein‐activated inwardly rectifying K+ channels. Nat Neurosci 2, 1091–1097. [DOI] [PubMed] [Google Scholar]
- Kofuji P, Davidson N & Lester HA (1995). Evidence that neuronal G‐protein‐gated inwardly rectifying K+ channels are activated by Gβγ subunits and function as heteromultimers. Proc Natl Acad Sci USA 92, 6542–6546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krapivinsky G, Gordon EA, Wickman K, Velimirovic B, Krapivinsky L & Clapham DE (1995). The G‐protein‐gated atrial K+ channel I KACh is a heteromultimer of two inwardly rectifying K+‐channel proteins. Nature 374, 135–141. [DOI] [PubMed] [Google Scholar]
- Krause RM, Buisson B, Bertrand S, Corringer PJ, Galzi JL, Changeux JP & Bertrand D (1998). Ivermectin: A positive allosteric effector of the α7 neuronal nicotinic acetylcholine receptor. Mol Pharmacol 53, 283–294. [DOI] [PubMed] [Google Scholar]
- Kubo Y, Reuveny E, Slesinger PA, Jan YN & Jan LY (1993). Primary structure and functional expression of a rat G‐protein‐coupled muscarinic potassium channel. Nature 364, 802–806. [DOI] [PubMed] [Google Scholar]
- Lesage F, Duprat F, Fink M, Guillemare E, Coppola T, Lazdunski M & Hugnot JP (1994). Cloning provides evidence for a family of inward rectifier and G‐protein coupled K+ channels in the brain. FEBS Lett 353, 37–42. [DOI] [PubMed] [Google Scholar]
- Lesage F, Guillemare E, Fink M, Duprat F, Heurteaux C, Fosset M, Romey G, Barhanin J & Lazdunski M (1995). Molecular properties of neuronal G‐protein‐activated inwardly rectifying K+ channels. J Biol Chem 270, 28660–28667. [DOI] [PubMed] [Google Scholar]
- Lewohl JM, Wilson WR, Mayfield RD, Brozowski SJ, Morrisett RA & Harris RA (1999). G‐protein‐coupled inwardly rectifying potassium channels are targets of alcohol action. Nat Neurosci 2, 1084–1090. [DOI] [PubMed] [Google Scholar]
- Liao YJ, Jan YN & Jan LY (1996). Heteromultimerization of G‐protein‐gated inwardly rectifying K+ channel proteins GIRK1 and GIRK2 and their altered expression in weaver brain. J Neurosci 16, 7137–7150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logothetis DE, Kurachi Y, Galper J, Neer EJ & Clapham DE (1987). The βγ subunits of GTP‐binding proteins activate the muscarinic K+ channel in heart. Nature 325, 321–326. [DOI] [PubMed] [Google Scholar]
- Lovell RA (1990). Ivermectin and piperazine toxicoses in dogs and cats. Vet Clin North Am Small Anim Pract 20, 453–468. [DOI] [PubMed] [Google Scholar]
- Lynagh T & Lynch JW (2010). A glycine residue essential for high ivermectin sensitivity in Cys‐loop ion channel receptors. Int J Parasitol 40, 1477–1481. [DOI] [PubMed] [Google Scholar]
- Lynagh T & Lynch JW (2012). Ivermectin binding sites in human and invertebrate Cys‐loop receptors. Trends Pharmacol Sci 33, 432–441. [DOI] [PubMed] [Google Scholar]
- McCavera S, Rogers AT, Yates DM, Woods DJ & Wolstenholme AJ (2009). An ivermectin‐sensitive glutamate‐gated chloride channel from the parasitic nematode Haemonchus contortus . Mol Pharmacol 75, 1347–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin RJ (1996). An electrophysiological preparation of Ascaris suum pharyngeal muscle reveals a glutamate‐gated chloride channel sensitive to the avermectin analogue, milbemycin d. Parasitology 112, 247–252. [DOI] [PubMed] [Google Scholar]
- Martin RJ & Pennington AJ (1989). A patch‐clamp study of effects of dihydroavermectin on Ascaris muscle. Br J Pharmacol 98, 747–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng XY, Liu S, Cui M, Zhou R & Logothetis DE (2016). The molecular mechanism of opening the helix bundle crossing (HBC) gate of a Kir channel. Sci Rep 6, 29399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata Y, Iwasaki H, Sasaki M, Inaba K & Okamura Y (2005). Phosphoinositide phosphatase activity coupled to an intrinsic voltage sensor. Nature 435, 1239–1243. [DOI] [PubMed] [Google Scholar]
- Murata Y & Okamura Y (2007). Depolarization activates the phosphoinositide phosphatase Ci‐VSP, as detected in Xenopus oocytes coexpressing sensors of PIP2 . J Physiol 583, 875–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida M, Cadene M, Chait BT & MacKinnon R (2007). Crystal structure of a Kir3.1‐prokaryotic Kir channel chimera. EMBO J 26, 4005–4015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noma A & Trautwein W (1978). Relaxation of the ACh‐induced potassium current in the rabbit sinoatrial node cell. Pflugers Arch 377, 193–200. [DOI] [PubMed] [Google Scholar]
- Reuveny E, Slesinger PA, Inglese J, Morales JM, Iniguez‐Lluhi JA, Lefkowitz RJ, Bourne HR, Jan YN & Jan LY (1994). Activation of the cloned muscarinic potassium channel by G protein βγ subunits. Nature 370, 143–146. [DOI] [PubMed] [Google Scholar]
- Robertson B (1989). Actions of anaesthetics and avermectin on GABAA chloride channels in mammalian dorsal root ganglion neurones. Br J Pharmacol 98, 167–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakata S, Hossain MI & Okamura Y (2011). Coupling of the phosphatase activity of Ci‐VSP to its voltage sensor activity over the entire range of voltage sensitivity. J Physiol 589, 2687–2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonrock B & Bormann J (1993). Activation of Cl– channels by avermectin in rat cultured hippocampal neurons. Naunyn Schmiedebergs Arch Pharmacol 348, 628–632. [DOI] [PubMed] [Google Scholar]
- Shan Q, Haddrill JL & Lynch JW (2001). Ivermectin, an unconventional agonist of the glycine receptor chloride channel. J Biol Chem 276, 12556–12564. [DOI] [PubMed] [Google Scholar]
- Sigel E & Baur R (1987). Effect of avermectin B1a on chick neuronal gamma‐aminobutyrate receptor channels expressed in Xenopus oocytes. Mol Pharmacol 32, 749–752. [PubMed] [Google Scholar]
- Silberberg SD, Li M & Swartz KJ (2007). Ivermectin interaction with transmembrane helices reveals widespread rearrangements during opening of P2X receptor channels. Neuron 54, 263–274. [DOI] [PubMed] [Google Scholar]
- Su Z, Brown EC, Wang W & MacKinnon R (2016). Novel cell‐free high‐throughput screening method for pharmacological tools targeting K+ channels. Proc Natl Acad Sci USA 113, 5748–5753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sui JL, Petit‐Jacques J & Logothetis DE (1998). Activation of the atrial KACh channel by the betagamma subunits of G proteins or intracellular Na+ ions depends on the presence of phosphatidylinositol phosphates. Proc Natl Acad Sci USA 95, 1307–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whorton MR & MacKinnon R (2011). Crystal structure of the mammalian GIRK2 K+ channel and gating regulation by G proteins, PIP2, and sodium. Cell 147, 199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yow TT, Pera E, Absalom N, Heblinski M, Johnston GA, Hanrahan JR & Chebib M (2011). Naringin directly activates inwardly rectifying potassium channels at an overlapping binding site to tertiapin‐Q. Br J Pharmacol 163, 1017–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeidner G, Sadja R & Reuveny E (2001). Redox‐dependent gating of G protein‐coupled inwardly rectifying K+ channels. J Biol Chem 276, 35564–35570. [DOI] [PubMed] [Google Scholar]
- Zemkova H, Tvrdonova V, Bhattacharya A & Jindrichova M (2014). Allosteric modulation of ligand gated ion channels by ivermectin. Physiol Res 63 (Suppl. 1), S215–224. [DOI] [PubMed] [Google Scholar]
- Zheng Y, Hirschberg B, Yuan J, Wang AP, Hunt DC, Ludmerer SW, Schmatz DM & Cully DF (2002). Identification of two novel Drosophila melanogaster histamine‐gated chloride channel subunits expressed in the eye. J Biol Chem 277, 2000–2005. [DOI] [PubMed] [Google Scholar]