

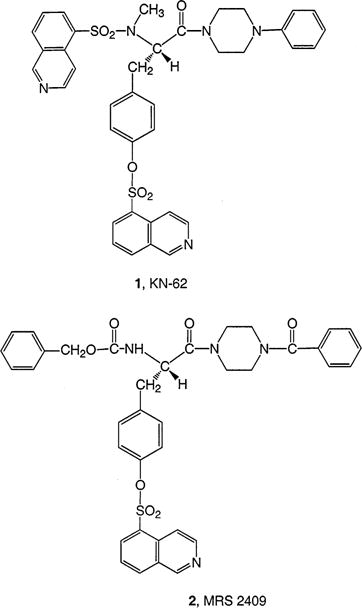
Abstract
Chemically funtionalized analogues of antagonists of the P2X7 receptor, an ATP-gated cation channel, were synthesized as tools for biophysical studies of the receptor. These functionalized congeners were intended for use in chemical conjugation with retention of biological potency. The antagonists were L-tyrosine derivatives, related to [N-benzyloxycarbonyl-O-(4-arylsulfonyl)-L-tyrosyl]benzoylpiperazine (such as MRS2409, 2). The analogues were demonstrated to be antagonists in an assay of human P2X7 receptor function, consisting of inhibition of ATP-induced K+ efflux in HEK293 cells expressing the recombinant receptor. The analogues were of the general structure R1-Tyr(OR2)-piperazinyl-R3, in which three positions (R1–R3) were systematically varied in structure through introduction of chemically reactive groups. Each of the three positions was designed to incorporate a 3- or 4-nitrophenyl group. The nitro groups were reduced using NaBH4–copper(II) acetylacetonate to amines, which were either converted to the isothiocyanate groups, as potential affinity labels for the receptor, or acylated, as models for conjugation. An alternate route to Nα-3-aminobenzyloxycarbonyl functionalization was devised. The various positions of functionalization were compared for effects on biological potency, and the R2 and R3 positions were found to be most amenable to derivatization with retention of high potency. Four dimeric permutations of the antagonists were synthesized by coupling each of the isothiocyanate derivatives to either the precursor amine or to other amine congeners. Only dimers linked at the R2-position were potent antagonists. In concentration–response studies, two derivatives, a 3-nitrobenzyloxycarbonyl derivative 18 and a 4-nitrotoluenesulfonate 26b, displayed IC50 values of roughly 100 nM as antagonists of P2X7 receptor-mediated K+ flux.
INTRODUCTION
P2X receptors are ligand-gated cation channels activated by ATP (adenosine 5′-triphosphate) and other purine nucleotides. Seven mammalian subtypes of P2X receptors (termed P2X1 through P2X7) have been cloned (1, 2). Each functional ion channel consists of an oligomeric assembly, probably a trimer. Both homo- and heterooligomerization seem to occur commonly; however the P2X7 subtype exists exclusively as a homomer, and the P2X6 subtype always occurs as a homomer. The cationic selectivity of the P2X receptors is Na+, K+ > Ca2+ (3).
The P2X7 receptor (formerly known as the P2Z receptor) is expressed primarily in the immune/inflammatory system (4–9), i.e., in blood cells (monocytes, macrophages, and lymphocytes) and in the brain on microglial cells, and in the salivary gland (10). Its activation is associated with apoptosis in the immune system and release of inflammatory cytokines, such as IL-1β (8, 9, 11). The P2X7 receptor is distinct from the other P2X subunits in that at high μM concentrations of agonists, it forms or activates a large pore in addition to a cation channel (12, 13). This pore increases permeability indiscriminately to molecules having MW ≤ 900, such as ethidium bromide, which is used as a marker for pore activity. The P2X7 receptor also has a long C-terminal segment which contributes to pore formation (13). 2′- and 3′-O-(4-benzoylbenzoyl)-ATP (BzATP) is among the most potent agonists at P2X7 receptors, but also has nanomolar potency at P2X1 receptors (14). Affinity labeling of the P2X7 receptor in mast cells has been carried out using [3H]-BzATP (15).
An objective of the present study was to design antagonist ligand probes based on tyrosine derivatives, to be used for biophysical characterization of the P2X7 receptor. The isoquinoline derivative of tyrosine 1-(N,O-bis[5-isoquinolinesulfonyl]-N-methyl-L-tyrosyl)-4-phenylpiperazine (1, KN-62), is a potent noncompetitive antagonist at P2X7 receptors in the submicromolar range (6,7,12). It is also an antagonist of Ca2+/calmodulin-dependent protein kinase II (CaMKII) in the micromolar range (16). We recently reported a series of potent P2X7 receptor antagonists derived from KN-62, of the general structure R1-Tyr(OR2)-piperazinyl-R3, in which three positions (R1, R2, and R3) were systematically varied (17). The derivative MRS 2409 ([N-benzyloxycarbonyl-O-[4-(isoquinolinylsulfonyl)]-L-tyrosyl]benzoylpiperazine), 2, in which R1 and R3 differed from the structure of KN-62, was among the most potent analogues prepared (IC50 of 200 nM, for release of K+ stimulated by 3 mM ATP in HEK293 cells expressing the human P2X7 receptor). We now extend the SAR (structure activity relationship) analysis in this series to include more varied functionalization of congeners (18–20) of MRS 2409, directed toward probing multiple sites on the antagonist molecules for sites of chain attachment that preserve the biological properties. This functionalization is intended for the purpose of cross-linking, either with the receptor and/or with reporter moieties such as fluorescent dyes or radioisotopes. Dimerization of the antagonists through various linkage points has also been carried out in order to explore the spatial relationship between possible multiple binding sites on the large extracellular loop (13, 21) of P2X7 receptor homomers.
MATERIALS AND METHODS
Chemical Synthesis
Protected tyrosine compounds are from Novabiochem (San Diego, CA). Other chemicals are from Aldrich (Milwaukee, WI). 1H NMR spectra were recorded using a Varian Gemini-300 spectrometer. High-resolution FAB (fast atom bombardment) mass spectra were taken with a JEOL SX102 spectrometer using nitrobenzoic acid as matrix. Elemental analysis was performed by Atlantic Microlab Inc. (Norcross, GA).
General Procedure for the Synthesis of 46, 51, 62
A mixture of Fmoc-Tyr-OH or Cbz-Tyr-OH (3.0 mmol), Boc-piperazine, or benzoyl-piperazine (3.0 mmol), Bis(2-oxo-3-oxoazolidinyl)phosphinic chloride (Bop-Cl) (0.77 g, 3.0 mmol), and CH2Cl2 (15 mL) was treated with Et3N (0.96 mL, 6.8 mmol) and stirred at room temperature for 1 h. The solvent was removed in vacuo, and the crude product was purified with silica gel chromatography eluting with methanol-chloroform (5:95) to furnish a white solid foam (yield 75–85%).
[N-Fmoc-L-tyrosyl]benzoylpiperazine (46)
1H NMR (CDCl3): δ 7.77 (d, J = 7.4 Hz, 2H), 7.59 (d, J = 7.4 Hz, 2H), 7.50–7.28 (m, 5H), 7.05 (d, J = 8.0 Hz, 2H), 6.75 (d, J = 8.0 Hz, 2H), 5.70 (d, J = 8.5 Hz, 1H), 4.84(b, 1H), 4.50–4.25 (m, 2H), 4.25–4.10 (t, J = 7.0 Hz, 1H), 3.80–2.80 (m, 10H).
[N-Fmoc-L-tyrosyl]-Boc-piperazine (51)
1H NMR (CDCl3): δ 7.77 (d, J = 7.3 Hz, 2H), 7.59 (d, J = 7.3 Hz, 2H), 7.40 (t, J = 7.3 Hz, 2H), 7.31 (t, J = 7.3 Hz, 2H), 7.04 (d, J = 7.7 Hz, 2H), 6.74 (d, J = 7.7 Hz, 2H), 5.88 (s, 1H), 5.72 (d, J = 8.4 Hz, 1H), 4.90–4.78 (m, 1H), 4.44–4.28 (m, 2H), 4.24–4.16 (t, J = 7.0 Hz, 1H), 3.60–2.80 (m, 10 H), 1.45 (s, 9H).
[N-Benzyloxycarbonyl-O-toluenesulfonyl-L-tyrosyl]pip-erazine (62)
1H NMR (CDCl3): δ 7.30–7.40 (m, 5H), 7.02 (d, J = 8.2 Hz, 2H), 6.72 (d, J = 8.2 Hz, 2H), 5.65 (d, J = 8.5 Hz, 1H), 5.45 (br s, 1H), 5.14–5.00 (ABq, J = 12.4 Hz, 2H), 4.90–4.75 (m, 1H), 3.60–2.80 (m, 10H), 1.45 (s, 9H).
General Procedure for the Synthesis of 23a, 23b, 47, 52a, 52b, 55
N-Substituted tyrosine (46, 51, or 62; 2.0 mmol) was dissolved in 10 mL of anhydrous CH2Cl2 and cooled to 0 °C. A solution of benzenesulfonyl chloride or related sulfonyl chloride (4.0 mmol), Et3N (0.3 mL, 2.2 mmol), DMAP (214 mg, 1.7 mmol), and CH2Cl2 (5.0 mL) was added dropwise and stirring continued for 30 min at 0 °C. TLC showed the completion of the reaction. The solvent was removed under reduced pressure. Column chromatography (silica gel, CH3OH:CHCl3 = 5:95) yielded a white solid foam (70–75% yield).
[N-Benzyloxycarbonyl-O-(3-nitrobenzenesulfonyl)-L-tyrosyl]-Boc-piperazine (23a)
1H NMR (CDCl3): δ 8.71 (t, J = 1.9 Hz, 1H), 8.52 (ddd, J = 1.1, 2.2, 8.2 Hz, 1H), 8.12 (m, 1H), 7.78 (t, J = 8.0 Hz, 1H), 7.40–7.30 (m, 5H), 7.14 (d, J = 8.5 Hz, 1H), 6.93 (m. 1H), 5.58 (d, J = 8.8 Hz, 1H), 5.14–5.00(ABq, J = 12.4 Hz, 2H), 4.88–4.77 (m, 1H), 3.60–2.84 (m, 10H), 1.44 (s, 9H). FAB–HRMS for C32H37N4O10S (MH+): calcd 669.2230, found 669.2231. Anal. (C32H36N4O10S·0.1H2O).
[N-Benzyloxycarbonyl-O-(4-nitrobenzenesulfonyl)-L-tyrosyl]-Boc-piperazine (23b)
1H NMR (CDCl3): δ 8.39 (d, J = 9.0 Hz, 2H), 8.02 (d, J = 9.1 Hz, 2H), 7.42–7.28 (m, 5H), 7.13 (d, J = 8.5 Hz, 2H), 6.90 (d, J = 8.5 Hz, 2H), 5.60 (d, J = 8.5 Hz, 1H), 5.14–5.00 (ABq, J = 13.7 Hz, 2H), 4.90–4.77 (m, 1H), 3.60–2.78 (m, 10H), 1.45 (s, 9H). FAB-MS: m/z (relative intensity) 669.1 (M + 1, 6), 613.1 (16), 91 (100). Anal. (C32H36N4O10S·0.1H2O).
[N-Fmoc-O-toluenesulfonyl-L-tyrosyl]benzoylpiperazine (47)
1H NMR (CDCl3): δ 7.86–7.64 (m, 4H), 7.60 (d, J = 7.7 Hz, 2H), 7.50–7.28 (m, 11H), 7.13 (d, J = 8.2 Hz, 2H), 6.84–6.88 (d, J = 8.2 Hz, 2H), 5.66 (d, J = 8.2 Hz, 1H), 4.82 (b, 1H), 4.50–4.25 (m, 2H), 4.25–4.10 (m, 1H), 3.8–2.7 (m, 10H), 2.45 (s, 3H).
[N-Fmoc-O-benzenesulfonyl-L-tyrosyl]-Boc-piperazine (52a)
1H NMR (CDCl3): δ 7.88–7.72 (m, 4H), 7.70–7.60 (m, 2H), 7.46–7.27 (m, 7H), 7.10 (d, J = 8.5 Hz, 2H), 6.90 (d, J = 8.5 Hz, 2H), 5.66 (d, J = 8.2 Hz, 1H), 4.90–4.70 (m, 1H), 4.50–4.30 (m, 2H), 4.25–4.10 (m, 1H), 3.8–2.7 (m, 10H), 1.45 (s, 9H).
[N-Fmoc-O-(4-nitrobenzenesulfonyl)-L-tyrosyl]-Boc-piperazine (52b)
1H NMR (CDCl3): δ 8.38 (d, J = 8.8 Hz, 2H), 8.02 (d, J = 8.8 Hz, 2H), 7.77 (d, J = 7.7 Hz, 2H), 7.56 (d, J = 7.4 Hz, 2H), 7.47–7.27 (m, 4H), 7.14 (d, J = 7.4 Hz, 2H), 6.91 (d, J = 8.5 Hz, 2H), 5.61 (d, J = 8.5 Hz, 1H), 4.90–4.70 (m, 1H), 4.50–4.28 (m, 2H), 4.26–4.10 (m, 1H), 3.58–2.70 (m. 10H), 1.45 (s, 9H).
[N-Fmoc-O-toluenesulfonyl-L-tyrosyl]-Boc-piperazine (55)
1H NMR (CDCl3): δ 7.76 (d, J = 7.1 Hz, 2H), 7.70 (d, J = 8.0 Hz, 2H), 7.57 (d, J = 6.9 Hz, 2H), 7.46–7.27 (m, 6H), 7.10 (d, J = 7.9 Hz, 2H), 6.90 (d, J = 7.8 Hz, 2H), 5.65 (d, J = 8.2 Hz, 1H), 4.85–4.70 (m, 1H), 4.50–4.25 (m, 2H), 4.24–4.15 (m, 1H), 3.80–2.70 (m, 10H), 2.43 (s, 3H), 1.45 (s, 9H).
General Procedure for the Synthesis of 17, 19, 20b, 25, 26a, 26b, 28, 31a, 31b, 33, 53a, 53b, 53c, 53d, 57a, 57b
A tyrosine derivative containing a free amine (20a, 24, 27, 32, 52a, 52b, 56, 58a, 58b, or 63; 3.0 mmol) was dissolved in CH2Cl2 (8.0 mL). DMAP (300 mg) was added, and the solution was cooled to 0 °C. A solution of a benzoyl chloride (4.2 mmol) (alternatively a benzyl chloroformate or acetyl chloride), Et3N (0.45 mL), and CH2Cl2 (5.0 mL) was added dropwise and stirring continued at 0 °C for 40 min. The solvent was removed, and the residue was washed with petroleum ether three times. The resulting crude product was purified by column chromatography (silica gel, CH3OH:CHCl3 = 5:95) to yield a white solid foam (80–90% yield).
[N-(4-Nitrobenzyloxycarbonyl)-O-toluenesulfonyl-L-tyrosyl]benzoylpiperazine (17)
1H NMR (CDCl3): δ 8.22 (d, J = 8.8 Hz, 2H), 7.73 (d, J = 8.2 Hz, 2H), 7.60–7.30 (m, 9H), 7.13 (d, J = 8.2 Hz, 2H), 6.93 (d, J = 8.5 Hz, 2H), 5.70 (d, J = 8.5 Hz, 1H), 5.26–5.10 (b, 2H), 4.92–4.70 (b, 1H), 3.90–2.70 (m, 10H), 2.46 (s, 3H). FAB-MS: m/z (relative intensity) 687.2 (M + 1, 35), 391.3 (70), 149.1 (100). Anal. (C35H34N4O9S·0.4EtOAc).
[N-(3-Nitrobenzyloxycarbonyl)-O-toluenesulfonyl-L-tyrosyl]benzoylpiperazine (19)
1H NMR (CDCl3): δ 8.22–8.16 (m, 2H), 7.72 (d, J = 8.5 Hz, 2H), 7.68–7.62 (m, 1H), 7.54 (t, J = 8.0 Hz, 1H), 7.46–7.30 (m, 7H), 7.13 (d, J = 8.5 Hz, 2H), 6.92 (d, J = 8.5 Hz, 2H), 5.70 (d, J = 8.5 Hz, 1H), 5.22–5.12 (m, 2H), 4.92–4.70 (b, 1H), 4.00–2.70 (m, 10H), 2.43 (s, 3H). FAB–HRMS for C35H35N4O9S (MH+): calcd 687.2125, found 687.2136.
[N-(3-Acetylaminobenzyloxycarbonyl)-O-toluenesulfonyl-L-tyrosyl]benzoylpiperazine (20b)
1H NMR (CDCl3): δ 7.71 (d, J = 8.5 Hz, 2H), 7.52–7.28 (m, 10H), 7.16–6.98 (m, 3H), 6.91 (d, J = 8.5 Hz, 2H), 5.88–5.52 (b, 1H), 5.05 (br s, 2H), 4.88–4.68 (b, 1H), 3.80–2.70 (m, 10H), 2.46 (s, 3H), 2.18 (s, 3H). FAB–HRMS for C37H39N4O8S (MH+): calcd 699.2489, found 699.2507.
[N-Benzyloxycarbonyl-O-(4-acetylamino)benzenesulfonyl-L-tyrosyl]-Boc-piperazine (25)
1H NMR (CDCl3): δ 7.72 (d, J = 8.8 Hz, 2H), 7.63 (d, J = 8.6 Hz, 2H), 7.40–7.28 (m, 5H), 7.10 (d, J = 8.2 Hz, 2H), 6.94 (d, J = 8.6 Hz, 2H), 5.64 (d, J = 8.5 Hz, 1H), 5.20–5.00 (ABq, J = 12.3 Hz, 2H), 4.90–4.70 (m, 1H), 4.00–2.70 (m, 10H), 2.23 (s, 3H), 1.47 (s, 9H). FAB–HRMS for C34H41N4O9S (MH+): calcd 681.2594, found 681.2610. Anal. (C34H40N4O9S).
[N-Benzyloxycarbonyl-O-(3-nitrobenzenesulfonyl)-L-tyrosyl]benzoylpiperazine (26a)
1H NMR (CDCl3): δ 8.76–8.64 (b, 1H), 8.58–8.30 (dq, J = 8.3, 1.1 Hz, 1H), 8.20–8.14 (m, 1H), 7.80 (t, J = 8.0 Hz, 1H), 7.48–7.28 (m, 10H), 7.16 (d, J = 8.5 Hz, 2H), 7.00–6.92 (m, 2H), 5.57 (d, J = 8.5 Hz, 1H), 5.14–5.02 (m, 2H), 4.92–4.70 (b, 1H), 3.80–2.70 (m, 10H). FAB–HRMS for C34H33N4O9S (MH+): calcd 673.1968, found 673.1981. Anal. (C34H32N4O9S).
[N-Benzyloxycarbonyl-O-(4-nitrobenzenesulfonyl)-L-tyrosyl]benzoylpiperazine (26b)
1H NMR (CDCl3): δ 8.40 (d, J = 8.8 Hz, 2H), 8.05 (d, J = 8.5 Hz, 2H), 7.48–7.28 (m, 10H), 7.16 (d, J = 8.2 Hz, 2H), 6.92 (d, J = 8.5 Hz, 2H), 5.76 (d, J = 8.5 Hz, 1H), 5.18–5.00 (m, 2H), 4.94–4.72 (b, 1H), 3.70–2.80 (m, 10H). Compound 26b (Rf = 0.85, CH3OH/CHCl3 = 1: 9) was shown to be pure by TLC.
[N-Benzyloxycarbonyl-O-(4-acetylamino)benzenesulfonyl-L-tyrosyl]benzoylpiperazine (28)
1H NMR (CDCl3): δ 7.72 (q, J = 9.1, 14.8 Hz, 4H), 7.50–7.30 (m, 10H), 7.10 (d, J = 7.7 Hz, 2H), 7.04–6.90 (b, 2H), 5.68–5.52 (b, 1H), 5.16–5.00 (ABq, J = 12.1, 16.2 Hz, 2H), 4.88–4.68 (b, 1H), 3.90–2.70 (m, 10H), 2.17 (s, 3H). FAB-MS: m/z (relative intensity) 685.4 (M + 1, 100), 641.4 (5), 91.1 (70).
[N-Benzyloxycarbonyl-O-toluenesulfonyl-L-tyrosyl]-(3-nitrobenzoyl) piperazine (31a)
1H NMR (CDCl3): δ 8.34–8.22 (m, 2H), 7.80–7.70 (m, 3H), 7.68–7.36 (m, 1H), 7.42–7.30 (m, 7H), 7.16 (d, J = 8.5 Hz, 2H), 6.96 (d, J = 8.8 Hz, 2H), 5.58 (d, J = 8.8 Hz, 1H), 5.26–5.00 (m, 2H), 4.90–4.70 (b, 1H), 4.00–2.70 (m, 10H), 2.45 (s, 3H). FAB-MS: m/z (relative intensity) 687.2 (M + 1, 40), 643.3 (10), 91.0 (90). FAB–HRMS (NOBA/CsI) for C35H34N4O9SCs [M + Cs]+: calcd 819.1101, found 819.1096.
[N-Benzyloxycarbonyl-O-toluenesulfonyl-L-tyrosyl]-(4-nitrobenzoyl)piperazine (31b)
1H NMR (CDCl3): δ 8.36–8.18 (m, 3H), 7.76 (d, J = 8.2 Hz, 2H), 7.56 (d, J = 8.5 Hz, 2H), 7.42–7.28 (m, 6H), 7.16 (d, J = 8.2 Hz, 2H), 6.96 (d, J = 8.0 Hz, 2H), 5.70–5.56 (m, 1H), 5.18–5.00 (m, 2H), 4.92–4.70 (b, 1H), 4.00–2.70 (m, 10H), 2.48 (s, 3H). FAB-MS: m/z (relative intensity) 687.2 (M + 1, 20), 643.3 (2), 91.0 (50). FAB–HRMS for C35H35N4O9S [MH]+: calcd 687.2125, found 687.2139. Anal. (C35H34N4O9S·0.2EtOAc).
[N-Benzyloxycarbonyl-O-toluenesulfonyl-L-tyrosyl]-(4-acetylaminobenzoyl) piperazine (33)
1H NMR (CDCl3): δ 7.72 (d, J = 8.2 Hz, 2H), 7.53 (d, J = 8.5 Hz, 2H), 7.45 (s, 1H), 7.40–7.28 (m, 9H), 7.13 (d, J = 8.2 Hz, 2H), 6.91 (d, J = 7.7 Hz, 2H), 5.80–5.56 (b, 1H), 5.20–5.00 (m, 2H), 4.90–4.72 (b, 1H), 3.90–2.70 (m, 10H), 2.45 (s, 3H), 2.19 (s, 3H). FAB-HRMS for C37H39N4O8S [MH]+: calcd 699.2489, found 699.2505. Compound 33 (Rf = 0.35, CH3OH/CHCl3 = 5: 95) was shown to be pure by TLC. Anal. (C37H38N4O8S·0.5EtOAc).
[N-Fmoc-O-benzenesulfonyl)-L-tyrosyl]benzoylpiperazine (53a)
1H NMR (CDCl3). δ 7.80–7.28 (m, 18H), 7.16 (d, J = 8.2 Hz, 2H), 6.93 (d, J = 8.2 Hz, 2H), 5.64 (d, J = 8.5 Hz, 1H), 4.83 (br s, 1H), 4.50–4.28 (m, 2H), 4.20 (m, 1H), 3.85–2.70 (m, 10H).
[N-Fmoc-O-(4-nitrobenzene)sulfonyl-L-tyrosyl]-(4-nitrobenzoyl)piperazine (53b)
1H NMR (CDCl3): δ 8.42 (d, J = 8.3 Hz, 2H), 8.27 (d, J = 7.7 Hz, 2H), 8.10 (d, J = 8.8 Hz, 2H), 7.78 (d, J = 7.7 Hz, 2H), 7.56 (m, 4H), 7.41 (t, J = 7.4 Hz, 2H), 7.36–7.27(m, 2H), 7.19 (d, J = 8.2 Hz, 2H), 6.98 (d, J = 7.4 Hz, 2H), 5.58 (d, J = 8.8 Hz, 1H), 4.83 (b, 1H), 4.50–4.26 (m, 2H), 4.19(m, 1H), 3.80–2.55 (m, 10H).
[N-Fmoc-O-benzenesulfonyl-L-tyrosyl]-(4-nitrobenzoyl)-piperazine (53c)
1H NMR (CDCl3): δ 8.40–8.16 (b, 2H), 7.90 (d, J = 7.7 Hz, 2H), 7.78 (d, J = 7.4 Hz, 2H), 7.70 (d, J = 7.1 Hz, 1H), 7.64–7.48 (m, 6H), 7.46–7.28 (m, 4H), 7.17 (d, J = 8.2 Hz, 2H), 7.06–6.90 (m, 2H), 5.75–5.50 (b, 1H), 4.90–4.70 (b, 1H), 4.50–4.30 (m, 2H), 4.25–4.10 (m, 1H), 4.00–2.70 (m, 10H).
[N-Fmoc-O-(4-nitrobenzene)sulfonyl-L-tyrosyl]benzoylpiperazine (53d)
1H NMR (CDCl3): δ 8.38 (d, J = 8.5 Hz, 2H), 8.05 (d, J = 8.2 Hz, 2H), 7.77 (d, J = 7.4 Hz, 2H), 7.56 (d, J = 7.4 Hz, 2H), 7.50–7.28 (m, 9H), 7.16 (d, J = 8.2 Hz, 2H), 6.94 (d, J = 8.2 Hz, 2H), 5.63 (d, J = 8.5 Hz, 1H), 4.90–4.47 (b, 1H), 4.50–4.25 (m, 2H), 4.25–4.10 (m, 1H), 3.80–2.70 (m, 10H).
[N-(4-Nitrobenzyloxycarbonyl)-O-toluenesulfonyl-L-tyrosyl]Boc-piperazine (57a)
1H NMR (CDCl3): δ 8.21 (d, J = 8.5 Hz, 2H), 7.76 (d, J = 8.2 Hz, 2H), 7.48 (d, J = 8.5 Hz, 2H), 7.36 (d, J = 8.2 Hz, 2H), 7.18 (d, J = 8.4 Hz, 2H), 7.02 (d, J = 8.5 Hz, 2H), 5.62(d, J = 8.5 Hz, 1H), 5.20 (ABq, J = 12.3, 15.6 Hz, 2H), 4.60–4.40 (m, 1H), 4.00–2.80 (m, 10H), 2.46 (s, 3H), 1.45 (s, 9H).
[N-Benzyloxycarbonyl-O-toluenesulfonyl-L-tyrosyl]Bocpiperazine (57b)
1H NMR (CDCl3): δ 7.69 (d, J = 7.9 Hz, 2H), 7.42–7.28 (m, 7H), 7.09 (d, J = 7.9 Hz, 2H), 6.88 (d, J = 7.9 Hz, 2H), 5.65 (d, J = 8.5 Hz, 1H), 5.07 (ABq, J = 12.6, 15.4 Hz, 2H), 4.90–4.76 (m, 1H), 3.60–2.70 (m, 10H), 2.45 (s, 3H), 1.44 (s, 9H).
General Procedure for the Synthesis of 3–16
The appropriate tyrosine derivative (48, 54a, 54b, 54c, or 54d; 1.0 mmol), Bop-Cl (1.0 mmol) and appropriate cinnamic acid derivative (1.0 mmol) were dissolved in CH2Cl2 (5.0 mL). The solution was treated with Et3N (2.0 mmol) and stirred at room temperature for 5 h. The solvent was removed, and the residue was purified using silica gel chromatography eluting with methanolchloroform (5:95) to get a solid foam (40–60% yield).
[N-(trans-Cinnamoyl)-O-benzenesulfonyl-L-tyrosyl]benzoylpiperazine (3)
1H NMR (CDCl3): δ 7.85 (d, J = 7.7 Hz, 2H), 7.74–7.60 (m, 2H), 7.60–7.30 (m, 11H), 7.17 (d, J = 8.5 Hz, 2H), 6.94 (d, J = 8.5 Hz, 2H), 6.58–6.46 (m, 1H), 6.40 (d, J = 15.7 Hz, 1H), 5.40–5.10 (b, 1H), 3.90–2.90 (m, 10H). FAB-MS: m/z (relative intensity) 624.3 (M + 1, 40), 434.2 (20), 191.2 (100), 131.1 (100), 105.0 (100). Compound 3 (Rf = 0.60, CH3OH/CHCl3 = 5: 95) was shown to be pure by TLC.
[N-(trans-2-Methoxycinnamoyl)-O-toluenesulfonyl-L-tyrosyl]benzoylpiperazine (4)
1H NMR (CDCl3): δ 7.72 (d, J = 8.1 Hz, 2H), 7.52–7.32 (m, 10H), 7.16–6.80 (m, 5H), 6.34 (d, J = 8.4 Hz, 1H), 6.00 (d, J = 15.7 Hz, 1H), 5.20–5.00 (m, 1H), 4.00–2.70 (m, 13H), 2.45 (s, 3H).
[N-(cis-2-Methoxycinnamoyl)-O-toluenesulfonyl-L-tyrosyl]-benzoylpiperazine (5)
1H NMR (CDCl3): δ 7.70 (d, J = 7.7 Hz, 2H), 7.50–7.30 (m, 10H), 7.14–6.82 (m, 5H), 6.34 (d, J = 8.5 Hz, 1H), 5.96 (d, J = 12.4 Hz, 1H), 5.20–5.00 (m, 1H), 4.00–2.60 (m, 13H), 2.46 (s, 3H).
[N-(trans-3-Chlorocinnamoyl)-O-toluenesulfonyl-L-tyrosyl]benzoylpiperazine (6)
1H NMR (CDCl3): δ 7.72 (d, J = 8.0 Hz, 2H), 7.61–7.29 (m, 11H), 7.16 (d, J = 8.5 Hz, 2H), 6.94 (d, J = 8.2 Hz, 2H), 6.57 (d, J = 8.3 Hz, 1H), 6.45–6.37 (d, J = 15.7 Hz, 1H), 5.37–5.07 (b, 1H), 4.00–2.70 (m, 10H), 2.46 (s, 3H).
[N-(trans-2,4,5-Trimethoxycinnamoyl)-O-toluenesulfonyl-L-tyrosyl]benzoylpiperazine (7)
1H NMR (CDCl3): δ 7.88 (s, 1H), 7.82 (s, 1H), 7.71 (d, J = 7.7 Hz, 2H), 7.56–7.30 (m, 5H), 7.17 (d, J = 8.2 Hz, 2H), 7.02–6.86 (m, 4H), 6.56 (d, J = 15.9 Hz, 1H), 6.43 (d, J = 8.3 Hz, 1H), 5.40–5.05 (b, 1H), 4.00–2.70 (m, 19H), 2.45 (s, 3H).
[N-(trans-4-Nitrocinnamoyl)-O-benzenesulfonyl-L-tyrosyl]-benzoylpiperazine (8)
1H NMR (CDCl3): δ 8.24 (d, J = 8.5 Hz, 2H), 7.86 (d, J = 8.0 Hz, 2H), 7.76–7.50 (m, 6H), 7.48–7.30 (m, 4H), 7.17 (d, J = 8.2 Hz, 2H), 6.96 (d, J = 8.5 Hz, 2H), 6.64 (d, J = 8.2 Hz, 1H), 6.52 (d, J = 15.9 Hz, 1H), 5.40–5.32 (b, 1H), 3.90–2.60 (m, 10H). FAB-MS: m/z (relative intensity) 669.2 (M + 1, 25), 251.1 (40), 119.0 (100). Compound 8 (Rf = 0.47, CH3OH/CHCl3 = 5: 95) was shown to be pure by TLC.
[N-(trans-Cinnamoyl)-O-(4-nitrobenzenesulfonyl)-L-tyrosyl]-benzoylpiperazine (9)
1H NMR (CDCl3): δ 8.40 (d, J = 8.5 Hz, 2H), 8.06 (d, J = 8.3 Hz, 2H), 7.70–7.30 (m, 10H), 7.20 (d, J = 8.2 Hz, 2H), 6.95 (d, J = 8.5 Hz, 2H), 6.52 (d, J = 8.2 Hz, 1H), 6.40 (d, J = 15.7 Hz, 1H), 5.40–5.34 (b, 1H), 3.90–2.80 (m, 10H). FAB-MS: m/z (relative intensity) 669.2 (M + 1, 20), 191.2 (85), 131.1 (100), 119.0 (75). Compound 9 (Rf = 0.59, CH3OH/CHCl3 = 5: 95) was shown to be pure by TLC.
[N-(trans-Cinnamoyl)-O-benzenesulfonyl-L-tyrosyl]-(4-nitrobenzoyl)piperazine (10)
1H NMR (CDCl3): δ 8.26 (d, J = 7.7 Hz, 2H), 7.89 (d, J = 7.7 Hz, 2H), 7.78–7.30 (m, 10H), 7.21 (d, J = 8.5 Hz, 2H), 7.00 (d, J = 7.4 Hz, 2H), 6.60–6.46 (m, 1H), 6.44–6.34 d, J = 15.7 Hz, 1H), 5.40–5.06 (b, 1H), 4.00–2.80 (m, 10H). FAB-MS: m/z (relative intensity) 669.2 (M + 1, 20), 434.2 (35), 131.1 (100). Compound 10 (Rf = 0.56, CH3OH/CHCl3 = 5: 95) was shown to be pure by TLC.
[N-(trans-4-Nitrocinnamoyl)-O-(4-nitrobenzenesulfonyl)-L-tyrosyl]-benzoylpiperazine (11)
1H NMR (CDCl3): δ 8.41 (d, J = 8.5 Hz, 2H), 8.24 (d, J = 8.5 Hz, 2H), 8.07 (d, J = 8.5 Hz, 2H), 7.74–7.58 (m, 3H), 7.50–7.34 (m, 4H), 7.20 (d, J = 8.5 Hz, 2H), 6.97 (d, J = 8.5 Hz, 2H), 6.64 (d, J = 8.0 Hz, 1H), 6.52 (d, J = 15.7 Hz, 1H), 5.40–5.16 (b, 1H), 3.84–2.80 (m, 10H). FAB-MS: m/z (relative intensity) 714.2 (M + 1, 2), 119.0 (100), 91.0 (50). Compound 11 (Rf = 0.50, CH3OH/CHCl3 = 5: 95) was shown to be pure by TLC.
[N-(trans-4-Nitrocinnamoyl)-O-benzenesulfonyl-L-tyrosyl]-(4-nitrobenzoyl)piperazine (12)
1H NMR (CDCl3): δ 8.36–8.14 (m, 4H), 7.90 (d, J = 7.4 Hz, 2H), 7.80–7.50 (m, 7H), 7.22 (d, J = 8.5 Hz, 2H), 7.10–6.92 (m, 2H), 6.72–6.40 (m, 2H), 5.40–5.08 (b, 1H), 4.00–2.80 (m, 10H). FAB-MS: m/z (relative intensity) 714.2 (M + 1, 10), 119.0 (100). Compound 12 (Rf = 0.47, CH3OH/CHCl3 = 5: 95) was shown to be pure by TLC.
[N-(trans-Cinnamoyl)-O-(4-nitrobenzenesulfonyl)-L-tyrosyl]-(4-nitrobenzoyl)piperazine (13)
1H NMR (CDCl3): δ 8.43 (d, J = 8.8 Hz, 2H), 8.28 (d, J = 8.2 Hz, 2H), 8.10 (d, J = 8.8 Hz, 2H), 7.70–7.46 (m, 4H), 7.44–7.34 (m, 3H), 7.23 (d, J = 8.5 Hz, 2H), 7.00 (d, J = 8.0 Hz, 2H), 6.49 (d, J = 8.2 Hz, 1H), 6.40 (d, J = 15.7 Hz, 1H), 5.40–5.16 (b, 1H), 3.90–2.80 (m, 10H). FAB-MS: m/z (relative intensity) 714.2 (M + 1, 5.5), 119.0 (100). Compound 13 (Rf = 0.56, CH3OH/CHCl3 = 5: 95) was shown to be pure by TLC.
[N-(trans-4-Nitrocinnamoyl)-O-(4-nitrobenzenesulfonyl)-L-tyrosyl]-(4-nitrobenzoyl)piperazine (14)
1H NMR (CDCl3): δ 8.43 (d, J = 8.8 Hz, 2H), 8.34–8.20(m, 4H), 8.10 (d, J = 8.8 Hz, 2H), 7.78–7.50 (m, 4H), 7.24 (d, J = 8.5 Hz, 2H), 7.01 (d, J = 7.7 Hz, 2H), 6.64 (d, J = 8.2 Hz, 1H), 6.54 (d, J = 15.6 Hz, 1H), 5.40–5.10 (b, 1H), 3.90–2.90 (m, 10H). FAB-MS: m/z (relative intensity) 759.2 (M + 1, 1.5), 119.0 (100). Compound 14 (Rf = 0.45, CH3OH/CHCl3 = 5: 95) was shown to be pure by TLC.
[N-Benzyloxycarbonyl-O-toluenesulfonyl-L-tyrosyl]-benzoylpiperazine (15)
1H NMR (CDCl3): δ 7.72 (d, J = 8.2 Hz, 2H), 7.48–7.28 (m, 12H), 7.12 (d, J = 8.2 Hz, 2H), 6.90 (d, J = 8.2 Hz, 2H), 5.61 (d, J = 8.5 Hz, 1H), 5.16–4.96 (m, 2H), 4.92–4.74 (m, 1H), 3.80–2.60 (m, 10H), 2.45 (s, 3H). Anal. (C35H35N3O7S·0.1 H2O).
[N-(2-Chlorobenzyloxycarbonyl)-O-toluenesulfonyl-L-tyrosyl]-benzoylpiperazine (16)
1H NMR (CDCl3): δ 7.72 (d, J = 7.7 Hz, 2H), 7.48–7.30 (m, 11H), 7.13 (d, J = 8.2 Hz, 2H), 6.92 (d, J = 8.2 Hz, 2H), 5.56 (d, J = 8.2 Hz, 1H), 5.28–5.10 (ABq, J = 13.1 Hz, 2H), 4.94–4.74 (m, 1H), 3.90–2.70 (m, 10H), 2.46 (s, 3H). Compound 16 (Rf = 0.64, CH3OH/CHCl3 = 5: 95) was shown to be pure by TLC.
General Procedure for the Synthesis of 20a, 24, 27, 32
To a solution of an aromatic nitro compound (19, 23b, 26b, or 31b; 2.0 mmol) in methanol (10 mL), sodium borohydride (10.0 mmol) was added and the solution stirred at room temperature for 5 min. Then copper(II) acetylacetonate was added and stirring continued for 1 h. TLC showed the completion of the reaction. The solvent was removed, and the residue was washed with petroleum ether two times. Then, the crude product was purified using silica gel chromatography eluting with methanol–chloroform (5:95) to provide a white solid foam (80–85% yield).
[N-(3-Aminobenzyloxycarbonyl)-O-toluenesulfonyl-L-tyrosyl]benzoylpiperazine (20a)
1H NMR (CDCl3): δ 7.72 (d, J = 8.5 Hz, 2H), 7.46–7.28 (m, 7H), 7.18–7.06 (m, 3H), 6.92 (d, J = 8.8 Hz, 2H), 6.74–6.58 (m, 3H), 5.57 (d, J = 9.3 Hz, 1H), 5.04–4.92 (m, 2H), 4.88–4.74 (b, 1H), 3.95–2.70 (m, 10H), 2.45 (s, 3H). FAB–MS: m/z (relative intensity) 657.3 (M + 1, 45), 658.3 (M + 2, 15), 85 (100). FAB–HRMS for C35H37N4O7S [M + H]+: calcd 657.2383, found 657.2385.
[N-Benzyloxycarbonyl-O-(4-aminobenzenesulfonyl)-L-tyrosyl]Boc-piperazine (24)
1H NMR (CDCl3): δ 7.53 (d, J = 8.5 Hz, 2H), 7.40–7.28 (m, 5H), 7.08 (d, J = 8.5 Hz, 2H), 6.92 (d, J = 8.4 Hz, 2H), 6.61 (d, J = 8.8 Hz, 2H), 5.61 (d, J = 8.5 Hz, 1H), 5.02–4.96 (ABq, J = 12.3, 16.0 Hz, 2H), 4.81 (b, 1H), 4.38 (br s, 2H), 3.80–2.60 (m, 10H), 1.45 (s, 9H). FAB-MS (NOBA): m/z (relative intensity) 639.3 (M + 1, 1), 661.3 (M + Na, 7), 583.2 (8), 539.3 (10). 91.0 (100). FAB-MS (NOBA/LiCl): m/z (relative intensity) 645.3 (M + Li, 15), 687.3 (M + Li+2Cl, 5).
[N-Benzyloxycarbonyl-O-(4-aminobenzenesulfonyl)-L-tyrosyl]benzoylpiperazine (27)
1H NMR (CDCl3): δ 7.55 (d, J = 8.5 Hz, 2H), 7.48–7.28 (m, 10H), 7.10 (d, J = 8.0 Hz, 2H), 6.94 (d, J = 8.0 Hz, 2H), 6.64 (d, J = 8.5 Hz, 2H), 5.63 (d, J = 8.8 Hz, 1H), 5.16–5.00 (ABq, J = 12.4, 15.9 Hz, 2H), 4.92–4.70 (b, 1H), 4.50–4.20 (b, 2H), 4.00–2.80 (m, 10H). FAB-MS: m/z (relative intensity) 643.4 (M + 1, 24), 391.4 (20), 251.2 (60), 91.1 (100). FAB–HRMS for C34H35N4O7S [M + H]+: calcd 643.2226, found 643.2244.
[N-Benzyloxycarbonyl-O-toluenesulfonyl-L-tyrosyl]-(4-aminobenzoyl)piperazine (32)
1H NMR (CDCl3): δ 7.70 (d, J = 8.2 Hz, 2H), 7.40–7.30 (m, 7H), 7.23 (d, J = 8.2 Hz, 2H), 7.12 (d, J = 8.5 Hz, 2H), 6.90 (d, J = 8.5 Hz, 2H), 6.70 (d, J = 8.4 Hz, 2H), 5.61 (d, J = 8.5 Hz, 1H), 5.12–5.02 (ABq, J = 12.3, 15.1 Hz, 2H), 4.88–4.76 (m, 1H), 3.89 (s, 2H), 3.90–2.80 (m, 10H), 2.45 (s, 3H). FAB-MS: m/z (relative intensity) 657.2 (M + 1, 95), 679.2 (M+ Na, 25), 120.1 (100), 91.0 (85). FAB–HRMS for C35H37N4O7S [M + H]+: calcd 657.2383, found 657.2396. Anal. (C35H36N4O9S·0.1H2O).
General Procedure for the Synthesis of 21, 29, 34
An amine compound (20a, 27, or 32; 0.08 mmol) was mixed with chloroform (2.0 mL), water (1.0 mL), and sodium bicarbonate (20 mg. 0.24 mmol) and stirred vigorously for 10 min. Thiophosgene (2.0 mmol) was added and stirring continued for 1 h. The solvent was removed, and the residue was washed two times with petroleum ether. The crude product was purified with a preparative TLC plate to provide a white solid foam (70–85% yield).
[N-(3-Isothiocyanato)benzyloxycarbonyl-O-toluenesulfonyl-L-tyrosyl]benzoylpiperazine (21)
1H NMR (CDCl3): δ 7.72 (d, J = 8.8 Hz, 2H), 7.46–7.30 (m, 8H), 7.24–7.08 (m, 5H), 6.92 (d, J = 8.8 Hz, 2H), 5.64 (d, J = 8.5 Hz, 1H), 5.05 (s, 2H), 4.90–4.72 (b, 1H), 4.00–2.70 (m, 10H), 2.45 (s, 3H). FAB–HRMS for C36H35N4O7S2 [M + H]+: calcd 699.1947, found 699.1948. Anal. (C36H34N4O7S2).
[N-Benzyloxycarbonyl-O-(4-isothiocyanato)benzenesulfonyl-L-tyrosyl]benzoylpiperazine (29)
1H NMR (CDCl3): δ 7.82 (d, J = 8.5 Hz, 2H), 7.48–7.28 (m, 12H), 7.14 (d, J = 8.5 Hz, 2H), 6.92 (d, J = 8.5 Hz, 2H), 5.59 (d, J = 8.5 Hz, 1H), 5.16–5.00 (ABq, J = 12.1, 14.8 Hz, 2H), 4.92–4.72 (b, 1H), 3.80–2.70 (m, 10H). FAB-HRMS for C35H33N4O7S2 [M + H]+: calcd 685.1791, found 685.1810.
[N-Benzyloxycarbonyl-O-toluenesulfonyl-L-tyrosyl]-(4-isothiocyanatobenzoyl)piperazine (34)
1H NMR (CDCl3): δ 7.42 (d, J = 8.5 Hz, 2H), 7.40–7.30 (m, 9H), 7.24 (d, J = 8.2 Hz, 2H), 7.14 (d, J = 8.5 Hz, 2H), 6.93 (d, J = 8.5 Hz, 2H), 5.60 (d, J = 8.5 Hz, 1H), 5.16–5.00 (ABq, J = 12.4, 15.4 Hz, 2H), 4.90–4.72 (b, 1H), 3.80–2.60 (m, 10H), 2.47 (s, 3H). FAB-MS (NOBA): m/z (relative intensity) 699.2 (M + 1, 22), 721.2 (M+ Na, 14), 91.1 (100). FAB–HRMS (CsI) for C36H34N4O7S2Cs [M + Cs]+: calcd 831.0923, found 831.0895. Anal. (C36H34N4O7S2·0.4EtOAc).
General Procedure for the Synthesis of 22, 30, 35a, 35b
1,4-Diaminobutane (100 mg, 1.1 mmol) or 1,2-diaminoethane (100 mg, 1.7 mmol) was added in the flask. Then, a solution of isothiocyanate compound (21 or 29 or 34) (0.01 mmol) and CH2Cl2 (0.1 mL) was added dropwise and stirring continued at room temperature for 1 h. The solvent was removed and the residue was washed two times with petroleum ether. The crude product was purified on a preparative TLC plate using methanol–chloroform (1:9) as mobile phase to afford a white solid foam (40–45% yield).
[N-3-{3-(4-Aminobutyl)thioureido}benzyloxycarbonyl-O-toluenesulfonyl-L-tyrosyl]benzoylpiperazine (22a)
1H NMR (CDCl3): δ 7.71 (d, J = 8.2 Hz, 2H), 7.48–7.30 (m, 10H), 7.18–7.10 (m, 2H), 7.08–7.00 (m, 1H), 6.90 (d, J = 8.2 Hz, 2H), 5.40–4.60 (m, 4H), 3.80–2.80 (m, 14H), 2.45 (s, 3H), 1.30–1.20 (m,4H). FAB-HRMS for C40H47N6O7S2 [M + H]+: calcd 787.2948, found 787.2957.
[N-Benzyloxycarbonyl-O-4-{3-(4-aminobutyl)thioureido} benzenesulfonyl-L-tyrosyl]benzoylpiperazine (30)
1H NMR (CDCl3): δ 7.80–7.56 (m, 4H), 7.54–7.30 (m, 10H), 7.20–6.90 (m, 4H), 5.64 (d, J = 8.5 Hz, 1H), 5.18–5.00 (ABq, J = 12.4, 15.0 Hz, 2H), 4.88–4.72 (b, 1H), 3.80–2.25 (m, 14H), 1.30–1.20 (m, 4H). FAB-HRMS (CsI) for C39H44N6O7S2Cs [M + Cs]+: calcd 905.1767, found 905.1771.
[N-Benzyloxycarbonyl-O-toluenesulfonyl-L-tyrosyl](4-{3-[(2-aminoethyl)thioureido})benzoylpiperazine (35a)
1H NMR (CDCl3): δ 7.71 (d, J = 8.0 Hz, 2H), 7.52–7.28 (m, 11H), 7.13 (d, J = 8.0 Hz, 2H), 6.91 (d, J = 8.0 Hz, 2H), 5.70 (d, J = 7.7 Hz, 1H), 5.16–4.86 (ABq, J = 12.4, 17.0 Hz, 2H), 4.90–4.72 (b, 1H), 3.84–2.56 (m, 14H), 2.45 (s, 3H). FAB-HRMS (CsI) for C38H42N6O7S2Cs [M + Cs]+: calcd 891.1611, found 891.1641.
[N-Benzyloxycarbonyl-O-toluenesulfonyl-L-tyrosyl](4-{3-[(4-aminobutyl)thioureido})benzoylpiperazine (35b)
1H NMR (CDCl3): δ 7.73 (d, J = 8.5 Hz, 2H), 7.48–7.28 (m, 11H), 7.15 (d, J = 8.2 Hz, 2H), 6.93 (d, J = 8.5 Hz, 2H), 5.61 (d, J = 9.1 Hz, 1H), 5.14–5.02 (ABq, J = 12.4, 15.9 Hz, 2H), 4.92–4.74 (b, 1H), 3.80–2.60 (m, 14H), 2.43 (s, 3H), 1.40–1.20 (m, 4H). FAB–HRMS (CsI) for C40H47N6O7S2 [M + H]+: calcd 787.2948, found 787.2919.
General Procedure for the Synthesis of 64
A solution of isothiocyanate compound 21 (20 mg, 0.029 mmol) in dry DMF (0.5 mL) was stirred under nitrogen for 4 days at 80 °C. The solvent was removed with nitrogen stream, and the residue was purified with preparative TLC (methanol: chloroform = 3:97) to afford a white solid (4.5 mg, 10% yield).
1,3-Bis{[3-N-benzyloxycarbonyl-O-toluenesulfonyl-L-tyrosyl]benzoylpiperazinethiourea (64)
1H NMR (CDCl3): δ 8.02 (b, 2H), 7.70 (d, J = 8.0 Hz, 4H), 7.50–7.30 (m, 20H), 7.22–7.02 (m, 6H), 6.90 (d, J = 8.2 Hz, 4H), 5.88 (b, 2H), 5.18–4.94 (m, 4H), 4.81 (b, 2H), 3.80–2.80 (m, 20H), 2.45 (s, 6H).). FAB-MS: m/z (relative intensity) 1377.3 (M + Na, 15), 1355.3 (M + H, 5), 261.2 (100). FAB-HRMS (CsI) for C71H70N8O14S3Cs [M + Cs]+: calcd 1487.3228, found 1487.3258. FAB-HRMS (CsI) for C71H69N8O14S3Cs2 {[(M − H)Cs]Cs}+: calcd 1619.2204, found 1619.2222.
General Procedure for the Synthesis of 65, 66, 67
Mixing equimolar amounts of isothiocyanate and amine component (Scheme 7) in dry DMF, and following the same synthetic procedure as for compound 64, the correspondence dimer was obtained in 5–10% yield.
Scheme 7.
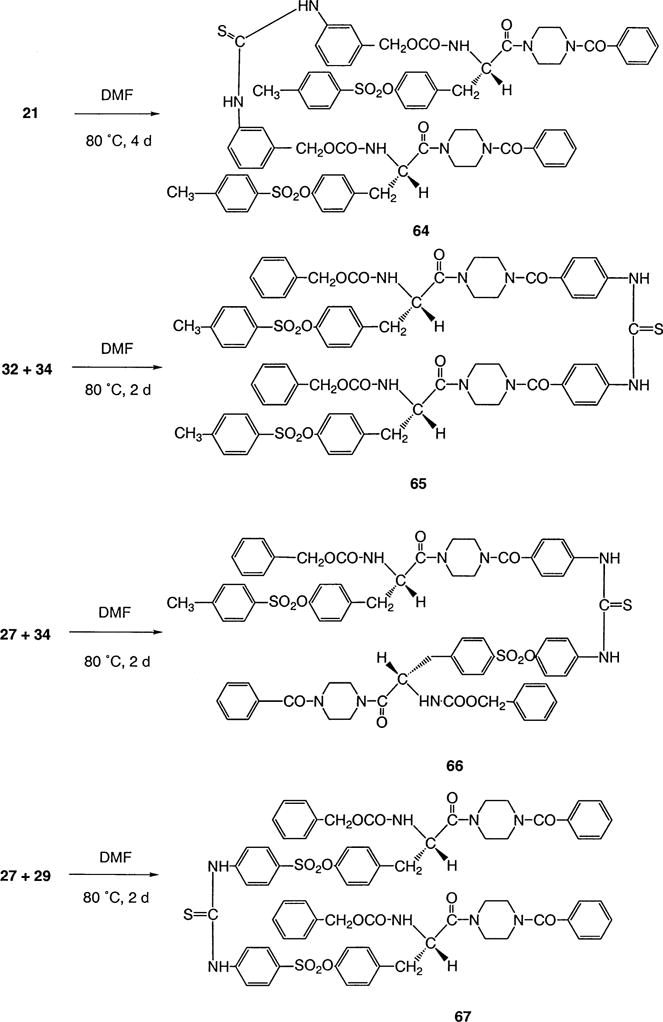
Dimerization of P2X7 Receptor Antagonists through Formation of Thiourea Linkages
1,3-Bis{[N-benzyloxycarbonyl-O-toluenesulfonyl-L-tyrosyl]-(benzo-4-yl)piperazine}thiourea (65)
1H NMR (CDCl3): δ 8.38 (b, 2H), 7.72 (d, J = 8.0 Hz, 4H), 7.50–7.28 (m, 20H), 7.24–7.06 (m, 6H), 6.94 (d, J = 8.2 Hz, 4H), 5.68–5.58 (m, 2H), 5.14–5.00 (m, 4H), 4.90–4.74 (b, 2H), 3.80–2.80 (m, 20H), 2.45 (s, 6H). FAB-MS: m/z (relative intensity) 1355.41 (M + H, 7), 1356.41 (M + 2H, 5.5), 1377.38 (M + Na, 1), 1393.35 (M + K, 2.5), 119.0 (100). FAB–HRMS (CsI) for C71H70O14N8S3Cs [M + Cs]+, calcd; 1487.3228; found, 1487.3210.
1-[(N-Benzyloxycarbonyl-O-(benzenesulfon-4-yl)-L-tyrosyl)benzoylpiperazine]-3-[(N-benzyloxycarbonyl-O-toluenesulfonyl-L-tyrosyl)(benzo-4-yl)piperazine]thiourea (66)
1H NMR (CDCl3): δ 7.80 (b, 2H), 7.72 (d, J = 8.2 Hz, 4H), 7.50–7.28 (m, 10H), 7.22–7.28 (m, 6H), 6.93 (d, J = 8.0 Hz, 4H), 5.60 (b, 2H), 5.16–4.98 (m, 4H), 4.80 (b, 2H), 3.80–2.80 (m, 20H), 2.45 (s, 3H). FAB-MS: m/z (relative intensity) 1341.4 (M + H, 5), 119.0 (100). FAB–HRMS (CsI) for C70H68O14N8S3Cs [M + Cs]+, calcd; 1473.3071; found, 1473.3105.
1,3-Bis{[N-benzyloxycarbonyl-O-(benzenesulfon-4-yl)-L-tyrosyl]benzoylpiperazine}thiourea (67)
1H NMR (CDCl3): δ 9.30 (b, 2H), 7.90–7.55 (m, 8H), 7.50–7.30 (m, 20H), 7.20–6.80 (m, 8H), 5.65 (d, J = 8.5 Hz, 2H), 5.25–4.90 (m, 4H), 4.80 (b, 2H), 3.80–2.50 (m, 20H). FAB–HRMS (CsI) for C69H66N8O14S3Cs [M + Cs]+: calcd 1459.2915, found 1459.2919.
Pharmacological Analysis. P2X7 Receptor Channel Activation
All experiments were performed using adherent HEK293 cells stably transfected with cDNA encoding the human P2X7 receptor. Adherent cells on 12-well polylysine-coated plates were incubated at 37 °C in 1 mL of physiological salt solution (125 mM NaCl, 5 mM KCl, 1 mM MgCl2, 1.5 mM CaCl2, 25 mM NaHEPES (pH 7.5), 10 mM D-glucose, 1 mg/mL BSA). Antagonists were added from 1000× stock solutions dissolved in DMSO. Cells were preincubated with antagonists for 15 min prior to stimulation for 10 min with 3 mM ATP (final concentration). Reactions were terminated by rapid aspiration of the extracellular medium in each well. The adherent cells in each well were then extracted overnight with 1 mL 10% HNO3. K+ content in these nitric acid extracts was assayed by atomic absorbance spectrophotometry. Duplicate or triplicate wells were run for all test conditions in each separate experiment, and the measured K+ contents were averaged. Antagonist function was measured by the percent inhibition of the K+ release triggered by 3 mM ATP in paired wells in the absence of antagonist.
RESULTS
Chemical Synthesis
The analogues consisted of L-tyrosine derivatives, of the general structure R1-Tyr-(OR2)-piperazinyl-R3 (Table 1), in which three variable regions were systematically varied in structure (compounds 3–35) through facile acylation reactions and other procedures. The lead compound MRS 2409 (17), 2, contained an isoquinolinyl sulfonate at the tyrosyl hydroxyl group. For the present study, this group was first substituted with a phenyl sulfonate ester, a structural simplification known to preserve affinity for the receptor (17). Each of the three regions (R1–R3) was probed systematically for possbile attachment sites, through synthesis alternating with biological evaluation, which consisted of screening at a single concentration (initially 3 μM). This process led to the identification and partial optimization of potent, functionalized P2X7 antagonists.
Table 1.
Antagonistic Effects of Tyrosine Derivatives on Function of Human P2X7 Receptors Expressed in HEK293 Cellsa
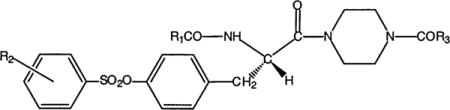
| |||||
|---|---|---|---|---|---|
| compound | R1 | R2 | R3 | % inhibition | n |
| Monomeric | |||||
| 3 | Ph-CH=CH (trans) | H | Ph | 62 ± 4 | 2 |
| 4 | 2-OCH3-Ph-CH=CH (trans) | 4-CH3 | Ph | 27 ± 2 | 2 |
| 5 | 2-OCH3-Ph-CH=CH (cis) | 4-CH3 | Ph | 17 ± 1 | 2 |
| 6 | 3-Cl-Ph-CH=CH (trans) | 4-CH3 | Ph | 57 ± 5 | 2 |
| 7 | 2,4,5-(OCH3)3-Ph-CH=CH (trans) | 4-CH3 | Ph | −2 ± 6 | 2 |
| 8 | 4-NO2-Ph-CH=CH (trans) | H | Ph | 43 ± 20 | 2 |
| 9 | Ph-CHdCH (trans) | 4-NO2 | Ph | 72 ± 4 | 3 |
| 10 | Ph-CHdCH (trans) | H | Ph-4-NO2 | 41 ± 1 | 2 |
| 11 | 4-NO2-Ph-CH=CH (trans) | 4-NO2 | Ph | 58 ± 1 | 2 |
| 12 | 4-NO2-Ph-CH=CH (trans) | H | Ph-4-NO2 | 30 ± 2 | 2 |
| 13 | Ph-CH=CH (trans) | 4-NO2 | Ph-4-NO2 | 43 ± 14 | 3 |
| 14 | 4-NO2-Ph-CH=CH (trans) | 4-NO2 | Ph-4-NO2 | 4 ± 9 | 2 |
| 15 | Ph-CH2O | 4-CH3 | Ph | 71 ± 21 | 2 |
| 16 | 2-Cl-Ph-CH2O | 4-CH3 | Ph | 43 ± 17 | 3 |
| 17 | 4-NO2-Ph-CH2O | 4-CH3 | Ph | 69 ± 25 | 3 |
| 18 | 3-NO2-Ph-CH2O | 4-CH3 | O-C(CH3)3 | 94 ± 28 | 3 |
| 19 | 3-NO2-Ph-CH2O | 4-CH3 | Ph | 72 ± 6 | 2 |
| 20a | 3-NH2-Ph-CH2O | 4-CH3 | Ph | 50 ± 6 | 2 |
| 20b | 3-CH3CONH–Ph-CH2O | 4-CH3 | Ph | 9 ± 13 | 2 |
| 21 | 3-SCN-Ph-CH2O | 4-CH3 | Ph | −4 ± 5 | 2 |
| 22 | 3-[H2N(CH2)4NH-CSNH]-Ph-CH2O | 4-CH3 | Ph | 17 ± 4 | 2 |
| 23a | Ph-CH2O | 3-NO2 | O-C(CH3)3 | 74 ± 2 | 2 |
| 23b | Ph-CH2O | 4-NO2 | O-C(CH3)3 | 30 ± 28 | 2 |
| 24 | Ph-CH2O | 4-NH2 | O-C(CH3)3 | 64 ± 17 | 2 |
| 25 | Ph-CH2O | 4-NHCOCH3 | O-C(CH3)3 | 64 ± 27 | 2 |
| 26a | Ph-CH2O | 3-NO2 | Ph | 75 ± 10 | 2 |
| 26b | Ph-CH2O | 4-NO2 | Ph | 78 ± 18 | 3 |
| 27 | Ph-CH2O | 4-NH2 | Ph | 56 ± 9 | 2 |
| 28 | Ph-CH2O | 4-NHCOCH3 | Ph | 31 ± 41 | 2 |
| 29 | Ph-CH2O | 4-NCS | Ph | 72 ± 1 | 2 |
| 30 | Ph-CH2O | 4-[H2N(CH2)4NH-CSNH]- | Ph | 80 ± 11 | 3 |
| 31a | Ph-CH2O | 4-CH3 | Ph-3-NO2 | 60 ± 16 | 2 |
| 31b | Ph-CH2O | 4-CH3 | Ph-4-NO2 | 61 ± 6 | 2 |
| 32 | Ph-CH2O | 4-CH3 | Ph-4-NH2 | 93 ± 3 | 2 |
| 33 | Ph-CH2O | 4-CH3 | Ph-4-NHCOCH3 | 45 ± 21 | 2 |
| 34 | Ph-CH2O | 4-CH3 | Ph-4-NCS | 30 ± 16 | 2 |
| 35a | Ph-CH2O | 4-CH3 | Ph-4-NHCS-NH(CH2)2NH2 | 14 + 19 | 2 |
| 35b | Ph-CH2O | 4-CH3 | Ph-4-NHCS-NH(CH2)4NH2 | 63 ± 16 | 2 |
| Dimeric | |||||
| linkage | |||||
| 64 | R1–R1 | −5 ± 4 | 2 | ||
| 65 | R3–R3 | 26 | 1 | ||
| 66 | R2–R3 | 61 ± 29 | 2 | ||
| 67 | R2–R2 | 72 ± 41 | 2 | ||
All experiments were performed using adherent HEK293 cells stably transfected with cDNA encoding the human P2X7 receptor. Cells were preincubated with antagonists (3 μM final concentration) for 15 min prior to stimulation for 10 min with 3 mM ATP (final concentration). Antagonist function was measured by the percent inhibition of the K+ release triggered by 3 mM ATP in paired wells in the absence of antagonist. Data points represent the mean ± SD of values obtained. 15, MRS 2427; 18, MRS 2464; 26b, MRS 2447; 30, MRS 2483; 32, MRS 2452; 35b, MRS 2484; 66, MRS 2454; 67, MRS 2455.
The synthetic routes for systematic substitution at three positions on the molecule are shown in Schemes 1–5. Subsequent functional group replacement beginning with nitro groups is shown in Scheme 6. Synthetic yields are listed in Materials and Methods. General probing of the SAR at each position was carried out. Scheme 1 indicates substitutions made at the R1 position, including both N-cinnamoyl (4–7) and N-benzyloxycarbonyl (15 and 16) derivatives. Each of the three ring-substitution positions shown in Table 1 (R1–R3) was designed for the initial incorporation of a nitro group (at the meta-position for R1, meta- or para-positions for R2 and R3). For a series of N-cinnamoyl derivatives (Scheme 2), multiple nitro groups (11–14) were also included. At R2 and R3 the nitro groups were introduced as shown in Schemes 3 and 4.
Scheme 1.
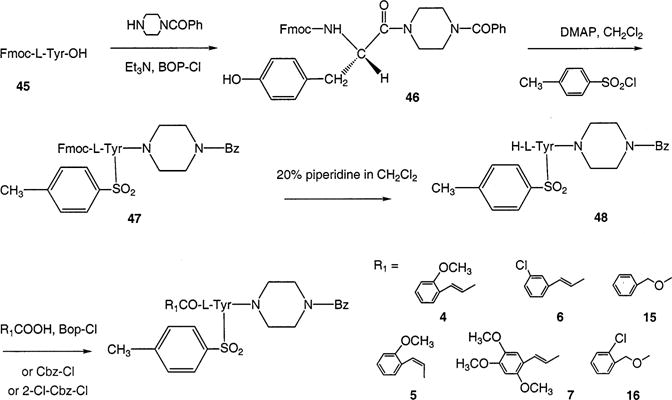
Synthesis of P2X7 Receptor Antagonists Consisting of L-Tyrosine Derivatives Differing in the Cinnamoyl or Urethane Substituent Present at the Nα Position
Scheme 5.
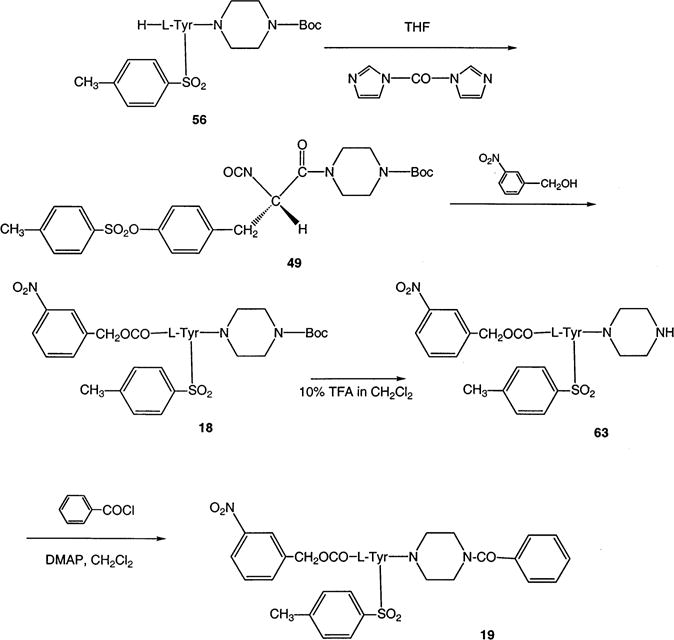
Synthesis of P2X7 Receptor Antagonists Consisting of L-Tyrosine Derivatives Containing 3-Nitro Substitution at the Nα-Benzyloxycarbonyl Substituent
Scheme 6.
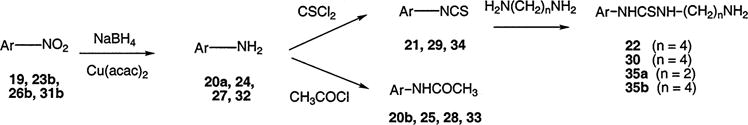
Functional Group Replacement in P2X7 Receptor Antagonists
Scheme 2.
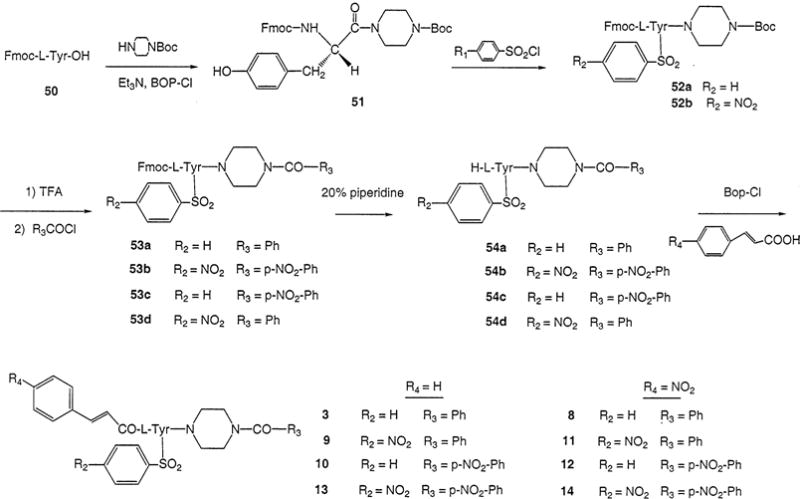
Synthesis of P2X7 Receptor Antagonists Consisting of L-Tyrosine Derivatives Containing a Nitro Substitution at Either the Nα-Cinnamoyl Substituent, the Aryl Sulfonate Group Present on the Tyrosyl Side Chain, or the Cα-Benzoylpiperazine Substituent
Scheme 3.
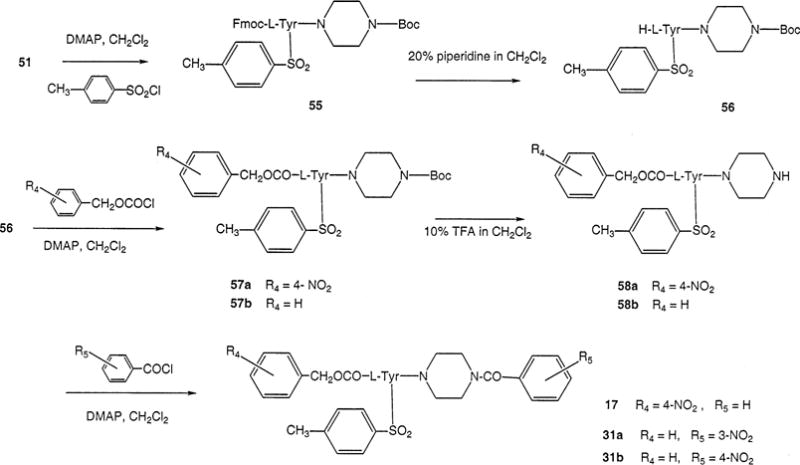
Synthesis of P2X7 Receptor Antagonists Consisting of L-Tyrosine Derivatives Containing a Nitro Substitution at the Nα-Benzyloxycarbonyl Substituent or of the Cα-Benzoylpiperazine Substituent
Scheme 4.
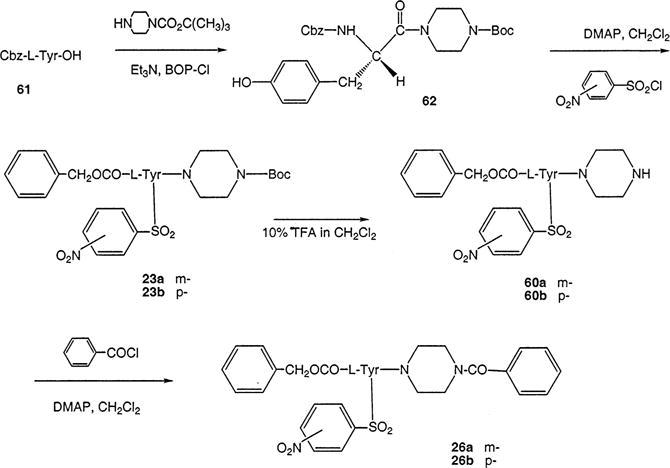
Synthesis of P2X7 Receptor Antagonists Consisting of L-Tyrosine Derivatives Containing a Nitro Substitution at the Aryl Sulfonate Group Present on the Tyrosyl Side Chain
The synthesis of N-(nitrobenzyloxycarbonyl)-L-tyrosine derivatives (Scheme 5) required a modification of the original route based on simple reduction of 4-nitro groups. Since the N-(4-aminobenzyloxycarbonyl)tyrosine derivative of 17 proved to be unstable, we sought to develop an alternate synthetic method, aimed instead at the 3-nitro derivative, 18. Usually, benzyl chloroformates are synthesized through the reaction of benzyl alcohol with phosgene (22). Here, an interesting alternative for the incorporation of N-(3-nitrobenzyloxycarbonyl) into tyrosine was the transformation of the α-amine of the tyrosyl moiety into an isocyanate group first with the aid of 1,1′-carbonyldiimidazole (23) to yield 49, and subsequent addition of the alcohol to yield 18.
The nitro groups at R1, R2, and R3 were directly reduced to amines (Scheme 6), which were either converted to the isothiocyanate groups, in 21, 29, and 34, as potential affinity labels for the receptor, or acylated, as models for conjugation. The isothiocyanate groups also reacted with alkyl diamines to produce the primary amine congeners 22, 30, and 35. Several methods were compared for reduction of the aromatic nitro compounds. Many reducing reagents have been used to reduce aromatic nitro compounds (24) with the most classic being zinc, tin, or iron in the presence of an acid. Other reagents include hydrazine, TiCl4–dialkyl telluride and sulfide (25). However, most of them lack the desired chemoselectivity over the functional groups that are present in our compounds such as amide, tert-butoxycarbonyl (Boc), 9-fluorenylmethoxycarbonyl (Fmoc), and benzyloxycarbonyl (Cbz). Though aromatic nitro compounds are seldom reduced by sodium borohydride in alcoholic solution, we found NaBH4–copper(II) acetylacetonate (26) to be the most effective and a mild catalyst for the reduction of nitro groups on these tyrosine derivatives. This reaction occurred nearly quantitatively as judged using TLC.
Four dimer permutations (64–67) were created by coupling each of the isothiocyanate derivatives to either the precursor amine or to other amine congeners (Scheme 7). These coupling reactions required elevated temperature and were carried out in moderate yield.
Biological Activity
The effects of substitution at R1, R2, and R3 on inhibition of P2X7 receptor-mediated ion flux (Table 1) were compared. Experiments were performed using adherent HEK293 cells stably transfected with cDNA encoding the human P2X7 receptor. Cells were preincubated with each antagonist at a fixed concentration (3 μM) prior to stimulation for 10 min with 3 mM ATP. The percent inhibition of the K+ release was the parameter used to indicate antagonist function.
The three structural regions for conjugation were compared in their effects on biological potency. Among N-cinnamoyl derivatives, more favorable potency was associated with the unsubstituted derivative 9 than with methoxy substitution. Multiple methoxy substitution, e.g., 7, eliminated the antagonistic effect at the P2X7 receptor. The 4-nitro substitution of the cinnamoyl group also reduced potency (cf. 14 vs 13). The Cbz derivative 15 was a potent antagonist of the human P2X7 receptor, and 4- or 3-nitro substitution preserved that property, as in 18 and 19. However, reduction and acetylation of the 3-nitro group, 20a and 20b, reduced potency. The corresponding isothiocyanate 21 and amine congener 22 were inactive or weakly potent, respectively.
Nitro subsitution of the tyrosyl side chain (R2 substituent), at the 3- or 4-position, as in benzoylpiperazine (at R3) derivatives 26a and 26b, respectively, maintained potency at the human P2X7 receptor. There was no difference in biological potency between 3- and 4-nitro derivatives. The corresponding t-Boc derivatives (at the R3 position, e.g., 23a and 23b) similarly antagonized the human P2X7 receptor. This antagonism was preserved when the 4-nitro group was reduced and acetylated (e.g. 24 and 25; 27 and 28). A primary amine congener linked at the R2 4-position, 30, was a potent antagonist of the human P2X7 receptor. The corresponding isothiocyanate 29 displayed considerable potency as a P2X7 receptor antagonist and warranted examination as an irreversible receptor inhibitor.
For the R3 substituent, 3- or 4-nitrobenzoyl subsitution, 31a and 31b, respectively, preserved antagonistic properties at the human P2X7 receptor. As for the R2 substituent, there was no difference in biological potency between 3- and 4-nitro derivatives. The P2X7 receptor antagonism was increased when the 4-nitro group was reduced but diminished upon acetylation (e.g., 32 and 33). An isothiocyanate 34 only weakly antagonized the effects of ATP at the P2X7 receptor. A primary amine congener containing four methylenes linked at the R2 4-position, 35b, was a potent antagonist of the human P2X7 receptor, while a shorter homologue 35a (two methylenes) antagonized weakly.
Full concentration–response curves provided a more precise means of comparison among some selected, potent compounds, including the 3-nitro-Cbz derivative 18, a 4-nitrotoluenesulfonate 26b, a primary amine congener 30, and 4-aminobenzoyl derivative 32 (Figure 1). Compounds 18 and 26b, were roughly as potent as 1 as P2X7 receptor antagonists, and appeared to have IC50 values in the 0.1 μM range. The other compounds appeared to have IC50 values in the 0.3 μM range (30) or > 0.3 μM (32).
Figure 1.
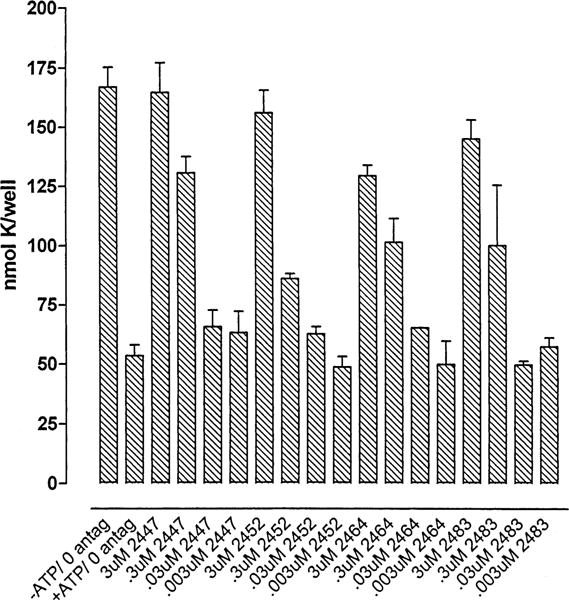
Effects of tyrosyl derivatives on P2X7 receptor-activation in hP2X7–HEK cells. Cells were preincubated with or without the indicated concentrations of selected antagonists (26b MRS2447; 32 MRS2452; 18 MRS2464; and 30 MRS2483) prior to stimulation with 3 mM ATP. Data points represent the mean (± SD) K+ contents from triplicate wells in a single experiment. IC50 values mentioned in the text are rough estimates from visual inspection of the concentration–response relationships. Hill coefficients were not determined, since previous studies have shown that the lead compound (KN-62) represses P2X7 receptor function via complex mechanisms that are not readily amenable to standard ligand-binding analyses.
Among antagonist dimers, only the R2–R3- and R2–R2-linked dimers, 66 and 67, respectively, displayed significant antagonistic properties at the P2X7 receptor.
DISCUSSION
The binding site for KN-62, 1, resides within the first 335 residues of the human P2X7 receptor, likely within the large extracellular loop (13, 21). While it is not feasible to model this binding interaction, due to lack of a high-resolution template for this ion channel and uncertainty about the oligomeric nature of the channel (27), chemical probes of the receptor may be very useful in studying the structure. The stoichiometry of association of subunits in P2X receptors has been studied suggesting a trimeric organization (2, 27, 28), but little is known about the correspondence of antagonist binding sites to the overall oligomeric ion channel structure. By functionalizing derivatives (17) of the widely used P2X7 antagonist KN-62 it may be possible to locate the binding site (in conjunction with mutagenesis studies). Mutagenesis studies of the P2X7 receptor aimed at structural and functional elucidation have already been carried out (29, 32). Peptide mapping of receptor digests (30) using electrophoretic or mass spectroscopic methods following affinity labeling of the receptor would also be useful in this regard.
Since there may be multiple binding sites on the P2X7 receptor homomer, it may be possible to detect the preferred orientation of covalently linked antagonist dimers that may occupy two distinct binding sites, based on divergent biological potencies for a variety of linkage combinations. Since the dimerization of the antagonists in the present study through the tyrosyl side chain, alone, provided retention of antagonist properties, it is likely that this region of the antagonist occurs in a portion of the binding site that is somewhat relaxed in its steric requirements. Whether the dimers 66 and/or 67 are able to bridge two putative binding sites on the P2X7 homomer will be the object of further studies. The species selectivity of the new functionalized congeners also should be investigated, since differences have been noted for KN-62 (33).
Receptor ligands that bridge two subunits of a multimeric receptor may also potentially be of use for tissue selectivity, if there are differences between tissues in the organization of subunits. Such differences have been demonstrated for native P2X7 receptors (34), which in peritoneal macrophage and bone marrow cells exist as a strongly bound multimeric complex, and in brain glia and/or astrocytes appear to form only as monomeric subunits.
In conclusion, we have systematically varied substituent groups in a series of tyrosyl analogues, resulting in analogues, among which are antagonists that are equipotent to the lead structure 1 and also contain chemical functional groups for linking purposes. Such functionalized congeners have been used effectively for the structural investigations of other purine receptors (18, 19, 31), including adenosine A1, A2A, and A3 receptors. Suitable sites and chemical approaches to cross-linking these antagonists have been found on the tyrosyl side chain (R2) and on the CR-substituent (R3). The more versatile region for functionalization appears to be the R2 substituent, since even dimerization through this substituent preserves the receptor binding property. We have prepared several amine functionalized congeners, that may potentially be conjugated to fluorescent probes for receptor characterization, or immobilized on a solid support for isolation of the receptor by affinity chromatography. Among P2X receptors, only the P2X1 subtype has so far been isolated in this manner (35).
Acknowledgments
We thank Gilead Sciences (Foster City, CA) for financial support to W. Z. Chen. This work was supported by NIH grant GM36387 to G.D. We thank Mr. Noel Whittaker and Mr. Victor Livengood for determination of mass spectra.
Abbreviations
- Bop-Cl
bis(2-oxo-3-oxazolidinyl)phosphinic chloride
- Boc
tert-butyloxycarbonyl
- BSA
bovine serum albumin
- Cbz
benzyloxycarbonyl
- DMAP
4-(dimethylamino)pyridine
- DMF
dimethylformamide
- DMSO
dimethyl sulfoxide
- EtOAc
ethyl acetate
- FAB
fast atom bombardment
- Fmoc
9-fluorenylmethyloxycarbonyl
- HEK
human embryonic kidney
- HEPES
N-(2-hydroxyethyl)piperazine-N′-(2-ethanesulfonic acid)
- HRMS
high-resolution mass spectrometry
- KN-62
1-[N,O-bis-(5-isoquinolinesulfonyl)-N-methyl-L-tyrosyl]-4-phenylpiperazine
- MRS 2409
[N-benzyloxycarbonyl-O-[4-(isoquinolinylsulfonyl)]-L-tyrosyl]benzoylpiperazine
- TFA
trifluoroacetic acid
- THF
tetrahydrofuran
- TLC
thin-layer chromatography
LITERATURE CITED
- 1.Fredholm BB, Abbracchio MP, Burnstock G, Dubyak GR, Harden TK, Jacobson KA, Schwabe U, Williams M. Toward a revised nomenclature for P1 and P2 receptors. Trends Pharm Sci. 1997;18:79–82. doi: 10.1016/s0165-6147(96)01038-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Khakh BS, Burnstock G, Kennedy C, King BF, North RA, Seguela P, Voigt M, Humphrey PP. International union of pharmacology. XXIV. Current status of the nomenclature and properties of P2X receptors and their subunits. Pharmacol Rev. 2001;53:107–118. [PubMed] [Google Scholar]
- 3.King BF. Cardiovascular Biology of Purines. Kluwer Academic Publishers; Norwell, MA: 1998. pp. 159–186. Ch. 10. [Google Scholar]
- 4.Collo G, Neidhart S, Kawashima E, Kosco-Vilbois M, North RA, Buell G. Tissue distribution of the P2 × 7 receptor. Neuropharmacology. 1997;36:1277–1283. doi: 10.1016/s0028-3908(97)00140-8. [DOI] [PubMed] [Google Scholar]
- 5.Ferrari D, Stroh C, Schulze-Osthoff K. P2X7/P2Z purinoreceptor-mediated activation of transcription factor NFAT in microglial cells. J Biol Chem. 1999;274:13205–13210. doi: 10.1074/jbc.274.19.13205. [DOI] [PubMed] [Google Scholar]
- 6.Humphreys BD, Rice J, Kertesy SB, Dubyak GR. Stress-activated protein kinase/JNK activation and apoptotic induction by the macrophage P2X7 nucleotide receptor. J Biol Chem. 2000;275:26792–26798. doi: 10.1074/jbc.M002770200. [DOI] [PubMed] [Google Scholar]
- 7.Gargett CE, Wiley JS. The isoquinoline derivative KN-62 a potent antagonist of the P2Z-receptor of human lymphocytes. Br J Pharmacol. 1997;120:1483–1490. doi: 10.1038/sj.bjp.0701081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ferrari D, La Sala A, Chiozzi P, Morelli A, Falzoni S, Girolomoni G, Idzko M, Dichmann S, Norgauer J, Di Virgilio F. The P2 purinergic receptors of human dendritic cells: identification and coupling to cytokine release. FASEB J. 2000;14:2466–2476. doi: 10.1096/fj.00-0031com. [DOI] [PubMed] [Google Scholar]
- 9.Solle M, Labasi J, Perregaux DG, Stam E, Petrushova N, Koller BH, Griffiths RJ, Gabel CA. Altered Cytokine Production in Mice Lacking P2X7 Receptors. J Biol Chem. 2001;276:125–132. doi: 10.1074/jbc.M006781200. [DOI] [PubMed] [Google Scholar]
- 10.Turner JT, Landon LA, Gibbons SJ, Talamo BR. Salivary gland P2 nucleotide receptors. Crit Rev Oral Biol Med. 1999;10:210–224. doi: 10.1177/10454411990100020701. [DOI] [PubMed] [Google Scholar]
- 11.Hu Y, Fisette PL, Denlinger LC, Guadarrama AG, Sommer JA, Proctor RA, Bertics PJ. Purinergic receptor modulation of lipopolysaccharide signaling and inducible nitric-oxide synthase expression in RAW 264.7 macrophages. J Biol Chem. 1998;273:27170–27175. doi: 10.1074/jbc.273.42.27170. [DOI] [PubMed] [Google Scholar]
- 12.Wiley S, Gargett CE, Zhang W, Snook MB, Jamieson GP. Partial agonists and antagonists reveal a second permeability state of human lymphocyte P2Z/P2X7 channel. Am J Physiol. 1998;275:C1224–C1231. doi: 10.1152/ajpcell.1998.275.5.C1224. [DOI] [PubMed] [Google Scholar]
- 13.Rassendren F, Buell GN, Virginio C, Collo G, North RA, Surprenant A. The permeabilizing ATP receptor, P2X7. Cloning and expression of a human cDNA. J Biol Chem. 1997;272:5482–5486. doi: 10.1074/jbc.272.9.5482. [DOI] [PubMed] [Google Scholar]
- 14.Bianchi BR, Lynch KJ, Touma E, Niforatos W, Burgard EC, Alexander KM, Park HS, Yu H, Metzger R, Kowaluk E, Jarvis MF, van Biesen T. Pharmacological characterization of recombinant human and rat P2X receptor subtypes. Eur J Pharmacol. 1999;376:127–138. doi: 10.1016/s0014-2999(99)00350-7. [DOI] [PubMed] [Google Scholar]
- 15.Erb L, Lustig KD, Ahmed AH, Gonzalez FA, Weisman GA. Covalent incorporation of 3′-O-(4-benzoyl)benzoyl-ATP into a P2 purinoceptor in transformed mouse fibroblasts. J Biol Chem. 1990;265:7424–7431. [PubMed] [Google Scholar]
- 16.Tokumitsu H, Chijiwa T, Hagiwara M, Mizutani A, Terasawa M, Hidaka H. KN-62, 1-[N, O-bis(5-isoquinolinesulfonyl)-N-methyl-L-tyrosyl]-4-phenylpiperazine, a specific inhibitor of Ca2+/calmodulin-dependent protein kinase II. J Biol Chem. 1990;265:4315–4320. [PubMed] [Google Scholar]
- 17.Ravi RG, Kertesy SB, Dubyak GR, Jacobson KA. Potent P2X7 receptor antagonists: Tyrosyl derivatives synthesized using a sequential parallel synthetic approach. Drug Devel Res. 2001;54:75–87. doi: 10.1002/ddr.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jacobson KA, Ukena D, Padgett W, Kirk KL, Daly JW. Molecular probes for extracellular adenosine receptors. Biochem Pharmacol. 1987;36:1697–1707. doi: 10.1016/0006-2952(87)90056-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boring DL, Ji XD, Zimmet J, Taylor KE, Stiles GL, Jacobson KA. Trifunctional agents as a design strategy for tailoring ligand properties: Irreversible inhibitors of A1 adenosine receptors. Bioconjugate Chem. 1991;2:77–88. doi: 10.1021/bc00008a002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karton Y, Baumgold J, Handen JS, Jacobson KA. Molecular probes for muscarinic receptors: Derivatives of the M1-antagonist telenzepine. Bioconjugate Chem. 1992;3:234–240. doi: 10.1021/bc00015a006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chessell IP, Simon J, Hibell AD, Michel AD, Barnard EA, Humphrey PPA. Cloning and functional characterization of the mouse P2X7 receptor. FEBS Lett. 1998;439:26–30. doi: 10.1016/s0014-5793(98)01332-5. [DOI] [PubMed] [Google Scholar]
- 22.Carter HE, Frank RL, Johnston HW. In: Organic Synthesis. Horning ED, editor. III. New York: Wiley Press, Coll; 1955. p. 167. [Google Scholar]
- 23.Staab HA, Benz W. Darstellung von isocyanaten. Angew Chem. 1961;73:66. [Google Scholar]
- 24.Yu C, Liu B, Hu L. Samarium (0) and 1, 1′-dioctyl-4, 4′-bipyridinium dibromide: a novel electron-transfer system for the chemoselective reduction of aromatic nitro groups. J Org Chem. 2001;66:919–924. doi: 10.1021/jo005666q. [DOI] [PubMed] [Google Scholar]
- 25.Suzuki H, Hanazaki Y. Titanium-mediated reduction of nitrobenzenes and benzil with dialkyl telluride. Chem Lett. 1986;4:549–550. [Google Scholar]
- 26.Hanaya K, Muramatsu T, Kudo K. Reduction of aromatic nitro-compounds to amines with sodium borohydride-copper (II) acetylacetonate. J Chem Soc, Perkin Trans. 1979;1 10:2409–2410. [Google Scholar]
- 27.Torres GE, Egan TM, Voigt MM. Heterooligomeric assembly of P2X receptor subunits. Specificities exist with regard to possible partners. J Biol Chem. 1999;274:6653–6659. doi: 10.1074/jbc.274.10.6653. [DOI] [PubMed] [Google Scholar]
- 28.Nicke A, Baumert HG, Rettinger J, Eichele A, Lambrecht G, Mutschler E, Schmalzing G. P2X1 and P2X3 receptors form stable trimers: a novel structural motif of ligand-gated ion channels. EMBO J. 1998;17:3016–3028. doi: 10.1093/emboj/17.11.3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim M, Jiang LH, Wilson HL, North RA, Surprenant A. Proteomic and functional evidence for a P2X7 receptor receptor signaling complex. EMBO J. 2001;20:6347–6358. doi: 10.1093/emboj/20.22.6347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kennedy AP, Mangum KC, Linden J, Wells JN. Covalent modification of transmembrane span III of the A1 adenosine receptor with an antagonist photoaffinity probe. Mol Pharmacol. 1996;50:789–798. [PubMed] [Google Scholar]
- 31.Barrington WW, Jacobson KA, Hutchison AJ, Williams M, Stiles GL. Identification of the A2 adenosine receptor binding subunit by photoaffinity cross-linking. Proc Natl Acad Sci USA. 1989;86:6572–6576. doi: 10.1073/pnas.86.17.6572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Klapperstuck M, Buttner C, Schmalzing G, Markwardt F. Functional evidence of distinct ATP activation sites at the human P2X7 receptor. J Physiol. 2001;534:25–35. doi: 10.1111/j.1469-7793.2001.00025.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Humphreys BD, Virginio C, Surprenant A, Rice J, Dubyak GR. Isoquinolines as antagonists of the P2X7 nucleotide receptor: high selectivity for the human versus rat receptor homologues. Mol Pharmacol. 1998;54:22–32. doi: 10.1124/mol.54.1.22. [DOI] [PubMed] [Google Scholar]
- 34.Kim M, Spelta V, Sim J, North RA, Surprenant A. Differential assembly of rat purinergic P2X7 receptor in immune cells of the brain and periphery. J Biol Chem. 2001;276:23262–23267. doi: 10.1074/jbc.M102253200. [DOI] [PubMed] [Google Scholar]
- 35.Chen L, Hardwick JP, McPhie P, Sitkovsky M, Jacobson KA. Purification and recognition of recombinant mouse P2X1 receptors expressed in a baculovirus system. Drug Devel Res. 2001;51:7–19. doi: 10.1002/1098-2299(20000901)51:1<7::AID-DDR2>3.0.CO;2-W. [DOI] [PMC free article] [PubMed] [Google Scholar]