

Abstract
Purpose
[18F]SP203 (3-fluoro−5-(2− (2-([18F]fluoromethyl)-thiazol−4-yl)ethynyl)benzonitrile) is an effective high-affinity and selective radioligand for imaging metabotropic 5 receptors (mGluR5) in human brain with PET. To provide a radioligand that may be used for more than one scanning session in the same subject in a single day, we set out to label SP203 with shorter-lived 11C (t1/2=20.4 min) and to characterize its behavior as a radioligand with PET in the monkey.
Methods
Iodo and bromo precursors were obtained by cross-coupling 2-fluoromethyl−4-((trimethylsilyl)ethynyl) −1,3-thia-zole with 3,5-diiodofluorobenzene and 3,5-dibromofluoroben-zene, respectively. Treatment of either precursor with [11C] cyanide ion rapidly gave [11C]SP203, which was purified with high-performance liquid chromatography. PET was used to measure the uptake of radioactivity in brain regions after injecting [11C]SP203 intravenously into rhesus monkeys at baseline and under conditions in which mGluR5 were blocked with 3-[ (2-methyl−1,3-thiazol−4-yl)ethynyl]pyridine (MTEP). The emergence of radiometabolites in monkey blood in vitro and in vivo was assessed with radio-HPLC. The stability of [11C]SP203 in human blood in vitro was also measured.
Results
The iodo precursor gave [11C]SP203 in higher radio-chemical yield (>98 %) than the bromo precursor (20–52 %). After intravenous administration of [11C]SP203 into three rhesus monkeys, radioactivity peaked early in brain (average 12.5 min) with a regional distribution in rank order of expected mGluR5 density. Peak uptake was followed by a steady decline. No radioactivity accumulated in the skull. In monkeys pretreated with MTEP before [11C]SP203 administration, radioactivity uptake in brain was again high but then declined more rapidly than in the baseline scan to a common low level. [11C]SP203 was unstable in monkey blood in vitro and in vivo, and gave predominantly less lipophilic radiometabolites. By contrast, [11C]SP203 was stable in human blood in vitro.
Conclusion
[11C]SP203 emulates [18F]SP203 with regard to providing a sizeable mGluR5-specific signal in monkey brain, and advantageously avoids troublesome accumulation of radioactivity in bone. Although [11C]SP203 is unsuitable for mGluR5 quantification in monkey brain, its evaluation as a PET radioligand for studying human brain mGluR5 is nevertheless warranted.
Keywords: Radioligand, PET, mGluR5, Carbon-11, SP203
Introduction
Metabotropic glutamate receptors (mGluRs) are members of the C family of G-protein-coupled receptors. mGluRs are proteins having a hydrophobic domain of seven transmembrane α-helices and are coupled through G proteins with phospholipase C or adenylate cyclase to modulate intracellular second messenger systems [1]. The endogenous ligand, l-glutamate, interacts with a site on the extracellular aminoterminal domain. Eight subtypes of mGluRs have been identified, and among them subtype 5 (mGluR5) has attracted high interest because of strong evidence of its possible involvement in several neuropsychiatric disorders, such as pain [2], anxiety [3], schizophrenia [4], substance abuse [5, 6], and fragile X syndrome [7]. Ligands targeting these receptors are potential drugs for treating these disorders [8]. Consequently, radioligands for imaging human brain mGluR5 with PET have value for neuropsychiatric research. They also have value for drug development as tools for measuring engagement and occupancy of brain mGluR5 by candidate drugs preceding major clinical trials.
We have developed a ligand with exceptionally high affinity and high selectivity for mGluR5 [9–12], namely 3-fluoro-5-(2-(2-(fluoromethyl)thiazol-4-yl)ethynyl)benzonitrile (1), which we named SP203. This ligand binds to an allosteric site on mGluR5 that is distinct from the binding site of glutamate itself. SP203 has been labeled in the fluoromethyl position in high yield with the positron-emitter 18F (t1/2 =109.7 min) to give the radioligand, [18F]SP203 ([18F]1, Fig. 1) [9]. [18F]SP203 is effective for imaging mGluR5 in human brain, giving high brain radioligand uptake and a high quantifiable specific signal [10–12]. Whereas [18F]SP203 is valuable for assessing brain mGluR5 density in individual human subjects, a radioligand labeled with shorter-lived 11C (t1/2=20.4 min) would be useful for assessing the effect of pharmacological challenges in healthy subjects, as is needed for example for assessing target engagement and occupancy by developmental drugs. Such subjects might be studied with a 11C-labeled ligand in a single day at baseline and after drug administration. Quantification of radioligand binding in brain may require arterial blood sampling to generate an arterial input function. In such cases, paired studies in a single day with a 11C-labeled ligand also have the advantage of requiring only a single arterial cannulation.
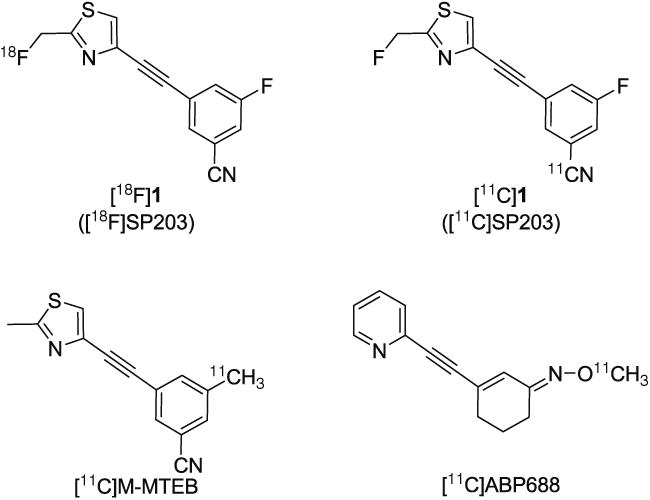
Fig. 1. Structures of radioligands [18F]1 and [11C]1, and of [11C]M-MTEB and [11C] ABP688.
[18F]SP203 shows marked species differences in metabolism [9–12]. In humans, although there is some radioactivity uptake in bone marrow, radiodefluorination of [18F]SP203 appears negligible [10, 11]. However, [18F]SP203 is extensively defluorinated to [18F]fluoride ion in the monkey. This defluorination results in high uptake of radioactivity in bone and skull, which detracts from successful measurement of mGluR5 density in nearby brain regions [9]. Extensive radiodefluorination of [18F]SP203 also occurs in the rat, and the mechanism has been established as involving the mercapturic acid pathway [13]. [18F]SP203 radiodefluorination in the rat and monkey would be eliminated by using a 11C label.
In this study, we aimed to prepare [11C]SP203 and to characterize its radioligand behavior in the monkey. We conclude that although [11C]SP203 is unsuitable for quantifying mGluR5 in the monkey, its further evaluation as a PET radioligand for studying brain mGluR5 in human subjects is nonetheless warranted.
Materials and methods
All chemicals and solvents were of analytical or high-performance liquid chromatography (HPLC) grade from Sigma-Aldrich (Milwaukee, WI) or Lancaster Synthesis (Windham, NH) and used without further purification. SP203 (1) and the bromo precursor for preparing [18F] SP203 [9, 14] were obtained from RTI International (Research Triangle Park, NC). 2-Fluoromethyl-4-((trimethylsilyl)e-thynyl)-1,3-thiazole (2) was prepared as described previously [9]. Yields of prepared compounds are given for chromatographically and spectroscopically pure materials. [18F]SP203 for injection into monkey was prepared by reaction of the bromomethyl analogue with cyclotron-produced [18F]fluorideion, as described previously [9]. 1H (400 MHz) and 13C (100 MHz) NMR spectra were recorded at room temperature on an Avance-400 spectrometer (Bruker, Billerica, MA). Chemical shifts are reported in δ (ppm) downfield from the signal for tetramethylsilane. Signals are quoted as s (singlet), d (doublet), or m (multiplet). High-resolution mass spectra (HRMS) were determined using a time-of-flight electrospray instrument (University of Illinois at Urbana; Champaign, IL). Melting points were determined using a Mel-temp melting point apparatus (Electrothermal; Fisher Scientific, Pittsburgh, PA). Mass spectra were acquired using a LCQ DECA LC-MS instrument (Thermo Finnigan; San Jose, CA) fitted with a reverse-phase liquid chromatography column (Luna, C18; 5 μm, 2 mm×150 mm; Phenomenex; Torrance, CA). New compounds were analyzed for chemical purity with HPLC on a Luna C18 column (10 μm, 4.6 mm×250 mm; Phenomenex) eluted at 2 mL/min with a gradient of a mixture of A and B, where A was aqueous ammonium formate (25 mM) and B a mixture of acetonitrile and aqueous ammonium formate (25 mM) (3:1 v/v). The gradient was started with 68 % B for 2 min, then increased to 78 % B for 20 min, and finally increased to 100 % B at 10 min. The column eluate was monitored for absorbance at 280 nm.
Chemistry
4-((3-Fluoro-5-iodophenyl)ethynyl)-2-(fluoromethyl)thiazole (5) 2-Fluoromethyl-4-((trimethylsilyl)ethynyl)-1,3-thiazole (2, 250 mg, 1.17 mmol), 3,5-diiodofluorobenzene (3, 490 mg, 1.41 mmol), CuI (15 mg, 79 μmol), tetra-kis(triphenylphosphine)palladium(0) (45 mg, 39 μmol), and triethylamine (0.8 mL) were added to 1,2 –dimethoxyethane (8 mL). Argon was bubbled into the solution while it was heated to 80 °C. Tetra-n-butylammonium fluoride (2 mmol; 1.0 M solution in tetrahydrofuran, THF, 2 mL) was added via syringe over 30 min. The resulting black reaction mixture was heated at 80 °C until thin-layer chromatography showed no starting material present (8–12 h). The reaction mixture was cooled to room temperature, filtered through Celite, and then evaporated to dryness. The resulting residue was dissolved in dichloromethane (25 mL) and washed with water (20 mL×2) and then brine (20 mL×2). The organic phase was dried over MgSO4 and evaporated under vacuum. The residue was purified by chromatography (silica gel; hexane-AcOEt, 80:20 to 60:40 v/v) to afford 5 (271 mg, 64 %) as a white powder; mp 161–162 °C; 1H-NMR (CDCl3): δ 8.13 (s, 1 H), 7.75 (s, 1 H), 7.53 (m, 1 H), 7.49 (m, 1 H), 5.65 (d, 2 H, J=46.7 Hz); 13C-NMR (CDCl3): δ 165.2 (d, J=25.2 Hz), 161.7 (d, J=27.2 Hz), 136.5, 131.7, 124.7 (d, J=1.94 Hz), 123.5 (d, J=9.82 Hz), 118.1 (d, J=25.1 Hz), 114.9 (d, J=10.2 Hz), 93.2, 90.2 (d, J= 3.7 Hz), 84.9 (d, J=31.9 Hz), 80.6 (d, J=171.2 Hz); HRMS: calculated for C12H7F2INS (M+ + H), 361.9312; found, 361.9311; HPLC: tR, 21.0 min; >99.5 % purity.
4-((3-Bromo-5-fluorophenyl)ethynyl)-2-(fluoromethyl)thiazole (6) The method described for the synthesis of 5 was applied to 2 (1.17 mmol) and 3,5-dibromofluorobenzene (4, 0.355 g, 1.4 mmol), and gave 6 as a beige powder (129 mg, 35 %); mp 142–144 °C; 1H-NMR (CDCl3): δ 7.70 (m, 1 H), 7.65 (s, 1 H), 7.48 (m, 1 H), 7.17 (m, 1 H), 5.65 (d, 2 H, J= 46.4 Hz); 13C-NMR (CDCl3): δ 165.1 (d, J=25.3 Hz), 162.0 (d, J=26.7 Hz), 136.5, 131.9, 126.8 (d, J=9.9 Hz), 124.5 (d, J= 1.85 Hz), 123.1 (d, J=11.1 Hz), 117.6 (d, J=25.1 Hz), 115.0 (d, J=10.2 Hz), 90.2, 84.9 (d, J=31.9 Hz), 80.6 (d, J= 170.8 Hz); HRMS: calculated for C12H7F2BrNS (M+ + H), 313.9445; found, 313.9452; HPLC: tR, 18.0 min; > 98.0 % purity.
Radiochemistry
Production of hydrogen [11 C]cyanide
Hydrogen [11C]cyanide was produced according to the method described by Iwata et al. [15]. No-carrier-added [11C]carbon dioxide was produced with a PETtrace cyclotron (GE Medical Systems; Milwaukee, WI), according to the 14N(p,α)11C reaction [16]. Thus, nitrogen gas (initial pressure 140 psi, volume 80 mL) containing 1 % oxygen was irradiated with a proton beam (15.9 MeV, 45 μA) for 50 min. The [11C]carbon dioxide (about 55 GBq) was then trapped on a molecular sieve (13×, 80–100 mesh, 0.55 g) in a stainless steel column at 40 °C while untrapped 13N activity was directed to waste. The [11C]carbon dioxide was eluted from the heated (350 °C) molecular sieve with nitrogen (250 mL/min), and then mixed with hydrogen (30 mL/min) and passed over heated (400 °C) nickel (Ni-3266; Engelhard Corp., Iselin, NJ) in a quartz column (13 mm OD×200 mm). The column effluent was passed through an oxygen scrubber (Oxisorb®; Sigma-Aldrich), mixed with anhydrous ammonia (20 mL/min, research grade; Matheson; Montgomeryville, PA) and passed over heated (950 °C) platinum wire (4.0 g, 0.2 mm OD×20 mm; Sigma-Aldrich) in a quartz column (9 mm OD×200 mm). The effluent gas delivered the [11C]hydrogen cyanide.
Synthesis of [11 C]SP203
Radiosyntheses were performed with a remotely controlled Synthia apparatus [17] in a lead-shielded hot-cell for radiation protection. Two procedures were used to prepare [11C]SP203, method A and method B, as described below. Method A was used for the monkey experiments in this work, and method B was subsequently developed as being suitable for producing [11C]SP203 for human use.
Method A Cyclotron-produced [11C]hydrogen cyanide (about 30 GBq) in nitrogen carrier gas was passed into a solution of K2CO3 (1 mg) and Kryptofix 2.2.2 (K 2.2.2; 4 mg) in THF (1.0 mL) at about 300 mL/min until the collected radioactivity reached a maximum. Then the reaction mixture was purged with nitrogen at 200 mL/min for 1 min while being heated to 80 °C. Precursor 5 or 6 (1 mg) and tetra-kis(triphenylphosphine)palladium(0) (1 mg) in THF (0.5 mL) were added to the reaction mixture which was then sealed and heated at 80 °C for 4 min. The reaction mixture was then injected onto a Luna C18 column (10 μm, 10 mm×250 mm; Phenomenex) eluted with a mixture (54:46 v/v) of acetonitrile and aqueous trifluoroacetic acid (0.1 %) at 6 mL/min, with the eluate monitored for radioactivity and for absorbance at 254 nm to separate [11C]SP203 (tR010.9 min). The radiochemical yield (percent incorporation of initial [11C]cyanide ion into [11C]SP203) was calculated from the radiochromatogram. The mobile phase was removed from the collected [11C]SP203 fraction by rotary evaporation at 80 °C. The radioactive residue was formulated for intravenous injection by dissolution in sterile saline for injection (20 mL, USP) containing ethanol (10 %v/v).
Method B Cyclotron-produced [11C]hydrogen cyanide (about 30 GBq) in nitrogen carrier gas was passed into a solution of KH2PO4 (2.5 mg) and K 2.2.2 (4.0 mg) in dimethyl sulfoxide (DMSO, 200 μL) at 300 mL/min until the collected radioactivity reached a maximum. Then the reaction mixture was purged with nitrogen at 200 mL/min for 1 min while the mixture was heated to 50 °C. Precursor 5 (0.6 mg) and tetra-kis(triphenylphosphine)palladium(0) (5.5 mg) in DMSO (0.2 mL) were added to the vial which was then sealed and heated at 80 °C for 4 min. The crude reaction mixture was diluted with water (3 mL) and subjected to HPLC as in Method A. The fraction containing [11C]SP203 was collected in HPLC-grade water (30 mL) and passed through a C18 Seppak cartridge. The cartridge was washed with water (10 mL). [11C]SP203 was then eluted from the cartridge with ethanol for injection (2.0 mL, USP), diluted with sodium chloride for injection (2.0 mL, USP), and passed through a sterile filter (0.22 μm, MP; Millipore) into a sterile vial containing sodium chloride for injection (0.9 %w/v, 16 mL).
Analysis of [11 C]SP203
The radiochemical purity, chemical purity, and specific activity of each batch of formulated [11C]SP203 were determined with HPLC on a Luna C18 column (10 μm, 4.6 mm OD×250 mm; Phenomenex) eluted at 2 mL/min with MeCN/10 mM HCOONH4 (60:40 v/v) with the column eluate monitored for radioactivity and absorbance at 310 nm ([11C]SP203, tR=5.7 min). The analytical method was calibrated for absorbance response (peak area) versus mass of injected SP203 to allow the amount of carrier SP203 in a known amount of [11C]SP203 to be measured, thereby giving the specific activity of the radioligand at the analysis time. This value was corrected for radioactive decay to the end of synthesis.
The identity of the [11C]SP203 was also confirmed by liquid chromatography–mass spectrometry of associated carrier.
The radiochemical stability of the formulated [11C]SP203 was checked by repeat HPLC analysis after the batch had been kept at room temperature for 60 min.
PET experiments in the monkey
All animal experiments were performed in accordance with the Guide for the Use of Laboratory Animals [18] and the National Institutes of Health Animal Care and Use Committee. For PET scanning, animals were immobilized with ketamine (10 mg/kg, i.v.) and maintained under anesthesia with 1.5 % isoflurane in oxygen via an endotracheal tube. Three male rhesus monkeys (Macaca mulatta; 9.9±5.3 kg) were each scanned twice with [11C]SP203 (96–215 MBq; specific activity at time of injection in baseline scans, 38–82 GBq/μmol; associated carrier in baseline scans, 0.13–0.76 nmol/kg) in a microPET Focus 220 scanner (Siemens Medical Solution; Knoxville, TN). The first scan was at baseline and the second at 30 min after treatment with the selective mGluR5 ligand, 3-[ (2-methyl-1,3-thiazol-4-yl)ethynyl]pyridine [19] (MTEP; 5 mg/kg, i.v.). The two injections of radioligand were separated by 3 h to allow radioactivity from the baseline experiment to decay away. One other male rhesus monkey (12.4 kg) was scanned twice in separate sessions approximately 1 month apart, first with [11C]SP203 (129 MBq; specific activity 35.7 GBq/μmol) and second with [18F]SP203 (86.5 MBq; specific activity 89.1 GBq/μmol). All scans were acquired in list mode for 90 min following radioligand injection and were histogrammed into dynamic frames of 6×30 s, 3×60 s, 2×120 s and 16×300 s. Images were reconstructed using Fourier rebinning plus two-dimensional filtered back-projection. Attenuation and scatter correction were applied for both datasets.
PET images were coregistered to a standardized MRI template [20] using a 12-parameter mutual information algorithm (FMRIB Software Library, Oxford, UK). A set of 34 predefined brain regions of interest from the MRI template were then applied to the coregistered PET image. Regional decay-corrected time–activity curves were obtained. Two lateral cerebellar nuclei were omitted from the region of interest taken for the cerebellum. The concentration of radioactivity in each region of interest was expressed as percent injected dose per milliliter.
All image analyses were performed using PMOD 3.0 (PMOD Technologies Ltd, Zurich, Switzerland).
Stability of [11C]SP203 in monkey and human blood in vitro
Fresh anticoagulated monkey whole blood (1 mL) was placed in a polypropylene Eppendorf tube along with formulated [11C]SP203 solution (680 kBq, 0.0175 nmol carrier, radiochemical purity 93 %, 4 μL) and incubated at room temperature. Incubate samples (50 μL) were taken at 5, 10, 20, and 30 min and immediately placed in tubes containing water (200 μL) that had been spiked with carrier SP203. Each tube was shaken well to lyse the blood cells. Acetonitrile (600 μL) was added to each mixture to precipitate protein and then water (100 μL) was added and mixed well. Each tube was counted in a γ-counter and then centrifuged at 10,000 g for 1 min. A sample of supernatant liquid was analyzed by radio-HPLC on a Novapak C18 column (100× 8 mm; Waters Corp., Milford, MA), equipped with a Radial-Pak® compression module (RCM-100) and sentry precolumn, and eluted with MeOH/H2O/Et3N (65/35/0.1 v/v/v) at 2.5 mL/min to measure the percentage of radioactivity present as unchanged radioligand. The precipitate was counted to allow calculation of the efficiency of radioactivity recovery into acetonitrile. The stability of [11C]SP203 in human blood was determined similarly.
Emergence of radiometabolites of [11C]SP203 in monkey plasma in vivo
The analytical method was based on that described previously [21]. Thus, a monkey (8.2 kg) was injected with [11C] SP203 (213 MBq) and an anticoagulated blood sample was drawn 10 min later. This blood sample was immediately centrifuged and the plasma (450 μL) was removed and deproteinized with acetonitrile (720 μL). The sample was mixed well and then water (100 μL) was added and mixed well. The sample was counted in the γ-counter and centrifuged at 10,000 g for 1 min. A sample of supernatant liquid was analyzed by radio-HPLC as in the preceding experiment to measure the percentage of radioactivity present as unchanged radioligand. The precipitate was counted to allow calculation of the efficiency of radioactivity recovery into acetonitrile.
Results
Chemistry
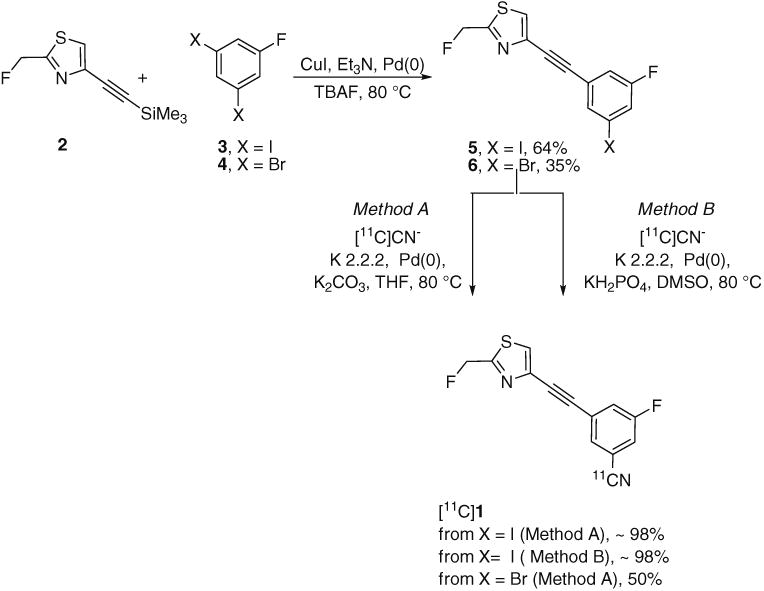
Treatment of the synthon 2 with 3,5-diiodofluorobenzene (3) or 3,5-dibromofluorobenzene (4) in the presence of tetra-kis(triphenylphosphine)palladium(0) readily gave 4-((3-fluoro-5-iodophenyl)ethynyl)-2-(fluoromethyl)thiazole (5) and 4-((3-fluoro-5-bromophenyl)ethynyl)-2-(fluorome-thyl)thiazole (6), respectively (Fig. 2). The yield of the Sonogashira cross-coupling reaction was higher from the iodoarene 3 (64 %, in 4 h) than from the bromoarene 4 (35 %, overnight).
Fig. 2. Radiosynthesis of [11C] 1 through 11C-cyanation of a bromo or iodo precursor.
Radiochemistry
When treated with [11C]cyanide ion according to Method A, the bromo precursor 6 gave [11C]SP203 in low to moderate decay-corrected radiochemical yields and with poor reproducibility (20–52 %). Treatment of the iodo precursor 5 with [11C]cyanide ion under similar reaction conditions (Method A) gave nearly quantitative decay-corrected radio-chemical yields of [11C]SP203. In the use of this iodo precursor for radioligand production, single-pass reverse-phase HPLC gave [11C]SP203 in useful amounts (0.74– 1.48 GBq; n=10) and in high radiochemical purity (> 98 %), in the absence of any significant chemical impurities. The specific activity of [11C]SP203 when ready for intravenous administration at 44±2 min from the end of radionuclide production was 105±33 GBq/μmol. This value is comparable to that reported for another radiotracer produced from hydrogen [11C]cyanide by similar labeling radiochemistry [22]. An even higher specific radioactivity might be attainable by generating the labeling agent, hydrogen [11C] cyanide, from cyclotron-produced [11C]methane instead, as used in this study, cyclotron-produced [11C]carbon dioxide [23]. HPLC analysis demonstrated that preparations of [11C] SP203 for injection were radiochemically stable.
Radiochemistry for the production of [11C]SP203 was subsequently modified as described under Method B. This method was not used for the PET experiments in this study, but was subsequently adopted for producing [11C]SP203 for administration to human subjects. The radiosynthesis from end of radionuclide production through to [11C]SP203 ready for administration also takes about 44 min. For 12 preparations, the average yield of [11C]SP203 of >97 % radiochemical purity and ready for administration was 3.97±1.14 GBq. The average specific activity of these preparations was 73.2± 19.9 GBq/μmol at the end of synthesis.
PET experiments in the monkey
Under baseline conditions, overall brain uptake of radioactivity after administration of [11C]SP203 to the monkeys was high, on average (n=3) reaching 0.069 % ID/mL in the striatum and 0.053 % ID/mL in the frontal cortex at about 12 min (Fig. 3). Radioactivity gradually washed out of all brain regions. In all three monkeys, the brain distribution of radioactivity at 90 min after administration of [11C]SP203 reflected the distribution of mGluR5 expected from in vitro autoradiography, which in the rhesus monkey [24] ranges from a high radioactivity concentration in the mGluR5-rich caudate (Bmaxabout 63 nM) to a low radioactivity concentration in the mGluR5-poor cerebellum (Bmaxabout 24 nM). Thus, the regional radioactivity concentrations ranked as follows: striatum > anterior cingulate ≈ frontal cortex > thalamus ≈ hippocampus ≈ temporal cortex > parietal cortex > occipital cortex ≈ cerebellum. In the three PET experiments in which the selective mGluR5 ligand MTEP was administered in high dose before [11C]SP203 injection, early brain radioactivity was again high but was followed by faster washout of radioactivity in all brain regions down to a low common level (Fig. 3).
Fig. 3.
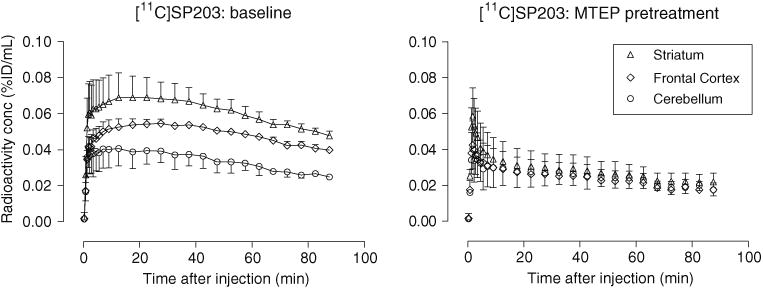
Average regional decay-corrected brain time–activity curves from three rhesus monkeys treated with [11C]SP203 intravenously under baseline conditions (left panel) and pretreatment conditions (MTEP, 5 mg/kg, i.v.; right panel). Time–activity curves for other brain regions at baseline (e.g., anterior cingulate, hippocampus, thalamus, temporal cortex, parietal cortex and occipital cortex) have been omitted for clarity, and lay between those for the frontal cortex and cerebellum. Time–activity curves for these other regions in the pretreatment study were very similar to that for the cerebellum. Error bars SD; where not visible SD is within the symbol size
Summed PET data acquired between 10 and 90 min after injection of [11C]SP203 at baseline also reflected the distribution of mGluR5, whereas the corresponding summed images from the MTEP-pretreatment experiments showed an almost uniform distribution of low radioactivity across the brain (Fig. 4).
Fig. 4.
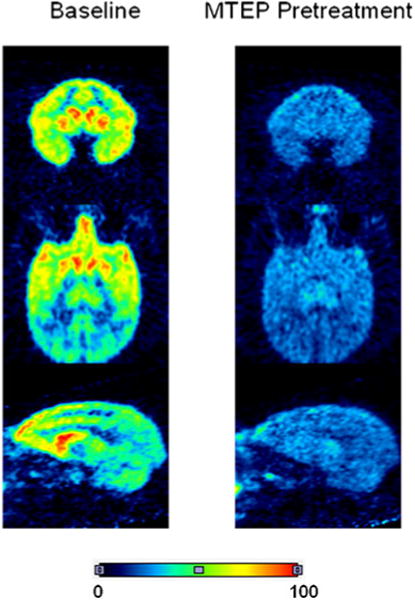
Summed axial, coronal and sagittal PET brain images (10–90 min) from a rhesus monkey scanned with [11C]SP203 under baseline conditions and pretreatment conditions (MTEP, 5 mg/kg, i.v.). The color scale is in arbitrary units of summed radioactivity concentration
Brain time–activity curves obtained at baseline in a single monkey with [18F]SP203 and with [11C]SP203 were very comparable (Fig. 5). However, unlike [18F]SP203, [11C] SP203 did not give steadily increasing uptake of radioactivity in the skull. The skull radioactivity concentration at 90 min after injection of [18F]SP203 was very high at about 0.05 % ID/mL, whereas the comparable value for [11C] SP203 was extremely low at 0.0027 % ID/mL.
Fig. 5.
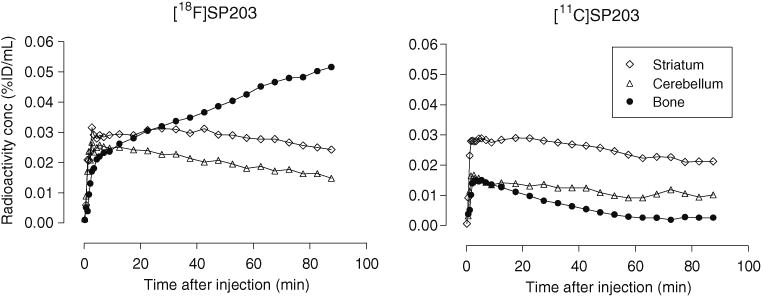
Decay-corrected brain time–activity curves at baseline after injection of [18F]SP203 (left panel) and [11C]SP203 (right panel) in the same monkey (radioligand injections were given about 1 month apart). No skull radioactivity uptake was seen for [11C]SP203
Although overall radioactivity uptake in the cerebellum was the lowest amongst all sampled regions after administration of [11C]SP203 at baseline, two bilateral small areas with relatively high radioactivity uptake were seen in the cerebellum of all four monkeys. These areas were not included in the region of interest for the cerebellum. In the pretreatment experiment, the uptake of radioactivity in these two small bilateral regions was reduced as in all other brain regions.
Stability of [11C]SP203 in monkey and human blood in vitro
Over 92 % of the radioactivity in incubates was extracted into acetonitrile. After incubation of [11C]SP203 in monkey blood at room temperature for 5, 10, 20 and 30 min in vitro, 11.5, 29.9, 48.5 and 60.9 % of the radioligand, respectively, had been converted into five radiometabolites, [11C]a-e, four of which eluted ahead of [11C]SP203 on reverse-phase HPLC (Fig. 6a). Only a minor radiometabolite ([11C]e) eluted after [11C]SP203. By contrast, [11C]SP203 was entirely stable in human blood under the same incubation conditions in vitro.
Fig. 6.
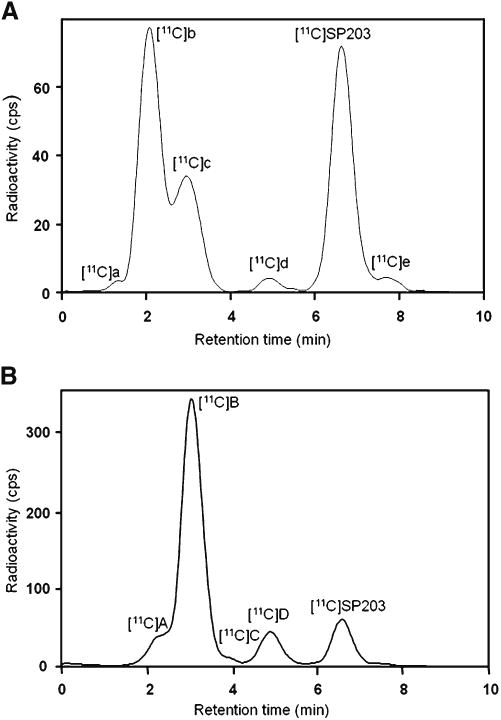
Reverse-phase radio-HPLC analysis of [11C]SP203 incubated with monkey blood for 30 min at room temperature (a) and of plasma taken 10 min after injection of the monkey with [11C]SP203 (b)
Emergence of radiometabolites of [11C]SP203 in monkey plasma in vivo
The efficiency of extraction of radioactivity from blood into acetonitrile was very high (>99 %). Reverse-phase radio-HPLC analysis of radioactivity extracted from plasma that had been sampled 10 min after injection of [11C]SP203 revealed the extensive formation of four radiometabolites, [11C]A-D, which all eluted ahead of [11C]SP203 (Fig. 6b).
Discussion
Chemistry and radiochemistry
The presence of an aryl nitrile group in SP203 raised the possibility of preparing [11C]SP203 through 11C-cyanation of a halo precursor [25]. The iodo precursor 5 was readily prepared in high yield and gave an almost quantitative radiochemical yield of [11C]SP203 in reaction with [11C] cyanide ion, unlike the bromo precursor 6 which was obtained in lower yield and gave a much lower and more variable radiochemical yield of [11C]SP203. Therefore, the iodo precursor 5 was much preferred for producing [11C] SP203. Method A was used to prepare the doses of [11C] SP203 used in this study. This method was subsequently modified (Method B) to prepare doses for injection into human subjects. The main advantage of Method B derives from the use of DMSO instead of THF as reaction solvent which leads to more reliable and efficient entrapment of hydrogen [11C]cyanide without excessive solvent loss. The radiochemical yield of [11C]SP203 from trapped hydrogen [11C]cyanide is not appreciably affected by the change of solvent, but the yields of formulated [11C]SP203 are increased substantially.
The (3-cyano-5-fluorophenyl)biaryl structural motif is generally recognized to confer high mGluR5 binding affinity [26]. Consequently, this motif appears in other mGluR5 PET radioligands, such as [11C]M-MTEB [24] and [18F]F-MTEB [24], and also in mGluR5 noncompetitive antagonists, known as negative allosteric modulators, that are being developed as drugs [26]. Therefore, the method we describe for labeling SP203 with 11C may have wider value for labeling mGluR5 ligands for PET investigations.
Characterization of [11C]SP203 behavior in monkey
In monkey, [11C]SP203 emulated [18F]SP203 as an mGluR5 radioligand [9] by giving high uptake of radioactivity in brain followed by relatively slow washout of radioactivity from all regions (Fig. 3). At late time-points during scanning, the brain distribution of radioactivity reflected the expected distribution of mGluR5, and a high proportion of this radioactivity represented mGluR5-specific binding, as shown by the effect of pretreatment with MTEP (Figs. 3 and 4). By consideration of the amounts of carrier injected into the monkeys, the levels of radioactivity attained in brain and the reported density of mGluR5 in the caudate of rhesus monkey brain [24], we estimate that on average brain receptor occupancy by carrier was low (<5 %) in these experiments.
Apart from the type and position of radiolabel, [11C]SP203 and [18F]SP203 have identical chemical structures. Hence, any differences seen in the kinetics and distribution of radioactivity from these two radioligands, when administered to the same living subject at similar doses of carrier SP203, must be attributable to the formation of different radiometabolite species. We have previously established that [18F]SP203 is rapidly metabolized in monkey in vivo, and also in monkey blood in vitro [9]. The high continuous uptake of radioactivity in monkey bone, including skull and mandible, after administration of [18F]SP203 is indicative of extensive radiodefluorination (Fig. 5). This likely occurs through a mercapturic acid pathway, as established for [18F]SP203 in the rat [13], in which the 18F is first displaced by nucleophilic attack to form a nonradioactive S-glutathione conjugate.
In the metabolism of [11C]SP203 by the mercapturic acid pathway, the generated S-glutathione conjugate would be a radiometabolite which would then be further transformed to a radioactive mercapturic acid derivative (Fig. 7). These two radiometabolites, since they are negatively charged at physiological pH, would not be expected to enter the brain easily. Incubation of [11C]SP203 with monkey blood at room temperature for 30 min in vitro resulted in several radiometabolites ([11C]a–d) which were predominantly more polar than parent [11C]SP203 (Fig. 6a), which has a moderate lipophilicity (measured logD 2.18 [27]). These observations are consistent with the previously reported instability of [18F]SP203 in monkey blood [9]. By contrast, [18F]SP203 is stable in human blood in vitro [9]; we verified that [11C] SP203 is similarly stable in human blood in vitro. Prompt reverse-phase radio-HPLC analysis of monkey plasma sampled at 10 min after administration of [11C]SP203 revealed the emergence of four radiometabolites ([11C]A-D). They all eluted earlier than parent radioligand (Fig. 6b), like the radiometabolites generated in vitro. These observations in vitro and in vivo appear consistent with the possible operation of the mercapturic acid pathway of metabolism.
Fig. 7.
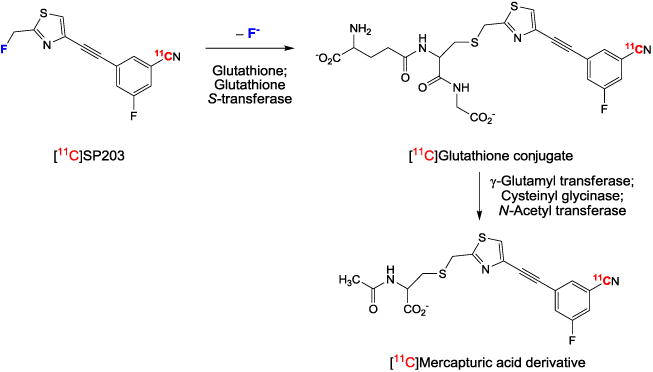
A major metabolic pathway predicted for [11C] SP203 in monkey, on the basis of known SP203 metabolism in the rat [13]
We have previously found that [18F]SP203 is not extensively metabolized by monkey or human brain tissue in vitro [9]. Therefore, [11C]SP203 is unlikely to generate extensive radiometabolites within monkey or human brain through the putative mercapturic acid pathway. We also note that S-glutathione conjugates, if formed within brain, might be expelled by the efflux transporter MRP1 [28].
Clearly, the use of [11C]SP203 instead of [18F]SP203 negated any accumulation of radioactivity in bone (Fig. 5), as was also clearly evident from the summed PET scans (Fig. 4). This raises the possibility of quantifying mGluR5 in monkey brain with [11C]SP203, either with a simplified reference tissue model (SRTM) or a kinetic compartmental model. However, difficulties with regard to achieving such quantification are apparent. Firstly, with regard to an SRTM approach, there is no clear option for a reference tissue. Monkey cerebellum cannot be used because a saturation binding study with the selective mGluR5 ligand [3H]M-MTEB has shown it to contain a low density of mGluR5 [24]. Secondly, with regard to a kinetic compartmental modeling approach, the instability of [11C]SP203 in monkey blood precludes accurate measurement of the required radioligand arterial input function. For this reason, no attempt was made to take such blood measurements in this study. Therefore, [11C]SP203, despite not giving an appreciable accumulation of radioactivity in the skull, remains unsuitable for rigorous quantitative measurements of brain mGluR5 density in the monkey. Nevertheless, the stability of [11C]SP203 in whole human blood in vitro renders accurate measurement of an arterial input function feasible for potential quantification of brain mGluR5 with compartmental modeling [10] or an equilibrium approach [12], as already shown for [18F]SP203. The moderate lipophilicity of SP203 also results in an appreciable but readily measurable human plasma free fraction (about 5.8 % [27]), where this is of interest for quantitative analysis of human PET data. The primary advantage of [11C]SP203 over [18F]SP203 would be to allow two injections of radioligand in one scanning session of a human subject, the first at baseline and the second after a pharmacological manipulation.
Among candidate 11C-labeled ligands previously developed for imaging brain mGluR5, only two, [11C]M-MTEB [24] and [11C]ABP688 [29–31] (Fig. 1), have shown promise by giving sizeable brain mGluR5-specific signals. [11C] M-MTEB has been studied in the rhesus monkey [24], and [11C]ABP688 in the baboon [30]. [11C]SP203 in the rhesus monkey gave higher brain uptake than [11C]M-MTEB in the monkey and also by estimation probably higher uptake than [11C]M-MTEB in the baboon [24]; other comparisons between the radioligands are difficult to make because of lack of detailed published data. As far as we are aware, [11C] ABP688 is the only 11C-labeled ligand that has been used to study mGluR5 in human brain [31]. In human, this radioligand has many favorable features, but also appears to have much lower peak brain uptake (estimated from reported data [31]) than radiolabeled SP203 [10].
Conclusion
[11C]SP203, although not easily applicable to PET quantification of brain mGluR5 in the monkey, has many promising characteristics and merits the detailed evaluation in human subjects that we are now undertaking and that will be published subsequently.
Acknowledgments
This research was supported by the Intramural Research Program of the National Institutes of Health (National Institute of Mental Health). We thank the National Institutes of Health Clinical PET Center (Chief: Dr. Peter Herscovitch) for the production of hydrogen [11C]cyanide and Dr. H. Umesha Shetty for mass spectrometry.
Footnotes
Conflicts of interest None.
Contributor Information
Fabrice G. Siméon, Molecular Imaging Branch, National Institute of Mental Health, National Institutes of Health, Building 10, Rm. B3 C346A, 10 Center Drive, Bethesda, MD 20892, USA
Jeih-San Liow, Molecular Imaging Branch, National Institute of Mental Health, National Institutes of Health, Building 10, Rm. B3 C346A, 10 Center Drive, Bethesda, MD 20892, USA.
Yi Zhang, Molecular Imaging Branch, National Institute of Mental Health, National Institutes of Health, Building 10, Rm. B3 C346A, 10 Center Drive, Bethesda, MD 20892, USA.
Jinsoo Hong, Molecular Imaging Branch, National Institute of Mental Health, National Institutes of Health, Building 10, Rm. B3 C346A, 10 Center Drive, Bethesda, MD 20892, USA.
Robert L. Gladding, Molecular Imaging Branch, National Institute of Mental Health, National Institutes of Health, Building 10, Rm. B3 C346A, 10 Center Drive, Bethesda, MD 20892, USA
Sami S. Zoghbi, Molecular Imaging Branch, National Institute of Mental Health, National Institutes of Health, Building 10, Rm. B3 C346A, 10 Center Drive, Bethesda, MD 20892, USA
Robert B. Innis, Molecular Imaging Branch, National Institute of Mental Health, National Institutes of Health, Building 10, Rm. B3 C346A, 10 Center Drive, Bethesda, MD 20892, USA
Victor W. Pike, Molecular Imaging Branch, National Institute of Mental Health, National Institutes of Health, Bethesda, MD 20892, USA
References
- 1.Conn PJ, Pin JP. Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol. 1997;37:205–37. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- 2.Zhu CZ, Wilson SG, Mikusa JP, Wismer CT, Gauvin DM, Lynch JJ, et al. Assessing the role of metabotropic glutamate receptor 5 in multiple nociceptive modalities. Eur J Pharmacol. 2004;506:107–18. doi: 10.1016/j.ejphar.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 3.Cosford NDP, Tehrani L, Roppe J, Schweiger E, Smith ND, Anderson J, et al. 3-[ (2-Methyl-1,3-thiazol-4-yl)ethynyl]-pyridine: a potent and highly selective metabotropic glutamate subtype 5 receptor antagonist with anxiolytic activity. J Med Chem. 2003;46:204–6. doi: 10.1021/jm025570j. [DOI] [PubMed] [Google Scholar]
- 4.Pietraszek M, Nagel J, Gravius A, Schafer D, Danysz W. The role of group I metabotropic glutamate receptors in schizophrenia. Amino Acids. 2007;32:173–8. doi: 10.1007/s00726-006-0319-9. [DOI] [PubMed] [Google Scholar]
- 5.Tessari M, Pilla M, Andreoli M, Hutcheson DM, Heidbreder CA. Antagonism at metabotropic glutamate 5 receptor inhibits nicotineand cocaine-taking behaviors and prevents nicotine-triggered relapse to nicotine seeking. Eur J Pharmacol. 2004;499:121–3. doi: 10.1016/j.ejphar.2004.07.056. [DOI] [PubMed] [Google Scholar]
- 6.Chiamulera C, Epping-Jordan MP, Zocchi A, Marcon C, Cottiny C, Tacconi S, et al. Reinforcing and locomotor stimulant effects of cocaine are absent in mGluR5 null mutant mice. Nat Neurosci. 2001;4:873–4. doi: 10.1038/nn0901-873. [DOI] [PubMed] [Google Scholar]
- 7.Maccarrone M, Rossi S, Bari M, De Chiara V, Rapino C, Musella A, et al. Abnormal mGlu5 receptor/endocannabinoid coupling in mice lacking FMRP and BC1 RNA. Neuropsychopharmacology. 2010;35:1500–9. doi: 10.1038/npp.2010.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gravius A, Pietraszek M, Dekundy A, Danysz W. Metabotropic glutamate receptors as therapeutic targets for cognitive disorders. Curr Topics Med Chem. 2010;10:187–206. doi: 10.2174/156802610790411018. [DOI] [PubMed] [Google Scholar]
- 9.Siméon FG, Brown AK, Zoghbi SS, Patterson VM, Innis RB, Pike VW. Synthesis and simple 18F-labeling of a high affinity 2-(fluo-romethyl)thiazole derivative ([18F]SP203) as a radioligand for imaging brain metabotropic glutamate subtype-5 receptors with PET. J Med Chem. 2007;50:3256–66. doi: 10.1021/jm0701268. [DOI] [PubMed] [Google Scholar]
- 10.Brown AK, Kimura Y, Zoghbi SS, Siméon FG, Liow JS, Kreisl WC, et al. Metabotropic glutamate subtype-5 (mGluR5) receptors quantified in human brain with a novel radioligand for positron emission tomography. J Nucl Med. 2008;49:2042–8. doi: 10.2967/jnumed.108.056291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kimura Y, Siméon FG, Hatazawa J, Mozley PD, Pike VW, Innis RB, et al. Biodistribution and radiation dosimetry of a positron emission tomography ligand, 18F-SP203, to image metabotropic glutamate subtype 5 (mGluR5) receptor in humans. Eur J Nucl Med Mol Imaging. 2010;37:1943–9. doi: 10.1007/s00259-010-1447-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kimura Y, Siméon FG, Zoghbi SS, Zhang Y, Hatazawa J, Pike VW, et al. Quantification of metabotropic glutamate receptor subtype 5 receptors in the brain by an equilibrium method using 18F-SP203. Neuroimage. 2012;59:2124–30. doi: 10.1016/j.neuroimage.2011.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shetty HU, Zoghbi SS, Siméon FG, Liow JS, Brown AK, Kannan P, et al. Radiodefluorination of 3-fluoro-5-(2-(2-([18F]fluorome-thyl)thiazol-4-yl)ethynyl)benzonitrile ([18F]SP203), a radioligand for imaging brain mGluR5 in vivo, occurs by glutathionylation in rat brain. J Pharmacol Exp Ther. 2008;327:727–35. doi: 10.1124/jpet.108.143347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gopinathan MB, Jin C, Rehder KS. A short and efficient synthesis of 3-{2-[2-(bromomethyl)thiazol-4-yl]ethynyl}-5-fluorobenzoni-trile: a precursor for PET radioligand [18F]SP203. Synthesis Stuttgart. 2009;12:1979–82. [Google Scholar]
- 15.Iwata R, Ido T, Takahashi T, Nakanishi H, Iida S. Optimization of [11C]HCN production and no-carrier-added [1-11C]amino acid synthesis. Appl Radial Isot. 1987;38:97–102. doi: 10.1016/0883-2889(87)90003-7. [DOI] [PubMed] [Google Scholar]
- 16.Christman DR, Finn RD, Karlström KI, Wolf AP. Production of ultra high activity 11C-labeled hydrogen-cyanide, carbon-dioxide, carbon-monoxide and methane via 14N(p, α)11C reaction. Int J Appl Radiat Isot. 1975;26:435–42. [Google Scholar]
- 17.Bjurling P, Reineck R, Westerberg G, Gee AD, Sutcliffe J, Långström B. Proceedings of the 6th Workshop on Targetry and Target Chemistry. TRIUMF. 1995:282–284. [Google Scholar]
- 18.Clark J, Baldwin R, Bayne K, Brown MJ, Gebhart GF, Gonder JC, et al. Guide for the care and use of laboratory animals. Washington, DC: Institute of Laboratory Animal Resources, National Research Council; 1996. [Google Scholar]
- 19.Cosford NDP, Tehrani L, Arruda J, King C, McDonald IA, Munoz B, et al. 3-[ (2-Methyl-1,3-thiazol-4-yl)ethynyl]pyridine (MTEP): design and synthesis of a potent and highly selective metabotropic glutamate subtype 5 (mGlu5) receptor antagonist with anxiolytic activity. Neuropharmacology. 2002;43:282–3. [Google Scholar]
- 20.Yasuno F, Brown AK, Zoghbi SS, Krushinski JH, Chernet E, Tauscher J, et al. The PET radioligand [11C]MePPEP binds reversibly and with high specific signal to cannabinoid CB1 receptors in nonhuman primate brain. Neuropsychopharmacology. 2008;33:259–69. doi: 10.1038/sj.npp.1301402. [DOI] [PubMed] [Google Scholar]
- 21.Zoghbi SS, Shetty UH, Ichise M, Fujita M, Imaizumi M, Liow JS, et al. PET imaging of the dopamine transporter with [18F]FECNT: a polar radiometabolite confounds brain radioligand measurements. J Nucl Med. 2006;47:520–7. [PubMed] [Google Scholar]
- 22.Airaksinen AJ, Andersson J, Truong P, Karlsson O, Halldin C. Radiosynthesis of [11C]ximelagatran via palladium catalyzed [11C] cyanation. J Label Compds Radiopharm. 2007;51:1–5. [Google Scholar]
- 23.Andersson J, Truong P, Halldin C. In-target produced [11C] methane: increased specific radioactivity. Appl Radiat Isot. 2009;67:106–10. doi: 10.1016/j.apradiso.2008.09.010. [DOI] [PubMed] [Google Scholar]
- 24.Hamill TG, Krause S, Ryan C, Bonnefous C, Govek S, Sieders TJ, et al. Synthesis, characterization, and first successful monkey imaging studies of metabotropic glutamate receptor subtype 5 (mGluR5) PET radiotracers. Synapse. 2005;56:205–16. doi: 10.1002/syn.20147. [DOI] [PubMed] [Google Scholar]
- 25.Andersson Y, Långström B. Transition metal-mediated reactions using [11C]cyanide in synthesis of 11C-labeled aromatic-compounds. J Chem Soc Perkin Trans. 1994;111:1395–400. [Google Scholar]
- 26.Lindsley CW, Bates BS, Menon UN, Jadhav SB, Kane AS, Jones CK, et al. (3-Cyano-5-fluorophenyl)biaryl negative allosteric modulators of mGlu5: discovery of a new tool compound with activity in the OSS mouse model of addiction. ACS Chem Neurosci. 2011;2:471–82. doi: 10.1021/cn100099n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zoghbi SS, Anderson KB, Jenko KJ, Luckenbaugh DA, Innis RB, Pike VW. On quantitative relationships between drug-like compound lipophilicity and plasma free fraction in monkey and human. J Pharm Sci. 2012;101:1028–39. doi: 10.1002/jps.22822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Okamura T, Kikuchi T, Okada M, Toramatsu C, Fukushi K, Takei M, et al. Noninvasive and quantitative assessment of the function of multidrug resistance-associated protein 1 in the living brain. J Cereb Blood Flow Metab. 2009;29:504–11. doi: 10.1038/jcbfm.2008.135. [DOI] [PubMed] [Google Scholar]
- 29.Ametamey SM, Kessler LJ, Honer M, Wyss MT, Buck A, Hintermann S, et al. Radiosynthesis and preclinical evaluation of 11C-ABP688 as a probe for imaging the metabotropic glutamate receptor subtype 5. J Nucl Med. 2006;47:698–705. [PubMed] [Google Scholar]
- 30.DeLorenzo C, Milak MS, Brennan KG, Kumar JSD, Mann JJ, Parsey RV. In vivo positron emission tomography imaging with [11C]ABP688: binding variability and specificity for the metabotropic glutamate receptor subtype 5 in baboons. Eur J Nucl Med Mol Imaging. 2011;38:1083–94. doi: 10.1007/s00259-010-1723-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ametamey SM, Treyer V, Streffer J, Wys MT, Schmidt M, Blagoev M, et al. Human PET studies of metabotropic glutamate receptor subtype 5 with 11C-ABP688. J Nucl Med. 2007;48:247–52. [PubMed] [Google Scholar]