

Abstract
In this review, we discuss how two evolutionarily conserved pathways at the interface of DNA replication and repair, template switching and break-induced replication, lead to the deleterious large-scale expansion of trinucleotide DNA repeats that cause numerous hereditary diseases. We highlight that these pathways, which originated in prokaryotes, may be subsequently hijacked to maintain long DNA microsatellites in eukaryotes. We suggest that the negative mutagenic outcomes of these pathways, exemplified by repeat expansion diseases, are likely outweighed by their positive role in maintaining functional repetitive regions of the genome such as telomeres and centromeres.
Keywords: break-induced replication, centromeres, DNA microsatellites, telomeres, template switching, trinucleotide repeats, replication fork stalling
Graphical abstract
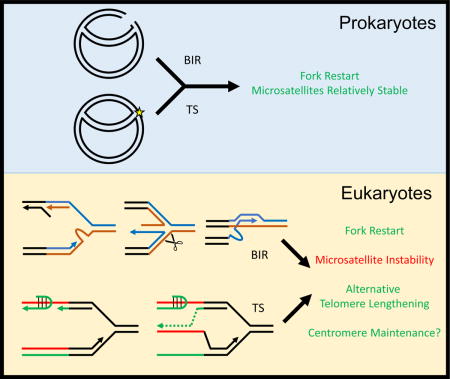
Break-induced replication (BIR) and template switching (TS) are conserved mechanisms of replication fork restart. They do not cause microsatellite instability in prokaryotes, but promote repeat expansion in eukaryotes. We suggest that TS and BIR persisted in eukaryotes despite their mutagenic potential because they help maintain long repetitive telomeres and centromeres.
Introduction
Repeat expansion diseases comprise a class of genetic disorders with a shared molecular cause: progressive lengthening of a simple DNA sequence repeat as the mutant allele passes through generations [1–4]. These simple DNA repeats, or microsatellites, can range from two to nine nucleotides in their unit lengths, though the majority affiliated with human diseases are trinucleotide repeats (TNR) (e.g. CAG/CTG). The overall tract length refers to the number of repeats that occur in tandem one after another and is highly polymorphic across organisms. In general, microsatellites comprise ~2% of the human genome, which is comparable to the protein-coding portion of the genome [5]. Thus, it is not the presence of long microsatellites in the genome per se, but rather rare instances when those microsatellites have expanded within functional genes that they pose a problem to human health. Disease pathology, in turn, often results from an expanded repeat acquiring dominant gain-of-function at the RNA and/or protein levels [6,7]. In addition, long repeat tracts can also lead to the repression of the genes in which they are located, typically by promoting heterochromatin formation [8].
Repeat expansion diseases are commonly neurological and/or developmental in nature. Huntington’s disease (HD), myotonic dystrophy type 1 (DM1), and several spinocerebellar ataxias (SCA) are caused by expanded (CAG)n/(CTG)n repeats. Other microsatellite sequences and their associated diseases include: (GAA)n in Friedreich’s ataxia (FA), (CGG)n in fragile X syndrome (FXS), (CCTG)n in myotonic dystrophy type 2 (DM2), (ATTCT)n in spinocerebellar ataxia type 10 (SCA10), and (GGGGCC)n in the most common form of inherited amyotrophic lateral sclerosis (ALS). For each disease, there is a threshold length below which the repeats do not expand [9]. However, when the number of repetitive units exceeds this threshold, the likelihood of further increases in length becomes greater. These DNA expansions frequently occur as the mutant allele is passed from parent to offspring, known as intergenerational transmission, and occasionally during the lifetime of an individual, known as somatic instability. Because the propensity to expand increases with increasing repeat length and longer repeat tracts are associated with more severe disease symptoms, these properties provide a biological explanation for the phenomenon of genetic anticipation, whereby the age of disease onset becomes apparent earlier in each successive generation and often with increased severity of symptoms.
The discovery in the 1990’s that expandable DNA repeats cause numerous genetic diseases provided an immediate motivation to elucidate the molecular mechanism of the expansion process. Disease-associated repeat sequences were found to form stable secondary structures such as imperfect hairpins (CAG, CTG, CGG, CCTG), G-quadruplexes (CGG, GGGGCC), and intramolecular triplexes/H-DNA (GAA) [1] (Figure 1 A–C). It was initially believed, therefore, that a unifying mechanism involving DNA secondary structures promotes repeat instability (i.e. both expansions and contractions). Slippage of repetitive strands during replicative DNA synthesis or ligation of repetitive 5′-flaps during Okazaki fragment maturation were discussed as possible mechanisms [10,11] (Figure 2A, B). These processes could generate expansions whose length is determined by the number of nucleotides in the hairpin-loop or flap structures, making them fairly small-scale (i.e. fewer than 60 nucleotides in the case of a long 5′ flap). However, the early assumption that all expandable DNA repeats always expand the same way and with the same step size has been subsequently amended owing to the realization that different molecular mechanisms contribute to the expansion process depending on the sequence of the repeat, the location of that sequence within a gene and the average length of disease-associated alleles.
Figure 1.
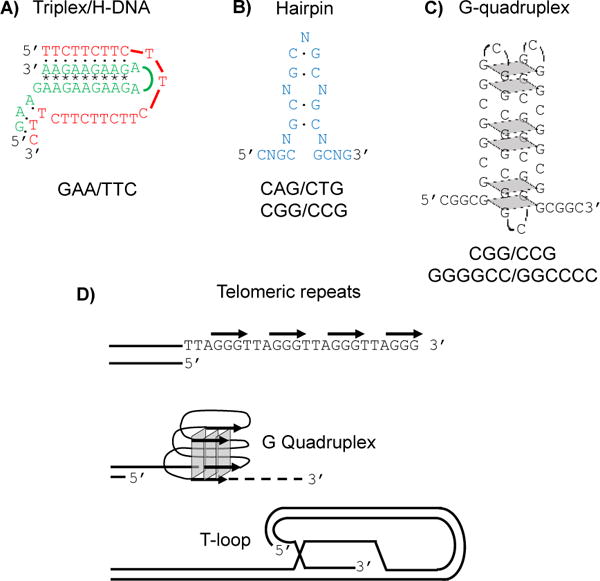
DNA structures formed by simple DNA repeats. A: H-DNA formed by GAA/TTC sequences. As shown, the homopurine (GAA) strand is donated to the triplex. Dots indicate Watson-Crick base pairing. Asterisks indicate reverse Hoogsteen base pairing. B: Imperfect hairpin formed by (CNG)n repeats. C: G-quadruplex formed by (CGG)n repeats. Grey squares represent hoogsteen basepairing between four guanines. D: The G-rich strand of telomeres can form G guadruplex structures. One possible example is shown here. The T-loop results from invasion of the single-stranded telomeric 3′-overhangs into the upstream telomeric duplex DNA.
Figure 2.
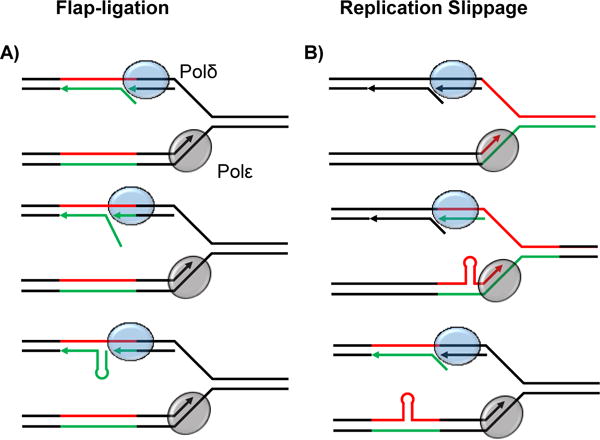
Mechanisms of small-scale repeat expansion. A: The flap ligation model of repeat expansion. The lagging strand polymerase (Polδ) performs displacement synthesis on the preceding Okazaki fragment leading to the formation of a 5′ flap. Formation of a hairpin structure within the flap precludes flap processing. Subsequent ligation of the hairpin flap to the adjacent Okazaki fragment results in a strand asymmetry that can lead to expansion upon the next round of replication. B: The replication slippage model of repeat expansion. The leading strand polymerase (Polε) slips within the repetitive sequence resulting in the formation of a hairpin on the nascent strand. This hairpin can be duplicated into an expansion upon the next round of replication. Slippage can occur on either the leading or lagging strands.
The length of expanded repeats at disease-associated loci differs depending on whether the sequence is protein-coding or not. The average length of the longest identified repetitive alleles at coding sequences across dozens of different trinucleotide repeat-associated disorders is around 60 repeats [12–14]. In contrast, the average of the longest disease-causing alleles identified for non-coding repeats is around 2,000 repeats [12,13]. Thus, it appears that repeat tracts >100 units in protein-coding areas of the genome experience negative selection, likely due to the fact these coding repeats are translated as toxic polyglutamine and polyalanine tracts [15]. The expansion of these shorter repetitive runs can be readily explained by strand slippage or 5′ flap ligation shown in Figure 2A, or iterative rounds of these events in dividing cells. In non-dividing cells, a model of “toxic oxidation cycles” has been proposed where oxidative damage at the CAG repeats is repaired by repeated rounds of base excision repair [16]. Expansions in this case are generated by strand slippage and 5′ flap-ligation during DNA repair synthesis.
In contrast, for repeat expansion diseases such as Friedreich’s ataxia and myotonic dystrophy, up to thousands of repeats at the disease gene locus are commonly observed [2,17]. This implies that distinct molecular mechanisms could be responsible for adding large numbers of repeats in a single step. This review concentrates on the insights made through investigating mechanisms of such large-scale expansion in budding yeast. We describe two pathways of large-scale repeat expansion involving either replicative template switching (TS) or break-induced replication (BIR). These mechanisms have evolved in prokaryotes to safely restart broken replication forks. We argue that while performing a similar function in the more complex eukaryotic genome, they inadvertently cause microsatellite instability. This begs the question of how such mechanisms persist in eukaryotic organisms given the deleterious consequences of microsatellite expansions within genes. We provide a possible answer by discussing the role of template switching and BIR in the maintenance of long telomeric and centromeric repeats, which are vital for the functioning of these structural chromosomal elements.
Molecular mechanisms that generate large-scale DNA repeat expansions
The emerging picture from two decades of repeat expansion studies in bacteria, yeast, Drosophila, mice, and cultured mammalian cells is that different molecular mechanisms contribute to repeat expansions depending on the sequence and starting length of the repeats, scale of expansion (small or large), developmental stage, and whether cells are mitotic or post-mitotic [18]. To specifically decipher the mechanisms of large-scale expansions, we developed two experimental systems in budding yeast to detect and analyze sizeable length increases for GAA/TTC, and CTG/CAG repeats [19–22]. These repeats were chosen since they are known to expand to over one thousand copies in Friedriech’s ataxia and myotonic dystrophy type 1 patients, respectively. To our surprise, the genetic control of large-scale expansions for CTG/CAG repeats appeared to be entirely distinct from GAA/TTC repeats [21]. Furthermore, genes that were previously implicated in small-scale expansions of CTG/CAG repeats did not appear to significantly contribute to large-scale events. We argue that the dramatic differences in secondary structures formed by the two repetitive sequences (i.e. triplex DNA versus DNA hairpins) arbitrate the preferred pathway for expansion.
Large-scale GAA repeat expansion proceeds via template switching
The duplication of the genome during S-phase can be disrupted by a wide-variety of replicative roadblocks, and multiple enzymatic pathways have evolved to resolve the subsequent replication fork stalling [23]. One such pathway, template switching, belongs to a class of DNA repair known as post replicative repair (PRR). The process of PRR is initiated when leading and lagging strand synthesis becomes “uncoupled”. This is often due to the stalling of one DNA polymerase at a site of DNA damage while the other polymerase and/or the replicative helicase continues progressing. When this uncoupling occurs, a stretch of single-stranded template DNA is exposed, activating the DNA damage response to halt the cell cycle and recruit the machinery necessary for fork restart [24,25]. The function of PRR is to allow replication to continue and then repair the lesion that caused the DNA polymerase to stall. PRR has two primary branches: translesion synthesis, which has been shown to not influence repeat length instability [26,27], and template switching (TS) (discussed below). Which pathway is chosen is determined by posttranslational modification of the replication clamp PCNA [28]. During TS, the polymerase-stalling lesion is bypassed when the newly synthesized strand (that has stalled) invades the nearby sister chromatid based on sequence homology and copies past the site of damage [29–31]. TS is considered to be a largely “error free” pathway because it uses a homologous template for damage bypass. When the leading strand polymerase is stalled, the lagging strand polymerase can jump forward initiating a new Okazaki fragment ahead of the stalled polymerase. To bypass the lesion, the stalled leading strand polymerase can then switch onto the nascent lagging strand as a template (Figure 3A). This scenario involves replication fork stalling and restart [32]. Alternatively, a stalled lagging strand polymerase might switch onto the nascent leading template to complete Okazaki fragment synthesis [33]. In this latter case, fork stalling and restart is not necessarily required as leading strand synthesis is not disrupted and discontinuous lagging strand synthesis can continue (Figure 3B).
Figure 3.
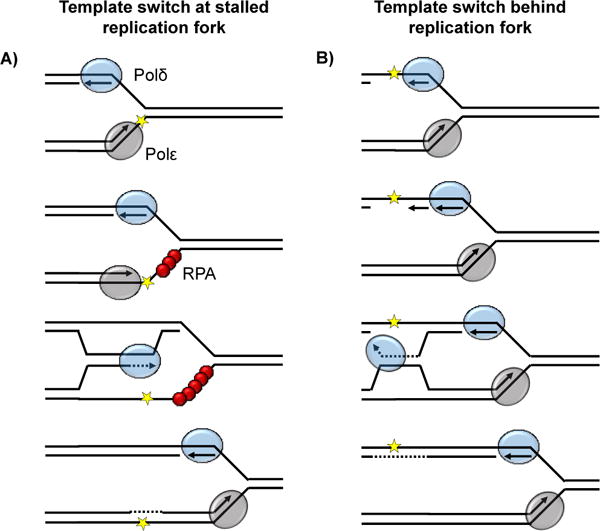
Two mechanisms of template switching. A: Template switching at a stalled replication fork occurs when the leading strand polymerase (Polε) encounters a lesion (yellow star) in the DNA template. Stalling of the leading strand polymerase leads to fork uncoupling as the lagging strand continues to be replicated. Fork stalling triggers the DNA damage response after replication protein A (RPA, red spheres) is recruited to single stranded DNA. Lesion bypass is primed by the nascent leading strand using Polδ and the nascent lagging strand as a homologous template. B: Template switching behind a replication fork is instigated by stalling of the lagging strand polymerase (Polδ) at a lesion in the DNA template (yellow star). The discontinuity of lagging strand replication allows fork progress to continue and template switching can occur without stalling using the nascent leading strand as a template.
Besides the role of TS in bypassing DNA damage during replication, we have found that it is a major contributor to large-scale expansions of GAA repeats responsible for Freidreich’s ataxia [19]. GAA repeats display several unique structural properties among expandable DNA repeats, which appear to constrain their expansion mode. First, unlike CAG or CGG repeats, GAA repeats only expand once they reach a threshold that is longer than the length of an Okazaki fragment (i.e. 60 repeats) [19,27]. Second, GAA repeats stall replication in a strictly orientation-dependent manner: when the GAA strand serves as the lagging strand template [19]. For other expandable DNA repeats, the orientation dependence of the replication stalling is less pronounced [34]. Thirdly, GAA repeats are uniquely capable of forming intramolecular triplexes [35]. These are three stranded DNA structures in which a portion of repetitive strand folds back to form a three-stranded complex with a repetitive duplex via hydrogen bonds known as Hoogsteen or reverse Hoogsteen [36]. Interestingly, triplexes (whether repetitive or not) seem to block fork progression in an orientation dependent manner [37]. It is also worth noting that the small-scale expansion of hairpin-forming CAG repeats is prevented by TS [38,39], in contrast to large-scale expansion of GAA repeats.
Detailed genetic analyses in budding yeast have led to a model of template switching at GAA repeats that takes into account the above traits. This model is predicated on two very important observations. First, since the rate at which GAA repeats expand changes ~ 2 fold when orientation is reversed but repeat-induced fork stalling is exquisitely orientation-dependent, then large-scale expansion of GAA repeats may not result from the replication fork stalling per se [19]. Second, since mutations that specifically influence lagging strand replication and template switching affect the rate and size of large-scale GAA expansion, then template switching at GAA repeats must be instigated by defective lagging strand synthesis [19,40,41]. Altogether, this has led to the model illustrated in Figure 4 [41].
Figure 4.
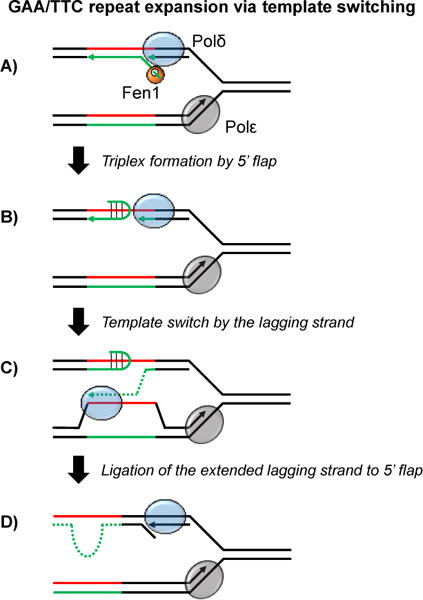
Proposed mechanism of large-scale GAA/TTC repeat expansion. Green = GAA repeats. Red = TTC repeats. A: Displacement synthesis by the lagging strand polymerase (Polδ) leads to the formation of a 5′ flap that can be processed by the flap endonuclease Fen1. B: In some circumstances (especially in the absence of Fen1) the displaced 5′ flap folds back, forming a triplex that blocks further synthesis by Polδ. C: Polδ switches template to the nascent leading strand in order to bypass the 5′ flap triplex and continue Okazaki fragment synthesis. D: Ligation of the extended Okazaki fragment to the folded back 5′ flap triplex resolves the template switching event and leads to a strand asymmetry that results in repeat expansion upon the next round of replication.
We hypothesize that template switching at GAA repeats is initiated when poorly processed 5′ flaps that arise during lagging strand replication form stable triplexes within an Okazaki fragment (Figure 4B) [41,42]. Such secondary structures would disrupt displacement synthesis and interrupt lagging strand replication, leading to fork uncoupling [43]. In our model, the invading strand during TS is a truncated Okazaki fragment that encountered the triplex structure (Figure 4C). If this invading sequence is repetitive, a nascent lagging strand that switches onto the nascent leading strand “out of register” can lead to subsequent expansion events (Figure 4C). The likelihood of this scenario would increase dramatically when the length of the GAA repeat as a whole surpasses the length of an Okazaki fragment. Importantly these expansions can result in a sequence up to double the length of the original repetitive run. Furthermore, changes to the length of Okazaki fragments will shift the scale of expansion events [27]. A strength of this model is that longer starting repeat lengths will result in longer repeat expansion events as (i) template switching is more likely overall for longer repeats and (ii) a longer donor sequence during template switching will result in a longer expansion. This could account for the pattern of genetic anticipation observed in human pedigrees. Because TS is tightly linked to DNA replication, it is likely that this mechanism of GAA expansion is limited to actively dividing cells.
Large-scale CAG expansion is the result of break-induced replication
In the template-switch model of large-scale expansion, the event that triggers expansion events is the disruption of lagging strand displacement synthesis by triplex formation during Okazaki fragment maturation. CAG/CTG repeats do not form triplexes, instead adopting stable hairpin structures (Figure 1B) [44]. Thus, it was unclear whether a TS mechanism could account for CAG/CTG microsatellite expansions as well. We have recently found that large-scale expansion of CAG repeats in yeast is driven by break-induced replication (BIR), rather than TS [21]. These results highlight the observation that different mechanisms of expansion appear to predominate depending on the repeat sequence and the scale of expansion.
BIR is a repair pathway uniquely suited for resolving one-ended double strand breaks (DSBs) [45,46]. Such breaks can result from replication forks encountering a single strand break (SSB) in the DNA backbone, which causes the fork to collapse. The incidence of single strand breaks (and thus the likelihood of fork collapse) is incredibly high. They occur not only from direct chemical damage to the DNA but also as intermediates in various DNA repair pathways. Thus, BIR and broken fork repair may be called into action quite frequently during the duplication of genomes [47].
BIR events are initiated when a one-ended DSB undergoes 5′-strand resection followed by invasion into a homologous sequence – either a sister chromatid, homologous chromosome or non-allelic homologous region of the genome. This newly invaded strand then serves as a primer for DNA synthesis using DNA polymerase delta – the lagging strand DNA polymerase in eukaryotes [48]. BIR synthesis is conducted within a migrating bubble known as a D-loop and requires specific helicases such as Pif1 and DNA polymerase subunits. BIR synthesis is conservative, not semi-conservative, in nature because discontinuous lagging strand synthesis is templated on the newly synthesized DNA that has been extruded during D-loop migration [49,50]. If BIR occurs during S phase, this conservative synthesis proceeds until it meets an oncoming replication fork, which has been termed broken fork repair in recent studies [47]. Remarkably, BIR events during G2 can lead to conservative synthesis all the way to the end of a homologous chromosome [51]. Though BIR can proceed for such long distances, it is prone to template switching [52]. Combined with the conservative duplication mechanism, DNA synthesis that is more prone to error, and the likelihood of non-allelic homologous invasion, BIR events can lead to massive loss of heterozygosity, clustered point mutations, translocations, and copy number changes [53–56]. Such mutational events have been linked to a wide-array of human diseases [57].
In the BIR model of CAG repeat expansion, the initiating event is replication fork stalling resulting from hairpin formation on the single stranded DNA template (Figure 5A), which is well documented [34,58,59]. Replication stalling at microsatellite repeats has been shown to cause reversal of the replication fork [60], which can isomerize to result in the formation of a four-way junction (Figure 5B). Endonucleolytic cleavage of this four-way junction can lead to a one-ended break (Figure 5C). Since the impetus for fork stalling and reversal was the repetitive CAG sequence, this sequence then serves as the platform for homology search. This allows for strand invasion within the corresponding CAG sequence on the sister chromatid (Figure 5D). Owing to the repetitive nature of both recipient and donor sequences, this invasion will readily occur out of register, especially if the invading single-stranded filament forms a hairpin-like structure (Figure 5D). Such out-of-register invasion and synthesis through the length of the repetitive sequence would then lead to large-scale expansion events (Figure 5E,F). Alternatively, multiple template-switching events, which commonly occur at the beginning of BIR, could be responsible for trinucleotide expansions in a fashion similar to template switching at GAA repeats [61–63]. BIR synthesis through the repeats proceeds as a migrating D-loop until it encounters a convergent replication fork. This encounter results in Holliday junction formation that must be resolved nucleolytically, completing the process of large-scale expansion in a single step. Similar to the template switch model of large-scale GAA expansion, this model can also explain genetic anticipation as longer repeats can promote longer expansion events.
Figure 5.
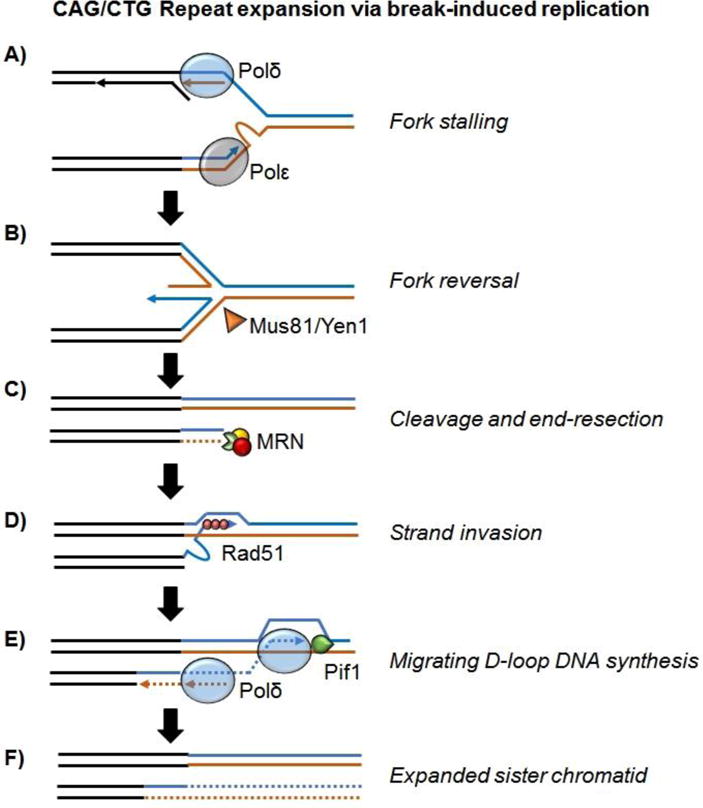
Proposed mechanism of large-scale CAG/CTG repeat expansion. Blue = CAG repeats. Brown = CTG repeats. A: CTG hairpin formation on the leading strand template stalls the replication fork by blocking Polε. B: Reversal of the replication fork leads to a four-way junction that can be recognized by the Holliday-junction endonucleases Mus81 and Yen1. C: Cleavage of the reversed fork leads to a one-ended double strand break that undergoes end section via the Mre11-Rad50-Nbs1/Xrs2 (MRN) complex. D: “Out-of-register” strand invasion between repetitive sequences mediated by Rad51 is encouraged by hairpin formation in the invading strand and primes break-induced replication (BIR) synthesis. E: BIR synthesis by Polδ proceeds as a migrating D-loop and is aided by Pif1 helicase through the repetitive tract until it encounters an oncoming replication fork at which point resolution requires cleavage by an endonuclease [17]. BIR synthesis is conservative as newly synthesized DNA extruded from the D-loop templates duplication of CTG repeats. F: Resolution of the BIR process results in repeat expansion in a single step.
In contrast to the template switch case, CAG expansions via BIR may also occur in non-dividing cells. This process would result from a two-ended DSB (not a reversed fork) being repaired via BIR, – a possibility currently studied by us and others. This is particularly important, as repeat expansions, including large-scale ones, have been detected in non-dividing somatic cells and may drive disease progression [64,65].
Functional roles of mechanisms responsible for large-scale repeat expansion
When considering the large-scale microsatellite expansions that can result from BIR and TS in eukaryotic organisms, it is important to keep in mind that these pathways arose in the context of a prokaryotic genome. Indeed, both pathways operate on stalled or collapsed replication forks in E. coli [66,67]. Notably, some of the parameters that encourage mutagenesis via these pathways in eukaryotes are quite different in prokaryotes. Take for example the fact that template switching in yeast leads to large-scale microsatellite instability only after a repetitive GAA sequence reaches a threshold longer than the size of an Okazaki fragment [19,27]. Okazaki fragments are much longer in prokaryotes (~1000 bp), and the likelihood of a repetitive sequence reaching such a length in a bacterial genome is negligible [68,69]. What about the fact that BIR can lead to significant rearrangements when it occurs between microhomologies on two different chromosomes? The E. coli version of BIR, recombination-dependent replication (RDR), utilizes specifically oriented Chi sites as replication restart sites, ensuring that the strand invasion step starts at non-repetitive 8 bp-long sequences to minimize likelihood of large deletion and insertion events [67,70]. Furthermore, BIR is not cell-cycle sequestered in eukaryotes, and events in G2 can lead to large-scale loss of heterozygosity [71]. In prokaryotes, DNA synthesis takes up the majority of the cell cycle and loss of heterozygosity is not a problem for the haploid genome [72].
Ultimately, the differences between prokaryotic and eukaryotic BIR and TS support the argument that these conserved pathways are poorly suited to maintaining accurate propagation of repetitive elements in eukaryotic genomes. Indeed, as we have already highlighted, both pathways are responsible for large-scale expansion of microsatellite sequences linked to human disease. Furthermore, many of the eukaryotic constraints on replication (e.g. shorter Okazaki fragments) and homology search (microhomology-mediated rather than Chi-mediated) encourage large-scale mutagenesis during TS and BIR. However, there are two critical functional regions of the eukaryotic genome that are repetitive in nature – telomeres and centromeres. In the next section we would like to highlight the idea that the persistence of TS and BIR in eukaryotes may be a result, in part, of their importance for duplicating these genomic elements.
Alternative lengthening of telomeres mimics large-scale microsatellite instability
The transition from storage of the genetic material in circular DNA molecules in prokaryotes to linear DNA molecules in eukaryotes resulted in what is known as the “end replication problem.” Since DNA polymerases operate in a unidirectional manner (5′ to 3′) and require RNA primers, copying the end of a linear DNA molecule and removing the primer sequence means the terminal portion of the lagging strand template is left uncopied [73,74]. A similar, albeit more complex mechanism also occurs during leading strand synthesis, which leads to the characteristic 3′ overhangs observed at the end of eukaryotic chromosomes [75]. Another dilemma caused by the linear nature of chromosomes is that DNA repair and recombination machineries must distinguish between DNA damage in the form of DSBs and double-stranded chromosomal ends. When this process fails, chromosomal ends are recognized as damage – halting the cell cycle and/or leading to chromosomal instability in the course of breakage-fusion-bridge cycles [76].
Most eukaryotic genomes rely on telomeres, special protein-bound DNA repeats at the chromosomal ends as a solution to both of the aforementioned problems. The sequence of telomeres is repetitive and varies slightly between eukaryotic organisms [77]. The majority (including humans) have (TTAGGG)n hexameric repeats or a slight variation thereof, with notable divergences being found in budding yeast (TG1–3)n and dipteran insects (including Drosophila melanogaster), which do not have telomeric repeats at all but instead protect chromosome ends using retrotransposition. Interestingly, a different mechanism of telomere maintenance in diptera does not seem to exmept them from using DNA recombination pathways to lengthen telomeres in the absence of retrotransposition [78].
Like most microsatellite sequences, telomeric repeats are capable of forming non-canonical DNA structures. They can form G-quadruplexes, four stranded DNA structures in which four pillars of guanines interact to form a stack (Figure 1D) [79]. They also form T-loops: lasso-like structures that result from an invasion of the single-stranded telomeric 3′-overhangs into the upstream telomeric duplex DNA (Figure 1D) [80]. Besides these structures, telomeres are protected from gratuitous double strand break repair by binding of numerous telomere-specific proteins forming the shelterin complex [76].
Owing to the end replication problem, each cycle of replication could lead to shortening of the telomere. When telomeres become too short, cells enter senescence and die [81]. Actively dividing cells, including most cancer cells, overcome this problem via the action of the telomerase complex [82,83], which increases the length of telomeres prior to DNA replication [81]. In dividing cells that lack telomerase activity, a backup mechanism of telomere lengthening known as alternative lengthening of telomeres (ALT) is responsible for maintaining telomere integrity. This pathway is active in a subset of cancer cells and represents a unique strategy for cell survival in a population of dividing cells that lack telomerase [84].
While it was widely believed that ALT occurs via canonical homologous recombination [85], it is now clear that it proceeds via BIR in several experimental systems. The key proteins implicated in BIR, such as Pol32, Mus81, Pif1 and Rad52 have all been linked to ALT [48,86–91]. It has been predicted that BIR extension of telomeres goes along the following pathway [45,86,92]. Telomeric repeats are hard to replicate owing to the formation of abnormal DNA structures and/or stable protein complexes, as is evident form fork stalling that is comparable to or even stronger than at other microsatellite sequences [93,94]. As discussed above, stalled forks can be readily converted into 5′ resected, one-ended DSBs via endonucleases involved in ALT [87,95]. Since telomeres are repetitive in nature, “out of register” strand invasion or multiple template-switching events can lead to lengthening of the repetitive sequence after BIR is complete. This directly parallels the mechanism of large-scale CAG expansion observed in yeast [21].
However, there are also important differences. BIR is specifically necessary in the case of ALT because telomeres are located at the ends of chromosomes [45]. A priori, fork stalling can be simply dealt with by the firing of a dormant origin(s) nearby. While this is indeed happening in the body of the chromosome [96] and even in the duplex parts of long telomeres [97], it can hardly solve the end-replication problem for short telomeres, as a dormant origin is unlikely to fire in a very short telomere. This reasoning might explain the preference of ALT for short telomeres [98]. Consequently, BIR becomes a necessary means for maintaining telomere length in cells lacking telomerase, in which telomeres are shortened. To an extent, this milieu mimics the situation in E. coli, where the presence of a single origin and set terminus make fork restart via RDR crucial for the completion of replication. Thus ALT represents an instance where prokaryotic repair mechanisms are necessary in the context of a linear eukaryotic genome.
There is also evidence of a role for TS in maintaining telomere sequences. We have found that knocking out genes involved in TS reduces the rate at which interstitial (meaning not at the chromosomal end) telomeric sequences expand [99]. Thus, TS may facilitate the maintenance of telomere length during or in addition to BIR. Supporting this notion, the critical TS factor Rad5 localizes to wild type telomeres in yeast to aid replication fork progression through full-length telomeres and prevent senescence [98]. Interestingly, the leading and lagging strand polymerases do not progress at the same rate through telomeric regions, leading to fork uncoupling [100]. Lagging strand synthesis through telomeres also suffers from poor processing of 5′ flaps [101,102], which could instigate template switching and repeat instability if telomeric flaps form fold-back structures (likely quadruplexes) that are hard to bypass in a fashion similar to the triplexes that may occur at GAA flaps (Figure 4B). That being said, the exact role of TS in telomere maintenance remains unclear and more studies are needed for its better understanding.
Is centromeric repeat instability similar to large-scale microsatellite expansion?
Many eukaryotic genomes also possess functional repetitive DNA within centromeres. Centromeric regions show greater sequence divergence amongst eukaryotic organisms than telomeres and are better defined by their specific chromatin modifications than any specific sequence [103]. These chromatin modifications allow centromeres to serve as the platform for kinetochore formation during mitosis and meiosis. Kinetochores are protein complexes that coordinate spindle formation and appropriate segregation of chromosomes. Dysfunction of the centromere/kinetochore apparatus results in numerical (e.g. trisomy) and structural (e.g. translocations) chromosome instability, which influences fertility and oncogenesis [104,105]. It is unclear whether the repetitive nature of centromeres is crucial to their function, yet most eukaryotic organisms contain long repetitive elements within their centromeres [106]. Unlike telomeres, the repetitive unit of most centromeric repeats is quite long (beyond microsatellite length) [106].
Human centromeres are composed primarily of alpha satellites – tandemly repeated sequences made up of 171bp units that are further organized into higher order arrays of repeating monomers [107,108]. Alpha satellite regions are flanked by pericentromeric human satellites of which several different varieties exist (I–III, beta, gamma) [109]. Human satellite II and III in particular are almost microsatellite-like. They are composed of a degenerate repeating TTCCA pentamer [110]. Coincidentally, expansion of a TTCCA microsatellite is responsible for the human disease SCA31 [111]. Centromeric and pericentromeric satellites are not known to form non-canonical DNA structures although the formation of slip-stranded structures, where a single strand loop is extruded from the duplex due to strand misalignment, is certainly possible.
Replication of centromeres is associated with chromosome fragility and double strand breaks, which may be due to the collapse of replication forks [112–114]. Pericentromeric and alpha satellite repeats evolve at a faster pace than the rest of the primate genome and also display length polymorphisms amongst humans, suggesting they are very prone to instability [106,115–117]. Studies in fission yeast (which have different centromere sequences but similar centromeric chromatin to humans) have identified several of the replication fork restart proteins, including Smc5/6 and Brc1, as crucial to duplicating and suppressing crossover recombination within centromeric regions (for a recent review see [118]). Mitotic recombination at centromeres in mammalian cells may be quite common and is also suppressed by centromere associated histone proteins and epigenetic silencing [119,120].
The exact mechanisms of centromere recombination are yet to be sorted out for alpha satellite arrays, but some key players have been identified. A recent study examining the proteins present during alpha satellite replication in Xenopus laevis egg extracts identified the recombination nucleases MUS81, MRE11 and RAD50 [121]. There is also specific enrichment for PARP1, an enzyme involved in restarting stalled or collapsed replication forks via recombination [122,123]. Extended BIR or fork uncoupling may explain the large stretches of ssDNA and/or increased positive supercoiling observed during centromere replication [121]. However, there are a wide array of questions that remain regarding centromere replication. Specifically, the mechanism of recombination at centromeres is yet to be elucidated and may provide crucial insight into fork restart within alpha satellites. We suspect BIR may play a role given the proteins that have been implicated thus far. Experiments in yeast have shown that BIR does not progress through the centromere when a distal homologous sequence is used to initiate the BIR event from a chromosome fragment [124]. However, the relevance of this observation to the situation of fork stalling within the centromere itself is not evident and requires further examination. There is also the question of whether human pericentromeric DNA sequences, which more closely resemble microsatellites, also recruit recombination machineries during replication.
Conclusion
For decades, biologists have struggled to understand the repetitive regions of the human genome. The discovery that expanded microsatellites are the genetic cause of dozens of human diseases has spurred a torrent of research on exactly how repeats go from subthreshold lengths to long alleles that cause disease. Our studies demonstrated two conserved mechanisms of large-scale repeat expansion – TS and BIR. Both originated in prokaryotic organisms, where they are much less harmful owing to crucial differences in DNA replication and recombination between pro- and eukaryotes.
An important question is why these pathways have persisted in eukaryotic organisms despite their high mutagenic potential. We speculate that part of the reason is the linear nature of eukaryotic chromosomes. This organization requires two critical functional elements – centromeres to appropriately segregate chromosomes during cell division and telomeres to maintain chromosomal ends. Both these elements are repetitive in nature and there is growing evidence to suggest that molecular mechanisms implicated in large-scale repeat expansion may also play a role in maintaining lengthy telomeres and centromeres. Thus, we would like to suggest that an answer to the question of why interstitial repetitive sequences have a tendency to get longer is that such lengthening is the deleterious flip-side of mechanisms crucial for the maintenance of a composite linear genome.
Acknowledgments
Research in the lab of S.M.M. is supported by NIH grants R01GM60987 and P01GM105473 and generous contribution from the White family. J.C.K. is supported by California State University San Marcos. We thank Alexander Shishkin, Kartik Shah, Yu Zhang and Kirill Lobachev for their contribution to the template-switch model for repeat expansions. We also thank Sue Lovett, Catherine Freudenreich and Anna Malkova for many helpful discussions of our models. Finally, we are indebted to John Tainer for getting us involved in studying the role of Fen1 in repeat expansions.
Abbreviations
- ALT
alternative lengthening of telomeres
- BIR
break-induced replication
- DSB
double strand break
- PRR
post replicative repair
- TS
template switching
Citations
- 1.Mirkin SM. Expandable DNA repeats and human disease. Nature. 2007;447:932–40. doi: 10.1038/nature05977. [DOI] [PubMed] [Google Scholar]
- 2.McMurray CT. Mechanisms of trinucleotide repeat instability during human development. Nature Reviews Genetics. 2010;11:786–99. doi: 10.1038/nrg2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.La Spada AR, Taylor JP. Repeat expansion disease: progress and puzzles in disease pathogenesis. Nat Rev Genet. 2010;11:247–58. doi: 10.1038/nrg2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schmidt MH, Pearson CE. Disease-associated repeat instability and mismatch repair. DNA Repair. 2016;38:117–26. doi: 10.1016/j.dnarep.2015.11.008. [DOI] [PubMed] [Google Scholar]
- 5.Treangen TJ, Salzberg SL. Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nat Rev Genet. 2011;13:36–46. doi: 10.1038/nrg3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang N, Ashizawa T. RNA toxicity and foci formation in microsatellite expansion diseases. Curr Opin Genet Dev. 2017;44:17–29. doi: 10.1016/j.gde.2017.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pearson CE. Repeat associated non-ATG translation initiation: one DNA, two transcripts, seven reading frames, potentially nine toxic entities! PLoS Genet. 2011;7:e1002018. doi: 10.1371/journal.pgen.1002018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kumari D, Lokanga R, Yudkin D, Zhao XN, et al. Chromatin changes in the development and pathology of the Fragile X-associated disorders and Friedreich ataxia. Biochimica et biophysica acta. 2012;1819:802–10. doi: 10.1016/j.bbagrm.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee DY, McMurray CT. Trinucleotide expansion in disease: why is there a length threshold? Curr Opin Genet Dev. 2014;26:131–40. doi: 10.1016/j.gde.2014.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kang S, Jaworski A, Ohshima K, Wells RD. Expansion and deletion of CTG repeats from human disease genes are determined by the direction of replication in E.coli. Nat Genet. 1995;10:213–8. doi: 10.1038/ng0695-213. [DOI] [PubMed] [Google Scholar]
- 11.Gordenin DA, Kunkel TA, Resnick MA. Repeat expansion–all in a flap? Nature genetics. 1997;16:116–8. doi: 10.1038/ng0697-116. [DOI] [PubMed] [Google Scholar]
- 12.Zhao XN, Usdin K. The Repeat Expansion Diseases: The dark side of DNA repair. DNA Repair (Amst) 2015;32:96–105. doi: 10.1016/j.dnarep.2015.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McMurray CT. Mechanisms of trinucleotide repeat instability during human development. Nat Rev Genet. 2010;11:786–99. doi: 10.1038/nrg2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Faux N. Single amino acid and trinucleotide repeats: function and evolution. Adv Exp Med Biol. 2012;769:26–40. doi: 10.1007/978-1-4614-5434-2_3. [DOI] [PubMed] [Google Scholar]
- 15.Mularoni L, Ledda A, Toll-Riera M, Alba MM. Natural selection drives the accumulation of amino acid tandem repeats in human proteins. Genome Res. 2010;20:745–54. doi: 10.1101/gr.101261.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kovtun IV, Liu Y, Bjoras M, Klungland A, et al. OGG1 initiates age-dependent CAG trinucleotide expansion in somatic cells. Nature. 2007;447:447–52. doi: 10.1038/nature05778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thornton CA. Myotonic dystrophy. Neurologic clinics. 2014;32:705–19, viii. doi: 10.1016/j.ncl.2014.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim JC, Mirkin SM. The balancing act of DNA repeat expansions. Current opinion in genetics & development. 2013;23:280–8. doi: 10.1016/j.gde.2013.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shishkin AA, Voineagu I, Matera R, Cherng N, et al. Large-scale expansions of Friedreich’s ataxia GAA repeats in yeast. Mol Cell. 2009;35:82–92. doi: 10.1016/j.molcel.2009.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shah KA, McGinty RJ, Egorova VI, Mirkin SM. Coupling transcriptional state to large-scale repeat expansions in yeast. Cell reports. 2014;9:1594–602. doi: 10.1016/j.celrep.2014.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim JC, Harris ST, Dinter T, Shah KA, et al. The role of break-induced replication in large-scale expansions of (CAG)n/(CTG)n repeats. Nat Struct Mol Biol. 2017;24:55–60. doi: 10.1038/nsmb.3334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shishkin AA, Voineagu I, Matera R, Cherng N, et al. Large-scale expansions of Friedreich’s ataxia GAA repeats in yeast. Molecular cell. 2009;35:82–92. doi: 10.1016/j.molcel.2009.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Branzei D, Foiani M. Maintaining genome stability at the replication fork. Nat Rev Mol Cell Biol. 2010;11:208–19. doi: 10.1038/nrm2852. [DOI] [PubMed] [Google Scholar]
- 24.Byun TS, Pacek M, Yee MC, Walter JC, et al. Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev. 2005;19:1040–52. doi: 10.1101/gad.1301205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huttner D, Ulrich HD. Cooperation of replication protein A with the ubiquitin ligase Rad18 in DNA damage bypass. Cell Cycle. 2008;7:3629–33. doi: 10.4161/cc.7.23.7166. [DOI] [PubMed] [Google Scholar]
- 26.Collins NS, Bhattacharyya S, Lahue RS. Rev1 enhances CAG.CTG repeat stability in Saccharomyces cerevisiae. DNA Repair (Amst) 2007;6:38–44. doi: 10.1016/j.dnarep.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 27.Shah KA, Shishkin AA, Voineagu I, Pavlov YI, et al. Role of DNA polymerases in repeat-mediated genome instability. Cell reports. 2012;2:1088–95. doi: 10.1016/j.celrep.2012.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Haracska L, Torres-Ramos CA, Johnson RE, Prakash S, et al. Opposing effects of ubiquitin conjugation and SUMO modification of PCNA on replicational bypass of DNA lesions in Saccharomyces cerevisiae. Mol Cell Biol. 2004;24:4267–74. doi: 10.1128/MCB.24.10.4267-4274.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang H, Lawrence CW. The error-free component of the RAD6/RAD18 DNA damage tolerance pathway of budding yeast employs sister-strand recombination. Proc Natl Acad Sci U S A. 2005;102:15954–9. doi: 10.1073/pnas.0504586102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Branzei D, Vanoli F, Foiani M. SUMOylation regulates Rad18-mediated template switch. Nature. 2008;456:915–20. doi: 10.1038/nature07587. [DOI] [PubMed] [Google Scholar]
- 31.Giannattasio M, Zwicky K, Follonier C, Foiani M, et al. Visualization of recombination-mediated damage bypass by template switching. Nat Struct Mol Biol. 2014;21:884–92. doi: 10.1038/nsmb.2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Branzei D. Ubiquitin family modifications and template switching. FEBS Lett. 2011;585:2810–7. doi: 10.1016/j.febslet.2011.04.053. [DOI] [PubMed] [Google Scholar]
- 33.Higuchi K, Katayama T, Iwai S, Hidaka M, et al. Fate of DNA replication fork encountering a single DNA lesion during oriC plasmid DNA replication in vitro. Genes Cells. 2003;8:437–49. doi: 10.1046/j.1365-2443.2003.00646.x. [DOI] [PubMed] [Google Scholar]
- 34.Viterbo D, Michoud G, Mosbach V, Dujon B, et al. Replication stalling and heteroduplex formation within CAG/CTG trinucleotide repeats by mismatch repair. DNA Repair (Amst) 2016;42:94–106. doi: 10.1016/j.dnarep.2016.03.002. [DOI] [PubMed] [Google Scholar]
- 35.Vetcher AA, Napierala M, Iyer RR, Chastain PD, et al. Sticky DNA, a long GAA.GAA.TTC triplex that is formed intramolecularly, in the sequence of intron 1 of the frataxin gene. J Biol Chem. 2002;277:39217–27. doi: 10.1074/jbc.M205209200. [DOI] [PubMed] [Google Scholar]
- 36.Frank-Kamenetskii MD, Mirkin SM. Triplex DNA structures. Annual Review of Biochemistry. 1995;64:65–95. doi: 10.1146/annurev.bi.64.070195.000433. [DOI] [PubMed] [Google Scholar]
- 37.Liu G, Myers S, Chen X, Bissler JJ, et al. Replication fork stalling and checkpoint activation by a PKD1 locus mirror repeat polypurine-polypyrimidine (Pu-Py) tract. J Biol Chem. 2012;287:33412–23. doi: 10.1074/jbc.M112.402503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Daee DL, Mertz T, Lahue RS. Postreplication repair inhibits CAG.CTG repeat expansions in Saccharomyces cerevisiae. Mol Cell Biol. 2007;27:102–10. doi: 10.1128/MCB.01167-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.House NC, Yang JH, Walsh SC, Moy JM, et al. NuA4 initiates dynamic histone H4 acetylation to promote high-fidelity sister chromatid recombination at postreplication gaps. Mol Cell. 2014;55:818–28. doi: 10.1016/j.molcel.2014.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang Y, Shishkin AA, Nishida Y, Marcinkowski-Desmond D, et al. Genome-wide screen identifies pathways that govern GAA/TTC repeat fragility and expansions in dividing and nondividing yeast cells. Mol Cell. 2012;48:254–65. doi: 10.1016/j.molcel.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tsutakawa S, Thompson M, Arvai A, Neil AJ, et al. Phosphate steering by Flap Endonuclease 1 promotes 5´-flap specificity and incision to prevent genome instability. Nat Commun. 2017 doi: 10.1038/ncomms15855. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Becker JR, Pons C, Nguyen HD, Costanzo M, et al. Genetic Interactions Implicating Postreplicative Repair in Okazaki Fragment Processing. PLoS Genet. 2015;11:e1005659. doi: 10.1371/journal.pgen.1005659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Krasilnikov AS, Panyutin IG, Samadashwily GM, Cox R, et al. Mechanisms of triplex-caused polymerization arrest. Nucleic Acids Res. 1997;25:1339–46. doi: 10.1093/nar/25.7.1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gacy AM, Goellner G, Juranic N, Macura S, et al. Trinucleotide repeats that expand in human disease form hairpin structures in vitro. Cell. 1995;81:533–40. doi: 10.1016/0092-8674(95)90074-8. [DOI] [PubMed] [Google Scholar]
- 45.Malkova A, Ira G. Break-induced replication: functions and molecular mechanism. Curr Opin Genet Dev. 2013;23:271–9. doi: 10.1016/j.gde.2013.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Anand RP, Lovett ST, Haber JE. Break-induced DNA replication. Cold Spring Harbor Perspectives in Biology. 2013;5:a010397. doi: 10.1101/cshperspect.a010397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mayle R, Campbell IM, Beck CR, Yu Y, et al. Mus81 and converging forks limit the mutagenicity of replication fork breakage. Science. 2015;349:742–7. doi: 10.1126/science.aaa8391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lydeard JR, Jain S, Yamaguchi M, Haber JE. Break-induced replication and telomerase-independent telomere maintenance require Pol32. Nature. 2007;448:820–3. doi: 10.1038/nature06047. [DOI] [PubMed] [Google Scholar]
- 49.Saini N, Ramakrishnan S, Elango R, Ayyar S, et al. Migrating bubble during break-induced replication drives conservative DNA synthesis. Nature. 2013;502:389–92. doi: 10.1038/nature12584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Donnianni RA, Symington LS. Break-induced replication occurs by conservative DNA synthesis. Proc Natl Acad Sci U S A. 2013;110:13475–80. doi: 10.1073/pnas.1309800110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Malkova A, Signon L, Schaefer CB, Naylor ML, et al. RAD51-independent break-induced replication to repair a broken chromosome depends on a distant enhancer site. Genes Dev. 2001;15:1055–60. doi: 10.1101/gad.875901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Smith CE, Llorente B, Symington LS. Template switching during break-induced replication. Nature. 2007;447:102–5. doi: 10.1038/nature05723. [DOI] [PubMed] [Google Scholar]
- 53.Sakofsky CJ, Malkova A. Break induced replication in eukaryotes: mechanisms, functions, and consequences. Critical reviews in biochemistry and molecular biology. 2017:1–19. doi: 10.1080/10409238.2017.1314444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sakofsky CJ, Roberts SA, Malc E, Mieczkowski PA, et al. Break-induced replication is a source of mutation clusters underlying kataegis. Cell reports. 2014;7:1640–8. doi: 10.1016/j.celrep.2014.04.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Anand RP, Tsaponina O, Greenwell PW, Lee CS, et al. Chromosome rearrangements via template switching between diverged repeated sequences. Genes Dev. 2014;28:2394–406. doi: 10.1101/gad.250258.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pardo B, Aguilera A. Complex chromosomal rearrangements mediated by break-induced replication involve structure-selective endonucleases. PLoS Genet. 2012;8:e1002979. doi: 10.1371/journal.pgen.1002979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hastings PJ, Ira G, Lupski JR. A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS Genet. 2009;5:e1000327. doi: 10.1371/journal.pgen.1000327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kerrest A, Anand RP, Sundararajan R, Bermejo R, et al. SRS2 and SGS1 prevent chromosomal breaks and stabilize triplet repeats by restraining recombination. Nat Struct Molec Biol. 2009;16:159–67. doi: 10.1038/nsmb.1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Samadashwily GM, Raca G, Mirkin SM. Trinucleotide repeats affect DNA replication in vivo. Nat Genet. 1997;17:298–304. doi: 10.1038/ng1197-298. [DOI] [PubMed] [Google Scholar]
- 60.Follonier C, Oehler J, Herrador R, Lopes M. Friedreich’s ataxia-associated GAA repeats induce replication-fork reversal and unusual molecular junctions. Nat Struct Mol Biol. 2013;20:486–94. doi: 10.1038/nsmb.2520. [DOI] [PubMed] [Google Scholar]
- 61.Kramara J, Osia B, Malkova A. Break-induced replication: an unhealthy choice for stress relief? Nat Struct Mol Biol. 2017;24:11–2. doi: 10.1038/nsmb.3361. [DOI] [PubMed] [Google Scholar]
- 62.Richard GF, Goellner GM, McMurray CT, Haber JE. Recombination-induced CAG trinucleotide repeat expansions in yeast involve the MRE11-RAD50-XRS2 complex. EMBO J. 2000;19:2381–90. doi: 10.1093/emboj/19.10.2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Richard GF, Paques F. Mini- and microsatellite expansions: the recombination connection. EMBO Rep. 2000;1:122–6. doi: 10.1093/embo-reports/kvd031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kovtun IV, Liu Y, Bjoras M, Klungland A, et al. OGG1 initiates age-dependent CAG trinucleotide expansion in somatic cells. Nature. 2007;447:447–52. doi: 10.1038/nature05778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Budworth H, Harris FR, Williams P, Lee DY, et al. Suppression of Somatic Expansion Delays the Onset of Pathophysiology in a Mouse Model of Huntington’s Disease. PLoS Genet. 2015;11:e1005267. doi: 10.1371/journal.pgen.1005267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Goldfless SJ, Morag AS, Belisle KA, Sutera VA, Jr, et al. DNA repeat rearrangements mediated by DnaK-dependent replication fork repair. Mol Cell. 2006;21:595–604. doi: 10.1016/j.molcel.2006.01.025. [DOI] [PubMed] [Google Scholar]
- 67.Kuzminov A. Collapse and repair of replication forks in Escherichia coli. Mol Microbiol. 1995;16:373–84. doi: 10.1111/j.1365-2958.1995.tb02403.x. [DOI] [PubMed] [Google Scholar]
- 68.Wu CA, Zechner EL, Marians KJ. Coordinated leading- and lagging-strand synthesis at the Escherichia coli DNA replication fork. I. Multiple effectors act to modulate Okazaki fragment size. J Biol Chem. 1992;267:4030–44. [PubMed] [Google Scholar]
- 69.Kassai-Jager E, Ortutay C, Toth G, Vellai T, et al. Distribution and evolution of short tandem repeats in closely related bacterial genomes. Gene. 2008;410:18–25. doi: 10.1016/j.gene.2007.11.006. [DOI] [PubMed] [Google Scholar]
- 70.Cockram CA, Filatenkova M, Danos V, El Karoui M, et al. Quantitative genomic analysis of RecA protein binding during DNA double-strand break repair reveals RecBCD action in vivo. Proc Natl Acad Sci U S A. 2015;112:E4735–42. doi: 10.1073/pnas.1424269112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Malkova A, Naylor ML, Yamaguchi M, Ira G, et al. RAD51-dependent break-induced replication differs in kinetics and checkpoint responses from RAD51-mediated gene conversion. Mol Cell Biol. 2005;25:933–44. doi: 10.1128/MCB.25.3.933-944.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kuzminov A. Recombinational repair of DNA damage in Escherichia coli and bacteriophage lambda. Microbiol Mol Biol Rev. 1999;63:751–813. doi: 10.1128/mmbr.63.4.751-813.1999. table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Watson JD. Origin of concatemeric T7 DNA. Nat New Biol. 1972;239:197–201. doi: 10.1038/newbio239197a0. [DOI] [PubMed] [Google Scholar]
- 74.Olovnikov AM. A theory of marginotomy. The incomplete copying of template margin in enzymic synthesis of polynucleotides and biological significance of the phenomenon. J Theor Biol. 1973;41:181–90. doi: 10.1016/0022-5193(73)90198-7. [DOI] [PubMed] [Google Scholar]
- 75.Soudet J, Jolivet P, Teixeira MT. Elucidation of the DNA end-replication problem in Saccharomyces cerevisiae. Mol Cell. 2014;53:954–64. doi: 10.1016/j.molcel.2014.02.030. [DOI] [PubMed] [Google Scholar]
- 76.Sfeir A, de Lange T. Removal of shelterin reveals the telomere end-protection problem. Science. 2012;336:593–7. doi: 10.1126/science.1218498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fulneckova J, Sevcikova T, Fajkus J, Lukesova A, et al. A broad phylogenetic survey unveils the diversity and evolution of telomeres in eukaryotes. Genome Biol Evol. 2013;5:468–83. doi: 10.1093/gbe/evt019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Melnikova L, Georgiev P. Enhancer of terminal gene conversion, a new mutation in Drosophila melanogaster that induces telomere elongation by gene conversion. Genetics. 2002;162:1301–12. doi: 10.1093/genetics/162.3.1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Biffi G, Tannahill D, McCafferty J, Balasubramanian S. Quantitative visualization of DNA G-quadruplex structures in human cells. Nat Chem. 2013;5:182–6. doi: 10.1038/nchem.1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Doksani Y, Wu JY, de Lange T, Zhuang X. Super-resolution fluorescence imaging of telomeres reveals TRF2-dependent T-loop formation. Cell. 2013;155:345–56. doi: 10.1016/j.cell.2013.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bodnar AG, Ouellette M, Frolkis M, Holt SE, et al. Extension of life-span by introduction of telomerase into normal human cells. Science. 1998;279:349–52. doi: 10.1126/science.279.5349.349. [DOI] [PubMed] [Google Scholar]
- 82.Shay JW, Bacchetti S. A survey of telomerase activity in human cancer. Eur J Cancer. 1997;33:787–91. doi: 10.1016/S0959-8049(97)00062-2. [DOI] [PubMed] [Google Scholar]
- 83.Barthel FP, Wei W, Tang M, Martinez-Ledesma E, et al. Systematic analysis of telomere length and somatic alterations in 31 cancer types. Nat Genet. 2017;49:349–57. doi: 10.1038/ng.3781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cesare AJ, Reddel RR. Alternative lengthening of telomeres: models, mechanisms and implications. Nat Rev Genet. 2010;11:319–30. doi: 10.1038/nrg2763. [DOI] [PubMed] [Google Scholar]
- 85.Tarsounas M, West SC. Recombination at mammalian telomeres: an alternative mechanism for telomere protection and elongation. Cell Cycle. 2005;4:672–4. doi: 10.4161/cc.4.5.1689. [DOI] [PubMed] [Google Scholar]
- 86.Roumelioti FM, Sotiriou SK, Katsini V, Chiourea M, et al. Alternative lengthening of human telomeres is a conservative DNA replication process with features of break-induced replication. EMBO Rep. 2016;17:1731–7. doi: 10.15252/embr.201643169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zeng S, Xiang T, Pandita TK, Gonzalez-Suarez I, et al. Telomere recombination requires the MUS81 endonuclease. Nat Cell Biol. 2009;11:616–23. doi: 10.1038/ncb1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Vasianovich Y, Harrington LA, Makovets S. Break-induced replication requires DNA damage-induced phosphorylation of Pif1 and leads to telomere lengthening. PLoS Genet. 2014;10:e1004679. doi: 10.1371/journal.pgen.1004679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lundblad V, Blackburn EH. An alternative pathway for yeast telomere maintenance rescues est1- senescence. Cell. 1993;73:347–60. doi: 10.1016/0092-8674(93)90234-h. [DOI] [PubMed] [Google Scholar]
- 90.Dewar JM, Lydall D. Pif1- and Exo1-dependent nucleases coordinate checkpoint activation following telomere uncapping. EMBO J. 2010;29:4020–34. doi: 10.1038/emboj.2010.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Dilley RL, Verma P, Cho NW, Winters HD, et al. Break-induced telomere synthesis underlies alternative telomere maintenance. Nature. 2016;539:54–8. doi: 10.1038/nature20099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Higa M, Fujita M, Yoshida K. DNA Replication Origins and Fork Progression at Mammalian Telomeres. Genes (Basel) 2017;8 doi: 10.3390/genes8040112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Anand RP, Shah KA, Niu H, Sung P, et al. Overcoming natural replication barriers: differential helicase requirements. Nucleic Acids Res. 2012;40:1091–105. doi: 10.1093/nar/gkr836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Maestroni L, Matmati S, Coulon S. Solving the Telomere Replication Problem. Genes (Basel) 2017;8 doi: 10.3390/genes8020055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cox KE, Marechal A, Flynn RL. SMARCAL1 Resolves Replication Stress at ALT Telomeres. Cell reports. 2016;14:1032–40. doi: 10.1016/j.celrep.2016.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Madireddy A, Kosiyatrakul ST, Boisvert RA, Herrera-Moyano E, et al. FANCD2 Facilitates Replication through Common Fragile Sites. Mol Cell. 2016;64:388–404. doi: 10.1016/j.molcel.2016.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Drosopoulos WC, Kosiyatrakul ST, Schildkraut CL. BLM helicase facilitates telomere replication during leading strand synthesis of telomeres. J Cell Biol. 2015;210:191–208. doi: 10.1083/jcb.201410061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Fallet E, Jolivet P, Soudet J, Lisby M, et al. Length-dependent processing of telomeres in the absence of telomerase. Nucleic Acids Res. 2014;42:3648–65. doi: 10.1093/nar/gkt1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Aksenova AY, Han G, Shishkin AA, Volkov KV, et al. Expansion of Interstitial Telomeric Sequences in Yeast. Cell reports. 2015;13:1545–51. doi: 10.1016/j.celrep.2015.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Moser BA, Subramanian L, Chang YT, Noguchi C, et al. Differential arrival of leading and lagging strand DNA polymerases at fission yeast telomeres. EMBO J. 2009;28:810–20. doi: 10.1038/emboj.2009.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Saharia A, Guittat L, Crocker S, Lim A, et al. Flap endonuclease 1 contributes to telomere stability. Current biology: CB. 2008;18:496–500. doi: 10.1016/j.cub.2008.02.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Parenteau J, Wellinger RJ. Accumulation of single-stranded DNA and destabilization of telomeric repeats in yeast mutant strains carrying a deletion of RAD27. Mol Cell Biol. 1999;19:4143–52. doi: 10.1128/mcb.19.6.4143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Allshire RC, Karpen GH. Epigenetic regulation of centromeric chromatin: old dogs, new tricks? Nat Rev Genet. 2008;9:923–37. doi: 10.1038/nrg2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Holubcova Z, Blayney M, Elder K, Schuh M. Human oocytes. Error-prone chromosome-mediated spindle assembly favors chromosome segregation defects in human oocytes. Science. 2015;348:1143–7. doi: 10.1126/science.aaa9529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zhang W, Mao JH, Zhu W, Jain AK, et al. Centromere and kinetochore gene misexpression predicts cancer patient survival and response to radiotherapy and chemotherapy. Nat Commun. 2016;7:12619. doi: 10.1038/ncomms12619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Melters DP, Bradnam KR, Young HA, Telis N, et al. Comparative analysis of tandem repeats from hundreds of species reveals unique insights into centromere evolution. Genome Biol. 2013;14:R10. doi: 10.1186/gb-2013-14-1-r10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Shepelev VA, Uralsky LI, Alexandrov AA, Yurov YB, et al. Annotation of suprachromosomal families reveals uncommon types of alpha satellite organization in pericentromeric regions of hg38 human genome assembly. Genom Data. 2015;5:139–46. doi: 10.1016/j.gdata.2015.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Willard HF. Chromosome-specific organization of human alpha satellite DNA. Am J Hum Genet. 1985;37:524–32. [PMC free article] [PubMed] [Google Scholar]
- 109.Plohl M, Mestrovic N, Mravinac B. Centromere identity from the DNA point of view. Chromosoma. 2014;123:313–25. doi: 10.1007/s00412-014-0462-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Frommer M, Prosser J, Tkachuk D, Reisner AH, et al. Simple repeated sequences in human satellite DNA. Nucleic Acids Res. 1982;10:547–63. doi: 10.1093/nar/10.2.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sato N, Amino T, Kobayashi K, Asakawa S, et al. Spinocerebellar ataxia type 31 is associated with “inserted” penta-nucleotide repeats containing (TGGAA)n. Am J Hum Genet. 2009;85:544–57. doi: 10.1016/j.ajhg.2009.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Li PC, Petreaca RC, Jensen A, Yuan JP, et al. Replication fork stability is essential for the maintenance of centromere integrity in the absence of heterochromatin. Cell reports. 2013;3:638–45. doi: 10.1016/j.celrep.2013.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Simi S, Simili M, Bonatti S, Campagna M, et al. Fragile sites at the centromere of Chinese hamster chromosomes: a possible mechanism of chromosome loss. Mutat Res. 1998;397:239–46. doi: 10.1016/s0027-5107(97)00219-4. [DOI] [PubMed] [Google Scholar]
- 114.Crosetto N, Mitra A, Silva MJ, Bienko M, et al. Nucleotide-resolution DNA double-strand break mapping by next-generation sequencing. Nat Methods. 2013;10:361–5. doi: 10.1038/nmeth.2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Davila-Rodriguez MI, Cortes Gutierrez EI, Cerda Flores RM, Pita M, et al. Constitutive heterochromatin polymorphisms in human chromosomes identified by whole comparative genomic hybridization. Eur J Histochem. 2011;55:e28. doi: 10.4081/ejh.2011.e28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Warren AC, Bowcock AM, Farrer LA, Antonarakis SE. An alpha satellite DNA polymorphism specific for the centromeric region of chromosome 13. Genomics. 1990;7:110–4. doi: 10.1016/0888-7543(90)90525-y. [DOI] [PubMed] [Google Scholar]
- 117.Mahtani MM, Willard HF. Pulsed-field gel analysis of alpha-satellite DNA at the human X chromosome centromere: high-frequency polymorphisms and array size estimate. Genomics. 1990;7:607–13. doi: 10.1016/0888-7543(90)90206-a. [DOI] [PubMed] [Google Scholar]
- 118.Forsburg SL, Shen KF. Centromere Stability: The Replication Connection. Genes (Basel) 2017;8 doi: 10.3390/genes8010037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Jaco I, Canela A, Vera E, Blasco MA. Centromere mitotic recombination in mammalian cells. J Cell Biol. 2008;181:885–92. doi: 10.1083/jcb.200803042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Giunta S, Funabiki H. Integrity of the human centromere DNA repeats is protected by CENP-A, CENP-C, and CENP-T. Proc Natl Acad Sci U S A. 2017;114:1928–33. doi: 10.1073/pnas.1615133114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Aze A, Sannino V, Soffientini P, Bachi A, et al. Centromeric DNA replication reconstitution reveals DNA loops and ATR checkpoint suppression. Nat Cell Biol. 2016;18:684–91. doi: 10.1038/ncb3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Bryant HE, Petermann E, Schultz N, Jemth AS, et al. PARP is activated at stalled forks to mediate Mre11-dependent replication restart and recombination. EMBO J. 2009;28:2601–15. doi: 10.1038/emboj.2009.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Dungey FA, Loser DA, Chalmers AJ. Replication-dependent radiosensitization of human glioma cells by inhibition of poly(ADP-Ribose) polymerase: mechanisms and therapeutic potential. Int J Radiat Oncol Biol Phys. 2008;72:1188–97. doi: 10.1016/j.ijrobp.2008.07.031. [DOI] [PubMed] [Google Scholar]
- 124.Morrow DM, Connelly C, Hieter P. “Break copy” duplication: a model for chromosome fragment formation in Saccharomyces cerevisiae. Genetics. 1997;147:371–82. doi: 10.1093/genetics/147.2.371. [DOI] [PMC free article] [PubMed] [Google Scholar]