

Abstract
Background
Werner syndrome is a progeroid disorder characterized by premature age-related phenotypes. Although it is well established that autosomal recessive mutations in the WRN gene is responsible for Werner syndrome, the molecular alterations that lead to disease phenotype remain still unidentified.
Results
To address whether epigenetic changes can be associated with Werner syndrome phenotype, we analysed genome-wide DNA methylation profile using the Infinium MethylationEPIC BeadChip in the whole blood from three patients affected by Werner syndrome compared with three age- and sex-matched healthy controls. Hypermethylated probes were enriched in glycosphingolipid biosynthesis, FoxO signalling and insulin signalling pathways, while hypomethylated probes were enriched in PI3K-Akt signalling and focal adhesion pathways. Twenty-two out of 47 of the differentially methylated genes belonging to the enriched pathways resulted differentially expressed in a publicly available dataset on Werner syndrome fibroblasts. Interestingly, differentially methylated regions identified CERS1 and CERS3, two members of the ceramide synthase family. Moreover, we found differentially methylated probes within ITGA9 and ADAM12 genes, whose methylation is altered in systemic sclerosis, and within the PRDM8 gene, whose methylation is affected in dyskeratosis congenita and Down syndrome.
Conclusions
DNA methylation changes in the peripheral blood from Werner syndrome patients provide new insight in the pathogenesis of the disease, highlighting in some cases a functional correlation of gene expression and methylation status.
Electronic supplementary material
The online version of this article (doi:10.1186/s13148-017-0389-4) contains supplementary material, which is available to authorized users.
Keywords: Werner syndrome, DNA methylation, Systemic sclerosis
Introduction
Werner syndrome (WS) is a rare adult premature ageing disease. Individuals affected by WS generally have a normal development until the third decade of life, when premature ageing phenotypes and symptoms begin to manifest including premature greying or loss of hair, bird-like faces, cataracts, sclerodermiform skin atrophy [1] and skin ulcers [2]. Because of apparent similar skin changes, WS is often misdiagnosed as systemic sclerosis (SSc) [3].
Particularly, WS represents an important part of the differential diagnosis in patients who present with scleroderma-like skin changes, as they share with SSc patients the main histological changes of skin. These include replacement of the subcutaneous tissue by hyalinized connective, hyalinized connective tissue in the lower dermis and hyalinization and formation of aneurysms in the dermal blood vessels [4, 5]. Otherwise, progeria, premature cataract, type 2 diabetes mellitus, sensorineural hearing loss, premature atherosclerosis and dyslipidemia in WS besides the absence of anti-scleroderma antibodies may be helpful for the differential diagnosis of WS [6]. WS is associated to an autosomal recessive mutation of Werner syndrome gene (WRN) [7, 8]; up today, more than 50 different disease-causing mutations in the WRN gene have been identified [8]. The WRN gene encodes a 180-kDa nuclear protein member of the RecQ subfamily of DNA and RNA helicases [8, 9] with an intrinsic 3′ to 5′ DNA helicase activity [10]. DNA helicases are involved in many aspects of DNA metabolism including transcription, replication and recombination [9]. Given its role as a 3′ to 5′ exonuclease and based on interactions between this protein and Ku70/80 heterodimer in DNA end processing, WRN plays a critical role in the repair of DNA double-strand breaks [11]. Additionally, recent studies suggest a role for WRN in maintaining DNA telomere stability [10]. It is well known that defects in telomere structure and/or function may have a strong impact on human health, leading to premature ageing and a variety of diseases [12, 13]. Overall, WRN protein is crucial in maintaining genome structure and integrity, and accordingly, WS patients exhibit early age-associated biomarkers like DNA damage accumulation and chromosomal instability [14]. Despite these evidences, the molecular bases of WS phenotype are still largely unknown.
Alterations in epigenetic patterns, in particular in DNA methylation profiles, could contribute to WS phenotype. DNA methylation, which consists in the addition of a methyl group to the cytosine in a CpG dinucleotide, is a key mechanism in development and differentiation, and profound changes in DNA methylation patterns occur during ageing and age-related diseases [15, 16]. At present, few studies assessed DNA methylation changes in WS. Heyn and colleagues evaluated genome-wide DNA methylation in lymphoblastoid cell lines (LCLs) derived from Epstein-Barr virus (EBV)-immortalized B cells of four WS patients and three healthy controls [7], observing profound DNA methylation changes in WS. More recently, a WRN promoter was found hypermethylated in naïve B lymphocytes and lymphoblastoid cell lines from two brothers carrying a novel homozygous WRN mutation. Finally, Maierhofer et al. applied Horvath’s epigenetic clock to a dataset including the whole blood from WS patients and age-matched controls, demonstrating that the disease is associated with an increase in epigenetic age [17].
In the present study, we analysed genome-wide DNA methylation in the peripheral whole blood from three WS patients and their age- and sex-matched controls using the Infinium MethylationEPIC BeadChip. Our results indicate that specific genome-wide alterations occur in WS, which partially overlap those that occur in diffuse and limited SSc [18].
Methods
Samples
Subjects were enrolled in the study “Early diagnosis of scleroderma and identification of predictor factors of disease development”. The study was approved by the Ethic Committee of ASL RM/B, and all participants signed the informed consent forms. WS was diagnosed according to clinical appearance and genetic testing. Two patients were siblings and carried the mutation c.2630+1G>A in the WRN coding sequence; the third patient carried the homozygous g.nt 77177 a>g mutation (GenBank acc. no. AY44237) in the WRN gene. They suffer of skin ulcers of suspected SSc derivation as they showed typical SSc skin signs being bound tightly to the underlying tissues and around joints causing an inability to pinch or lift the skin up (Fig. 1a) and calcinosis cutis, a type of calcinosis which characterized scleroderma skin ulcers [19–21] (Fig. 1b). Serum was collected, and anti-nuclear antibodies (ANAs) were analysed using indirect immunofluorescence (IIF) assays on ANA-HEp-2 cells (A. Menarini Diagnostics) according to the manufacturer’s protocol. Anti-extractable nuclear antigen (ENA) antibodies [anti-Sjögren’s syndrome A (SS-A), anti-Sjögren’s syndrome B (SS-B), anti-Smith (SM), anti-ribonucleoprotein (RNP), anti-topoisomerase I (SCL-70) and anti-histidyl-tRNA synthetase (JO-1)] were also dosed using chemiluminescence assay (Zenit RA autoimmunity, A. Menarini Diagnostics) according to the manufacturer’s protocol. These autoantibodies, considered as SSc diagnostic markers, were undetectable in selected patients (data not shown). SSc patients were also subjected to nailfold video capillaroscopy (NVC). Capillaroscopic alterations and ectasias compatible with diagnosis of acrocyanosis were detected in all three patients (Fig. 1c). Peripheral blood was collected from the three patients.
Fig. 1.
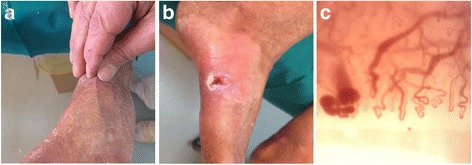
SSc typical skin signs in WS patients. a Skin unable to be lifted in pliers and/or pinched. b Cutaneous ulcer with calcinosis. c Capillaroscopic alterations and ectasias
Genome-wide DNA methylation analysis
Genomic DNA was extracted from whole blood using the Duplica® Blood DNA Kit (EuroClone) on Duplica® Prep Platform (EuroClone). DNA was bisulfite-converted using the EZ DNA Methylation-Gold Kit (Zymo Research) and analysed using the Infinium HumanMethylationEPIC BeadChip (Illumina) following the manufacturer’s instructions. Signal intensity files were extracted using the minfi Bioconductor package. Normalization was performed using the functional normalization function implemented in minfi. DNA methylation data are available at the NCBI Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE100825.
DNA methylation differences between WS patients and healthy controls were assessed using two different approaches: (1) we used analysis of variance (ANOVA) to find out whether there was any statistically significant difference between WS patients and healthy controls when we consider differentially methylated positions (DMPs), that is single-CpG sites whose methylation differs between the groups under investigation. DMPs with a non-adjusted P value < 0.001 were retained as significantly differentially methylated. (2) We applied multivariate analysis of variance (MANOVA) on sliding windows including three adjacent CpG sites as described in Bacalini et al. [15] in order to identify statistically significantly differentially methylated regions (DMRs), that is regions in which multiple adjacent CpG sites differ between the two groups under investigation. This approach was applied only to the CpG probes mapping in CpG-rich regions (CpG islands, shores and shelves) associated to a gene. DMRs with a non-adjusted P value < 0.001 and in which at least two adjacent CpG sites had a minimum absolute DNA methylation difference of 0.3 were retained as significant. Although a 0.15 threshold for mean DNA methylation difference is often suggested [22], here we preferred to use a more stringent criterion, given the small sample size, in order to provide a short, but more likely reproducible, list of DMRs.
Infinium450k datasets GSE42865 and GSE75310 were downloaded from the GEO database. GSE42865 includes DNA methylation data from the LCL of three Hutchinson-Gilford progeria syndrome patients, four WS patients and three healthy patients, together with peripheral blood mononuclear cells from three healthy subjects and naïve B cells from three healthy subjects. We compared the four WS patients and the three LCL controls by ANOVA. GSE75310 includes DNA methylation data from four dyskeratosis congenita (DKC) patients. To investigate DNA methylation differences in DKC, we performed ANOVA using as controls the same four GEO samples (GSM796678, GSM796674, GSM796667 and GSM796671) used in the original paper [23].
Pathway enrichment
Pathway analysis of differentially methylated genes and gene clusters was performed with the publicly available tool Enrichr (http://amp.pharm.mssm.edu/Enrichr) that provides access to various gene set libraries [24, 25]. Enrichr currently contains annotated gene sets from 102 gene set libraries organized in eight categories. Details of the gene set libraries in Enrichr can be found in previously published studies [24, 25]. Enrichment analysis checks whether the input set of genes significantly overlaps with annotated gene sets. Each gene set within the Enrichr database is associated with a functional term or an enrichment term such as a pathway, cell line or disease. The output of Enrichr is ranked lists of terms, one list for each gene set library. The most highly ranked enrichment terms for the user’s input gene list provide knowledge about the input list. We used the publicly Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway gene sets to identify the relatively enriched biological pathways and then extracted the enriched gene sets for each pathway. We considered pathways as enriched if their P value was lower than 0.05 and ranked them using the combined score (CS). CS is a multiplication of the P value computed using Fisher’s exact test with the z-score of the deviation from the expected rank. In particular, c is equal to log (p) × z where c is the combined score, p is the P value computed using Fisher’s exact test and z is the z-score computed to assess the deviation from the expected rank.
Differential gene expression analysis
In order to perform differential expression analysis, we used shinyGEO (http://gdancik.github.io/shinyGEO/) [26], a Web-based tool that allows a user to download the expression and sample data from a GEO dataset, select a gene of interest and perform a survival or differential expression analysis using the available data [27]. For both analyses, shinyGEO is able to generate R code, ensuring that all analyses are reproducible. The tool is developed using shiny, a Web-based application framework for R, a language for statistical computing and graphics. Fold change (FC) and P value are calculated by two-sample Student’s t test for differential expression.
Results
We used the Infinium MethylationEPIC BeadChip to compare whole blood genome-wide DNA methylation profiles between three WS patients and three age- and sex-matched controls (CTRs). We conducted principal component analysis to identify major traits of variation between samples (Additional file 1: Figure S1). No clear separation between WS patients and CTRs was observed along the first component (which explained 32% of the variance) or the second component (which explained 51% of the remaining variance).
ANOVA identified 1125 DMPs that distinguished WS patients from CTRs (non-adjusted P value < 0.001) (Fig. 2a, Additional file 2, Table 1). Of these, 511 probes (mapping in 382 genic regions) were hypermethylated and 614 probes (mapping in 416 genic regions) were hypomethylated in WS patients compared to CTRs. Several DMPs showed large DNA methylation differences between the two groups, with 87/511 hypermethylated and 110/614 hypomethylated probes having mean methylation differences larger than 0.15 [22]. The volcano plot in Fig. 2a shows many CpG sites with non-adjusted P values lower than 0.001 but having low DNA methylation differences between the groups. This behaviour might be related to the small number of analysed WS samples.
Fig. 2.
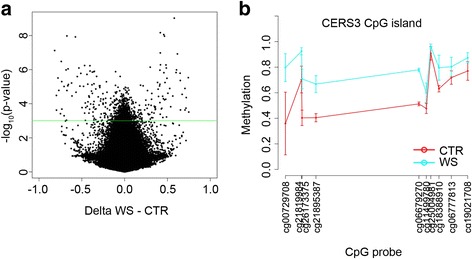
DNA methylation alterations in Werner syndrome. a Volcano plot of DMPs between WS patients and CTRs. The difference between mean DNA methylation values in WS patients and in CTRs is plotted on the x-axis, while the non-adjusted P value for ANOVA between the two groups is on the y-axis (− 1 × log10 scale). The green line corresponds to a non-adjusted P value of 0.001. b DNA methylation profile of the CpG island located in the CERS3 gene
Table 1.
List of the top 20 DMPs
| Probe | CHR | MAPINFO | Gene | CpG island name | Relation with respect to the CpG island | Non-adjusted P value |
|---|---|---|---|---|---|---|
| cg15294279 | 3 | 174,842,010 | NAALADL2 | – | – | 9.48E−10 |
| cg26845082 | 3 | 13,555,664 | – | – | – | 6.75E−09 |
| cg23432430 | 12 | 125,538,377 | – | chr12:125534060-125534527 | S_Shelf | 8.75E−09 |
| cg00597723 | 5 | 158,691,793 | UBLCP1 | chr5:158690013-158690541 | S_Shore | 1.05E−08 |
| cg13956086 | 7 | 2,434,521 | – | – | – | 1.08E−08 |
| cg16995742 | 2 | 237,992,612 | COPS8 | chr2:237994004-237994876 | N_Shore | 1.19E−08 |
| cg17779733 | 22 | 49,589,242 | – | – | – | 7.55E−08 |
| cg06052372 | 16 | 83,967,808 | – | – | – | 1.04E−07 |
| cg23928292 | 12 | 21,815,474 | – | – | – | 1.19E−07 |
| cg10360725 | 8 | 144,139,316 | – | – | – | 3.31E−07 |
| cg14782559 | 6 | 33,131,893 | COL11A2 | chr6:33129291-33129718 | S_Shelf | 5.19E−07 |
| cg18673341 | 7 | 22,481,962 | STEAP1B | – | – | 5.28E−07 |
| cg26822175 | 22 | 27,018,010 | CRYBA4 | – | – | 5.71E−07 |
| cg08161337 | 22 | 45,814,116 | RIBC2 | – | – | 5.86E−07 |
| cg22664298 | 5 | 128,795,827 | ADAMTS19 | chr5:128795503-128797417 | Island | 7.16E−07 |
| cg13885829 | 1 | 17,482,041 | – | – | – | 7.98E−07 |
| cg07584620 | 1 | 2,265,881 | MORN1 | chr1:2266007-2266432 | N_Shore | 8.00E−07 |
| cg20757478 | 6 | 31,012,262 | – | – | – | 8.99E−07 |
| cg17628377 | 5 | 180,121,337 | – | – | – | 1.03E−06 |
| cg15865722 | 11 | 68,860,657 | – | – | – | 1.03E−06 |
Next, we performed a gene set enrichment analysis in order to examine in silico the biological functions of DMPs in WS patients compared to CTR samples. To this aim, we searched for the overrepresented biological pathways associated with the differentially methylated genes, using as input for the Enrichr tool the list of genes associated with DMPs. Enrichr’s combined score, a combination of the P value and z-score, was considered in order to prioritize enriched pathways (see “Methods” for more details). As detailed in the Enrichr manuscript [24], the combined score provides a compromise between both methods (P value and z-score) and, in several benchmarks, has reported the best ranking when compared with the other scoring schemes.
We took into account the most highly ranked enriched pathways according to the combined score while we considered significantly overrepresented pathways, those showing a P value < 0.05. Results for enriched biological pathways are shown in Table 2. The hypermethylated genes in KEGG pathway enrichment analysis results were associated with the enriched pathways with P value < 0.05, including “glycosphingolipid biosynthesis (HSA-00603)”, “FoxO signalling pathway (HSA-04068)” and “insulin signalling pathway (HSA-04910)”. The hypomethylated genes were associated with the enriched pathways with P value < 0.05, including “PI3K-Akt signalling (HSA-04151)” and “focal adhesion (HSA-04510)”.
Table 2.
Gene set enrichment analysis details are reported for DPMs (hypo- and hypermethylated genes) and DMRs
| Enriched pathways | Pathway ID | Overlap | P value | Combined score | Overlapping genes | |
|---|---|---|---|---|---|---|
| Hypermethylated probes | Glycosphingolipid biosynthesis | HSA-00603 | 3/14 | 0.001 | 2.29 | ST3GAL1, GBGT1, ST3GAL2 |
| FoxO signal pathway | HSA-04068 | 8/133 | 0.003 | 2.60 | MAPK10, USP7, AKT2, STAT3, PTEN, FOXO3, SKP2, GABARAP | |
| Insulin signalling pathway | HSA-04910 | 8/139 | 0.004 | 2.49 | MAPK10, PTPN1, SHC2, AKT2, PRKAK1B, FASN, TSC2, CRKL | |
| Hypomethylated probes | PI3K-Akt signalling | HSA_04151 | 16/341 | 0.006 | 3.46 | CSF-1R, CDKN1B, TNXB, VWF, LAMA1, FLT4, THBS1, PTK2, LPAR5, PPP2R2B, PPP2R2D, MAPK1, COL6A6, FGFR1, BCL2L1, ITGA9 |
| Focal adhesion | HSA_04510 | 12/202 | 0.01 | 3.26 | MAPK10, TNXB, VWF, ROCK2, LAMA1, FLT4, PXN, MAPK1, COL6A6, THBS1, PTK2, ITGA9 | |
| DMRs | Sphingolipid metabolism | HSA_00600 | 2/120 | 0.003 | 4.54 | CERS3, CERS1 |
| Sphingolipid signalling pathway | HSA_047071 | 1/47 | 0.001 | 5.36 | CERS3 |
Overlap indicates the number of hits from the differentially methylated gene sets compared to the KEGG gene set library, while “overlapping genes” column contains names of these hits. Differentially expressed genes between normal and WS fibroblasts in Cheung HH dataset [30] are reported. Enriched pathways were selected based on the P value. Combined score is a multiplication of the P value computed using Fisher’s exact test with the z-score of the deviation from the expected rank (see “Methods” for details)
Finally, we also used the pipeline reported by Bacalini et al. [15] (see “Methods” for details) to identify differentially methylated regions (DMRs) between WS patients and CTRs. We found 27 DMRs with a non-adjusted P value < 0.001 (Additional file 3, Additional file 4, Table 3), 20 of which were clearly hypermethylated in WS patients compared to CTRs. Even these 27 DMRs (corresponding to 37 genes) were employed in the Enrichr pathway analysis approach, revealing “sphingolipid metabolism (HSA-00600)” and “sphingolipid signalling pathway (HSA-04071)” among the most enriched pathways with P value < 0.05. Both pathways contain CERS3 and CERS1 genes, which are two members of the ceramide synthase family.
Table 3.
List of the 27 DMRs
| CHR | CpG island name | Relation with respect to the CpG island | Gene | Non-adjusted P value |
|---|---|---|---|---|
| 18 | chr18:30349690-30352302 | S_Shore | KLHL14 | 1.70E−05 |
| 12 | chr12:50297580-50297988 | S_Shore | FAIM2 | 0.00013152 |
| 6 | chr6:33396050-33396296 | S_Shelf | SYNGAP1 | 0.000168391 |
| 5 | chr5:110074605-110075223 | N_Shore | SLC25A46 | 0.000215085 |
| 22 | chr22:46366726-46368726 | S_Shore | WNT7B | 0.000233192 |
| 17 | chr17:46620367-46621373 | S_Shore | HOXB2; HOXB-AS1 | 0.000244681 |
| 10 | chr10:5930914-5932389 | N_Shore | ANKRD16; FBXO18 | 0.000261161 |
| 3 | chr3:9811466-9811736 | S_Shore | CAMK1 | 0.000281325 |
| 2 | chr2:129075197-129077639 | Island | HS6ST1 | 0.000292046 |
| 15 | chr15:71145995-71146820 | N_Shore | LARP6; LRRC49 | 0.000302033 |
| 15 | chr15:101084428-101085178 | Island | CERS3 | 0.000341282 |
| 6 | chr6:143999154-143999667 | N_Shore | PHACTR2 | 0.000393635 |
| 6 | chr6:166755812-166756510 | S_Shore | LOC100289495; SFT2D1 | 0.000423645 |
| 4 | chr4:146540053-146540656 | N_Shore | MMAA | 0.000441415 |
| 17 | chr17:20059028-20060060 | N_Shore | SPECC1 | 0.000444431 |
| 6 | chr6:31939730-31940559 | S_Shore | DXO; STK19 | 0.000488824 |
| 15 | chr15:99791328-99792042 | N_Shore | TTC23; LRRC28 | 0.0004949 |
| 19 | chr19:19006031-19007546 | S_Shore | GDF1; CERS1 | 0.000509929 |
| 22 | chr22:20134462-20134705 | S_Shelf | LOC388849 | 0.000585452 |
| 19 | chr19:19624954-19627258 | Island | NDUFA13; TSSK6 | 0.000650774 |
| 1 | chr1:160990718-160991225 | N_Shore | F11R | 0.000652612 |
| 6 | chr6:33266302-33267582 | N_Shore | RGL2 | 0.000759464 |
| 8 | chr8:117886284-117887319 | S_Shore | RAD21-AS1; RAD21; MIR3610 | 0.000773036 |
| 19 | chr19:35491151-35492020 | N_Shore | GRAMD1A | 0.000798561 |
| 17 | chr17:4692249-4693977 | N_Shore | GLTPD2 | 0.000853965 |
| 4 | chr4:9783035-9784960 | Island | DRD5 | 0.000911924 |
| 4 | chr4:1396291-1401730 | Island | NKX1-1 | 0.000928038 |
To assess if the epigenetic pattern that we described in our WS cohort was reproducible in other WS DNA methylation datasets, we compared our results with those available from a publicly available dataset (GEO accession ID: GSE42865). GSE42865 includes DNA methylation data from LCL from four WS patients and three CTRs, measured using the previous version of the microarray, the Infinium450k BeadChip [7]. Of the 1125 DMPs identified in our analysis, 581 are included in the Infinium450k design. Of these, only two probes were differentially methylated between LCLs of WS patients and CTRs with a comparable statistical threshold (non-adjusted P value < 0.001), while 30 probes were found differentially methylated, considering a less stringent threshold (non-adjusted P value < 0.05) (Additional file 2). To investigate the reason of this low reproducibility of the data, we focused on the DMPs mapping in the genes resulting from the pathway enrichment analysis that are present in both the microarray versions. We included in this analysis also the probes mapping in CERS1 and CERS3 DMRs. As shown in Additional file 5, DNA methylation in LCL showed a larger variation in WS and/or CTR subjects, possibly as a consequence of the immortalization process [28]. Accordingly, DNA methylation differences were observed between LCL and naïve B cells (Additional file 5). It is therefore plausible that low overlapping between the GSE42865 dataset and ours is related to the different sources of genomic DNA (LCL vs whole blood). Despite these differences, we noted that several CpG sites showed the same trend in DNA methylation changes between WS patients and CTRs, in particular within the CERS3 DMR and in VWF and FOXO3 genes.
Furthermore, we compared WS-related epigenetic changes with those occurring in DKC, a premature ageing disease associated to impaired telomere maintenance. We exploited a publicly available Infinium450k dataset (GEO accession ID: GSE75310) performed on the whole blood from four DKC subjects [23]. Of the 581 WS-DMPs included in the Infinium450k design, 14 were differentially methylated in DKC with a comparable threshold (non-adjusted P value < 0.001), but only three of them showed a concordant DNA methylation change in the two diseases (Additional file 2). Despite this, we noted that the list of WS-DMPs included three CpG sites (cg27111250, cg10129063 and cg27639662) mapping in the S_Shore of the chr4:81109887-81110460 CpG island in the PR domain containing eight (PRDM8) genes and hypermethylated in WS patients compared to CTRs. This is of particular interest, as PRDM8 is hypermethylated in DKC, aplastic anaemia (AA) [23] and Down syndrome [29], although in a different region (spanning from the CpG island chr4:81118137-81118603 to the CpG island chr4:81119095-81119391).
Unfortunately, RNA was not available from the samples included in our cohort and it was not possible to assess the expression of the genes identified as differentially methylated. To overcome this limitation, we exploited the GEO database in order to get more insight into the relationship between DNA methylation changes and WS features. We selected the genes emerged from the pathway enrichment analysis, and we checked their expression in a dataset including ten WS and ten CTR skin fibroblasts (GEO accession ID: GSE48761) [30]. Enrichment analysis checks whether an input set of genes significantly overlaps with annotated gene sets. We found 47 genes overlapping with the annotated gene set from KEGG pathways (see the overlap column in Table 1), and 22 out of 47 of these genes were differentially expressed between WR patients and CTRs (P value < 0.05), as reported in Fig. 3 and Additional file 6: Figure S2. The relationship between DNA methylation and expression changes is reported in Additional file 7. Focusing on the DMRs, HS6ST1 was hypermethylated in the whole blood from WS patients and downregulated in WS fibroblasts, while both CERS1 and CERS3 were hypermethylated in the whole blood from WS patients and upregulated in WS fibroblasts. Interestingly, we also noted that PRDM8 gene expression is downregulated in WS fibroblasts (data not shown), like it was previously reported in the whole blood from DKC patients [30]. Although further investigation is needed, possibly on the same biological specimen from the same subject, these data underline an association between deregulation of DNA methylation and gene expression in healthy donors and patients.
Fig. 3.
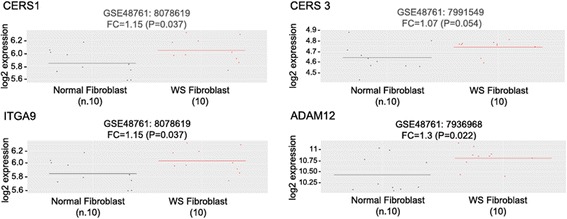
Differential expression of CERS1, CERS3, ITGA9 and ADAM12 genes based on the publicly available dataset on WS fibroblast analyses
Discussion
In this study, we analysed genome-wide DNA methylation in the peripheral blood from three WS subjects and three age- and sex-matched controls, identifying 1125 DMPs and 27 DMRs. Pathway enrichment analysis on the list of hypermethylated and hypomethylated DMPs and on the list of DMR provided interesting hints on the epigenetic alterations in WS patients, possibly related with the disease phenotype. Bioactive sphingolipids have been suggested to play a role in ageing and cellular senescence, and in WS, lipid profile is often abnormal [31]. Moreover, glycosphingolipids are elevated during ageing in the mouse brain and liver as well as in human fibroblasts obtained from elderly individuals [32]. On the other hand, longevity is associated with genetic variation in insulin-FOXO pathways [33] and the signalling pathway connecting insulin and FoxO transcription factors provides the most compelling example for a conserved genetic pathway at the interface between ageing and cancer. FoxO proteins are a subgroup of the forkhead family of transcription factors, which regulate the expression of genes involved in several physiological events including apoptosis, cell cycle control, glucose metabolism, oxidative stress resistance and longevity [34, 35]. In response to insulin or growth factors, the protein is phosphorylated by Akt, downstream of PI3K. This constitutes a signal to export FoxO from the nucleus to the cytoplasm, thereby decreasing the expression of FoxO target genes [34, 36]. Dysregulation of the insulin/PI3K/Akt pathway is implicated in several human diseases including cancer, diabetes, cardiovascular diseases and neurological diseases [37, 38]. Insulin signalling pathway regulates ageing in many organisms, ranging from simple invertebrates to mammals, including human [36]. Notably, the enrichment of hypermethylated probes in insulin and FoxO signalling pathways is also consistent with the clinical and metabolic evidences that relate WS to disorders of lipid and liver function [39].
The hypomethylated probes were enriched in PI3K-Akt signalling (HSA-04151) and focal adhesion (HSA-04510) pathways. Even these results underline the importance of deregulated methylation positions we detected. In fact, the role of the PI3K/Akt/FoxO signalling in longevity appears to be well conserved across species [40] and senescence-related morphological alteration is one of the main features of the senescent phenotype and it is deeply dependent to changes of cellular structural determinants in terms of their levels and activities. These determinants included integrins, focal adhesion complexes and small Rho GTPases [41].
Importantly, the alteration of the above-mentioned pathways in WS was fully supported when we correlated gene sets detected as differentially methylated and a publicly available gene expression dataset on WS fibroblasts including ten WS and ten CTR [30]. Indeed, several of the differentially methylated genes belonging to the enriched pathways showed altered RNA expression in fibroblasts from WS patients compared to healthy controls (Fig. 3, Additional file 6: Figure S2).
The small overlap between WS and DKC DMPs suggests that the two progeroid diseases show distinct patterns of epigenetic alterations. However, the PRDM8 gene resulted hypermethylated in both WS and DKC, although in different CpG sites, suggesting that epigenetic remodelling in different premature ageing syndromes can converge on the same gene.
Interestingly, the gene ITGA9, which encodes for an alpha integrin, was previously reported to be hypomethylated and overexpressed in fibroblasts from patients with diffuse and limited SSc compared to fibroblasts from healthy controls [18]. Although in our dataset WS affected the methylation of a distinct CpG site in the body of ITGA9 (cg22345769, which is not assessed in the study by Altorok et al. [18], as included in the Infinium MethylationEPIC and not in the Infinium450k microarray), it is noteworthy that the methylation and mRNA level of the same gene is altered in both the diseases. In fact, in our bioinformatics analysis, we detected a fold change equal to 1.15 for the differential expression of ITGA9 (P value = 0.037). Furthermore, we found that another gene is implicated in cell-cell and cell-matrix interactions, ADAM12, that resulted hypomethylated both in WS and in diffuse and limited SSc [18] and overexpressed (fold change equal to 1.3, P value = 0.022) in WS with respect to CTR when we analysed the GSE48761 dataset [30] (Fig. 3). ADAM12 is involved in the process of fibrosis through enhancing TGF-β signalling pathway [42, 43]. Of note, the peripheral blood from WS patients shared the same hypomethylated CpG site in the body of ADAM12 with SSc fibroblasts.
Finally, CERS3 was shown to exhibit differentially methylated status between WS patients and CTRs. A distinct expression profile was also found between normal and WS fibroblasts in the GSE48761 dataset (Fig. 3) [30]. This gene regulates sphingolipid synthesis and is involved in the synthesis of ceramides with ultra-long-chain acyl moieties (ULC-Cers), playing an important role in creating a barrier for the epidermis from the environment [44]. A mutation in CERS3 is responsible of autosomal recessive congenital ichthyosis [45], and interestingly, scleroderma-like changes have been described in different clinical variants of ichthyosis [46]. All these data are of particular interest as WS has some features that are typical SSc signs like skin sclerosis and calcification and ankle ulcerations [47, 48]. Indeed, skin sclerosis is a hallmark of SSc and skin thickening represents the definitive diagnostic criterion of SSc in the vast majority of cases [19–21, 49]. Common to SSc subjects, WS patients show SSc-like skin involvement, calcinosis and skin ulcers; in particular, ulcers are present in 50% of WS patients and are generally the main cutaneous symptom [50, 51]. Anyway, the possibility for WS patients to be diagnosed as SSc was avoided as, based on 2013 ACR/EULAR classification criteria for SSc disease [52], serological analyses performed on subjects enrolled in the present study for anti-ANA and anti-ENA (anti-SS-A, anti-SS-B, anti-SM, anti-RNP, anti-SCL-70 and anti-JO-1) antibodies resulted negative. The presence of serum autoantibodies directed to multiple intracellular antigens is considered the serological hallmark of SSc [53].
The emerging connection in WS and SSc epigenetic alterations that we observed in this study could be related to the fact that the WS patients that we analysed for genome-wide DNA methylation were recruited in the framework of a study on SSc. Although we cannot exclude a priori this hypothesis, it is worth to note that (1) the CpG sites cg17287034 in the ADAM12 gene and cg06679270 in the CERS3 gene showed the same DNA methylation trend in the GSE42865 dataset and in ours (we cannot check ITGA9 because the probe is missing in the Infinium450k design) (Additional file 5) and (2) ITGA9, ADAM12 and CERS3 showed gene expression changes between WS and normal fibroblasts according to the GSE48761 dataset. As in both the publicly available studies WS patients were not selected on the basis of a SSc-like phenotype, we can suggest that SSc-related epigenetic changes are an intrinsic characteristic of WS. Although further studies should confirm our findings, they could be particularly interesting since recent studies highlight that another progeroid disease, the Hutchinson-Gilford progeria syndrome, is associated with SSc-like skin changes [54, 55] and that SSc-associated fibrosis is considered as an accelerated ageing phenotype [56].
Conclusions
In summary, we identified for the first time genome-wide DNA methylation changes in the peripheral blood from WS patients, providing important new insight in the pathogenesis of the diseases and emphasizing the potential role of DNA methylation changes in Werner disorder.
Additional files
Principal Component Analysis for the DNA methylation levels of the probes included in the InfiniumEPIC beadchip in WS and CTR. (TIFF 1825 kb)
List of DMPs between WS and CTR. (XLS 531 kb)
DNA methylation profiles of the 27 DMRs between WS and CTR. For each DMR, the title of the plot reports the name of the gene/genes in which the DMR maps, the not-adjusted P value of the ANOVA comparison between WS and CTR, and the length of the DMR in base pairs. Below the plot, the name of the CpG island and the position of the DMR respect to the CpG island are reported. (PDF 58 kb)
List of DMRs between WS and CTR. (XLS 60 kb)
Boxplots of DNA methylation values of the CpG probes in the genes belonging to the enriched pathways in naïve B cells, CTR LCL, WS LCL from the GSE42865 dataset and in CTR and WS whole blood samples assessed in this study. The brown dots correspond to the individual samples. For each CpG probe, the title of the plot reports the name of the CpG probe, the name of the gene/genes in which the probe maps and, if present, the name of the CpG island and the position of the probe respect to the CpG island. (PDF 41 kb)
Differential expression of several genes belonging to the enriched pathways resulted based on publicly available dataset on WS fibroblasts analyses. (TIFF 2607 kb)
Relationship between DNA methylation of DMPs and DMRs in the present dataset and RNA expression in the GSE48761 dataset. (XLS 68 kb)
Acknowledgements
Not applicable.
Funding
This study was supported by the European Union’s Seventh Framework Programme [grant number 602757 (“HUMAN: Health and the understanding of Metabolism, Aging and Nutrition”)] and by the European Union’s H2020 Project [grant number 634821 (“PROPAG-AGEING: The continuum between healthy ageing and idiopathic Parkinson Disease within a propagation perspective of inflammation and damage: the search for new diagnostic, prognostic and therapeutic targets”) and JPco-fuND (“ADAGE: Alzheimer’s Disease pathology within the ageing physiology”)].
Availability of data and materials
All data generated or analysed during this study are included in this published article [and its Additional files].
Abbreviations
- DKC
Dyskeratosis congenita
- DMPs
Differentially methylated positions
- DMRs
Differentially methylated regions
- LCLs
Lymphoblastoid cell lines
- NVC
Nailfold video capillaroscopy
- SSc
Systemic sclerosis
- WRN
Werner syndrome gene
- WS
Werner syndrome
Authors’ contributions
TG, MGB, AM, CF, PG, DR and FB designed the research. TG managed the biobank and performed the clinical laboratory analyses. MGB performed the statistical analysis of the data. DR performed the bioinformatics analysis and wrote the paper. AM recruited the patients, performed the NVC and wrote the paper. TG, MGB, DG, SP and DBAM performed the experiments. PG, CF, AS, FB, TG, AC and MGB analysed and discussed the data. TG and MGB were the major contributors in writing the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Subjects were enrolled in the study “Early diagnosis of scleroderma and identification of predictor factors of disease development”. All participants signed the informed consent forms. The study was approved by the Ethic Committee of ASL RM/B, Prot.054/CE-RMB.
Consent for publication
Consent for publication was obtained from the patient.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
T. Guastafierro and MG Bacalini have shared first name.
P. Garagnani and F. Bondanini have co-senior authorship.
Electronic supplementary material
The online version of this article (doi:10.1186/s13148-017-0389-4) contains supplementary material, which is available to authorized users.
Contributor Information
T. Guastafierro, Email: tiziana.guastafierro@gmail.com
D. Raimondo, Email: domenico.raimondo@uniroma1.it
References
- 1.Takemoto M, Mori S, Kuzuya M, Yoshimoto S, Shimamoto A, Igarashi M, Tanaka Y, Miki T, Yokote K. Diagnostic criteria for Werner syndrome based on Japanese nationwide epidemiological survey. Geriatr Gerontol Int. 2013;13:475–481. doi: 10.1111/j.1447-0594.2012.00913.x. [DOI] [PubMed] [Google Scholar]
- 2.Coppede F. The epidemiology of premature aging and associated comorbidities. Clin Interv Aging. 2013;8:1023–1032. doi: 10.2147/CIA.S37213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goto M, Okawa-Takatsuji M, Aotsuka S, Nakai H, Shimizu M, Goto H, Shimamoto A, Furuichi Y. Significant elevation of IgG anti-WRN (RecQ3 RNA/DNA helicase) antibody in systemic sclerosis. Mod Rheumatol. 2006;16:229–234. doi: 10.3109/s10165-006-0496-0. [DOI] [PubMed] [Google Scholar]
- 4.Bes C, Vardi S, Guven M, Soy M. Werner’s syndrome: a quite rare disease for differential diagnosis of scleroderma. Rheumatol Int. 2010;30:695–698. doi: 10.1007/s00296-009-0982-8. [DOI] [PubMed] [Google Scholar]
- 5.Capell BC, Tlougan BE, Orlow SJ. From the rarest to the most common: insights from progeroid syndromes into skin cancer and aging. J Invest Dermatol. 2009;129:2340–2350. doi: 10.1038/jid.2009.103. [DOI] [PubMed] [Google Scholar]
- 6.Hettema ME, Zhang D, de Leeuw K, Stienstra Y, Smit AJ, Kallenberg CG, Bootsma H. Early atherosclerosis in systemic sclerosis and its relation to disease or traditional risk factors. Arthritis Res Ther. 2008;10:R49. doi: 10.1186/ar2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heyn H, Moran S, Esteller M. Aberrant DNA methylation profiles in the premature aging disorders Hutchinson-Gilford progeria and Werner syndrome. Epigenetics. 2013;8:28–33. doi: 10.4161/epi.23366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Muftuoglu M, Oshima J, von Kobbe C, Cheng WH, Leistritz DF, Bohr VA. The clinical characteristics of Werner syndrome: molecular and biochemical diagnosis. Hum Genet. 2008;124:369–377. doi: 10.1007/s00439-008-0562-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhu X, Zhang G, Kang L, Guan H. Epigenetic regulation of werner syndrome gene in age-related cataract. J Ophthalmol. 2015;2015:579695. doi: 10.1155/2015/579695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Edwards DN, Machwe A, Chen L, Bohr VA, Orren DK. The DNA structure and sequence preferences of WRN underlie its function in telomeric recombination events. Nat Commun. 2015;6:8331. doi: 10.1038/ncomms9331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rossi ML, Ghosh AK, Bohr VA. Roles of Werner syndrome protein in protection of genome integrity. DNA Repair (Amst) 2010;9:331–344. doi: 10.1016/j.dnarep.2009.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burla R, Carcuro M, Raffa GD, Galati A, Raimondo D, Rizzo A, La Torre M, Micheli E, Ciapponi L, Cenci G, et al. AKTIP/Ft1, a new shelterin-interacting factor required for telomere maintenance. PLoS Genet. 2015; 11:e1005167. [DOI] [PMC free article] [PubMed]
- 13.Burla R, Carcuro M, Torre ML, Fratini F, Crescenzi M, D’Apice MR, Spitalieri P, Raffa GD, Astrologo L, Lattanzi G, et al. The telomeric protein AKTIP interacts with A- and B-type lamins and is involved in regulation of cellular senescence. Open Biol. 2016;6 [DOI] [PMC free article] [PubMed]
- 14.Jovanovic SV, Clements D, MacLeod K. Biomarkers of oxidative stress are significantly elevated in Down syndrome. Free Radic Biol Med. 1998;25:1044–1048. doi: 10.1016/S0891-5849(98)00137-3. [DOI] [PubMed] [Google Scholar]
- 15.Bacalini MG, Boattini A, Gentilini D, Giampieri E, Pirazzini C, Giuliani C, Fontanesi E, Remondini D, Capri M, Del Rio A, et al. Erratum: A meta-analysis on age-associated changes in blood DNA methylation: results from an original analysis pipeline for Infinium 450k data. Aging (Albany NY) 2016;8:831. doi: 10.18632/aging.100915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bacalini MG, D’Aquila P, Marasco E, Nardini C, Montesanto A, Franceschi C, Passarino G, Garagnani P, Bellizzi D. The methylation of nuclear and mitochondrial DNA in ageing phenotypes and longevity. Mech Ageing Dev. 2017. doi:10.1016/j.mad.2017.01.006. [DOI] [PubMed]
- 17.Maierhofer A, Flunkert J, Oshima J, Martin GM, Haaf T, Horvath S. Accelerated epigenetic aging in Werner syndrome. Aging (Albany NY) 2017;9:1143–1152. doi: 10.18632/aging.101217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Altorok N, Tsou PS, Coit P, Khanna D, Sawalha AH. Genome-wide DNA methylation analysis in dermal fibroblasts from patients with diffuse and limited systemic sclerosis reveals common and subset-specific DNA methylation aberrancies. Ann Rheum Dis. 2015;74:1612–1620. doi: 10.1136/annrheumdis-2014-205303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Silver RM. Clinical aspects of systemic sclerosis (scleroderma) Ann Rheum Dis. 1991;50(Suppl 4):854–861. doi: 10.1136/ard.50.Suppl_4.854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haustein UF. Systemic sclerosis-scleroderma. Dermatol Online J. 2002;8:3. [PubMed] [Google Scholar]
- 21.van den Hoogen F, Khanna D, Fransen J, Johnson SR, Baron M, Tyndall A, Matucci-Cerinic M, Naden RP, Medsger TA, Jr, Carreira PE, et al. classification criteria for systemic sclerosis: an American College of Rheumatology/European League Against Rheumatism Collaborative Initiative. Ann Rheum Dis. 2013;72:1747–1755. doi: 10.1136/annrheumdis-2013-204424. [DOI] [PubMed] [Google Scholar]
- 22.Du P, Zhang X, Huang CC, Jafari N, Kibbe WA, Hou L, Lin SM. Comparison of beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics. 2010;11:587. doi: 10.1186/1471-2105-11-587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weidner CI, Lin Q, Birkhofer C, Gerstenmaier U, Kaifie A, Kirschner M, Bruns H, Balabanov S, Trummer A, Stockklausner C, et al. DNA methylation in PRDM8 is indicative for dyskeratosis congenita. Oncotarget. 2016;7:10765–10772. doi: 10.18632/oncotarget.7458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, Ma’ayan A. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics. 2013;14:128. doi: 10.1186/1471-2105-14-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016;44:W90–W97. doi: 10.1093/nar/gkw377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dumas J, Gargano MA, Dancik GM. shinyGEO: a web-based application for analyzing gene expression omnibus datasets. Bioinformatics. 2016;32:3679–3681. doi: 10.1093/bioinformatics/btw519. [DOI] [PubMed] [Google Scholar]
- 27.Barrett T, Troup DB, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH, Sherman PM, et al. NCBI GEO: archive for functional genomics data sets—10 years on. Nucleic Acids Res. 2011;39:D1005–D1010. doi: 10.1093/nar/gkq1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grafodatskaya D, Choufani S, Ferreira JC, Butcher DT, Lou Y, Zhao C, Scherer SW, Weksberg R. EBV transformation and cell culturing destabilizes DNA methylation in human lymphoblastoid cell lines. Genomics. 2010;95:73–83. doi: 10.1016/j.ygeno.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 29.Bacalini MG, Gentilini D, Boattini A, Giampieri E, Pirazzini C, Giuliani C, Fontanesi E, Scurti M, Remondini D, Capri M, et al. Identification of a DNA methylation signature in blood cells from persons with Down syndrome. Aging (Albany NY) 2015;7:82–96. doi: 10.18632/aging.100715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cheung HH, Liu X, Canterel-Thouennon L, Li L, Edmonson C, Rennert OM. Telomerase protects werner syndrome lineage-specific stem cells from premature aging. Stem Cell Reports. 2014;2:534–546. doi: 10.1016/j.stemcr.2014.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oshima J, Martin GM, Hisama FM. Werner syndrome. 1993.
- 32.Hernandez-Corbacho MJ, Jenkins RW, Clarke CJ, Hannun YA, Obeid LM, Snider AJ, Siskind LJ. Accumulation of long-chain glycosphingolipids during aging is prevented by caloric restriction. PLoS One. 2011;6:e20411. doi: 10.1371/journal.pone.0020411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brunet A. Aging and the control of the insulin-FOXO signaling pathway. Med Sci (Paris) 2012;28:316–320. doi: 10.1051/medsci/2012283021. [DOI] [PubMed] [Google Scholar]
- 34.Greer EL, Brunet A. FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene. 2005;24:7410–7425. doi: 10.1038/sj.onc.1209086. [DOI] [PubMed] [Google Scholar]
- 35.Eijkelenboom A, Burgering BM. FOXOs: signalling integrators for homeostasis maintenance. Nat Rev Mol Cell Biol. 2013;14:83–97. doi: 10.1038/nrm3507. [DOI] [PubMed] [Google Scholar]
- 36.Greer EL, Brunet A. FOXO transcription factors in ageing and cancer. Acta Physiol (Oxf) 2008;192:19–28. doi: 10.1111/j.1748-1716.2007.01780.x. [DOI] [PubMed] [Google Scholar]
- 37.Peltier J, O’Neill A, Schaffer DV. PI3K/Akt and CREB regulate adult neural hippocampal progenitor proliferation and differentiation. Dev Neurobiol. 2007;67:1348–1361. doi: 10.1002/dneu.20506. [DOI] [PubMed] [Google Scholar]
- 38.Adams HH, Hibar DP, Chouraki V, Stein JL, Nyquist PA, Renteria ME, Trompet S, Arias-Vasquez A, Seshadri S, Desrivieres S, et al. Novel genetic loci underlying human intracranial volume identified through genome-wide association. Nat Neurosci. 2016;19:1569–1582. doi: 10.1038/nn.4398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Murata K, Nakashima H. Clinical and metabolic studies on Werner’s syndrome: with special reference to disorders of lipid and liver function. Adv Exp Med Biol. 1985;190:285–304. doi: 10.1007/978-1-4684-7853-2_13. [DOI] [PubMed] [Google Scholar]
- 40.Hay N. Interplay between FOXO, TOR, and Akt. Biochim Biophys Acta. 2011;1813:1965–1970. doi: 10.1016/j.bbamcr.2011.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cho KA, Ryu SJ, Oh YS, Park JH, Lee JW, Kim HP, Kim KT, Jang IS, Park SC. Morphological adjustment of senescent cells by modulating caveolin-1 status. J Biol Chem. 2004;279:42270–42278. doi: 10.1074/jbc.M402352200. [DOI] [PubMed] [Google Scholar]
- 42.Atfi A, Dumont E, Colland F, Bonnier D, L’Helgoualc’h A, Prunier C, Ferrand N, Clement B, Wewer UM, Theret N. The disintegrin and metalloproteinase ADAM12 contributes to TGF-beta signaling through interaction with the type II receptor. J Cell Biol. 2007;178:201–208. doi: 10.1083/jcb.200612046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Taniguchi T, Asano Y, Akamata K, Aozasa N, Noda S, Takahashi T, Ichimura Y, Toyama T, Sumida H, Kuwano Y, et al. Serum levels of ADAM12-S: possible association with the initiation and progression of dermal fibrosis and interstitial lung disease in patients with systemic sclerosis. J Eur Acad Dermatol Venereol. 2013;27:747–753. doi: 10.1111/j.1468-3083.2012.04558.x. [DOI] [PubMed] [Google Scholar]
- 44.Jennemann R, Rabionet M, Gorgas K, Epstein S, Dalpke A, Rothermel U, Bayerle A, van der Hoeven F, Imgrund S, Kirsch J, et al. Loss of ceramide synthase 3 causes lethal skin barrier disruption. Hum Mol Genet. 2012;21:586–608. doi: 10.1093/hmg/ddr494. [DOI] [PubMed] [Google Scholar]
- 45.Radner FP, Marrakchi S, Kirchmeier P, Kim GJ, Ribierre F, Kamoun B, Abid L, Leipoldt M, Turki H, Schempp W, et al. Mutations in CERS3 cause autosomal recessive congenital ichthyosis in humans. PLoS Genet. 2013, 9:e1003536. [DOI] [PMC free article] [PubMed]
- 46.Giacomin MF, Franca CM, Oliveira ZN, Machado MC, Sallum AM, Silva CA. Generalized morphea in a child with harlequin ichthyosis: a rare association. Rev Bras Reumatol Engl Ed. 2016;56:82–85. doi: 10.1016/j.rbr.2014.05.004. [DOI] [PubMed] [Google Scholar]
- 47.Brown WT, Kieras FJ, Houck GE, Jr, Dutkowski R, Jenkins EC. A comparison of adult and childhood progerias: Werner syndrome and Hutchinson-Gilford progeria syndrome. Adv Exp Med Biol. 1985;190:229–244. doi: 10.1007/978-1-4684-7853-2_10. [DOI] [PubMed] [Google Scholar]
- 48.Imura H, Nakao Y, Kuzuya H, Okamoto M, Yamada K. Clinical, endocrine and metabolic aspects of the Werner syndrome compared with those of normal aging. Adv Exp Med Biol. 1985;190:171–185. doi: 10.1007/978-1-4684-7853-2_6. [DOI] [PubMed] [Google Scholar]
- 49.Valentini G, D’Angelo S, Della Rossa A, Bencivelli W, Bombardieri S. European Scleroderma Study Group to define disease activity criteria for systemic sclerosis. IV. Assessment of skin thickening by modified Rodnan skin score. Ann Rheum Dis. 2003;62:904–905. doi: 10.1136/ard.62.9.904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lok C, Ruto F, Labeille B, Pietri J, Denoeux JP. Leg ulcers in Werner’s syndrome. Report of one case. J Mal Vasc. 1991;16:381–382. [PubMed] [Google Scholar]
- 51.Yeong EK, Yang CC. Chronic leg ulcers in Werner’s syndrome. Br J Plast Surg. 2004;57:86–88. doi: 10.1016/j.bjps.2003.10.011. [DOI] [PubMed] [Google Scholar]
- 52.Mohan C, Assassi S. Biomarkers in rheumatic diseases: how can they facilitate diagnosis and assessment of disease activity? BMJ. 2015, 351:h5079. [DOI] [PMC free article] [PubMed]
- 53.Kayser C, Fritzler MJ. Autoantibodies in systemic sclerosis: unanswered questions. Front Immunol. 2015;6:167. doi: 10.3389/fimmu.2015.00167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Madej-Pilarczyk A, Rosinska-Borkowska D, Rekawek J, Marchel M, Szalus E, Jablonska S, Hausmanowa-Petrusewicz I. Progeroid syndrome with scleroderma-like skin changes associated with homozygous R435C LMNA mutation. Am J Med Genet A. 2009;149A:2387–2392. doi: 10.1002/ajmg.a.33018. [DOI] [PubMed] [Google Scholar]
- 55.Zhang S, Zhang K, Jiang M, Zhao J. Hutchinson-Gilford progeria syndrome with scleroderma-like skin changes due to a homozygous missense LMNA mutation. J Eur Acad Dermatol Venereol. 2016;30:463–465. doi: 10.1111/jdv.12840. [DOI] [PubMed] [Google Scholar]
- 56.Luckhardt TR, Thannickal VJ. Systemic sclerosis-associated fibrosis: an accelerated aging phenotype? Curr Opin Rheumatol. 2015;27:571–576. doi: 10.1097/BOR.0000000000000219. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Principal Component Analysis for the DNA methylation levels of the probes included in the InfiniumEPIC beadchip in WS and CTR. (TIFF 1825 kb)
List of DMPs between WS and CTR. (XLS 531 kb)
DNA methylation profiles of the 27 DMRs between WS and CTR. For each DMR, the title of the plot reports the name of the gene/genes in which the DMR maps, the not-adjusted P value of the ANOVA comparison between WS and CTR, and the length of the DMR in base pairs. Below the plot, the name of the CpG island and the position of the DMR respect to the CpG island are reported. (PDF 58 kb)
List of DMRs between WS and CTR. (XLS 60 kb)
Boxplots of DNA methylation values of the CpG probes in the genes belonging to the enriched pathways in naïve B cells, CTR LCL, WS LCL from the GSE42865 dataset and in CTR and WS whole blood samples assessed in this study. The brown dots correspond to the individual samples. For each CpG probe, the title of the plot reports the name of the CpG probe, the name of the gene/genes in which the probe maps and, if present, the name of the CpG island and the position of the probe respect to the CpG island. (PDF 41 kb)
Differential expression of several genes belonging to the enriched pathways resulted based on publicly available dataset on WS fibroblasts analyses. (TIFF 2607 kb)
Relationship between DNA methylation of DMPs and DMRs in the present dataset and RNA expression in the GSE48761 dataset. (XLS 68 kb)
Data Availability Statement
All data generated or analysed during this study are included in this published article [and its Additional files].