

Abstract
Proper neural circuit formation requires the precise regulation of neuronal migration, axon guidance, and dendritic arborization. Mutations affecting the function of the transmembrane glycoprotein dystroglycan cause a form of congenital muscular dystrophy that is frequently associated with neurodevelopmental abnormalities. Despite its importance in brain development, the role of dystroglycan in regulating retinal development remains poorly understood. Using a mouse model of dystroglycanopathy (ISPDL79*) and conditional dystroglycan mutants of both sexes, we show that dystroglycan is critical for the proper migration, axon guidance, and dendritic stratification of neurons in the inner retina. Using genetic approaches, we show that dystroglycan functions in neuroepithelial cells as an extracellular scaffold to maintain the integrity of the retinal inner limiting membrane. Surprisingly, despite the profound disruptions in inner retinal circuit formation, spontaneous retinal activity is preserved. These results highlight the importance of dystroglycan in coordinating multiple aspects of retinal development.
SIGNIFICANCE STATEMENT The extracellular environment plays a critical role in coordinating neuronal migration and neurite outgrowth during neural circuit development. The transmembrane glycoprotein dystroglycan functions as a receptor for multiple extracellular matrix proteins and its dysfunction leads to a form of muscular dystrophy frequently associated with neurodevelopmental defects. Our results demonstrate that dystroglycan is required for maintaining the structural integrity of the inner limiting membrane (ILM) in the developing retina. In the absence of functional dystroglycan, ILM degeneration leads to defective migration, axon guidance, and mosaic spacing of neurons and a loss of multiple neuron types during retinal development. These results demonstrate that disorganization of retinal circuit development is a likely contributor to visual dysfunction in patients with dystroglycanopathy.
Keywords: axon, dendrite, dystroglycan, extracellular matrix, migration, retina
Introduction
The precise lamination of neurons is critical for establishing proper connectivity in the developing nervous system. The retina is organized in three cellular layers: the outer nuclear layer (ONL), containing rod and cone photoreceptors; the inner nuclear layer (INL), containing horizontal cells, bipolar cells, and amacrine cells; and the ganglion cell layer (GCL), containing retinal ganglion cells (RGCs) and displaced amacrine cells (Bassett and Wallace, 2012). Two synaptic lamina form postnatally: the outer plexiform layer (OPL), which contains synapses among photoreceptors, bipolar cells, and horizontal cells; and the inner plexiform layer (IPL), which contains synapses among bipolar cells, amacrine cells, and RGCs. The molecular cues that direct the laminar positioning of neurons and the stratification of their processes within the synaptic layers remain poorly understood.
Unlike the cerebral cortex, where many neurons migrate along the radial glia scaffold, retinal migration does not require contact between neurons and neuroepithelial cells (Reese, 2011). RGCs, bipolar cells, and photoreceptors migrate by nuclear translocation through a basally directed process. Basal process contact with the inner limiting membrane (ILM) is critical for the polarization and migration of RGCs (Randlett et al., 2011). The ILM is enriched with extracellular matrix (ECM) proteins including laminins, collagen IV, and perlecan (Taylor et al., 2015; Varshney et al., 2015). Mutations in specific laminins (Lamα1, Lamβ2, and Lamγ3) or the laminin receptor β1-integrin disrupt formation of the ILM and organization of the GCL (Edwards et al., 2010; Pinzón-Duarte et al., 2010; Gnanaguru et al., 2013; Riccomagno et al., 2014). How laminins and other ECM proteins are initially organized in the ILM and how the ILM directs the organization of the retina remain unclear.
In addition to β1-integrin, the transmembrane glycoprotein dystroglycan functions as a receptor for laminins and other ECM proteins through its extracellular α-subunit. Dystroglycan connects to the actin cytoskeleton through the intracellular domain of its transmembrane β-subunit, which is part of the dystrophin glycoprotein complex (Moore and Winder, 2010). Mutations disrupting the glycosylation of dystroglycan affect its binding to laminin G-domain-containing ECM proteins and lead to a form of congenital muscular dystrophy termed dystroglycanopathy (Taniguchi-Ikeda et al., 2016). The most severe forms, Muscle–Eye–Brain disease and Walker–Warburg Syndrome, are accompanied by cortical malformation (type II lissencephaly), cerebellar abnormalities, and retinal dysplasias (Dobyns et al., 1989).
Brain malformations in dystroglycanopathies reflect the critical role that dystroglycan plays in maintaining the architecture of the neuroepithelial scaffold (Moore et al., 2002; Myshrall et al., 2012). Focal regions of retinal dysplasia have also been observed in mouse models of dystroglycanopathy, with ectopic cells protruding through the ILM (Takeda et al., 2003; Lee et al., 2005; Satz et al., 2008; Chan et al., 2010; Takahashi et al., 2011). In Xenopus, morpholino depletion of dystroglycan results in micropthalmia, degeneration of the ILM, and abnormal positioning of photoreceptors, bipolar cells, and RGCs (Lunardi et al., 2006). However, several important questions regarding the role of dystroglycan in mammalian retinal development remain unaddressed. First, what is the underlying cause of retinal dysplasia in dystroglycanopathy? Second, is dystroglycan required for the proper migration and lamination of specific subtypes of retinal neurons? Third, how does the loss of dystroglycan affect axon guidance, dendritic stratification, and mosaic spacing in the retina? Finally, how do retinal dysplasias in models of dystroglycanopathy affect the function of the retina?
Here, using multiple genetic models, we identify a critical role for dystroglycan in multiple aspects of retinal development. We show that dystroglycan within the neuroepithelium is critical for maintaining the structural integrity of the ILM. We provide in vivo evidence that dystroglycan's maintenance of the ILM is required for proper neuronal migration, axon guidance, formation of synaptic lamina, and the survival of multiple retinal neuron subtypes. Surprisingly, spontaneous retinal activity appears unperturbed despite the dramatic disruption in inner retinal development. Together, these results provide critical insight into how dystroglycan directs the proper functional assembly of retinal circuits.
Materials and Methods
Experimental design and statistical analysis.
Mice were maintained on mixed genetic backgrounds and were used regardless of sex. All phenotypic analysis was conducted with n ≥ 3 animals obtained from at least 2 different litters of mice. Statistical analysis was performed using JMP Pro version 13.0 software (SAS Institute). Comparison between two groups was analyzed using a Student's t test. Comparison between two or more groups was analyzed using a two-way ANOVA and Tukey's post hoc test. Comparison of retinal wave parameters was analyzed using a Wilcoxon rank-sum test. The significance threshold was set at 0.05 for all statistical tests (*p < 0.05; **p < 0.01; ***p < 0.001).
Animals.
Animal procedures were approved by Oregon Health & Science University Institutional Animal Care and Use Committee and conformed to the National Institutes of Health's Guide for the Care and Use of Laboratory Animals. Animals were killed by administration of CO2. The day of vaginal plug observation was designated as embryonic day 0 (e0) and the day of birth in this study was designated as postnatal day 0 (P0). The generation and genotyping protocols for ISPDL79*/L79* (Wright et al., 2012), DGF/F (Moore et al., 2002), and DGβcyt (Satz et al., 2009) mice have been described previously. The presence of the Cre allele in Sox2Cre (Hayashi et al., 2002), Six3Cre (Furuta et al., 2000), Isl1Cre (Yang et al., 2006), and NestinCre mice (Tronche et al., 1999) was detected by generic Cre primers. ISPD+/L79*, DGβcyt/+, and DGF/+;Six3Cre or DGF/+ age-matched littermates were used as controls.
Tissue preparation and immunohistochemistry.
Embryonic retinas were left in the head and fixed overnight at 4°C in 4% PFA and washed in PBS for 30 min. Heads were equilibrated in 15% sucrose overnight and flash frozen in optimal cutting temperature medium. Postnatal retinas were dissected out of the animal and the lens was removed from the eye cup. Intact retinas were fixed at room temperature for 30 min in 4% PFA. Retinas were washed in PBS for 30 min and equilibrated in a sequential gradient of 10%, 20%, and 30% sucrose overnight. Tissue was sectioned on a cryostat at 16–25 μm. Tissue sections were blocked in a PBS solution containing 2% normal donkey serum and 0.2% Triton X-100 for 30 min and incubated in primary antibody overnight at 4°C. Sections were washed for 30 min and incubated in secondary antibody in a PBS solution containing 2% normal donkey serum for 2–4 h. Sections were incubated in DAPI to stain nuclei for 10 min, washed for 30 min, and mounted using Fluoromount medium. The source and concentration of all antibodies used in this study are listed in Table 1.
Table 1.
Antibodies used throughout the study
| Target | Host species | Dilution | Company/origin | Catalog no. | RRID |
|---|---|---|---|---|---|
| 2H3 | Mouse | 1:1000 | Developmental Studies Hybridoma Bank | 2h3 | AB_531793 |
| β-Dystroglycan | Mouse | 1:500 | Developmental Studies Hybridoma Bank | MANDAG2(7D11) | AB_2618140 |
| β-Dystroglycan | Rabbit | 1:100 | Santa Cruz Biotechnology | sc-28535 | AB_782259 |
| Calbindin | Rabbit | 1:10,000 | Swant | CB 38 | AB_10000340 |
| Calretinin | Rabbit | 1:10,000 | Swant | CG 1 | AB_2619710 |
| ChAT | Goat | 1:500 | Millipore | AB144P-200UL | AB_11214092 |
| Chx10 | Goat | 1:500 | Santa Cruz Biotechnology | sc-21690 | AB_2216006 |
| Cleaved Caspase-3 | Rabbit | 1:500 | Cell Signaling Technology | 9661S | AB_2341188 |
| Collagen IV | Goat | 1:250 | Southern Biotechnology | 1340-01 | AB_2082646 |
| Glutamine Synthetase | Mouse | 1:1000 | BD Biosciences | 610517 | AB_397879 |
| GlyT1 | Rabbit | 1:800 | Gift from Dr. David Pow | ||
| IB4 | 1:250 | Life Technologies | I21411 | AB_2314662 | |
| L1 | Rat | 1:500 | Millipore | MAB5272 | AB_2133200 |
| Laminin | Rabbit | 1:1000 | Sigma-Aldrich | L9393 | AB_477163 |
| MGluR6 | Sheep | 1:100 | gift from Dr. Catherine Morgans | ||
| Perlecan | Rat | 1:500 | Millipore | MAB1948P | AB_10615958 |
| PH3 | Rat | 1:250 | Abcam | ab10543 | AB_2295065 |
| PKC | Mouse | 1:500 | Sigma-Aldrich | P5704 | AB_477375 |
| RBPMS | Guinea pig | 1:500 | PhosphoSolutions | 1832-RBPMS | AB_2492226 |
| Recoverin | Rabbit | 1:200 | Millipore | AB5585 | AB_2253622 |
| Ribeye/Ctbp2 | Mouse | 1:1000 | BD Biosciences | 612044 | AB_399431 |
| Secretagogin | Rabbit | 1:4000 | Biovendor | rd181120100 | AB_2034060 |
| VGAT | Rabbit | 1:500 | Synaptic Systems | 131-003 | AB_887869 |
| VGLUT1 | Guinea pig | 1:500 | Millipore | AB5905 | AB_2301751 |
Whole-mount retinal staining.
Postnatal retinas were dissected out of the animal and the lens was removed from the eye cup. Intact retinas were fixed at room temperature for 30 min in 4% PFA. Retinas were incubated in primary antibody diluted in PBS solution containing 2% normal donkey serum and 0.2% Triton for 2 d at 4°C. Retinas were washed in PBS for 1 d and incubated in secondary antibody diluted in PBS solution containing 2% normal donkey serum for 2 d at 4°C, washed for 1 d in PBS, and mounted using Fluoromount medium.
Microscopy.
Imaging was performed on a Zeiss Axio Imager M2 upright microscope equipped with an ApoTome.2. Imaging of synapses was performed on a Zeiss Elyra PS.1 with LSM 710 laser-scanning confocal Super-Resolution Microscope with AiryScan. Imaging of retinal waves was performed on a Nikon TiE inverted microscope with full environmental chamber equipped with a Yokogawa CSU-W1 spinning disk confocal unit.
Quantification of cell number and mosaic spacing.
For each experiment, three to four locations per retina at the midpoint of each lobe were sampled. Cell counts of horizontal cells (calbindin), apoptotic cells (Cleaved Caspase-3, P0), and starburst amacrine cells (ChAT) were obtained from 500 × 500 μm images and quantified in Fiji software. Cell counts of apoptotic cells (Cleaved Caspase-3, e16) and ganglion cells (RNA binding protein with multiple splicing, RPBMS) were obtained from 250 × 250 μm images and quantified in Fiji software. Analysis of retinal mosaics (calbindin, ChAT) were conducted on 500 × 500 μm images by measuring the X–Y coordinates for each cell and Voronoi domains were calculated in Fiji software and nearest neighbor measurements calculated with WinDRP. Cell counts of proliferating cells (PH3 positive, e13, e16) were done by counting the number of positive cells in midretinal 20 μm sections.
Live-cell imaging and analysis.
Retinas from P1 DGF/+;Six3Cre;R26-LSL-GCaMP6f and DGF/−;Six3Cre;R26-LSL-GCaMP6f mice were dissected into chilled Ames' medium (Sigma-Aldrich) buffered with sodium bicarbonate and bubbled with carbogen gas (95% O2, 5% CO2). The retinas were dissected out of the eye cup, mounted RGC side up on cellulose membrane filters (Millipore), and placed in a glass-bottom Petri dish containing Ames' medium. A platinum harp was used to stabilize the filter paper during imaging. Imaging was performed at 30°C using a 10 × 0.45 Plan Apo Air objective with a field of 1664 by 1404 μm with a 3 Hz imaging timeframe. The field was illuminated with a 488 nm laser. Three to 4 retinal fields were imaged per retina, each field of retina was imaged for a 2 min time series using a 300 ms exposure, and each field was sampled 3–5 times per imaging session.
Thirty representative control and 30 representative mutant time series were selected randomly for analysis. Only waves that initiated and terminated within the imaging field were used for analysis. To measure wave area, movies were viewed manually using Fiji software frame by frame to determine the start and end frame of a wave. A Z-projection for maximum intensity was used to create an image with the entire wave and the boundary of the wave was traced manually to determine the area. Wave area per time was calculated by dividing the area of the wave by the duration in seconds of the wave. Any wave lasting <2 s was not used in analysis, consistent with previous studies (Blankenship et al., 2009).
Results
Dystroglycan is required for ILM integrity
Dystroglycan plays a critical role in the developing cortex, where it anchors radial neuroepithelial end feet to the basement membrane along the pial surface. In the absence of functional dystroglycan, disruptions in the cortical basement membrane and detachment of neuroepithelial endfeet lead to profound neuronal migration phenotypes (Moore et al., 2002; Myshrall et al., 2012). In the adult retina, dystroglycan is present in blood vessels, in RGCs, at ribbon synapses in the OPL, and at the ILM, which serves as a basement membrane that separates the neural retina from the vitreous space (Montanaro et al., 1995; Omori et al., 2012). However, the role of dystroglycan in regulating neuronal migration, axon guidance, or dendritic stratification of specific cell types during retinal development has not been examined in a comprehensive manner. To address this open question, we first examined the expression pattern of dystroglycan in the developing retina. Using immunohistochemistry, we observed dystroglycan expression along radial processes that span the width of the retina and its selective enrichment at the ILM from age e13 through birth P0 (Fig. 1A). These processes are likely a combination of neuroepithelial cells and the basal process of migrating RGCs. Loss of staining in retinas from an epiblast-specific dystroglycan conditional knock-out (DGF/−;Sox2Cre) confirmed the specificity of this expression pattern (Fig. 1B).
Figure 1.
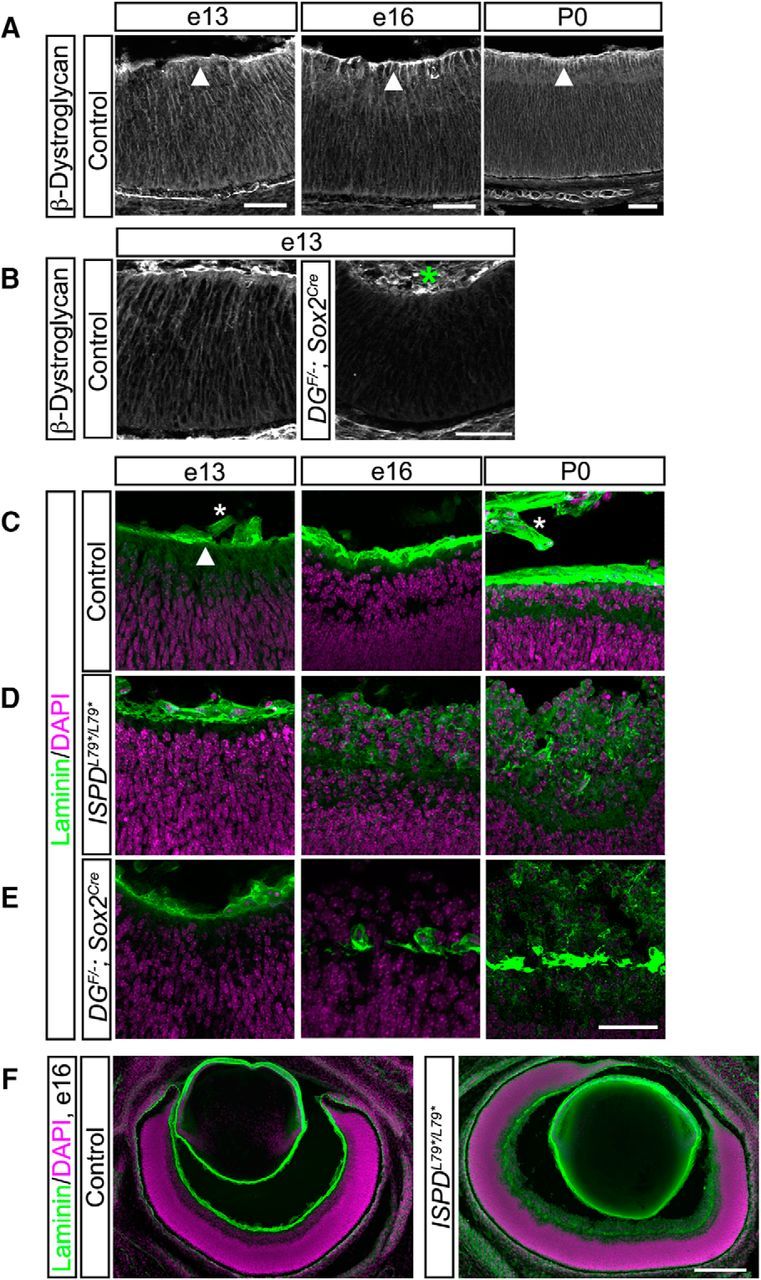
The ILM undergoes progressive degeneration in the absence of functional dystroglycan. A, Dystroglycan (β-DG) is expressed throughout the developing retina, with an enrichment at the ILM. B, Dystroglycan expression is lost in DGF/−;Sox2Cre mice. C–E, The initial assembly of the ILM (laminin, green) occurs normally in the absence of functional dystroglycan in ISPDL79*/L79* (D) and DGF/−;Sox2Cre (E) retinas. The ILM in ISPDL79*/L79* and DGF/−;Sox2Cre retinas undergoes progressive degeneration at e16 (middle) and P0 (right) and retinal neurons migrate into the vitreous. F, ILM in ISPDL79*/L79* retinas undergoes degeneration (right) that is present across the entire span of the retina at e16. Arrowheads indicate ILM; asterisks indicate nonspecific staining of blood vessels. Scale bars: A–E, 50 μm; F, 200 μm.
The first step in the assembly of basement membranes is the recruitment of laminin polymers to the cell surface by sulfated glycolipids, followed by the stabilization of laminin polymers by transmembrane receptors (Yurchenco, 2011). To determine whether dystroglycan is required for the initial formation of the ILM during retinal development, we used two complementary genetic models. ISPDL79*/L79* mutants, previously identified in a forward genetic screen, lack the mature glycan chains required for dystroglycan to bind ligands such as laminin and are a model for severe dystroglycanopathy (Wright et al., 2012). DGF/−;Sox2Cre conditional mutants lack dystroglycan in epiblast-derived tissues, including all retinal tissue, and were used to confirm that phenotypes observed in ISPDL79*/L79* mice are dystroglycan dependent. The enrichment of laminin at the ILM appeared normal in early retinal development at e13 in both ISPDL79*/L79* and DGF/−;Sox2Cre mutants (Fig. 1C–E), indicating that dystroglycan is not required for the initial formation of the ILM. However, at e16, we observed a loss of laminin staining and degeneration of the ILM across the entire surface of the retina in ISPDL79*/L79* and DGF/−;Sox2Cre mutants (Fig. 1D–F). The loss of ILM integrity in ISPDL79*/L79* and DGF/−;Sox2Cre mutants was accompanied by the inappropriate migration of retinal neurons, resulting in the formation of an ectopic layer of neurons protruding into the vitreous space.
After the initial polymerization of laminin on cell surfaces, additional ECM proteins bind and cross-link the nascent basement membrane to increase its stability and complexity. Examination of ISPDL79*/L79* and DGF/−;Sox2Cre mutant retinas at P0 revealed a loss of the ECM proteins collagen IV (red) and perlecan (green), coinciding with the disruptions in laminin (purple) (Fig. 2A–C). These data suggest that, although dystroglycan is not required for the initial formation of the ILM, it is essential for its maturation and maintenance. Furthermore, dystroglycan is critical for the ILM to function as a structural barrier to prevent the ectopic migration of neurons into the vitreous space.
Figure 2.

Dystroglycan is required to localize ECM proteins at the ILM. A, Multiple ECM proteins, including laminin (purple), collagen IV (red), and perlecan (green), localize to the ILM in controls. B, C, Localization of ECM proteins is disrupted in the ILM in the absence of functional dystroglycan in ISPDL79*/L79* (B) and DGF/−;Sox2Cre (C) retinas at P0. Arrowheads indicate ILM; asterisks indicate blood vessels. Scale bar, 50 μm.
Dystroglycan is required for vascular and optic fiber layer development
The hyaloid vasculature in the embryonic retina normally regresses as astrocytes and the retinal vascular plexus emerge through the optic nerve head beginning around birth (Fruttiger, 2007). Previous studies have found that defects in ILM integrity disrupt the emergence and migration of astrocytes and the retinal vasculature (Lee et al., 2005; Edwards et al., 2010; Takahashi et al., 2011; Tao and Zhang, 2016). In agreement with these findings, we observe that, at embryonic ages in ISPDL79*/L79* mutant retinas, the hyaloid vasculature becomes embedded within the ectopic retinal neuron layer at e16 (Fig. 3A) and fails to regress at P0 (Fig. 3B). In addition, the emergence of the retinal vasculature and astrocytes is stunted in ISPDL79*/L79* mutants (Fig. 3B).
Figure 3.

Dystroglycan regulates normal retinal vasculature development. A, IB4-labeled hyaloid vasculature is present in the vitreous adjacent to the GCL in control retinas (left), but is embedded within ectopic cell clusters ISPDL79*/L79* (right) retinas at e16. B, Flat-mount retinas at P0 show the emergence of the primary vascular plexus (IB4, green) and astrocytes (GFAP, purple) in controls (left). In ISPDL79*/L79* retinas (right), the emergence of astrocytes and the primary vascular plexus into the retina is delayed (arrow) and there is a persistence of hyaloid vasculature. Scale bars: A, 50 μm; B, 100 μm.
We have shown previously that the organization of the basement membrane by dystroglycan provides a permissive growth substrate for axons in the developing spinal cord (Wright et al., 2012). In addition, contact with laminin in the ILM stabilizes the leading process of newly generated RGCs to direct the formation of the nascent axon (Randlett et al., 2011). These axons remain in close proximity to the ILM as they extend centrally toward the optic nerve head, forming the optic fiber layer. Therefore, we investigated whether the disruptions in the ILM affected the guidance of RGC axons in ISPDL79*/L79* and DGF/−;Sox2Cre mutants. At e13, axons in both control and mutant retinas formed a dense and continuous network in the basal retina directly abutting the ILM (Fig. 4A). In contrast, at e16 (Fig. 4B) and P0 (Fig. 4C), RGC axons in ISPDL79*/L79* and DGF/−;Sox2Cre mutants were disorganized, exhibiting both defasciculation (asterisks) and hyperfasciculation (arrowheads).
Figure 4.
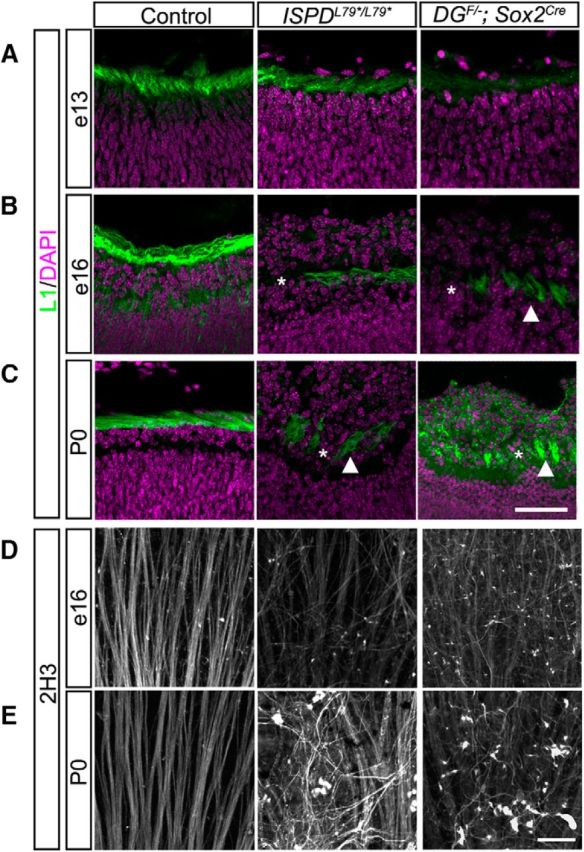
Dystroglycan is required for intraretinal axon guidance. A, L1-positive axons in the optic fiber layer (OFL) initially appear normal in ISPDL79*/L79* (middle) and DGF/−;Sox2Cre retinas (right) at e13. B, C, As the ILM degenerates in ISPDL79*/L79* and DGF/−;Sox2Cre retinas at e16 (B) and P0 (C), axons hyperfasciculate (arrowhead) and exhibit defasciculation (asterisk) within the OFL. D, E, Flat-mount preparations from ISPDL79*/L79* and DGF/−;Sox2Cre retinas at e16 (D) and P0 (E) show progressive disruption of axon tracts (Neurofilament, 2H3). Scale bar, 50 μm.
To gain further insight into the specific defects that occur in RGC axons, we used a flat-mount retina preparation. Regardless of their location in the retina, all RGCs orient and extend their axons toward the center of the retina, where they exit the retina through the optic nerve head (Bao, 2008). In control retinas at e16 (Fig. 4D, left) and P0 (Fig. 4E, left), axons traveled toward the optic nerve head in fasciculated, nonoverlapping bundles. In contrast, we frequently observed defasciculated RGC axons that grew in random directions without respect to their orientation to the optic nerve head in both ISPDL79*/L79* and DGF/−;Sox2Cre mutants. Together, these data show that proper growth and guidance of RGC axons to the optic nerve head requires dystroglycan to maintain an intact ILM as a growth substrate.
Dystroglycan is required for axonal targeting, dendritic lamination, and cell spacing in the postnatal retina
Our results in ISPDL79*/L79* and DGF/−;Sox2Cre mutants demonstrate that dystroglycan is required for ILM integrity and to prevent the ectopic migration of neurons into the vitreous (Fig. 1). However, the specific neuronal subtypes affected in models of dystroglycanopathy are unknown and the role of dystroglycan in regulating postnatal aspects of retinal development has not been examined. The synaptic layers of the retina develop postnatally, with tripartite synapses among the photoreceptors, bipolar cells, and horizontal cells forming in the OPL and synapses among bipolar cells, amacrine cells, and RGCs forming in the IPL. The development of these synaptic layers requires the precise stratification of both axons and dendrites that occurs between P0 and P14. Because ISPDL79*/L79* and DGF/−;Sox2Cre mutant mice exhibit perinatal lethality, we deleted dystroglycan selectively from the early neural retina using a Six3Cre driver (Furuta et al., 2000; Fig. 5A).
Figure 5.
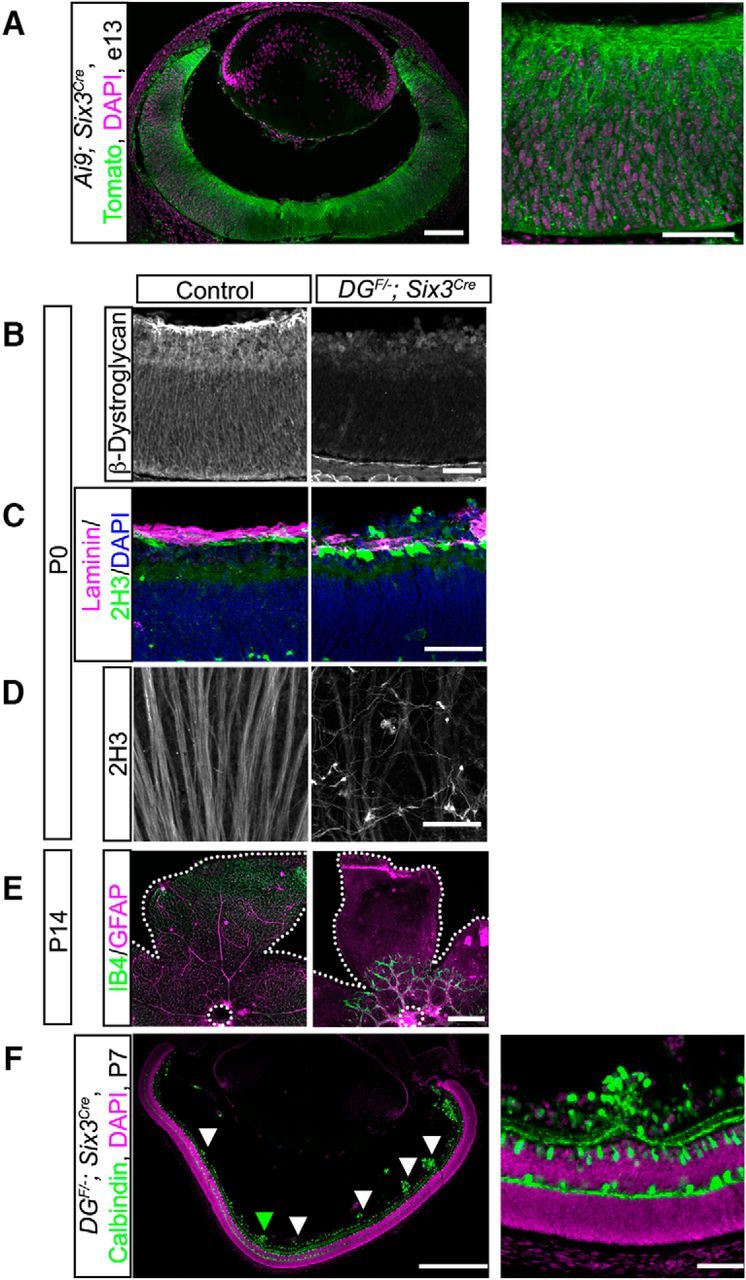
Conditional deletion of dystroglycan in the developing retina results in migration and axon guidance defects. A, Recombination pattern of Rosa26-lox-stop-lox-TdTomato;Ai9 reporter (green) by Six3Cre shows expression throughout the retina and in axons at e13. B, Dystroglycan protein expression is lost in DGF/−;Six3Cre mice. C, D, DGF/−;Six3Cre (right) mice exhibit ILM degeneration (top, purple, laminin) and abnormal axonal fasciculation and guidance (top, green, bottom, 2H3). E, Primary vascular plexus (IB4, green) and astrocytes (GFAP, purple) migrate from the optic nerve head (dashed circle) to the edge of the retina (dashed line) in control retinas at P14 (right). Vascular and astrocyte migration is stunted in DGF/−;Six3Cre retinas. F, Focal migration defects (arrowheads) in P7 DGF/−;Six3Cre retinas are present across the entire span of the retina. Green arrowhead indicates high-magnification image in F (right). Scale bars: A, left panel, 100 μm, right panel 50 μm; B–D, 50 μm; E, 500 μm; F, left panel 500 μm, right panel 50 μm.
Analysis of DGF/−;Six3Cre retinas confirmed that dystroglycan protein was lost along the neuroepithelial processes and at the ILM (Fig. 5B). We next confirmed that DGF/−;Six3Cre mice recapitulated the retinal phenotypes identified in ISPDL79*/L79* and DGF/−;Sox2Cre mice. We observed a degeneration of the ILM (laminin, purple) accompanied by ectopic migration of neurons into the vitreous (Fig. 5C), abnormal fasciculation and guidance of RGC axons (Fig. 5D), and defective emergence and migration of astrocytes and the vascular plexus (Fig. 5E). Although fully penetrant, the ILM degeneration and neuronal migration defects in DGF/−;Six3Cre mice were milder than in DGF/−;Sox2Cre mice, exhibiting a patchiness that was distributed across the retina (Fig. 5F). The defects in DGF/−;Six3Cre mice contrast the finding that conditional deletion of dystroglycan with NestinCre does not affect the overall structure of the retina (Satz et al., 2009). We find that this difference is likely due to the onset and pattern of Cre expression because recombination of a Cre-dependent reporter (Rosa26-lox-stop-lox-TdTomato;Ai9) occurred earlier and more broadly in Six3Cre mice than in NestinCre mice (Fig. 5A and data not shown).
DGF/−;Six3Cre mice are healthy and survive into adulthood, allowing us to examine the role of dystroglycan in postnatal retinal development. We analyzed DG;Six3Cre retinas at P14, when migration is complete and the laminar specificity of axons and dendrites has been established (Reese 2011). The overall architecture of the ONL appeared unaffected by the loss of dystroglycan in DGF/−;Six3Cre mice and cell body positioning of photoreceptors appeared similar to controls (Fig. 6A). Within the INL, the laminar positioning of rod bipolar cell bodies (Fig. 6B), cone bipolar cell bodies (Fig. 6C), horizontal cells (calbindin; Fig. 6E, arrows), and Müller glia cell bodies (Fig. 6H) and the targeting of their processes to the OPL appeared normal in DGF/−;Six3Cre mutants. However, bipolar cell axons that are normally confined to the synaptic layers in the IPL and Müller glia processes that are normally concentrated at the ILM both extended aberrant projections into the ectopic clusters (Fig. 6B,C,H).
Figure 6.

Disrupted postnatal circuit formation in the inner retina of dystroglycan mutants. A, Photoreceptors (recoverin) have normal lamination in P14 DGF/−;Six3Cre retinas (right). B, C, Cell bodies of bipolar cells (PKC, B; SCGN, C) exhibit normal lamination patters, whereas their axons extend into ectopic cellular clusters in the GCL. D–G, Abnormal cellular lamination and disruptions in dendritic stratification of multiple amacrine and RGC types is observed in DGF/−;Six3Cre retinas. D–G, ChAT labels starburst amacrine cells (D); calbindin (E) and calretinin (F) label amacrine and ganglion cells; and GlyT1 labels glycinergic amacrine cells (G). H, Müller glia (glutamine synthetase) cell bodies are normally positioned, whereas their inner retinal processes extend into ectopic cellular clusters. Arrowheads indicate axons or cell bodies in ectopic clusters; arrows indicate horizontal cell layer. Scale bar, 50 μm.
In contrast to the normal laminar architecture of the outer retina, the inner retina was disorganized in DGF/−;Six3Cre mutants. Subsets of amacrine and ganglion cells labeled by ChAT (starburst amacrine cells; Fig. 6D), calbindin (Fig. 6E), and calretinin (Fig. 6F) that are normally confined to the INL and GCL were present in the ectopic clusters that protrude into the vitreous space. Glycinergic amacrine cells (GlyT1; Fig. 6G), the cell bodies of which are normally found in a single layer within the INL, were also present within ectopic clusters. Compared with the OPL, which appeared grossly normal, dendritic stratification within the IPL in DGF/−;Six3Cre mutant retinas was disrupted. The laminated dendritic strata appeared expanded (Fig. 6D), fragmented (Fig. 6D–F), and occasionally lacked an entire lamina (Fig. 6E). These defects were restricted to regions of the retina where ectopic neuronal clusters were present, whereas regions of the DGF/−;Six3Cre mutant retina with normal cellular migration and lamination also had normal dendritic stratification (Fig. 5F). These results demonstrate that the ectopic clusters consisted of multiple subtypes of amacrine cells and ganglion cells that normally reside in the INL and GCL and that the disorganization of the dendritic strata are likely secondary to the cell migration defects.
Over the course of retinal development, multiple cell types, including horizontal cells and amacrine cells, develop mosaic spacing patterns that ensure that cells maintain complete and nonrandom coverage over the surface of the retina (Wässle and Riemann, 1978). This final mosaic pattern is established by both the removal of excess cells through apoptosis and the lateral dispersion of “like-subtype” cells via homotypic avoidance mechanisms (Kay et al., 2012; Li et al., 2015). To determine whether the defects in establishing proper laminar positioning of retinal subtypes in DGF/−;Six3Cre mutants extends to mosaic spacing, we performed nearest neighbor analysis. For horizontal cells, which exhibit normal lamination in DGF/−;Six3Cre mutant retinas, we observed a small, but significant reduction in the number of cells (two-way ANOVA, p < 0.0001, Tukey's HSD post hoc test, ***p < 0.0001; Fig. 7A,B). Despite the reduction in horizontal cell number, nearest neighbor curves between controls and mutants are the same shape, indicating that horizontal cell mosaics are maintained in dystroglycan mutants (Fig. 7A,B).
Figure 7.

Dystroglycan is required for mosaic cell spacing in the GCL. A, B, Horizontal cells (calbindin) in flat-mount P14 adult retinas have reduced cellular density, but normal mosaic cell spacing curves (nearest neighbor analysis, p < 0.0001, two-way ANOVA, Tukey's HSD post hoc test, ***p < 0.0001, n = 20 samples from 5 control retinas, 18 samples from 5 mutant retinas). C, D, Mosaic cell spacing of starburst amacrine cells (ChAT) in the INL is normal (nearest neighbor analysis, two-way ANOVA, Tukey's HSD post hoc test, *p = 0.0494, n = 10 samples from 3 control retinas, 10 samples from 3 mutant retinas), whereas the ectopic clustering of starburst amacrine cells in the GCL results in a significant disruption of mosaic spacing (nearest neighbor analysis, p < 0.0001, two-way ANOVA, Tukey's HSD post hoc test, ***p < 0.0001, n = 10 samples from 3 control retinas, 10 samples from 3 mutant retinas). Scale bar, 100 μm.
ChAT-positive starburst amacrine cells are present in two distinct lamina that form mosaic spacing patterns independent from one another. Consistent with this, ChAT-labeled cells in the INL showed normal mosaic cell spacing (two-way ANOVA, Tukey's HSD post hoc test, *p = 0.0494; Fig. 7C,D, top). In contrast, the GCL contained prominent ChAT-positive clusters that corresponded to the ectopic protrusions that extend into the vitreous, resulting in a decrease in cell spacing as determined by nearest neighbor analysis (p < 0.0001, Tukey's HSD post hoc test, ***p < 0.0001, two-way ANOVA; Fig. 7C, D, bottom). These results demonstrate that laminar migration defects in DGF/−;Six3Cre mutants degrade the mosaic spacing of cells in the GCL and that contact with an intact ILM is likely required for the proper lateral dispersion of these cells.
Deletion of dystroglycan leads to a loss of photoreceptors, horizontal cells, and ganglion cells
During development, normal physiological apoptotic cell death during the first 2 postnatal weeks plays an important role in retinal maturation (Young, 1984). This process is critical for establishing the proper numbers and spacing of some subtypes of cells across the mature retina, as well as removing cells that fail to connect to appropriate synaptic targets (Braunger et al., 2014). Degeneration of the ILM during development can lead to a reduction in the number of ganglion cells and previous analysis of dystroglycanopathy mutants has noted thinning of the retina (Halfter et al., 2005; Lee et al., 2005; Satz et al., 2008; Chan et al., 2010; Takahashi et al., 2011). In agreement with these results, we observed that the retinas of DGF/−;Six3Cre mutants are thinner (Figs. 6, 8). However, the specific cell types affected by the loss of dystroglycan in the retina are unknown.
Figure 8.

Retinal thinning in dystroglycan mutants. A–D, P14 DGF/−;Six3Cre retinas show decreased retinal thickness (DAPI, p = 0.0039, t test, n = 7 control, 7 mutant), a decreased thickness of the photoreceptor layer (C; Recoverin, green, p = 0.0163, t test, n = 7 control, 7 mutant), and no change in thickness of the bipolar cell layer (D; Chx10, purple, p > 0.05, t test, n = 7 control, 7 mutant). Starburst amacrine cell density is normal in both the INL (E; p > 0.05, t test, n = 10 samples from 3 control retinas, 10 samples from 3 mutant retinas) and GCL (F; p > 0.05, t test, n = 10 samples from 3 control retinas, 10 samples from 3 mutant retinas), whereas horizontal cells show a slight reduction in cell density in P14 DGF/−;Six3Cre retinas (G; p = 0.0112, t test, n = 20 samples from 5 control retinas, 18 samples from 5 mutant retinas). H, I, Ganglion cell density (RNA binding protein with multiple splicing, RBPMS) is reduced by ∼50% in P14 DGF/−;Six3Cre retinas (p < 0.0001, t test, n = 12 samples from 3 control retinas, 12 samples from 3 mutant retinas). Scale bar, 50 μm.
To investigate the mechanism by which loss of dystroglycan contributes to retinal thinning, we began by measuring the distance between the edges of the inner and outer retina in control and DGF/−;Six3Cre mutants by DAPI staining and found that there was a significant reduction in overall retinal thickness by ∼20% in mutants (t test, p = 0.0039; Figs. 8A, blue, 8B). We next investigated which specific cell types contribute to retinal thinning. In the outer retina, the thickness of the photoreceptor layer (recoverin; t test, p = 0.0163; Figs. 8A, green, 8C) was reduced by ∼20% and the density of horizontal cells had a small yet significant reduction in DGF/−;Six3Cre mutants (calbindin; t test, p = 0.0112; Fig. 8G). In contrast, the thickness of the bipolar cell layer (Chx10; t test, p > 0.05; Figs. 8A, purple, 8D) was normal. In the inner retina, there was a 50% reduction in the density of ganglion cells (t test, p < 0.0001; Fig. 8H,I), whereas the density of ChAT-positive starburst amacrine cells in both the INL and GCL was normal in DGF/−;Six3Cre mutants (t test, p > 0.05; Fig. 8E,F). Therefore, a reduction in the number of photoreceptors, horizontal cells and RGCs contribute to the overall thinning of DGF/−;Six3Cre retinas.
To determine whether the reduction in photoreceptors, horizontal cells, and RGCs in the absence of dystroglycan was due to defects in proliferation of retinal progenitors, we examined phosphohistone H3 (PH3) staining at embryonic ages. PH3-positive mitotic progenitors were localized along the apical surface of the retina and were present at the normal number in ISPDL79*/L79* mutants at e13 and e16 and (t test, p > 0.05; Fig. 9A,B). To determine whether the reduced number of neurons in mutant retinas was due to increased apoptosis, we quantified the number of caspase-3-positive cells. At e13, we observed no difference between in the number of caspase-3-positive cells in ISPDL79*/L79* mutants and controls (data not shown). In contrast, there was a significant increase in caspase-3-positive cells in ISPDL79*/L79* mutants at e16 (t test, p < 0.0001; Fig. 9D) and P0 (t test, p < 0.0001; Fig. 9C,D) that was restricted to the GCL. Similarly, we observed an increased number of cleaved caspase-3-positive cells in the GCL in DGF/−;Six3Cre mutants at P0 (t test, p = 0.0279; Fig. 9C,D). These results led us to conclude that the loss of RGCs in DGF/−;Six3Cre mutants is due to increased apoptotic cell death.
Figure 9.

Loss of dystroglycan results in increased developmental cell death. A, Immunohistochemistry for mitotic cells (PH3) at e13 (top) and e16 (bottom) shows normally positioned mitotic retinal progenitor cells adjacent to the RPE. B, Quantification of mitotic cells shows no difference between control and ISPDL79*/L79* mutants (p > 0.05, t test, n = 4 control and 4 mutant retinas at e13, p > 0.05, t test, n = 4 control and 4 mutant retinas at e16). C, D, Immunohistochemistry for cleaved caspase-3 in a flat-mount preparation of P0 DGF/−;Six3Cre retinas shows an increase in apoptotic cells (p = 0.0279, t test, n = 18 samples from 6 control retinas, 17 samples from 6 mutant retinas). D, Quantification of cleaved caspase-3-positive cells shows an increase in apoptotic cells at e16 (p < 0.0001, t test, n = 18 samples from 6 control retinas, 18 samples from 8 mutant retinas) and P0 (C, D; p < 0.0001, t test, n = 18 samples from 6 control retinas, 15 samples from 5 mutant retinas) between control (left) and ISPDL79*/L79* (middle) retinas. Scale bar, 50 μm.
Dystroglycan functions non-cell-autonomously as an extracellular scaffold in the developing retina
We next sought to provide mechanistic insight into how dystroglycan regulates retinal development in vivo. In the cerebral cortex, the loss of dystroglycan results in breaches of the pial basement membrane and detachment of neuroepithelial endfeet from the pial surface, depriving neurons of a migratory scaffold. In addition, the cortical basement membrane defects cause the mispositioning of Cajal–Retzius cells, which are the source of Reelin that regulates somal translocation of neurons as they detach from the neuroepithelial scaffold (Nakagawa et al., 2015). Deletion of dystroglycan specifically from postmitotic cortical neurons does not result in a migration phenotype (Satz et al., 2010), supporting a model in which the cortical migration phenotypes arise due to disrupted interactions between the basement membrane and neuroepithelial scaffold. In contrast to the cerebral cortex, the basal migration of RGCs does not involve contact with the neuroepithelial scaffold. Instead, newly born RGCs migrate via somal translocation using an ILM-attached basal process that eventually becomes the nascent axon (Randlett et al., 2011; Icha et al., 2016). Dystroglycan's expression in ILM-attached basal processes (Fig. 1A) and RGCs (Montanaro et al., 1995) and the restriction of neuronal migration and axon guidance defects to the GCL raise the possibility that dystroglycan could be functioning cell-autonomously in the basal processes of newly born RGCs. To test this possibility, we generated DGF/−;Isl1Cre conditional knock-outs. Islet1 is expressed in the majority of ganglion cells as they differentiate from the retinal progenitor pool (Pan et al., 2008). Analysis of Isl1Cre mice at e13 confirmed recombination occurs in the majority of newly born ganglion cells, but not in neuroepithelial progenitors (Fig. 10A). Interestingly, dystroglycan protein expression in DGF/−;Isl1Cre mutants was not altered compared with controls, suggesting that RGCs do not provide a significant source of dystroglycan at the ILM (Fig. 10B). Examination of DGF/−;Isl1Cre mutants indicated that deletion of dystroglycan selectively from RGCs did not affect ILM integrity (Fig. 10C). Neuronal migration (Fig. 10C,E,F), axon guidance (Fig. 10C,D), and the stratification of dendrites in the IPL (Fig. 10E,F) all appeared normal in DGF/−;Isl1Cre mutants. These results demonstrate that dystroglycan is not required within RGCs themselves during retinal development.
Figure 10.
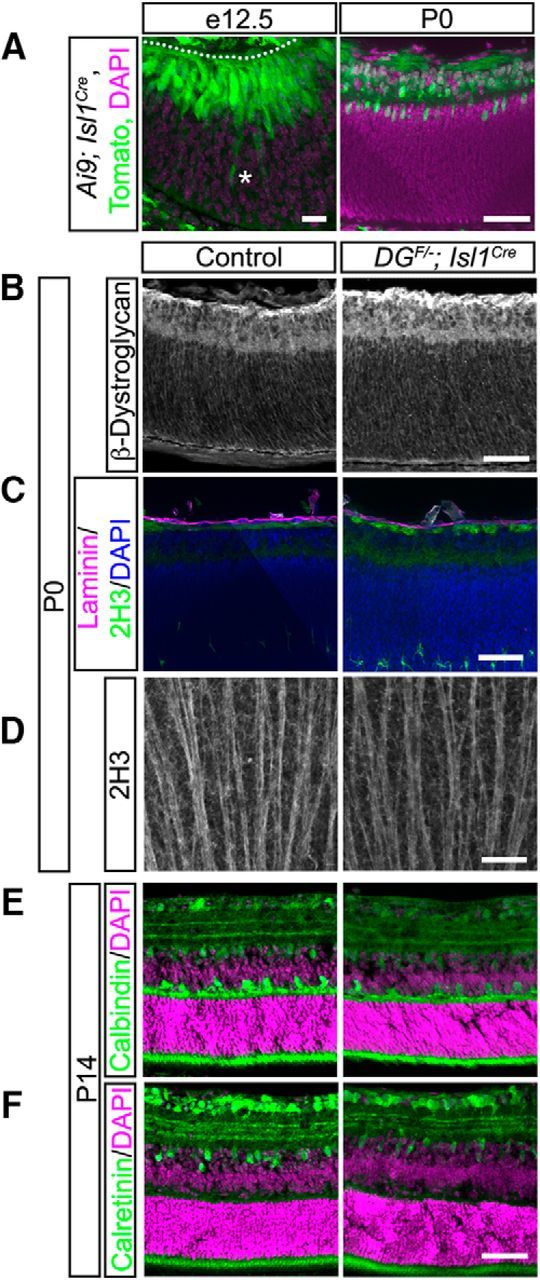
Dystroglycan is not required within RGCs for their migration and axon outgrowth. A, Recombination pattern of Rosa26-lox-stop-lox-TdTomato;Ai9 reporter (green) by Isl1Cre by shows expression the majority of differentiated ganglion cells at e12.5 (left) and P0 (right). B, Dystroglycan expression at the ILM is unchanged between control and DGF/−;Isl1Cre retinas. C, ILM (laminin, purple) and axons in the OFL (2H3, green) in DGF/−;Isl1Cre retinas appear similar to control. D, Flat-mount preparations show normal axon fasciculation (2H3) in DGF/−;Isl1Cre retinas. E, F, DGF/−;Isl1Cre (right) retinas (P14) have normal cellular lamination and dendritic stratification. Dashed line indicates ILM. Asterisk indicates a differentiated ganglion cell body that is still migrating toward the ILM. Scale bars: A, left, 20 μm; A, right, B–F, 50 μm.
Dystroglycan consists of two subunits that can play distinct roles in the overall function of the protein. The extracellular α-subunit is heavily glycosylated and functions as an extracellular scaffold by binding to ECM components such as laminin. The β-subunit contains a transmembrane and intracellular domain and can bind directly to the intracellular scaffolding protein dystrophin and other modifiers of the actin cytoskeleton, as well as initiate intracellular signaling cascades (Moore and Winder, 2010). The intracellular domain of dystroglycan is required for the localization of dystrophin to the ILM and mice lacking the intracellular domain of dystroglycan (DG−/βcyt; Satz et al., 2009) or the predominant retinal isoform of dystrophin (Mdx3Cv; Blank et al., 1999) have abnormal scotopic electroretinograms, suggesting a defect in retinal function. Although these mice do not have any disruptions in the ILM or gross malformations in the retina, whether dystroglycan signaling through dystrophin is required for neuronal migration, axon guidance, or dendritic stratification has not been examined. Consistent with the original report, examination of the ILM and overall architecture of the retina is normal in DG−/βcyt mice (Fig. 11A). In addition, we find that neuronal migration, axon guidance, and stratification of dendritic lamina are unaffected in DG−/βcyt mice (Fig. 11A–D). Therefore, intracellular signaling, including through dystrophin, is not required for these aspects of retinal development. Together with our results in DGF/−;Isl1Cre mutants, these findings indicate that dystroglycan primarily functions within neuroepithelial cells as an extracellular scaffold to regulate the structural integrity of the ILM. The progressive degeneration of the ILM then leads to secondary defects including aberrant migration, axon guidance, and dendritic stratification that primarily affect the inner retina.
Figure 11.
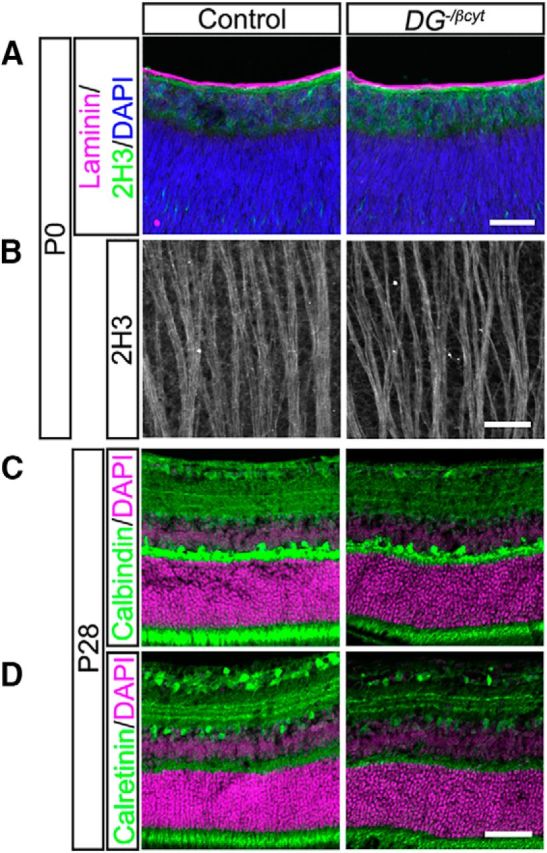
Intracellular signaling domain of dystroglycan is not required for proper retinal development. A, ILM integrity (laminin, purple), neuronal migration (DAPI, blue), and axon outgrowth (2H3, green) all appear normal in mice lacking the intracellular domain of dystroglycan (DG−/βcyt) at P0. B, Flat-mount preparations showing normal axon fasciculation (2H3) in DG−/βcyt retinas. C, D, DG−/βcyt retinas (P28) have normal cellular lamination and dendritic stratification. Scale bar, 50 μm.
Dystroglycan is dispensable for the generation of spontaneous retinal waves
One of the critical functions for laminar targeting in neural circuit development is to ensure that the axons and dendrites of appropriate cell types are in physical proximity to one another during synaptogenesis. In addition to regulating the laminar positioning of neurons in the cortex and retina, dystroglycan is required for the development of a subset of inhibitory synapses in the brain (Früh et al., 2016). Therefore, we investigated the possibility that the loss of dystroglycan disrupts synapse formation in the retina. Previous studies have shown that ribbon synapses in the OPL are dysfunctional in the absence of dystroglycan or its ligand, pikachurin (Sato et al., 2008). These synapses are normal at the resolution of light microscopy, but electron microscopy reveals that dystroglycan and pikachurin are required for the insertion of bipolar cell dendrite tips into ribbon synapse invaginations (Omori et al., 2012). Consistent with these results, we found that presynaptic and postsynaptic markers for ribbon synapses are present in DGF/−;Six3Cre mutants (Fig. 12A). In the inner retina, markers for excitatory synapses (VGLUT1; Fig. 12B) and inhibitory synapses (VGAT; Fig. 12C) were present in the IPL and were also present in the mislocalized ectopic cell clusters that protrude into the vitreous (asterisks). This finding is similar to a recent study in which mice lacking the Cas family of intracellular adaptor proteins express synaptic markers localized to aberrant neuronal ectopia that protrude into the vitreous (Riccomagno et al., 2014). These results suggest that mislaminated neurons in the retina are still able to recruit synaptic partners despite their abnormal location.
Figure 12.

Synaptic markers are present in the retinas of dystroglycan mutants. A, Markers for outer retinal ribbon synapses (Ribeye, presynaptic and mGluR6, postsynaptic) appear structurally normal in the absence of dystroglycan. The density of excitatory (VGLUT1) (B) and inhibitory (VGAT) (C) presynaptic markers appear similar to control in the inner retinas of DGF/−;Six3Cre mutants. Synapses are also present within ectopic clusters (asterisks). Scale bar, 50 μm wide view; 10 μm enlarged view.
The presence of synaptic markers in ectopic neuronal clusters in the inner retina does not guarantee normal function of these neurons. Recording synaptic activity in RGCs in response to light stimuli in DGF/−;Six3Cre mutants is not feasible due to the requirement for dystroglycan at photoreceptor ribbon synapses. We instead analyzed retinal waves, which are spontaneous bursts of activity that propagate across the retina before eye opening and are independent of light stimulation. During early postnatal development, these waves are initiated by acetylcholine (ACh) release from starburst amacrine cells and propagate along the starburst amacrine cell network before transmission to RGCs (Xu et al., 2016). In DGF/−;Six3Cre mutants, ChAT-positive starburst amacrine cells are present in normal numbers and, whereas they are normally localized and mosaically spaced in the INL, they are disorganized in the GCL. Therefore, we expected that these defects might affect the propagation of retinal waves through the starburst amacrine cell and RGC network. Because disruptions in the ILM in DGF/−;Six3Cre mutants would lead to unequal bulk loading of cell-permeable calcium indicators, we used the genetically encoded calcium indicator GCaMP6f crossed onto the DG;Six3Cre line to visualize retinal waves.
Retinal waves in control DGF/+;Six3Cre;R26-LSL-GCaMP6f retinas at P1–P2 were robust and had similar spatiotemporal features (area, rate of propagation, refractory period) to waves measured using cell-permeable calcium indicators (Arroyo and Feller, 2016). Consistent with previous reports, there is a broad distribution of wave area in control retinas (Fig. 13A,C, Movie 1). Neighboring waves do not overlap with one another, but rather tile the retinal surface during the 2 min imaging period. To our surprise, retinal waves were present and appeared grossly normal in DGF/−;Six3Cre;R26-LSL-GCaMP6f mutants (Fig. 13B, Movie 2). Waves in control and mutant retinas exhibited a similar distribution in wave area (Fig. 13C) and the average wave area showed no statistical difference. The rate of wave propagation showed a similar distribution between controls and mutants (Fig. 13D). The average rate of wave propagation showed a small, but statistically significant, decrease (Wilcoxon rank-sum test, p = 0.0493). Therefore, despite the dramatic disorganization of ChAT-positive starburst amacrine cells in the GCL of DGF/−;Six3Cre mutants, the generation and propagation of retinal waves persisted.
Figure 13.

Dystroglycan is dispensable for the generation and propagation of retinal waves. A, B, At P1, waves propagate normally across the retina in DGF/+;Six3Cre;R26-LSL-GCaMP6f (A) and DGF/−;Six3Cre;R26-LSL-GCaMP6f mice (B). C, Distributions of wave areas show no difference between controls and mutants (Wilcoxon rank-sum test, p > 0.05). D, Wave propagation rate is slightly slower in mutant retinas (Wilcoxon rank-sum test, p = 0.0493). Wave parameters were calculated from 142 control waves obtained from six retinas from three control mice and 173 mutant waves obtained from eight retinas from five mutant mice. Arrowheads indicate the initiation site of a retinal wave. Scale bar, 500 μm. Time is displayed in milliseconds.
Retinal waves in DGF/+;Six3Cre;R26-LSL-GCaMP6f mice. Shown are 15 frames/s for a total of 21 frames.
Retinal waves in DGF/−;Six3Cre;R26-LSL-GCaMP6f mice. Shown are 15 frames/s for a total of 21 frames.
Discussion
Although defects in retinal structure and function are observed in both human patients and mouse models of dystroglycanopathy, the mechanism of dystroglycan function in the mammalian retina and the consequence of its loss on specific cell types are poorly understood (Takeda et al., 2003; Lee et al., 2005; Satz et al., 2008; Satz et al., 2009; Chan et al., 2010; Takahashi et al., 2011). Our study establishes a critical role for dystroglycan in maintaining the integrity of the ILM, which is required for intraretinal axon guidance and establishing laminar architecture and mosaic spacing in the inner retina. Mechanistically, we show that dystroglycan functions non-cell-autonomously as an extracellular scaffold because mice with selective deletion of dystroglycan from postmitotic RGCs (DG;Isl1Cre) and mice lacking the intracellular domain of dystroglycan (DG−/βcyt) appear phenotypically normal. Despite the dramatic disruptions in cellular lamination in the GCL and dendritic stratification in the IPL in DG;Six3Cre mutants, dystroglycan appears dispensable for the formation of synapses and the generation of spontaneous, light-independent activity in the retina.
Requirement for dystroglycan at the ILM during retinal development
Using two genetic models for the complete loss of functional dystroglycan (ISPDL79* and DG;Sox2Cre), we demonstrated that, after the ILM forms, it degenerates rapidly in the absence of dystroglycan. These results suggest that dystroglycan is not required for the formation of the nascent ILM, but rather plays a critical role in maintaining ILM structure as it expands to accommodate the growing retina. Recruitment of laminin is a critical early step in the assembly of the ILM and several laminin mutants have similar disruptions in ILM integrity (Edwards et al., 2010; Pinzón-Duarte et al., 2010; Gnanaguru et al., 2013). In contrast to Xenopus, in which depletion of dystroglycan results in degeneration of both the ILM in the inner retina and Bruch's membrane in the outer retina, Bruch's membrane is unaffected by the loss of dystroglycan or isoforms of Laminin (Lunardi et al., 2006; Pinzón-Duarte et al., 2010). Mice lacking β-1 Integrin in the retina exhibit a similar defect to dystroglycan mutants, raising the possibility that these laminin receptors may be functionally redundant (Riccomagno et al., 2014). Surprisingly, however, mice in which both dystroglycan and β-1-Integrin are deleted from the retina (DG;ItgB1;Six3Cre) still formed an ILM at e13 and the subsequent degeneration of the ILM was indistinguishable from DG;Six3Cre mutants (R.C., K.M.W., unpublished observations). Therefore, sulfated glycolipids alone are likely sufficient for the recruitment of laminin during the initial formation of the ILM.
ILM is required for neuronal migration and axon outgrowth in the retina
In the developing cortex, the loss of functional dystroglycan leads to the degeneration of the radial glia processes and ectopic clustering of reelin-secreting Cajal–Retzius neurons, which are thought to be the principal drivers of structural brain defects in dystroglycanopathies (Myshrall et al., 2012; Nakagawa et al., 2015; Booler et al., 2016). Retinal neurons do not migrate along the neuroepithelial scaffold and there is no cue analogous to reelin to signal termination of migration, suggesting that, whereas dystroglycan functions primarily in neuroepithelial cells in the retina, the functional implications are distinct from its role in cortical neuroepithelial cells. In the cortex of dystroglycan-deficient mice, the organization of neurons across all lamina is affected, whereas within the retina, migration defects are restricted to amacrine cells and RGCs in the inner retina. In contrast, migration defects in dystroglycan-deficient Xenopus retinas are more widespread and also affect outer retinal neurons, likely reflecting the requirement for dystroglycan at both the ILM and Bruch's membrane (Lunardi et al., 2006). What is the driving force behind the selective localization of amacrine cells and RGCs to the ectopic clusters that protrude into the vitreous in the mammalian retina? Although the elimination of RGCs does not affect the lamination of other neurons, other neurons will organize themselves around mislocalized RGCs, resulting in an overall disorganization of retinal lamination (Wang et al., 2001; Kay et al., 2004; Icha et al., 2016). In dystroglycan mutants, RGCs that encounter the degenerating ILM and inappropriately migrate into the vitreous may then actively recruit later-born neurons such as amacrine cells to inappropriate locations.
The establishment of retinal mosaics requires tangential migration that is regulated by short-range interactions between immature neurites of neighboring cells (Galli-Resta et al., 1997; Reese et al., 1999; Galli-Resta et al., 2002; Huckfeldt et al., 2009). A key feature of this process is that it requires homotypic cells to be localized within the same lamina and cells in which mosaic spacing is disrupted are no longer restricted to a 2D plane (Fuerst et al., 2008; Kay et al., 2012). In dystroglycan mutants, the laminar organization and mosaic spacing is normal in horizontal cells and INL starburst amacrine cells, but disrupted in starburst amacrine cells in the GCL. This defect is not due to dystroglycan functioning within starburst amacrine cells because mosaic spacing was normal in DG;Isl1Cre mutant retinas (data not shown). Rather, the selective defects in mosaic spacing of GCL starburst amacrine cells suggests that this is likely a consequence of disrupting the 2D organization of the GCL. Alternatively, mosaic spacing of cells within the GCL may require cues present in the ILM for their tangential dispersion.
Basement membranes are highly dynamic structures that contain pro-axon-growth ECM molecules such as laminins and collagens and also regulate the distribution of secreted axon guidance cues (Halfter et al., 1987; Chai and Morris, 1999; Xiao et al., 2011; Wright et al., 2012). A number of secreted cues direct intraretinal axon guidance of RGCs. Deletion of Netrin (Deiner et al., 1997) specifically affects exit of RGC axons through the optic nerve head and deletion of Slits (Thompson et al., 2006) or Sfrps (Marcos et al., 2015) leads to the invasion of RGC axons into the outer retina. In contrast, the randomized growth and defasciculation of axons that we observed in ISPDL79* and DG;Six3Cre mutant retinas is more consistent with defects observed upon deletion of adhesion receptors (Bastmeyer et al., 1995; Brittis et al., 1995; Ott et al., 1998). These results suggest that dystroglycan primarily functions to organize the ILM as a substrate for axonal adhesion.
Loss of retinal neurons in the absence of dystroglycan
Although previous studies of mouse models of dystroglycanopathy have consistently noted retinal thinning, it was unclear which retinal cell types were affected. Our comprehensive analysis found that lack of dystroglycan led to reductions in photoreceptor layer thickness, horizontal cell number, and RGC number (Fig. 8). Analysis of e13, e16, and P0 retinas from ISPDL79* and DG;Six3Cre mutants indicated that the loss of RGCs was not due to altered proliferation of retinal progenitors, but rather was primarily due to increased apoptosis of cells in the GCL that preceded and extended into the normal window of developmental apoptotic cell death. This increase in apoptotic cell death did not persist in adult (P56) DG;Six3Cre mutants (data not shown), suggesting that it was likely restricted to the developing retina. Why are RGCs lost at such a high rate in DG;Six3Cre mutants whereas the displaced amacrine cell number in the GCL remains normal? One possibility is that RGCs are affected selectively because they are the only cell type to project out of the retina. Indeed, we observed profound guidance defects of RGCs at the optic chiasm in ISPDL79* and DG;Six3Cre mutants (R.C., K.M.W., unpublished observations). This result is similar to Isl1- and Brn3b-deficient mice, in which axon growth defects at the optic chiasm precede an increase in RGC apoptosis (Gan et al., 1999; Pan et al., 2008). The death of RGCs with axons that fail to reach retinorecipient regions of the brain is consistent with the need for target-derived factors to support their survival, although the identity of these factors remains elusive.
Although we did not observe increased caspase-3 reactivity in photoreceptors or horizontal cells at P0, it is possible that the loss of cells occurred gradually during the first 2 postnatal weeks. Dystroglycan is required for the proper formation of ribbon synapses among photoreceptors, horizontal cells, and bipolar cells in the OPL (Sato et al., 2008; Omori et al., 2012). Therefore, the loss of appropriate synaptic contact in the absence of dystroglycan may lead to the elimination of a proportion of photoreceptors and horizontal cells.
Persistence of retinal waves in the absence of dystroglycan
The defects in lamination and dendritic stratification of starburst amacrine cells in DG;Six3Cre mutants led us to hypothesize that this would affect their ability to generate retinal waves. These waves are initiated by the spontaneous activity of starburst amacrine cells independent of light stimuli, allowing us to circumvent the requirement for dystroglycan in proper transmission at ribbon synapses. Contrary to our expectations, retinal waves were present and propagated normally in DG;Six3Cre mutants (Fig. 13). The persistence of retinal waves even in the context of disrupted cellular organization supports the model that these waves are the product of volume release of ACh from starburst amacrine cells that can trigger extrasynaptic responses in cells that are not physically connected (Ford et al., 2012). Therefore, the relatively normal organization of INL starburst amacrine cells may be sufficient to overcome the disorganization of GCL starburst amacrine cells.
Conclusion
Retinal dysplasia and optic nerve hypoplasia are frequently observed in patients with severe forms of dystroglycanopathy (Manzini et al., 2008). Using multiple mouse models, we demonstrate that dystroglycan is required for multiple aspects of retinal development. We show that dystroglycan functions within neuroepithelial cells in the retina to regulate the structural integrity of a basement membrane (the ILM), which is required for the coordination of neuronal migration, axon guidance, and dendritic stratification in the inner retina. In addition, we find that there is a significant loss of photoreceptors, horizontal cells, and almost 50% of RGCs due to increased apoptotic cell death. Our data suggest that the disorganization of the inner retina resulting from degeneration of the ILM is a key contributor to visual impairment in dystroglycanopathies.
Footnotes
This work was supported by the National Institutes of Health (Grants R01-NS091027 (K.M.W.), the Whitehall Institute (K.M.W.), the Medical Research Foundation of Oregon (K.M.W.), the National Science Foundation Graduate Research Fellowship Program (R.C.), a LaCroute Neurobiology of Disease Fellowship (R.C.), a Tartar Trust Fellowship (R.C.), the National Institute of Neurological Disorders and Stroke (Grant P30-NS061800 to the Oregon Health & Science University Advanced Light Microscopy Core), and the Paul D. Wellstone Muscular Dystrophy Cooperative Research Center (Grant 1U54NS053672 to K.P.C.). K.P.C. is an investigator of the Howard Hughes Medical Institute. We thank Patrick Kerstein and Kylee Rosette for their technical assistance; Marla Feller and Franklin Caval-Holme for advice on visualizing and analyzing retinal waves; W. Rowland Taylor and Teresa Puthussery and members of their laboratories for antibodies and technical advice; David Pow for the GlyT1 antibody; Catherine Morgans for the mGluR6 antibody, Stefanie Kaech Petrie and the Oregon Health & Science University Advanced Light Microscopy Core for assistance with confocal imaging; and David Ginty, Alex Kolodkin, Martin Riccomagno, Randall Hand, and members of the Campbell and Wright laboratories for discussion throughout the course of this study and comments on the manuscript.
The authors declare no competing financial interests.
References
- Arroyo DA, Feller MB (2016) Spatiotemporal features of retinal waves instruct the wiring of the visual circuitry. Front Neural Circuits 10:54. 10.3389/fncir.2016.00054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao ZZ. (2008) Intraretinal projection of retinal ganglion cell axons as a model system for studying axon navigation. Brain Res 1192:165–177. 10.1016/j.brainres.2007.01.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassett EA, Wallace VA (2012) Cell fate determination in the vertebrate retina. Trends Neurosci 35:565–573. 10.1016/j.tins.2012.05.004 [DOI] [PubMed] [Google Scholar]
- Bastmeyer M, Ott H, Leppert CA, Stuermer CA (1995) Fish E587 glycoprotein, a member of the L1 family of cell adhesion molecules, participates in axonal fasciculation and the age-related order of ganglion cell axons in the goldfish retina. J Cell Biol 130:969–976. 10.1083/jcb.130.4.969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blank M, Koulen P, Blake DJ, Kröger S (1999) Dystrophin and beta-dystroglycan in photoreceptor terminals from normal and mdx3Cv mouse retinae. Eur J Neurosci 11:2121–2133. 10.1046/j.1460-9568.1999.00636.x [DOI] [PubMed] [Google Scholar]
- Blankenship AG, Ford KJ, Johnson J, Seal RP, Edwards RH, Copenhagen DR, Feller MB (2009) Synaptic and extrasynaptic factors governing glutamatergic retinal waves. Neuron 62:230–241. 10.1016/j.neuron.2009.03.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booler HS, Williams JL, Hopkinson M, Brown SC (2016) Degree of Cajal-Retzius cell mislocalization correlates with the severity of structural brain defects in mouse models of dystroglycanopathy. Brain Pathol 26:465–478. 10.1111/bpa.12306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braunger BM, Demmer C, Tamm ER (2014) Programmed cell death during retinal development of the mouse eye. Adv Exp Med Biol 801:9–13. 10.1007/978-1-4614-3209-8_2 [DOI] [PubMed] [Google Scholar]
- Brittis PA, Lemmon V, Rutishauser U, Silver J (1995) Unique changes of ganglion cell growth cone behavior following cell adhesion molecule perturbations: a time-lapse study of the living retina. Mol Cell Neurosci 6:433–449. 10.1006/mcne.1995.1032 [DOI] [PubMed] [Google Scholar]
- Chai L, Morris JE (1999) Heparan sulfate in the inner limiting membrane of embryonic chicken retina binds basic fibroblast growth factor to promote axonal outgrowth. Exp Neurol 160:175–185. 10.1006/exnr.1999.7195 [DOI] [PubMed] [Google Scholar]
- Chan YM, Keramaris-Vrantsis E, Lidov HG, Norton JH, Zinchenko N, Gruber HE, Thresher R, Blake DJ, Ashar J, Rosenfeld J, Lu QL (2010) Fukutin-related protein is essential for mouse muscle, brain and eye development and mutation recapitulates the wide clinical spectrums of dystroglycanopathies. Hum Mol Genet 19:3995–4006. 10.1093/hmg/ddq314 [DOI] [PubMed] [Google Scholar]
- Deiner MS, Kennedy TE, Fazeli A, Serafini T, Tessier-Lavigne M, Sretavan DW (1997) Netrin-1 and DCC mediate axon guidance locally at the optic disc: loss of function leads to optic nerve hypoplasia. Neuron 19:575–589. 10.1016/S0896-6273(00)80373-6 [DOI] [PubMed] [Google Scholar]
- Dobyns WB, Pagon RA, Armstrong D, Curry CJ, Greenberg F, Grix A, Holmes LB, Laxova R, Michels VV, Robinow M (1989) Diagnostic criteria for Walker-Warburg syndrome. Am J Med Genet 32:195–210. 10.1002/ajmg.1320320213 [DOI] [PubMed] [Google Scholar]
- Edwards MM, Mammadova-Bach E, Alpy F, Klein A, Hicks WL, Roux M, Simon-Assmann P, Smith RS, Orend G, Wu J, Peachey NS, Naggert JK, Lefebvre O, Nishina PM (2010) Mutations in Lama1 disrupt retinal vascular development and inner limiting membrane formation. J Biol Chem 285:7697–7711. 10.1074/jbc.M109.069575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford KJ, Félix AL, Feller MB (2012) Cellular mechanisms underlying spatiotemporal features of cholinergic retinal waves. J Neurosci 32:850–863. 10.1523/JNEUROSCI.5309-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Früh S, Romanos J, Panzanelli P, Bürgisser D, Tyagarajan SK, Campbell KP, Santello M, Fritschy JM (2016) Neuronal dystroglycan is necessary for formation and maintenance of functional CCK-positive basket cell terminals on pyramidal cells. J Neurosci 36:10296–10313. 10.1523/JNEUROSCI.1823-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fruttiger M. (2007) Development of the retinal vasculature. Angiogenesis 10:77–88. 10.1007/s10456-007-9065-1 [DOI] [PubMed] [Google Scholar]
- Fuerst PG, Koizumi A, Masland RH, Burgess RW (2008) Neurite arborization and mosaic spacing in the mouse retina require DSCAM. Nature 451:470–474. 10.1038/nature06514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuta Y, Lagutin O, Hogan BL, Oliver GC (2000) Retina- and ventral forebrain-specific Cre recombinase activity in transgenic mice. Genesis 26:130–132. [PubMed] [Google Scholar]
- Galli-Resta L, Resta G, Tan SS, Reese BE (1997) Mosaics of islet-1-expressing amacrine cells assembled by short-range cellular interactions. J Neurosci 17:7831–7838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galli-Resta L, Novelli E, Viegi A (2002) Dynamic microtubule-dependent interactions position homotypic neurones in regular monolayered arrays during retinal development. Development 129:3803–3814. [DOI] [PubMed] [Google Scholar]
- Gan L, Wang SW, Huang Z, Klein WH (1999) POU domain factor Brn-3b is essential for retinal ganglion cell differentiation and survival but not for initial cell fate specification. Dev Biol 210:469–480. 10.1006/dbio.1999.9280 [DOI] [PubMed] [Google Scholar]
- Gnanaguru G, Bachay G, Biswas S, Pinzón-Duarte G, Hunter DD, Brunken WJ (2013) Laminins containing the beta2 and gamma3 chains regulate astrocyte migration and angiogenesis in the retina. Development 140:2050–2060. 10.1242/dev.087817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halfter W, Reckhaus W, Kröger S (1987) Nondirected axonal growth on basal lamina from avian embryonic neural retina. J Neurosci 7:3712–3722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halfter W, Willem M, Mayer U (2005) Basement membrane-dependent survival of retinal ganglion cells. Invest Ophthalmol Vis Sci 46:1000–1009. 10.1167/iovs.04-1185 [DOI] [PubMed] [Google Scholar]
- Hayashi S, Lewis P, Pevny L, McMahon AP (2002) Efficient gene modulation in mouse epiblast using a Sox2Cre transgenic mouse strain. Mechanisms of Development 119:S97–S101. 10.1016/S0925-4773(03)00099-6 [DOI] [PubMed] [Google Scholar]
- Huckfeldt RM, Schubert T, Morgan JL, Godinho L, Di Cristo G, Huang ZJ, Wong RO (2009) Transient neurites of retinal horizontal cells exhibit columnar tiling via homotypic interactions. Nat Neurosci 12:35–43. 10.1038/nn.2236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Icha J, Kunath C, Rocha-Martins M, Norden C (2016) Independent modes of ganglion cell translocation ensure correct lamination of the zebrafish retina. J Cell Biol 215:259–275. 10.1083/jcb.201604095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay JN, Roeser T, Mumm JS, Godinho L, Mrejeru A, Wong RO, Baier H (2004) Transient requirement for ganglion cells during assembly of retinal synaptic layers. Development 131:1331–1342. 10.1242/dev.01040 [DOI] [PubMed] [Google Scholar]
- Kay JN, Chu MW, Sanes JR (2012) MEGF10 and MEGF11 mediate homotypic interactions required for mosaic spacing of retinal neurons. Nature 483:465–469. 10.1038/nature10877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Kameya S, Cox GA, Hsu J, Hicks W, Maddatu TP, Smith RS, Naggert JK, Peachey NS, Nishina PM (2005) Ocular abnormalities in Large(myd) and Large(vls) mice, spontaneous models for muscle, eye, and brain diseases. Mol Cell Neurosci 30:160–172. 10.1016/j.mcn.2005.07.009 [DOI] [PubMed] [Google Scholar]
- Li S, Sukeena JM, Simmons AB, Hansen EJ, Nuhn RE, Samuels IS, Fuerst PG (2015) DSCAM promotes refinement in the mouse retina through cell death and restriction of exploring dendrites. J Neurosci 35:5640–5654. 10.1523/JNEUROSCI.2202-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunardi A, Cremisi F, Dente L (2006) Dystroglycan is required for proper retinal layering. Dev Biol 290:411–420. 10.1016/j.ydbio.2005.11.044 [DOI] [PubMed] [Google Scholar]
- Manzini MC, Gleason D, Chang BS, Hill RS, Barry BJ, Partlow JN, Poduri A, Currier S, Galvin-Parton P, Shapiro LR, Schmidt K, Davis JG, Basel-Vanagaite L, Seidahmed MZ, Salih MA, Dobyns WB, Walsh CA (2008) Ethnically diverse causes of Walker-Warburg syndrome (WWS): FCMD mutations are a more common cause of WWS outside of the Middle East. Hum Mut 29:E231–E241. 10.1002/humu.20844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcos S, Nieto-Lopez F, Sandonìs A, Cardozo MJ, Di Marco F, Esteve P, Bovolenta P (2015) Secreted frizzled related proteins modulate pathfinding and fasciculation of mouse retina ganglion cell axons by direct and indirect mechanisms. J Neurosci 35:4729–4740. 10.1523/JNEUROSCI.3304-13.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montanaro F, Carbonetto S, Campbell KP, Lindenbaum M (1995) Dystroglycan expression in the wild type and mdx mouse neural retina: synaptic colocalization with dystrophin, dystrophin-related protein but not laminin. J Neurosci Res 42:528–538. 10.1002/jnr.490420411 [DOI] [PubMed] [Google Scholar]
- Moore CJ, Winder SJ (2010) Dystroglycan versatility in cell adhesion: a tale of multiple motifs. Cell Commun Signal 8:3. 10.1186/1478-811X-8-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore SA, Saito F, Chen J, Michele DE, Henry MD, Messing A, Cohn RD, Ross-Barta SE, Westra S, Williamson RA, Hoshi T, Campbell KP (2002) Deletion of brain dystroglycan recapitulates aspects of congenital muscular dystrophy. Nature 418:422–425. 10.1038/nature00838 [DOI] [PubMed] [Google Scholar]
- Myshrall TD, Moore SA, Ostendorf AP, Satz JS, Kowalczyk T, Nguyen H, Daza RA, Lau C, Campbell KP, Hevner RF (2012) Dystroglycan on radial glia end feet is required for pial basement membrane integrity and columnar organization of the developing cerebral cortex. J Neuropathol Exp Neurol 71:1047–1063. 10.1097/NEN.0b013e318274a128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa N, Yagi H, Kato K, Takematsu H, Oka S (2015) Ectopic clustering of Cajal-Retzius and subplate cells is an initial pathological feature in Pomgnt2-knockout mice, a model of dystroglycanopathy. Sci Rep 5:11163. 10.1038/srep11163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omori Y, Araki F, Chaya T, Kajimura N, Irie S, Terada K, Muranishi Y, Tsujii T, Ueno S, Koyasu T, Tamaki Y, Kondo M, Amano S, Furukawa T (2012) Presynaptic dystroglycan-pikachurin complex regulates the proper synaptic connection between retinal photoreceptor and bipolar cells. J Neurosci 32:6126–6137. 10.1523/JNEUROSCI.0322-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott H, Bastmeyer M, Stuermer CA (1998) Neurolin, the goldfish homolog of DM-GRASP, is involved in retinal axon pathfinding to the optic disk. J Neurosci 18:3363–3372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan L, Deng M, Xie X, Gan L (2008) ISL1 and BRN3B co-regulate the differentiation of murine retinal ganglion cells. Development 135:1981–1990. 10.1242/dev.010751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinzón-Duarte G, Daly G, Li YN, Koch M, Brunken WJ (2010) Defective formation of the inner limiting membrane in laminin beta2- and gamma3-null mice produces retinal dysplasia. Invest Ophthalmol Vis Sci 51:1773–1782. 10.1167/iovs.09-4645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randlett O, Poggi L, Zolessi FR, Harris WA (2011) The oriented emergence of axons from retinal ganglion cells is directed by laminin contact in vivo. Neuron 70:266–280. 10.1016/j.neuron.2011.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reese BE. (2011) Development of the retina and optic pathway. Vision Res 51:613–632. 10.1016/j.visres.2010.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reese BE, Necessary BD, Tam PP, Faulkner-Jones B, Tan SS (1999) Clonal expansion and cell dispersion in the developing mouse retina. Eur J Neurosci 11:2965–2978. 10.1046/j.1460-9568.1999.00712.x [DOI] [PubMed] [Google Scholar]
- Riccomagno MM, Sun LO, Brady CM, Alexandropoulos K, Seo S, Kurokawa M, Kolodkin AL (2014) Cas adaptor proteins organize the retinal ganglion cell layer downstream of integrin signaling. Neuron 81:779–786. 10.1016/j.neuron.2014.01.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato S, Omori Y, Katoh K, Kondo M, Kanagawa M, Miyata K, Funabiki K, Koyasu T, Kajimura N, Miyoshi T, Sawai H, Kobayashi K, Tani A, Toda T, Usukura J, Tano Y, Fujikado T, Furukawa T (2008) Pikachurin, a dystroglycan ligand, is essential for photoreceptor ribbon synapse formation. Nat Neurosci 11:923–931. 10.1038/nn.2160 [DOI] [PubMed] [Google Scholar]
- Satz JS, Barresi R, Durbeej M, Willer T, Turner A, Moore SA, Campbell KP (2008) Brain and eye malformations resembling Walker-Warburg syndrome are recapitulated in mice by dystroglycan deletion in the epiblast. J Neurosci 28:10567–10575. 10.1523/JNEUROSCI.2457-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satz JS, Philp AR, Nguyen H, Kusano H, Lee J, Turk R, Riker MJ, Hernández J, Weiss RM, Anderson MG, Mullins RF, Moore SA, Stone EM, Campbell KP (2009) Visual impairment in the absence of dystroglycan. J Neurosci 29:13136–13146. 10.1523/JNEUROSCI.0474-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satz JS, Ostendorf AP, Hou S, Turner A, Kusano H, Lee JC, Turk R, Nguyen H, Ross-Barta SE, Westra S, Hoshi T, Moore SA, Campbell KP (2010) Distinct functions of glial and neuronal dystroglycan in the developing and adult mouse brain. J Neurosci 30:14560–14572. 10.1523/JNEUROSCI.3247-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi H, Kanesaki H, Igarashi T, Kameya S, Yamaki K, Mizota A, Kudo A, Miyagoe-Suzuki Y, Takeda S, Takahashi H (2011) Reactive gliosis of astrocytes and Muller glial cells in retina of POMGnT1-deficient mice. Mol Cell Neurosci 47:119–130. 10.1016/j.mcn.2011.03.006 [DOI] [PubMed] [Google Scholar]
- Takeda S, Kondo M, Sasaki J, Kurahashi H, Kano H, Arai K, Misaki K, Fukui T, Kobayashi K, Tachikawa M, Imamura M, Nakamura Y, Shimizu T, Murakami T, Sunada Y, Fujikado T, Matsumura K, Terashima T, Toda T (2003) Fukutin is required for maintenance of muscle integrity, cortical histiogenesis and normal eye development. Hum Mol Genet 12:1449–1459. 10.1093/hmg/ddg153 [DOI] [PubMed] [Google Scholar]
- Taniguchi-Ikeda M, Morioka I, Iijima K, Toda T (2016) Mechanistic aspects of the formation of alpha-dystroglycan and therapeutic research for the treatment of alpha-dystroglycanopathy: a review. Mol Aspects Med 51:115–124. 10.1016/j.mam.2016.07.003 [DOI] [PubMed] [Google Scholar]
- Tao C, Zhang X (2016) Retinal proteoglycans act as cellular receptors for basement membrane assembly to control astrocyte migration and angiogenesis. Cell Rep 17:1832–1844. 10.1016/j.celrep.2016.10.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor L, Arnér K, Engelsberg K, Ghosh F (2015) Scaffolding the retina: the interstitial extracellular matrix during rat retinal development. Int J Dev Neurosci 42:46–58. 10.1016/j.ijdevneu.2015.03.002 [DOI] [PubMed] [Google Scholar]
- Thompson H, Camand O, Barker D, Erskine L (2006) Slit proteins regulate distinct aspects of retinal ganglion cell axon guidance within dorsal and ventral retina. J Neurosci 26:8082–8091. 10.1523/JNEUROSCI.1342-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tronche F, Kellendonk C, Kretz O, Gass P, Anlag K, Orban PC, Bock R, Klein R, Schütz G (1999) Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat Genet 23:99–103. 10.1038/12703 [DOI] [PubMed] [Google Scholar]
- Varshney S, Hunter DD, Brunken WJ (2015) Extracellular matrix components regulate cellular polarity and tissue structure in the developing and mature retina. J Ophthalmic Vis Res 10:329–339. 10.4103/2008-322X.170354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SW, Kim BS, Ding K, Wang H, Sun D, Johnson RL, Klein WH, Gan L (2001) Requirement for math5 in the development of retinal ganglion cells. Genes Dev 15:24–29. 10.1101/gad.855301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wässle H, Riemann HJ (1978) The mosaic of nerve cells in the mammalian retina. Proc R Soc Lond B Biol Sci 200:441–461. 10.1098/rspb.1978.0026 [DOI] [PubMed] [Google Scholar]
- Wright KM, Lyon KA, Leung H, Leahy DJ, Ma L, Ginty DD (2012) Dystroglycan organizes axon guidance cue localization and axonal pathfinding. Neuron 76:931–944. 10.1016/j.neuron.2012.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao T, Staub W, Robles E, Gosse NJ, Cole GJ, Baier H (2011) Assembly of lamina-specific neuronal connections by slit bound to type IV collagen. Cell 146:164–176. 10.1016/j.cell.2011.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu HP, Burbridge TJ, Ye M, Chen M, Ge X, Zhou ZJ, Crair MC (2016) Retinal wave patterns are governed by mutual excitation among starburst amacrine cells and drive the refinement and maintenance of visual circuits. J Neurosci 36:3871–3886. 10.1523/JNEUROSCI.3549-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Cai CL, Lin L, Qyang Y, Chung C, Monteiro RM, Mummery CL, Fishman GI, Cogen A, Evans S (2006) Isl1Cre reveals a common Bmp pathway in heart and limb development. Development 133:1575–1585. 10.1242/dev.02322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young RW. (1984) Cell death during differentiation of the retina in the mouse. J Comp Neurol 229:362–373. 10.1002/cne.902290307 [DOI] [PubMed] [Google Scholar]
- Yurchenco PD. (2011) Basement membranes: cell scaffoldings and signaling platforms. Cold Spring Harb Perspect Biol 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Retinal waves in DGF/+;Six3Cre;R26-LSL-GCaMP6f mice. Shown are 15 frames/s for a total of 21 frames.
Retinal waves in DGF/−;Six3Cre;R26-LSL-GCaMP6f mice. Shown are 15 frames/s for a total of 21 frames.