

Abstract
An emerging group of high grade renal cell carcinomas (RCCs), particularly carcinomas arising in the hereditary leiomyomatosis renal cell carcinoma syndrome (HLRCC), show fumarate hydratase (FH) gene mutation and loss of function. Based on similar cytomorphology and clinicopathologic features between these tumors and cases described as tubulocystic carcinomas with poorly differentiated foci of infiltrative adenocarcinoma (TC-PD), we hypothesized a relationship between these entities. First, 29 RCCs with morphology of TC-PD were identified retrospectively and assessed for FH expression and aberrant succination (2SC) by immunohistochemistry (IHC), with targeted next generation sequencing (NGS) of 409 genes—including FH—performed on a subset. The 29 TC-PD RCCs included 21 males and 8 females, aged 16-86 years (median 46), with tumors measuring 3-21 cm (median 9) arising in the right (n=16) and left (n=13) kidneys. Family history or stigmata of HLRCC were identified in only 3 (12%). These tumors were aggressive, with 79% showing perinephric extension, nodal involvement in 41%, and metastasis in 86%. Of these, 16 (55%) demonstrated loss of FH by IHC (14/14 with positive 2SC). In contrast, 5 (17%) showed a wild type immunoprofile of FH+/2SC-. An intriguing group of 8 (28%) showed variable FH± positivity, but with strong/diffuse 2SC+. NGS revealed 8 cases with FH mutations, including 5 FH-/2SC+ and 3 FH±/2SC+ cases, but none in FH+/2SC- cases. Secondly, we retrospectively reviewed the morphology of two well-characterized cohorts of RCCs with FH-deficiency determined by IHC or sequencing (n=23 and n=9), unselected for TC-PD pattern, identifying the TC-PD morphology in 10 (31%). We conclude that RCCs with TC-PD morphology are enriched for FH deficiency, and we recommend additional work up, including referral to genetic counseling, for prospective cases. Additionally, based on these and other observations, we propose the term “FH-deficient RCC” as a provisional term for tumors with a combination of suggestive morphology and immunophenotype but where genetic confirmation is unavailable upon diagnosis. This term will serve as a provisional nomenclature that will enable triage of individual cases for genetic counseling and testing, while designating these cases for prospective studies of their relationship to HLRCC.
Keywords: renal cell carcinoma, tubulocystic carcinoma of the kidney, HLRCC; fumarate hydratase; fumarate hydratase-deficient RCC; 2SC
Introduction
Tubulocystic carcinoma of the kidney was likely first recognized as the “Bellinien epithelioma” or collecting duct carcinoma by Pierre Masson in the 1950s and as “low-grade collecting duct carcinoma” in the third series AFIP fascicle (1). Based on experience with a cohort of such cases and observation of their distinctive morphology and relatively favorable prognosis, in 2004 some of us proposed “tubulocystic carcinoma of the kidney” as a separate class of renal cell carcinoma (RCC), reporting 31 cases in 2009 (2). Tubulocystic carcinoma was formally included as a distinct entity in the International Society of Urological Pathology (ISUP) Vancouver Classification of Renal Neoplasia (3) and the recent 4th Edition World Health Organization Classification of kidney tumors (4). Tumors present grossly as a complex cystic mass characteristically in male patients (M:F ratio of ∼7:1) during their seventh decade. These tumors demonstrate a consistent morphology of variably cystically dilated tubules, admixed with a background of fibrous stroma, and lined by markedly atypical cells with eosinophilic cytoplasm and high grade nuclei with prominent nucleoli (ISUP nucleolar grade 3). Belied by their high grade nuclear features, most reported tumors have been low stage with only rare reports of clinical progression and aggressive behavior (2, 5-9).
Recently, three cases of RCC with the morphologic pattern of tubulocystic carcinoma, with concomitant areas of a high grade, infiltrative adenocarcinoma reminiscent of collecting duct carcinoma, were reported by two of the current authors and termed “tubulocystic carcinoma with poorly differentiated foci” (10). In each of these cases, there was an extensive, classic tubulocystic carcinoma-like morphology, involving between 30 and 90% of the tumors. However, other morphologic patterns were present, with two of three showing at least focal papillary morphology and all three demonstrating variable proportions of infiltrative high grade tubular adenocarcinoma with collecting duct-type features, invading perinephric adipose in two. A very recent report provides two further examples of such cases, emphasizing their aggression (11), and illustrative of the wisdom of the Vancouver Classification's recommendation not to designate tumors with such a mixed features as tubulocystic carcinoma (3).
In recent years, we have increasingly recognized in our consultation and hospital based practices a number of cases showing this morphologic pattern. While we have diagnosed these cases as renal cell carcinoma, unclassified, per contemporary recommendations, providing additional descriptive commentary about the morphology, some of our cases demonstrated intriguing high grade nuclear features. In particular, we believed that we were observing nucleolar forms reminiscent of the prominent, viral inclusion-like nucleolus with perinucleolar halo described previously as the cytomorphologic hallmark of renal cell carcinomas arising in the setting of hereditary leiomyomatosis renal cell carcinoma syndrome (HLRCC-RCCs) (12).
HLRCC syndrome is caused by germline mutation of fumarate hydratase (FH), resulting in a highly penetrant (>90%) uterine and cutaneous leiomyomatosis with variably penetrant (estimated 5-20%) high grade RCC with papillary or collecting duct-like morphology (13). More recently an expanded histologic spectrum of HLRCC-RCC has been described (12), and some of us have even reported cases where areas resembling tubulocystic carcinoma were admixed with tubulopapillary and papillary patterns (14). Importantly, HLRCC-RCCs are exceptionally aggressive tumors, often presenting at high stage, underscoring the importance of their recognition (12, 14). Thus recent reports propose immunohistochemical adjuncts for screening for HLRCC-RCCs, including staining for FH protein expression, which may be lost in a majority of cases (15-18) and for aberrant succination of nuclear and cytoplasmic proteins (S-(2-succino)-cysteine, 2SC), which is induced upon loss of FH function (14, 17, 19, 20).
The suggestive overlapping nuclear findings between many of our index TC-PD cases and HLRCC-RCCs led us to study this entity in greater detail, with an eye to clarifying the nosologic and molecular relationships between these tumors. This, we undertook via complementary studies of cases selected for TC-PD morphology on one hand, and by retrospective morphologic review among bona fide FH-deficient carcinomas, searching for TC-PD morphology, on the other. Significantly, we identified a group of tumors within the aforementioned morphologic spectrum but lacking family history or syndromal evidence of HLRCC despite FH deficiency. Since there is no appropriate terminology to designate tumors (whether showing TC-PD or the other reported patterns) demonstrating this immunophenotype in the contemporary classification schema for RCC (3), we have proposed the term FH-deficient RCC (21) for provisional use, pending definitive workup for germline FH mutation.
Methods
Cases and Cohorts
This study is a retrospective clinicopathologic, morphologic, immunohistochemical, and molecular review of cases of tubulocystic carcinoma with poorly differentiated foci (TC-PD). The first cohort is composed of cases identified, based on TC-PD morphology alone, retrospectively from pathology database searches of the contributing institutions and upon review of cases of high grade distal nephron adenocarcinomas (HDNAs) assembled in an international collaborative effort, approved by the Institutional Review Boards of contributing institutions. Medical and surgical pathologic records were reviewed to tabulate deidentified demographic and clinicopathologic data. The criterion employed for TC-PD cases was defined morphologically as tumor with at least some component of classic tubulocystic carcinoma with adjacent or admixed high grade, infiltrative, tubular/tubulopapillary, solid, or cribriform adenocarcinoma. Classic tubulocystic carcinoma morphology was defined as previously reported (2) and described above. All cases were reviewed by multiple authors (SCS/CO/DS/MBA).
The second cohort of cases studied is composed of two published, well characterized cohorts of cases with FH-deficient status (n=23) (18) or HLRCC-RCC (n=9) (14). These previously reported cases were reviewed to determine whether any TC-PD morphology were apparent, at least focally. For cases where TC-PD morphology was identified upon this review, updated clinical, morphologic (as described below), and molecular data were tabulated (by KT/AJG and Y-BC/SKT/VER, respectively).
Review of Gross and Microscopic Tumor Morphology
For the first cohort, using gross description and photographs available for a subset of cases, pathologic parameters including size, focality and location within the kidney (medullary versus cortical), were tabulated, as was the gross impression of the tumor, whether circumscribed and/or infiltrative, whether or not it was cystic, and whether necrosis was grossly evident. The presence of a number of microscopic architectural patterns was evaluated. These included patterns of 1) infiltrative adenocarcinoma with desmoplastic stroma (collecting duct carcinoma-like); 2) multinodular infiltrative papillary pattern (variably sized invasive papillary nodules with intervening desmoplastic reaction; 3) tubular, tubulopapillary, and papillary pattern; 4) non-glandular pattern, including solid sheets, cords, variably sized nests, and rhabdoid areas; 5) intracystic papillary growth; 6) cribriform morphology; 7) conventional tubulocystic carcinoma-like morphology; and 8) yolk sac carcinoma-like pattern with reticular architecture. These patterns were considered for each case, and quantitated as primary if involving ≥50% of the tumor, secondary, if involving a substantial but lesser component of the morphology (10%-49%), or focal (≤10%). Presence or absence of coagulative tumor cell necrosis was evaluated for each case, as was the nucleolar grade, both as defined by the ISUP Vancouver recommendations for prognostic parameters (22). Finally, the specific nuclear feature of a large nucleus with very prominent inclusion-like orangiophilic or eosinophilic nucleolus with perinucleolar halo (viral-like inclusion) was additionally scored for each case, including if present focally (≤10% cells) or diffusely.
Again, for the first cohort of new cases, immunohistochemistry for FH was performed by use of clone J-13, Santa Cruz Biotechnology, essentially as reported previously (19, 20), but at dilution of 1:1000 or at 1:100 under CLIA-validated IHC testing protocols performed by two of our IHC labs (a subset of our cases were studied by both with identical results). Our 2SC staining protocol has been reported previously (14). The FH staining was visually scored qualitatively as negative or positive when compared to internal positive control (endothelial cells), excepting a subset of cases showing weak, variable FH staining that was at least focally positive, which were designated FH±. The 2SC staining assessed for intensity (1+ to 3+) and staining pattern (nuclear and cytoplasmic vs. cytoplasmic only), though only 3+ intensity nucleocytoplasmic staining was interpreted as positive, also as reported previously (14). Reflective of experience of ours (14, 20, 23) and others (15-17, 19) with relation of FH and 2SC immunophenotype to FH mutational status, we classified the immunophenotypes observed as FH+2SC- as “FH-retained”, FH-2SC+ as “FH-deficient”, and FH±2SC+ as “FH-suspicious”.
Molecular Studies
Tumors from the first cohort identified based on TC-PD morphology where archival materials were available were also studied via multiplexed PCR-based NGS, with IRB approval, using the Ion Torrent Comprehensive Cancer Panel as we have reported previously (24-26). First, if technically possible based on available archival material, conventional tubulocystic carcinoma-like areas and poorly differentiated areas were carefully separately macrodissected after review of paired H&E slides (SAT). In cases where these morphologies could not be separated, tumor was carefully macrodissected from normal tissues, and the percentage tumor content estimated. Four cases of conventional tubulocystic carcinoma without poorly differentiated areas were also analyzed for comparison. Briefly, NGS was performed on isolated DNA from each sample using the Comprehensive Cancer Panel, which targets ∼16k amplicons including the complete coding sequence of 409 cancer-related genes. Libraries were created using the Ion Ampliseq Library Kit 2.0 and barcoded for multiplex analysis, with templates prepared using the Ion PI Template OT2 200 Kit v3 on the Ion One Touch 2, with sequencing on the Ion Torrent Proton Sequencer using P1 chips and the Ion PI Sequencing 200 Kit v3. Data analysis was performed exactly as described (26), using Torrent Suite (4.2.0), the Coverage Analysis Plug-in (v4.0-r73765), and in-house validated annotation, filtering, and prioritization pipelines to identify high confidence, prioritized somatic mutations, with the exception of requiring called variants to have variant allele frequencies (FAO/FDP) >10% and all FH variants were prioritized if otherwise meeting our usual criteria. To identify copy number alterations (CNAs), normalized, GC-content-corrected, total read counts per amplicon for each sample were divided by those from a composite normal sample yielding a copy number ratio for each amplicon. Gene-level copy number estimates were determined by taking the coverage-weighted mean of the per-probe ratios, with expected error determined by the probe-to-probe variance, as we have reported previously (24-27).
Results
Clinical and Gross Features of TC-PD
Tubulocystic carcinoma with poorly differentiated (TC-PD) morphology was studied by complementary approaches in two cohorts. The first was by retrospective review of cases identified in database searches for TC-PD morphology among cases encountered in the surgical pathology practices of the authors and contributed in an international collaborative consortium studying high grade adenocarcinomas arising in the distal nephron (HDNA) (n=29). These cases arose in 21 males and 8 females, for an overall M:F ratio of 2.6. The patients included 14 (48%) of Caucasian, 2 (7%) of Asian, and 5 (17%) of reported African descent (no data for 8, 28%). Ages ranged from 16 to 86 years, with a median age of 46 years. The tumors were unilateral in all cases, arising in in the right kidney (n=16) and left kidney (n=13) (see Figure 1A-B). No case had personal or family history of HLRCC, nor any clinical consideration of such, in any available medical records. Three cases, however, had findings, identified retrospectively upon review of the kidney tumors, compatible with or suggestive of an association with HLRCC. Case 4 had a first degree relative with metastatic RCC, respectively, while case 13 had a first degree relative with multiple cutaneous leiomyomas. Case 7 had no family history but had numerous uterine leiomyomas described radiologically as a “remarkably myomatous uterus”.
Figure 1. Clinical and Gross Features of TC-PD.
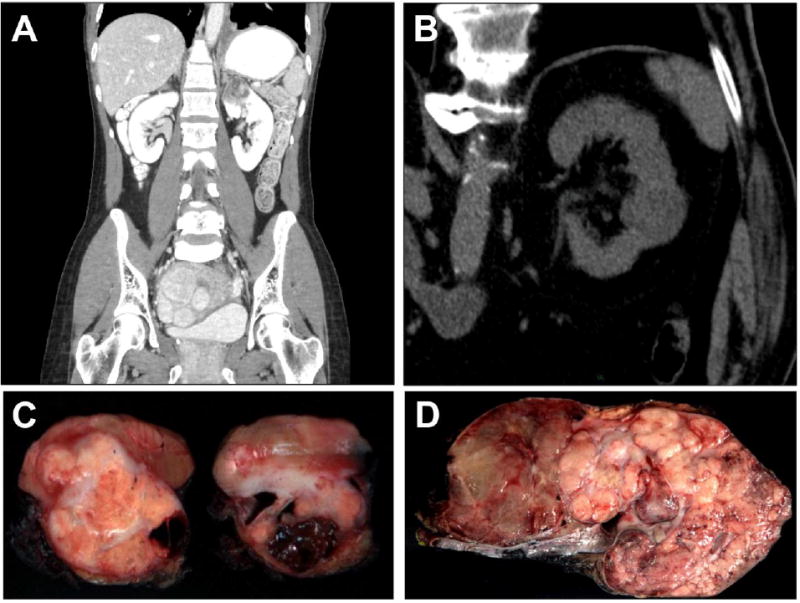
A. Contrast abdominal CT scan in the coronal plane of case TC-PD 17 demonstrates a 4 cm solid and cystic tumor in the upper pole of the left kidney. The lower left lung demonstrates pleural-based nodularity and effusion, which proved to be metastatic disease, while uterine leiomyomatosis was quite prominent in this case, which was FH-deficient immunohistochemically. B. Non-contrast abdominal CT scan of case TC-PD 7 demonstrates a lesion in the posterior of interpolar left kidney which proved to be a TC-PD with FH-retained immunophenotype. C. Gross photograph of representative cross sections of the nephrectomy specimen from case TC-PD 17, which shows a solid (predominant on left section) and cystic (predominant on right section) tumor appearing based in the medulla. D. Gross cut section of case TC-PD 3, where tumor is seen diffusely infiltrative of the entire kidney. The areas to the left show some features of the multicystic so-called “bubble wrap” appearance described for conventional tubulocystic carcinoma.
Grossly, the tumors ranged in greatest dimension from 3 to 21cm, with a median of 9 cm. Among cases where the growth pattern of the tumor was described grossly (n=26), these were described equally as circumscribed (50%) or infiltrative/poorly defined (50%), with a subset (23%) including additional description of a cystic or multicystic gross appearance (see Figure 1C-D). Of 27 cases where the gross pattern of involvement was detailed by anatomic compartment of the kidney, 26% were predominantly cortically based, 19% were predominantly medullary based, and 56% involved both, often diffusely. From the standpoint of staging, correlating gross and microscopic impressions in 28 cases, only a subset were confined to the kidney (21%, stages pT1-2), while 79% were pT3a-4, including gross and/or microscopic invasion of the renal vein in 9 of 27 cases (33%). Lymph nodes were sampled in 22 cases and were involved by carcinoma in 9 (41%). At presentation, metastasis was apparent in 9 cases, while it was observed during follow-up in an additional 10 cases over a median 9 months of available follow-up (total 19/22 cases with available data), including frequent liver, lung and bone metastasis (in 52%, 52%, and 42% of cases with metastasis, respectively). Table 1 summarizes the clinical features of these cases.
Table 1. Clinicopathologic Features of TC-PD RCCs.
| Case # | Classification∼ | Age [y] | Sex | Side | HLRCC?# | Size [cm] | pT Stage& | Node Status$ | Metastasis@ | Progression [mo]ˆ | Status% | Follow-Up (mo) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TC-D 1 | FH-D | 18 | M | R | - | 10 | - | - | - | - | - | - |
| TC-D 2 | FH-D | 20 | M | L | No | 12.5 | pT3b | Neg | Li | 1 | DOD | 25 |
| TC-D 3 | FH-D | 25 | M | R | No | 15.5 | pT3a | Neg | Li, Lu, Bn, Adr | P | DOD | 8 |
| TC-D 4 | FH-D | 40 | M | L | Fam, RCC | 3.7 | pT3a | - | Li | P | AWD | 2 |
| TC-D 5 | FH-D | 42 | M | L | No | 5 | pT3a | I | Bn | P | AWD | 16 |
| TC-D 6 | FH-D | 42 | F | R | No | 6 | pT3a | R | Li, R | P | DOD | 2 |
| TC-D 7 | FH-D | 42 | F | L | UM | 4.1 | pT3a | Neg | Lu, E, CW | P | DOD | 3 |
| TC-D 8 | FH-D | 44 | M | R | No | 10.5 | pT3a | Neg | - | - | NED | 2 |
| TC-D 9 | FH-D | 45 | F | R | No | 5.5 | pT3 | Neg | Lu, Li, R, Bn, M, O | 10 | DOD | 43 |
| TC-D 10 | FH-D | 46 | M | L | No | 8.5 | pT3a | A | Bn, Lu, Li, T, A | P | DOD | - |
| TC-D 11 | FH-D | 46 | M | R | No | 10 | pT3c | Neg | Li | 4 | DOD | 25 |
| TC-D 12 | FH-D | 52 | M | L | - | 12 | pT4 | Neg | - | - | LTF | - |
| TC-D 13 | FH-D | 53 | M | R | Fam, MCL | 6 | pT3a | C | - | - | LTF | 4 |
| TC-D 14 | FH-D | 59 | M | L | No | 16 | pT2b | Neg | None | - | LTF | - |
| TC-D 15 | FH-D | 61 | M | L | - | 14 | pT2 | Neg | Li, Bn, C | 2 | AWD | 68 |
| TC-D 16 | FH-D | 67 | M | R | No | 10 | pT3a | R | Li, Lu | 6 | DOD | 6 |
| TC-D 17 | FH-D* | 34 | F | L | No | 3.7 | pT1a | - | Lu | 4 | AWD | 10 |
| TC-D 18 | FH-D* | 40 | F | L | No | 6 | pT1b | Neg | - | - | DNOS | - |
| TC-D 19 | FH-D* | 71 | M | R | No | 8 | pT3a | U | - | - | DOD | 16 |
| TC-D 20 | FH-S | 16 | F | R | No | 6.5 | pT1b | Neg | None | - | NED | 126 |
| TC-D 21 | FH-S | 35 | M | L | No | 14 | pT3 | A | Adr, Bn, E | P | DOD | 9 |
| TC-D 22 | FH-S | 50 | M | R | No | 21 | pT2b | Neg | Lu | 8 | DOD | 10 |
| TC-D 23 | FH-S | 53 | M | R | No | 13 | pT3 | Neg | P, R | - | AWD | 5 |
| TC-D 24 | FH-S | 53 | F | L | No | 3.3 | pT3a | - | - | - | AWD | 2 |
| TC-D 25 | FH-R | 70 | M | R | No | 11.7 | pT3a | - | Adr | P | AWD | 0 |
| TC-D 26 | FH-R | 71 | M | R | No | 8.5 | pT3a | U | Lu, Li | P | AWD | 0 |
| TC-D 27 | FH-R | 74 | M | L | No | 4.5 | pT3a | - | Lu , Bn | - | LTF | - |
| TC-D 28 | FH-R | 80 | F | R | No | 5.5 | pT3a | A | Lu , Bn | 8 | DOD | 15 |
| TC-D 29 | FH-R | 86 | M | R | No | 3 | pT3a | - | None | 0 | NED | 7 |
Abbreviations: ‘-’, no data; TC-PD, tubulocystic carcinoma with poorly differentiated foci; FH, fumarate hydratase; RCC, renal cell carcinoma; IHC, immunohistochemistry;
Classification – Final integrating IHC and molecular findings; FH-D, FH-deficient RCC; FH-S, FH-suspicious RCC; FH-R, FH-retained RCC, * indicates IHC suspicious case reclassified as FH-D based on NGS findings
HLRCC: includes any evidence of HLRCC, whether stigmata apparent clinically (UM-uterine leiomyomatosis) or family history (Fam), RCC, renal cell carcinoma; MCL, multiple cutaneous leiomomas) reported, see Table 3.
pTNM: Classification by AJCC 2010 TNM Staging System
Lymph Node Metastasis: nodal apparent at surgical staging; -, no data; Neg, negative; I, positive iliac node; R, positive retroperitoneal node; AC, positive aortic/caval lymph node; S, sinus; Re, positive “regional” lymph node; U, positive unspecified node; lymph node metastasis evolving during follow-up is included only in “Metastatc Site”
Metastatic Site: metastatic sites apparent either at presentation or evolving during follow-up; Li, liver; Lu, lung; Bn, bone; R, retroperitoneal node; E, malignant effusion; CW, chest wall; M, mediastinal node; O, ovary; T, thoracic LN; Ab, abdominal LN; C, cervical LN; Sp, spleen; P, pancreas;
Progression: time to first metastatic progression, months; P, metastatic at presentation
Status: last available status at followup; DOD, dead of disease; NED, no evidence of disease; LTF, lost to follow-up; AWD, alive with disease; DNOS, dead, not otherwise specified
Microscopic Features of TC-PD
Microscopically, these cases demonstrated variable components of morphology as described for tubulocystic carcinoma of kidney in all cases. In 11 cases (38%), tubulocystic areas represented the predominant pattern and involved over half of the tumor; in 10 cases (34%) tubulocystic morphology was a significant but secondary morphology (10-49% of the tumor); in 8 cases (28%) it was focal. Figure 2A-D presents four cases, showing a representative range of tubulocystic versus poorly differentiated morphology at low power. Overall, after tubulocystic morphology (primary or secondary pattern in 72% of cases), the most common predominant patterns presented included tubular or tubulopapillary pattern (21%) and non-glandular (solid/sheet-like, nested, cord-like, rhabdoid, 17%). Less frequent were patterns of multinodular infiltrative papillary and infiltrative tubular adenocarcinoma with desmoplasia (collecting duct carcinoma-like) patterns predominant in two cases each (each 7%).
Figure 2. TC-PD at low power, with variable proportions of tubulocystic and poorly differentiated components.
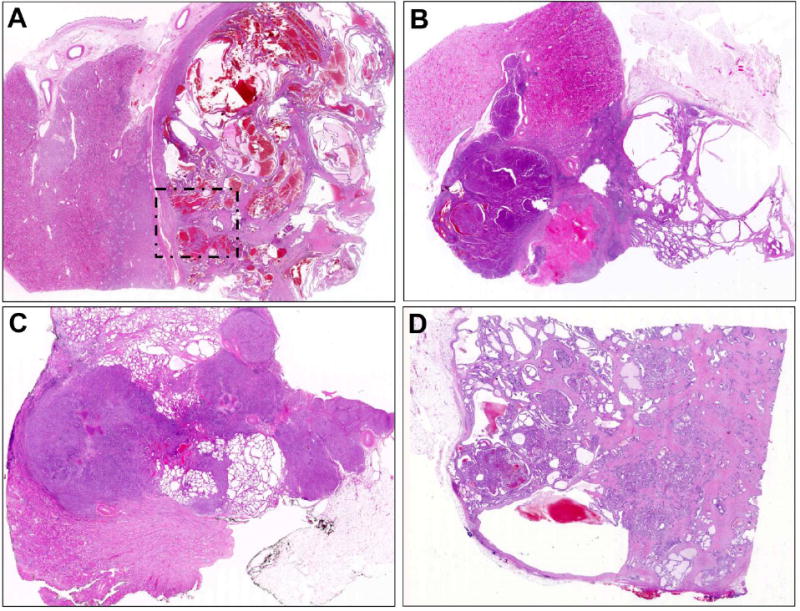
A. Case TC-PD 18 demonstrates predominantly well circumscribed tubulocystic morphology, in this case ∼90% over all sections reviewed, with only focal dedifferentiation apparent in the area indicated (detailed Figure 7B-D). B. TC-PD 29 demonstrated ∼50% tubulocystic pattern (right) with ∼50% solid and dense tubular growth across all sections reviewed. C. TC-PD 27 demonstrated overall ∼30% tubulocystic morphology, seen here above and below the predominant pattern of collecting duct carcinoma-like infiltrative glandular growth with desmoplasia. D. Case TC-PD 16 demonstrated only focal (∼10%) tubulocystic morphology (apparent upper left and Figure 3A) with predominant poorly differentiated patterns of infiltrative glands with desmoplasia, solid/nested, and cribriform growth.
Variable non-tubulocystic morphologies were present. Patterns present more than focally (>10%), in order of prevalence, included tubular/tubulopapillary morphology (48%), non-glandular sheets, nests, or cords (41%), cribriform growth (28%), infiltrative adenocarcinoma with desmoplasia (17%), intracystic papillary growth (14%), and multinodular infiltrative papillary (10%). Figures 3-5 and Supplemental Figures S1-S2 illustrate cases demonstrating a full range of morphologic findings for TC-PD cases. In terms of prognostic features that have been associated with aggressive behavior in other types of RCC, coagulative tumor necrosis was seen in eight cases (28%); Supplemental Figures S3A-B. ISUP nucleolar grade was 3 in 82%, grade 4 in the remaining cases. Cytologically all TC-PD cases were remarkable for prominent nucleoli with perinucleolar halos of the kind described previously as the cytomorphologic hallmark of HLRCC-RCC (12), including focally in 10 (34%) or diffusely in the remaining 19 (66%) (see Supplemental Figures S3C-D). Table 2 summarizes the gross and morphologic features of these TC-PD cases.
Figure 3. TC-PD with FH-deficient immunophenotype.
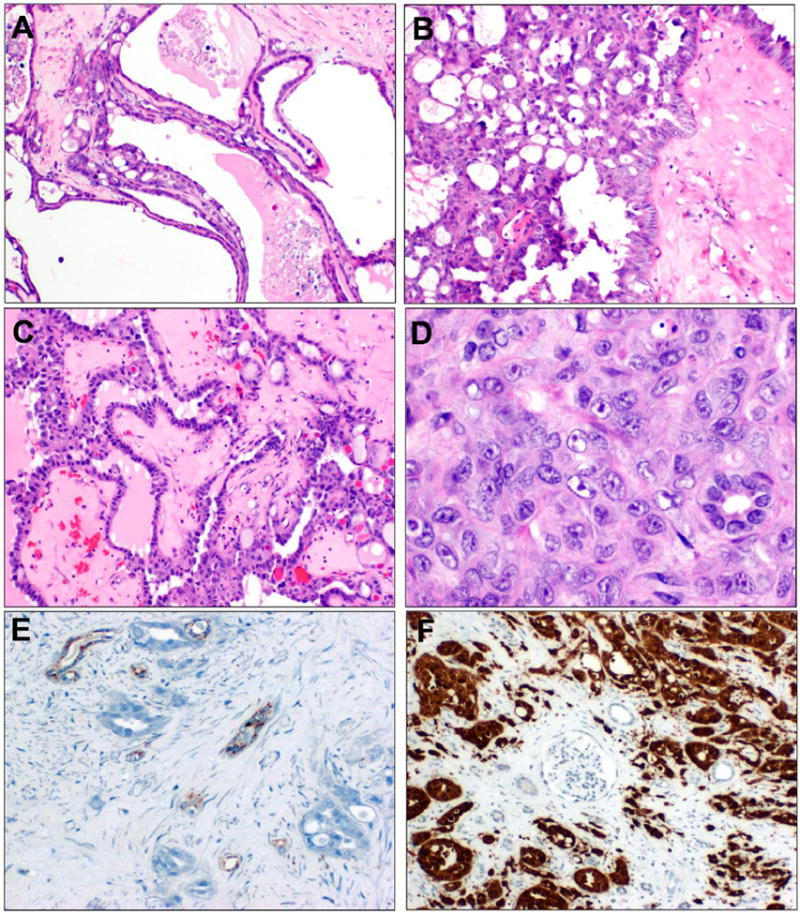
Case TC-PD 7 also demonstrated more focal TC-PD morphology, with these areas seeming to give rise to infiltrative glands with desmoplasia (A). More prevalent growth patterns included cribriform growth (B), foci of intracystic papillary growth with hyalinized cores (C), and solid areas, with prominent nucleoli and examples of focal perinucleolar clearing (D). By immunohistochemistry, this carcinoma was negative for FH (panel E, note internal positive control entrapped tubules) and diffusely positive for 2SC in nucleocytoplasmic manner (panel F, note entrapped internal negative control glomerulus and tubules).
Figure 5. TC-PD with FH-suspicious immunophenotype.
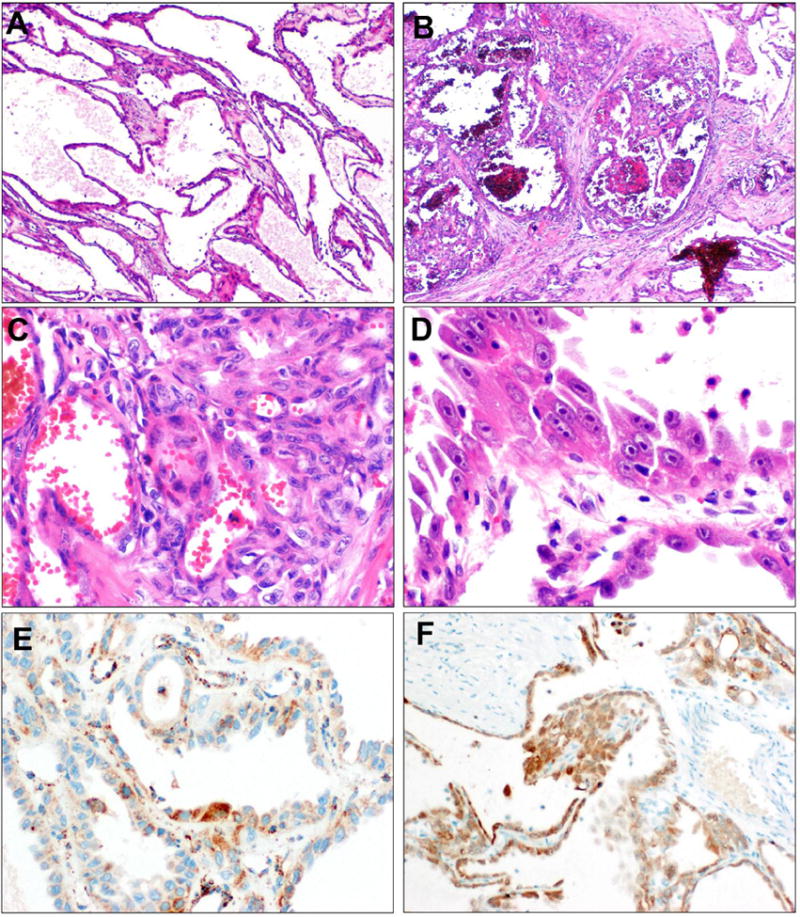
TC-PD 18 demonstrates predominant tubulocystic morphology (A). However, focally, areas of greater epithelial complexity than allowable in tubulocystic carcinoma are apparent (B). In these areas, nested cribriform growth is apparent (C). Cytologically, prominent nucleoli with perinuclear clearing are evident (D). FH immunostain was at least weakly positive in the malignant epithelium, with scattered patches of more intense positivity. (E). 2SC, however, showed strong nucleocytoplasmic positivity (F). This case harbored homozygous R233H mutations of FH, and was reclassified as FH-deficient.
Table 2. Gross, Morphologic, IHC and Molecular Findings for TC-PD RCCs.
| Final Label | Age | Sex | Classification∼ | Gross# | Siteˆ | TC-Proportion& | Nucleoli% | Necrosis$ | Sarcomatoid$ | ISUP Grade$ | FH IHC© | 2SC IHC© | FH Mutation |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TC-D 1 | 18 | M | FH-D | 1, 3 | - | 1 | 2 | 0 | 0 | 4 | lost | - | - |
| TC-D 2 | 20 | M | FH-D | 2 | C,M | 1 | 2 | 1 | 0 | 3 | lost | pos (3+) | - |
| TC-D 3 | 25 | M | FH-D | 1 | C,M | 3 | 1 | 0 | 0 | 3 | lost | pos (3+) | homozygous Y251fs |
| TC-D 4 | 40 | M | FH-D | 1,3 | C | 3 | 2 | 0 | 0 | 3 | lost | pos (3+) | homozygous K80fs |
| TC-D 5 | 42 | M | FH-D | 2 | C,M | 3 | 2 | 0 | 0 | 3 | lost | pos (3+) | - |
| TC-D 6 | 42 | F | FH-D | 2 | C,M | 2 | 2 | 0 | 0 | 3 | lost | pos (3+) | homozygous R101X |
| TC-D 7 | 42 | F | FH-D | 2 | C,M | 1 | 2 | 0 | 0 | 3 | lost | pos (3+) | focal deletion |
| TC-D 8 | 44 | M | FH-D | 1 | C,M | 3 | 2 | 0 | 0 | 4 | lost | pos (3+) | - |
| TC-D 9 | 45 | F | FH-D | 2 | C,M | 1 | 2 | 0 | 0 | 3 | lost | pos (3+) | - |
| TC-D 10 | 46 | M | FH-D | 2,3 | C,M | 1 | 1 | 0 | 0 | 3 | lost | pos (3+) | - |
| TC-D 11 | 46 | M | FH-D | 2 | C,M | 1 | 2 | 1 | 0 | 3 | lost | pos (3+) | - |
| TC-D 12 | 52 | M | FH-D | - | M | 1 | 2 | 2 | 1 | 4 | lost | pos (3+) | - |
| TC-D 13 | 53 | M | FH-D | 1,3 | M | 2 | 1 | 0 | 0 | 3 | lost | pos (3+) | - |
| TC-D 14 | 59 | M | FH-D | 2 | C,M | 2 | 2 | 1 | 0 | 3 | lost | pos (3+) | - |
| TC-D 15 | 61 | M | FH-D | 1, 3 | C,M | 2 | 2 | 0 | 0 | 3 | lost | - | - |
| TC-D 16 | 67 | M | FH-D | 2 | C,M | 1 | 2 | 0 | 0 | 3 | lost | pos (3+) | K230R |
| TC-D 17 | 34 | F | FH-D* | 2 | M | 2 | 2 | 0 | 0 | 4 | weak | pos (3+) | V279F |
| TC-D 18 | 40 | F | FH-D* | 1 | C | 3 | 1 | 0 | 0 | 3 | weak | pos (3+) | R233H |
| TC-D 19 | 71 | M | FH-D* | - | M | 2 | 1 | 0 | 0 | 3 | weak | pos (3+) | heterozygous N188T, H318Y |
| TC-D 20 | 16 | F | FH-S | 1 | M | 3 | 1 | 0 | 0 | 3 | weak | pos (3+) | Hypermutated |
| TC-D 21 | 35 | M | FH-S | 2 | C,M | 3 | 2 | 0 | 0 | 3 | weak | pos (3+) | - |
| TC-D 22 | 50 | M | FH-S | 2 | C | 2 | 2 | 0 | 0 | 3 | weak | pos (3+) | - |
| TC-D 23 | 53 | M | FH-S | 2 | C,M | 3 | 2 | 0 | 0 | 4 | weak | pos (3+) | - |
| TC-D 24 | 53 | F | FH-S | 1 | C | 2 | 2 | 0 | 0 | 3 | weak | pos (3+) | - |
| TC-D 25 | 70 | M | FH-R | 1 | C | 2 | 1 | 2 | 0 | 3 | retained | neg (1+) | - |
| TC-D 26 | 71 | M | FH-R | 1 | C,M | 3 | 2 | 0 | 0 | 3 | retained | neg (1+) | - |
| TC-D 27 | 74 | M | FH-R | - | - | 2 | 1 | 1 | 0 | 3 | retained | neg (1+) | None |
| TC-D 28 | 80 | F | FH-R | 1 | C | 3 | 1 | 1 | 1 | 3 | retained | neg (1+) | None |
| TC-D 29 | 86 | M | FH-R | 1,3 | C | 3 | 1 | 1 | 0 | 3 | retained | neg (1+) | - |
Abbreviations: ‘-’, no data; TC-PD, tubulocystic carcinoma with poorly differentiated foci; FH, fumarate hydratase; RCC, renal cell carcinoma; IHC, immunohistochemistry; HLRCC, hereditary leiomyomatosis renal cell carcinoma syndrome; met., metastatic;
Classification – Final integrating IHC and molecular findings; FH-D, FH-deficient RCC; FH-S, FH-suspicious RCC; FH-R, FH-retained RCC,
indicates IHC suspicious case reclassified as FH-D based on NGS findings
Gross: gross appearance described as 1, circumscribed; 2, infiltrative/ill-defined; 3, cystic/multicystic
Site: anatomic localization on the kidney; M, predominantly medullary; C, predominantly cortical, C,M, involving both cortex and medulla
TC-Proportion: extent of tubulocystic pattern; 1, focal <10%; 2, non-focal 10-50%; 3, predominant >50%.
Nucleoli: degree of HLRCC-like prominent, viral inclusion-like nucleoli with perinuclear halo; 1, focal; 2 diffuse.
Necrosis (0, absent; 1, present <50%, 2, present >50%), sarcomatoid transformation (0, absent; 1, present), and ISUP grade (3, grade 3; 4, grade 4) per 2012 ISUP Vancouver Classification recommended criteria.
Immunohistochemistry for FH evaluated as retained if expressed equivalent to internal control endothelium, weak, decreased, or lost if negative. IHC for 2SC was deemed positive if strong, diffuse (3+) and nucleocytoplasmic, 2+ if weaker, or negative (0-1+) as reported previously (Chen et al. Am J Surg Pathol 2014).
Immunophenotypic Studies
All cases were studied by immunohistochemistry for FH, and 27/29 for 2SC. Overall, we observed that there were three distinct immunophenotypes. The predominant immunophenotype was the characteristic “FH-deficient” immunophenotype of FH-/2SC+ such that sixteen cases (55%) showed complete loss of FH (Figure 3E-F, Supplemental Figure S1E-F). This was accompanied by induction of strong, diffuse 3+ nucleocytoplasmic 2SC positivity in 14/14 (100%) of these FH- cases. In contrast, five cases (17%) showed the FH+/2SC- immunophenotype characteristic of RCC with retained FH function (Figure 4E-F) similar to four examples of conventional tubulocystic carcinoma studied for comparison. Third, a total of 8 cases (28%) showed retained but weak FH expression, with convincing, diffuse, intense 3+ nucleocytoplasmic 2SC positivity, which we deemed suspicious for FH mutation, Figure 5E-F, Supplemental Figure S2E-F. In all cases, the immunostaining pattern and intensity of FH and 2SC was the same in tubulocystic and poorly differentiated components of the same tumor.
Figure 4. TC-PD with FH-retained immunophenotype.
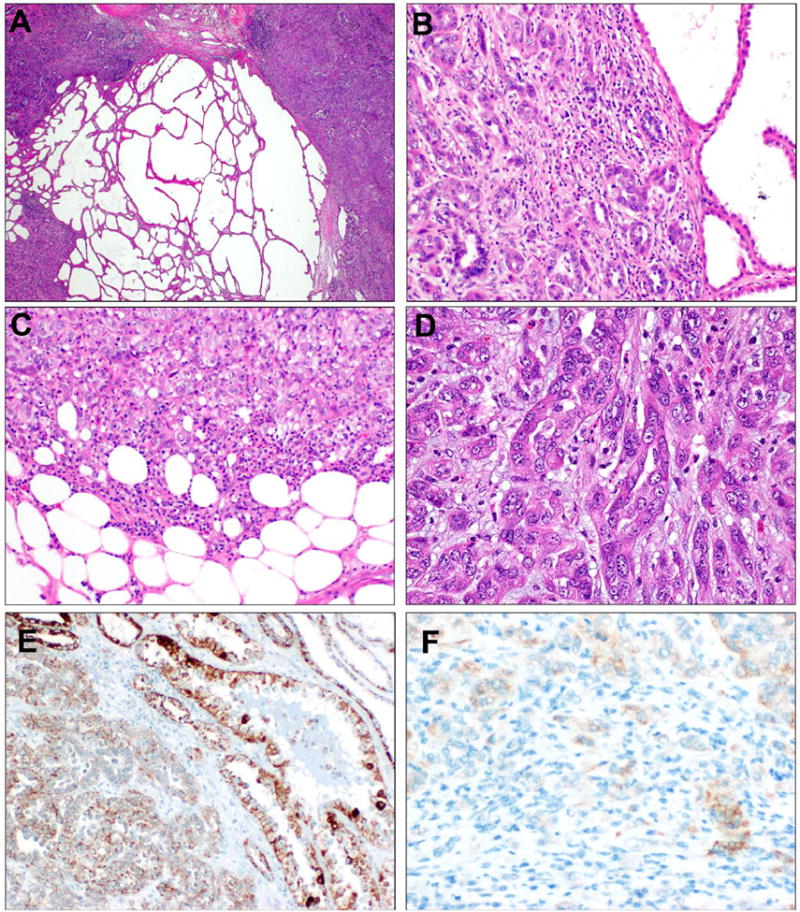
Case TC-PD 27 showed areas of tubulocystic growth directly juxtaposed to dense, cellular areas of poorly differentiated morphology (A). On higher power, the infiltrative glandular morphology can be seen directly juxtaposed to tubulocystic areas (B). At the periphery, solid and nested patterns are seen invading into perinephric adipose (C). Most of the tumor showed infiltrative, collecting duct carcinoma-like glands seen embedded in inflamed desmoplastic stroma (D). This case showed diffuse expression of FH by IHC (E). 2SC showed only focal, weak cytoplasmic staining (1+, F), which is interpreted as negative for this stain.
Some but not all clinicopathologic features of TC-PD differed significantly with respects to immunophenotype, whether FH-retained, FH-suspicious, or FH-deficient (including the cases with additional evidence of HLRCC). Most notably, comparing distributions of ages between these three groups, we observed significantly different distributions (p=0.003 Kruskal-Wallis), such that the FH-suspicious and FH-deficient cases had significantly lower median ages (each 45y) than the FH-retained cases (74y). While laterality (p=0.5 χ2), tumor size (p=0.4, Kruskal-Wallis), and proportion of tubulocystic morphology (p=0.06, χ2) did not differ significantly in distribution by immunophenotypic group. The only significantly different morphologic feature was that a higher proportion of FH-retained cases demonstrated focal rather than diffuse presence of inclusion-like nucleoli with perinucleolar halos (4/5) than did FH-suspicious (3/8) or FH-deficient (3/16), p=0.04, acknowledging the cohort size. Nodal status (p=0.34) and metastatic progression (p=0.98) did not differ between groups.
Molecular Analysis of TC-PD
To assess for coding mutations and copy number alterations in 409 cancer related genes, including FH, we performed multiplexed PCR Ampliseq-based NGS on a total of 20 tumor samples. These constituted 16 samples from 11 TC-PD cases, which included five cases where paired tubulocystic and poorly differentiated components could be dissected and analyzed separately for comparison, and six cases where intimately admixed tubulocystic and poorly differentiated areas could not be separately analyzed. Four additional “conventional” tubulocystic carcinomas were also studied. Considering all rare FH variants (whether deleterious or not) and all high-confidence prioritized non-synonymous variants identified by our validated analysis pipelines (24, 25, 27), we identified a total of 8 cases with FH alterations, including focal copy number deletion, and missense and frameshift mutations. In all cases where paired tubulocystic and poorly differentiated components could be separately dissected and analyzed, the same mutations were identified at comparable variant allele frequencies in both components. The five cases showing the FH-/2SC+, FH-deficient immunophenotype that were assessed by NGS demonstrated the following FH mutations: one case with focal, high level two-copy deletion of the FH gene (spanning all FH amplicons, Figure 6A), one K230R homozygous mutation, one R101X homozygous mutation, and two cases with homozygous frameshifts, one at K80 and one at Y251.
Figure 6. Molecular findings in TC-PD by next generation sequencing (NGS).
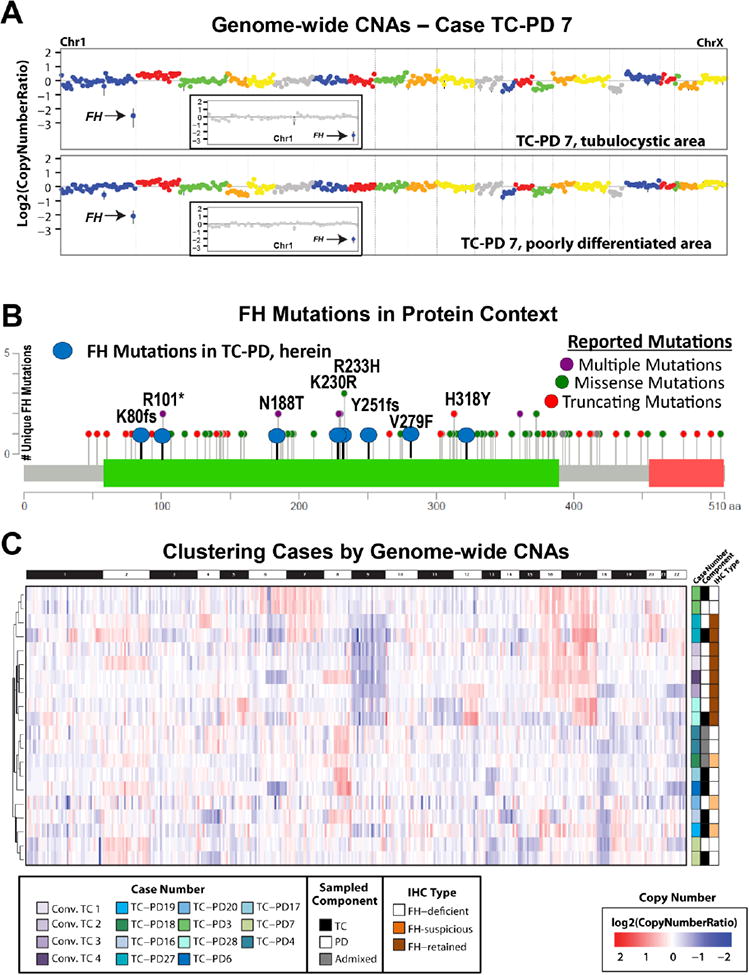
A. Focal, high level gains or losses were rare in our cohort, but included one case, TC-PD 7, which showed a focal homozygous FH deletion on chromosome 1 (inset), present in both the tubulocystic and poorly differentiated areas. B. Mutations identified in FH in TC-PD cases were well within the spectrum of that described previously for FH; here MutationMapper (cBioPortal (48)) was used to plot all previously reported unique FH mutations from the FH mutation database (37) (purple, green, and red, as indicated), while mutations reported herein are indicated in blue. All mutations we identified were in the lyase domain (green). C. Hierarchical clustering based on inferred genome-wide copy number was performed for all TC-PD samples (shades of green and blue, indicated, with pairing indicated by black, white or grey) and conventional tubulocystic carcinomas (purple), identified two main branches in the dendrogram, correlating mostly with IHC type (shades of brown). Conventional tubulocystic carcinomas clustered in a subcluster with one FH-retained TC-PD.
Among four cases analyzed that showed the FH-suspicious immunophenotype, FH±/2SC+, we observed FH mutations in three; one case showed heterozygous N188T and H318Y mutations, another case harbored a V279F homozygous mutations, and a third case harbored a R233H homozygous mutation. The latter R233H case also demonstrated a second prioritized, high confidence somatic CREBBP R1341X mutation (heterozygous). Based on the documentation of the FH mutations, we reclassified these FH-suspicious cases as FH-deficient RCC (see cases TC-PD 17-19, cases numbered as per Tables 1-2). The fourth FH±/2SC+ case analyzed demonstrated a “hypermutator” phenotype, exhibiting thousands of mutations, including non-specifically, mutations of FH. All identified FH mutations are shown in relation to the exon structure of FH in Figure 6B.
Two cases of TC-PD that showed a FH-retained FH+/2SC- immunophenotype were among those studied by NGS. Neither case harbored FH mutations. One case showed no prioritized variants in any of the 409 genes studied; the other case demonstrated a prioritized 2bp frameshifting deletion at residue 832 of ARID1A. Four cases of “conventional” tubulocystic carcinoma were also studied by NGS; none showed FH mutations. Two of these did not show any prioritized non-synonymous variants among all genes studied, while one showed a 7bp deletion at codon 4509 of KMT2C and one other showed a 16bp deletion at codon 888 of KDM5C and a R337H mutation of TP53. Table 2 summarizes the immunophenotypic and sequencing findings for each of these TC-PD cases.
Finally, we used the NGS data to infer genome wide copy number changes in these cases. As shown in Figure 6C, unsupervised hierarchical clustering based on copy number profiling identified four clusters of tumors. The first major branch separated the cases by immunophenotype FH-deficient and FH-suspicious (but now all known to harbor FH mutations as above) versus FH-retained TC-PD and conventional tubulocystic carcinomas, with the exception of one case, TC-PD 3. Secondary branching of the dendrogram revealed four groups, one cluster containing the four conventional tubulocystic carcinomas, and paired low and high grade components from an FH-retained TC-PD 28. These tumors were characterized by chromosome 9 losses and gains of chromosomes 17 and 16, with chromosome 16 gains observed only in the conventional tubulocystic carcinomas. A second cluster comprised of low and high grade components from two TC-PDs (one each with and without retained FH), also showed gains of chromosome 16 and 17, but also harbored gains of chromosome 7. The other two clusters, consisting entirely of FH-deficient or FH-suspicious TC-PD, lacked the above alterations and showed more variable alterations, most frequently losses of chromosome 18 and gain of chromosome 8q. Focal, high level gains or losses were rare in our cohort, but included FH deletions in the paired components of TC-PD 7 (described above), a focal two copy 6p loss (encompassing EPHA7 and MAP3K7) in both the low and high grade components of TC-PD 3 (FH-deficient), and a focal two copy loss of CDKN2A exclusively in the high-grade component of TC-PD 27 (FH-retained).
TC-PD Morphology Identified Retrospectively Among Tumors with FH deficiency
Lastly, we used the reverse experimental design to study the relationship between TC-PD morphology and FH-deficient RCC in a second cohort composed of two previously reported RCCs with FH-deficiency (n=23, and n=9) (14, 18), reasoning that if TC-PD morphology were associated with FH-deficiency, then the pattern of tubulocystic carcinoma with poorly differentiated areas might be recognized retrospectively. Among the 23 cases of FH-deficient RCC reported by Trpkov et al., TC-PD morphology was seen in 7 (30%), while of the 9 cases of HLRCC reported by Chen et al. TC-PD was apparent in 3 (33%). Supplementary Table 1 presents these cases with updated clinical, morphologic, and molecular features.
Discussion
The association of multiple cutaneous and uterine leiomyomas (MCUL) was first reported by Reed et al. (28). Subsequently, building on findings of a group of such families also affected by an aggressive RCC, the association of this type of RCC with MCUL was reported in 2001, renamed HLRCC (29), and mapped to a locus on the long arm of chromosome 1 shown to be the locus of FH (30). HLRCC demonstrates among affected males and females a highly penetrant (75-100%, average onset at 25 years) phenotype of multiple painful cutaneous leiomyomas, particularly on the face, torso and extremities. Among affected females multiple uterine leiomyomas are also highly penetrant, often requiring surgery at a young age, reviewed recently (13, 31). Adrenocortical adenomas have been reported in ∼10% (13, 32). Classic reports of the morphology of the less penetrant (<5-20%, depending on the series (13, 33)) RCCs that arise in these patients described them as “type 2 papillary” or collecting duct carcinoma-like RCCs, though their wider morphologic spectrum, including papillary, tubulopapillary, tubular and solid examples, and distinctive cytologic features, including large viral inclusion-like nucleolus with perinucleolar halo were first detailed in a series by Merino et al.(12). While the morphology, tumor size, and age of onset observed varied greatly, most important was the clinicopathologic observation of a ubiquitously aggressive course, with frequent metastasis and high stage presentation. Recent reports emphasize the potential to use immunohistochemistry for FH itself or 2SC (biochemical consequence of FH dysfunction) for identification of such cases (17, 18, 20).
We believe that our experience reported herein, further characterizing RCC cases which we have descriptively labeled as tubulocystic carcinoma with poorly differentiated foci (TC-PD, as reported previously (10)), lends novel understanding regarding their aggressive behavior by identifying FH-deficient immunophenotype (and/or FH mutations detected among tumors) among a significant subset. Also, for at least the subset of FH-deficient cases, our findings argue against the potential relationship with conventional papillary renal cell carcinoma postulated in the original description of TC-PD (10). Noting the cytomorphologic hallmark described in HLRCC-RCCs—prominent inclusion-like nucleoli and perinucleolar halo (12)—in several cases encountered in our practice, we retrospectively identified a cohort of TC-PD, employing contemporary FH and 2SC immunohistochemistry to characterize the immunophenotype. Our findings suggest that well over half of these cases represent FH-deficient tumors, demonstrating the FH-/2SC+ immunophenotype and FH mutations in all such cases tested. This central finding is further supported by the observation of cases with TC-PD morphology identified from two, independent, previously reported cohorts of RCCs with FH deficiency (by immunohistochemistry and/or molecular studies) that were identified from among high grade unclassified RCCs and unclassified RCCs with papillary features (14, 18). We interpret these findings as strong support for the premise that the TC-PD pattern, observed presenting sporadically by not only us but also by at least two other groups (10, 11), represents in most cases a morphologic variation of RCC with FH deficiency.
We posit that when encountered prospectively, RCCs with TC-PD morphology, at the very minimum, deserve recommendation of additional workup with consideration first of FH-deficient status, and then, if identified, for HLRCC with referral for genetic counseling. This observation regarding TC-PD pattern is also consistent with prior descriptions of the morphologic range of cases reported with FH-deficiency. While the morphologic tour de force provided by Merino et al. (12) in their report of 40 tumors did not use the term “tubulocystic” to describe HLRCC-RCC cases (indeed “tubulocystic carcinoma of the kidney” had barely been reported prior to this study) we do note that they reported 21/40 cases with at least a minor cystic component. Another very recent report also identifies one example of an RCC with predominant tubulocystic pattern, with FH mutation (34), again, very consistent with our observations. It is tempting also to consider our TC-PD finding in light of prior findings from a mouse model, employing a kidney-specific biallelic FH knockout strategy, which observed onset of clonal, proliferative cysts thought to arise from the collecting ducts and thick ascending limb of the loop of Henle (35). Indeed, a recent case report of an HLRCC patient seems to describe an apparently benign multicystic lesion the kidney contralateral to an HLRCC-RCC, contemplating a precursor lesion (36).
Much less clarity, then, regards the relationship of our FH-deficient TC-PD cases to syndromal HLRCC. Overall the lower median age seen among the FH-deficient carcinomas and suspicious FH±2SC+, is at least consistent with consideration of a component of syndromal neoplasia. Remarkably, nonetheless, in the cohort of 29 TC-PDs, only 3 cases demonstrated any clinical evidence suggestive of or compatible with HLRCC, including one with family history of multiple cutaneous leiomyomas, one with family history of RCC, and one with concurrent, extensive uterine leiomyomas. The mutations that we observed in the subset of FH-deficient cases studied are entirely within the spectrum of germline mutations seen in HLRCC, including some involving residues reported mutated in HLRCC previously, while others are as yet unreported. We note with regards to the novel variants observed that the argument has been made that most missense mutations of FH are likely pathogenic, given the high degree of conservation of the protein across taxa (37). We also emphasize that these were detected from mutational profiling of tumors rather than the germline, which is the “gold standard” for diagnosis of this syndrome.
We do note that some of our prior experience supports the idea that HLRCC-RCC cases may present apparently sporadically, yet in fact represent bona fide germline HLRCC (14). Similarly, other investigators have recently found that a renal tumor may be the first and only manifestation of HLRCC (with germline FH gene mutation) (38). We also acknowledge that the penetrance of uterocutaneous leiomyomatosis and RCC in HLRCC might be even more variable across different geographic or ethnic groups than in those reported previously (13). However, we would also highlight emerging data with regards to purely somatic FH mutations. Somatic FH mutation was previously considered to be quite rare, at least among uterine leiomyomas (13, 39, 40), but emerging data with contemporary sequencing technologies suggest this may not be quite so. Indeed, a very recent cohort identified FH mutations in ∼1% of leiomyomas presenting sporadically, among a testable subset of which all were confirmed not to be germline (41). In tandem, and more a propos to RCC, are the data from the recently reported TCGA cohort of papillary RCCs, identifying a CpG island methylator phenotype (a molecular hallmark consistent with FH deficiency) in 9/60 “type 2” papillary renal cell carcinomas, among which five tumors showed “germline or somatic mutation of FH” and among all of which were noted “decreased expression of FH mRNA” (42). In light of these emerging findings, we are even more suspicious that somatic mutation of FH (or other mechanisms) might represent a way to attain the FH-deficient phenotype; indeed, one of our cases showing patchy loss of FH (FH±/2SC+) and showing an unusual hypermutator molecular phenotype characterized by innumerable mutations of all genes studied (including FH, though such might represent an epiphenomenon) would seem to be at least one example thereof. In this particular case, the mutator phenotype was in some ways similar to that seen in neoplasms with dysfunctional mismatch repair such as arise in Lynch Syndrome, though MLH1, MSH2, MLH6, and PMS2 expression were all retained by immunohistochemistry, data not shown.
This latter case raises the issue of tumors with the immunophenotype of FH±/2SC+, which some of us have designated as “equivocal” or “indeterminate” in prior reports (18). Our experience with these cases, which include 25% of the present cohort, leads us to be quite suspicious and designate cases with this immunophenotype and appropriate cytomorphology as suspicious for FH-deficient RCC; indeed, three of the four FH±/2SC+ cases studied by NGS exhibited FH mutations, presumably that result in retained expression of a dysfunctional or hypomorphic allele. Of these H318Y and R233H have been reported previously; N188T is novel, though the closely related N188S variant has been reported; V279F is novel but adjacent to residues previously reported mutated (37). Prior experience with FH versus 2SC immunostain suggests that the two perform similarly, perhaps 2SC with greater sensitivity but less specificity. For example, some of us have reported in abstract form that of 14 genetically confirmed HLRCC-RCC cases, 12 (86%) exhibited loss of FH expression while all 14 showed induction of nucleocytoplasmic 2SC expression (20). Importantly, some of us have reported previously that cytoplasmic (but not nucleocytoplasmic) 2SC positivity may be seen in high grade unclassified and papillary RCCs (14), which might be prospectively difficult to interpret. Overall, our assessment is that use of both markers would be ideal, emphasizing the caveats that retained FH staining does not exclude mutation resulting in expression of a dysfunctional protein and that the full spectrum of 2SC positivity remains unclear. Notably, even many targeted sequencing protocols currently in use would not detect the FH deletions reported in a subset of HLRCCs. Certainly, we emphasize that at present 2SC is only available in a research setting, while FH, for which a monoclonal antibody exists, is becoming increasingly available in CLIA-compliant laboratories. Either way, we recommend inclusion of commentary suggesting genetic counseling and germline sequencing in any cases where morphologic, immunophenotypic, or clinical settings are unclear.
Lastly, we observed a subset of FH+/2SC- cases that show a “wild-type” immunophenotype with respects to FH status. Our observation that there exists a distinct subset of cases with no molecular or immunophenotypic evidence implicating dysfunction in FH or HLRCC suggests that this morphology can occur due to lesions in or alterations in other cellular pathways. Indeed, at least among one of the FH+/2SC- TC-PD cases, we observed mutation of an FH-unrelated gene, ARID1A, known to be significantly mutated in ovarian clear cell carcinoma (∼50%) and gastric adenocarcinoma (∼30%) (43), and among a frequently mutated SWI/SNF gene cluster in clear cell RCC (44) and urothelial carcinoma (mutated in ∼25%) (45).
In summary, we find that the TC-PD morphologic pattern in RCC is strongly associated with FH-deficient status. Importantly, from the standpoint of prospective practice in surgical pathology, these cases most often are not encountered with clinical evidence HLRCC family history or stigmata, even on close questioning and examination. Despite this fact, genetic counseling to rule out germline mutation should be recommended, as it is likely that a significant proportion will be associated with germline FH mutation. In any case, the remarkable aggressiveness of disease seen in this cohort emphasizes the importance of recognition, diagnostic workup, and interdisciplinary consideration of the genetic implications to patient families. While this aspect raises intriguing questions regarding whether renal cell carcinomas which are FH-deficient might occur outside of the hereditary setting through somatic FH mutation, epigenetic mechanisms, or otherwise, resolution of this question will require greater experience with these tumors, even beyond the morphologic spectrum of TC-PD. We also characterized an infrequent but real subset of tumors with TC-PD morphology that appear unrelated to FH-deficiency and tend to arise in somewhat older individuals.
Based on preliminary observations on a subset of the present cohort (and experience with RCC cases even without the TC-PD pattern (18)) we proposed at the 2015 USCAP meeting (23) use of the provisional diagnostic term, FH-deficient RCC, for tumors with suggestive morphology and FH-2SC+ immunophenotype but where HLRCC stigmata/history cannot be reliably ascertained and genetic testing is urged. We believe that this diagnostic term, when rigorously applied based on both distinctive histopathological features and increasingly reliable immunohistochemistry, allows triage for further work up or establishment of HLRCC by genetic testing. It is also adaptable to emerging technologies such as NGS-based tumor mutation profiling, which we suspect will also be used to identify FH-deficient RCCs with some frequency going forward. It also allows designation of cases that might represent apparently sporadic forms, and standardizes analogous terminology between FH-deficient RCC and SDH-deficient RCC (46, 47), two settings where a constellation of characteristic morphology and immunophenotype are employed to establish diagnosis of tumors strongly associated with hereditary disease and where referral for genetic counseling is urged. Finally, prospective studies of cases recognized as FH-deficient RCC with consent for comprehensive comparative constitutional and tumor molecular profiling and family genetic counseling would enable ascertainment of the relative prevalence of apparent germline versus somatic mutations, addressing one of the principal outstanding questions for this interesting group of tumors.
Supplementary Material
Case TC-PD 16 (see also Figure 2D) demonstrated only focal tubulocystic morphology involving ∼10% of the tumor (A), though in such areas hobnailed cells with high nucleolar grade line variable sized tubules as appropriately expected (B). Poorly differentiated morphology was extensive in this case, including areas of dense tubulopapillary growth (C) and areas of solid and cordlike growth (D). This case was FH- by immunohistochemistry (panel E, lower half, with scattered FH-positive entrapped normal tubules and normal kidney above). This case was positive for 2SC (panel F, same field as depicted in E, showing diffuse, strong nucleocytoplasmic 2SC positivity in the carcinoma), consistent with homozygous K230 mutations detected by NGS.
TC-PD 19 demonstrates a large area of tubulocystic pattern with a cellular poorly differentiated zone to the lower left (A). In some areas the dense tubular morphology seemed to overrun even the tubulocystic component (B). In other areas, the architecture was more papillary, reminiscent of that described for HLRCC (C), while the nucleoli and perinucleolar clearing were quite prominent multifocally (D, high power inset of C). FH was diffusely positive in both the internal control normal kidney and the tumor (E, same field as A) while 2SC was diffusely positive in the tumor (F, same fields as A and E). This suspicious case harbored heterozygous N188T and H318Y mutations of FH, and was reclassified as FH-deficient.
A subset of nine cases demonstrated coagulative tumor necrosis per ISUP Vancouver criteria (A-B). Prominent nucleoli with perinucleolar clearing were variably apparent in these cases, generally more diffuse in those associated with the FH-deficient immunophenotype. However, nucleolar forms in the range described previously for HLRCC-RCCs were identifiable at least focally in all cases, including these two, which showed FH-retained immunophenotype (C-D).
Acknowledgments
A preliminary version of this study was presented as an oral paper at the March 2015 United States and Canadian Academy of Pathology Meeting in Boston, MA. The authors would like to thank Ann Bialik for technical assistance with immunohistochemistry.
Conflicts of Interest and Source of Funding: S.A.T. had a prior sponsored research agreement with and has received travel support from Thermo Fisher Scientific. S.A.T is supported by the A. Alfred Taubman Medical Research Institute. O.H. acknowledges support of the Charles University Research Fund (project number P36) and by the project CZ.1.05/2.1.00/03.0076 from European Regional Development Fund.
References
- 1.Murphy WM, Beckwith JB, Farrow GM, et al. Tumors of the kidney, bladder, and related urinary structures. Washington, D.C.: Available from the American Registry of Pathology, Armed Forces Institute of Pathology; 1994. [Google Scholar]
- 2.Amin MB, MacLennan GT, Gupta R, et al. Tubulocystic carcinoma of the kidney: clinicopathologic analysis of 31 cases of a distinctive rare subtype of renal cell carcinoma. The American journal of surgical pathology. 2009;33:384–392. doi: 10.1097/PAS.0b013e3181872d3f. [DOI] [PubMed] [Google Scholar]
- 3.Srigley JR, Delahunt B, Eble JN, et al. The International Society of Urological Pathology (ISUP) Vancouver Classification of Renal Neoplasia. The American journal of surgical pathology. 2013;37:1469–1489. doi: 10.1097/PAS.0b013e318299f2d1. [DOI] [PubMed] [Google Scholar]
- 4.Moch H, Humphrey PA, Ulbright TM, Reuter VE, editors. WHO Classifications of Tumours of the Urinary System and Male Genital Organs. Lyon: IARC Press; 2016. [DOI] [PubMed] [Google Scholar]
- 5.MacLennan GT, Cheng L. Tubulocystic carcinoma of the kidney. The Journal of urology. 2011;185:2348–2349. doi: 10.1016/j.juro.2011.03.043. [DOI] [PubMed] [Google Scholar]
- 6.Tran T, Jones CL, Williamson SR, et al. Tubulocystic renal cell carcinoma is an entity that is immunohistochemically and genetically distinct from papillary renal cell carcinoma. Histopathology. 2016;68:850–857. doi: 10.1111/his.12840. [DOI] [PubMed] [Google Scholar]
- 7.Osunkoya AO, Young AN, Wang W, et al. Comparison of gene expression profiles in tubulocystic carcinoma and collecting duct carcinoma of the kidney. The American journal of surgical pathology. 2009;33:1103–1106. doi: 10.1097/PAS.0b013e3181a13e7b. [DOI] [PubMed] [Google Scholar]
- 8.Yang XJ, Zhou M, Hes O, et al. Tubulocystic carcinoma of the kidney: clinicopathologic and molecular characterization. The American journal of surgical pathology. 2008;32:177–187. doi: 10.1097/PAS.0b013e318150df1d. [DOI] [PubMed] [Google Scholar]
- 9.Zhou M, Yang XJ, Lopez JI, et al. Renal tubulocystic carcinoma is closely related to papillary renal cell carcinoma: implications for pathologic classification. The American journal of surgical pathology. 2009;33:1840–1849. doi: 10.1097/PAS.0b013e3181be22d1. [DOI] [PubMed] [Google Scholar]
- 10.Al-Hussain TO, Cheng L, Zhang S, et al. Tubulocystic carcinoma of the kidney with poorly differentiated foci: a series of 3 cases with fluorescence in situ hybridization analysis. Human pathology. 2013;44:1406–1411. doi: 10.1016/j.humpath.2012.11.015. [DOI] [PubMed] [Google Scholar]
- 11.Zhao M, Teng X, Ru G, et al. Tubulocystic renal cell carcinoma with poorly differentiated foci is indicative of aggressive behavior: clinicopathologic study of two cases and review of the literature. Int J Clin Exp Pathol. 2015;8:11124–11131. [PMC free article] [PubMed] [Google Scholar]
- 12.Merino MJ, Torres-Cabala C, Pinto P, et al. The morphologic spectrum of kidney tumors in hereditary leiomyomatosis and renal cell carcinoma (HLRCC) syndrome. The American journal of surgical pathology. 2007;31:1578–1585. doi: 10.1097/PAS.0b013e31804375b8. [DOI] [PubMed] [Google Scholar]
- 13.Schmidt LS, Linehan WM. Hereditary leiomyomatosis and renal cell carcinoma. Int J Nephrol Renovasc Dis. 2014;7:253–260. doi: 10.2147/IJNRD.S42097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Udager AM, Alva A, Chen YB, et al. Hereditary leiomyomatosis and renal cell carcinoma (HLRCC): a rapid autopsy report of metastatic renal cell carcinoma. The American journal of surgical pathology. 2014;38:567–577. doi: 10.1097/PAS.0000000000000127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Llamas-Velasco M, Requena L, Kutzner H, et al. Fumarate hydratase immunohistochemical staining may help to identify patients with multiple cutaneous and uterine leiomyomatosis (MCUL) and hereditary leiomyomatosis and renal cell cancer (HLRCC) syndrome. J Cutan Pathol. 2014;41:859–865. doi: 10.1111/cup.12396. [DOI] [PubMed] [Google Scholar]
- 16.Castro-Vega LJ, Buffet A, De Cubas AA, et al. Germline mutations in FH confer predisposition to malignant pheochromocytomas and paragangliomas. Hum Mol Genet. 2014;23:2440–2446. doi: 10.1093/hmg/ddt639. [DOI] [PubMed] [Google Scholar]
- 17.Joseph NM, Solomon DA, Frizzell N, et al. Morphology and Immunohistochemistry for 2SC and FH Aid in Detection of Fumarate Hydratase Gene Aberrations in Uterine Leiomyomas From Young Patients. The American journal of surgical pathology. 2015;39:1529–1539. doi: 10.1097/PAS.0000000000000520. [DOI] [PubMed] [Google Scholar]
- 18.Trpkov K, Hes O, Agaimy A, et al. Fumarate Hydratase-deficient Renal Cell Carcinoma Is Strongly Correlated With Fumarate Hydratase Mutation and Hereditary Leiomyomatosis and Renal Cell Carcinoma Syndrome. The American journal of surgical pathology. 2016;40:865–875. doi: 10.1097/PAS.0000000000000617. [DOI] [PubMed] [Google Scholar]
- 19.Martinek P, Grossmann P, Hes O, et al. Genetic testing of leiomyoma tissue in women younger than 30 years old might provide an effective screening approach for the hereditary leiomyomatosis and renal cell cancer syndrome (HLRCC) Virchows Arch. 2015;467:185–191. doi: 10.1007/s00428-015-1783-y. [DOI] [PubMed] [Google Scholar]
- 20.Chen YB, Kong MX, Bialik A, et al. Hereditary Leiomyomatosis and Renal Cell Carcinoma (HLRCC)-Associated Renal Cancer: A Comparison of Fumarate Hydratase (FH) and S-(2-Succino)-Cysteine (2SC) Immunohistochemistry as Ancillary Tools. Mod Pathol. 2015;28 [Google Scholar]
- 21.Smith SC, Trpkov K, Mehra R, et al. Is Tubulocystic Carcinoma With Dedifferentiation a form of HLRCC/Fumarate Hydratase-Deficient RCC? Mod Pathol. 2015;(supplement) [Google Scholar]
- 22.Delahunt B, Cheville JC, Martignoni G, et al. The International Society of Urological Pathology (ISUP) grading system for renal cell carcinoma and other prognostic parameters. The American journal of surgical pathology. 2013;37:1490–1504. doi: 10.1097/PAS.0b013e318299f0fb. [DOI] [PubMed] [Google Scholar]
- 23.Smith SC, Trpkov K, Mehra R, et al. Is Tubulocystic Carcinoma With Dedifferentiation a form of HLRCC/Fumarate Hydratase-Deficient RCC? Mod Pathol. 2015;(suppl 2s):260A. [Google Scholar]
- 24.Grasso C, Butler T, Rhodes K, et al. Assessing copy number alterations in targeted, amplicon-based next-generation sequencing data. The Journal of molecular diagnostics : JMD. 2015;17:53–63. doi: 10.1016/j.jmoldx.2014.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Warrick JI, Hovelson DH, Amin A, et al. Tumor evolution and progression in multifocal and paired non-invasive/invasive urothelial carcinoma. Virchows Arch. 2015;466:297–311. doi: 10.1007/s00428-014-1699-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kadakia KC, Tomlins SA, Sanghvi SK, et al. Comprehensive serial molecular profiling of an “N of 1” exceptional non-responder with metastatic prostate cancer progressing to small cell carcinoma on treatment. J Hematol Oncol. 2015;8:109. doi: 10.1186/s13045-015-0204-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grasso CS, Cani AK, Hovelson DH, et al. Integrative molecular profiling of routine clinical prostate cancer specimens. Ann Oncol. 2015;26:1110–1118. doi: 10.1093/annonc/mdv134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reed WB, Walker R, Horowitz R. Cutaneous leiomyomata with uterine leiomyomata. Acta Derm Venereol. 1973;53:409–416. [PubMed] [Google Scholar]
- 29.Launonen V, Vierimaa O, Kiuru M, et al. Inherited susceptibility to uterine leiomyomas and renal cell cancer. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:3387–3392. doi: 10.1073/pnas.051633798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tomlinson IP, Alam NA, Rowan AJ, et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nature genetics. 2002;30:406–410. doi: 10.1038/ng849. [DOI] [PubMed] [Google Scholar]
- 31.Adeniran AJ, Shuch B, Humphrey PA. Hereditary Renal Cell Carcinoma Syndromes: Clinical, Pathologic, and Genetic Features. The American journal of surgical pathology. 2015;39:e1–e18. doi: 10.1097/PAS.0000000000000562. [DOI] [PubMed] [Google Scholar]
- 32.Shuch B, Ricketts CJ, Vocke CD, et al. Adrenal nodular hyperplasia in hereditary leiomyomatosis and renal cell cancer. The Journal of urology. 2013;189:430–435. doi: 10.1016/j.juro.2012.07.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Menko FH, Maher ER, Schmidt LS, et al. Hereditary leiomyomatosis and renal cell cancer (HLRCC): renal cancer risk, surveillance and treatment. Fam Cancer. 2014;13:637–644. doi: 10.1007/s10689-014-9735-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ulamec M, Skenderi F, Zhou M, et al. Molecular Genetic Alterations in Renal Cell Carcinomas With Tubulocystic Pattern: Tubulocystic Renal Cell Carcinoma, Tubulocystic Renal Cell Carcinoma With Heterogenous Component and Familial Leiomyomatosis-associated Renal Cell Carcinoma. Clinicopathologic and Molecular Genetic Analysis of 15 Cases. Appl Immunohistochem Mol Morphol. 2015 doi: 10.1097/PAI.0000000000000213. [DOI] [PubMed] [Google Scholar]
- 35.Pollard PJ, Spencer-Dene B, Shukla D, et al. Targeted inactivation of fh1 causes proliferative renal cyst development and activation of the hypoxia pathway. Cancer Cell. 2007;11:311–319. doi: 10.1016/j.ccr.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 36.Ristau BT, Kamat SN, Tarin TV. Abnormal Cystic Tumor in a Patient with Hereditary Leiomyomatosis and Renal Cell Cancer Syndrome: Evidence of a Precursor Lesion? Case Rep Urol. 2015;2015:303872. doi: 10.1155/2015/303872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bayley JP, Launonen V, Tomlinson IP. The FH mutation database: an online database of fumarate hydratase mutations involved in the MCUL (HLRCC) tumor syndrome and congenital fumarase deficiency. BMC Med Genet. 2008;9:20. doi: 10.1186/1471-2350-9-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gardie B, Remenieras A, Kattygnarath D, et al. Novel FH mutations in families with hereditary leiomyomatosis and renal cell cancer (HLRCC) and patients with isolated type 2 papillary renal cell carcinoma. Journal of medical genetics. 2011;48:226–234. doi: 10.1136/jmg.2010.085068. [DOI] [PubMed] [Google Scholar]
- 39.Barker KT, Bevan S, Wang R, et al. Low frequency of somatic mutations in the FH/multiple cutaneous leiomyomatosis gene in sporadic leiomyosarcomas and uterine leiomyomas. Br J Cancer. 2002;87:446–448. doi: 10.1038/sj.bjc.6600502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vaidya S, Shaik NA, Latha M, et al. No evidence for the role of somatic mutations and promoter hypermethylation of FH gene in the tumorigenesis of nonsyndromic uterine leiomyomas. Tumour Biol. 2012;33:1411–1418. doi: 10.1007/s13277-012-0391-6. [DOI] [PubMed] [Google Scholar]
- 41.Harrison WJ, Andrici J, Maclean F, et al. Hydratase-deficient Uterine Leiomyomas Occur in Both the Syndromic and Sporadic Settings. The American journal of surgical pathology. 2015 doi: 10.1097/PAS.0000000000000573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Linehan WM, Spellman PT, Ricketts CJ, et al. Comprehensive Molecular Characterization of Papillary Renal-Cell Carcinoma. N Engl J Med. 2015 doi: 10.1056/NEJMoa1505917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu JN, Roberts CW. ARID1A mutations in cancer: another epigenetic tumor suppressor? Cancer Discov. 2013;3:35–43. doi: 10.1158/2159-8290.CD-12-0361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cancer Genome Atlas Research N. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013;499:43–49. doi: 10.1038/nature12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cancer Genome Atlas Research N. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature. 2014;507:315–322. doi: 10.1038/nature12965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Williamson SR, Eble JN, Amin MB, et al. Succinate dehydrogenase-deficient renal cell carcinoma: detailed characterization of 11 tumors defining a unique subtype of renal cell carcinoma. Mod Pathol. 2014 doi: 10.1038/modpathol.2014.86. [DOI] [PubMed] [Google Scholar]
- 47.Gill AJ, Hes O, Papathomas T, et al. Succinate Dehydrogenase (SDH)-deficient Renal Carcinoma: A Morphologically Distinct Entity: A Clinicopathologic Series of 36 Tumors From 27 Patients. The American journal of surgical pathology. 2014;38:1588–1602. doi: 10.1097/PAS.0000000000000292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Case TC-PD 16 (see also Figure 2D) demonstrated only focal tubulocystic morphology involving ∼10% of the tumor (A), though in such areas hobnailed cells with high nucleolar grade line variable sized tubules as appropriately expected (B). Poorly differentiated morphology was extensive in this case, including areas of dense tubulopapillary growth (C) and areas of solid and cordlike growth (D). This case was FH- by immunohistochemistry (panel E, lower half, with scattered FH-positive entrapped normal tubules and normal kidney above). This case was positive for 2SC (panel F, same field as depicted in E, showing diffuse, strong nucleocytoplasmic 2SC positivity in the carcinoma), consistent with homozygous K230 mutations detected by NGS.
TC-PD 19 demonstrates a large area of tubulocystic pattern with a cellular poorly differentiated zone to the lower left (A). In some areas the dense tubular morphology seemed to overrun even the tubulocystic component (B). In other areas, the architecture was more papillary, reminiscent of that described for HLRCC (C), while the nucleoli and perinucleolar clearing were quite prominent multifocally (D, high power inset of C). FH was diffusely positive in both the internal control normal kidney and the tumor (E, same field as A) while 2SC was diffusely positive in the tumor (F, same fields as A and E). This suspicious case harbored heterozygous N188T and H318Y mutations of FH, and was reclassified as FH-deficient.
A subset of nine cases demonstrated coagulative tumor necrosis per ISUP Vancouver criteria (A-B). Prominent nucleoli with perinucleolar clearing were variably apparent in these cases, generally more diffuse in those associated with the FH-deficient immunophenotype. However, nucleolar forms in the range described previously for HLRCC-RCCs were identifiable at least focally in all cases, including these two, which showed FH-retained immunophenotype (C-D).