

Abstract
Objective:
Although individual neonatal epilepsy syndromes are rare, as a group they represent a sizable subgroup of neonatal seizure etiologies. We evaluated the profile of neonatal epilepsies in a prospective cohort of newborns with seizures.
Methods:
Consecutive newborns with seizures were enrolled in the Neonatal Seizure Registry (an association of 7 US children's hospitals). Treatment and diagnostic testing were at the clinicians' discretion. Neonates with seizures related to epileptic encephalopathies (without structural brain abnormalities), brain malformations, or benign familial epilepsies were included in this analysis.
Results:
Among 611 consecutive newborns with seizures, 79 (13%) had epilepsy (35 epileptic encephalopathy, 32 congenital brain malformations, 11 benign familial neonatal epilepsy [BFNE], 1 benign neonatal seizures). Twenty-nine (83%) with epileptic encephalopathy had genetic testing and 24/29 (83%) had a genetic etiology. Pathogenic or likely pathogenic KCNQ2 variants (n = 10) were the most commonly identified etiology of epileptic encephalopathy. Among 23 neonates with brain malformations who had genetic testing, 7 had putative genetic etiologies. Six infants with BFNE had genetic testing; 3 had pathogenic KCNQ2 variants and 1 had a pathogenic KCNQ3 variant. Comorbid illnesses that predisposed to acute symptomatic seizures occurred in 3/35 neonates with epileptic encephalopathy vs 10/32 with brain malformations (p = 0.03). Death or discharge to hospice were more common among newborns with brain malformations (11/32) than those with epileptic encephalopathy (3/35, p = 0.01).
Conclusions:
Neonatal epilepsy is often due to identifiable genetic causes. Genetic testing is now warranted for newborns with epilepsy in order to guide management and inform discussions of prognosis.
Although most seizures in the neonatal period reflect acute, acquired brain dysfunction, a substantial minority of newborns with seizures have a neonatal-onset epilepsy. Early differentiation of acute symptomatic seizures from neonatal-onset epilepsies has important therapeutic and prognostic implications since the evaluation and long-term management of neonatal epilepsies are distinct from those of acute symptomatic seizures. Many neonatal epilepsy syndromes have a genetic etiology1,2 and some may be amenable to targeted therapy.3 For example, recent studies have shown that oral carbamazepine, a sodium channel blocker seldom used in newborns, is safe and effective in KCNQ2-related neonatal epilepsies, even in the context of status epilepticus.4,5 Other syndromes, such as pyridoxine-dependent epilepsy, have clearly defined and specific treatments.6,7 In addition, the prognosis of many neonatal epilepsies is unfavorable, with the notable exception of benign familial neonatal epilepsy (BFNE).2
Since neonatal seizures are relatively rare,8,9 and neonatal epilepsies are even less common, effective clinical research on this topic requires multicenter collaboration. We aimed to determine the proportion of newborns who have neonatal-onset epilepsy, rather than acute symptomatic seizures, among all newborns who present with seizures to tertiary care children's hospitals. Further, we aimed to identify the clinical characteristics, etiologies, and initial treatments prescribed for a contemporary prospective cohort of newborns with neonatal epilepsies who were treated at 1 of 7 US tertiary children's hospitals.
METHODS
Standard protocol approvals, registrations, and patient consents.
The Neonatal Seizure Registry is an association of 7 US neonatal neurology programs with level IV neonatal intensive care units that were selected based on their adherence to the American Clinical Neurophysiology Society (ACNS) guideline on neonatal EEG monitoring.10 Each center obtained institutional review board approval and a waiver of informed consent was granted for this study.
Patients.
Every newborn with a clinical or EEG diagnosis of seizures treated at 1 of the 7 participating tertiary children's hospitals between January 2013 and November 2015 was prospectively enrolled in the Neonatal Seizure Registry. The clinical profile of a subset of newborns with acute symptomatic seizures has been presented previously.11 Prospective, systematic medical record review was conducted by a research assistant and an investigator at each study site. In addition, the Registry principal investigator (H.C.G.) reviewed a random sample of 5 charts from each center to insure data integrity and consistent data abstraction across sites.
Newborns were considered to have neonatal epilepsy, and included in the present analysis, if the study site investigator indicated that they had any of the following: (1) a clinical diagnosis of neonatal epileptic encephalopathy (no developmental brain abnormality, history incompatible with benign neonatal seizures or BFNE, and lack of primary acute symptomatic etiology), (2) a congenital brain malformation as the seizure etiology (e.g., lissencephaly, focal malformation of cortical development, holoprosencephaly), or (3) a clinical diagnosis of BFNE or benign neonatal seizures. Neonates with epilepsy and a comorbid illness that predisposed to acute symptomatic seizures were included.
Evaluation (e.g., EEG, neuroimaging, and genetic testing) and treatment (including medication selection, dosing, and duration of treatment) were conducted at the discretion of the clinical teams and were not mandated by a research protocol. Data regarding evaluation for etiology, treatment, and hospital outcomes (e.g., death or discharge to home) are presented.
Counseling and consent for genetic testing were conducted by the treating clinicians and the available clinical result reports were evaluated by the investigators. Single nucleotide variants and small indels were identified using commercial gene panels or Sanger sequencing. Whole exome sequencing was used in a subset of cases. Copy number variations were identified using genomic single nucleotide polymorphism arrays or comprehensive genomic hybridization arrays. The variants detected were queried against the Exome Variant Server database (evs.gs.washington.edu) in order to determine their prevalence in large control populations. Polyphen-2 software (genetics.bwh.harvard.edu/pph2) was used to determine in silico likelihood of pathogenicity. Clinical significance of variants and inheritance patterns were queried using the ClinVar (ncbi.nlm.nih.gov/clinvar) and OMIM (ncbi.nlm.nih.gov/omim) databases.
EEG monitoring was performed according to the ACNS guideline.10 Seizure burden, defined through continuous video-EEG monitoring, is presented as rare (<7 recorded seizures), many isolated (≥7 recorded seizures), frequent recurrent, and status epilepticus (>50% of any 60-minute EEG epoch composed of seizures). Seizure semiology and EEG background classification were extracted from clinical EEG monitoring reports. The EEG background was assigned to a category by the site investigators (normal, mildly abnormal, moderately abnormal, severely abnormal), according to a published classification rubric.12
Analysis.
Descriptive statistics are presented and results of χ2, Fisher exact tests, t tests, and Kruskal-Wallis tests are reported. A p value <0.05 was considered significant. Analyses were conducted using SAS (SAS Institute Inc., Cary, NC).
RESULTS
Between January 2013 and November 2015, 611 newborns were enrolled in the Neonatal Seizure Registry. Among these newborns, 79 (13%) had neonatal epilepsies (table 1). These included 35 with neonatal epileptic encephalopathy syndromes, 32 with congenital brain malformations, 11 with BFNE, and 1 with benign neonatal seizures (tables 2 and 3).
Table 1.
Demographic and clinical profiles of 79 newborns with neonatal-onset epilepsy syndromes, compared with 531 with acute symptomatic neonatal seizures
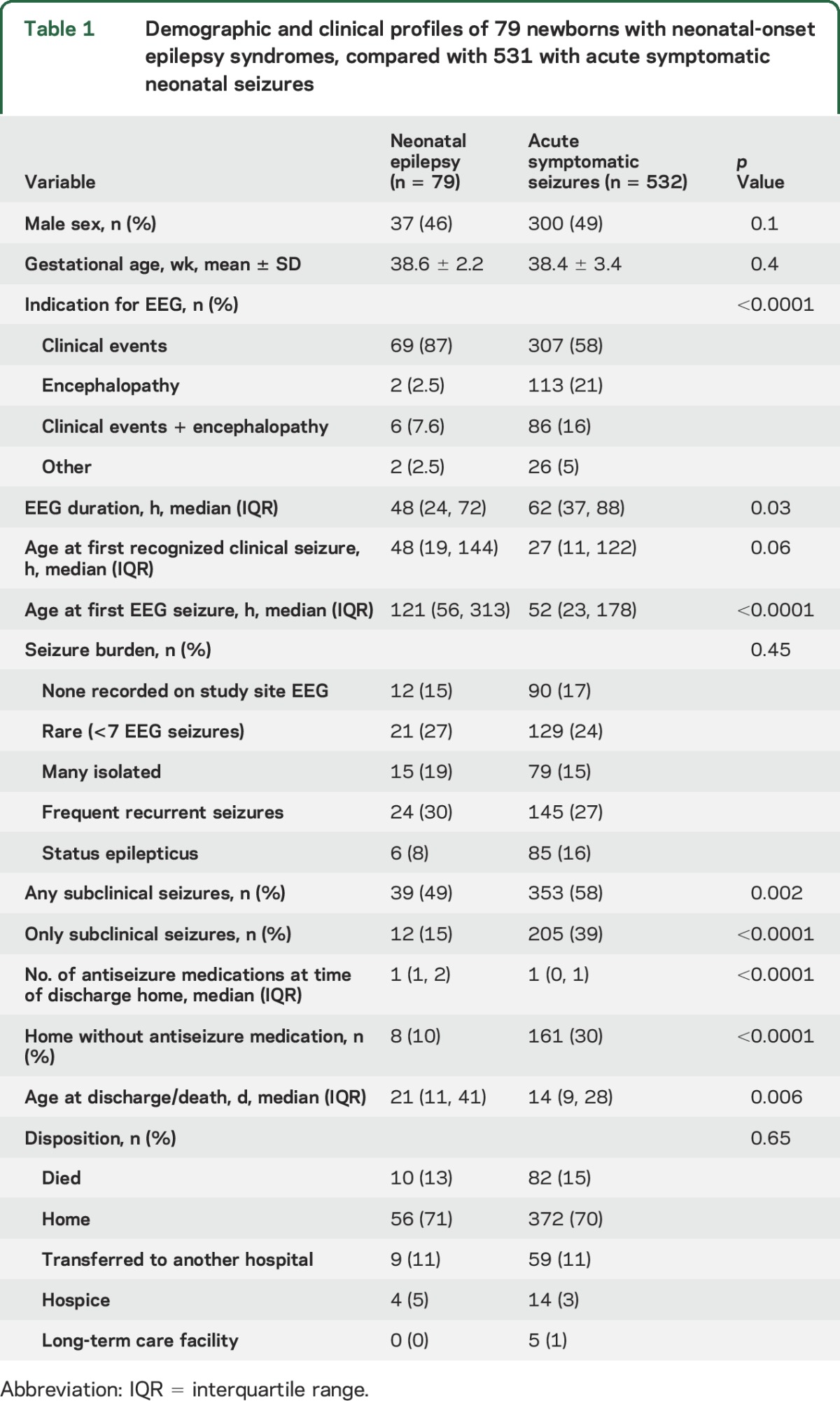
Table 2.
Genetic testing results for newborns with neonatal epilepsy syndromes in a cohort of consecutive patients from US neonatal intensive care units
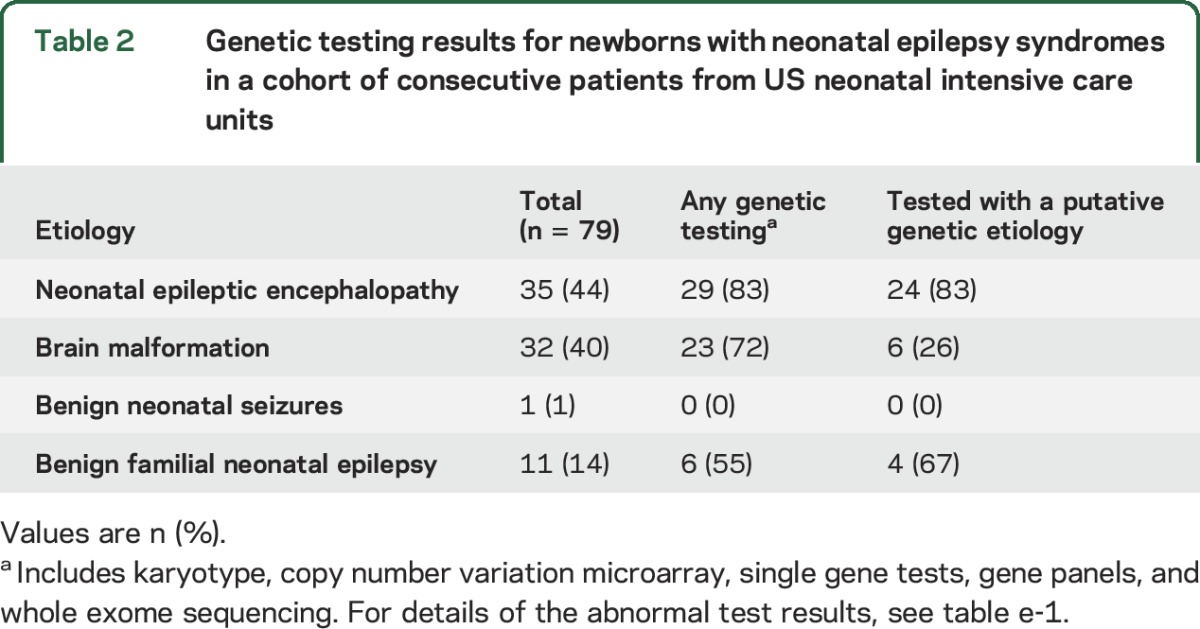
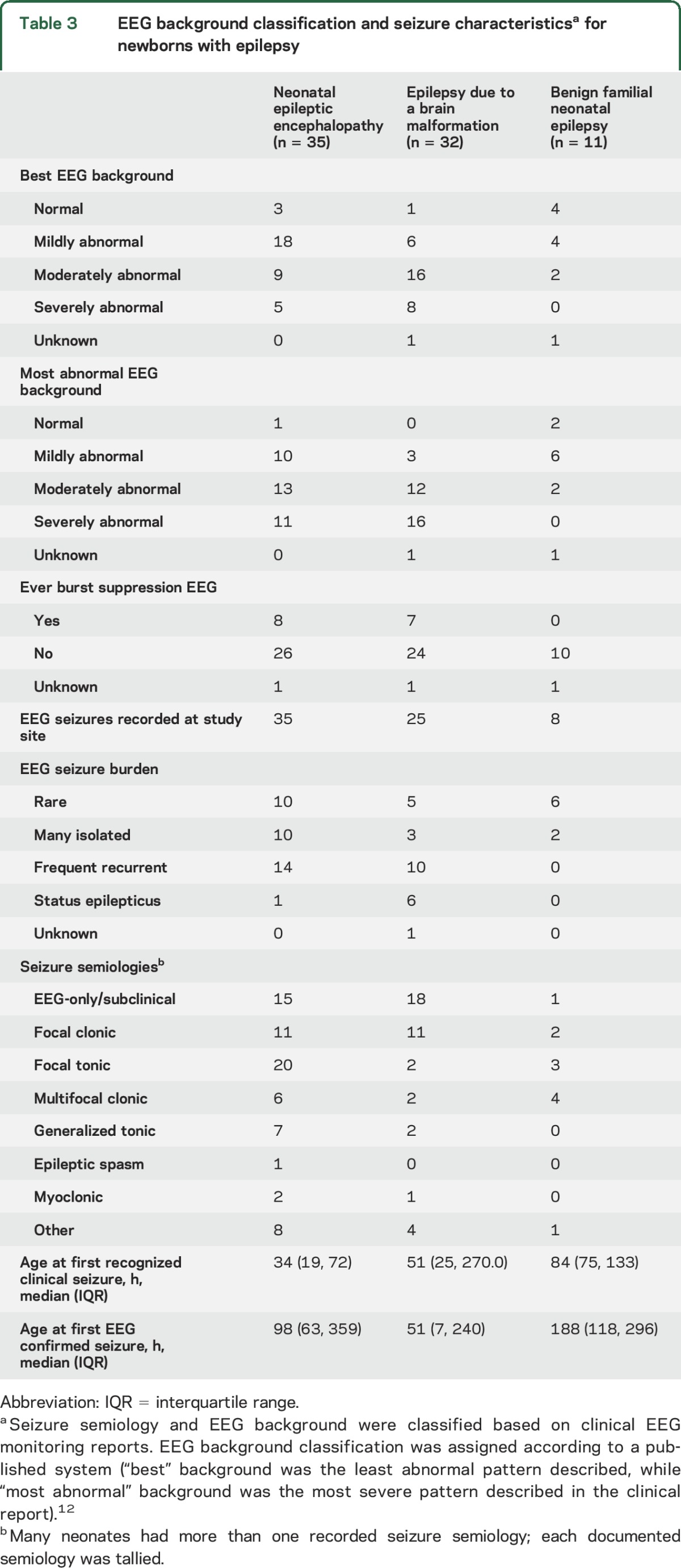
Table 3.
EEG background classification and seizure characteristicsa for newborns with epilepsy
There was no difference in gestational age, sex distribution, seizure burden, or final disposition between neonates with epilepsy and those whose seizures were due to an acute symptomatic etiology (table 1). EEG monitoring was most commonly initiated due to clinical events concerning for seizures for newborns with epilepsy (n = 69 [87%]), while those with acute symptomatic seizures were most often monitored to identify possible subclinical (EEG-only) seizures in the setting of neonatal encephalopathy (n = 307 [58%]). Newborns with neonatal epilepsy were less likely than those with acute symptomatic seizures to have any subclinical seizures (39/79 vs 353/532, p = 0.002) or to have exclusively subclinical seizures (12/79 vs 205/532, p < 0.0001). The duration of EEG monitoring was shorter for neonates with epilepsy (median 48 hours) compared with those monitored for acute symptomatic seizures (median 62 hours, p = 0.03).
Neonatal epileptic encephalopathies.
Thirty-five newborns (20 male, estimated gestational age [EGA] 39.4 ± 1.2 weeks) had neonatal epileptic encephalopathy syndromes (tables 2 and 3). Four had a documented clinical diagnosis of Ohtahara syndrome and one had early myoclonic epilepsy. The others were not assigned a classic epilepsy syndrome by their treating clinicians. Among the 31 for whom the information was documented, 22 received at least one dose of pyridoxine, folinic acid, or pyridoxyl-5-phosphate, although none was ultimately diagnosed with vitamin-dependent epilepsy.
Most (29/35 [83%]) had genetic testing performed on a clinical basis. The majority of those tested had a genetic etiology identified (24/29 [83%]). Of these, pathogenic or likely pathogenic variants in KCNQ2 were the most common (n = 10, of whom 1 also had a 15q11.2 deletion). Three infants had SCN2a variants, and 1 each had variants in SLC13A5, STXBP1, KCNT1, GDLC, CDKL5, CHD7, MBD5, and TSC2 genes. One newborn had variants in both SCN1a and GRIN2B; 1 had variants in COQ4 as well as MARS and SLC2A8; and 1 had a large deletion at 16p13.11. Details of the genetic test results are presented in table e-1 at Neurology.org.
Clinicians most commonly ordered an epilepsy gene panel (15 of 23 completed gene panels were diagnostic of a genetic epilepsy). Chromosomal microarray was completed for 12 neonates and revealed the cause of the epilepsy in 2. Whole exome sequencing was diagnostic for 2 of the 3 individuals for whom it was performed (1 with a KCNQ2 variant and 1 with a SLC13A5 variant). Single gene testing was obtained for 9 newborns, and 5 confirmed the suspected diagnosis (2 KCNQ2, 1 TSC2, 1 GDLC, 1 CDH7). Two infants had comorbid sepsis and 1 had hypocalcemia.
Three of these newborns died during the neonatal admission and 3 others were transferred to a different hospital for continued care. The remaining 29 (83%) survived to discharge home. These infants were prescribed a median of 1 antiseizure medication at the time of discharge (range 0–6). Antiseizure medications at the time of discharge to home or transfer to a different hospital included phenobarbital (23 [72%]), levetiracetam (14 [44%]), topiramate (8 [25%]), phenytoin (5 [18%]), pyridoxine (5 [13%]), folinic acid (4 [12%]), carbamazepine (3 [9%]), oxcarbazepine (2 [6%]), clobazam (1 [3%]), acetazolamide (1 [3%]), and pyridoxal-5-phosphate (1 [3%]).
Epilepsy related to congenital brain malformations.
Thirty-two newborns (12 male, EGA 37.5 ± 2.7 weeks) had epilepsy due to congenital brain malformations. The most common malformations were neuronal migration abnormalities (5 lissencephaly, including 1 with associated congenital muscular dystrophy; 3 polymicrogyria; 3 focal cortical dysplasia; 2 schizencephaly, including 1 with associated polymicrogyria; 1 pachygyria; 1 subependymal gray matter heterotopia). Two had hemimegalencephaly, 5 had Dandy-Walker malformations, 3 had holoprosencephaly, and 1 had septo-optic dysplasia. Fifteen had no extracerebral congenital anomalies, while 17 had a range of congenital abnormalities (including 7 congenital heart disease, 2 myelomeningocele, 1 tracheal-esophageal fistula, 1 omphalocele, and 2 congenital glaucoma). Ten (31%) also had a comorbid acute symptomatic seizure etiology (3 hypoxic-ischemic encephalopathy, 2 parenchymal hemorrhages, 2 subdural hematoma, 1 CNS infection, and 2 comorbid peroxisomal disorders, 1 of whom also had sepsis). Comorbid acute illnesses that predisposed to acute symptomatic seizures were more common among newborns with brain malformations than those with epileptic encephalopathies (10/32 vs 3/35, p = 0.02).
Twenty-three newborns with brain malformations had recorded genetic testing results (table 2 and table e-1), and 6 of these had a defined genetic diagnosis (1 each had TUBA1A, DCX, SETBP1, LAMB1, Emmanuel syndrome, and 15q11.1 duplication). Twenty infants had a chromosomal microarray (1 with abnormalities thought to explain the epilepsy: 15q11.1 duplication). A gene panel was diagnostic in 3 of 4 infants (TUBA1A, DCX, LAMB1). Single gene testing was also diagnostic for 1 of 2 infants (SETBP1). Whole exome sequencing was performed for 2 patients; the result was negative for 1 infant and revealed homozygosity at 7q21.11 to q31.33, which encompasses RELN, and might explain the brain malformation and epilepsy for 1 infant.
None of the neonates with brain malformation was given a clinical diagnosis of Ohtahara syndrome or early myoclonic epilepsy by the treating clinicians.
Eight neonates with epilepsy related to brain malformations did not have seizures recorded at the study center; 1 of these had documented EEG seizures at a referring hospital and the others had seizures diagnosed based on clinical semiologies prior to EEG monitoring (4 focal multifocal clonic, 2 bilateral tonic, 1 focal clonic, 4 other). Additional EEG and seizure semiology data are presented in table 3.
Neonates with epilepsy related to brain malformations were more likely to die (n = 7) or to be discharged to hospice care (n = 4) than those with epileptic encephalopathies (n = 3 died and none discharged to hospice; p = 0.01). Six were transferred to a local hospital for continued care. The 15 who survived to discharge home were prescribed a median of 2 antiseizure medications at the time of discharge (range 0–7). Medications at the time of discharge to home or transfer to a different hospital for ongoing care included phenobarbital (17 [81%]), levetiracetam (9 [43%]), topiramate (5 [24%]), oxcarbazepine (2 [10%]), carbamazepine (1 [5%]), clobazam (1 [5%]), phenytoin (1 [5%]), and lacosamide (1 [5%]).
Benign familial neonatal epilepsy.
Only 1 neonate had benign neonatal seizures (no genetic testing performed), whereas 11 were diagnosed with BFNE. Among the 11 newborns with BFNE (5 male, EGA 38.9 ± 1.9 weeks), 3 had no recorded EEG seizures (all 3 had clinical events of focal or multifocal clonic seizures along with focal or bilateral tonic seizures). All of the neonates with EEG seizures had rare or isolated seizures; none had frequent recurrent seizures or status epilepticus (table 3). Compared with neonates who had brain malformations or epileptic encephalopathy, those with BFNE had lower seizure burden (p < 0.0001).
Six of 11 with BFNE had genetic testing. This included 2 single gene tests (both revealed pathogenic KCNQ2 variants) and 4 gene panels (1 positive for a pathogenic KCNQ2 variant and 1 for a pathogenic KCNQ3 variant). One neonate had both a gene panel and whole exome sequencing, neither of which was diagnostic (table 2 and table e-1).
All of these newborns survived to discharge home and all were prescribed chronic antiseizure medication at the time of hospital discharge. Medications at the time of discharge to home included phenobarbital monotherapy (8 [73%]), carbamazepine monotherapy (3 [27%]), levetiracetam monotherapy (1 [9%]), oxcarbazepine monotherapy (1 [9%]), and levetiracetam and oxcarbazepine (1 [9%]).
DISCUSSION
Neonatal-onset epilepsies cause an important minority of neonatal seizures and present major diagnostic and therapeutic challenges. Our data indicate that identifiable genetic etiologies are responsible for many neonatal epilepsies; more than three-quarters of those who underwent testing had abnormalities identified. In addition, neonatal seizures due to epilepsy presented most often as clinically apparent events and required more than one antiseizure medication, as opposed to the more common subclinical (EEG-only) seizures recorded for newborns with acute symptomatic etiologies. Notably, nearly one-third of newborns with brain malformations as their primary seizure etiology had coexisting illnesses that also predisposed them to acute symptomatic seizures (hypoxic-ischemic encephalopathy, infection); this highlights the need for comprehensive diagnostic evaluations for all newborns with seizures.
Neonatal-onset epilepsies have a broad spectrum of phenotypes that ranges from relatively benign to much more severe. Genetic etiologies are being discovered for a growing proportion of these disorders, and this has helped to elucidate their molecular mechanisms.13 We demonstrate here that newborns with epilepsy very often have an identifiable genetic etiology. In the past, genetic testing could provide a diagnosis but did not alter treatment for most individuals; however, the role of genetic testing as a guide for treatment strategies is expanding. Several recent studies3,4,14 have highlighted the importance of accurate phenotype definition and seizure characterization in the neonatal period in order to expedite early diagnosis. Advances in diagnostic testing raise the important possibility of targeted treatment designed to improve seizure control and potentially improve long-term neurodevelopment. Finally, genetic testing may provide important long-term prognostic information for families to guide overall short- and long-term management, and to inform recurrence risk for future pregnancies.
Our multicenter prospective study gathered clinical data from the neonatal hospitalization of more than 600 consecutive newborns with seizures. Yet not every neonatal epilepsy syndrome or etiology was represented in this cohort. For example, there were no identified cases of vitamin-responsive epilepsy, even though more than two-thirds of neonates with epileptic encephalopathy received trials of pyridoxine, folinic acid, or pyridoxal-5-phosphate. This underscores the need for extensive multicenter collaborations for the study of rare neonatal epilepsies.
The classic neonatal epileptic encephalopathy syndromes15 were rarely identified by name in these patients' medical records. Only 4 were diagnosed with Ohtahara syndrome and just one with early myoclonic encephalopathy by their treating clinicians. This may be related to improving identification of the genetic underpinnings of many neonatal epilepsies such that newborns were given a specific genetic diagnosis (e.g., KCNQ2 encephalopathy) rather than the broader epileptic encephalopathy syndrome diagnosis.
All 7 study sites have level IV neonatal intensive care units and provide state-of-the-art neuromonitoring and clinical epilepsy care. All sites follow the ACNS guidelines for neonatal EEG monitoring, which includes conventional video-EEG throughout cooling and rewarming for neonates treated with therapeutic hypothermia for hypoxic-ischemic encephalopathy.10 This might explain why the duration of EEG monitoring was longer among neonates with acute symptomatic seizures than in those with epilepsy (hypoxic-ischemic encephalopathy was the most common cause of acute symptomatic seizures in this cohort11). In addition, neonates with epilepsy were less likely to have subclinical seizures than were those with acute symptomatic etiologies. This also likely contributed to the difference in EEG monitoring duration between the groups, since all centers monitor the majority of neonates until both electroclinical and subclinical (EEG-only) seizures have resolved for at least 24 hours.
A strength of our work is that we present real-world clinical data regarding a large number of newborns with epilepsy. Our study has some limitations related to its clinically based design. The study sites did not use uniform guidelines for diagnostic evaluations or treatment of seizures. All newborns with epilepsy had a brain MRI study, but the interpretation was performed locally by a pediatric neuroradiologist and confirmed by the study investigators rather than by a central neuroradiologist. EEG background characteristics and seizure assessments were extracted from clinical reports by the study investigators rather than by a central electroencephalographer. Genetic testing was undertaken at the clinicians' discretion and within the constraints of local policies regarding insurance reimbursement. Therefore, the approach to genetic testing was not consistent across study sites and the specific details of detected genetic variants were not uniformly available. Parental testing was not always obtained and this complicates the interpretation of some patients' results. It is possible that increased use of genetic testing would have identified an even greater number of neonates with a genetic basis for their epilepsy. It is also possible that, in time, variants that are currently determined to be of uncertain significance will be reclassified as pathogenic or as benign.
The gene discovery efforts of the last decade, particularly for the early-onset epileptic encephalopathies, have shown that genetic variants play a major etiologic role and should often be considered in the differential diagnosis of seizures in newborns and infants.16,17 However, many unresolved questions remain. The electroclinical presentation of some neonatal-onset epilepsies is not yet well-defined since many patients are only diagnosed in retrospect, when documented clinical and EEG findings related to the neonatal period may be scarce. In addition, variants in a single gene (e.g., KCNQ2) can be associated with both relatively benign and severe epilepsies. Yet our data suggest that a focused effort to increase access to clinical genetic testing for newborns with epilepsy may have far-reaching implications since a putative genetic etiology was found for three-quarters of newborns who were tested. Targeted treatments could eventually be provided to those with genetically proven etiologies and early effective treatment may improve the developmental trajectory for infants with severe early-life epilepsy.
Supplementary Material
ACKNOWLEDGMENT
The authors thank the research assistants at each study site for their work on this project: Manogna Manne, Stephanie Rau, Kari Harris, Emily Thorn, Catherine Clark, and Jessica Landers.
GLOSSARY
- ACNS
American Clinical Neurophysiology Society
- BFNE
benign familial neonatal epilepsy
- EGA
estimated gestational age
Footnotes
Supplemental data at Neurology.org
Editorial, page 880
Contributor Information
Collaborators: Neonatal Seizure Registry, Ann Marie Bergin, Dennis Dlugos, Donna M. Ferriero, Faye Silverstein, and Kevin Staley
AUTHOR CONTRIBUTIONS
Dr. Shellhaas co-conceived the study, acquired data, analyzed the data, and wrote the first draft of the manuscript. Dr. Wusthoff acquired data, assisted with interpretation of the results, and provided critical revision of the manuscript for important intellectual content. Dr. Tsuchida acquired data, assisted with interpretation of the results, and provided critical revision of the manuscript for important intellectual content. Dr. Glass co-conceived the study, acquired data, assisted with interpretation of the results, and provided critical revision of the manuscript for important intellectual content. Dr. Chu acquired data, assisted with interpretation of the results, and provided critical revision of the manuscript for important intellectual content. Dr. Massey acquired data and provided critical revision of the manuscript for important intellectual content. Dr. Soul acquired data, assisted with interpretation of the results, and provided critical revision of the manuscript for important intellectual content. Dr. Wiwattanadittakul acquired data and provided critical revision of the manuscript for important intellectual content. Dr. Abend acquired data, assisted with interpretation of the results, and provided critical revision of the manuscript for important intellectual content. Dr. Cilio co-conceived the study, reviewed and analyzed all genetic testing results, assisted with interpretation of the results, and provided critical revision of the manuscript for important intellectual content.
STUDY FUNDING
Supported by a grant from the Pediatric Epilepsy Research Foundation.
DISCLOSURE
R. Shellhaas receives research support from NIH, the American Sleep Medicine Foundation, the Pediatric Epilepsy Research Foundation, the Patient-Centered Outcomes Research Institute, and the University of Michigan Department of Pediatrics & Communicable Diseases; receives royalties from UpToDate; and serves as a consultant for the Epilepsy Study Consortium. C. Wusthoff receives research support from the Patient-Centered Outcomes Research Institute. T. Tsuchida receives research support from NIH. H. Glass receives research support from NIH, the Patient-Centered Outcomes Research Institute, the Pediatric Epilepsy Research Foundation, and the Cerebral Palsy Alliance. C. Chu receives research support from NIH. S. Massey reports no disclosures. J. Soul receives research support from NIH and receives honoraria from UpToDate. N. Wiwattanadittakul reports no disclosures. N. Abend receives research support from NIH. M. Cilio reports no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Yamamoto H, Okumura A, Fukuda M. Epilepsies and epileptic syndromes starting in the neonatal period. Brain Dev 2011;33:213–220. [DOI] [PubMed] [Google Scholar]
- 2.Grinton BE, Heron SE, Pelekanos JT, et al. Familial neonatal seizures in 36 families: clinical and genetic features correlate with outcome. Epilepsia 2015;56:1071–1080. [DOI] [PubMed] [Google Scholar]
- 3.Numis AL, Angriman M, Sullivan JE, et al. KCNQ2 encephalopathy: delineation of the electroclinical phenotype and treatment response. Neurology 2014;82:368–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pisano T, Numis AL, Heavin SB, et al. Early and effective treatment of KCNQ2 encephalopathy. Epilepsia 2015;56:685–691. [DOI] [PubMed] [Google Scholar]
- 5.Sands TT, Balestri M, Bellini G, et al. Rapid and safe response to low-dose carbamazepine in neonatal epilepsy. Epilepsia 2016;57:2019–2030. [DOI] [PubMed] [Google Scholar]
- 6.Van Hove JL, Lohr NJ. Metabolic and monogenic causes of seizures in neonates and young infants. Mol Genet Metab 2011;104:214–230. [DOI] [PubMed] [Google Scholar]
- 7.Mastrangelo M, Celato A, Leuzzi V. A diagnostic algorithm for the evaluation of early onset genetic-metabolic epileptic encephalopathies. Eur J Paediatric Neurol 2012;16:179–191. [DOI] [PubMed] [Google Scholar]
- 8.Ronen GM, Penney S, Andrews W. The epidemiology of clinical neonatal seizures in Newfoundland: a population-based study. J Pediatr 1999;134:71–75. [DOI] [PubMed] [Google Scholar]
- 9.Saliba RM, Annegers FJ, Waller DK, Tyson JE, Mizrahi EM. Incidence of neonatal seizures in Harris county, Texas, 1992–1994. Am J Epidemiol 1999;150:763–769. [DOI] [PubMed] [Google Scholar]
- 10.Shellhaas RA, Chang T, Tsuchida T, et al. The American Clinical Neurophysiology Society's guideline on continuous electroencephalography monitoring in neonates. J Clin Neurophysiol 2011;28:611–617. [DOI] [PubMed] [Google Scholar]
- 11.Glass HC, Shellhaas RA, Wusthoff CJ, et al. Contemporary profile of seizures in neonates: a prospective cohort study. J Pediatr 2016;174:98–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shellhaas RA, Clancy RR. Characterization of neonatal seizures by conventional and single channel EEG. Clin Neurophysiol 2007;118:2156–2161. [DOI] [PubMed] [Google Scholar]
- 13.Nabbout R, Dulac O. Genetics of early-onset epilepsy with encephalopathy. Nat Rev Neurol 2012;8:129–130. [DOI] [PubMed] [Google Scholar]
- 14.Bearden D, Strong A, Ehnot J, DiGiovine M, Dlugos D, Goldberg EM. Targeted treatment of migrating partial seizures in infancy with quinidine. Ann Neurol 2014;76:457–461. [DOI] [PubMed] [Google Scholar]
- 15.Berg AT, Berkovic SF, Brodie MJ, et al. Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE Commission on Classification and Terminology, 2005–2009. Epilepsia 2010;51:676–685. [DOI] [PubMed] [Google Scholar]
- 16.Epi4K Consortium, Epilepsy Phenome/Genome Project. De novo mutations in epileptic encephalopathies. Nature 2013;501:217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Trump N, McTague A, Brittain H, et al. Improving diagnosis and broadening the phenotypes in early-onset seizure and severe developmental delay disorders through gene panel analysis. J Med Genet 2016;53:310–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.