


Abstract
To assess whether there are universal rules that govern amino acid–base recognition, we investigate hydrogen bonds, van der Waals contacts and water-mediated bonds in 129 protein–DNA complex structures. DNA–backbone interactions are the most numerous, providing stability rather than specificity. For base interactions, there are significant base–amino acid type correlations, which can be rationalised by considering the stereochemistry of protein side chains and the base edges exposed in the DNA structure. Nearly two-thirds of the direct read-out of DNA sequences involves complex networks of hydrogen bonds, which enhance specificity. Two-thirds of all protein–DNA interactions comprise van der Waals contacts, compared to about one-sixth each of hydrogen and water-mediated bonds. This highlights the central importance of these contacts for complex formation, which have previously been relegated to a secondary role. Although common, water-mediated bonds are usually non-specific, acting as space-fillers at the protein–DNA interface. In conclusion, the majority of amino acid–base interactions observed follow general principles that apply across all protein–DNA complexes, although there are individual exceptions. Therefore, we distinguish between interactions whose specificities are ‘universal’ and ‘context-dependent’. An interactive Web-based atlas of side chain–base contacts provides access to the collected data, including analyses and visualisation of the three-dimensional geometry of the interactions.
INTRODUCTION
Recognition of a specific nucleotide sequence by a DNA-binding protein is determined by the atomic interactions between the amino acids of the latter and the nucleotides of the former. While numerous studies introducing protein–DNA structures have gone a long way to explaining the basis of specificity in individual or highly-related complexes, no simple rules have been found for a universal or generic recognition code that adequately explains observations for all proteins.
The first step towards rationalising such a code was taken by Seeman et al. (1), who identified hydrogen-bonding atoms on DNA base edges (Fig. 1) and used them to predict possible amino acid–base pairings. They proposed a scheme whereby certain nucleotides could be recognised by particular amino acid side chains and reasoned that greater specificity was more likely through interactions in the major groove rather than the minor groove. Their findings were compared with interactions observed in tRNA complexes, which were the only available protein–nucleic acid structures at the time.
Figure 1.

Schematic diagrams of bases and their pairings in a DNA molecule. Arrows mark the accessible hydrogen-bonding positions, pointing towards acceptors and away from donors. Major groove access to the bases is shown at the bottom of each diagram and minor groove access at the top. (A) Base pairing between adenine (A) and thymine (T), showing the two hydrogen bonds made between them. (B) Base pairing between guanine (G) and cytosine (C), showing the three hydrogen bonds made. The atoms are labelled according to the numbering system in PDB format. The zigzag lines represent the sugar–phosphate groups to which bases are attached.
Preliminary studies of amino acid–base contacts in protein–DNA complexes were conducted by Pabo and Sauer (2) and later by Matthews (3). These studies were restricted by the small number of high-resolution structures available and were largely confined to descriptions of interactions in the context of the complex they came from. Later work has placed greater emphasis on finding common interactions through systematic examination of different proteins and has added a quantitative element to the analysis. Pabo and Sauer (4) summarised the interactions in eight complexes and highlighted the most frequently found amino acid–base contacts, such as those with purine bases. Suzuki (5) inspected 20 complexes to demonstrate how the occurrence of different interactions may be explained by using stereochemical rules and, most recently, Mandel-Gutfreund et al. (6) confirmed the existence of significant interdependence between amino acid–base pairs in the interactions of 28 complex structures.
All these studies have concluded that while there appear to be favoured interactions, the specificity for entire DNA sequences can rarely be explained by one-to-one correspondences between amino acids and bases. The consensus is that DNA-binding varies substantially between protein families, and that at present no simple code can adequately describe the recognition of target sites on nucleic acids (7).
Here we present a new study of protein–DNA interactions at an atomic level. For the first time, the roles of van der Waals contacts and water-mediated bonds are examined; while these interactions have been studied for individual structures, they have been largely neglected in global studies of protein–DNA structures. Therefore, conclusions about their roles have been mostly anecdotal. We also expand the analysis of hydrogen bonds from simple one-to-one amino acid–base interactions towards complex interaction networks involving multiple base steps. We anticipate that these interactions are indispensable to the overall specificity of protein–DNA complexes.
Finally, with over four times the number of protein–DNA structures at our disposal, we are able to draw firm conclusions about amino acid–base interactions that provide universal specificity in all complexes. At the end, a Web-based ‘Atlas of Side Chain-Base Contacts’ is introduced.
MATERIALS AND METHODS
The following procedure was used to construct the datasets of hydrogen bonds, van der Waals contacts and water-mediated interactions. (i) Protein–DNA complex structures in the Protein Data Bank (PDB) (8,9) were identified. (ii) The complexes were structurally classified and aligned according to the protein structure. (iii) All interactions between the proteins and DNA in these structures were calculated. (iv) Lists of non-homologous interactions were produced by eliminating identical protein–DNA interactions made by equivalent amino acid positions in homologous protein structures. The details of each step are given below. We also describe the method used to calculate theoretical distributions of protein–DNA interactions.
Structural dataset
Protein–DNA complex structures solved by X-ray crystallography to a resolution of higher than 3.0 Å were obtained from the March 1998 release of the PDB (Table 1). The complexes were defined as any structure containing one or more protein chains and at least one double-stranded DNA of >4 bp in length. From this set we excluded structures containing single- and quadruple-stranded DNA. This resulted in a structural dataset of 129 protein–DNA complexes. Included were 11 homodimeric complexes whose asymmetric unit contained only half the structure; the full co-ordinate files for these entries were obtained from the Nucleic Acids Database (NDB) (10).
Table 1. The 129 protein–DNA complexes structures identified by their PDB codes.
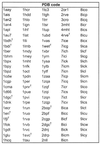 |
A full table with the protein name, source, resolution and structural classification is available at http://www.biochem.ucl.ac.uk/~nick/aa-base/.
+Homodimers that only contain half the structure and (nw) structures that do not contain water molecules.
The PDB entries were classified using a two-tier hierarchy, first according to structural features present in the proteins (e.g. containing the helix–turn–helix DNA-binding motif) and secondly by their taxonomy. Classification at the first level was performed manually by visual inspection of the proteins in RasMol and from the literature. This gave eight groups in all. At the second level, the DNA recognition domains were classified into homologous families by comparing their structures in pairs using the Secondary Structure Alignment Program (SSAP; 11). The program returns a score of 100 for identical proteins, and >80 for protein pairs that are structurally homologous; proteins were automatically assigned to the same family if they scored above this cut-off. More distantly related proteins with scores of >70 were also placed in the same family if they perform similar biological functions (12). This gave a total of 33 families. Finally, multiple structural alignments were produced for each structural family using the CORA program suite (13). Prior to conducting the alignments, proteins were broken down into their constituent DNA-binding domains. In most dimers, each domain comprises distinct subunits and the structure simply needed to be separated into the constituent chains. However in proteins such as the ββα-zinc fingers, a chain can contain several binding domains and therefore the subunits were separated into the appropriate segments, which are listed at http://www.biochem.ucl.ac.uk/~nick/aa-base/ (this includes lists of all proteins used in this analysis, the relative ASAs for amino acids and DNA base and backbone groups, tables detailing single, bidentate and complex interactions using protein main chain atoms, and schematic diagrams of all complex interactions).
Calculation of interactions
Hydrogen bonds and van der Waals contacts in a structure were calculated using the program HBPLUS (14). To identify hydrogen bonds, the program finds all proximal donor (D) and acceptor (A) atom pairs that satisfy specified geometrical criteria for bond formation. Theoretical hydrogen atom (H) positions are then calculated for those donor atoms that fit the criteria and bonds are calculated between the hydrogen and acceptor atoms. The criteria used for the current study are: H–A distance <2.7 Å, D–A distance <3.35 Å, D–H–A angle >90° and H–A–AA angle >90°, where AA is the atom attached to the acceptor. All atoms not involved in hydrogen bonds but separated by <3.9 Å were considered to be interacting through van der Waals contacts.
The program was run on the PDB structures in the dataset and all protein–DNA bonds and contacts were extracted from the HBPLUS output files using GROW, a program to extract protein–ligand interactions (15). This resulted in a total of 2575 hydrogen bonds, 1733 water-mediated hydrogen bonds and 11 472 van der Waals contacts.
Datasets of non-homologous interactions
In any statistical study of proteins and their interactions, it is common to use a set of structurally non-homologous proteins by selecting a representative from each family (16). This is to eliminate any bias towards proteins with a large number of structures in the PDB, for example DNA polymerase-β. There are two concerns with using such an approach here. First, there are few protein families and using only representatives would leave a very small dataset on which to conduct a statistical analysis. Secondly, properties unique to particular proteins within a family are eliminated by removing their structures from the dataset. This is especially important in the current study; many structures are solved for homologous proteins bound to different DNA sequences and using only a single representative would result in loss in diversity of interactions shown by all the complexes.
In this study, we devised a filtering procedure to maximise the use of all complex structures. The multiple alignments for each structural family were scanned and the interactions at each position were inspected. If more than two aligned structures used equivalent atoms from the same amino acid type to interact with equivalent atoms from the same base type or backbone group, only the interaction from the highest resolution structure was retained and the others discarded. A further filter was applied to van der Waals contacts. If an amino acid was involved in a hydrogen bond to the DNA, all contacts from atoms in the residue were excluded from the dataset. However, for DNA bases hydrogen-bonded to the protein, van der Waals contacts from atoms in the base were included. The process resulted in removal of 4497 contacts. The resulting filtered datasets consisted of 1111 hydrogen bonds, 821 water-mediated hydrogen bonds and 3576 van der Waal contacts. As 12 out of the total 129 structures do not contain water molecules (Table 1), care must be taken in comparing the data for water-mediated bonds with those for the other two interaction types.
Comparison of interaction datasets
Figure 2 allows comparison of the datasets of the filtered interactions just described, the datasets of all interactions, prior to filtering, and the interactions datasets obtained by using non-homologous structures (i.e. one representative per homologous family).
Figure 2.

A comparison of the number of interactions in the filtered datasets of hydrogen bonds, water-mediated bonds and van der Waals contacts (solid lines). The number of interactions in the unfiltered and ‘one representative per family’ datasets for hydrogen bonds are also shown (dotted lines). The effect of varying the resolution cut-off for the structures is depicted.
The unfiltered datasets contain many more interactions than the other two. At a cut-off of 3.0 Å resolution, the unfiltered hydrogen bond dataset contains 2575 interactions compared to 1111 in the filtered and 545 in the non-homologous structures dataset. Similar trends are also observed for van der Waals contacts and water-mediated bonds. It is clear that a large number of identical interactions have been removed during the filtering process, but all the unique interactions that would have been otherwise lost are actually retained.
The plot also demonstrates the effect of varying the resolution cut-off on dataset sizes. The total number of protein–DNA interactions falls rapidly with increased stringency in the quality of structures. The size of the hydrogen bonds dataset almost halves when lowering the cut-off from 3.0 Å resolution (1111 bonds) to 2.5 Å (610 bonds). At 2.0 Å, the cut-off used in a study of protein side-chain interactions (17), only 175 hydrogen bonds remain. A similar decrease is observed for the datasets of van der Waals contacts and water-mediated bonds. For the study to be viable, we required the largest dataset possible without over-compromising the quality of the structures, and so a cut-off of 3.0 Å was chosen. In making this decision we carefully considered the validity of the interactions, particularly in the lower resolution structures. As mentioned above, during the filtering procedure, we ensured that the representative interaction was always selected from the best quality structure. Furthermore, comparisons between family members show that the interactions made in higher resolution structures are almost always maintained in the lower resolution structures, as long as the same amino acid and bases are presented.
Expected distributions of interactions
Below we introduce distributions of the three interaction types classified according to the participating amino acid and DNA bases or backbone. In order to determine whether these distributions reflect a preference for certain amino acid–DNA component interactions, it was important to compare the observed distribution with that expected in a random docking between any protein and nucleic acid. The expected distribution was calculated as the product of the relative accessible surface areas (ASA) of each amino acid type and DNA component. For van der Waals contacts, the accessibility of all atoms was considered, but for hydrogen and water-mediated bonds, only the accessibilities of polar atoms were used.
The average amino acid composition of a protein surface was computed for 119 non-homologous monomeric proteins (http://www.biochem.ucl.ac.uk/~nick/aa-base/) using the program NACCESS (18). The proteins were selected from a set of homologous superfamily representatives (H-level) in the April 1998 release of the CATH database (19). From these, monomers were identified on the basis of quaternary structure assignments in their corresponding SWISS-PROT (20) or Protein Quaternary Structure (PQS) database entries (21). The total or polar ASA of each amino acid was summed for all proteins and the relative composition was calculated as a percentage of the total (http://www.biochem.ucl.ac.uk/~nick/aa-base/). The average surface composition of a DNA molecule was determined in a similar fashion (http://www.biochem.ucl.ac.uk/~nick/aa-base/): ASAs of base and backbone groups were calculated for the nucleic acids bound in the complex structures used for this study.
RESULTS
As described in the Materials and Methods, protein–DNA interactions (in terms of hydrogen bonds, van der Waals contacts and water-mediated hydrogen bonds) were calculated for 129 complex structures from the PDB using the program HBPLUS (14). To minimise any bias towards proteins with multiple PDB entries (e.g. polymerase-β), interactions that are repeated in structurally related complexes were removed from the dataset. This filtering procedure resulted in non-homologous datasets of 1111 hydrogen bonds, 3576 van der Waals contacts and 821 water-mediated bonds, which are the subject of our current analysis.
Hydrogen bonds
Table 2 shows the numbers of hydrogen bonds observed between the 20 amino acids and the four DNA bases. Also shown are the hydrogen bonds made between the amino acids and the DNA backbone (subdivided into the sugar and phosphate parts). In parentheses are the numbers of hydrogen bonds that would be expected from purely random dockings of amino acids to DNA (see the Materials and Methods for calculation). The χ2-test is used to evaluate the degree of divergence between the observed and expected numbers of hydrogen bonds: the P-value (P) gives the probability that the observed number of bonds is as expected. As the χ2-test requires an expected value of >4, some entries are pooled according to similar amino acids to give combined P-values (Pcomb): arginine and lysine, asparagine and glutamine, and aspartate and glutamate. The remaining protein residues that could not be combined sensibly were not tested.
Table 2. Distribution of hydrogen bonds according to the participating amino acid and DNA base or backbone group.
 |
The amino acids are shown in the left-hand column and the bases, sugar and phosphate groups along the top. Amino acids and bases are ordered by the number of interactions that they make. The expected number of bonds from random protein–DNA dockings is in parentheses (see the Materials and Methods) and the χ2-test is used to evaluate the degree of divergence between the observed and expected numbers. Entries that diverge from the expected distribution with P > 0.9999 are in bold. As the χ2-test requires an expected value of >4, some entries are pooled according to amino acids with similar side chains: arginine and lysine, asparagine and glutamine, and aspartate and glutamate. Other amino acids that could not be combined sensibly were not tested.
First we examined hydrogen bonds with the DNA backbone. These are not usually implicated in specificity, but the two-thirds contribution to the dataset highlights their importance in stabilising protein–DNA complexes. The interactions are independent of the DNA sequence (P > 0.9999) underlining their non-specific nature, but interestingly, there are about 100 fewer interactions than anticipated [ratio observed:expected (Roe) = 0.9]. This suggests a preferential binding of amino acids to the DNA bases as compared to the backbone.
Although phosphate bonds may have a role for indirect read-out by recognising variations in DNA structure, there is no reason for a correspondence between the amino acid type and underlying base sequence. Changes in local DNA structure are often dependent on the physical environment of the nucleic acid, as well as its nucleotide sequence, and such recognition processes depend more on structural complementarity than the presence of particular amino acid side chains.
355 hydrogen bonds are made with the DNA bases. Guanine has the highest ratio of observed to expected hydrogen bonds; 183 hydrogen bonds observed, with only 69.4 expected, giving Roe = 2.6 and P > 0.9999. This might be expected given that guanine exposes the greatest number of potential hydrogen-bonding atoms on the base edges. However, the other bases do not have Roe values that reflect their hydrogen-bonding capabilities in the same way. The decreasing order of hydrogen-bonding potential is adenine, cytosine, thymine, yet their corresponding Roe values are 0.9, 1.7 and 1.3, respectively. This apparent anomaly will be discussed below.
On the protein side, the polar and charged residues play a central role. Of the amino acids that make the largest number of hydrogen bonds, arginine (Roe = 5.8), lysine (Roe = 1.5), serine (Roe = 1.5) and threonine (Roe = 1.4) exceed the anticipated number of bonds (P < 0.001 for all), while asparagine (Roe = 1.0) and glutamine (Roe = 1.2) interact as expected (P > 0.72 and P > 0.12, respectively). Acidic residues, aspartate and glutamate, are used sparingly (P < 0.0001), presumably because of the unfavourable electrostatic interaction between the side chain and DNA phosphate groups. Of the non-polar amino acids, only glycine makes a significant number of interactions but Roe is still only 0.5; the larger side chains of the remaining hydrophobic residues hinder access of main chain atoms to the DNA and few interactions are produced.
Favoured amino acid–base hydrogen bonds
There are clear preferences for particular pairings of amino acids and bases. Arginine and lysine strongly favour guanine (Pcomb < 0.0001, Roe = 24.5 and 4.2) and largely account for the abundance of hydrogen-bond interactions with this base. To a lesser extent, asparagine and glutamine prefer adenine (Pcomb < 0.0001, Roe = 3.0 and 4.2). The combinations are by no means exclusive, and these amino acids also interact with other base types, albeit less often. For example, arginine also makes a larger than expected number of interactions with thymine and adenine (Pcomb < 0.001 and 0.0001).
Further patterns include the slight affinity of serine and histidine for guanine (P-value unavailable) and of the acidic amino acids for cytosine (Pcomb < 0.001). Closer inspection reveals that the latter are mostly found in methyltransferase complexes where the base has been excised into the catalytic centre of the protein. As the bond is impossible without severe distortion of the DNA molecule, it cannot be considered to provide universal amino acid–base specificity.
While serine and threonine contribute a large number of bonds, >80% of their interactions are with the DNA backbone, well above the average 67% displayed by other amino acids. The short side chains have limited access to bases and therefore generally contribute to stability rather than specificity.
Hydrogen bond geometries
In order to rationalise the preference for certain amino acid–base pairs, entries were classified into (i) single interactions where one hydrogen bond is found between an amino acid and base, (ii) bidentate interactions in which there are two or more bonds with a base or base pair, and (iii) complex bonds where an amino acid interacts with more than one base step simultaneously.
The distribution of bond types is summarised in Table 3. Of the 355 hydrogen bonds with bases, 131 are in single, 120 in bidentate and 121 in complex interactions. Seventeen entries are part of complex bonds that contact one of the bases through a bidentate interaction and are counted in both categories. The numbers demonstrate that a substantial number (63.1%) of hydrogen bonds with bases are involved in the more complicated interaction networks.
Table 3. The number of hydrogen bonds that participate in each interaction type.
 |
*Seventeen bonds belong to both bidentate and complex interactions.
Single interactions
Many of the amino acid–base combinations that are possible through single bonds are observed at least once (Table 4). While numbers are too small to show definite trends, it is clear that many of the preferences highlighted previously in the overall distribution are not displayed by single interactions.
Table 4. The distribution of single hydrogen bonds according to the participating amino acid and DNA base.
 |
The numbers of interactions are given in each cell.
Bidentate interactions
Bidentate interactions are those where two or more hydrogen bonds are made with a base or base pair. To achieve this interaction, amino acids must possess more than one hydrogen-bonding atom. While a combination of main and side chain atoms could also be employed, it is only observed in four examples due to the geometrical constraints of binding in the DNA groove. Table 5 summarises the interactions that are made using the protein side chains; a list of all bidentate interactions, including the main chain atoms, is provided at http://www.biochem.ucl.ac.uk/~nick/aa-base/. Here we combine interactions with single bases and those that span across base pairs as they are equivalent in terms of recognising a base step. Multiple interactions to single base atoms, such as O2 on cytosine, are not considered as they do not increase specificity. Bifurcated interactions, where a hydrogen atom is shared between two bonds, are not included either. Potential interactions are listed in the first column of Table 5 and are determined on the basis of possible donor and acceptor atom combinations on the amino acid and base.
Table 5. Possible and observed bidentate and complex interactions that are made using amino acid side chains.
 |
The first column lists protein residues that can participate in interactions, classified according to the hydrogen-bonding atoms on the side chains. The second and third columns give the base or base pairs that can potentially participate in bidentate interactions and the examples that are actually observed. - denotes a base pair. The fourth to ninth columns show the possible base combinations for complex interactions in the major and minor grooves, and the interactions that are observed with stacked bases and diagonally-positioned bases. Stacked bases are separated by (.) and diagonally-positioned bases by (\).
The observed interactions are listed in the third column of Table 5 and are depicted in Figure 3. Starting with multiple donors, amino acids that can donate two or more hydrogen bonds, the Arg–G pair is most common (29 examples, 64 hydrogen bonds); here the number of hydrogen bonds is not exactly double the number of examples because of cross-over interactions. There are two possible interaction conformations: the end-on approach interacts with one or both of the distal nitrogen atoms (Fig. 3A–C, 27 examples) and the side-on approach uses the Nɛ and Nη1 atoms (Fig. 3D, two examples). The preference for the first over the second is probably due to the easier access of arginine side chain as probes compared to extending along the groove floor. Lysine performs an analogous interaction to the end-on conformation by placing the Nξ atom between the guanine acceptor atoms (Fig. 3E, eight examples). Acceptor + donor residues can both accept and donate hydrogen bonds. Here glutamine (Fig. 3G, eight examples) and asparagine (Fig. 3F, six examples) interact with the major groove edge of adenine using the distal nitrogen and oxygen atoms. In one example, glutamine binds the guanine N2 and N3 atoms in the minor groove (Fig. 3H). Multiple acceptors, amino acids that can accept two or more bonds, do not participate in bidentate interactions as no base pair presents more than one donor atom on a single face.
Figure 3.

Schematic diagrams of bidentate interactions. Arrows are drawn between interacting atoms and point from the donor to the acceptor. The number of examples of each type of interaction is given in parentheses. (A–D) Arg–G interactions, (E) Lys–G, (F) Asn–A, (G) Gln–A and (H) Gln–G, (I–K) Arg–A:T, (L) Lys–G:C and (M) ThrA:T. The amino acid main chains are shown as dotted lines.
Just five interactions span a base pair (Fig. 3I–M). Given the possibilities of such interactions, very few are actually observed. These usually involve base atoms that are bonded to each other, for example N6-O4 in an A-T pair. Therefore, the remaining hydrogen-bonding orbitals point in opposing directions, making it difficult to interact from a single side chain.
Apart from the single example of threonine, there are no instances of bidentate interactions by other amino acid types. Serine, threonine and cysteine are generally too small for effective contact in the DNA grooves. Threonine has the added hindrance of the methyl group and similar steric reasons apply to tyrosine.
Complex interactions
Complex interactions are those where a protein residue binds more than one base step simultaneously. As with bidentate interactions, amino acids that can be used are generally restricted to those with side chains that are capable of multiple hydrogen bonds. Although there are examples of combined use of both main and side chain atoms, ∼75% of the interactions use only the latter. Complex bonds can be broadly classified into two types depending on the relative positioning of the interacting bases. In the first, nucleotides belong to the same strand, and are stacked directly above one another; these bases are denoted with a full point between them (e.g. G.G). In the second, bases belong in different strands and are situated diagonally to each other; these are separated by a backslash (e.g. G\G). For diagonal positioning, bases are required to be 5′ of each other with respect to their own strands; owing to the helical nature of DNA, the 3′-bases are too far apart for simultaneous interactions.
Table 5 summarises the possible and observed complex bonds using just the side chain atoms. Details of complex interactions involving main chain atoms are available at http://www.biochem.ucl.ac.uk/~nick/aa-base/. Of 43 examples, 25 are with stacked bases, 16 with diagonally positioned pairs and two combine both. Thirty-two examples are in the major groove. All interactions are to adjacent steps except in the Pit-1 POU homeodomain structure (1au7), which skips a base step (Fig. 4ii). Figures 4 and 5 show schematic diagrams of the side chain interactions.
Figure 4.

Schematic diagrams of complex interactions with stacked bases. The coloured boxes represent the major or minor groove base edges labelled with potential hydrogen-bonding atoms. Amino acid names are given at the bottom of each diagram, along with the number of examples and PDB structures in which they are found. Arrows are drawn between interacting atoms and point from the donor to the acceptor. Complex interactions are by (A) multiple donor amino acids in the (i) DNA major groove and (ii) minor groove, (B) by multiple acceptor and (C) acceptor + donor amino acids.
Figure 5.

Schematic diagrams of complex interactions with diagonally-positioned bases. Complex interactions are by (A) multiple donor amino acids in the (i) major groove and (ii) minor groove, and by (B) acceptor + donor amino acids.
Multiple donor amino acids require bases that display at least one acceptor atom each. These appear to be the most versatile and a total of 25 complex bonds are made, mostly involving arginine or lysine. Looking at interactions with stacked bases, the G.G step is recognised on five occasions, G.T on three and A.G on one (Fig. 4). For diagonally-positioned bases, T\G is bound seven times, A\C twice, T\T and T\A once each (Fig. 5).
Multiple acceptor amino acids have a more limited choice of interacting bases, and this is reflected in the small number of observed interactions. There are only two complex bonds, both to stacked bases (Fig. 4): aspartate binds A.C in the RAP1 complex structure (1ign) and glutamate binds C.A in the Max protein (1hlo). In both, the carboxylate atoms span the base steps. Acceptor + donor amino acids have a wider selection of base combinations: T\C is recognised three times, and G\C, C.A and A.A once each (Figs 4 and 5).
It is impractical to enumerate all possible interactions using the main chain atoms as there are countless possibilities. A total of 10 interactions are found (see http://www.biochem.ucl.ac.uk/~nick/aa-base/): seven with stacked bases, one with diagonal bases and two with a combination of both.
Summary of single, bidentate and complex interactions
For single interactions, the data show very few amino acid–base preferences. Exceptions are arginine, serine and histidine. Closer inspection of Arg–G interactions reveals that 25 out of 26 cases are from lower resolution structures that have narrowly missed the bonding criteria for bidentate interactions. For interactions with cytosine and thymine, the apparent affinity is due to the ability of the base O2 atoms to accept two hydrogen bonds. Turning to serine and histidine, while unable to act as multiple donors in bidentate interactions, both produce many bifurcated interactions with guanine. Therefore, these interactions appear to provide some basis of specificity in a comparable manner to bidentate bonds. In summary, single interactions do not inherently confer specificity, although there may be particular complexes for which the interaction is essential. In other words, the degree of specificity of a single interaction depends on the context in which it is made.
Bidentate interactions provide an economical way of increasing the bond energy per amino acid–base pair. More important, however, is the increased specificity in the recognition of the DNA sequence. This is demonstrated by the Arg–G interaction in the major groove. With a single bond interaction, arginine interacts with either the O6 or N7 atom on guanine, but not both. As the N7 atom also exists on adenine, protein residues interacting with only this atom will not distinguish between the two purine bases. In a bidentate bond, arginine interacts with both O6 and N7 atoms, therefore specifically recognising guanine.
The reason that arginine is used most frequently is explained by three factors: (i) the length of the side chain, (ii) the capacity to interact in different conformations, and (iii) the ability to produce good hydrogen-bonding geometries. Lysine and glutamine also possess long side chains, however, they can only interact in one configuration. Additionally, lysine can only use a single side chain atom for binding, and the hydrogen bonds are less likely to resemble the ideal geometry than for arginine (John Mitchell, personal communication).
In summary, bidentate interactions are central to the recognition of single base positions along the nucleic acid. They are independent of protein family, and account for the universal specificity of arginine and lysine for guanine, and asparagine and glutamine for adenine. Although other amino acid types are also capable of similar interactions, they are not used frequently. Few interactions are made across a base pair.
Finally, complex interactions extend the concept of simultaneous bonds further. By binding with more than one base step, amino acids are able to recognise short DNA sequences. While these interactions are partly dependent on the conformation of the DNA molecule, those involving just the side chain can be considered to confer universal specificity. Of the 59 possible combinations, 24 are observed. Here, the limiting factor has been the amount of data available: as many occur only once, we are unable to explain which interactions constitute true preferences. Nevertheless, the interactions of five base combinations are found in multiple protein families, and the list is expected to grow as the data increase. In contrast, the few interactions that involve main chain atoms are clearly affected by the protein conformation found in the protein–DNA complex and so are not generic.
van der Waals contacts
The most important observation for van der Waals contacts is the fact that they comprise 64.9% of all protein–DNA interactions. As for the hydrogen bonds, interactions with the DNA backbone are most prominent (Table 6); the total of 2775 contacts is almost exactly as expected (P > 0.25, Roe = 1.0) and can be entirely attributed to the relative ASA of the backbone groups. Although there are a significant number of contacts with the sugar group, interactions with the phosphate group still dominate due to their high exposure on the DNA surface.
Table 6. Distribution of van der Waals contacts according to the participating amino acid and DNA component.
 |
The layout is as for Table 2.
Interactions with bases differ from the hydrogen bond distribution; thymine (Roe = 1.0) interacts most, then adenine (Roe = 1.1), guanine (Roe = 1.0) and cytosine (Roe = 1.0). The theoretical distribution shows that these figures can be explained by the relative ASA of each base type (P > 0.2 for all).
Turning to the protein, five residues, arginine (Roe = 1.3), threonine (Roe = 1.4), phenylalanine (Roe = 3.3), isoleucine (Roe = 2.1), histidine (Roe = 1.4) and cysteine (Roe = 2.2) surpass the expected number of contacts (P < 0.001 for each). Four amino acid types, glutamate (Roe = 0.4), aspartate (Roe = 0.4), alanine (Roe = 0.7) and leucine (Roe = 0.7) interact less often than anticipated (P < 0.001 for each). The contacts of other important residues, lysine (Roe = 0.9), glutamine (Roe = 1.1), serine (Roe = 1.1) and glycine (Roe = 1.2) are as expected (P > 0.05). As the usage of protein residues resembles that of the hydrogen bond distribution, we investigated whether the dataset is swamped by potential hydrogen-bond pairs that missed the stricter geometrical criteria. However, examination of the contacts at 2.0 Å resolution discounted this possibility for most amino acids (see below).
The affinity of phenylalanine, proline and histidine for many base types is explained by their ability to produce extensive ring-stacking interactions in structures with suitably deformed DNA, for example the TATA box-binding protein complexes (e.g. 1ytb). Where no base is exposed, the side chains are commonly positioned with the plane of the ring facing the DNA, therefore maximising the contact surface area. Analogous interactions were reported for side chain–side chain interactions within protein structures (22). Phenylalanine and proline also intercalate between adjacent base steps (e.g. integration host factor complex, 1ihf), usually resulting in severe kinks in the DNA structure. In a unique example, two leucines from separate protein subunits are jointly used to intercalate in the purine repressor complex (e.g. 1wet). Although tyrosine and tryptophan should be capable of similar interactions, few contacts are produced as they are not frequently found in DNA-binding sites (23).
Surprisingly, cysteine has a high propensity to contact the DNA backbone. The interactions are mostly found in zinc-coordinating proteins (e.g. 1aay) where the amino acid binds the metal ion and is situated near the DNA. The side chain is a weak donor and the bonding geometry suggests possible hydrogen bonds with the phosphate group.
Four amino acids produce less than the expected number of interactions (glutamate, alanine, leucine, aspartate). The distributions of the two acidic residues are due to unfavourable electrostatic interactions with the DNA and those of alanine and leucine are due to the shortness of their side chains.
Favoured amino acid–base contacts
Although less marked than for hydrogen bonds, Table 6 draws our attention to a few favoured amino acid–base contacts. Care must be taken in interpreting these observations as a single amino acid–base pair may produce up to five or six van der Waals contacts each. Arginine displays an affinity for guanine (P < 0.0001, Roe = 2.3), glutamine for adenine (P < 0.0001, Roe = 2.5) and thymine (P < 0.0001, Roe = 1.3), threonine for thymine (P < 0.0001, Roe = 2.3), and phenylalanine, histidine and proline for adenine (P < 0.0001, Roe = 2.6).
Most Arg–G and Gln–A interactions involve pairs that have just missed the hydrogen-bonding criteria. The preference for thymine by threonine is because of the methyl–methyl contact and possibly a weak hydrogen bond between the hydroxyl group and base O4 atom. The lack of a methyl on serine probably explains its lower position in the table. The interactions the aromatic protein residues make with multiple base types as opposed to a one-to-one interaction were explained above.
In summary, van der Waals contacts do not generally confer sequence specificity. The preferences displayed by polar residues are due to their favoured hydrogen-bonding interactions, and the only favoured pairings through van der Waals contacts are those of threonine and the aromatic residues. The remainder of the distribution in Table 6 can be explained by the random dockings between proteins and DNA. However, the 75% contribution to all protein–DNA interactions highlights their importance in forming these complexes.
Water-mediated bonds
Water-mediated bonds are nearly as common as direct hydrogen bonds, and make up 14.9% of all protein–DNA interactions. As 12 structures in the dataset lack water molecules, this figure almost certainly underestimates the contribution of these interactions.
The distribution of interactions resembles that for direct hydrogen bonds (Table 7). Just over 70% of bonds are with the DNA backbone (Roe = 0.9), mostly the phosphate group, and interactions with purine bases (Roe = 1.4) are more common than with pyrimidine bases (Roe = 1.5). Polar and charged amino acids are frequently used: arginine (Roe = 3.9), lysine (Roe = 1.2), asparagine (Roe = 1.2), glutamine (Roe = 1.2), serine (Roe = 1.4) and threonine (Roe = 1.3). In contrast to other interaction types, glutamate and aspartate make significant contributions, presumably because of their ability to interact at a distance. Of the hydrophobic residues, alanine (Roe = 0.6) and glycine (Roe = 0.4) with small side chains, partake in bonds using their main chain atoms.
Table 7. Distribution of water-mediated bonds according to the participating amino acid and DNA component.
 |
The layout is as for Table 2.
Although there are small peaks in the distribution—for example arginine (Roe = 7.8) and lysine (Roe = 2.5) interact readily with guanine, asparagine with adenine (Roe = 2.3) and guanine (Roe = 2.6), and glutamate with cytosine (Roe = 5.0)—the preferences are not as strong as for the direct bonds. The important difference between water-mediated bonds and other interaction types is the fact that they do not manifest their specificity in a one-to-one relationship between the amino acid and nucleotide. For a water to be bridging there must be at least one interaction with the protein and one with the DNA. Since water has two donors and two acceptors, tetrahedrally oriented, just two hydrogen bonds will not be specific because the water can potentially rotate to present either a donor or acceptor to the base. Therefore, specificity only arises when the water makes more than two hydrogen bonds simultaneously.
Of the 525 distinct water molecules in the dataset, 154 interact with bases. Only 32 of these participate in three or more hydrogen bonds, indicating that most are used as space fillers for stability. This does not exclude the possibility of context-dependent specificity by water molecules, however, and a well-documented example is the use of a bridging water molecule in the Trp repressor–operator complex, 1trr (24).
Three-dimensional distributions of interacting atoms
Figures 6 and 7 depict three-dimensional diagrams of the spatial distributions of hydrogen bonds and van der Waals contacts around the four base types. Each diagram displays all interacting protein atoms superposed about the central base, regardless of the amino acid they originate from. Protein and base atoms that interact with each other are identified by the same colour.
Figure 6.

Three-dimensional diagrams of the spatial distribution of hydrogen-bonding atoms around bases. The interacting atoms are superposed about a central base. Protein and DNA atoms that interact with each other are identified by the same colour; those in the major groove are coloured red through green and in the minor groove green through blue. Distributions around (A) adenine, (B) guanine, (C) thymine and (D) cytosine are shown in two orientations: (i) facing onto the planes of the bases from the 3′-end and (ii) from the base-pairing edges.
Figure 7.

Three-dimensional distributions of van der Waals-contacting atoms around bases. Diagrams are for (A) adenine, (B) guanine, (C) thymine and (D) cytosine from (i) above the planes of the bases (3′-end) and (ii) the base-pairing edges.
The distributions of interacting atoms reflect the overall geometry of the bases and their accessibilities via the two grooves. Most protein atoms belong to large clusters in the major groove or smaller ones in the minor groove, although some clusters are poorly defined because of sparse populations. On the whole, distributions are more confined for hydrogen bonds because of their directional nature and dependence on atom type. Of interest is the correspondence between the distributions and the protein residues from which the atoms originate. In line with the universal specificities discussed earlier, particular amino acid types concentrate around particular base atoms in the major groove. These include asparagine and glutamine atoms around adenine (Fig. 6A), arginine, lysine, serine and histidine around guanine (Fig. 6B), and threonine about thymine (Fig. 7C). Base atoms that are not implicated in specificity interact with many different amino acid types.
In order to fully appreciate the interactions that we have discussed, we have developed a Web-based Atlas of Amino Acid-Base Interactions (R.A.Laskowski et al., manuscript in preparation), based on the Atlas of Protein Side Chain Interactions published by Singh and Thornton (17). The Web site (www.biochem.ucl.ac.uk/bsm/sidechains/) allows users to inspect the distributions of amino acid side chains around each base type interactively using RasMol (25) and provides a detailed geometrical analysis of the interactions between all side chain–base pairs, including the separation distance, angles defining the spatial disposition of side chains with respect to the base, and an inter-planar angle defining the relative orientation of the two. Separate entries have been created for hydrogen bonds and van der Waals contacts of each amino acid–base pair, giving a statistical analysis of a total of 160 distributions.
DISCUSSION
In this paper, we have investigated the interactions between protein residues and DNA to see whether there are generic rules that govern direct recognition of base sequences by proteins. Three types of interactions, hydrogen bonds, water-mediated bonds and van der Waals contacts, were analysed in a wide range of protein–DNA complexes.
We found that two-thirds of all protein–DNA interactions involve van der Waals contacts, compared to about one-sixth each for hydrogen bonds and water-mediated bonds. Our study therefore highlights the importance of van der Waals contacts in complex formation, which have been relegated to a secondary role until now. For all interaction types, over two-thirds of contacts are made with the sugar–phosphate backbone of the DNA. Because these do not directly depend on the underlying DNA sequence, most protein–DNA interactions can be said to stabilise the complex or aid the indirect read-out of the bases through recognition of the DNA structure.
Our main interest has been with the base contacts and the effect they have on sequence specificity. These are mostly in the major groove and there are significant differences between the interaction types. The universal rules of recognition between amino acid side chains and bases are summarised in Table 8.
Table 8. Summary of the universal preferences of interactions by protein side chains and DNA bases.
 |
Amino acids are grouped by similarity in preferences and listed alongside bases that are recognised. A brief description of the mode of recognition is provided.
Two-thirds of hydrogen bonds with bases are involved in bidentate and complex interactions, and provide the greatest specificity. In line with the main conclusions of Suzuki (5) and Mandel-Gutfreund et al. (6), the hydrogen bond distribution clearly demonstrates that particular amino acid–base pairs are favoured: in particular arginine, lysine, serine and histidine with guanine; and asparagine and glutamine with adenine (Table 8). As first suggested by Seeman et al. (1), the observation is explained by the formation of bidentate or bifurcated bonds with more than one base atom. These interactions are found across different protein families and therefore can be considered to universally recognise single base steps in a DNA sequence. Complex interactions extend the concept of increasing specificity through multiple bonds and recognise short DNA sequences by contacting several base steps simultaneously. These were originally described by Suzuki (5) and we now find them to occur frequently. As the dataset is still relatively small, it is not yet clear whether equivalent amino acid–base combinations are common for different DNA-binding proteins. However, the data suggest that, as long as the DNA is relatively undistorted, complex interactions should represent a generic form of DNA sequence recognition and we expect them to play an important role in providing specificity.
van der Waals contacts, which make up almost 75% of all protein–DNA interactions, largely correspond to the random docking of proteins and DNA, although some interactions were observed more frequently than expected (Table 8). Most obvious are the preferences of threonine for thymine through methyl–methyl contacts, and phenylalanine and proline for adenine and thymine owing to the large surface area provided by the aromatic rings. Interactions by polar amino acids (e.g. Arg–G) mainly involve side chains that just missed the hydrogen-bonding criteria. In general, van der Waals contacts do not display significant preferences and are mostly used to stabilise the complex.
The distribution of water-mediated hydrogen bonds is roughly comparable to that for direct hydrogen bonds. The most notable difference is the more extensive use of aspartate and glutamate, for which the unfavourable electrostatic charge is minimised by interacting via an intermediate water. In order for interactions to be specific, water molecules must satisfy three or more bonds, a condition that is met by only a small proportion of the dataset. Therefore, although they are almost as common as direct hydrogen bonds, we suggest that water-mediated bonds are mostly used as gap-fillers in the protein–DNA interface.
From the strength of preferences displayed by the data, we conclude that the notion of ‘universal’ or ‘generic’ specificity through favourable one-to-one or one-to-many amino acid–base contacts is reasonable. However, we also note that there are many single hydrogen bonds, van der Waals contacts and water-mediated bonds in combinations other than those deemed favourable. Many of these bonds are used for stability, but some are clearly essential for specificity in particular complexes. We term this type of recognition ‘context-dependent’ and the specificity provided is not universal to all protein–DNA complexes. The complications of identifying and isolating such interactions from those that provide universal specificity make predictions of protein–DNA contacts very difficult without structural data. However, given prior knowledge of the complex structure, the preferences summarised in Table 8 can be used to highlight the specific interactions and interpret the data in a predictive manner such as anticipating the effect of amino acid mutations.
References
- 1.Seeman N.C., Rosenberg,J.M. and Rich,A. (1976) Sequence-specific recognition of double helical nucleic acids by proteins. Proc. Natl Acad. Sci. USA, 73, 804–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pabo C.O. and Sauer,R.T. (1982) Protein–DNA recognition. Annu. Rev. Biochem., 53, 293–321. [DOI] [PubMed] [Google Scholar]
- 3.Matthews B.W. (1988) No code for recognition. Nature, 335, 294–295. [DOI] [PubMed] [Google Scholar]
- 4.Pabo C.O. and Sauer,R.T. (1992) Transcription factors: structural families and principles of DNA recognition. Annu. Rev. Biochem., 61, 1053–1095. [DOI] [PubMed] [Google Scholar]
- 5.Suzuki M. (1994) A framework for the DNA–protein recognition code of the probe helix in transcription factors: the chemical and stereochemical rules. Structure, 2, 317–326. [DOI] [PubMed] [Google Scholar]
- 6.Mandel-Gutfreund Y., Schueler,O. and Margalit,H. (1995) Comprehensive analysis of hydrogen bonds in regulatory protein–DNA complexes: in search of common principles. J. Mol. Biol., 253, 370–382. [DOI] [PubMed] [Google Scholar]
- 7.Smith T.L. (1998) Secret code. Nat. Struct. Biol., 5, 100. [DOI] [PubMed] [Google Scholar]
- 8.Bernstein F.C., Koetzle,T.F., Williams,G.J., Meyer,E.E.,Jr, Brice,M.D., Rodgers,J.R., Kennard,O., Shimanouchi,T. and Tasumi,M. (1977) The Protein Data Bank: a computer-based archival file for macromolecular structures. J. Mol. Biol., 112, 535–542. [DOI] [PubMed] [Google Scholar]
- 9.Berman H.M., Westbrook,J., Feng,Z., Gilliland,G., Bhat,T.N., Weissig,H., Shindyalov,I.N. and Bourne,P.E. (2000) The Protein Data Bank. Nucleic Acids Res., 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Berman H.M., Olson,W.K., Beveridge,D.L., Westbrook,J., Gelbin,A., Demeny,T., Hsieh,S.H., Srinivasan,A.R. and Schneider,B. (1992) The Nucleic Acid Database. A comprehensive relational database of three-dimensional structures of nucleic acids. Biophys. J., 63, 751–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Orengo C.A. and Taylor,W.R. (1996) SSAP: sequential structure alignment program for protein structure comparison. Methods Enzymol., 266, 617–635. [DOI] [PubMed] [Google Scholar]
- 12.Orengo C.A., Flores,T.P., Taylor,W.R. and Thornton,J.M. (1993) Identification and classification of protein fold families. Protein Eng., 6, 485–500. [DOI] [PubMed] [Google Scholar]
- 13.Orengo C.A. (1999) CORA-topological fingerprints for protein structural families. Protein Sci., 8, 699–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McDonald I.K. and Thornton,J.M. (1994) Satisfying hydrogen bonding potential in proteins. J. Mol. Biol., 238, 777–793. [DOI] [PubMed] [Google Scholar]
- 15.Milburn D., Laskowski,R.A. and Thornton,J.M. (1998) Sequences annotated by structure: a tool to facilitate the use of structural information in sequence analysis. Protein Eng., 11, 855–859. [DOI] [PubMed] [Google Scholar]
- 16.Jones S. and Thornton,J.M. (1996) Principles of protein–protein interactions. Proc. Natl Acad. Sci. USA, 93, 13–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Singh J. and Thornton,J.M. (1992) Atlas of Protein Side-Chain Interactions. IRL Press, Oxford, Vols I and II.
- 18.Hubbard S.J. (1992). NACCESS: a program to calculate atomic and residue accessibilities. University College, London.
- 19.Orengo C.A., Pearl,F.M., Bray,J.E., Todd,A.E., Martin,A.C., Lo Conte,L. and Thornton,J.M. (1999) The CATH Database provides insights into protein structure/function relationships. Nucleic Acids Res., 27, 275–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bairoch A. and Apweiler,R. (2000) The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res., 28, 45–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Henrick K. and Thornton,J.M. (1998) PQS: a protein quaternary structure file server. Trends Biochem. Sci., 23, 358–361. [DOI] [PubMed] [Google Scholar]
- 22.Singh J. and Thornton,J.M. (1985) The interaction between phenylalanine rings in proteins. FEBS Lett., 191, 1–6. [Google Scholar]
- 23.Jones S., van Heyningen,P., Berman,H.M. and Thornton,J.M. (1999) Protein–DNA interactions: a structural analysis. J. Mol. Biol., 287, 877–896. [DOI] [PubMed] [Google Scholar]
- 24.Lawson C.L. and Carey,J. (1993) Tandem binding in crystals of a Trp repressor/operator half-site complex. Nature, 366, 178–182. [DOI] [PubMed] [Google Scholar]
- 25.Sayle R.A. and Milner-White,E.J. (1995) RasMol: biomolecular graphics for all. Trends Biochem. Sci., 20, 374. [DOI] [PubMed] [Google Scholar]