

 A conceptually new transformation of allylic alcohols to Z-fluoroalkenes was realized through the cleavage of a non-activated carbon–carbon bond..
A conceptually new transformation of allylic alcohols to Z-fluoroalkenes was realized through the cleavage of a non-activated carbon–carbon bond..
Abstract
An unconventional cleavage of an unstrained carbon–carbon bond in allylic alcohols can be induced by the use of N-fluorobenzenesulfonimide (NFSI) under catalyst-free conditions. By using this simple procedure, a wide range of functionalized Z-fluoroalkenes can be accessed in high yield and selectivity from cyclic and acyclic allylic alcohols.
Introduction
The incorporation of fluorine in pharmaceuticals and agrochemicals has become common practice due to the attractive properties that fluorine can provide.1 Accordingly, fluorination chemistry has been extensively explored in the last few decades to deliver fluorinated compounds with various substitution patterns.2 Despite the great progress achieved for catalytic Csp3–F bond formation3 as well as aryl fluoride synthesis,4 significant limitations still exist in the preparation of certain structures such as fluoroalkenes that are highly valuable as peptide isosteres and building blocks.5 The preparation of fluoroalkenes through direct C–F formation requires the use of sensitive organometallic reagents or intermediates (such as alkenyl lithium) and is thus limited in substrate scope.6 Alternatively, classical olefination reactions using fluorine-containing reagents have been extensively explored;7 the control of olefin geometry in these methods, however, has remained an unsolved challenge. Only very recently the synthesis of 1,2-disubstituted Z-fluoroalkenes was achieved through catalytic cross metathesis.8 We present here a highly efficient and operationally simple method that can deliver functionalized trisubstituted Z-fluoroalkenes from readily available allylic alcohols with excellent stereoselectivity.
Electrophilic fluorination of enolate intermediates and alkenes has proven to be a highly successful strategy for C–F bond formation.3 By utilizing allylic alcohols as the substrate, a tandem metal-catalyzed isomerization to the enolate followed by fluorination has been reported to prepare α-fluoroketones.9 Alternatively, fluorination of an alkene moiety followed by semi-pinacol rearrangement was realized to prepare β-fluoroketones (Scheme 1a).10 Related to this, the fluorination induced ring opening of cyclopropanols was also reported recently to deliver β-fluoroketones using Ag/Cu or photo-catalysis (Scheme 1b).11 We have discovered a general and catalyst-free fluorination of allylic alcohols simply by reacting the substrate with commercial NFSI open to air (Scheme 1c). In particular, a conceptually new transformation of allylic alcohols to Z-fluoroalkenes was realized through the cleavage of a non-activated carbon–carbon bond. It is noteworthy that carbon–carbon bond activation has been actively investigated as a new strategy in organic synthesis, with the majority of systems involving a transition metal-mediated opening of strained C–C bonds.12 While examples of the activation of unstrained C–C bonds in carbonyl derivatives are known under transition metal- or photocatalytic conditions,13 our method represents an unprecedented reactivity of allylic alcohols, in which the non-activated C–C bond undergoes cleavage induced by NFSI. The stereoselective formation of fluoroalkenes also renders this system a valuable tool in chemical synthesis.
Scheme 1. Fluorination of allylic alcohols and cyclopropanols.

Results and discussion
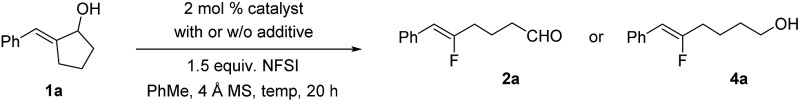
In the past few years, our laboratory has been exploring catalytic enantioselective redox-neutral processes for the preparation of important chiral entities.14 During our investigation of a Rh-catalyzed isomerization of allylic alcohol 1a to the corresponding α-benzyl cyclopentanone (Scheme 2),14e as a mechanistic probe we attempted the synthesis of 3a by trapping the proposed enolate intermediate with an electrophilic fluorinating reagent such as NFSI. To our great surprise, no 3a could be accessed under this set of conditions and a completely unexpected product 2a was formed instead. This transformation involves the cleavage of an unstrained carbon–carbon (C–C) bond in a five-membered ring and delivers synthetically versatile fluoroalkene 2a as a single Z-isomer (determined by the J H–F value as well as 2D NMR; see the ESI† for details). Intrigued by this discovery, we decided to explore this transformation in detail.
Scheme 2. Discovery of fluoroalkene synthesis from allylic alcohols.

As summarized in Table 1, a series of transition metal complexes based on Rh, Ir, Ru, etc. were examined first. Interestingly, all these catalysts led to the formation of 2a in similar yields of 58–60% (entries 1–3). The addition of ligands such as triphenylphosphine had no effect at all (results not shown). When the reaction was carried out by simply mixing 1a and NFSI at ambient temperature in the absence of any catalyst, a 61% yield was obtained for 2a (entry 4). Clearly this intriguing C–C bond cleavage of allylic alcohols proceeds under transition metal-free conditions. The use of other fluorinating reagents such as Selectfluor provided no conversion to 2a at all (entry 5). The effect of various bases as the additive was examined next, which led to reduced reaction efficiency (e.g. use of K2CO3 in entry 6). Considering the important precedent on photo-catalyzed fluorination reactions from the Lectka group,13b we tested the effect of light in our reaction by carrying out the same reaction in the dark. The same reaction efficiency was obtained, thus excluding the possibility of photocatalysis (entry 7). When Ag- or Cu-based salts that are known to promote single electron transfer were examined, no improvement was observed for our reaction either (entries 8–9).11
Table 1. Optimization of fluoroalkene synthesis a .
| ||||
| Entry | Catalyst | F+ (equiv.) | T (°C) | Yield b (%) |
| 1 | [Rh(cod)2]BF4 | NFSI (1.5) | 23 | 58 |
| 2 | [Ir(cod)Cl]2 | NFSI (1.5) | 23 | 60 |
| 3 | [RuCl2(p-cymeme)]2 | NFSI (1.5) | 23 | 59 |
| 4 | — | NFSI (1.5) | 23 | 61 |
| 5 | — | Selectfluor | 23 | <5 |
| 6 | K2CO3 | NFSI (1.5) | 23 | 55 |
| 7 | In the dark | NFSI (1.5) | 23 | 60 |
| 8 | Ag2CO3 | NFSI (1.5) | 23 | 54 |
| 9 | CuCl | NFSI (1.5) | 23 | 50 |
| 10 | — | NFSI (1.5) | 40 | 66 |
| 11 | — | NFSI (2.0) | 40 | 70 |
| 12 | — | NFSI (2.0) | 40 | 82 (4a) c |
aThe reactions were carried out with 0.2 mmol of 1a and 100 mg of a 4 Å molecular sieve in 2 mL of toluene open to air using commercially available NFSI.
bIsolated yields.
cThe isolated yield of alcohol 4a after the reduction of the aldehyde.
To further optimize the reaction, the loading of NFSI and the reaction temperature were varied. The use of 2.0 equiv. of NFSI at 40 °C proved to be optimal (entries 10–11). Overall, a simple and effective procedure that involves simple mixing of the substrate and NFSI with gentle heating proved to be the most effective. Finally, a more convenient procedure was adopted that included an in situ reduction of 2a using NaBH4; the corresponding primary alcohol 4a could be isolated in a high yield of 82% (entry 12). The scale-up of this reaction proved to be straightforward as well. A similar yield of 78% was obtained for 4a on a 5 mmol scale (Scheme 3).
Scheme 3. Scope of fluorination-induced ring opening of cyclic allylic alcohols. See the ESI† for the detailed procedure. The product Z/E ratios were determined by crude NMR. All yields are isolated yields. a 5 mmol scale reaction.

With the optimal conditions in hand, we moved on to explore the scope of this ring-opening fluorination reaction. For the ring-opening of cyclopentanols (n = 1), various substituted aryl groups can be well-tolerated (Scheme 3a). A wide range of fluoroalkenes bearing electron-deficient (4b–4e), electron-neutral (4f–g) and electron-rich (4h) substitutions at the para-position of the arene were prepared in good to excellent yield. Reactive functionalities such as ester or cyano groups could be well tolerated to produce 4i–4j in good yield as single isomers. meta-Substituted arenes such as 4k were produced similarly well. For ortho-substituted substrates (4l–4n), only moderate yields were obtained probably due to the steric hindrance of these substrates. Naphthyl- and heterocycle-substituted fluoro-alkenes (4o–4r) could also be prepared in reasonable yields. While alkyl-substituted allylic alcohols failed to produce the desired products, alkenyl- and alkynyl-substituted substrates underwent fluorination smoothly to produce fluoro-substituted diene 4s and enyne 4t. Such highly functionalized fluoroalkenes are accessed for the first time and could prove highly valuable as building blocks in chemical synthesis.
In addition to cyclopentanols, this fluorination method is also applicable to the opening of different sized rings, including 4, 6, 7, and 8-membered substrates (Scheme 3b). In this way a series of fluoroalkenes bearing a formyl or alcohol functionality at different distances can be accessed conveniently, although the efficiency gradually drops for longer tethers (4u–4x). In addition to the generation of simple linear products, substitution at different positions in the cycloalkanol substrates (such as 1y and 1z) was well-tolerated to produce α- or β-branched fluoroalkene-containing alcohols 4y and 4z with excellent Z-selectivity (Scheme 3c).
Finally, double fluorination of bis-allylic alcohol 5 also proceeded smoothly to produce bis-fluoroalkene 6 in moderate yield as a single isomer (Scheme 3d). This transformation, however, failed to produce ketone products by the reaction of tertiary allylic alcohols related to 1a. Although tertiary alcohols are more popular substrates in carbon–carbon bond activation, in our case the greater steric hindrance in these substrates is likely the culprit for this lack of reactivity.
The success of opening large-ring substrates suggested that acyclic substrates might undergo a similar transformation as well. Substituted acyclic allylic alcohols were examined next. As shown in Scheme 4a, this type of substrate underwent the same C–C bond cleavage to produce trisubstituted fluoroalkenes,7,8 while producing benzaldehyde as the side product. It is noteworthy that the alternative semi-pinacol rearrangement product (Scheme 1b) was not observed at all. The efficiency of this process, however, leaves much room for further optimization. While 8a bearing an electron-rich aryl substituent was obtained in a good yield of 70%, most fluoroalkenes were produced in yields ranging from 14–33%. The stereoselectivity of this process, however, is uniformly high to produce the fluoroalkenes as a single Z-isomer. Related fluoroalkenes of the same substitution pattern have been popular targets in medicinal chemistry15 and our method represents an alternative method for their synthesis with unparalleled stereoselectivity.
Scheme 4. Scope of fluorination of acyclic allylic and benzylic alcohols.

In addition, this fluorination of allylic alcohols can be extended to benzylic alcohols (Scheme 4b). A proof-of-principle reaction involves the fluorination of indole-substituted benzylic alcohol 9, which produced 3-fluoroindole 10 in 24% yield. This greatly expanded the scope of this fluorination method and further application is currently under investigation.
It is worth noting that an aryl (or sp2-hybridized) substituent on the alkene moiety of the allylic alcohol substrates is required for the synthesis of fluoroalkenes as shown in Schemes 3 and 4. When allylic alcohols bearing an alkyl substituent were examined (11 or 12), an intriguing and complete change in reactivity was observed (Scheme 5). In the case of cyclopentanols 11, fluorination under identical conditions led to the formation of β-fluoroketones 13a–c in moderate to good yields, which should be formed via β-fluorination followed by a hydride migration. In contrast, the acyclic allylic alcohols 12 underwent β-fluorination followed by a more classical semi-pinacol rearrangement to deliver β-fluoroaldehydes 14a–f in uniformly good yields.10 While these β-fluoroketones 13 and 14 are valuable compounds on their own, their synthesis also provided important insight into the mechanism of this catalyst-free fluorination of allylic alcohols. More intriguingly, a small amount of fluoroalkene 8a was obtained during the synthesis of 14e and 14f. The formation of 8a from 12, a formal cleavage of the alkene moiety of the substrate, led us to hypothesize the involvement of a key oxetane intermediate in the fluoroalkene synthesis.
Scheme 5. Fluorination of alkyl-substituted allylic alcohols. See the ESI† for the detailed procedure. The product d.r.s were determined by crude NMR. All yields are isolated yields. a The reaction was carried out at 80 °C.

In an effort to better understand the limitations in the fluorination of acyclic allylic alcohols, the reaction of 7g was carried out on a larger scale and the product mixture was analyzed carefully (Scheme 6a). In addition to the desired fluoroalkene 8g, β-fluoroaldehyde 17 was isolated in a low yield, indicating that two fluorination pathways were in competition. More interestingly, a small amount of oxetane 15 and the alternative Z-fluoroalkene 16 was also isolated, which could be linked to the formation of 8a from the fluorination of 12e/12f as shown in Scheme 5.
Scheme 6. Preliminary mechanistic studies and the proposed reaction pathway.

When oxetane 15 was subjected to the fluorination conditions (Scheme 6b), however, only a slow conversion to 16 was observed; no formation of 8g could be detected. We hypothesized that the conversion of 15 to 16 likely proceeded through a zwitterionic intermediate and a carbocation is formed preferentially on the more electron-rich benzylic position, which explained why 8g was not formed. This observation also indicated that oxetane formation is likely a side reaction pathway in the fluoroalkene synthesis instead of being an intermediate for the formation of 8g or other fluoroalkenes shown in Scheme 4.
Based on the above observations and taking all of the types of fluorination products into consideration, we propose the general mechanism for fluorination of allylic alcohols using NFSI as shown in Scheme 6c. For the substrates bearing an aryl substituent (or alkenyl and alkynyl as in 5s and 5t), the electrophilic fluorination generates a zwitterion II, in which the formation of a more stable benzylic carbocation serves as the driving force for this regio-selectivity. Fragmentation of II then leads to the formation of fluoroalkenes. Oxetane formation (III) is a side pathway of this reaction, and oxetane could undergo fragmentation through zwitterion II or the alternative species to deliver the more electron-rich fluoroalkene product (e.g., 16).16 For the substrates bearing an alkyl substituent, on the other hand, the electrophilic fluorination takes place at the β-position to generate zwitterion IV. Again, the stability of the carbocation is likely the determining factor. A migration (of either a hydride or aryl group) will then follow to deliver the more traditional β-fluorocarbonyl products.
The products from this reaction can be used as versatile building blocks to prepare various organofluorine compounds (Scheme 7). In addition to the reduction of 2a to yield alcohol 4a, reductive amination or Wittig olefination of 2a proceeded smoothly in one pot with fluorination to yield amine 18 or enone 19 in good yields (Scheme 7a). The alcohol functionality in 4a could also be converted to ester or bromide functionalities as in 20 and 21 without optimization (Scheme 7b). More importantly, the fluoroalkene moiety has proven to be a synthetically versatile building block to access a diverse range of fluoro-containing compounds.17 As representative examples, hydrogenation and dibromination of 4a were carried out to produce alkyl fluoride 22 and multiple-halogen-containing 23.
Scheme 7. Derivatization of fluoroalkenes.

Conclusions
We have discovered a general fluorination of allylic alcohols and in particular, a conceptually new and practical method to access functionalized Z-fluoroalkenes with good to excellent geometry control. This operationally simple procedure involves the reaction of readily available allylic alcohols and NFSI open to air with gentle heating to produce the versatile functionalized fluoroalkenes. Current efforts in our laboratory are focused on the application of this method to the preparation of other valuable fluorinated compounds.
Acknowledgments
We are grateful for the generous financial support from the Singapore National Research Foundation (NRF Fellowship R-143-000-477-281) and the Ministry of Education (MOE) of Singapore (R-143-000-613-112).
Footnotes
References
- For selected recent reviews, see: ; (a) Müller K., Faeh C., Diederich F. Science. 2007;317:1881. doi: 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]; (b) Purser S., Moore P. R., Swallow S., Gouverneur V. Chem. Soc. Rev. 2008;37:320. doi: 10.1039/b610213c. [DOI] [PubMed] [Google Scholar]; (c) Cametti M., Crousse B., Metrangolo P., Milani R., Resnati G. Chem. Soc. Rev. 2012;41:31. doi: 10.1039/c1cs15084g. [DOI] [PubMed] [Google Scholar]
- (a) Furuya T., Kamlet A. S., Ritter T. Nature. 2011;473:470. doi: 10.1038/nature10108. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Liang T., Neumann C. N., Ritter T. Angew. Chem., Int. Ed. 2013;52:8214. doi: 10.1002/anie.201206566. [DOI] [PubMed] [Google Scholar]; (c) Brooks A. F., Topczewski J. J., Ichiishi N., Sanford M. S., Scott P. J. H. Chem. Sci. 2014;5:4545. doi: 10.1039/C4SC02099E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For a selected recent review, see: Yang X., Wu T., Phipps R. J., Toste F. D., Chem. Rev., 2015, 115 , 826 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- For a selected recent review, see: Campbell M. G., Ritter T., Chem. Rev., 2015, 115 , 612 . [DOI] [PubMed] [Google Scholar]
- For a selected review on fluoroalkenes, see: ; (a) Couve-Bonnaire S., Cahard D., Pannecoucke X. Org. Biomol. Chem. 2007;5:1151. doi: 10.1039/b701559c. [DOI] [PubMed] [Google Scholar]; (b) Burton D. J., Yang Z.-Y., Qiu W. Chem. Rev. 1996;96:1641. doi: 10.1021/cr941140s. [DOI] [PubMed] [Google Scholar]; (c) van Steenis J. H., der Gen A. V. J. Chem. Soc., Perkin Trans. 1. 2002:2117. [Google Scholar]; (d) Landelle G., Bergeron M., Turcotte-Savard M.-O., Paquin J.-F. Chem. Soc. Rev. 2011;40:2867. doi: 10.1039/c0cs00201a. [DOI] [PubMed] [Google Scholar]
- Yang M.-H., Matikonda S. S., Altman R. A., Org. Lett., 2013, 15 , 3894 , , and references therein . [DOI] [PubMed] [Google Scholar]
- For selected reviews on fluoroalkene synthesis via olefination, see: ; (a) Zajc B., Kumar R. Synthesis. 2010:1822. doi: 10.1055/s-0029-1218789. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Pfund E., Lequeux T., Gueyrard D. Synthesis. 2015;47:1534. [Google Scholar]; (c) Zhao Y., Jiang F., Hu J. J. Am. Chem. Soc. 2015;137:5199. doi: 10.1021/jacs.5b02112. [DOI] [PubMed] [Google Scholar]; (d) Zhang W., Huang W., Hu J. Angew. Chem., Int. Ed. 2009;48:9858. doi: 10.1002/anie.200905077. [DOI] [PubMed] [Google Scholar]
- Koh M. J., Nguyen T. T., Zhang H., Schrock R. R., Hoveyda A. H. Nature. 2016;531:459. doi: 10.1038/nature17396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahlsten N., Martin-Matute B. Chem. Commun. 2011;47:8331. doi: 10.1039/c1cc12653a. [DOI] [PubMed] [Google Scholar]
- (a) Wang B., Tu Y. Q. Acc. Chem. Res. 2011;44:1207. doi: 10.1021/ar200082p. [DOI] [PubMed] [Google Scholar]; (b) Romanov-Michailidis F., Guénée L., Alexakis A. Angew. Chem., Int. Ed. 2013;52:9266. doi: 10.1002/anie.201303527. [DOI] [PubMed] [Google Scholar]
- (a) Zhao H., Fan X., Yu J., Zhu C. J. Am. Chem. Soc. 2015;137:3490. doi: 10.1021/jacs.5b00939. [DOI] [PubMed] [Google Scholar]; (b) Bloom S., Bume D. D., Pitts C. R., Lectka T. Chem.–Eur. J. 2015;21:8060. doi: 10.1002/chem.201501081. [DOI] [PubMed] [Google Scholar]; (c) Ren S., Feng C., Loh T.-P. Org. Biomol. Chem. 2015;13:5105. doi: 10.1039/c5ob00632e. [DOI] [PubMed] [Google Scholar]
- For selected reviews, see: ; (a) Dong G., Topics in Current Chemistry, Springer, 2014. [DOI] [PubMed] [Google Scholar]; (b) Marek I., Masarwa A., Delaye P.-O., Leibeling M. Angew. Chem., Int. Ed. 2015;54:414. doi: 10.1002/anie.201405067. [DOI] [PubMed] [Google Scholar]; (c) Souillart L., Cramer N. Chem. Rev. 2015;115:9410. doi: 10.1021/acs.chemrev.5b00138. [DOI] [PubMed] [Google Scholar]
- For a selected review, see: ; (a) Chen F., Wang T., Jiao N. Chem. Rev. 2014;114:8613. doi: 10.1021/cr400628s. [DOI] [PubMed] [Google Scholar]; (b) Pitts C. R., Bloom M. S., Bume D. D., Zhang Q. A., Lectka T. Chem. Sci. 2015;6:5225. doi: 10.1039/c5sc01973g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Zhang Y., Lim C.-S., Sim D. S. B., Pan H.-J., Zhao Y. Angew. Chem., Int. Ed. 2014;53:1399. doi: 10.1002/anie.201307789. [DOI] [PubMed] [Google Scholar]; (b) Rong Z.-Q., Zhang Y., Chua R. H. B., Pan H.-J., Zhao Y. J. Am. Chem. Soc. 2015;137:4944. doi: 10.1021/jacs.5b02212. [DOI] [PubMed] [Google Scholar]; (c) Pan H.-J., Ng T. W., Zhao Y. Chem. Commun. 2015;51:11907. doi: 10.1039/c5cc03399c. [DOI] [PubMed] [Google Scholar]; (d) Yang L.-C., Wang Y.-N., Zhang Y., Zhao Y. ACS Catal. 2017;7:93. [Google Scholar]; (e) Liu T.-L., Ng T. W., Zhao Y. J. Am. Chem. Soc. 2017;139:3643. doi: 10.1021/jacs.7b01096. [DOI] [PubMed] [Google Scholar]
- Alloatti D., Giannini G., Cabri W., Lustrati I., Marzi M., Ciacci A., Gallo G., Tinti M. O., Marcellini M., Riccioni T., Guglielmi M. B., Carminati P., Pisano C. J. Med. Chem. 2008;51:2708. doi: 10.1021/jm701362m. [DOI] [PubMed] [Google Scholar]
- (a) Ludwig J. R., Zimmerman P. M., Gianino J. B., Schindler C. S. Nature. 2016;533:374. doi: 10.1038/nature17432. [DOI] [PubMed] [Google Scholar]; (b) Ma L., Li W., Xi H., Bai X., Ma E., Yan X., Li Z. Angew. Chem., Int. Ed. 2016;55:10410. doi: 10.1002/anie.201604349. [DOI] [PubMed] [Google Scholar]
- For selected examples, see: ; (a) Engman M., Diesen J. S., Paptchikhine A., Andersson P. G. J. Am. Chem. Soc. 2007;129:4536. doi: 10.1021/ja0686763. [DOI] [PubMed] [Google Scholar]; (b) Wong O. A., Shi Y. J. Org. Chem. 2009;74:8377. doi: 10.1021/jo901553t. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.