

Abstract
IKKβ plays a central role in the canonical NF-kB pathway, which has been extensively characterized. The role of IKKα in the noncanonical NF-kB pathway, and indeed in the canonical pathway as a complex with IKKβ, is less well understood. One major reason for this is the absence of chemical tools designed as selective inhibitors for IKKα over IKKβ. Herein, we report for the first time a series of novel, potent, and selective inhibitors of IKKα. We demonstrate effective target engagement and selectivity with IKKα in U2OS cells through inhibition of IKKα-driven p100 phosphorylation in the noncanonical NF-kB pathway without affecting IKKβ-dependent IKappa-Bα loss in the canonical pathway. These compounds represent the first chemical tools that can be used to further characterize the role of IKKα in cellular signaling, to dissect this from IKKβ and to validate it in its own right as a target in inflammatory diseases.
Introduction
Nuclear factor-κB (NF-κB) transcription factors are central coordinators of the innate and adaptive immune response and play key roles in cancer development and progression.1,2 NF-κBs also have a major role in controlling the ability of both preneoplastic and malignant cells to resist apoptosis and support tumor angiogenesis and invasiveness.1,2 The signaling pathways that mediate the activation of the different NF-κB complexes are therefore attractive targets for new chemotherapeutic interventions.
The NF-κB pathways, which are regulated by the inhibitory κB kinases (IKKs), are elevated when homeostasis is disrupted. This is represented by an increase in constitutive IKKα/β activity leading to enhanced NF-κB expression and subsequent nuclear localization. The IKKs are upstream regulators of the NF-κBs, which exist as either homo- or heterodimers bound to inhibitory kappa Bs (IκB’s).1,2 The activation of these IKK complexes dictates the phosphorylation, targeted ubiquitination, and proteolytic removal of IκBs in the canonical pathway and the phosphorylation and processing of high molecular weight NF-κB proteins (p100) in the noncanonical pathway.1,2 This in turn allows NF-κB complexes to translocate to the nucleus and bind specific promoter regions of their targeted genes. Studies3,4 have indicated that IKKα and IKKβ play key but divergent roles in the regulation of global NF-κB signaling and many aspects of cellular transcription. IKKβ controls the canonical pathway via activation of p65 RelA–p50 heterodimers,5−7 and its inhibition leads to a reduction in pro-inflammatory gene expression in several cell types. This is relevant to cancer because several pro-inflammatory species associated with tumor development and progression are encoded by genes regulated through the IKKβ-NF-κB axis.3,4,6 IKKα has been shown to have a minor role in the canonical pathway4,6 but is pivotal in the noncanonical pathway, catalyzing the phosphorylation and proteolytic processing of p100 NF-κB2 which in turn liberates distinct NF-κB p52/RelB dimers and initiates transcription of a specific subset of genes. IKKα and IKKβ have specific cellular functions,3,8,9 and the selective inhibition of one isoform over the other may provide a useful and novel therapeutic strategy in cancer and inflammatory diseases.
Over the past 15 years, many inhibitors of IKKβ have been reported,10−13 primarily toward developing clinical agents to treat inflammatory conditions such as asthma. However, recent studies suggest there may be significant toxicity and side effects associated with IKKβ inhibition, including the development of inflammatory skin disease and sensitization of colonic epithelium to a range of insults.14 In addition, IKKβ knockout mice display severe liver dysfunction.15 Intestinal and liver toxicity have also been an issue in several clinical trials of IKKβ inhibitors which may further limit their clinical applications. Some IKKα inhibitors have been described in the patent literature but with little detail regarding activity and specificity.16 Asamitsu et al.17 reported that the natural product, noraristeromycin (NAM), inhibits IκBα phosphorylation and degradation upon TNFα stimulation and prevents p65 phosphorylation through selective IKKα inhibition. However, the pharmacodynamic readouts used were reporters for both IKKβ and IKKα activity in cells and do not focus on specific biomarkers of the IKKα-controlled noncanonical pathway such as p100 phosphorylation and subsequent processing to p52.
Given the growing evidence that IKKα has an important role in a number of cancers,18−20 selective IKKα inhibitors are required in order to fully understand and validate its role in cancer development and progression, particularly in prostate,19,21 breast,22−24 and pancreatic25−27 cancers. Herein we describe the design, synthesis, and evaluation of a series of 4-substituted 2-amino-5-cyanopyrrolo[2,3-d]pyrimidines as part of our program to develop isoform selective IKKα inhibitors. We present a comparison of the kinase domains of IKKα and IKKβ based on molecular dynamics simulations to explore differences in conformational flexibility that would enable small molecule inhibitors to discriminate between the two isoforms. Finally, we report the first example of IKKα-selective compounds that recapitulate activity in cells against isoform-related pharmacodynamic readouts.
Results and Discussion
Strategy for Discriminating between IKKα and IKKβ
To date, no group has been able to successfully crystallize IKKα and report a high resolution structure of sufficient detail to guide structure-based inhibitor design. To explore differences between the two IKK isoforms, we built a homology model of the IKKα kinase domain based on the crystal structure ofIKKβ (chain B, residues 1–309, PDB entry 4KIK),28 keeping the inhibitor (KSA700 in the pdb file) and water molecules found within 6 Å of the protein–inhibitor complex (Figure 1). Both IKK kinase domains were solvated and then subjected to extended molecular dynamics, with an average structure generated for the last 21 ns (IKKα) or 26 ns (IKKβ) and subsequently minimized.
Figure 1.

Sequence alignment for the kinase domains of IKKα and IKKβ (4KIK_chainB) showing 61% of identical residues (colored in turquoise), a further 14% similar residues (polar for polar, hydrophobic for hydrophobic; in light blue), and 25% nonsimilar residues (white).
When superimposing the presimulated structures of both IKK isoforms, it was striking to see that regions making up the ATP-binding pocket were essentially identical (Figure 2, left). However, analysis of descriptors of motion extracted from their MD trajectories such as residual fluctuation revealed dynamic differences between the two isoforms that could potentially be exploited in an inhibitor design program.
Figure 2.

(left) Minimized average structure of IKKβ highlighting residues that are identical to (turquoise), similar to (light blue), or different from (white) IKKα. The ATP analogue marks the ATP binding site and is surrounded by turquoise residues. (right) Residual fluctuations of IKKα (black line) and IKKβ (blue line) arising during the MD simulations. Several areas were found to be more flexible in IKKα (red underline).
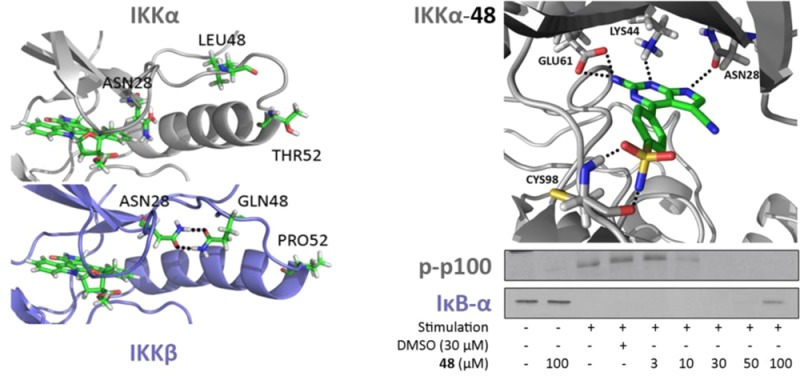
Residual fluctuations obtained from the MD trajectories highlighted areas of the IKKs that acted differently during the simulations (Figure 2, right). Overall, the two isoforms behaved very similarly but IKKα appeared more flexible in several key areas around the ATP binding site, particularly at the G-loop (residues 22–27) above the site entrance and the loop located just adjacent to the hinge (residues 155–159 in IKKα (VGGKI) and residues 156–160 in IKKβ (GEQRL)). Two residues could account for the differences observed with the G-loop: Pro52 and Gln48 in IKKβ (Thr52 and Leu48 in IKKα) induce a tension at the tip of the first α-helix through proline’s intrinsic structure and the engagement of the glutamine side chain in a reciprocal H-bond dimer arrangement with the side chain amide of Asn28 (Figure 3). This asparagine is located at the end of the G-loop, and its interaction with Gln48 restrained the G-loop movement by more than 1 Å in IKKβ when compared with IKKα. A different dynamic behavior was observed with IKKα, as the equivalent residues do not impose restraints on the G-loop movements: Thr52 has no rigid turn restriction like Pro52 in IKKβ, and the side chain of Leu48 seeks a hydrophobic environment and will not engage in H-bond formation with Asn28, leaving the side chain of this latter residue free to make interactions in the ATP binding site of IKKα (in contrast to being sequestered by Gln48 as in IKKβ) (Figure 3). The other significant difference was related to the VGGKI loop (residues 155–159) in IKKα (residues GEQRL: 156–160 in IKKβ). In this case, one residue is responsible for the change observed in residual fluctuation: Lys158 in IKKα is replaced by Arg159 in IKKβ. The slightly longer arginine side chain and its bifurcate H-bonding capacity forms a reciprocal H-bond dimer with the side chain carboxylate of Asp128 in IKKβ that was maintained throughout the simulation, while the equivalent lysine in IKKα never engaged in a strong interaction with the equivalent Asp127 in IKKα (Figure 4). This interaction in IKKβ is responsible for tethering the 156–160 loop to α-helix 3 (Asp128 is located in the middle of this helix), thus reducing its flexibility compared with IKKα.
Figure 3.

Superimposition of IKKα (white) and IKKβ (blue) highlighting the differences near/in the ATP binding site (marked by the staurosporine analogue as a stick model) between the two isoforms. The expanded area shows equivalent residues in IKKα (Asn28, Leu48, and Thr52) and IKKβ (Asn28, Gln48, and Pro52) engaged in different interactions/structural effects, resulting in Asn28 being available to interact with putative ligands in the binding pocket of IKKα but not IKKβ.
Figure 4.

(left) Loop conformation located below the active site in IKKα (white) and IKKβ (blue) and its relationship with α-helix 3 residue Asp127 (IKKα)/Asp128 (IKKβ). In IKKβ, Arg159 makes a reciprocal hydrogen bond dimer interaction with Asp128, whereas in IKKα Lys158 has no close interactions with Asp127. (right) Side chain amine nitrogen (Lys158 (IKKα)/Arg159 (IKKβ)) to side chain acid oxygen (Asp127 (IKKα)/Asp128 (IKKβ)) distance throughout the equilibrated phase of the simulation.
With respect to putative inhibitor binding, the implications for isoform selectivity of these two sets of differences in the ATP site are 2-fold: first, it should be possible to design small molecules that target the free Asn28 side chain amide presented at the back of the IKKα pocket that is otherwise engaged in IKKβ (Figure 3), and second, because the ATP binding pocket has greater flexibility in IKKα, it has the potential to accommodate larger substituents, particularly below the G-loop.
Initial Hit Identification
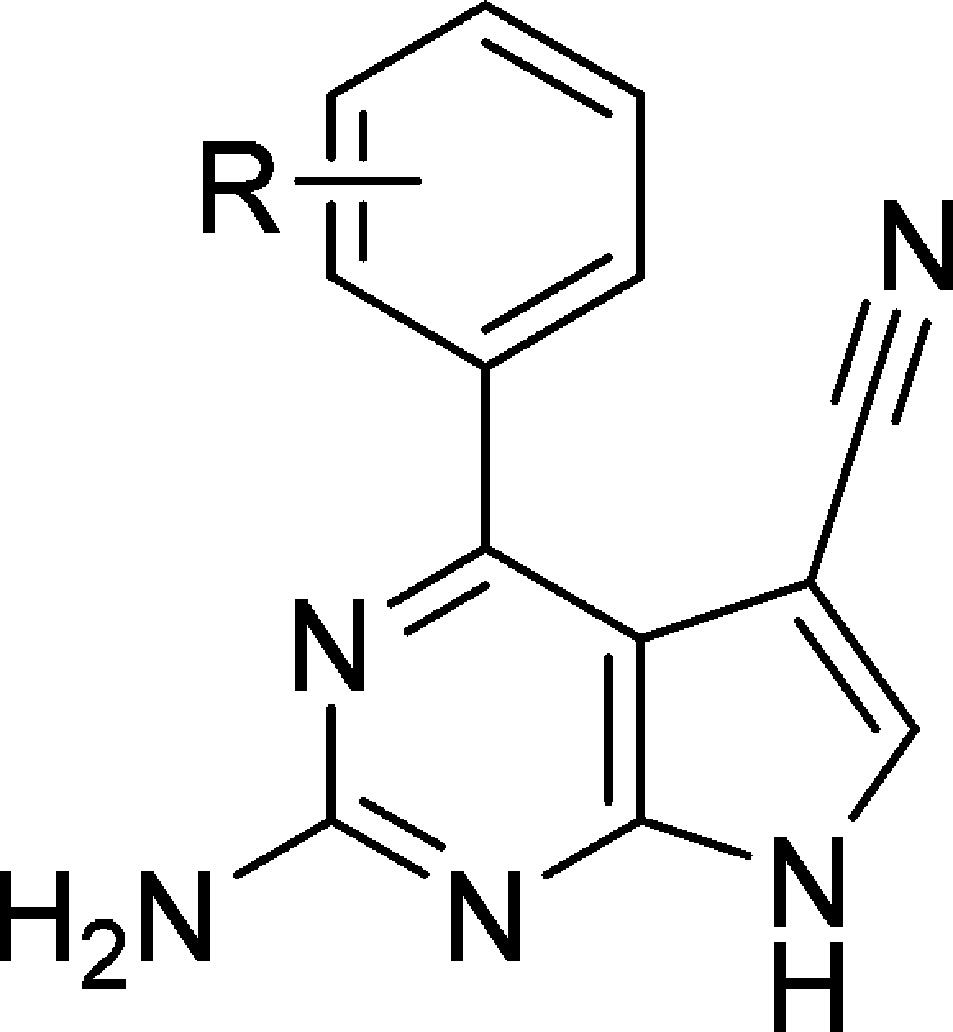
To identify hits that could be developed to exploit the differences between the two isoforms, we screened our in-house compound library compiled of fragments designed to target the common hinge-binding motif found in protein kinases. Using a DELFIA kinase assay kit with minor modifications29 to measure IKKα and IKKβ inhibitory activity, we identified 2-amino-4-chloro-5-cyanopyrrolo[2,3-d]pyrimidine 4 as our primary hit (Table 1). On the basis of the assumption that the 2-aminopyrrolo[2,3-d]pyrimidine core of compound 4 was binding at the hinge of IKKα, we varied substitution at the 4-position widely. Differences in bulk and polar functionality were incorporated to exploit the greater flexibility in the IKKα ATP binding site and the position of the Asn28 side chain at the back of the pocket.
Table 1. Biochemical Inhibitory Data for the N4-Substituted 2-Amino-5-cyanopyrrolo[2,3-d]pyrimidinesa.


Ki values are expressed in μM units and are the result of three independent experiments.
Chemistry
The general route used to prepare N4-substituted 2-amino-5-cyanopyrrolo[2,3-d]pyrimidines for assessment began with the preparation of 1 using a published procedure.301 was then protected as either its diacetyl derivative 2 or pivaloyl derivative 3, which were subsequently converted to the aryl chlorides 4 and 5 (Scheme 1) and used to prepare an initial series of compounds (4–31) (Table 1) by nucleophilic aromatic substitution with a range of primary and secondary amines. The second series of aryl cross coupled compounds exploring the 4-phenyl 2-amino-5-cyanopyrrolo[2,3-d]pyrimidines was prepared by Suzuki–Miyaura couplings (43–62) with 5. In general, the use of the N-pivalamide 5 gave higher yields of the final compounds compared to the unprotected form 4.
Scheme 1.

Reagents and conditions: (a) Ac2O, DMF, 150 °C, 4 h; (b) PivCl, pyridine, 85 °C; (c) POCl3, DMA; (d) amine, Et3N, 1,4-dioxane 200 °C (μW) 20 min; (e) amine, Et3N, n-BuOH, reflux, 16 h then KOH, EtOH, 80 °C, 20 h; (f) boronic acid/ester, Pd(dppf)Cl2, KOAc, H2O/dioxane, 110 °C 16 h.
To explore polar functionality further in the N4-substituted 2-amino-5-cyanopyrrolo[2,3-d]pyrimidines, a series of amino alcohol derivatives (23–31) was prepared as analogues of 16. Compounds 23–29 were prepared from commercially available amines using the procedure detailed in Scheme 1. To incorporate 4-substituents with more than one hydroxyl group in the cyclopentane ring as in the putative IKKα selective inhibitor NAM,17 we prepared the diol 30 and triol 31.31 The synthesis of 30 began with bromination32 of cyclopentene 32 to produce the unstable halide 33, which was immediately treated with N,N-dibenzylamine. The resulting allylic amine 34 underwent syn-dihydroxylation to afford the diol 35(33) with excellent diastereoselectivity, which was then protected as the dibenzoate 36. The amine was subsequently deprotected by hydrogenation to yield 37, which was coupled with 5. Global deprotection gave the desired final product 30 (Scheme 2).
Scheme 2.

Reagents and conditions: (a) NBS, (PhCO2)2, CCl4, 90 °C, 1 h; (b) NH(Bn)2, CCl4, rt, 12 h, 70%; (c) OsO4, NMO, acetone/H2O, rt, 4 h, 71%, 96:4 dr; (d) BzCl, pyridine, 0 °C to rt, 24 h, 84%; (e) H2 (1 atm), Pd(OH)2, rt, 16 h, 98% (f) 5, Et3N, n-BuOH, reflux, 16 h then KOH, EtOH 80 °C, 20 h, 42%.
To prepare the triol 31, the allyl acetate 38 was reacted with sodium di-tert-butyl iminodicarbonate34,35 to produce 39, which was oxidized to the syn-diol 40 followed by benzoate protection of the hydroxyl functionalities to give 41 and removal of the BOC groups under acidic conditions to yield the amine 42. This was then coupled with 5 followed by basic hydrolysis to remove the protecting groups to give the desired final product 31 (Scheme 3).
Scheme 3.

Reagents and conditions: (a) Pd(PPh3)4, PPh3, NaH, NH(BOC)2, THF/DMF, 50 °C 24 h, 42%; (b) OsO4, NMO, acetone/H2O, rt, 24 h, 89%; (c) BzCl, pyridine, 0 °C to rt, 17 h, 74%; (d) 4 N HCl in 1,4-dioxane, 0 °C to rt, 16 h, 76%; (e) 7, Et3N, nBuOH, reflux, 16 h then KOH, EtOH 80 °C, 20 h, 33%.
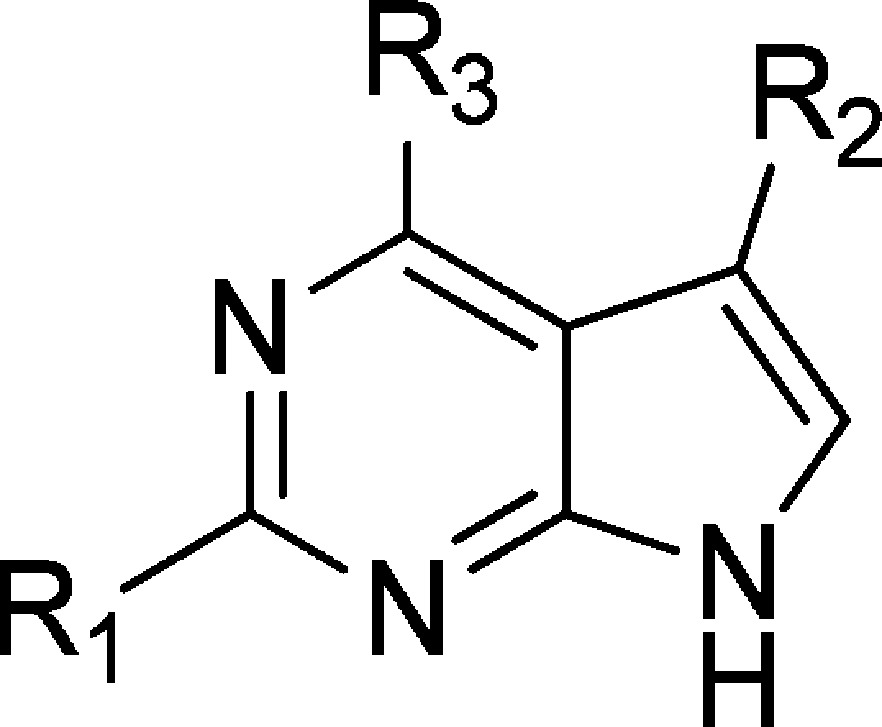
An obvious progression from this series was to replace the bulky saturated ring with a phenyl ring bearing a selection of polar o-, m-, or p-substituents. This series was prepared by reacting 5 with a variety of boronic acids or esters under Suzuki–Miyaura coupling conditions to afford the products 43–62 (Scheme 1; Table 2).
Table 2. Biochemical Inhibitory Data for the 4-Phenyl 2-Amino-5-cyanopyrrolo[2,3-d]pyrimidine Seriesa.

Ki values are expressed in μM units and are the result of three independent experiments.
To assess the importance of the 2-amino and 5-cyano groups for activity and selectivity, we purchased (63) or prepared compounds (64–71) without either or both substituents (Scheme 4, Table 3).
Scheme 4.

Reagents and conditions: (a) alkylamine, Et3N, 1,4-dioxane 200 °C (μW) 20 min; (b) boronic acid/ester, Pd(dppf)Cl2, KOAc, H2O/dioxane, 110 °C, 16 h.
Table 3. Biochemical Inhibitory Data for the 4-Substituted Pyrrolo[2,3-d]pyrimidines With and Without the 2-Amino and/or 5-Cyano groupsa.

Ki values are expressed in μM units and are the result of three independent experiments.
Structure–Activity Relationship Studies
In the N4-substituted series (Table 1) several compounds had similar Ki values against IKKα compared to the initial hit 4. For example, compounds with aliphatic substituents retained potency and selectivity against IKKα and had the potential to be improved through further derivatization. Inhibitory activity for the secondary amines (7, 8, and 9) suggests the presence of a restricted lipophilic pocket in IKKα, which could be responsible for a 5-fold increase in potency from the methylamine analogue 7 to the cyclohexylamine derivative 9. However, the inactivity of the compound with the bulkier cyclohexylmethylamino group (10) suggested that this lipophilic pocket was limited in size. In contrast, the more planar phenyl and benzyl analogues (11 and 14) were found to be less active than 9, and the insertion of polar substituents in the para position of the aniline group produced no significant improvement in activity (12 and 13). Conversely, the insertion of polar groups in more flexible side chains was tolerated (16), which suggested the presence of a polar region adjacent to the lipophilic pocket.
Although compounds with a piperidyl side chain (17 and 18) were found to be active, other tertiary amines at position 4 were not tolerated (19–22), which could be due to additional heteroatoms not being accepted in a lipophilic area of the binding directly proximal to position 4 of the pyrrolo[2,3-d]pyrimidine scaffold. By contrast, the activity of 16 suggested that a flexible ethyl chain is able to direct the heteroatom away from this lipophilic region and possibly engage an adjacent polar region more effectively.
Removal of the hydrogen bond donor (HBD) from the 4-amino substituent by replacing the secondary cyclohexylamino group in 9 with the tertiary N-methylcyclohexylamine (10) abrogated activity. However, the activity of piperidyl analogue 17 suggests that a HBD at the 4-position is not an absolute requirement for inhibition of IKKα. The inactivity of compound 10 can be explained by rotation of the N-methyl (and hence the cyclohexyl group) substituent orthogonal to the pyrrolo[2,3-d]pyrimidine moiety to reduce steric clash with the 5-cyano substituent and generate a conformation that is no longer compatible with the binding site.
On the basis of the activity of the 4-hydroxyethylamino derivative 16, we prepared a second series of amino alcohol derivatives (Table 1). Extending the alkyl chain of 16 from ethyl to propyl was detrimental to activity (23), whereas the cyclic analogues produced more interesting results. The (R)-pyrrolidin-3-ol enantiomer 25 was active and selective against IKKα, whereas the (S)-enantiomer 24 was inactive, suggesting the directionality of the hydroxyl group had a major influence on activity and selectivity. On the other hand, the (R)- and (S)-pyrrolidin-2-yl methanol enantiomers 26 and 27 shared the same activity and selectivity against IKKα, implying that free rotation around the methyl alcohol is sufficient to allow equivalent interactions.
The trans-aminocyclohexanol derivative 28 was also active and selective against IKKα, but the cyclohexylamino ethyl alcohol 29, designed as a hybrid of compounds 8 and 16, proved to be inactive. Like compound 10, this is probably due to the aliphatic ring and the ethyl chain being twisted 90° away from the nitrile axis to adopt a more energetically favored conformation that is incompatible with the binding site. The diol 30 was active and selective but did not offer significant improvement. The triol 31, prepared as an analogue of NAM, proved to be as active as 30 but less selective. Overall, no significant improvement in binding or selectivity was gained by the introduction of more than one hydroxyl group in this series of secondary cyclic amine derivatives.
The direct attachment of an aromatic ring to position 4 of the pyrrolo[2,3-d]pyrimidine scaffold exemplified by compound 43 retained the potency and selectivity of the original hit 4 (Table 2). In general, compounds with para substituents in the phenyl ring had better activity against IKKα and produced our first nanomolar IKKα inhibitors in 47 and 48. Although activity against IKKβ was evident for these two compounds, their higher potency for IKKα ensured significant isoform selectivity was maintained. Compounds with meta substituents were low micromolar inhibitors of IKKα (for example 53, 56, and 57) but were equipotent against IKKβ, apart from 55, which was inactive against both isoforms. As with compounds 10 and 28, the inactivity of the ortho-substituted derivatives in this series can be attributed to enforced conformational rotation of the phenyl ring to a more orthogonal relationship with the pyrrolo[2,3-d]pyrimidine scaffold, which introduces steric clashes in the binding site. This is less pronounced with an o-fluoro substituent and, consequently, 59 had similar potency to the unsubstituted 4-phenyl derivative 43.
Compound 48 (proprietary code SU909), with a para-primary sulfonamide, represents the most potent IKKα inhibitor reported to date that has significant selectivity over IKKβ, although a limited kinase profile using 40 kinases representative of the kinome identified off-target effects (>80% inhibitory activity) with other kinases albeit at 10 μM (Aurora A, CaMK1, CHK2, CK1, GSK3, MEK1, and PKC: Figure 5).
Figure 5.

Percent residual activity of 40 kinases challenged with compound 48 at 10 μM.
To explore the role of the primary sulfonamide, the 4-methyl sulfone analogue 49 and the secondary sulfonamides 50 and 51 were prepared. Notably, 49 and 50 had reduced potency against IKKα, but both were more active against IKKβ, reversing the isoform selectivity for the first time in this series. Similarly, when the orientation of the sulfonamide with respect to the phenyl ring (52) was reversed, a reduction in potency against IKKα occurred but an improvement against IKKβ was again evident, inverting isoform selectivity.
The absence of a 5-cyano group in 48 (compound 64) reduced activity against IKKα more than 30-fold (Table 3), although there was a slight improvement against IKKβ. Compound 65, which lacked both the 5-cyano and the 2-amino substituent, had lower activity against both isoforms. Compound 66, without the 2-amino group but with the 5-cyano, attenuated IKKβ activity but did not re-establish the potency against IKKα seen for 48. The importance of the 5-cyano group for activity against IKKα was further demonstrated by preparing analogues of selected active N4-substituted exemplars from Table 1: removal of the 5-cyano substituent produced inactive compounds against both isoforms (67–69), and its replacement with other electron-withdrawing substituents such as a trifluoromethyl (70) or a carboxamide (71) moiety also attenuated all IKKα inhibitory activity (Scheme 4, Table 3). Furthermore, compound 63, which represents the original hit compound (4) without the 5-cyano group, was also inactive against IKKα (Table 3).
Overall, our SAR data suggest that the 5-cyano substituent is essential for IKKα activity in this series of compounds but must be combined with a 2-amino group to promote selectivity. To enhance potency for IKKα, a 4-phenyl group bearing a specific polar para-substituent appears to be crucial.
Docking Studies
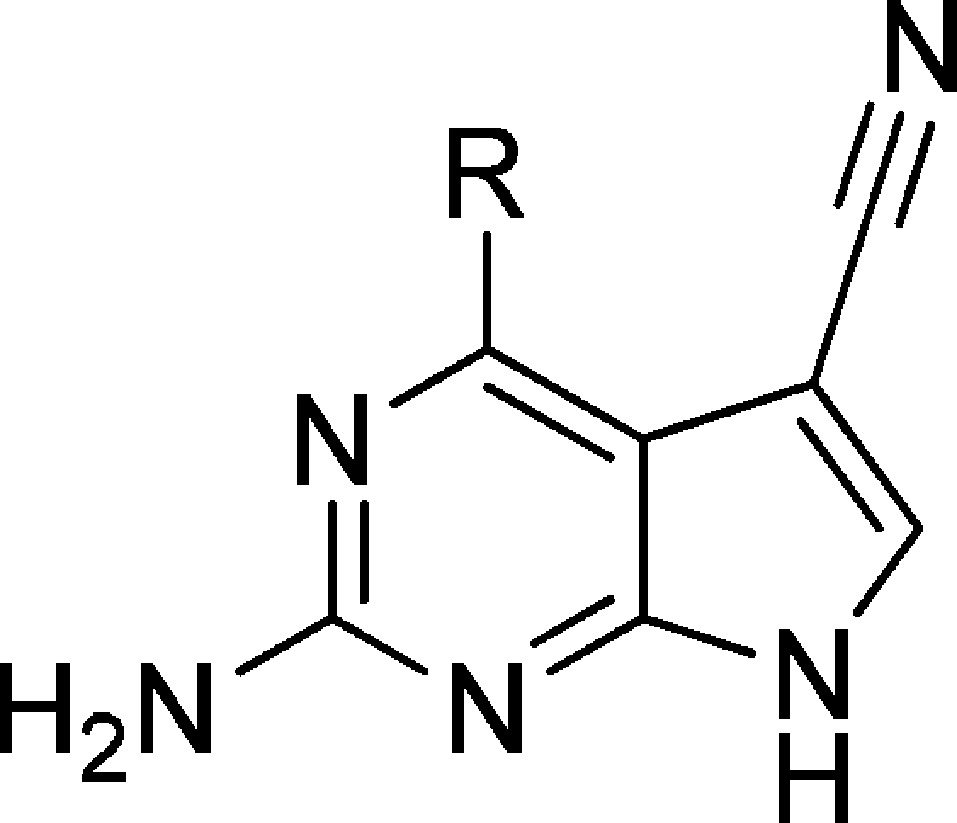
To explain the general SAR profile observed with our series and to relate these to the differences between the two IKK isoforms that our MD studies had revealed, we performed docking studies using GOLD.36 A limited flexibility was enabled in the side chains of specific residues that had shown significant fluctuation in the MD trajectories, namely Asn28, Val29, and Lys44. In the co-crystal structure of IKKβ28 that had formed the basis of our simulations, the staurosporine analogue interacts though H-bonds with the backbone groups of GK+1 (Glu97) and GK+3 (Cys99) in the conserved hinge region. Initially, no attempt was made to direct the ligands to interact specifically with the hinge region in our docking simulations; the only requirement was to occupy the ATP binding site. This first docking study was performed to find a common binding mode that could explain the IKKα inhibitory activity and selectivity reported for exemplars from our two series. To explore this, compounds bearing different hydrophobic substituents were selected for their similar potency and selectivity (e.g., 4, 9, and 43). The poses where the 2-aminopyrrolo[2,3-d]pyrimidine scaffold interacted with the hinge region by H-bonds were studied in detail and compared to similar hinge-binding protein kinase inhibitors.37 For example, aminopurines are known to adopt two different binding modes in interactions with the hinge region of CDK2 via three hydrogen bonds.38 However, in similar poses for our compounds in IKKα, the analogous triplet of hydrogen bonds was poorly supported by the IKKα hinge region in terms of bond distances and bond angles. Not surprisingly, similar poses were found when the same compounds were docked with IKKβ, which is due to the high isoform homology and subtle differences in topography in this hinge region of the ATP binding site. These poses could not account for the selectivity reported in our assays and suggested that this was not the binding mode responsible for conveying such discriminatory activity between the isoforms. However, a binding mode where the 2-aminopyrrolo[2,3-d]pyrimidine scaffold interacted with the back pocket of the IKKα active site explained the potency and selectivity displayed by our compounds more effectively. Parts A–C of Figure 6 illustrate how compounds 4, 9, and 43 were predicted to interact with IKKα, specifically targeting the carboxylate group of Glu61, the ammonium group of Lys44 and, most significantly, the side chain carbonyl group of Asn28 via four H-bonds. In IKKβ, because Asn28 is involved in a dimeric H-bond interaction with Gln48 revealed by our simulations (Figure 3), which also changes the position of Glu61, the triple H-bond interaction observed in IKKα is lost. We therefore propose that the aminopyrrolo[2,3-d]pyrimidine scaffold is anchored to the back of the ATP site specifically targeting Asn28 in IKKα, which accounts for the observed selectivity. In this orientation, the importance of the 5-cyano group when binding with IKKα can also be explained. Examination of the trajectory from an MD simulation of 47 revealed a water molecule present for the majority of the run that formed a three-centered hydrogen bond bridge between the nitrile, the side chain carboxylate of Asp102, and the backbone NH of the same residue (Figure 6D). Overall, binding to IKKα appears to be facilitated through four centers on the 2-amino-5-cyanopyrrolo[2,3-d]pyrimidine scaffold with four residues in the ATP binding site: Glu61, Lys44, Asn28, and Asp102. A similar binding orientation with IKKβ is less likely because the two of the equivalent sites are less accessible (Asn28 and Glu61), and this would account for the lack of activity in this isoform.
Figure 6.

Docked poses of 4, 9, 43, and 47 (A–D, respectively) binding to the back of the ATP pocket in IKKα, where the 2-aminopyrrolo[2,3-d]pyrimidine motif forms H-bonds (shown in green) with the side chain carboxylate of Glu61, the side chain ammonium group of Lys44, and the side chain carbonyl group of Asn28. No equivalent poses were identified when these compounds were docked with IKKβ. (D) An MD simulation of 47 revealed an additional interaction with a molecule of water that formed an H-bond bridging interaction between the 5-cyano group, the side chain of Asp102, and the backbone NH of the same residue.
Although the poses described for compounds 4, 9, and 43 showed no interaction with the hinge region, the equivalent poses for the nanomolar inhibitors 47 and 48 had significant H-bond interactions with the GK+3 residue via their p-hydroxymethyl and p-sulfonamide substituents (Figures 6D and 7). The equivalent binding pose in IKKβ did not feature the hydrogen bonds HB1 and HB2 due to local structural differences observed between the kinase isoforms and could explain the lower affinity of 48 for IKKβ. Interestingly, the presence of the methyl group in the secondary sulfonamide 50 compromised its ability to form an H-bond with the HBA carbonyl group of GK+3 because of a steric clash with the hinge region, thus reducing affinity for IKKα. The sulfonyl group could still interact with the NH of GK+3, which explains why 50 and the sulfone 49 both have similar potency against IKKα.
Figure 7.

(left) Docked pose of 48. As before, the 2-aminopyrrolo[2,3-d]pyrimidine moiety engages with the back of the ATP pocket in IKKα and the sulfonamide group generates additional H-bonds with the main chain carbonyl and NH of GK+3 that produce nanomolar activity. (right) Schematic representation of the proposed binding mode in the IKKα-ATP binding site showing key interactions for compound 48.
The reason for both 49 and 50 also having greater inhibitory activity against IKKβ that reverses isoform selectivity is because the polar side chain of Asn28 in IKKβ rotates out of the back pocket to H-bond with Gln48 (Figure 3). When 49 and 50 were docked in the IKKβ structure, they commonly adopted a flipped pose, where the 2-aminopyrrolo[2,3-d]pyrimidine engaged with the GK+1 and +3 residues at the hinge through H-bonding, while the sulfonyl oxygen and the 5-cyano group H-bonded with K44 in the back pocket (Figure 8). Binding to IKKβ by 49 and 50 in this orientation is facilitated by moving the polar Asn28 from the immediate vicinity, creating a cleft that can more easily accommodate the hydrophobic methyl moiety of 49 and 50. A similar reversal of selectivity was seen with 64, which lacks the 5-cyano substituent of 48. The drop in potency against IKKα can be attributed to the absence of a water-bridging H-bond interaction with Asp102 but also to the removal of the conformationally restraining effect of the 5-cyano group on the adjacent 4-phenyl ring which would normally optimize alignment of the p-sulfonamide H-bonding interaction with GK+3 in the hinge. The enhanced affinity for IKKβ by 64 can again be explained by the adoption of a flipped pose when binding to this isoform, which is enabled by a more flexible p-sulfonamide that can H-bond more effectively with Lys44. Just as importantly, this flexibility allows the HBD of the sulfonamide group to access the less accessible Glu41 at the back of IKKβ pocket.
Figure 8.

(left) Docked pose of 50 in IKKβ. Unlike IKKα, the aminopyrrolo[2,3-d]pyrimidine moiety has flipped and H-bonds to GK+1 and GK+3. The rotation of Asn28 out of the pocket to H-bond with Glu48 allows the sulfonyl group to H-bond to Lys44 and accommodates the lipophilic methyl group in the cleft opened up by this rotation. (right) Schematic representation of the proposed binding mode in the IKKβ-ATP binding site showing key interactions for compound 50.
In summary, in line with our SAR data and the hypothesis derived from our MD simulation studies, we suggest that nanomolar potency against IKKα with selectivity over IKKβ can be accomplished by having molecules that can bind to the hinge region of the ATP binding site and to the Asn28 residue at the back of the pocket that is available in IKKα but not in IKKβ (Figure 8). Furthermore, inhibitory potency against IKKα can be enhanced when compounds can form H-bonding interactions with the three residues in the back pocket (Glu41, Lys44, Asn28) and the GK+3 and Asp102 residues in the hinge.
Demonstrating IKKα Activity and Selectivity Is Recapitulated in Cells
The objective of the cell-based assessment was to characterize whether the two most active compounds against IKKα (47 and 48) recapitulated their activity in a cellular environment. IKKα but not IKKβ plays a role in the regulation of the noncanonical NF-κB cascade which initially involves phosphorylation of p100, followed by p100 processing to p52 to regulate a number of genes that contribute to and promote cellular growth.39 More recent studies have shown that overactivation of the noncanonical NF-κB pathway in a variety of cellular settings, e.g., prostate19,21 and pancreas,25−27 is inherently associated with cell survival and proliferation. The U2OS osteosarcoma cell line is representative of these proliferative characteristics and is dependent on constitutively activated IKKα signaling;40 it was therefore selected as the system to demonstrate perturbation of IKKα activity in the cellular environment.
Pretreatment of U2OS cells with increasing concentration of 48 and 47 resulted in concentration-dependent inhibition of FCS-stimulated phosphorylation of p100 (Figures 9a and 10a). The IC50 values for 48 and 47 were calculated as 8.8 and 13.9 μM, respectively. Two IKKβ-dependent readouts were selected to assess selectivity over IKKβ. First, TNFα stimulates IκBα degradation through activation of IKKβ and if IKKβ is inhibited, then a band corresponding to the IκBα protein should be evident by Western analysis. Second, IKKβ phosphorylates p65 on Ser536, which can also be assessed by Western blot analysis. Only at the highest concentration (100 μM) of 48 was IκBα degradation and p65 (Ser536) phosphorylation inhibited (Figure 9b). Even at this high concentration, these effects were not apparent following pretreatment with 47 (Figure 10b).
Figure 9.

Effect of compound 48 on (a) FCS-stimulated noncanonical and (b) TNF-α-induced canonical NF-kB activation in U2OS cells. Cells were pretreated with 48 1 h prior to stimulation with FCS (10%) for 4 h or TNF-α (10 ng/mL) for 30 min. Whole cell lysate were prepared, separated by SDS-PAGE, and assessed for (a) inhibition of p100 phosphorylation (Ser866/870) and (b) IkB-α and p-p65 (Ser536) status. (c) Blots were quantified, and the IC50 value for inhibition of p100 phosphorylation was determined (48 IC50 = 5.8 μM). The results are representative of three independent experiments.
Figure 10.

Effect of compound 47 on (a) FCS-stimulated noncanonical and (b) TNF-α-induced canonical NF-kB activation in U2OS cells. Cells were pretreated with 47 1 h prior to stimulation with FCS (10%) for 4 h or TNF-α (10 ng/mL) for 30 min. Whole cell lysate were prepared, separated by SDS-PAGE, and assessed for (a) inhibition of p100 phosphorylation (Ser866/870) and (b) IkB-α and p-p65 (Ser536) status. (c) Blots were quantified, and the IC50 value for inhibition of p-p100 was determined (47 IC50 = 19.1 μM). The results are representative of three independent experiments.
To provide further evidence for cellular activity, mouse embryonic fibroblasts lacking the IKKβ kinase subunit were utilized to investigate the potency of both compounds against agonist-stimulated IKKα-regulated NF-κB transcriptional activation. Cells were infected with an adenovirus encoding an NF-κB-luciferase promoter gene. NF-κB transcriptional activity driven by IKKα was stimulated specifically using IL-1β,41 and the ability of the compounds to inhibit this response was assessed. At maximal concentrations of 50–100 μM, 48 abolished transcriptional activity compared to 47 which inhibited activity by approximately 81% (% stim: Il-1β+DMSO = 100%, 48+Il-1β = −40.9 ± 4.6%, 47+IL-1β = 18.79 ± 7.2%, both **P < 0.01) (Table 4). Both 47 and 48 demonstrated concentration-dependent inhibitory effects on FCS-induced phosphorylation of p100 by IKKα. As IKKα plays a lesser role in the canonical NF-κB cascade,42 particularly the rapid and transient stimulated degradation of IκBα, selective IKKα inhibitors should not affect TNF-α induced IκBα degradation and p65 phosphorylation. Compound 47 did not affect either agonist stimulated IκBα degradation or p65 phosphorylation, while 48 only had a small effect on both markers at the highest concentration tested (100 μM). Taken together, these data demonstrate that 47 and 48 are the first examples of IKKα-selective compounds that inhibit agonist-stimulated NF-κB signaling of the noncanonical pathway at much lower concentrations than the canonical pathway.
Table 4. Il-1β-Stimulated NF-κB Transcriptional Activation in IKKα–/– MEFsa.
| treatment | % stimulation |
|---|---|
| IL-1β + DMSO | 100 |
| Il-1β+47 (100 μM) | –40.9 ± 4.6** |
| Il-1β+48 (100 μM) | 18.79 ± 7.2** |
IKKβ–/– MEFs infected with Adv.NF-kB.luc were grown to near confluency and rendered quiescent by serum deprivation for 24 h. Cells were then pre-treated for 1 h with increasing concentrations of compounds 48 or 47 prior to stimulation with IL-1β (10 ng/mL) for a further 6 h. Cells were assayed for NF-κB-linked luciferase reporter activity, as outlined in the methods section. Values (RLUs) were collated and converted to percentage stimulation (of positive control = IL-1β + DMSO). Each value represents the mean ± SEM from at least four independent experiments, and data was quantified using one way ANOVA, with Dunnett’s post-test. *P < 0.05. 88P < 0.01 compared to IL-1β+DMSO alone.
Conclusion
There are many reported inhibitors of IKKβ, primarily because it was considered a viable target in inflammatory disease. Designing inhibitors of IKKα that are selective over IKKβ has to this date not been reported despite the former isoform now being recognized as a potential target in a number of cancers. One reason for this is the high sequence-homology in the ATP-binding site of the two isoforms and the absence of any high-resolution crystal structure of IKKα to guide structure-based inhibitor design.
By employing molecular dynamics simulations on a homology model of IKKα based on IKKβ, we identified key dynamic differences at the ATP-binding site and exploited these to design the first selective inhibitors of IKKα. Our compounds demonstrated target engagement with IKKα-related pharmacodynamic markers and nonengagement with IKKβ markers in cells. These compounds therefore represent the first chemical tools that can be used to further characterize the role of IKKα in cellular signaling, to dissect this from the very similar IKKβ isoform and to validate IKKα in its own right as a target in cancers, such as prostate, breast, and pancreatic cancer.
The discovery of a 2-aminopyrrolo[2,3-d]pyrimidine chemical series also provides valuable information with respect to SAR for IKKα inhibitory activity. Substituents at position 4 of the pyrrolo[2,3-d]pyrimidine scaffold allowed for diversification, with IKKα selectivity achievable through the introduction of groups that specifically target residues in its binding site. However, further significant improvements in activity through aliphatic 4-amino substituents appear to be unlikely. Modifying the 4-phenyl substituent offers a more promising route toward generating compounds with improved activity and selectivity. Using this strategy, we are currently developing more potent IKKα-inhibitors that display functional outputs in a number of cell lines, all of which will be reported in due course.
Experimental Section
IKKα Homology Model
Building a homology model of IKKα: the kinase domain of IKKβ (chain B, residues 1–309, PDB entry 4KIK.28 was used as a template to build the kinase domain of IKKα, keeping the inhibitor (KSA700 in the pdb file) and waters found within 6 Å of the protein–inhibitor complex. The residue alignment (Figure Mod1) and homology building were performed using Discovery Studio 3.1 (Accelrys Inc., San Diego, USA) after missing residues D174, Q175, and G176 were added in an extended conformation.
Both IKK kinase domains were subjected to molecular dynamics using the AMBER12 simulation software. The inhibitor present (residue name KSA700) was kept and parametrized using antechamber and charges calculated and fitted using the AM1-BCC scheme. The two systems were placed in a periodic octahedral box and solvated with TIP3P water with outer edges 6 Å in each direction from the closest solute atom. The systems were then neutralized and physiological salt conditions applied (∼150 mM) by adding 24 Cl– and 29 Na+ ions to the IKKα system and 24 Cl– and 30 Na+ ions to the IKKβ system. The AMBER ff12SB was applied to all protein atoms, while gaff was used for the ligand. Parameters for the phosphoserine residues were taken from Craft and Legge.43 Before the MD production phase, minimization and equilibration (to reach 310 K) were performed in two stages as described.44 The NPT ensemble was used at 310 K until the systems had stabilized for at least 20 ns (50 and 100 ns simulation time for IKKβ and IKKα, respectively). All MD steps used the SHAKE algorithm45 with a 2 fs time-step and a 10 Å cutoff for long-range electrostatic interactions. An average structure was generated (using ptraj within the AMBER suite) for the last 21 ns (IKKα) or 26 ns (IKKβ) and subsequently minimized in three steps, with the solvent, ions, and hydrogen atoms initially minimized while the protein and inhibitor were restrained by 100 kcal mol–1 Å–2. The restraint was then removed from the protein side chain atoms, and finally the whole system was allowed to minimize until a derivative of 0.1 kcal mol–1 Å–2 was achieved. These structures were then utilized for further docking studies with the GOLD software.
General Experimental
Solvents (reagent grade or better) were purchased from Sigma-Aldrich or Fischer Scientific. Anhydrous solvents where purchased from Sigma-Aldrich. Deuterated solvents were purchased from Sigma-Aldrich. Chemicals (95% purity or above) were purchased from Acros Organics, Alfa Aesar, Apollo, Fluorochem, or Sigma-Aldrich. Solvents and chemicals were used as received without further purification or treatment.
Oxygen- or moisture-sensitive reactions were carried out under a nitrogen atmosphere.
Microwave reactions were performed with a Biotage Initiator system. High absorbance was selected for polar solvents and normal absorbance was selected for nonpolar solvents.
The progress of the reactions was monitored on Merck 60F254 TLC plates. Spots were visualized by irradiation with ultraviolet light (254/366 nm) or KMnO4, ninhydrin, or phosphomolibdic acid (PMA) TLC stains.
Column chromatography was performed with a Biotage SP4 system; cartridge size and eluent specified in the corresponding experiments (% is referring to the most polar solvent in the mixture), using silica gel as the stationary phase (particle size 0.040–0.063 mm, Merck or Fisher Scientific).
Specific rotations were measured in a PerkinElmer polarimeter 341 apparatus at 20 °C and a wavelength of 589 nm (sodium D line) in DMSO UV spectrophotometric analysis grade.
1H and 13C NMR data were recorded on either a JEOL ECX-400 (400 MHz) or Bruker Avance3/DPX400 (400 MHz) spectrometers at 400.0 and 100.6 MHz, respectively. Chemical shifts (δ) are expressed in parts per million (ppm) coupling constants (J) are in hertz (Hz). Chemical shifts (δ) are reported relative to TMS (δ = 0 ppm) and/or referenced to the solvent in which they were measured. All measurements were carried out at 298 K (except when stated). Abbreviations used in the description of NMR data are as follows: app, apparent; s, singlet; bs, broad singlet; d, doublet; t, triplet; q, quartet; p, pentuplet; m, multiplet.
HR-MS was conducted using a Thermo Scientific Exactive Orbitrap mass analyzer. LR-MS was conducted using a ThermoQuest Finnigan LCQ Duo instrument. GC-MS was conducted using a ThermoQuest Finnigan Polaris Q instrument.
Final compounds tested in the kinase inhibition assay possessed a purity of ≥95% by HPLC analysis (unless stated otherwise) conducted using an Agilent Technologies 1220 series system (methods A, B, C, and D). Column: Agilent Eclipse XDB C18 4.6 mm ID × 250 mm (5 μm) 80 Å. Flow rate: 1 mL/min. Detector: 254 nm. Sample volume: 10 μL. Mobile phase: (method A) 15% MeCN in H2O (3 min), 15–90% MeCN in H2O (12 min) followed by equilibration/blank run; (method B) 5% MeCN in H2O (3 min), 5–100% MeCN in H2O (14 min), 100% MeCN in H2O (5 min), 100–5% MeCN in H2O (5 min), 5% MeCN in H2O (5 min) followed by blank run; (method C) 5% MeCN + 5 mM NH4Ac in H2O + 5 mM NH4Ac (3 min), 5–100% MeCN + 5 mM NH4Ac in H2O + 5 mM NH4Ac (14 min), 100% MeCN + 5 mM NH4Ac in H2O + 5 mM NH4Ac (5 min), 100–5% MeCN in H2O (5 min), 5% MeCN + 5 mM NH4Ac in H2O + 5 mM NH4Ac (5 min) followed by blank run. Method D was conducted in a Dionex UltiMate 3000 LC system. Column: ACE 3 C8 3 mm ID × 50 mm. Mobile phase: 5–95% MeCN + 0.1% HCO2H in H2O + 0.1% HCO2H (24 min), 95–5% MeCN + 0.1% HCO2H in H2O + 0.1% HCO2H (1 min), 5% MeCN + 0.1% HCO2H in H2O + 0.1% HCO2H (5 min). Flow rate: 0.4 mL/min. Detector: 254 nm. Sample volume: 10 μL.
2-Amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidine-5-carbonitrile (1)
Methyl formate (18.0 mL, 17.48 g, 291.4 mmol) in toluene (8 mL) was added at 0 °C to a stirred suspension of NaOMe (14.30 g, 264.9 mmol) in toluene (200 mL). This was followed by dropwise addition of chloroacetonitrile (16.8 mL, 20.00 g, 264.9 mmol) in toluene (60 mL) over 1 h. The reaction mixture was stirred for 3 h followed by addition of H2O (150 mL). The organic layer was separated, and the aqueous layer was acidified to pH 5 using 6 M HCl and subsequently extracted with EtOAc (3 × 100 mL). The organic layers were combined and dried over MgSO4 and concentrated in vacuo (40 °C, 70 mbar). The dark residue was suspended in H2O (60 mL) and added to a solution of NaOAc (16.39 g, 199.8 mmol) and 2,6-diaminopyrimidin-4(3H)-one (12.00 g, 95.2 mmol) in H2O (200 mL) (previously stirred at 100 °C until complete dissolution). The reaction was refluxed for 16 h. After cooling to room temperature, the suspension was filtered and washed with H2O (2 × 20 mL), acetone (2 × 10 mL), and Et2O (2 × 40 mL) to yield 1 (10.11 g, 60%) as a light-tan solid. 1H NMR (400 MHz, DMSO-d6) δ: 6.37 (bs, 2H), 7.58 (s, 1H), 10.70 (bs, 1H), 11.95 (bs, 1H). 13C NMR (100 MHz, DMSO-d6) δ: 86.0, 99.2, 116.3, 128.2, 158.0, 152.1, 154.2. 3
N-(7-Acetyl-5-cyano-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-2- yl)acetamide (2)
A suspension of 1 (1.18 g, 6.74 mmol) in acetic anhydride (18 mL) and dry DMF (10 mL) was heated at 150 °C for 4 h. Solvents were evaporated in vacuo, and the residue was triturated with Et2O (2 × 10 mL) to yield 2 (1.65 g, 95%) as a brown solid. 1H NMR (400 MHz, DMSO-d6) δ: 2.22 (s, 3H), 2.86 (s, 3H), 8.41 (s, 1H), 11.73 (bs, 1H), 12.20 (bs, 1H). 13C NMR (100 MHz, DMSO-d6) δ: 24.5, 26.0, 90.5, 105.1, 114.2, 129.8, 148.9, 149.3, 155.7, 168.5, 174.4.
N-(5-Cyano-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-2-yl)pivalamide (3)
1 (7.33 g, 28.30 mmol) was stirred in dry pyridine (60 mL) at 60 °C for 1 h. Upon formation of a homogeneous suspension, the mixture was cooled to 0 °C and treated dropwise with pivaloyl chloride (10.5 mL, 10.23 g, 84.87 mmol). The suspension was then stirred at 85 °C for 2 h. After cooling to room temperature, the resulting suspension was neutralized with 33% ammonia in H2O and left to stand at 4 °C for 16 h. The suspension was filtered off, washed with H2O (10 mL), dried, and then triturated with Et2O (2 × 10 mL) to afford 3 (6.97 g, 64%) as a pale-brown solid. 1H NMR (400 MHz, DMSO-d6) δ: 1.25 (s, 9H), 7.93 (s, 1H), 11.00 (bs, 1H), 12.11 (bs, 1H) and 12.65 (bs, 1H). 13C NMR (100 MHz, DMSO-d6) δ: 27.0, 40.7, 86.9, 103.7, 115.9, 130.9, 149.1, 149.1, 156.4, 181.7.
2-Amino-4-chloro-7H-pyrrolo[2,3-d]pyrimidine-5-carbonitrile (4)
POCl3 (5.2 mL, 8.57 g, 55.9 mmol) was added dropwise to a suspension of 2 (1.61 g, 6.21 mmol) and N,N-dimethylaniline (1.2 mL, 1.13 g, 9.32 mmol) in dry MeCN (10 mL). The reaction mixture was heated at 100 °C for 6 h. After cooling to room temperature, the mixture was placed in an ice bath and neutralized with saturated aqueous Na2CO3. The suspension was filtered off, washed with H2O (10 mL), dried, and then triturated with Et2O (2 × 10 mL) to afford 6 (1.44 g) as a brown solid. Chromatographic purification (manual column, solvent system: MeOH/EtOAc; gradient 0% 50 mL, 0–25% 200 mL, 25% 100 mL) yielded a pure analytical sample of 6. 1H NMR (400 MHz, DMSO-d6) δ: 6.95 (bs, 2H), 8.11 (s, 1H), 12.52 (bs, 1H). 13C NMR (100 MHz, DMSO-d6) δ: 83.5, 106.5, 115.4, 134.5, 151.5, 155.0, 160.8. HRMS (ESI) calculated for for C7H5N5Cl [M + H]+ 194.0228, found 194.0229. HPLC tR = 5.56 min (method D).
N-(4-Chloro-5-cyano-7H-pyrrolo[2,3-d]pyrimidin-2-yl)pivalamide (5)
A suspension of 3 (6.82 g, 26.31 mmol), N,N-dimethylaniline (14 mL, 13.39 g, 110.48 mmol), and triethylbenzylammonium chloride (2.93 g, 13.15 mmol) in dry MeCN (100 mL) was treated dropwise with POCl3 (24.5 mL, 40.33 g, 263.05 mmol). The reaction mixture was refluxed for 1 h, allowed to cool down, and concentrated in vacuo. The resulting dark oil was cautiously treated with ice and was set to pH = 5 using 33% ammonia in H2O. The aqueous layer was extracted with EtOAc (4 × 100 mL), and the combined organic layers were dried over MgSO4 and concentrated in vacuo. The residue was triturated with Et2O (3 × 30 mL), MeOH (2 × 20 mL), and Et2O (2 × 20 mL) to give 5 (2.56 g, 35%) as a light-tan solid. 1H NMR (400 MHz, DMSO-d6) δ: 1.22 (s, 9H), 8.50 (s, 1H), 10.27 (bs, 1H), 13.35 (bs, 1H). 13C NMR (100 MHz, DMSO-d6) δ: 27.4, 40.3, 84.1, 111.8, 115.1, 138.2, 151.4, 153.5, 153.8, 176.5.
2,4-Diamino-7H-pyrrolo[2,3-d]pyrimidine-5-carbonitrile (6)
Pyrimidine-2,4,6-triamine (800 mg, 6.39 mmol) was added to a solution of NaOAc (1.10 g, 13.43 mmol) in distilled water (20 mL) and stirred at 50 °C until near total dissolution. A solution of 2-chloro-3-oxopropanenitrile (794 mg, 7.67 mmol) in H2O (10 mL) was added dropwise over 1 h to the reaction mixture, then stirred 16 h at 50 °C, and subsequently, the mixture was refluxed for 1 h. After cooling to room temperature, the reaction was cooled to 0 °C for 2 h. The obtained black solid was boiled in MeOH (4 mL) and washed with hot MeOH (2 × 4 mL). The remaining solid was refluxed in H2O (5 mL) and washed with hot H2O (5 mL) to yield 34 (91 mg, 8%) as a dark solid. 1H NMR (400 MHz, DMSO-d6) δ: 5.89 (bs, 2H), 6.17 (bs, 2H), 7.68 (s, 1H), 11.82 (bs, 1H). 13C NMR (100 MHz, DMSO-d6) δ: 82.5, 94.6, 117.4, 129.4, 154.3, 157.8, 161.7. HRMS (ESI) calculated for for C7H5N6 [M – H]− 173.0581, found 173.0581.
2-Amino-4-(methylamino)-7H-pyrrolo[2,3-d]pyrimidine-5-carbonitrile (7) (Procedure A*)
5 (100 mg, 0.52 mmol), methylamine (8 M solution in EtOH) (53 mg, 0.20 mL, 1.70 mmol), and N,N-dimethylaniline (94 mg, 0.1 mL, 0.77 mmol) were suspended in dry 1,4-dioxane (2 mL) and heated for 20 min at 200 °C in the microwave reactor. After cooling to room temperature, the reaction mixture was suspended in MeOH, adsorbed on silica gel, and concentrated in vacuo*. Chromatographic purification (Biotage SP4, 10 g cartridge, solvent system: MeOH/EtOAc; gradient 0% 4CV, 0–5% 10CV, 5% 4CV) yielded 7 (20 mg, 21%) as a pale-cream solid. 1H NMR (400 MHz, DMSO-d6) δ: 2.92 (d, J = 4.6 Hz, 3H), 5.94 (bs, 2H), 6.05 (q, J = 4.6 Hz, 1H), 7.67 (s, 1H), 11.73 (bs, 1H). 13C NMR (100 MHz, DMSO-d6) δ: 28.3, 82.3, 94.8, 117.4, 129.1, 153.6, 157.3, 161.5. HRMS (ESI) calculated for for C8H9N6 [M + H]+ 189.0883, found 189.0883. HPLC tR = 4.09 min (method A).
2-Amino-4-((cyclopropylmethyl)amino)-7H-pyrrolo[2,3-d]pyrimidine-5-carbonitrile (8) (Procedure B)
5 (0.109 g, 0.39 mmol) and cyclopropanemethylamine (0.105 mL, 1.21 mmol) in 1,4-dioxane (2 mL, anhydrous) in a microwave vial was heated to 200 °C for 20 min. The resulting solution was then diluted with EtOH (6 mL) and KOH (2 pellets) was added and the reaction mixture was heated to 90 °C for 20 h. The reaction mixture was then concentrated under reduced pressure, the residue was suspended in water, and the mixture was pH adjusted to pH 5.5, extracted into EtOAc, dried over MgSO4, and concentrated under reduced pressure. The resulting solid was triturated with Et2O and filtered to afford the title compound as an off-white solid (0.032 g, 0.14 mmol, 36%). 1H NMR (DMSO-d6): δ 0.29 (m, 2H), 0.44 (m, 2H), 1.16 (m, 1H), 3.32 (m, 2H), 5.90 (t, J = 5.4 Hz, 1H), 5.96 (s, 2H), 7.69 (s, 1H), 11.82 (br s, 1H). HRMS (ESI) calculated for C11H13N6 229.1194, found 229.1196.
2-Amino-4-(cyclohexylamino)-7H-pyrrolo[2,3-d]pyrimidine-5-carbonitrile (9)
Prepared according to procedure A. Chromatographic purification (Biotage SP4, 10 g cartridge, solvent system: MeOH/EtOAc; gradient 0% 4CV, 0–2% 12CV) yielded 11 (35 mg, 26%) as a pale-cream solid. 1H NMR (400 MHz, DMSO-d6) δ: 1.22–1.28 (m, 1H), 1.30–1.37 (m, 4H), 1.54–1.57 (m, 1H), 1.67–1.70 (m, 2H), 1.91–1.93 (m, 2H), 4.01–4.08 (m, 1H), 5.32 (d, J = 8.0 Hz, 1H), 5.92 (bs, 2H), 7.66 (s, 1H), 11.80 (bs, 1H). 13C NMR (100 MHz, DMSO-d6) δ: 24.8, 25.7, 32.8, 48.3, 81.8, 94.6, 117.7, 128.7, 153.6, 156.0, 161.6. HRMS (ESI) calculated for for C13H17N6 [M + H]+ 257.1509, found 257.1504. HPLC tR = 11.09 min (method A).
2-Amino-4-(cyclohexylmethylamino)-7H-pyrrolo[2,3-d]pyrimidine-5-carbonitrile (10)
Prepared according to procedure A. Chromatographic purification (Biotage SP4, 10 g cartridge, solvent system: EtOAc/Hex; gradient 25% 4CV, 25–100% 10CV, 100% 6CV) yielded 9 (27 mg, 19%) as a pale-cream solid. 1H NMR (400 MHz, DMSO-d6) δ: 0.91–1.01 (m, 2H), 1.13–1.23 (m, 3H), 1.54–1.65 (m, 2H), 1.68–1.74 (m, 4H), 3.32 (m, 2H), 5.75 (t, J = 5.7 Hz, 1H), 5.94 (bs, 2H), 7.68 (s, 1H), 11.82 (bs, 1H). 13C NMR (100 MHz, DMSO-d6) δ: 26.0, 26.7, 31.0, 38.0, 46.4, 82.0, 94.8, 117.6, 128.9, 153.6, 157.0, 161.6. HRMS (ESI) calculated for for C14H17N6 [M – H]− 269.1520, found 269.1519. HPLC tR = 11.83 min (method A).
2-Amino-4-(phenylamino)-7H-pyrrolo[2,3-d]pyrimidine-5-carbonitrile (11)
Prepared according to procedure A. Chromatographic purification (Biotage SP4, 10 g cartridge, solvent system: EtOAc/Hex; gradient 25% 4CV, 25–100% 10CV, 100% 6CV) yielded 11 (37 mg, 29%) as a pale-yellow solid. 1H NMR (400 MHz, DMSO-d6) δ: 6.20 (bs, 2H), 7.00–7.04 (m, 1H), 7.30–7.33 (m, 2H), 7.68–7.70 (m, 2H), 7.68–7.70 (m, 1H), 8.03 (bs, 1H), 11.86 (bs, 1H). 13C NMR (100 MHz, DMSO-d6) δ: 82.8, 96.0, 117.3, 120.8, 122.8, 129.2, 130.3, 140.5, 154.5, 154.6, 161.2. HRMS (ESI) calculated for for C13H9N6 [M – H]− 249.0894, found 249.0891. HPLC tR = 9.85 min (method A).
4-((2-Amino-5-cyano-7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino)benzoic acid (12)
5 (0.103 g, 0.37 mmol) and methyl-4-aminobenzoate (0.166 g, 1.1 mmol) in 1,4-dioxane (2 mL, anhydrous) in a 2–5 mL microwave vial was heated to 200 °C for 20 min. Ethanol (4 mL) was added to the reaction mixture and KOH (1 pellet) was added and the mixture heated to 80 °C for 24 h. The reaction mixture was concentrated under reduced pressure, water was added, and the mixture was pH adjusted to pH 5 with acid. The resulting white precipitate was filtered under reduced pressure and dried in an oven to afford the title compound as a white solid (0.0135 g, 0.05 mmol, 13%). 1H NMR (DMSO-d6): δ 6.33 (br s, 2H), 7.88 (m, 5H), 8.57 (s, 1H), 12.08 (br s, 1H), 12.61 (br s, 1H). HRMS (ESI) calculated for C14H11O2N6 295.0938, found 295.0938.
4-((2-Amino-5-cyano-7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino)benzenesulfonamide (13)
Prepared according to procedure B. The remaining solid was triturated with cold H2O (2 × 2 mL), filtered, dried, and then triturated with Et2O (2 × 3 mL) to yield 13 as an off-white solid (0.0096 g, 0.03 mmol, 8%). 1H NMR (DMSO-d6): δ 6.63 (br s, 2H), 7.55 (br s, 2H), 7.80 (m, 4H), 8.13 (s, 1H), 12.38 (br s, 1H). HRMS (ESI) calculated for C13H12O2N7S 330.0768, found 330.0768.
2-Amino-4-(benzylamino)-7H-pyrrolo[2,3-d]pyrimidine-5-carbonitrile (14)
Prepared according to procedure A. Chromatographic purification (Biotage SP4, 10 g cartridge, solvent system: EtOAc/Hex; gradient 80% 4CV, 80–100% 4CV, 100% 13 4C,; then MeOH/EtOAc, gradien: 0–10% 4CV) yielded 14 (53 mg, 39%) as a light-tan solid. 1H NMR (400 MHz, DMSO-d6) δ: 4.69 (d, J = 5.9 Hz, 1H), 5.96 (bs, 2H), 6.48 (t, J = 5.9 Hz, 1H), 7.23–7.38 (m, 5H), 7.71 (s, 1H), 11.85 (bs, 1H). 13C NMR (100 MHz, DMSO-d6) δ: 43.7, 82.2, 94.6, 117.4, 127.2, 127.9, 128.89, 129.4, 140.7, 153.9, 156.6, 161.5. HRMS (ESI) calculated for C14H11N6 [M – H]− 263.10507, found 263.10489. HPLC tR = 10.05 min (method A).
2-Amino-4-(2-morpholinoethylamino)-7H-pyrrolo[2,3-d]pyrimidine-5-carbonitrile (15)
Prepared according to procedure A. Chromatographic purification (Biotage SP4, 10 g cartridge, solvent system: MeOH/EtOAc; gradient 0% 4CV, 0–10% 12CV, 10% 6CV) yielded 15 (23 mg, 31%) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ: 2.42–2.46 (m, 4H), 2.54–2.57 (m, 2H), 3.50–3.54 (m, 2H), 3.61–3.63 (m, 4H), 5.98 (bs, 2H), 6.17 (t, J = 4.7 Hz, 1H), 7.69 (s, 1H), 11.82 (bs, 1H). 13C NMR (100 MHz, DMSO-d6) δ: 36.9, 53.5, 56.7, 66.8, 82.1, 94.8, 117.4, 128.9, 153.6, 156.5, 161.7. HRMS (ESI) calculated for C13H16ON7 [ M– H]− 286.1422, found 286.1421. HPLC tR = 3.25 min (method B).
2-Amino-4-(2-hydroxyethylamino)-7H-pyrrolo[2,3-d]pyrimidine-5-carbonitrile (16)
Prepared according to procedure A. Chromatographic purification (Biotage SP4, 10 g cartridge, solvent system: MeOH/EtOAc; gradient 0% 6CV, 0–10% 12CV, 10% 6CV) yielded 16 (16 mg, 24%) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ: 3.52–3.58 (m, 4H), 4.86 (t, J = 4.6 Hz, 1H), 5.89 (t, J = 5.1 Hz, 1H), 5.97 (bs, 2H), 7.29 (s, 1H), 11.83 (bs, 1H). 13C NMR (100 MHz, DMSO-d6) δ: 42.9, 60.2, 82.0, 94.6, 117.3, 129.0, 153.5, 156.7, 161.3. HRMS (ESI) calculated for C9H10ON6 [M + H]+ 219.0989, found 219.0987. HPLC tR = 2.79 min (method A).
2-Amino-4-(piperidin-1-yl)-7H-pyrrolo[2,3-d]pyrimidine-5-carbonitrile (17)
Prepared according to procedure B. The remaining solid was triturated with cold H2O (2 × 2 mL), filtered, dried, and then triturated with Et2O (2 × 3 mL) to yield 17 (43 mg, 49%) as a pale-yellow solid. 1H NMR (400 MHz, DMSO-d6) δ: 1.62–1.65 (m, 6H), 3.52–3.57 (m, 4H), 6.02 (bs, 2H), 7.83 (s, 1H), 11.95 (bs, 1H). 13C NMR (100 MHz, DMSO-d6) δ: 24.7, 26.00, 49.2, 84.1, 95.9, 118.2, 131.1, 155.6, 159.8, 160.6. HRMS (ESI) calculated for C12H16N6 [M + H]+ 243.1353, found 243.1349. HPLC tR = 9.63 min (method A).
1-(2-Amino-5-cyano-7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidine-4-sulfonamide (18)
5 (105 mg, 0.38 mmol), piperidine-4-sulfonic acid amide hydrochloride (139 mg, 0.85 mmol) and triethylamine (0.16 mL, 1.15 mmol) in 1,4-dioxane (2.5 mL) was degassed under nitrogen prior to being irradiated with microwaves at 200 °C for 20 min. Once cooled to room temperature, the reaction mixture was diluted with EtOH (8 mL) and KOH (2 pellets) were added and the reaction mixture was heated to 90 °C for 22 h. The reaction mixture was then concentrated under reduced pressure, water was added, and the mixture was adjusted to pH 5.5 with 1 M HCl. This was then extracted into EtOAc, and the organics were dried over MgSO4, filtered, and concentrated under reduced pressure. The crude material was then purified by column chromatography (using 100% hexane–100% EtOAc as eluent) to afford the title compound 18 as a white solid (26.1 mg, 0.08 mmol, 21%). 1H NMR (DMSO-d6): δ 1.73 (m, 2H), 2.09 (d, J = 8.4 Hz, 2H), 3.00 (t, J = 9.4 Hz, 2H), 3.11 (m, 1H), 4.32 (d, J = 10.4 Hz, 2H), 6.06 (br s, 2H), 6.80 (br s, 2H), 7.85 (s, 1H), 11.99 (br s, 1H). HRMS (ESI) calculated for C12H14O2N7S 320.0935, found 320.0938.
2-Amino-4-morpholino-7H-pyrrolo[2,3-d]pyrimidine-5-carbonitrile (19)
Prepared according to procedure A. Chromatographic purification (Biotage SP4, 10 g cartridge, solvent system: MeOH/EtOAc; gradient 0% 4CV, 0–2% 10CV) yielded 21 (40 mg, 32%) as a pale-cream solid. 1H NMR (400 MHz, DMSO-d6) δ: 3.53–3.55 (m, 4H), 3.73–3.75 (m, 4H), 6.13 (bs, 2H), 7.88 (s, 1H), 11.78 (bs, 1H). 13C NMR (100 MHz, DMSO-d6) δ: 49.0, 66.5, 83.7, 96.2, 118.3, 131.5, 155.7, 159.8, 160.7. HRMS (ESI) calculated for C11H11ON6 [M – H]− 243.0999, found 243.0997. HPLC tR = 6.84 min (method A).
2-Amino-4-(4-methylpiperazin-1-yl)-7H-pyrrolo[2,3-d]pyrimidine-5-carbonitrile (20)
Prepared according to procedure B. The remaining solid was triturated with cold H2O (2 × 2 mL), filtered, dried, and then triturated with Et2O (2 × 3 mL) to yield 20 (63 mg, 68%) as a pale-yellow solid. 1H NMR (400 MHz, DMSO-d6) δ: 2.23 (s, 3H), 2.45 (app t, J = 4.8 Hz, 4H), 3.55 (app t, J = 4.8 Hz, 4H), 6.09 (bs, 2H), 7.86 (s, 1H), 11.96 (bs, 1H). 13C NMR (100 MHz, DMSO-d6) δ: 27.5, 48.0, 54.9, 83.8, 95.9, 118.1, 131.3, 155.6, 159.5, 160.5. HRMS (ESI) calculated for C12H14N7 [M – H]− 256.1316, found 256.1317. HPLC tR = 1.75 min (method C).
2-Amino-4-(4-phenylpiperazin-1-yl)-7H-pyrrolo[2,3-d]pyrimidine-5-carbonitrile (21)
Prepared according to procedure B. The remaining solid was triturated with cold H2O (2 × 2 mL), filtered, dried, and then triturated with Et2O (2 × 3 mL) to yield 21 (83 mg, 85%) as a pale-yellow solid. 1H NMR (400 MHz, DMSO-d6) δ: 3.29 (app t, J = 4.8 Hz, 4H), 3.71 (app t, J = 4.8 Hz, 4H), 6.06 (bs, 2H), 6.79–6.83 (m, 1H), 6.99–7.01 (m, 2H), 7.22–7.24 (m, 2H), 7.87 (s, 1H), 12.04 (bs, 1H). 13C NMR (100 MHz, DMSO-d6) δ: 48.3, 48.7, 83.2, 96.7, 116.1, 118.7, 119.7, 129.6, 132.9, 151.5, 156.5, 159.7, 160.5. HRMS (ESI) calculated for C17H18N7 [M + H]+ 320.1618, found 320.1614. HPLC tR = 11.09 min (method A).
2-Amino-4-(4-(2-hydroxyethyl)piperazin-1-yl)-7H-pyrrolo[2,3-d]pyrimidine-5-carbonitrile (22)
Prepared according to procedure B. The remaining solid was suspended in MeOH, adsorbed on silica gel, and concentrated in vacuo. Chromatographic purification (Biotage SP4, 10 g cartridge, solvent system: 5% NH4OH in MeOH/EtOAc; gradient 10% 4CV, 10–25% 10CV, 25% 4CV) yielded product X (51 mg, 40%,) a gum which was triturated with acetone (2 × 1 mL) to afford 22 (72 mg, 70%) as a pale-yellow solid. 1H NMR (400 MHz, DMSO-d6) δ: 2.46 (app t, J = 6.2 Hz, 2H), 2.56–2.58 (m, 4H), 3.53–3.58 (m, 6H), 4.42 (bs, 1H), 6.04 (bs, 2H), 7.84 (s, 1H), 11.94 (bs, 1H). 13C NMR (100 MHz, DMSO-d6) δ: 47.6, 52.9, 58.5, 60.2, 83.3, 95.3, 117.6, 130.7, 155.1, 158.9, 160.0. HRMS (ESI) calculated for C13H16ON7 [M – H]− 286.1422, found 286.1422. HPLC tR = 7.24 min (method C).
2-Amino-4-(3-hydroxypropylamino)-7H-pyrrolo[2,3-d]pyrimidine-5-carbonitrile (23)
Prepared according to procedure B. The remaining solid was triturated with cold H2O (2 × 2 mL), filtered, dried, and then triturated with Et2O (2 × 3 mL) to yield 23 (62 mg, 74%) as a pale-cream solid. 1H NMR (400 MHz, DMSO-d6) δ: 1.70–1.76 (m, 2H), 3.48–3.55 (m, 4H), 4.66 (t, J = 4.3 Hz, 1H), 5.95 (bs, 2H), 6.08 (t, J = 5.1 Hz, 1H), 7.68 (s, 1H), 11.82 (bs, 1H). 13C NMR (100 MHz, DMSO-d6) δ: 32.6, 38.5, 59.6, 82.2, 94.7, 117.3, 129.1, 153.6, 156.8, 161.5. HRMS (ESI) calculated for C10H12NO [M – H]− 231.1000, found 231.1001. HPLC tR = 3.66 min (method A).
(R)-2-Amino-4-(3-hydroxypyrrolidin-1-yl)-7H-pyrrolo[2,3-d]pyrimidine-5-carbonitrile (24)
5 (100 mg, 0.36 mmol), (3R)-3-hydroxypyrrolidine (0.2 mL, 2.48 mmol), and TEA (0.15 mL, 1.13 mmol) in dioxane (2.5 mL) was heated in the microwave at 200 °C for 20 min. The reaction mixture was then diluted with EtOH (8 mL) and KOH (2 pellets) added and heated at 90 °C for 24 h. The reaction mixture was concentrated at reduced pressure, water (5 mL) added, acidified to pH 5.7 (3 M HCl), and extracted with EtOAc (10 mL × 3). The organic fractions were combined, dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure to give the crude product. The crude was triturated with Et2O to afforded 24 (28 mg, 0.11 mmol, 33%). 1H NMR (500 MHz, DMSO-d6): d 1.85–1.94 (m, 1H), 1.94–2.04 (m, 1H), 3.56–3.64 (m, 1H), 3.70–3.85 (m, 3H), 4.39 (s, 1H), 4.99 (br s, 1H), 5.81 (s, 2H), 7.79 (s, 1H), 11.88 (br s, 1H). 13C NMR (125 MHz, DMSO-d6): d 33.0, 46.7, 57.3, 68.7, 83.4, 93.7, 118.6, 130.9, 154.7, 155.5, 159.8. HRMS: calculated for C11H13ON6 245.1145 [M + H]+, found 245.1143.
(R)-2-Amino-4-(3-hydroxypyrrolidin-1-yl)-7H-pyrrolo[2,3-d]pyrimidine-5-carbonitrile (25)
Prepared according to procedure A. Chromatographic purification (Biotage SP4, 10 g cartridge, solvent system: MeOH/EtOAc; gradient 0% 6CV, 0–10% 12CV, 10% 6CV) yielded 25 (10 mg, 13%) as a light-brown solid; [α]D20 −21.5o (c = 0.37, DMSO). 1H NMR (400 MHz, DMSO-d6) δ: 1.85–1.92 (m, 1H), 1.94–2.03 (m, 1H), 3.58–3.61 (m, 1H), 3.70–3.83 (m, 3H), 4.36–4.41 (m, 1H), 5.01 (d, J = 3.4 Hz, 1H), 5.84 (bs, 2H), 7.80 (s, 1H), 11.84 (bs, 1H). 13C NMR (100 MHz, DMSO-d6) δ: 33.6, 47.3, 57.9, 69.3, 84.0, 94.3, 119.2, 131.5, 155.3, 156.0, 160.4. HRMS (ESI) calculated forC11H11ON6 [M – H]− 243.1000, found 243.1001. HPLC tR = 7.85 min (method C).
(R)-2-Amino-4-(2-(hydroximethyl)pyrrolidin-1-yl)-7H-pyrrolo[2,3-d]pyrimidine-5-carbonitrile (26)
Prepared according to procedure B. The residue was suspended in MeOH, adsorbed on silica gel, and concentrated in vacuo. Chromatographic purification (Biotage SP4, 10 g cartridge, solvent system: MeOH/DCM; gradient 0% 4CV, 0–5% 4CV, 5% 4CV, 5–10% 4CV, 10% 4CV) yielded 26 (62 mg, 67%) as fluffy off-white solid; [α]D20 −96.3o (c = 0.55, DMSO). 1H NMR (400 MHz, DMSO-d6) δ: 1.83–1.89 (m, 1H), 1.90–1.96 (m, 2H), 2.02–2.10 (m, 1H), 3.52–3.63 (m, 2H), 3.70–3.76 (m, 1H), 3.86–3.91 (m, 1H), 4.46–4.51 (m, 1H), 4.77 (t, J = 5.0 Hz, 1H), 5.81 (bs, 2H), 7.81 (d, J = 1.0 Hz, 1H), 11.89 (bs, 1H). 13C NMR (100 MHz, DMSO-d6) δ: 24.6, 27.5, 51.1, 60.4, 62.3, 84.3, 94.5, 119.3, 131.6, 155.4, 156.2, 160.2. HRMS (ESI) calculated forC12H15ON6 [M + H]+ 259.1302, found 259.1299. HPLC tR = 7.26 min (method A).
(S)-2-Amino-4-(2-(hydroximethyl)pyrrolidin-1-yl)-7H-pyrrolo[2,3-d]pyrimidine-5-carbonitrile (27)
Prepared according to procedure B. The residue was suspended in MeOH, adsorbed on silica gel, and concentrated in vacuo. Chromatographic purification (Biotage SP4, 10 g cartridge, solvent system: MeOH/DCM; gradient 0% 4CV, 0–5% 4CV, 5% 4CV, 5–10% 4CV, 10% 4CV) yielded 27 (68 mg, 73%) as fluffy off-white solid; [α]D20 −93.8o (c = 0.54, DMSO). 1H NMR (400 MHz, DMSO-d6) δ: 1.83–1.89 (m, 1H), 1.90–1.96 (m, 2H), 2.02–2.10 (m, 1H), 3.52–3.63 (m, 2H), 3.70–3.76 (m, 1H), 3.86–3.91 (m, 1H), 4.46–4.51 (m, 1H), 4.77 (t, J = 5.0 Hz, 1H), 5.81 (bs, 2H), 7.81 (d, J = 1.0 Hz, 1H), 11.89 (bs, 1H). 13C NMR (100 MHz, DMSO-d6) δ: 24.6, 27.5, 51.1, 60.4, 62.3, 84.3, 94.5, 119.3, 131.6, 155.4, 156.2, 160.2. HRMS (ESI) calculated for C12H15ON6 [M + H]+ 259.1302, found 259.1298. HPLC tR = 7.26 min (method A).
2-Amino-4-((1R,4R)-4-hydroxycyclohexylamino)-7H-pyrrolo[2,3-d]pyrimidine-5-carbonitrile (28)
Prepared according to procedure B. The remaining solid was suspended in MeOH, adsorbed on silica gel, and concentrated in vacuo. Chromatographic purification (Biotage SP4, 10 g cartridge, solvent system: MeOH/EtOAc; gradient 0% 6CV, 0–15% 16CV, 15% 8CV) yielded a beige solid which was triturated with MeOH (0.5 mL) to yield 28 (32 mg, 35%) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ: 1.26–1.34 (m, 2H), 1.41–1.52 (m, 2H), 1.83–1.88 (m, 2H), 1.92–1.98 (m, 2H), 3.41–3.49 (m, 1H), 3.97–4.08 (m, 1H), 6.67 (bs, 1H), 7.31 (bs, 2H), 7.85 (s, 1H), 12.68 (bs 1H). 13C NMR (100 MHz, DMSO-d6) δ: 30.2, 34.2, 49.5, 68.4, 84.3, 94.6, 116.2, 130.5, 147.1, 158.0, 160.4. HRMS (ESI) calculated for C13H15ON6 [M + H]+ 271.1313, found 271.1316. HPLC tR = 6.54 min (method A).
2-Amino-4-(cyclohexyl(2-hydroxyethyl)amino)-7H-pyrrolo[2,3-d]pyrimidine-5-carbonitrile (29)
Prepared according to procedure B. The remaining solid was suspended in MeOH, adsorbed on silica gel, and concentrated in vacuo. Chromatographic purification (Biotage SP4, 10 g cartridge, solvent system: MeOH/DCM; gradient 0% 4CV, 0–5% 4CV, 5% 4CV, 5–10% 4CV, 10% 4CV) yielded 29 (51 mg, 40%) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ: 1.05–1.14 (m, 1H), 1.36–1.45 (m, 2H), 1.53–1.61 (m, 3H), 1.71–1.77 (m, 4H), 3.44–3.48 (m, 2H), 3.54–3.49 (m, 1H), 4.16–4.21 (m, 1H), 4.55 (t, J = 5.4 Hz, 1H), 5.91 (bs, 2H), 7.84 (s, 1H), 11.95 (bs, 1H). 13C NMR (100 MHz, DMSO-d6) δ: 25.4, 25.7, 31.1, 44.9, 58.9, 60.5, 84.2, 95.4, 118.2, 131.5, 155.8, 158.7, 160.1. HRMS (ESI) calculated forC15H19ON6 [M – H]− 299.1626, found 299.1628. HPLC tR = 9.46 min (method A).
2-Amino-4-((1RS,2SR,3RS)-2,3-dihydroxycyclopentylamino)-7H-pyrrolo[2,3-d]pyrimidine-5-carbonitrile (30)
5 (80 mg, 0.29 mmol), 37 (70 mg, 0.35 mmol), and Et3N (0.04 mL, 29 mg, 0.29 mmol) were suspended in n-BuOH (4 mL) and refluxed for 16 h. After cooling to room temperature, EtOH (4 mL) and KOH (3 pellets) were added and the reaction mixture was heated at 80 °C for 20 h. The mixture was neutralized using 6 M HCl and subsequently concentrated in vacuo. The remaining solid was suspended in MeOH, adsorbed on silica gel, and concentrated in vacuo. Chromatographic purification (Biotage SP4, 50g cartridge, solvent system: MeOH/CHCl3; gradient 0% 4CV, 0–15% 14CV, 15% 4CV) yielded 30 (33 mg, 42%) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ: 1.29–1.38 (m, 1H), 1.47–1.55 (m, 1H), 1.87–1.96 (m, 1H), 2.18–2.27 (m, 1H), 3.72–3.75 (m, 1H), 3.89–3.93 (m, 1H), 4.28–4.36 (m, 1H), 4.39 (d, J = 3.3 Hz, 1H), 4.94 (d, J = 5.5 Hz, 1H), 5.66 (d, J = 7.1 Hz, 1H), 5.99 (bs, 2H), 7.69 (s, 1H), 11.83 (bs, 1H). 13C NMR (100 MHz, DMSO-d6) δ: 27.0, 28.8, 55.4, 70.6, 77.8, 81.5, 94.2, 116.9, 128.4, 153.0, 156.5, 160.8. HRMS (ESI) calculated for C12H13O2N6 [M + H]+ 273.1105, found 273.1107. HPLC tR = 4.47 min (method A).
2-Amino-4-((1R,2S,3R,4S)-2,3,4-trihydroxycyclopentylamino)-7H-pyrrolo[2,3-d]pyrimidine-5-carbonitrile (31)
5 (70 mg, 0.25 mmol), 42 (146 mg, 0.30 mmol), and Et3N (0.08 mL, 61 mg, 0.60 mmol) were suspended in n-BuOH (4 mL) and refluxed for 16 h. After cooling to room temperature, EtOH (4 mL) and KOH (3 pellets) were added and the reaction mixture was heated at 80 °C for 20 h. The mixture was neutralized using 6 M HCl and subsequently concentrated in vacuo. The remaining solid was suspended in MeOH, adsorbed on silica gel, and concentrated in vacuo. Chromatographic purification (Biotage SP4, 10 g cartridge, solvent system: 5% NH4OH in MeOH/CHCl3; gradient 2% 6CV, 2–15% 12CV, 10% 15CV) yielded a beige solid which was further triturated with MeOH (0.2 mL) and Et2O (2 mL) to yield 31 (24 mg, 33%) as a white solid; [α]D20 −25.7° (c = 0.12, DMSO). 1H NMR (400 MHz, DMSO-d6) δ: 1.23–1.29 (m, 1H), 2.5–2.551 (m, 1H), 3.67–3.68 (m, 1H), 3.80–3.81 (m, 1H), 3.91–3.93 (m, 1H), 4.30–4.36 (m, 1H), 4.58 (d, J = 3.6 Hz, 1H), 4.91 (bs, 1H), 4.92 (bs, 1H), 5.70 (d, J = 7.5 Hz, 1H), 5.99 (bs, 2H), 7.69 (s, 1H), 11.83 (bs, 1H). 13C NMR (100 MHz, DMSO-d6) δ: 37.8, 55.2, 74.6, 76.8, 77.6, 94.1, 117.0, 128.5, 129.5, 153.1, 156.2, 160.9. HRMS (ESI) calculated forC12H15O3N6 [M + H]+ 291.1200, found 291.1189. HPLC tR = 6.40 min (method B).
3-(N,N-Dibenzylamino)cyclopent-1-ene (34)
A mixture of cyclopentene (12.3 mL, 9.18 g, 0.135 mol), NBS (6.01 g, 33.71 mmol), and benzoyl peroxide (70%, 163 mg, 0.67 mmol) in CCl4 (21 mL) was heated at reflux for 1 h. The reaction mixture was cooled to 0 °C, filtered through a pad of Celite (eluent CCl4), and solvent and cyclopentene were distilled off in vacuo. The residue was dissolved in CCl4 (30 mL), cooled to 0 °C, and N,N-dibenzylamine (16.2 mL, 16.60 g, 84.28 mmol) was added to the crude solution of bromide 33. The mixture then warmed to room temperature and stirred for 30 min. The reaction mixture was then filtered, heated to 40 °C, and stirred at this temperature for 1 h, then filtered and stirred at room temperature for 16 h. The mixture was then filtered and concentrated in vacuo. Chromatographic purification (Biotage SP4, 100 g cartridge, solvent system: 10% Et2O in Hex/Hex; gradient 0% 4CV, 0–10% 10CV) yielded 34 (6.23 g, 70%) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ: 1.78–1.93 (m, 2H), 2.22–2.29 (m, 1H), 2.34–2.41 (m, 1H), 3.43 (d, J = 13.8 Hz, 2H), 3.64 (d, J = 13.8 Hz, 2H), 4.03–4.05 (m, 1H), 5.75–5.76 (m, 1H), 5.86–5.89 (m, 1H), 7.19–7.23 (m, 2H), 7.27–7.31 (m, 4H), 7.37–7.39 (m, 4H). 13C NMR (100 MHz, CDCl3) δ: 23.4, 31.9, 54.5, 66.1, 126.8, 128.3, 128.8, 132.1, 133.3, 140.8.
(1RS,2SR,3RS)-3-(Dibenzylamino)cyclopentane-1,2-diol (35)
A solution of OsO4 in H2O (4% w/v, 0.32 mL, 13 mg, 0.05 mmol) was added to a stirred solution of 51 (1.31 g, 4.98 mmol) and NMO (1.33 g, 14.94 mmol) in acetone/H2O (4:1, 35 mL), and the resultant mixture was stirred at room temperature for 4 h. Saturated aq Na2SO3 (5 mL) was then added, and the solution was stirred for an additional 30 min. Acetone was evaporated in vacuo, H2O (10 mL) was added, and the aqueous layer was extracted with DCM (3 × 20 mL). The organic layer was adsorbed on silica gel and concentrated in vacuo. Chromatographic purification (Biotage SP4, 50 g cartridge, solvent system: EtOAc/Hex; gradient 10% 4CV, 10–20% 6CV, 20% 4CV, 20–60% 6CV; 60% 6CV) yielded 35 (1.06 g, 72%) as a white solid. 1H NMR (400 MHz, CDCl3) δ: 1.52–1.75 (m, 2H), 1.83–1.96 (m, 2H), 2.28 (bs, 1H), 2.37 (bs, 1H), 3.27 (app q, J = 8.4 Hz, 1H), 3.57 (d, J = 13.9 Hz, 2H), 3.78 (d, J = 13.9 Hz, 2H), 3.92 (app dd, J = 8.2, 5.2 Hz, 1H), 3.99–4.05 (m, 1H), 7.21–7.25 (m, 2H), 7.29–7.32 (m, 4H), 7.35–7.37 (m, 4H). 13C NMR (100 MHz, CDCl3) δ: 19.6, 29.0, 55.0, 65.1, 71.1, 74.6, 127.1, 128.4, 128.7, 139.9. HRMS (ESI) calculated for C19H24O2N [M + H]+ 298.1802, found 298.1798.
(1RS,2SR,3RS)-3-(Dibenzylamino)cyclopentane-1,2-diyl-dibenzoate (36)
Benzoyl chloride (1.62 mL, 1.96 g, 13.92 mmol) was added dropwise to a stirred solution of 35 (1.03 g, 3.48 mmol) in dry pyridine (30 mL) at 0 °C, left to warm to room temperature, and stirred for 1 day. Pyridine was evaporated in vacuo, and the residue was suspended in Et2O (40 mL), cooled to 0 °C, followed by addition of saturated aqueous NaHCO3 (10 mL). The organic layer was then washed with saturated aqueous NaHCO3 (3 × 20 mL) and subsequently adsorbed on silica gel and concentrated in vacuo. Chromatographic purification (Biotage SP4, 50 g cartridge, solvent system: 10% Et2O in Hex/Hex; gradient 0% 3CV, 0–10% 3CV, 10% 4CV, 10–20% 4CV, 20% 4CV, 20–40% 6CV) yielded 36 (1.45 g, 84%) as a white solid. 1H NMR (400 MHz, CDCl3) δ: 1.75–1.84 (m, 1H), 1.88–1.96 (m, 1H), 2.01–2.10 (m, 1H), 2.18–2.26 (m, 1H), 3.67 (d, J = 13.8 Hz, 2H), 3.77 (d, J = 13.8 Hz, 2H), 3.78–3.86 (m, 1H), 5.54 (dt, J = 5.0, 4.6 Hz, 1H), 5.67 (dd, J = 8.0, 5.0 Hz, 1H), 7.20–7.40 (m, 14 H), 7.49 (m, 1H), 7.55 (m, 1H), 7.76 (dd, J = 8.4, 1.3 Hz, 2H), 7.94 (dd, J = 8.4, 1.3 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ: 20.7, 27.6, 54.7, 62.2, 73.3, 74.0, 126.9, 128.3, 128.3, 128.4, 128.7, 129.6, 129.8, 130.1, 130.2, 132.9, 133.0, 139.7, 165.7, 165.9. HRMS (ESI) calculated for C33H32O4N [M + H]+ 506.2326, found 506.2321.
(1RS,2SR,3RS)-3-Aminocyclopentane-1,2-diyl dibenzoate (37)
Pd(OH)2/C (20% w/w, 700 mg) was added to a vigorously stirred solution of 36 (1.42 g, 2.81 mmol) in EtOH/EtOAc (5:1, 60 mL), and the resultant suspension was stirred at room temperature under H2 (1 atm) for 16 h. The suspension was filtered through a Celite pad (eluent MeOH) and concentrated in vacuo. The residue was triturated with Et2O (2 × 20 mL) to give 37 (908 mg, 98%) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ: 1.73–1.82 (m, 1H), 1.93–2.02 (m, 1H), 2.32–2.43 (m, 2H), 3.93 (app dt, J = 8.0, 7.6 Hz, 1H), 5.49 (dd, J = 7.5, 5.1 Hz, 1H), 5.59–5.63 (m, 1H), 7.42–7.49 (m, 4H), 7.61–7.66 (m, 2H), 7.86–7.89 (m, 2H), 8.38 (bs, 2H). 13C NMR (100 MHz, DMSO-d6) δ: 25.2, 27.7, 53.4, 73.2, 76.6, 129.1, 129.2, 129.6, 129.7, 129.8, 129.9, 134.1, 134.1, 165.4, 165.5. HRMS (ESI) calculated for C19H20O4N [M + H]+ 326.1387, found 326.1382.
Di-tert-butyl-[(1R,4S)-4-hydroxycyclopent-2-en-1-yl]imidodicarbonate (39)
A suspension of sodium di-tert-butyl-iminodicarboxylate, previously prepared by reaction of di-tert-butyl-iminodicarboxylate (1.195 g, 5.50 mmol) with NaH (60% suspension in mineral oil) (132 mg, 5.50 mmol) in dry THF (18 mL), was cannulated to a room temperature solution of (1R,4S)-4-hydroxycyclopent-2-enyl acetate (521 mg, 3.67 mmol), PPh3 (144 mg, 0.55 mmol), and Pd(PPh3)4 (636 mg, 0.55 mmol) in dry THF/DMF (1:1) (16 mL). The reaction mixture was heated at 50 °C for 1 day, then diluted with MeOH (10 mL) and dry loaded on to silica. Chromatographic purification (Biotage SP4, 50 g cartridge, solvent system: EtOAc/Hex; gradient 0% 4CV, 0–20% 10CV, 20% 6CV) yielded 39 (463 mg, 42%) as a colorless oil that solidifies upon standing to afford a white solid. 1H NMR (400 MHz, CDCl3) δ: 1.48 (s, 18H), 1.83 (dt, J = 15.1, 2.3 Hz, 1H), 2.68 (ddd, J = 15.1, 9.3, 7.9 Hz, 1H), 3.20 (bs, 1H), 4.57–4.61 (m, 1H), 5.09 (app dq, J = 9.3, 2.3 Hz, 1H), 5.74 (dd, J = 5.4, 2.3 Hz, 1H), 6.04 (dt, J = 5.4, 2.3 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ: 28.1, 38.9, 60.3, 75.8, 82.9, 131.5, 136.9, 153.4.
Di-tert-butyl-[(1R,2S,3R,4S)-2,3,4-trihydroxycyclopentyl] imidodicarbonate (40)
A solution of OsO4 in H2O (4% w/v, 0.06 mL, 3 mg, 0.01 mmol) was added to a solution of 39 (120 mg, 0.40 mmol) and NMO (141 mg, 1.20 mmol) in a 4:1 mixture acetone/H2O (10 mL). The solution was stirred at room temperature for 1 day. Saturated aqueous Na2SO3 (5 mL) was added, and the reaction mixture was stirred for 30 min. Acetone was removed in vacuo, and the aqueous layer was extracted with EtOAc (4 × 20 mL). The organic layer was adsorbed on silica gel and concentrated in vacuo. Chromatographic purification (Biotage SP4, 10 g cartridge, solvent system: EtOAc/DCM; gradient 20% 6CV, 20–80% 8CV, 80% 8CV) yielded 40 (71 mg, 53%) as a colorless oil that solidifies upon standing to afford a white solid. 1H NMR (400 MHz, CDCl3) δ: 1.49 (s, 18H), 1.79–1.85 (m, 1H), 2.53–2.62 (m, 1H), 2.91 (bs, 3H), 3.98–3.99 (m, 1H), 4.01–4.04 (m, 1H), 4.39–4.45 (m, 1H), 4.55–4.58 (m, 1H). 13C NMR (100 MHz, CDCl3) δ: 28.0, 34.1, 61.9, 74.9, 75.1, 77.3, 83.6, 153.7. HRMS (ESI) calculated for C15H27O7NNa [M + Na]+ 356.1680, found 356.1682.
(1S,2R,3S,4R)-4-(Bis(tert-butoxycarbonyl)amino)cyclopentane-1,2,3-triyl tribenzoate (41)
Benzoyl chloride (0.33 mL, 397 mg, 2.83 mmol) was added dropwise to a stirred solution of 40 (235 mg, 0.71 mmol) in dry pyridine (6 mL) at 0 °C and left to warn to room temperature and stirred for 1 day. Pyridine was evaporated in vacuo, and the residue was partitioned between Et2O (40 mL) and saturated aqueous NaHCO3 (10 mL). The organic layer was separated and washed with saturated aqueous NaHCO3 (3 × 20 mL) and subsequently adsorbed on silica gel and concentrated in vacuo. Chromatographic purification (Biotage SP4, 50 g cartridge, solvent system: Et2O/Hex; gradient 0% 6CV, 0–20% 10CV, 20% 6CV) yielded 41 (338 mg, 74%) as a colorless oil that solidified upon standing to afford a white solid. 1H NMR (400 MHz, CDCl3) δ: 1.51 (s, 18H), 2.21–2.29 (m, 1H), 2.75–2.82 (m, 1H), 5.00–5.07 (m, 1H), 5.58–5.64 (m, 1H), 5.98–6.05 (m, 2H), 7.29–7.34 (m, 4H), 7.41–7.51 (m, 3H), 7.54–7.63 (m, 2H), 7.90–7.93 (m, 4H), 8.08–8.13 (m, 2H). 13C NMR (100 MHz, CDCl3) δ: 28.1, 30.3, 58.3, 73.7, 74.3, 75.6, 83.3, 128.3, 128.4, 128.5, 129.4, 129.4, 129.6, 129.7, 129.8, 129.9, 133.1, 133.1, 133.2, 152.5, 165.3, 165.4, 166.0. HRMS (ESI) calculated for C36H39O10NNa [M + Na]+ 668.2463, found 668.2463.
(1S,2R,3S,4R)-4-Aminocyclopentane-1,2,3-triyl-tribenzoate Hydrochloride (42)
41 (297 mg, 0.46 mmol) was placed in an oven-dry round-bottom flask, purged with N2, cooled to 0 °C, and subsequently treated with 4 M HCl in 1,4-dioxane (10 mL). The mixture was left to warm to room temperature, stirred for 16 h, and concentrated in vacuo. The residue was triturated with Et2O (5 × 1 mL) to yield 42 (168 mg, 76%) as a white solid. 1H NMR (400 MHz, CDCl3) δ: 2.01–2.09 (m, 1H), 2.97–3.04 (m, 1H), 4.07–4.14 (m, 1H), 5.51–5.56 (m, 1H), 5.73–5.76 (m, 1H), 5.82–5.83 (m, 1H), 7.43–7.49 (m, 4H), 7.55–7.59 (m, 2H), 7.63–7.73 (m, 3H), 7.87–7.92 (m, 4H), 8.03–8.05 (m, 2H), 8.66 (bs, 3H). 13C NMR (100 MHz, CDCl3) δ: 31.6, 51.6, 73.6, 74.6, 74.8, 129.0, 129.1, 129.2, 129.2, 129.3, 129.4, 129.8, 129.9, 129.9, 134.2, 164.9, 165.2, 165.4. HRMS (ESI) calculated forC26H24O6N [M + H]+ 446.1598, found 446.1599.
2-Amino-4-phenyl-7H-pyrrolo[2,3-d]pyrimidine-5-carbonitrile (43)
5 (96 mg 0.35 mmol), phenyl boronic acid (73 mg, 0.60 mmol), and Pd(PPh3)4 (0.041 g, 0.047 mmol) were suspended in a mixture of IPA/H2O (3:1.5 mL) and degassed. t-Butylamine (0.19 mL, 1.81 mmol) was added, and the mixture was irradiated at 160 °C for 40 min. The resulting solution was diluted with EtOH (8 mL) and KOH (2 pellets) were added and the mixture was heated to 90 °C for 20 h. The reaction mixture was then concentrated under reduced pressure, and the resulting mixture was suspended in water, pH adjusted to pH 5.3, extracted into EtOAc (2 × 100 mL), and dried over MgSO4. The resulting solid was purified by column chromatography (100% hexane–100% ethyl acetate) and triturated with diethyl ether to afford the title compound 43 as a light-brown solid (4.9 mg, 0.0208 mmol, 6.0%). 1H NMR (400 Hz, DMSO-d6): δ 6.63 (br s, 2H), 7.54–7.55 (m, 3H), 7.78–7.80 (m, 2H), 8.12 (s, 1H), 12.40 (br s, 1H). HRMS (ESI) calculated for C13H9N5 [M + H]+ 236.0931, found 236.0930.
2-Amino-4-(4-hydroxyphenyl)-7H-pyrrolo[2,3-d]pyrimdine-5-carbonitrile (44)
5 (115 mg, 0.42 mmol), 4-hydroxyphenyl boronic acid (92 mg, 0.67 mmol), and Pd(PPh3)4 (49 mg, 0.042 mmol) were suspended in a mixture of IPA/H2O (3:1.5 mL) and degassed. t-Butylamine (0.18 mL, 1.71 mmol) was added, and the mixture was irradiated at 160 °C for 40 min. The reaction mixture was then concentrated under reduced pressure, and the resulting solid was triturated with methanol, filtered, and dried. EtOH (9 mL) was added KOH (2 pellets) and the mixture heated to 90 °C for 48 h. The reaction mixture was concentrated under reduced pressure, water was added, and the mixture was pH adjusted to pH 6 and extracted into EtOAc. The organic layer was dried over MgSO4 and concentrated under reduced pressure to afford the title compound 44 as a brown solid 10.1 mg, 0.040 mmol, 67%). 1H NMR (400 Hz, DMSO-d6): δ 6.51 (br s, 2H), 6.89 (d, 2H, J = 8.4 Hz), 7.67 (d, 2H, J = 8.8 Hz), 8.08 (s, 1H), 9.90 (s, 1H), 12.28 (br s, 1H). HRMS (ESI) calculated for C13H9N5O 252.0880 [M + H]+, found 252.0880. HPLC: tR = 8.62 min (70.46%).
2-Amino-4-(4-fluorophenyl)-7H-pyrrolo[2,3-d]pyrimidine-5-carbonitrile 945)
5 (100 mg 0.31 mmol), Pd-118 (20 mg, 0.03 mmol), t-butylamine (160 μL, 1.55 mmol), and 4-fluorophenylboronic acid (69 mg, 0.5 mmol) were suspended in a 2:1 mixture i-PrOH/H2O (4.5 mL) and were irradiated with microwaves at 160 °C for 40 min. Once cooled to room temperature, the reaction mixture was concentrated under reduced pressure. To this was added EtOH (5 mL) and KOH (2 pellets), and the reaction mixture was refluxed at 90 °C for 20 h. The mixture was then concentrated under reduced pressure, and the crude material was then suspended in H2O and adjusted to pH 5 with 1 M HCl. The solution was then extracted with EtOAc (2 × 50 mL); the organic layer was adsorbed on silica gel and concentrated under reduced pressure. Chromatographic purification (EtOAc/Hex: 50% 4CV, 50–75% 6CV, 75% 4CV, 75–100% 7CV, 100% 6CV) yielded a beige solid that was triturated with Et2O to afford the title compound 45 as a beige solid (13 mg, 0.05 mmol, 17%). 1H NMR (400 MHz, DMSO-d6): δ 6.62 (s, 2H), 7.36 (t, J = 8.3 Hz, 2H), 7.83 (t, J = 7.7 Hz, 2H), 8.12 (s, 1H), 12.36 (s, 1H). HRMS: calculated for C13H9N5F [M + H]+ 254.0837, found 254.0831.
4-(2-Amino-5-cyano-7H-pyrrolo[2,3-d]pyrimdin-4-yl)benzamide (46)
5 (101 mg, 0.36 mmol), 4-aminocarbonyl phenylboronic acid (96 mg, 0.58 mmol), and Pd(PPh3)4 (40 mg, 0.035 mmol) were suspended in a mixture of IPA/H2O (3:1.5 mL) and degassed. t-Butylamine (0.20 mL, 1.90 mmol) was added, and the mixture was irradiated at 160 °C for 40 min. The resulting solution was diluted with EtOH (6 mL) and KOH (2 pellets) were added and the mixture was heated to 90 °C for 30 h. The reaction mixture was then concentrated under reduced pressure, and the resulting mixture was suspended in water, pH adjusted to pH 5.2, extracted into EtOAc (2 × 100 mL), and dried over MgSO4. The mixture was then concentrated under reduced pressure, triturated with diethyl ether and methanol, and filtered. The solid and filtrate were combined and purified by column chromatography (100% hexane–100% ethyl acetate). The isolated compound was triturated with diethyl ether, filtered under reduced pressure, and dried to afford the title compound 46 as an off-white solid (6 mg, 0.022 mmol, 5.9%). 1H NMR (400 Hz, DMSO-d6): δ 6.68 (s, 2H), 7.51 (br s, 1H), 7.85 (d, 2H, J = 9.2 Hz), 8.40 (d, 2H, J = 10.4 Hz), 8.14 (br s, 1H), 8.16 (s, 1H), 12.40 (br s, 1H). HRMS (ESI) calculated for C14H8N6O [M + H]+ 277.0843, found 277.0844. HPLC: tR = 7.94 min (method A) (76.01%).
2-Amino-4-(4-hydroxymethyl)phenyl-7H-pyrrolo[2,3-d]pyrimidine-5-carbonitrile (47)
5 (103 mg, 0.37 mmol), 4-hydroxymethyl)phenyl boronic acid (99 mg, 0.65 mmol), and Pd-118 (47 mg, 0.072 mmol) were suspended in a mixture of IPA/H2O (3:1.5 mL) and degassed. t-Butylamine (0.20 mL, 1.90 mmol) was added, and the mixture was irradiated at 160 °C for 40 min. The resulting solution was diluted with EtOH (8 mL) and KOH (2 pellets) were added and the mixture was heated to 90 °C for 20 h. The reaction mixture was then concentrated under reduced pressure, and the resulting mixture was suspended in water, pH adjusted to pH 5.3, extracted into EtOAc (2 × 100 mL), and dried over MgSO4. The resulting solid was purified by column chromatography (100% hexane–100% ethyl acetate) to afford the title compound 47 as an off-white solid (28.5 mg, 0.108 mmol, 29%). 1H NMR (400 Hz, DMSO-d6): δ 4.61 (d, 2H, J = 5.6 Hz), 5.34 (t, 1H, J = 5.8 Hz), 6.60 (br s, 2H), 7.47 (d, 2H, J = 7.6 Hz), 7.77 (d, 2H, J = 8.0 Hz), 8.11 (s, 1H), 12.34 (br s, 1H). HRMS (ESI) calculated for C14H11N5O [M + H]+ 264.0891, found 264.0891. HPLC: tR = 8.45 min (method A) (94.35%).
4-(2-Amino-5-cyano-7H-pyrrolo[2,3-d]pyrimidin-4-yl)benzenesulfonamide (48)
5 (100.6 mg, 0.36 mmol), 4-sulphamoylbenzene boronic acid (124 mg 0.62 mmol), Pd-118 (43 mg 0.066 mmol) were suspended in a mixture of IPA/H2O (3:1.5 mL) and degassed. t-Butylamine (0.195 mL, 1.86 mmol) was added and the mixture was irradiated at 160 °C for 40 min. The resulting solution was diluted with EtOH (6 mL) and KOH (2 pellets) were added and the mixture was heated to 90 °C for 20 h. The reaction mixture was then concentrated under reduced pressure and the resulting mixture was suspended in water, pH adjusted to pH 5.5, extracted into EtOAc (2 × 100 mL) and dried over MgSO4. The resulting solid was purified by column chromatography (100% hexane–100% ethyl acetate) and triturated with diethyl ether to afford the title compound 48 as an off-white solid (19.1 mg, 0.0608 mmol, 17%). 1H NMR (400 Hz, DMSO-d6): δ 6.70 (br s, 2H), 7.54 (br s, 2H), 7.97 (br s, 4H), 8.17 (s, 1H), 12.45 (br s, 1H). HRMS (ESI) calculated for C13H8N6O2S [M + H]+ 313.0513, found 313.0517. HPLC: tR = 8.33 min (method A) (94.29%).
2-Amino-4-(4-(methylsulfonyl)phenyl)-7H-pyrrolo[2,3-d]pyrimidine-5-carbonitrile (49)
5 (104 mg, 0.38 mmol), (4-methylsulfonyl)benzene boronic acid (128 mg, 0.64 mmol), and Pd-118 (25 mg, 0.038 mmol) were suspended in a mixture of IPA/H2O (3:1.5 mL) and degassed. t-Butylamine (0.22 mL, 2.09 mmol) was added, and the mixture was irradiated at 160 °C for 40 min. The resulting solution was diluted with EtOH (8 mL) and KOH (2 pellets) were added and the mixture was heated to 90 °C for 22 h. The reaction mixture was then concentrated under reduced pressure, and the resulting mixture was suspended in water, pH adjusted to pH 5.7, extracted into EtOAc (2 × 100 mL), and dried over MgSO4. The resulting solid was purified by column chromatography (100% hexane–100% ethyl acetate), triturated with diethyl ether, filtered, and dried to afford the title compound 49 as a pale-yellow solid (54.8 mg, 0.175 mmol, 47%). 1H NMR (500 Hz, DMSO-d6): δ 3.30 (br s, 3H), 6.67 (br s, 2H), 8.03 (d, 2H, J = 8.5 Hz), 8.09 (d, 2H, J = 8.5 Hz), 8.17 (s, 1H), 12.45 (br s, 1H). HRMS (ESI) calculated for C14H10N5O2S [M + H]+ 312.0561, found 312.0564. HPLC: tR = 8.97 min (method A).
N-(4-(2-Amino-5-cyano-7H-pyrrolo[2,3-d]pyrimidin-4-yl)phenyl)methanesulfonamide (50)
5 (0.103 g, 0.37 mmol), 4-(methylsulfonyl)aminobenzene boronic acid (0.121 g, 0.56 mmol), Pd-118 (0.027 g, 0.041 mmol) were suspended in a mixture of IPA/H2O (3:1.5 mL) and degassed. t-Butylamine (0.20 mL, 1.90 mmol) was added, and the mixture was irradiated at 160 °C for 40 min. The resulting solution was diluted with EtOH (8 mL) and KOH (2 pellets) were added and the mixture was heated to 90 °C for 22 h. The reaction mixture was then concentrated under reduced pressure, and the resulting mixture was suspended in water, pH adjusted to pH 5.7, extracted into EtOAc (2 × 100 mL), and dried over MgSO4. The resulting solid was purified by column chromatography (100% hexane–100% ethyl acetate), triturated with diethyl ether, filtered, and dried to afford the title compound 50 as a pale-yellow solid (0.0399 g, 0.122 mmol, 32.7%). 1H NMR (400 Hz, DMSO-d6): δ 3.08 (s, 3H), 6.55 (br s, 2H), 7.34 (d, 2H, J = 8.8 Hz), 7.78 (d, 2H, J = 8.4 Hz), 8.10 (s, 1H), 10.08 (br s, 1H), 12.33 (br s, 1H). HRMS (ESI) calculated for C14H11N6O2S [M + H]+ 327.0670, found 327.0673. HPLC: tR = 8.92 min (method A) (85.80%).
4-(2-Amino-5-cyano-7H-pyrrolo[2,3-d]pyrimidin-4-yl)-N-(p-tolyl)benzenesulfonamide (51)
5 (100 mg, 0.36 mmol), 4-(N-p-tolylsulfamoyl)phenylboronic acid (168 mg, 0.58 mmol), and Pd-118 (23 mg, 0.03 mmol) in IPA/H2O (3:1.5 mL) was degassed with nitrogen. t-Butylamine (0.19 mL, 1.8 mmol) was added, and the mixture was heated in the microwave at 160 °C for 40 min. The reaction mixture was then diluted with EtOH (4 mL), KOH (2 pellets) added and heated at 90 °C for 24 h. The reaction mixture was concentrated under reduced pressure, and the resulting mixture was suspended in water (5 mL), acidified to pH 5.7 (3 M HCl) and extracted with EtOAc (10 mL × 3). The organic fractions were combined, dried over anhydrous MgSO4, filtered and concentrated under reduced pressure to give the crude product. Purification by flash column chromatography (silica gel with EtOAc (75–100%) in hexane afforded the title compound 51 as an off-white solid (0.005 g, 0.01 mmol, 3%). 1H NMR (400 Hz, DMSO-d6): 2.18 (s, 3H), 6.69 (br s, 2H), 6.98 (d, 2H, J = 8.4 Hz), 7.03 (d, 2H, J = 8.4 Hz), 7.84 (d, 2H, J = 8.4 Hz), 7.88 (d, 2H, J = 8.4 Hz), 8.14 (s, 1H), 10.20 (s, 1H), 12.43 (br s, 1H). HRMS: calculated for C20H15O2N6S [M + H]+ 403.0983, found 403.0986.
4-(2-Amino-5-cyano-7H-pyrrolo[2,3-d]pyrimidin-4-yl)-N-methylbenzenesulfonamide (52)
5 (115 mg, 0.42 mmol), 4-((methylamino)sulphonyl)benzene boronic acid (134 mg 0.62 mmol), and Pd-118 (32 mg, 0.049 mmol) were suspended in a mixture of IPA/H2O (3:1.5 mL) and degassed. t-Butylamine (0.22 mL, 2.09 mmol) was added, and the mixture was irradiated at 160 °C for 40 min. The resulting solution was diluted with EtOH (8 mL) and KOH (2 pellets) were added and the mixture was heated to 90 °C for 22 h. The reaction mixture was then concentrated under reduced pressure, and the resulting mixture was suspended in water, pH adjusted to pH 5.5, extracted into EtOAc (2 × 100 mL), and dried over MgSO4. The resulting solid was purified by column chromatography (100% hexane–100% ethyl acetate), triturated with diethyl ether, filtered, and dried to afford the title compound 52 as a pale-yellow solid (47.2 mg, 0.144 mmol, 35%). 1H NMR (400 Hz, DMSO-d6): δ 2.46 (d, 3H, J = 4.8 Hz), 6.71 (br s, 2H), 7.60 (q, 1H, J = 4.9 Hz), 7.92 (d, 2H, J = 8.0 Hz), 7.98 (d, 2H, J = 8.4 Hz), 8.16 (s, 1H), 12.45 (br s, 1H). HRMS (ESI) calculated for C14H11N6O2S [M + H]+ 327.0670, found 327.0674. HPLC: tR = 9.24 min (method A) (82.18%).
2-Amino-4-(3-hydroyphenyl)-7H-pyrrolo[2,3-d]pyrimidine-5-carbonitrile (53)
5 (113 mg, 0.41 mmol), 3-hydroxyphenyl boronic acid (95 mg, 0.69 mmol), and Pd-118 (33 mg 0.051 mmol) were suspended in a mixture of IPA/H2O (3:1.5 mL) and degassed. t-Butylamine (0.22 mL, 2.09 mmol) was added, and the mixture was irradiated at 160 °C for 40 min. The resulting solution was diluted with EtOH (8 mL) and KOH (2 pellets) were added and the mixture was heated to 90 °C for 22 h. The reaction mixture was then concentrated under reduced pressure, and the resulting mixture was suspended in water, pH adjusted to pH 5.5, extracted into EtOAc (2 × 100 mL), and dried over MgSO4. The resulting solid was purified by column chromatography (100% hexane–100% ethyl acetate) to afford the title compound 53 as an off-white solid (12 mg, 0.048 mmol, 11.7%). 1H NMR (400 Hz, DMSO-d6): δ 6.58 (br s, 2H), 6.91 (d, 1H, J = 8.0 Hz), 7.15–7.19 (m, 2H), 7.31 (t, 1H, J = 7.6 Hz), 8.09 (s, 1H), 9.62 (s, 1H), 12.33 (br s, 1H). HRMS (ESI) calculated for C13H7N5O [M + H]+ 250.0734, found 250.0737. HPLC: tR = 8.75 min (method A).
2-Amino-4-(3-fluorophenyl)-7H-pyrrolo[2,3-d]pyrimidine-5-carbonitrile (54)
5 (104 mg, 0.38 mmol), 3-fluorobenzene boronic acid (85 mg, 0.61 mmol), and Pd(PPh3)4 (45 mg, 0.039 mmol) were suspended in a mixture of IPA/H2O (3:1.5 mL) and degassed. t-Butylamine (0.16 mL, 1.52 mmol) was added, and the mixture was irradiated at 160 °C for 40 min. The reaction mixture was then concentrated under reduced pressure, and the resulting solid was triturated with methanol, filtered, and dried to give the intermediate compound as an off-white solid (28.3 mg, 0.084 mmol, 22%). To EtOH (10 mL) was added KOH (2 pellets) and the mixture heated to 90 °C for 20 h. The reaction mixture was then concentrated under reduced pressure and suspended in water. The mixture was then pH adjusted to pH 5.8 and extracted into EtOAc (2 × 100 mL), dried over MgSO4, and concentrated under reduced pressure. The resulting solid was triturated with diethyl ether, filtered, and dried to afford the title compound 54 as an off-white solid (12.2 mg, 0.048 mmol, 58%). 1H NMR (400 Hz, DMSO-d6): δ 6.67 (br s, 2H), 7.39 (t, 1H, J = 7.5 Hz), 7.55–7.63 (m, 3H), 8.15 (s, 1H), 12.40 (br s, 1H). HRMS (ESI) calculated for C13H8N5F [M + H]+ 254.0837, found 254.0833. HPLC: tR = 10.42 min (method A) (90.05%).
3-(2-Amino-5-cyano-7H-pyrrolo[2,3-d]pyrimdin-4-yl)benzamide (55)
5 (112 mg, 0.40 mmol), 3-(aminocarbonyl)phenyl boronic acid (108 mg, 0.65 mmol), and Pd-118 (37 mg, 0.056 mmol) were suspended in a mixture of IPA/H2O (3:1.5 mL) and degassed. t-Butylamine (0.21 mL, 2.0 mmol) was added, and the mixture was irradiated at 160 °C for 40 min. The resulting solution was diluted with EtOH (8 mL) and KOH (2 pellets) were added and the mixture was heated to 90 °C for 24 h. The reaction mixture was then concentrated under reduced pressure, and the resulting mixture was suspended in water, pH adjusted to pH 5.5, extracted into EtOAc (2 × 100 mL), and dried over MgSO4. The resulting solid was purified by column chromatography (100% hexane–100% ethyl acetate–5% methanol/ethyl acetate) to afford the title compound 55 as a yellow solid (34.2 mg, 0.123 mmol, 30%).1H NMR (400 Hz, DMSO-d6): δ 6.67 (br s, 2H), 7.45 (br s, 1H), 7.61 (t, 1H, J = 7.6 Hz), 7.91 (d, 1H, J = 7.6 Hz), 8.01–8.05 (m, 2H), 8.14 (s, 1H), 8.30 (br s, 1H), 12.45 (br s, 1H). HRMS (ESI) calculated for C14H9N5O [M + H]+ 277.0843, found 277.0846. HPLC: tR = 8.17 min (method A) (46.30%).
2-Amino-4-(3-(hydroxymethyl)phenyl)-7H-pyrrolo[2,3-d]pyrimidine-5-carbonitrile (56)
5 (103 mg, 0.37 mmol), 3-(hydroxymethyl)phenyl boronic acid (94 mg, 0.62 mmol), and Pd-118 (53 mg, 0.081 mmol) were suspended in a mixture of IPA/H2O (3:1.5 mL) and degassed. t-Butylamine (0.20 mL, 1.90 mmol) was added, and the mixture was irradiated at 160 °C for 40 min. The resulting solution was diluted with EtOH (8 mL) and KOH (2 pellets) were added and the mixture was heated to 90 °C for 20 h. The reaction mixture was then concentrated under reduced pressure, and the resulting mixture was suspended in water, pH adjusted to pH 5.5, extracted into EtOAc (2 × 100 mL), and dried over MgSO4. The resulting solid was purified by column chromatography (100% hexane–100% ethyl acetate), triturated with diethyl ether, filtered, and dried to afford the title compound 56 as a yellow solid (30.5 mg g, 0.115 mmol, 31%). 1H NMR (400 Hz, DMSO-d6): δ 4.60 (d, 2H, J = 4.4 Hz), 5.28 (t, 1H, J = 5.2 Hz), 6.60 (br s, 2H), 7.49 (br s, 2H), 7.65–7.66 (m, 1H), 7.74 (s, 1H), 8.11 (s, 1H), 12.20 (br s, 1H). HRMS (ESI) calculated for C14H9N5O [M + H]+ 264.0891, found 264.0894. HPLC: tR = 8.62 min (method A).
3-(2-Amino-5-cyano-7H-pyrrolo[2,3-d]pyrimidin-4-yl)benzenesulfonamide (57)
5 (102 mg g, 0.37 mmol), 3-(aminosulphonyl)benzene boronic acid (122 mg, 0.61 mmol), and Pd-118 (24 mg, 0.037 mmol) were suspended in a mixture of IPA/H2O (3:1.5 mL) and degassed. t-Butylamine (0.20 mL, 1.90 mmol) was added, and the mixture was irradiated at 160 °C for 40 min. The resulting solution was diluted with EtOH (8 mL) and KOH (2 pellets) were added, and the mixture was heated to 90 °C for 24 h. The reaction mixture was then concentrated under reduced pressure, and the resulting mixture was suspended in water, pH adjusted to pH 5.7, extracted into EtOAc (2 × 100 mL), and dried over MgSO4. The resulting solid was purified by column chromatography (100% hexane–100% ethyl acetate) to afford the title compound 57 as a pale solid (43.2 mg, 0.138 mmol, 38%). 1H NMR (400 Hz, DMSO-d6): δ 6.74 (br s, 2H), 7.44 (br s, 2H), 7.74 (t, 1H, J = 7.8 Hz), 8.00 (t, 2H, J = 7.2 Hz), 8.17 (s, 1H), 8.23 (br s, 1H), 12.45 (br s, 1H). HRMS (ESI) calculated for C13H9N6S [M + H]+ 313.0513, found 313.0516. HPLC: tR = 8.44 min (method A) (93.47%).
2-Amino-4-(2-hydroxyphenyl)-77H-pyrrolo[2,3-d]pyrimidine-5-carbonitrile (58)
5 (117 mg, 0.36 mmol), 2-hydroxybenzene boronic acid (94 mg, 0.68 mmol), and Pd-118 (26 mg, 0.040 mmol) were suspended in a mixture of IPA/H2O (3:1.5 mL) and degassed. t-Butylamine (0.20 mL, 1.90 mmol) was added, and the mixture was irradiated at 160 °C for 40 min. The resulting solution was diluted with EtOH (8 mL) and KOH (2 pellets) were added and the mixture was heated to 90 °C for 24 h. The reaction mixture was then concentrated under reduced pressure, and the resulting mixture was suspended in water, pH adjusted to pH 5.3, extracted into EtOAc (2 × 100 mL), and dried over MgSO4. The mixture was then concentrated under reduced pressure, triturated with diethyl ether, and purified by column chromatography (100% hexane–100% ethyl acetate). The resulting solid was triturated with diethyl ether, filtered under reduced pressure, and dried to afford the title compound 58 as a bright-yellow solid (30.7 mg, 0.122 mmol, 34%). 1H NMR (400 Hz, DMSO-d6): δ 6.99 (t, 1H, J = 7.5 Hz), 7.07 (d, 1H, J = 8.0 Hz), 7.46 (d of t, 1H, J = 1.5, 7.0 Hz), 7.56 (d of d, 1H, J = 2.0, 1.5 Hz), 8.36 (s, 1H). HRMS (ESI) calculated for C15H10N2O2 [M + H]+ 250.0737, found 250.0735. HPLC: tR = 10.11 min (method A).
2-Amino-4-(2-fluorophenyl)-7H-pyrrolo[2,3-d]pyrimidine-5-carbonitrile (59)
5 (100 mg, 0.31 mmol), Pd-118 (20 mg, 0.03 mmol), t-butylamine (160 μL, 1.55 mmol), and 2-fluorophenylboronic acid (69 mg, 0.5 mmol) were suspended in a 2:1 mixture i-PrOH/H2O (4.5 mL) and were irradiated with microwaves at 160 °C for 40 min. Once cooled to room temperature, the reaction mixture was concentrated under reduced pressure. To this was added EtOH (5 mL) and KOH (2 pellets) and the reaction mixture was refluxed t at 90 °C for 20 h. The mixture was then concentrated under reduced pressure, and the crude material was then suspended in H2O and adjusted to pH 5 with 1 M HCl. The solution was then extracted with EtOAc (2 × 50 mL); the organic layer was adsorbed on silica gel and concentrated under reduced pressure. Chromatographic purification (EtOAc/Hex: 50% 4CV, 50–75% 6CV, 75% 4CV, 75–100% 7CV, 100% 6CV) yielded a beige solid that was triturated with Et2O to afford the title compound 59 and as a beige solid (16 mg g, 0.06 mmol, 20%). 1H NMR (400 MHz, DMSO-d6): δ 6.68 (s, 2H), 7.35 (t, J = 8.3 Hz, 2H), 7.58 (t, J = 7.7 Hz, 2H), 8.06 (s, 1H), 12.34 (s, 1H). HRMS (ESI) calculated for C13H9N5F [M + H]+ 254.0837 found 254.0831.
2-(2-Amino-5-cyano-7H-pyrrolo[2,3-d]pyrimidin-4-yl)benzamide (60)
5 (104 mg, 0.32 mmol), 2-(aminocarbonyl)benzne boronic acid (87 mg, 0.53 mmol), and Pd-118 (21 mg, 0.032 mmol) were suspended in a mixture of IPA/H2O (3:1.5 mL) and degassed. t-Butylamine (0.18 mL, 1.71 mmol) was added, and the mixture was irradiated at 160 °C for 40 min. The resulting solution was diluted with EtOH (8 mL) and KOH (2 pellets) were added and the mixture was heated to 90 °C for 24 h. The reaction mixture was then concentrated under reduced pressure, and the resulting mixture was suspended in water, pH adjusted to pH 5.3, extracted into EtOAc (2 × 100 mL), and dried over MgSO4. The mixture was then concentrated under reduced pressure, triturated with methanol, and purified by column chromatography (100% hexane–10% methanol/ethyl acetate). The resulting solid was recrystallized from dichloromethane and diethyl ether and dried to afford the title compound 60 as a bright-yellow solid (3.5 mg, 0.0126 mmol, 3.9%). 1H NMR (400 Hz, DMSO-d6): δ 6.48 (br s, 2H), 7.23 (s, 1H), 7.40–7.42 (m, 1H), 7.51–7.60 (m, 2H), 7.71 (d, 1H, J = 2.2 Hz), 7.73 (br s, 1H), 7.96 (s, 1H), 12.20 (br s, 1H). HRMS (ESI) calculated for C14H9N6O [M + H]+ 277.0843, found 277.0851. HPLC: tR = 7.86 min (method A) (88.64%).
2-Amino-4-(2-(hydroxymethyl)phenyl)-7H-pyrrolo[2,3-d]pyrimidine-5-carbonitrile (61)
5 (110 mg, 0.40 mmol), 2-hydroxymethylbenzne boronic acid (133 mg, 0.87 mmol), and Pd-118 (31 mg, 0.048 mmol) were suspended in a mixture of IPA/H2O (3:1.5 mL) and degassed. t-Butylamine (0.21 mL, 2.0 mmol) was added, and the mixture was irradiated at 160 °C for 40 min. Organics were combined and concentrated under reduced pressure. The resulting solid was triturated with diethyl ether, filtered, and dried to afford the title compound 61 as an off-white solid (51 mg g, 0.193 mmol, 49%). 1H NMR (400 Hz, DMSO-d6): δ 4.55 (d, 2H, J = 5.6 Hz), 5.09 (t, 1H, J = 5.8 Hz), 6.64 (br s, 2H), 7.37–7.41 (m, 2H), 7.48 (d of t, 1H, J = 2.0, 7.2 Hz), 7.61 (d, 1H, J = 7.6 Hz), 8.05 (s, 1H), 12.31 (br s, 1H). HRMS (ESI) calculated for C14H10N5O [M + H]+ 264.0891, found 264.0891.
2-(2-Amino-5-cyano-7H-pyrrolo[2,3-d]pyrimidin-4-yl)benzenesulfonamide (62)
5 (130 mg, 0.405 mmol), 2-(sulfamoylphenyl)boronic acid (130 mg, 0.65 mmol), and Pd-118 (28 mg, 0.043 mmol) was suspended in a mixture of IPA/H2O (3 mL:1.5 mL), and the mixture was degassed under nitrogen. t-Butylamine (0.25 mL, 2.4 mmol) was added, and the reaction mixture was degassed under nitrogen. The reaction mixture was irradiated with microwaves at 160 °C for 40 min. Once cooled to room temperature, the mixture was diluted with EtOH (8 mL) and KOH (2 pellets) added. The reaction mixture was heated to 95 °C for 20 h. The reaction mixture was then concentrated under reduced pressure. The residue was suspended in water, and the mixture was pH adjusted to pH 5.5 and extracted with ethyl acetate and concentrated under reduced pressure. The resulting solid was triturated with diethyl ether and filtered off. Further purification was required, and this was carried out using HPLC to afford the title compound 62 (6.8 mg). 1H NMR (400 MHz, DMSO-d6): δ 6.82 (br s, 2H), 7.16 (br s, 2H), 7.62 (m, 1H), 7.73 (m, 2H), 8.04 (m, 1H), 8.06 (d, J = 5.0 Hz, 1H), 12.39 (br s, 1H). HRMS (ESI) calculated for C13H11N6O2S [M + H]+ 315.0659, found 315.0656.
4-(2-Amino-7H-pyrrol[2,3-d]pyrimidin-4-yl)benzenesulfonamide (64)
4-Chloro-7H-pyrrolo[2,3-d]pyrimdin-2-amine (70 mg, 0.42 mmol), 4-sulphamoylbenzne boronic acid (133 mg, 0.66 mmol), and Pd-118 (37 mg, 0.057 mmol) were suspended in a mixture of IPA/H2O (3:1.5 mL) and degassed. t-Butylamine (0.22 mL, 2.09 mmol) was added, and the mixture was irradiated at 160 °C for 40 min. Once cooled to room temperature the reaction mixture was diluted with ethyl acetate and concentrated under reduced pressure. The resulting solid was purified by column chromatography (100% hexane–100% ethyl acetate), triturated with diethyl ether, filtered, and dried to afford the title compound 64 as a pale-yellow solid (23.7 mg, 0.082 mmol, 20%). 1H NMR (400 Hz, DMSO-d6): δ 6.23 (br s, 2H), 6.61 (d, 1H, J = 2.0 Hz), 7.17 (d, 1H, J = 2.8 Hz), 7.48 (br s, 2H), 7.98 (d, 2H, J = 8.4 Hz), 8.2 (d, 2H, J = 8.4 Hz), 11.36 (br s, 1H). HRMS (ESI) calculated for C12H11N5O2S [M + H]+ 290.0706, found 290.0703. HPLC: tR = 8.45 min (method A).
4-(7H-Pyrrolo[2,3-d]pyrimidin-4-yl)benzenesulfonamide (65)
6-Chloro-7-deazapurine (233 mg, 1.45 mmol), 4-sulfamoylbenzene boronic acid (408 mg, 2.03 mmol), and 2-amino-2-methylpropane (0.61 mL, 5.80 mmol) were dissolved in 2-propanol (7.5 mL) and water (3.75 mL), and the solution degassed with argon. Dichloro[1,1′ bis(di-tert-butylphosphino)]ferrocene palladium(II) (94.4 mg, 0.145 mmol) was added, and the mixture was heated at 160 °C for 40 min in a microwave. The reaction mixture was concentrated under reduced pressure and purified by flash column chromatography (silica gel with EtOAc (100%)–50% MeOH/EtOAc) to give the crude product. Trituration of the crude product with DCM/MeOH (9:1) gave the title compound 65 as a yellow solid (155 mg, 0.57 mmol, 39%). 1H NMR (400 MHz, DMSO-d6): 6.95 (d, 1H, J = 2.0 Hz), 7.52 (s, 2H), 7.27 (dd, 1H, J = 2.8, 2.0 Hz), 8.04 (d, 2H, J = 8.4 Hz), 8.37 (d, 2H, J = 8.4 Hz), 8.89 (s, 1H), 12.41 (br s, 1H). HRMS (ESI) calculated for C12H10N4O2S [M + H]+ 275.0603, found 275.0597.
4-(5-Cyano-7H-pyrrolo[2,3-d]pyrimidin-4-yl)benzenesulfonamide (66)
4-Chloro-7H-pyrrolo[2,3-d]pyrimidine-5-carbonitrile (95 mg, 0.53 mmol), 4-sulfamoylphenylboronic acid (171 mg, 0.85 mmol), and Pd-118 (35 mg, 0.05) was suspended in a mixture of IPA/H2O (3 mL:1.5 mL), and the mixture was degassed under nitrogen. t-Butylamine (0.28 mL, 2.7 mmol) was added, and the reaction mixture was degassed under nitrogen. The reaction mixture was irradiated with microwaves at 160 °C for 1 h. The reaction mixture was cooled to room temperature, and the crude material was suspended in methanol, adsorbed onto silica gel, and concentrated under reduced pressure. Purification was achieved using column chromatography (100% hexane–50/50 hexane/ethyl acetate–100% ethyl acetate) to afford the title compound 66 as a beige solid (76 mg, 0.25 mmol, 48%). 1H NMR (400 MHz, DMSO-d6): δ 7.58 (s, 2H), 8.03 (d, J = 8.0 Hz, 2H), 8.08 (d, J = 8.3 Hz, 2H), 8.73 (s, 1H), 9.06 (s, 1H), 13.52 (s, 1H). HRMS (ESI) calculated for C13H10O2N5S [M + H]+ 300.0550, found 300.0546.
4-(Piperidin-1-yl)-7H-pyrrolo[2,3-d]pyrimidin-2-amine (67)
4-Chloro-7H-pyrrolo[2,3-d]pyrimidin-2-amine (80 mg, 4.76 mmol), NEt3 (0.2 mL, 1.43 mmol), and piperidine (0.15 mL, 1.52 mmol) was suspended in 1,4-dioxane (2.5 mL), which was degassed under nitrogen. The reaction mixture was irradiated with microwaves at 200 °C for 20 min. The resulting mixture was diluted with EtOAc (10 mL) and concentrated under reduced pressure. Purification by column chromatography (100% EtOAc–10% MeOH/EtOAc) followed by trituration with diethyl ether and filtration afforded the title compound 67 as a white solid (8 mg, 0.04 mmol, 0.8%).1H NMR (400 Hz, DMSO-d6): δ 1.56 (m, 4H), 1.64 (m, 2H), 3.75 (t, J = 5.0 Hz, 4H), 5.49 (br s, 2H), 6.31 (dd, J = 2.0, 3.5 Hz, 1H), 6.72 9dd, J = 2.5, 3.5 Hz, 1H), 10.80 (br s, 2H). HRMS (ESI) calculated for C11H16N5 [M + H]+ 218.1400, found 218.1398.
N4-Cyclohexyl-7H-pyrrolo[2,3-d]pyrimidine-2,4-diamine (68)
To 2-amino-3H-pyrrolo[2,3-d]pyrimidin-4(7H)-one (110 mg, 0.73 mmol), BOP (44 mg, 1.00 mmol), and DBU (0.22 mL, 1.47 mmol) in DMF/DMSO (1:1, 4 mL) was added cyclohexylamine (0.25 mL, 2.19 mmol) at room temperature. The reaction mixture was stirred at room temperature for 4 d then heated to 60 °C for 2 h. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The resulting residue was triturated with water and extracted into EtOAc. The organic layer was concentrated under reduced pressure to afford the title compound 68 as an yellow solid (50 mg, 0.22 mmol, 30%). 1H NMR (400 Hz, DMSO-d6): δ 1.14 (m, 1H), 1.35 (m, 4H), 1.63 (m, 2H), 1.76 (m, 2H), 1.90 (m, 2H), 6.49 (br s, 2H), 6.63 (s, 1H), 6.85 (s, 1H), 7.36 (br s, 1H), 11.71 (br s, 1H). LRMS (ESI): calculated for C12H17N5 [M + H]+ 232.29, found 232.27.
(1R,4R)-4-(2-Amino-7H-pyrrolo[2,3-d]pyrimidin-4-ylamino)cyclohexanol (69)
Prepared according to procedure B using 4-chloro-7H-pyrrolo[2,3-d]pyrimidin-2-amine. Chromatographic purification (Biotage SP4, 10 g cartridge, solvent system: MeOH/DCM; gradient 5% 4CV, 0–20% 10CV, 20% 4CV) yielded 69 (94 mg, 64%) as a fluffy white solid; mp 178–180 °C (d). 1H NMR (400 MHz, DMSO-d6) δ: 1.20–1.36 (m, 4H), 1.84–1.90 (m, 4H), 3.37–3.44 (m, 1H), 3.91–4.01 (m, 1H), 4.54 (d, J = 4.4 Hz, 1H), 5.38 (bs, 2H), 6.31–6.32 (m, 1H), 6.59–6.60 (m, 1H), 6.63 (bs, 1H), 10.57 (bs, 1H). 13C NMR (100 MHz, DMSO-d6) δ: 31.4, 35.0, 48.0, 69.1, 96.5, 99.4, 117.0, 153.2, 156.6, 160.3. HRMS (ESI) calculated for C12H18ON5 [M + H]+ 248.1506, found 248.1504. HPLC tR = 7.40 min (method C).
N4-Cyclohexyl-5-(trifluoromethyl)-7H-pyrrolo[2,3-d]pyrimidine-2,4-diamine (70)
Prepared according to procedure A using 4-chloro-5-(trifluoromethyl)-7H-pyrrolo[2,3-d]pyrimidin-2-amine (100 mg, 0.42 mmol). Chromatographic purification (Biotage SP4, 10 g cartridge, solvent system: EtOAc/Hex; gradient 15% 4CV; 15–75% 10CV; 75% 6CV) yielded a brown oil. The oil was dissolved in Et2O (1 mL) and poured dropwise into Hex (6 mL). The precipitate was filtered off to yield 70 (30 mg, 24%) as a light-brown solid; mp 202–204 °C (d). 1H NMR (400 MHz, DMSO-d6) δ: 1.11–1.20 (m, 3H), 1.25–1.37 (m, 2H), 1.58–1.62 (m, 1H), 1.70–1.74 (m, 2H), 1.88–1.91 (m, 2H), 3.70–3.80 (m, 1H), 6.91 (d, J = 7.3 Hz, 1H), 7.09 (bs, 2H), 7.99 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ: 25.3, 25.9, 33.1, 49.0, 59.3, 100.0, 105.5, 121.8, 123.9, 147.6, 151.6. HRMS (ESI) calculated for C13H16N5F3 [M – H]− 298.1285, found 298.1283. HPLC tR = 10.76 min (method A).
2-Amino-4-((1R,4R)-4-hydroxycyclohexylamino)-7H-pyrrolo[2,3-d]pyrimidine-5-carboxamide (71)
5 (100 mg, 0.56 mmol) and trans-4-aminocyclohexanol (124 mg, 1.08 mmol) were suspended in n-BuOH (4 mL) and refluxed for 16 h. After cooling to room temperature H2O (2 mL) and KOH (2 pellets) were added and the reaction mixture was heated for 40 min at 150 °C in the microwave oven. The mixture was neutralized using 6 M HCl and concentrated in vacuo. The residue was triturated with H2O (4 mL) and dried in vacuo. The remaining solid was then triturated with Et2O (2 × 10 mL) and dried in vacuo to afford 71 (91 mg, 87% yield) as a beige solid; mp >300 °C (d). FTIR (neat): 3441.8, 3352.4, 3130.0, 2938.5, 2854.5, 1628.5, 1593.6, 1507.5, 1451.01, 1417.06 cm–1. 1H NMR (400 MHz, DMSO-d6) δ: 1.18–1.32 (m, 4H), 1.83–1.85 (m, 2H), 1.95–1.98 (m, 2H), 3.43–3.53 (m, 1H), 3.88–3.96 (m, 1H), 4.54 (d, J = 2.6 Hz, 1H), 5.56 (bs, 2H), 7.00 (bs, 1H), 7.50 (s, 1H), 7.65 (bs, 1H), 9.37 (d, J = 7.7 Hz, 1H), 11.16 (bs, 1H). 13C NMR (100 MHz, DMSO-d6) δ: 30.7, 33.9, 47.4, 68.4, 95.0, 111.1, 122.1, 154.0, 157.0, 161.1, 168.0. HRMS (ESI) calculated for C13H19O2N6 [M + H]+ 291.1564, found 291.1563. HPLC tR = 6.64 min (method C).
IKK Assays
IKKα and IKKβ inhibitory activity was determined using a dissociation enhanced ligand fluorescent immunoassay (DELFIA) based on the protocol of HTScan IKKβ kinase assay (Cell Signaling Technology, USA).
Recombinant IKKα or IKKβ, 37 nM (Millipore, Dundee, UK), was incubated with IκB-α (Ser32) (New England Biolabs, Hitchin, UK), biotinylated peptide substrate (0.375 μM), and 40 μM ATP in assay buffer (40 mM Tris-HCl (pH 7.5), 20 mM MgCl2, EDTA 1 mM, DTT 2 mM, and BSA 0.01 mg/mL) in a V-well 96-well plate in the presence and absence of test compound. The assay plate was incubated for 60 min at 30 °C, after which the kinase reaction was quenched by the addition of 50 mM EDTA, pH 8. The resulting mixture was transferred to a streptavidin-coated 96-well plate (PerkinElmer, Beaconsfield, UK) and incubated for 1 h at 30 °C to immobilize the substrate peptide. After three washes with wash buffer (0.01 M phosphate buffered saline (PBS), 0.05% Tween-20, pH 7.4), a primary antibody against the phosphorylated substrate (phospho-IκB-α) (Ser32/36) (5A5) mouse mAb (New England Biolabs, Hitchin, UK) (1:1000 dilution with 1% bovine serum albumin (BSA) in wash buffer) was added and incubated at 37 °C for 2 h.
After a further three washes, a secondary europiated antibody (Eu–N1 labeled antimouse IgG, (PerkinElmer, Beaconsfield, UK) diluted 1:500 in 1% BSA/wash buffer) was added and incubated at 30 °C for 30 min. After a further five washes, DELFIA enhancement solution (PerkinElmer, Beaconsfield UK) was added and allowed to incubate for 10 min at room temperature, protected from light, to facilitate the chemifluorescent detection. The relative fluorescence units (RFU) signal were measured on a Wallac Victor 1420 multilabel counter (PerkinElmer, Beaconsfield, UK) in time-resolved fluorescence mode. The counter was set at an excitation wavelength of 340 nm with a 400 μs delay before detecting emitted light at 615 nm. The apparent Ki of the phosphorylated substrate was calculated for each compound using the Cheng–Prusoff equation.
Cell Culture
U2OS cells were cultured with McCoy’s 5A modified medium and MEF were cultured with DEME medium containing 10% (v/v) FCS and medium was changed every 2 d thereafter until cells became confluent. Cells were incubated at 37 °C in humidified air with 5% CO2 and rendered quiescent by serum deprivation for 24 h prior to stimulation in serum-free medium.
Western Blot Analysis
Whole cell lysates were prepared from U2OS and status of IκBα, p65, p-p65 (Ser536), and p-p100 (Ser 866/870) assessed by Western blotting as described in Mackenzie et al. (2007).46
NFκB Transcriptional Activity Assays
An adenoviral vector encoding NFκB-luciferase (Adv.NFκB-Luc) was purchased from Vector Biolabs (University of Pennsylvania, Philadelphia, US). Large-scale production of high titer recombinant adenovirus was performed by routine methods.47 IKKβ knockout mouse embryonic fibroblasts (MEFs; kindly provided by Prof. I. Verma, UCSD, USA) were counted when approximately 60–70% confluent in T75 cm2 flasks and infected with adenovirus up to 300 pfu/cell–1 for 24 h in 10% DMEM. Cells were then lifted from the flasks and plated into 96-well clear bottom, black luciferase plates and allowed to settle for 24 h before serum starved overnight. Cells were then stimulated with the appropriate agonists for the indicated times and reactions terminated by addition of lysis buffer containing 0.2 mM luciferin substrate. Relative light units (RLU) were measured using a Trilux microbeta counter (luminometer).
Kinase Profiling
Kinase profiling of compound 48 was outsourced to Millipore, specifically the Kinase Profiler Customer Service for single concentration studies using duplicate assay points.
Acknowledgments
Financial support was provided by Cancer Research UK through grant no. C7599/A9336 and Prostate Cancer UK, grant no. PG12-27
Glossary
Abbreviations Used
- CDK2
cyclin dependent kinase 2
- DELFIA
dissociation-enhanced lanthanide fluorescence immunoassay
- FCS
fecal calf serum
- GK
gatekeeper
- G-loop
glycine-rich loop
- IkBα
inhibitory kappa B α
- IKK
inhibitory kappa B kinase
- IL-1β
interleukin-1β
- MD
molecular dynamics
- MEF
mouse embryonic fibroblasts
- NAM
noraristeromycin
- NF-kB
nuclear factor kappa-light-chain-enhancer of activated B cells
- RLU
relative light units
- TNFα
tumor necrosis factor α
- U2OS
human bone osteosarcoma cells
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.7b00484.
IKKα homology model (PDB)
IKKα with compound 4(PDB)
IKKα with compound 9(PDB)
IKKα with compound 43(PDB)
IKKα with compound 47(PDB)
IKKα with compound 48(PDB)
IKKα with compound 50(PDB)
Molecular formula strings with associated IKK biochemical inhibitory data in SMILES format (CSV)
The inhibitory kinome profile for compound 48 against 40 kinases (XLS)
The authors declare no competing financial interest.
Supplementary Material
References
- Karin M. Nuclear factor-kappa B in cancer development and progression. Nature 2006, 441, 431–436. 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- Xiao C. C.; Ghosh S.. NF-kappa B, an evolutionarily conserved mediator of immune and inflammatory responses. In Mechanisms of Lymphocyte Activation and Immune Regulation X: Innate Immunity, Gupta S.; Paul W. E., Steinman R., Eds.; Springer: Boston, MA, 2005; Vol. 560, pp 41–45. [DOI] [PubMed] [Google Scholar]
- Häcker H.; Karin M. Regulation and function of IKK and IKK-related kinases. Science’s STKE 2006, 2006, re13. 10.1126/stke.3572006re13. [DOI] [PubMed] [Google Scholar]
- Xiao G. T.; Rabson A. B.; Young W.; Qing G. L.; Qu Z. X. Alternative pathways of NF-kappa B activation: A double-edged sword in health and disease. Cytokine Growth Factor Rev. 2006, 17, 281–293. 10.1016/j.cytogfr.2006.04.005. [DOI] [PubMed] [Google Scholar]
- Hu Y. L.; Baud V.; Oga T.; Kim K. I. I.; Yoshida K.; Karin M. IKKalpha controls formation of the epidermis independently of NF-kappa B. Nature 2001, 410, 710–714. 10.1038/35070605. [DOI] [PubMed] [Google Scholar]
- Karin M.; Greten F. R. NF kappa B: Linking inflammation and immunity to cancer development and progression. Nat. Rev. Immunol. 2005, 5, 749–759. 10.1038/nri1703. [DOI] [PubMed] [Google Scholar]
- Sizemore N.; Lerner N.; Dombrowski N.; Sakurai H.; Stark G. R. Distinct roles of the IkappaB kinase alpha and beta subunits in liberating nuclear factor kappa B (NF-kappa-B) from IkappaB and in phosphorylating the p65 subunit of NF-kappa B. J. Biol. Chem. 2002, 277, 3863–3869. 10.1074/jbc.M110572200. [DOI] [PubMed] [Google Scholar]
- Park K. J.; Krishnan V.; O’Malley B. W.; Yamamoto Y.; Gaynor R. B. Formation of an IKK alpha-dependent transcription complex is required for estrogen receptor-mediated gene activation. Mol. Cell 2005, 18, 71–82. 10.1016/j.molcel.2005.03.006. [DOI] [PubMed] [Google Scholar]
- Wen D. Y.; Nong Y. H.; Morgan J. G.; Gangurde P.; Bielecki A.; DaSilva J.; Keaveney M.; Cheng H.; Fraser C.; Schopf L.; Hepperle M.; Harriman G.; Jaffee B. D.; Ocain T. D.; Xu Y. J. A selective small molecule IkappaB kinase beta inhibitor blocks nuclear factor kappa B-mediated inflammatory responses in human fibroblast-like synoviocytes, chondrocytes, and mast cells. J. Pharmacol. Exp. Ther. 2006, 317, 989–1001. 10.1124/jpet.105.097584. [DOI] [PubMed] [Google Scholar]
- Burke J. R.; Pattoli M. A.; Gregor K. R.; Brassil P. J.; MacMaster J. F.; McIntyre K. W.; Yang X. X.; Iotzova V. S.; Clarke W.; Strnad J.; Qiu Y. P.; Zusi F. C. BMS-345541 is a highly selective inhibitor of IkappaB kinase that binds at an allosteric site of the enzyme and blocks NF-kappa B-dependent transcription in mice. J. Biol. Chem. 2003, 278, 1450–1456. 10.1074/jbc.M209677200. [DOI] [PubMed] [Google Scholar]
- Kim B. H.; Lee J. Y.; Seo J. H.; Lee H. Y.; Ryu S. Y.; Ahn B. W.; Lee C. K.; Hwang B. Y.; Han S. B.; Kim Y. Artemisolide is a typical inhibitor of IkappaB kinase beta targeting cysteine-179 residue and down-regulates NF-kappa B-dependent TNF-alpha expression in LPS-activated macrophages. Biochem. Biophys. Res. Commun. 2007, 361, 593–598. 10.1016/j.bbrc.2007.07.069. [DOI] [PubMed] [Google Scholar]
- Podolin P. L.; Callahan J. F.; Bolognese B. J.; Li Y. H.; Carlson K.; Davis T. G.; Mellor G. W.; Evans C.; Roshak A. K. Attenuation of murine collagen-induced arthritis by a novel, potent, selective small molecule inhibitor of IkappaB kinase 2, TPCA-1 (2-(aminocarbonyl)amino-5-(4-fluorophenyl)-3-thiophenecarboxamide), occurs via reduction of proinflammatory cytokines and antigen-induced T cell proliferation. J. Pharmacol. Exp. Ther. 2005, 312, 373–381. 10.1124/jpet.104.074484. [DOI] [PubMed] [Google Scholar]
- Strnad J.; Burke J. R. IkappaB kinase inhibitors for treating autoimmune and inflammatory disorders: potential and challenges. Trends Pharmacol. Sci. 2007, 28, 142–148. 10.1016/j.tips.2007.01.005. [DOI] [PubMed] [Google Scholar]
- Pasparakis M. Regulation of tissue homeostasis by NF-kappa B signalling: implications for inflammatory diseases. Nat. Rev. Immunol. 2009, 9, 778–788. 10.1038/nri2655. [DOI] [PubMed] [Google Scholar]
- Li Q. T.; Van Antwerp D.; Mercurio F.; Lee K. F.; Verma I. M. Severe liver degeneration in mice lacking the IkappaB kinase 2 gene. Science 1999, 284, 321–325. 10.1126/science.284.5412.321. [DOI] [PubMed] [Google Scholar]
- Llona-Minguez S.; Baiget J.; Mackay S. P. Small-molecule inhibitors of IκB kinase (IKK) and IKK-related kinases. Pharm. Pat. Anal. 2013, 2, 481–498. 10.4155/ppa.13.31. [DOI] [PubMed] [Google Scholar]
- Asamitsu K.; Yamaguchi T.; Nakata K.; Hibi Y.; Victoriano A. F. B.; Imai K.; Onozaki K.; Kitade Y.; Okamoto T. Inhibition of human immunodeficiency virus type 1 replication by blocking IkappaB Kinase with noraristeromycin. J. Biochem. 2008, 144, 581–589. 10.1093/jb/mvn104. [DOI] [PubMed] [Google Scholar]
- Palayoor S. T.; Youmell M. Y.; Calderwood S. K.; Coleman C. N.; Price B. D. Constitutive activation of IkappaB kinase alpha and NF-kappa B in prostate cancer cells is inhibited by ibuprofen. Oncogene 1999, 18, 7389–7394. 10.1038/sj.onc.1203160. [DOI] [PubMed] [Google Scholar]
- Ammirante M.; Luo J. L.; Grivennikov S.; Nedospasov S.; Karin M. B-cell-derived lymphotoxin promotes castration-resistant prostate cancer. Nature 2010, 464, 302–U187. 10.1038/nature08782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Neriah Y.; Karin M. Inflammation meets cancer, with NF-kappa B as the matchmaker. Nat. Immunol. 2011, 12, 715–723. 10.1038/ni.2060. [DOI] [PubMed] [Google Scholar]
- Luo J. L.; Tan W.; Ricono J. M.; Korchynskyi O.; Zhang M.; Gonias S. L.; Cheresh D. A.; Karin M. Nuclear cytokine-activated IKKalpha controls prostate cancer metastasis by repressing Maspin. Nature 2007, 446, 690–694. 10.1038/nature05656. [DOI] [PubMed] [Google Scholar]
- Hao L.; Rizzo P.; Osipo C.; Pannuti A.; Wyatt D.; Cheung L. W. K.; Sonenshein G.; Osborne B. A.; Miele L. Notch-1 activates estrogen receptor-alpha-dependent transcription via IKKalpha in breast cancer cells. Oncogene 2010, 29, 201–213. 10.1038/onc.2009.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y. X.; Luo J. L.; Karin M. I kappa B kinase a kinase activity is required for self-renewal of ErbB2/Her2-transformed mammary tumor-initiating cells. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 15852–15857. 10.1073/pnas.0706728104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y. X.; Bonizzi G.; Seagroves T. N.; Greten F. R.; Johnson R.; Schmidt E. V.; Karin M. IKKalpha provides an essential link between RANK signaling and cyclin D1 expression during mammary gland development. Cell 2001, 107, 763–775. 10.1016/S0092-8674(01)00599-2. [DOI] [PubMed] [Google Scholar]
- Liou G. Y.; Doppler H.; Necela B.; Krishna M.; Crawford H. C.; Raimondo M.; Storz P. Macrophage-secreted cytokines drive pancreatic acinar-to-ductal metaplasia through NF-kappa B and MMPs. J. Cell Biol. 2013, 202, 563–577. 10.1083/jcb.201301001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storz P. Targeting the alternative NF-kappa B pathway in pancreatic cancer: a new direction for therapy?. Expert Rev. Anticancer Ther. 2013, 13, 501–504. 10.1586/era.13.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doppler H.; Liou G. Y.; Storz P. Downregulation of TRAF2 mediates NIK-induced pancreatic cancer cell proliferation and tumorigenicity. PLoS One 2013, 8, e53676. 10.1371/journal.pone.0053676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S. P.; Misquitta Y. R.; Olland A.; Johnson M. A.; Kelleher K. S.; Kriz R.; Lin L. L.; Stahl M.; Mosyak L. Crystal Structure of a human IkappaB Kinase beta asymmetric dimer. J. Biol. Chem. 2013, 288, 22758–22767. 10.1074/jbc.M113.482596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrestha S.; Bhattarai B. R.; Cho H.; Choi J. K.; Cho H. PTP1B inhibitor ertiprotafib is also a potent inhibitor of IkappaB kinase beta (IKK-beta). Bioorg. Med. Chem. Lett. 2007, 17, 2728–2730. 10.1016/j.bmcl.2007.03.001. [DOI] [PubMed] [Google Scholar]
- Klepper F.; Polborn K.; Carell T. Robust synthesis and crystal-structure analysis of 7-cyano-7-deazaguanine (PreQ(0) base) and 7-(aminomethyl)-7-deazaguanine (PreQ(1) base). Helv. Chim. Acta 2005, 88, 2610–2616. 10.1002/hlca.200590201. [DOI] [Google Scholar]
- Llona-Minguez S.; Mackay S. P. Stereoselective synthesis of carbocyclic analogues of the nucleoside Q precursor (PreQ(0)). Beilstein J. Org. Chem. 2014, 10, 1333–1338. 10.3762/bjoc.10.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler K.; Schenck G.; Krockow E. W.; Siebert A.; Wenz A.; Weber H. The synthesis of cantharidin. Justus Liebigs Ann. Chem. 1942, 551, 1–79. 10.1002/jlac.19425510102. [DOI] [Google Scholar]
- Blades K.; Donohoe T. J.; Winter J. J. G.; Stemp G. A syn selective dihydroxylation of cyclic allylic trichloroacetamides using catalytic osmium tetroxide. Tetrahedron Lett. 2000, 41, 4701–4704. 10.1016/S0040-4039(00)00714-0. [DOI] [Google Scholar]
- Lam P. Y. S.; Clark C. G.; Li R. H.; Pinto D. J. P.; Orwat M. J.; Galemmo R. A.; Fevig J. M.; Teleha C. A.; Alexander R. S.; Smallwood A. M.; Rossi K. A.; Wright M. R.; Bai S. A.; He K.; Luettgen J. M.; Wong P. C.; Knabb R. M.; Wexler R. R. Structure-based design of novel guanidine/benzamidine mimics: Potent and orally bioavailable factor Xa inhibitors as novel anticoagulants. J. Med. Chem. 2003, 46, 4405–4418. 10.1021/jm020578e. [DOI] [PubMed] [Google Scholar]
- Orsini P.; Menichincheri M.; Vanotti E.; Panzeri A. Highly efficient synthesis of 5-benzyl-3-aminoindazoles. Tetrahedron Lett. 2009, 50, 3098–3100. 10.1016/j.tetlet.2009.04.024. [DOI] [Google Scholar]
- Jones G.; Willett P.; Glen R. C. Molecular recognition of receptor-sites using a genetic algorithm with a description of desolvation. J. Mol. Biol. 1995, 245, 43–53. 10.1016/S0022-2836(95)80037-9. [DOI] [PubMed] [Google Scholar]
- Ghose A. K.; Herbertz T.; Pippin D. A.; Salvino J. M.; Mallamo J. P. Knowledge based prediction of ligand binding modes and rational inhibitor design for kinase drug discovery. J. Med. Chem. 2008, 51, 5149–5171. 10.1021/jm800475y. [DOI] [PubMed] [Google Scholar]
- Carbain B.; Paterson D. J.; Anscombe E.; Campbell A. J.; Cano C.; Echalier A.; Endicott J. A.; Golding B. T.; Haggerty K.; Hardcastle I. R.; Jewsbury P. J.; Newell D. R.; Noble M. E. M.; Roche C.; Wang L. Z.; Griffin R. J. 8-Substituted O-6-Cyclohexylmethylguanine CDK2 inhibitors: Using structure-based inhibitor design to optimize an alternative binding mode. J. Med. Chem. 2014, 57, 56–70. 10.1021/jm401555v. [DOI] [PubMed] [Google Scholar]
- Huang W. C.; Hung M. C. Beyond NF-kappa B activation: nuclear functions of IkappaB kinase alpha. J. Biomed. Sci. 2013, 20, 3. 10.1186/1423-0127-20-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leopizzi M.; Cocchiola R.; Milanetti E.; Raimondo D.; Politi L.; Giordano C.; Scandurra R.; Scotto d’Abusco A. IKKα inibition by a glucosamine derivative enhances Maspin expression in osteosarcoma cell line. Chem.-Biol. Interact. 2017, 262, 19–28. 10.1016/j.cbi.2016.12.005. [DOI] [PubMed] [Google Scholar]
- Solt L. A.; Madge L. A.; Orange J. S.; May M. J. Interleukin-1-induced NF-kappa B activation is NEMO-dependent but does not require IKK beta. J. Biol. Chem. 2007, 282, 8724–8733. 10.1074/jbc.M609613200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denk A.; Goebeler M.; Schmid S.; Berberich I.; Ritz O.; Lindemann D.; Ludwig S.; Wirth T. Activation of NF-kappa B via the I kappa B kinase complex is both essential and sufficient for proinflammatory gene expression in primary endothelial cells. J. Biol. Chem. 2001, 276, 28451–28458. 10.1074/jbc.M102698200. [DOI] [PubMed] [Google Scholar]
- Craft J. W.; Legge G. B. An AMBER/DYANA/MOLMOL phosphorylated amino acid library set and incorporation into NMR structure calculations. J. Biomol. NMR 2005, 33, 15–24. 10.1007/s10858-005-1199-0. [DOI] [PubMed] [Google Scholar]
- Talapatra S. K.; Anthony N. G.; Mackay S. P.; Kozielski F. Mitotic kinesin Eg5 overcomes inhibition to the phase I/II clinical candidate SB743921 by an allosteric resistance mechanism. J. Med. Chem. 2013, 56, 6317–6329. 10.1021/jm4006274. [DOI] [PubMed] [Google Scholar]
- Ryckaert J. P.; Ciccotti G.; Berendsen H. J. C. Numerical-integration of cartesian equations of motion of a system with constraints - molecular dynamics of N-alkanes. J. Comput. Phys. 1977, 23, 327–341. 10.1016/0021-9991(77)90098-5. [DOI] [Google Scholar]
- MacKenzie C. J.; Ritchie E.; Paul A.; Plevin R. IKK alpha and IKK beta function in TNF alpha-stimulated adhesion molecule expression in human aortic smooth muscle cells. Cell. Signalling 2007, 19, 75–80. 10.1016/j.cellsig.2006.06.001. [DOI] [PubMed] [Google Scholar]
- Nicklin S. A.; Baker A. H.. Simple methods for preparing recombinant adenoviruses for high-efficiency transduction of vascular cells. In Vascular Disease: Molecular Biology and Gene Therapy Protocols; Baker A. H., Ed.; Humana Press: Totowa, NJ, 1999; pp 271–283. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.