


Abstract
Measurement of steady-state rates of unwinding of double-stranded oligonucleotides by helicases is hampered due to rapid reannealing of the single-stranded DNA products. Including an oligonucleotide in the reaction mixture which can hybridize with one of the single strands can prevent reannealing. However, helicases bind to single-stranded DNA, therefore the additional oligonucleotide can sequester the enzyme, leading to slower observed rates for unwinding. To circumvent this problem, the oligonucleotide that serves as a trap was replaced with a strand of peptide nucleic acid (PNA). Fluorescence polarization was used to determine that a 15mer PNA strand does not bind to the bacteriophage T4 Dda helicase. Steady-state kinetic parameters of unwinding catalyzed by Dda were determined by using PNA as a trapping strand. The substrate consisted of a partial duplex with 15 nt of single-stranded DNA and 15 bp. In the presence of 250 nM substrate and 1 nM Dda, the rate of unwinding in the presence of the DNA trapping strand was 0.30 nM s–1 whereas the rate was 1.34 nM s–1 in the presence of the PNA trapping strand. PNA prevents reannealing of single-stranded DNA products, but does not sequester the helicase. This assay will prove useful in defining the complete kinetic mechanism for unwinding of oligonucleotide substrates by this helicase.
INTRODUCTION
DNA helicases are ubiquitous enzymes that use the energy from ATP hydrolysis to unwind double-stranded (ds) DNA (1–3). These enzymes play a vital role in DNA metabolism by providing single-stranded (ss) DNA during replication, recombination and repair. Defects in DNA helicases have been implicated in human genetic disorders such as Werner syndrome, Bloom Syndrome and Xeroderma pigmentosum (4). The kinetic mechanism for ATP coupled DNA unwinding has not been clearly elucidated for any member of this important class of enzymes, although progress has been made towards this goal in some systems (5–9).
The standard helicase assay utilizes a partially ds oligonucleotide substrate that is radiolabeled on one of the strands. Native polyacrylamide gel electrophoresis is used to resolve the ssDNA reaction products from duplex substrates. The ss products generated by a helicase can spontaneously hybridize to reform the substrate. The observed rate for hybridization is dependent on the concentration of ssDNA; therefore, unwinding rates can be determined under conditions in which the substrate concentration is held relatively low. For example, successful experiments have been performed in which the helicase concentration was in excess of substrate concentration when substrate was kept in the low nanomolar range (5,10–13). The rate of hybridization of products is slow under these conditions, and does not influence the rate of unwinding. However, the available biochemical assays are inadequate for the collection of data under steady-state conditions, due to the reannealing problem inherent to the unwinding assay (1). In the steady state, the substrate concentration exceeds that of the enzyme, therefore the rate of reannealing of the products can have a significant bearing on the observed rate of product formation (14). A ‘trapping strand’ can be included in the reaction mixture to prevent reannealing of ssDNA products. The trapping strand must be complementary to one of the ssDNA products, and must be added in excess of the substrate concentration. However, the presence of the trapping strand contributes an additional complication for measuring reaction velocities due to binding of the enzyme by the ssDNA trap. Hence, measurement of helicase-catalyzed DNA unwinding under steady-state conditions using oligonucleotide substrates has not been straightforward.
Peptide nucleic acid (PNA) is a polymer that has a neutral, pseudo-peptide backbone. It is made up of the normal purine and pyrimidine bases linked together via a N-(2-aminoethyl) glycine backbone. PNA readily hybridizes with DNA and RNA according to Watson–Crick base pairing rules (15). Due to its hybridization properties, PNA has been used as an antisense agent (16) and for the study of enzymes that manipulate nucleic acid, such as RNA polymerases (17), helicases (13,18,19) and DNA polymerases (20). PNA–DNA hybrids were reported to inhibit NS3, an RNA helicase from hepatitis C virus (19) and UL9, a DNA helicase from herpes simplex virus (18). However, the bacteriophage T4 Dda helicase was found to unwind DNA–PNA hybrids at similar rates as DNA–DNA hybrids (13). Here, we report a new application for PNA as a trapping strand to prevent reannealing of the products of helicase-catalyzed unwinding. Dda helicase is reportedly involved in early events in bacteriophage T4 replication (21) and is known to bind to other T4 proteins such as the recombinase, uvsX and the ssDNA binding protein, gp32 (22). Dda is a 5′→3′ helicase and a member of helicase superfamily I. Recent evidence suggests that it can function as a monomer during DNA unwinding (23). Apparent steady-state kinetic constants for unwinding by Dda were determined using PNA as a trap for ssDNA products. These methods will be useful for defining the complete kinetic mechanism for DNA unwinding catalyzed by Dda.
MATERIALS AND METHODS
Materials
PEP and PK/LDH were purchased from Sigma. HEPES, ATP and acrylamide were from Fisher. [γ-32P]ATP was purchased from PerkinElmer Life Sciences. T4 polynucleotide kinase was purchased from New England Biolabs. Oligonucleotides were purchased from Operon Technologies. Oligonucleotides were purified by preparative polyacrylamide gel electrophoresis and 5′-radiolabeled (13). PNA was prepared, purified by reverse-phase HPLC and characterized as described (24). Fluorescein labeling of PNA was performed on the resin by adding 5-(and 6-)carboxyfluorescein succinimidyl ester (Molecular Probes) at a ratio of 3 mg/µmol PNA in 30:1 DMF and DIPEA, respectively. PNA was heated to 65°C for 10 min prior to use. Dda helicase was over-expressed and purified as described (25,26).
Fluorescence polarization assay
Interaction between Dda and fluorescein-labeled 15mer DNA (F-DNA), fluorescein-labeled 15mer PNA (F-PNA) and blunt-end duplex F-DNA:PNA was followed by fluorescence polarization. Binding buffer consisted of 25 mM HEPES (pH 7.5), 2 mM β-mercaptoethanol, 0.1 mg/ml BSA, 0.1 mM EDTA (pH 8.0) and 10 mM KOAc. A series of 1 ml samples with either 0.1 nM F-DNA, F-PNA or F-DNA:PNA in binding buffer were incubated for 10 min at 25°C with Dda. The change in polarization (mP) was measured using a Beacon Fluorescence Polarization System (PanVera). The change in polarization was fit to a hyperbola to determine Kd values.
Helicase substrate and trapping strands
The sequence of the 30:15mer substrate used for unwinding assays with Dda was as follows: 5′-CTGACTGCGATCCGACTGTCCTGCATGATG-3′; 3′-GACAGGACGTACTAC-5′. A solution of gel-purified 30mer DNA (10 nmol) and gel-purified 15mer DNA complement (15 nmol) was prepared in 10 mM HEPES (pH 7.5) and 1 mM EDTA. The 15mer was added in 1.5-fold excess to ensure the formation of the 30:15mer, which can be separated readily from the free 15mer by PAGE. The mixture of 30:15mer was heated to 95°C for 10 min using a heat block and allowed to slow cool to room temperature by turning off the power to the heat block. The 30:15mer was separated from the free 15mer by non-denaturing PAGE (Hoefer Vertical Slab Gel Unit). The purified 30:15mer was excised from the preparative slab gel after identification by UV shadowing. The purified 30:15mer was electroeluted from the gel slice (ELUTRAP), desalted on a SepPak C18 cartridge (Waters) and lyophilized (Savant SpeedVac) (13). The 30:15mer was suspended in 100 µl of 10 mM HEPES (pH 7.5)/1 mM EDTA, and quantified by A260 using calculated extinction coefficients (13). To generate end-labeled, partial dsDNA substrate (30:15mer), 10 pmol of 5′-radiolabeled 30mer was added to 10 pmol of the 15mer. The radiolabeled DNA was used to spike the unlabeled 30:15mer stock solution. The mixture of labeled and unlabeled oligonucleotides was annealed by heating to 95°C for 10 min, then slow cooling.
The sequences of 15mer trapping strands used in the unwinding assay were as follows: 5′-CTGTCCTGCATGATG-3′ (DNA) and NH2-gly-CTGTCCTGCATGATG-lys (PNA).
Hybridization of DNA and PNA measured by stopped-flow absorbance
The hybridization rate of the 15mer PNA trapping strand with a complementary 15mer DNA strand was measured using an SX.18MV stopped-flow reaction analyzer (Applied Photophysics). PNA and DNA were dissolved in 25 mM HEPES (pH 7.5) with 5 mM Mg(OAc)2 and loaded into the two syringes of the instrument. After rapid mixing, the reaction was observed by measuring the reduction in absorbance at 260 nm at 25°C. Rates were measured at several concentrations of PNA (4, 5, 7.5, 10 and 12.5 µM) and a fixed concentration of 15mer DNA (0.5 µM). The data were fit to a single exponential and the resulting observed rates were plotted as a function of PNA concentration to obtain the second-order rate constant for hybridization. For comparison, hybridization of the DNA strand of identical sequence to the 15mer PNA was investigated under the same conditions.
DNA unwinding assays
The unwinding assays were performed at 25°C in reaction buffer which had a final concentration of 25 mM HEPES (pH 7.5), 2 mM β-mercaptoethanol, 0.1 mg/ml BSA, 10 U/ml phosphoenolpyruvate kinase, 15.4 U/ml lactate dehydrogenase, 0.1 mM EDTA (pH 8.0), 4 mM PEP (pH 7.5) and 10 mM KOAc unless stated otherwise. Dda was diluted into 25 mM HEPES (pH 7.5), 50 mM NaCl, 2 mM β-mercaptoethanol, 1 mM EDTA, 20% glycerol and 0.1 mg/ml BSA prior to conducting experiments.
Dda (1 nM final concentration) was incubated with varying concentrations of substrate (30:15mer) in the reaction buffer for 2 min at 25°C. The reaction was initiated by addition of an equal volume of reaction buffer containing a final concentration of 5 mM Mg(OAc)2, 5 mM ATP and varying concentrations of trapping strand. Aliquots were withdrawn at varying times and added to an equal volume of quencher (200 mM EDTA). Non-denaturing gel loading buffer (0.1% bromophenol blue, 0.1% xylene cyanol in 6% glycerol) was added to each sample and reaction products were separated on a 20% native polyacrylamide gel. Radioactivity in ssDNA and dsDNA was determined by using a PhosphorImager and ImageQuant software (Molecular Dynamics). The quantity of radioactivity of substrates and products was used to determine the quantity of ssDNA as described (27). Under conditions in which the substrate concentration was low (0.01–0.05 µM) the unwinding assays were performed at 25°C using a Kintek rapid chemical quench flow instrument (Kintek, Inc., Austin, TX) as described (13). In these experiments, the trapping strand was introduced along with the ATP and Mg(OAc)2, and the reaction was quenched with 200 mM EDTA. For experiments in which the concentration of ATP was varied, the unwinding assays were carried out with 500 nM 30:15mer substrate in reaction buffer with 10 mM KOAc. Apparent kinetic constants Km and Vmax were determined by fitting the data to the Michaelis–Menten equation using the program KaleidaGraph.
RESULTS AND DISCUSSION
Rationale
Towards the goal of understanding the mechanism of the Dda helicase, the steady-state kinetic parameters for DNA unwinding were examined. Helicase-catalyzed unwinding of oligonucleotide substrates produces ssDNA that can spontaneously hybridize to reform the substrate (Fig. 1A). During the reaction, formation of product due to helicase activity equilibrates with the formation of substrate due to hybridization, making steady-state parameters for the unwinding reaction difficult to determine (1,14). The annealing reaction can be prevented by including an oligonucleotide trapping strand in the reaction which can bind one of the ssDNA products. However, the trapping strand can also sequester the helicase and slow the observed steady-state unwinding rates. We sought to develop a method in which hybridization of ssDNA products could be prevented, while not sequestering the helicase. A DNA mimic, PNA, was chosen for this purpose. PNA is able to bind to DNA via Watson–Crick base pairing interactions, but does not contain a deoxyribose-phosphate backbone, which has been shown to be important for helicase–DNA interactions (Fig. 1B). We have shown that PNA serves as an efficient trap for unwinding reactions by annealing to ssDNA products without binding to Dda.
Figure 1.


(A) Schematic illustration of Dda-catalyzed unwinding of a DNA substrate. Presence of either DNA or PNA trapping strand in the reaction prevents the spontaneous reannealing of single-stranded DNA products. (B) Structures of the DNA and PNA backbone.
Dda does not bind to the PNA trapping strand
To determine if Dda would be sequestered by the PNA trapping strand, fluorescence polarization was used to look for binding. Fluorescence polarization has previously been used to study the interaction of Dda with a 30:15mer partial DNA duplex (23) as well as to study the interaction of the hepatitis C virus helicase, NS3, with a 30:15mer partial DNA duplex and 30:15mer partial DNA–PNA duplex (19). Dda bound to 0.1 nM F-DNA with a Kd value of 0.52 ± 0.05 nM (Fig. 2). Under identical conditions, no increase in fluorescence polarization was observed with F-PNA, indicating that Dda does not bind tightly to PNA (Fig. 2). This suggests that the PNA trap should not sequester Dda in an unwinding assay as does a ssDNA trap. When used as a trapping strand, the 15mer PNA is expected to hybridize to a 15mer ssDNA, which forms due to unwinding of the 30:15mer substrate (Fig. 1A). In order to determine whether the PNA–DNA hybrid will bind to the helicase, F-DNA was hybridized to the 15mer PNA and was titrated with Dda. No increase in fluorescence polarization was observed up to 100 nM enzyme, indicating that Dda does not interact strongly with the blunt-end F-DNA:PNA (Fig. 2). Thus, neither PNA nor the DNA:PNA hybrid would be expected to sequester Dda during unwinding.
Figure 2.

Binding between Dda and F-DNA, F-PNA or blunt-end duplex F-DNA:PNA was monitored by fluorescence polarization. Dda bound to 0.1 nM F-DNA with Kd = 0.52 ± 0.05 nM (squares). No interaction was observed between 0.1 nM F-PNA (circles) or 0.1 nM F-DNA:PNA blunt-end duplex (diamonds). Binding data was fit to a hyperbola to determine the Kd value.
PNA hybridization with DNA
The rate of hybridization of PNA to the complementary DNA sequence was determined by using stopped-flow absorbance spectroscopy. PNA was rapidly mixed with a complementary DNA, and the change in absorbance at 260 nm was measured (Fig. 3). The observed rate of annealing under psuedo first-order conditions was measured at several concentrations of PNA, and the rates were plotted as a function of PNA concentration to obtain the second-order rate constant for hybridization (Fig. 3C). PNA was found to hybridize to the DNA at a rate of 8.6 × 105 M–1 s–1 whereas a 15mer DNA of identical sequence hybridized to the complementary DNA with a rate of 1.95 × 106 M–1 s–1. Thus, the rate of hybridization of the 15mer PNA was similar to that of a 15mer DNA. The hybridization results suggest that the concentration of PNA trapping strand should be >20-fold in excess of the concentration of ssDNA product in order to trap 90% or more of the product.
Figure 3.

Hybridization rates of DNA or PNA with complementary DNA as determined by stopped-flow absorbance. (A) The 15mer DNA (4 µM) was mixed with the complementary strand of DNA (0.5 µM) and the change in absorbance at 260 nm was measured. The line is the fit of the data to a single exponential. (B) The 15mer PNA (4 µM) was mixed with the complementary strand of DNA and the change in absorbance at 260 nm was measured. (C) Plot of observed hybridization rates at several concentrations of DNA (circles) or PNA (squares). The slope of the linear fit to each data set was used to obtain the observed second order rate constant for hybridization. 15mer DNA hybridized to its complementary DNA sequence at a rate of 1.95 × 106 M–1 s–1 whereas the 15mer PNA hybridized to the DNA at a rate of 8.6 × 105 M–1 s–1.
PNA functions as a trapping strand in helicase-catalyzed unwinding of 30:15mer
In order to assess the utility of PNA as a trapping strand in helicase-catalyzed unwinding of oligonucleotide substrates, experiments were performed in the absence of a trapping strand and in the presence of either PNA or DNA trapping strand. Figure 4 illustrates the analysis of reaction products by 20% native polyacrylamide gel electrophoresis. A 30:15mer served as substrate, and the ssDNA product (30mer) was well resolved from the substrate on the gel. The ratio of substrate and product was determined by using a PhosphorImager, and the data are plotted in Figure 5. Initially, conditions were chosen in which substrate (250 nM) was in excess of enzyme (1 nM), and the trapping strand (3 µM) was in the quench solution. Under these conditions, product formation was non-linear, whereby ssDNA formed over the initial time frame of the reaction and reached a plateau when spontaneous hybridization of the product reached equilibrium with unwinding of substrate (14). Because of this, little increase in product was observed after 60 s (Fig. 5, squares). The addition of a DNA trapping strand (3 µM) in the reaction mixture led to less product formation in the initial linear phase than that observed in the absence of trapping strand, presumably due to binding of the helicase by the trap (Fig. 5B, circles). Over time the product formation with a DNA trap exceeds the product formation in the absence of a trapping strand (Fig. 5A). In the presence of the PNA trapping strand (3 µM), product formation was greater than in the absence of the trap or presence of DNA trap (Fig. 5, diamonds). Unwinding in the presence of a PNA trap produced complete turnover of the substrate (Fig. 5A). The unwinding rates in the presence of the PNA trap and the DNA trap were 1.43 nM s–1 and 0.30 nM s–1, respectively, under these conditions. To ensure that strand separation in the presence of the PNA trap was not due to spontaneous melting of the substrate, a mock unwinding experiment with 3 µM PNA + 30:15mer was performed in the absence of Dda, and no strand-separation was observed (Fig. 4D). Similarly, a control experiment with Dda + 30:15mer + 3 µM PNA in the absence of ATP resulted in no unwinding (data not shown).
Figure 4.
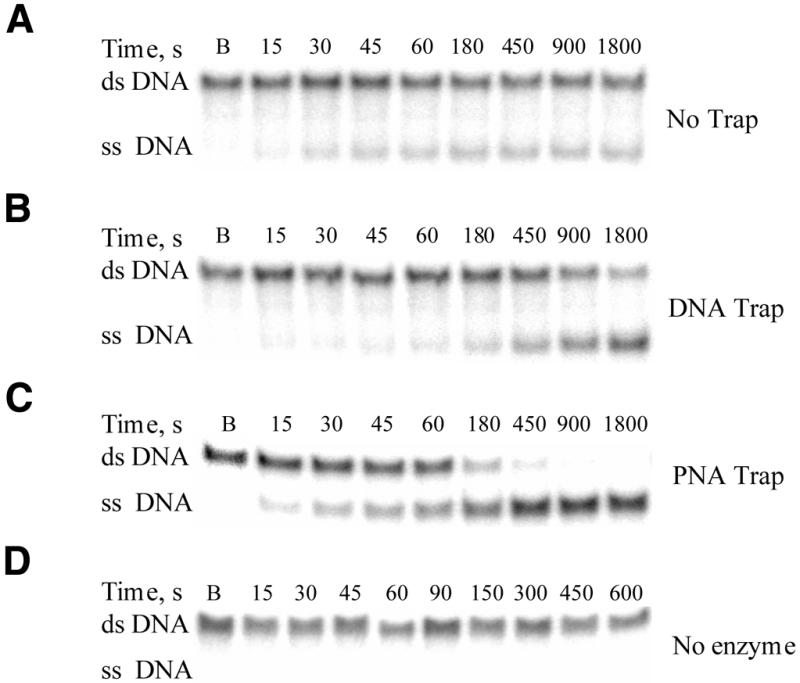
Analysis of Dda-catalyzed unwinding of 30:15mer by native polyacrylamide gel electrophoresis. Dda (1 nM) was incubated with the 30:15mer (250 nM) and the reaction was initiated by mixing with ATP (5 mM) and Mg(OAc)2 (5 mM). Aliquots were withdrawn at various times, and the reaction was quenched with EDTA. A blank sample, B, was obtained by first mixing an aliquot of ATP + Mg(OAc)2 with quench, and then adding an equivalent aliquot of Dda + 30:15mer. (A) Phosphorimage of the products from Dda-catalyzed unwinding in which a 15mer DNA trapping strand was introduced with the quencher. (B) Phosphorimage of the products from the unwinding reaction in which the 15mer DNA trapping strand (3 µM) was included in the reaction mixture. (C) Phosphorimage of the products from the unwinding reaction in which the 15mer PNA trapping strand (3 µM) was included in the reaction mixture. (D) Phosphorimage of a control experiment with 3 µM PNA, 30:15mer and no enzyme.
Figure 5.

Unwinding of the 30:15mer (250 nM) by Dda (1 nM). The data from Figure 4 are plotted. The reaction was carried out in the absence of a trapping strand (squares), in the presence of a DNA trapping strand (circles) or in the presence of a PNA trapping strand (diamonds). (A) Complete unwinding was readily observed in the presence of the PNA trap. Without trap in the reaction, product formation reached a plateau prior to complete unwinding. (B) Expanded view of the linear phase from (A). The data in (B) were fit to a linear function to obtain unwinding rates. The rate of unwinding was 0.30 nM s–1 in the presence of the DNA trapping strand and 1.43 nM s–1 in the presence of the PNA trapping strand.
Trapping efficiency at varying PNA concentrations
In order to determine the concentration range over which PNA could serve as an effective trap, unwinding assays were performed at varying PNA concentrations in the presence of 250 nM substrate and 1 nM Dda. The reactions were carried out in the presence of an equivalent, 0.4-, 4-, 12-, 24- and 36-fold (0.25, 0.1, 1, 3, 6 and 9 µM, respectively) molar excess concentration of PNA over 30:15mer (Fig. 6). PNA concentrations from 1 to 9 µM serve as a trapping strand with similar efficiency under these conditions. For the linear phase, the unwinding rates at different PNA concentrations were 0.18, 0.99, 1.70, 1.43, 1.53 and 1.56 nM s–1 at 0.1, 0.25, 1, 3, 6 and 9 µM PNA, respectively. Thus, even though the lowest, effective concentration of PNA (1 µM) was only 4-fold greater than the concentration of substrate (250 nM), it was capable of serving as an efficient trap. The concentration of ssDNA product that exists at any time during the progress of the reaction is much lower than the total substrate concentration (Fig. 6). At PNA concentrations equivalent to or below the concentration of substrate, complete product formation did not occur (Fig. 6B). Reproducibility of the assay was determined by measuring unwinding rates at 250 nM substrate with 3 µM PNA in triplicate, resulting in an average unwinding rate of 1.34 nM s–1 and a standard deviation of 0.17 nM s–1.
Figure 6.

Dda-catalyzed unwinding in the presence of varying concentration of PNA trapping strand. (A) Unwinding of the 30:15mer (250 nM) by Dda (1 nM) in the presence of 0.1 µM (filled squares), 0.25 µM (open circles), 1 µM (filled diamonds), 3 µM (open squares), 6 µM (filled circles) and 9 µM (open diamonds) PNA trapping strand. (B) Expanded view of the linear phase from (A). Rates of unwinding at 0.1, 0.25, 1, 3, 6 and 9 µM PNA were 0.18, 0.99, 1.70, 1.43, 1.53 and 1.56 nM s–1, respectively.
Determination of the kinetic constants for Dda catalyzed unwinding of 30:15mer under steady-state conditions
Unwinding assays under conditions in which the helicase concentration is in excess over the substrate concentration have been reported. If the substrate concentration is ∼1–2 nM, then a trapping strand need not be included in the reaction mixture because hybridization occurs very slowly (10). Under steady-state conditions, unwinding is more difficult to measure, due to annealing of ssDNA during the unwinding reaction. Use of a PNA trap prevents hybridization of ssDNA without sequestering the helicase, making it possible to measure steady-state rates of unwinding. However, one of the strands is not trapped by PNA, and this strand can bind to the helicase leading to product inhibition. Product inhibition can be taken into account when developing complete kinetic mechanisms for DNA unwinding by measuring the affinity of Dda for the single-stranded product.
To illustrate the utility of the PNA trapping strand, the apparent Km for ATP during Dda-catalyzed unwinding was measured. The rate of unwinding was measured as a function of ATP concentration at 500 nM 30:15mer and 1 nM Dda (Fig. 7). The concentration of ATP required to reach one-half the maximum unwinding velocity under conditions of low salt (10 mM KOAc) was 92 ± 19 µM. This value is similar to the Km reported previously (130 µM) in which ATP hydrolysis served as the endpoint of the assay (28). Subsequent unwinding assays were performed at a saturating concentration of ATP (5 mM). The PNA trapping strand concentration in the assay was 3 µM and the substrate concentration was varied. The apparent Km for 30:15mer was 208 ± 54 nM in the presence of 150 mM KOAc (Fig. 8). At lower salt concentration (10 mM KOAc), the apparent Km for 30:15mer was 65 ± 9 nM, suggesting that the affinity of the enzyme for the substrate increases with lower salt concentrations (Fig. 8). The Vmax of the reaction was slightly increased in the presence of low salt (Fig. 8B; Table 1). Presteady-state experiments will be required to evaluate the important steps in the kinetic mechanism that contribute to the observed steady-state parameters.
Figure 7.

Unwinding of 30:15mer (500 nM) by Dda (1 nM) at varying ATP concentrations in the presence of the PNA trapping strand (3 µM). The line through the data represents the best fit to a hyperbola. The concentration of ATP required for half-maximal DNA unwinding was 92 ± 19 µM whereas the maximum velocity for unwinding under these conditions was 1.22 ± 0.06 nM s–1.
Figure 8.

Steady-state unwinding of the 30:15mer by Dda in the presence of a PNA trapping strand. Varying concentrations of the DNA substrate were incubated with 1 nM Dda and a saturating concentration of ATP (5 mM) in the presence of 3 µM PNA trapping strand. Plots are shown for unwinding at 150 mM KOAc (A) and 10 mM KOAc (B). In all the plots, the line through the data points represents the fit to the Michaelis–Menten equation using KaleidaGraph. Kinetic constants are listed in Table 1.
Table 1. Steady-state kinetic parameters for DNA unwinding by Dda.
|
Km (nM) |
Vmax (nM s–1) |
| 208 ± 54a | 1.24 ± 0.10a |
| 65 ± 9b | 1.67 ± 0.06b |
Rates were determined as described in Materials and Methods. Standard errors are reported from the numerical fit of the data.
a150 mM KOAc.
b10 mM KOAc.
Fluorescence-based continuous assays have been used to study unwinding of dsDNA by Escherichia coli RecBCD helicase (29,30). The continuous assay measures the decrease in intrinsic fluorescence of SSB protein upon binding to ssDNA generated by helicase unwinding of duplex DNA. However, RecBCD is a highly processive helicase (28), whereas Dda is a relatively non-processive helicase (14,28). Elucidating the detailed kinetic mechanism of unwinding of dsDNA substrates by Dda will require the application of both the pre-steady-state and steady-state experiments. Therefore, we sought to develop an assay that would utilize oligonucleotide substrates, yet be amenable to steady-state conditions.
PNA binds to DNA according to Watson–Crick base pairing rules; therefore, we used PNA as a trapping strand in steady-state unwinding assays with Dda. Including PNA in the reaction prevents reannealing of ssDNA products formed by the helicase. PNA hybridizes to its complementary sequence during the unwinding reaction. It is possible that the trapping strand may hybridize to partially unwound intermediates, thereby contributing to product formation. However, the fact that the same rates are observed at varying concentrations of PNA suggests that partially unwound intermediates are not trapped as products. Because Dda does not bind to PNA, the observed rates reflect the activity of the enzyme towards the substrate rather than the trapping strand. Using this assay we have determined apparent steady-state kinetic constants for Dda-catalyzed unwinding of a 30:15mer substrate. This is an important and necessary step in enumerating the overall kinetic mechanism for unwinding of DNA by this enzyme. Measurement of unwinding parameters in the steady state also forms the framework for designing pre-steady unwinding experiments that are currently in progress.
Acknowledgments
ACKNOWLEDGEMENT
This work was supported by research grant GM59400 (K.D.R.) from the National Institutes of Health.
References
- 1.Lohman T.M. and Bjornson,K.P. (1996) Mechanisms of helicase-catalyzed DNA unwinding. Annu. Rev. Biochem., 65, 169–214. [DOI] [PubMed] [Google Scholar]
- 2.Hall M.C. and Matson,S.W. (1999) Helicase motifs: the engine that powers DNA unwinding. Mol. Microbiol., 35, 867–877. [DOI] [PubMed] [Google Scholar]
- 3.Soultanas P. and Wigley,D.B. (2000) DNA helicases: ‘inching forward’. Curr. Opin. Struct. Biol., 10, 124–128. [DOI] [PubMed] [Google Scholar]
- 4.Ellis N.A. (1997) DNA helicases in inherited human disorders. Curr. Opin. Genet. Dev., 7, 354–363. [DOI] [PubMed] [Google Scholar]
- 5.Ali J.A. and Lohman,T.M. (1997) Kinetic measurement of the step size of DNA unwinding by Escherichia coli UvrD helicase. Science, 275, 377–380. [DOI] [PubMed] [Google Scholar]
- 6.Walstrom K.M., Dozono,J.M., Robic,S. and von Hippel,P.H. (1997) Kinetics of the RNA–DNA helicase activity of Escherichia coli transcription termination factor Rho. 1. Characterization and analysis of the reaction. Biochemistry, 36, 7980–7992. [DOI] [PubMed] [Google Scholar]
- 7.Walstrom K.M., Dozono,J.M. and von Hippel,P.H. (1997) Kinetics of the RNA–DNA helicase activity of Escherichia coli transcription termination factor Rho. 2. Processivity, ATP consumption and RNA binding. Biochemistry, 36, 7993–8004. [DOI] [PubMed] [Google Scholar]
- 8.Jankowsky E., Gross,C.H., Shuman,S. and Pyle,A.M., (2000) The DExH protein NPH-II is a processive and directional motor for unwinding RNA. Nature, 403, 441–451. [DOI] [PubMed] [Google Scholar]
- 9.Dillingham M.S., Wigley,D.B. and Webb,M.R. (2000) Demonstration of unidirectional single-stranded DNA translocation by PcrA helicase: measurement of step size and translocation speed. Biochemistry, 39, 205–212. [DOI] [PubMed] [Google Scholar]
- 10.Amaratunga M. and Lohman,T.M. (1993) Escherichia coli Rep helicase unwinds DNA by an active mechanism. Biochemistry, 32, 6815–6820. [DOI] [PubMed] [Google Scholar]
- 11.Raney K.D., Carver,T. and Benkovic,S.J. (1996) Stoichiometry and DNA unwinding by the bacteriophage T4 41:59 helicase. J. Biol. Chem. 271, 14074–14081. [DOI] [PubMed] [Google Scholar]
- 12.Washington M.T. and Patel,S.S. (1998) Increased DNA unwinding efficiency of bacteriophage T7 DNA helicase mutant protein 4A′/E348K. J. Biol. Chem. 273, 7880–7887. [DOI] [PubMed] [Google Scholar]
- 13.Tackett A.J., Morris,P.D., Dennis,R.D., Goodwin,T.E. and Raney,K.D. (2001) Unwinding of unnatural substrates by a DNA helicase. Biochemistry, 40, 543–548. [DOI] [PubMed] [Google Scholar]
- 14.Raney K.D., Sowers,L.D., Millar,D.P. and Benkovic,S.J. (1994) A fluorescence-based assay for monitoring helicase activity. Proc. Natl Acad. Sci. USA, 91, 6644–6648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Egholm M., Buchardt,O., Christensen,L., Behrens,C., Freier,S.M., Driver,D.A., Begg,R.H., Kim,S.K., Norden,B. and Nielsen,P.E. (1993) PNA hybridizes to complementary oligonucleotides obeying the Watson–Crick hydrogen-bonding rules. Nature, 365, 566–568. [DOI] [PubMed] [Google Scholar]
- 16.Hanvey J.C., Peffer,N.C., Bisi,J.E., Thomsom,S.A., Cadilla,R., Josey,J.A., Ricca,D.J., Hassman,C.F., Bonham,M.A., Au,K.G. et al. (1992) Antisense and antigene properties of peptide nucleic acids. Science, 258, 1481–1485. [DOI] [PubMed] [Google Scholar]
- 17.Nielsen P.E., Egholm,M. and Buchart,O. (1994) Sequence specific transcription arrest by PNA bound to the template strand. Gene, 149, 139–145. [DOI] [PubMed] [Google Scholar]
- 18.Bastide L., Boehmer,P.E., Villani,G. and Lebleu,B. (1999) Inhibition of DNA-helicase by peptide nucleic acids. Nucleic Acids Res., 27, 551–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tackett A.J., Wei,L., Cameron,C.E. and Raney,K.D. (2001) Unwinding of nucleic acids by HCV NS3 helicase is sensitive to the structure of the duplex. Nucleic Acids Res., 29, 565–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lutz M.J., Benner,S.A., Hein,S., Breipohl,G. and Uhlmann,E. (1997) Recognition of uncharged polyamide-linked nucleic acid analogs by DNA polymerases and reverse transcriptases. J. Am. Chem. Soc., 119, 3177–3178. [Google Scholar]
- 21.Gauss P., Park,K., Spencer,T.E. and Hacker,K.J. (1994) DNA helicase requirements for DNA replication during bacteriophage T4 infection. J. Bacteriol ., 176, 1667–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Formosa T., Burke,R.L. and Alberts,B.M. (1983) Affinity purification of T4 bacteriophage proteins essential for DNA replication and genetic recombination. Proc. Natl Acad. Sci. USA, 80, 2442–2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morris P.D., Tackett,A.J., Babb,K., Nanduri,B., Chick,C., Scott,J. and Raney,K.D. (2001) Evidence for a functional monomeric form of the bacteriophage T4 Dda helicase. J. Biol. Chem., in press. [DOI] [PubMed] [Google Scholar]
- 24.Goodwin T.E., Holland,R.D., Lay,J.O. and Raney,K.D. (1998) A simple procedure for solid-phase synthesis of peptide nucleic acids with N-terminal cysteine. Bioorg. Med. Chem. Lett., 8, 2231–2234. [DOI] [PubMed] [Google Scholar]
- 25.Raney K.D. and Benkovic,S.J. (1995) Bacteriophage T4 Dda helicase translocates in a unidirectional fashion on single-stranded DNA. J. Biol. Chem., 270, 22236–22242. [DOI] [PubMed] [Google Scholar]
- 26.Hacker K.J. and Alberts,B.M. (1992) Overexpression, purification, sequence analysis and characterization of the T4 bacteriophage Dda DNA helicase. J. Biol. Chem., 267, 20674–20681. [PubMed] [Google Scholar]
- 27.Amaratunga M. and Lohman,T.M. (1993) Escherichia coli Rep helicase unwinds DNA by an active mechanism. Biochemistry, 32, 6815–6820. [DOI] [PubMed] [Google Scholar]
- 28.Jongeneel C.V., Formosa,T. and Alberts,B.M. (1984) Purification and characterization of the bacteriophage T4 dda protein. J. Biol. Chem., 259, 12925–12932. [PubMed] [Google Scholar]
- 29.Eggleston A.K., Rahim,N.A. and Kowalczykowski,S.C. (1996) A helicase assay based on the displacement of fluorescent, nucleic acid-binding ligands. Nucleic Acids Res., 24, 1179–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Roman L.J. and Kowalczykowski,S.C. (1989) Characterization of the helicase activity of the E. coli RecBCD enzyme using a novel helicase assay. Biochemistry, 28, 2863–2873. [DOI] [PubMed] [Google Scholar]