

Abstract
Neurodegenerative diseases are characterized by progressive dysfunction and loss of neurons associated with depositions of pathologically altered proteins showing hierarchical involvement of brain regions. The role of astrocytes in the pathogenesis of neurodegenerative diseases is explored as contributors to neuronal degeneration or neuroprotection pathways, and also as potential mediators of the transcellular spreading of disease‐associated proteins. Protein astrogliopathy (PAG), including deposition of amyloid‐β, prion protein, tau, α‐synuclein, and very rarely transactive response DNA‐binding protein 43 (TDP‐43) is not unprecedented or unusual in neurodegenerative diseases. Morphological characterization of PAG is considered, however, only for the neuropathological diagnosis and classification of tauopathies. Astrocytic tau pathology is seen in primary frontotemporal lobar degeneration (FTLD) associated with tau pathologies (FTLD‐Tau), and also in the form of aging‐related tau astrogliopathy (ARTAG). Importantly, ARTAG shares common features with primary FTLD‐Tau as well as with the astroglial tau pathologies that are thought to be hallmarks of a brain injury‐related tauopathy known as chronic traumatic encephalopathy (CTE). Supported by experimental observations, the morphological variability of PAG might reflect distinct pathogenic involvement of different astrocytic populations. PAG might indicate astrocytic contribution to spreading or clearance of disease‐associated proteins, however, this might lead to astrocytic dysfunction and eventually contribute to the degeneration of neurons. Here, we review recent advances in understanding ARTAG and other related forms of PAG.
Keywords: A‐beta, alpha‐synuclein, ARTAG, astrocyte, neurodegeneration, PAG, prion, prion protein, protein astrogliopathy, tau, TDP‐43
Introduction: Astrocytes and Neurodegeneration
Neurodegenerative diseases (NDD) comprise disorders thought to affect predominantly neurons. Based on the anatomical distribution of malfunctioning and dying neurons, the clinical symptomatology and prognosis varies considerably. In contrast to neurons, astrocytes, as well as oligodendrocytes, have been long considered as side players or bystanders in the pathogenesis of NDD. Recent studies, however, highlighted an underestimated spectrum of astrocytic functions 128, 148, 163 that might be considered as contributory to the development of neuronal dysfunction and degeneration thereby implicating astroglial responses in mechanisms of diverse NDD and brain aging 34.
In addition to the loss of neurons in NDD, most NDD are characterized by the presence of inclusions, such as Lewy bodies, neurofibrillary tangles (NFTs), or Pick bodies, that have been detected in neurons in neurodegenerative conditions more than a century ago. The introduction of silver staining methods showing positivity for example of NFTs and Pick bodies 4 indicated that these bodies include components with altered physicochemical properties. Indeed, the finding that altered proteins are central in the pathogenesis of neurodegenerative conditions lead to the introduction of immunohistochemical (IHC) methods applying specific antibodies against different modifications of proteins, complemented by ultrastructural examinations and biochemistry. These revealed an unexpected plethora of various intra‐ and extracellular protein depositions. Argyrophilic inclusions have been described in glial cells as well: first in oligodendroglia as glial cytoplasmic inclusions morphologically unifying different clinical forms of multiple system atrophy (MSA) 127 followed soon by the elucidation of astroglial inclusions 5 such as tufted astrocytes 48, 124, 170, thorn‐shaped astrocytes 55, 124, and astrocytic plaques 32, 105. However, in contrast to the oligodendroglial Papp–Lantos bodies in MSA, the recognition of argyrophilic inclusions in astrocytes was inspired first by tau IHC observations.
The next highly significant milestone for NDD research was the introduction of the concept of cell‐to‐cell spreading of pathological disease proteins; this has been discussed mostly for neurons leading to hierarchical involvement of anatomical regions in the human brain 18, 45, 61. However, it has been recognized that astrocytes also appear early in the pathogenesis and might also internalize different pathological disease proteins 41, 70, 77, 90, 95. Therefore, the role of astrocytes is now explored not only as players in neuronal degeneration or neuroprotection pathways, but also as potential participants in the transcellular spreading of disease‐associated proteins. In this review, we summarize the spectrum of protein depositions in astrocytes, termed here protein astrogliopathy (PAG), in various neurodegenerative conditions and aging.
The relevance of PAG for the Classification of Neurodegenerative Diseases
Classification of NDD includes three levels: (i) clinical: reflecting the anatomical involvement of the disease process; (ii) protein‐based: indicating that specific disease proteins with various physiological functions show conformational change and biochemical modifications related to distinct groups of NDD; and (iii) cellular and subcellular pathology: implying that pathological disease protein deposits involve neurons or glial cells, furthermore, which of their subcellular compartment, or whether these are found extracellularly 68. NDD are more frequently associated with aging. Various gene alterations with distinct protein products are associated with rare disease forms; however, the most frequent NDD of the adult or aging brain involve six proteins: amyloid‐β (Aβ), prion protein (PrP), microtubule‐associated protein tau, α‐synuclein (α‐syn), transactive response (TAR) DNA‐binding protein 43 (TDP‐43), and FET proteins, which include the fused in sarcoma (FUS), Ewing's sarcoma RNA‐binding protein 1 (EWSR1), and TATA‐binding protein‐associated factor 15 (TAF15) 68.
For the molecular pathologic classification (i.e., protein and cellular distribution) mostly morphological criteria are used, although genetic abnormalities and biochemical modifications are also considered in the assessment of NDD 68. Alzheimer disease (AD) is characterized by extracellular Aβ deposits known as amyloid plaques and intracellular tau aggregates known as NFTs. The distribution of NFTs shows stages in the progressive accumulation of this tau pathology, which also include plaque‐associated tau positive neurites and neuropil threads distributed diffusely in the gray matter neuropil 15, while the distribution of different morphologies of parenchymal Aβ deposits are defined as occurring in progressive phases stereotypically involving ever wider regions of cortex followed by deep gray structures, brainstem and eventually cerebellum in the most advanced stages of AD 157. Intracellular Aβ deposits, including those in astrocytes, are discussed but not included in the neuropathological classification of AD, and the significance of these intracellular species of Aβ are uncertain 30. Classification of prion diseases is based on the etiology and molecular subtyping; the latter includes a gene polymorphism (codon 129 of the prion protein gene) and biochemical examination of the size of the proteinase kinase (PK)‐resistant core of the abnormal prion protein (PrP); IHC demonstrates mostly extracellular deposits and astrocytes are not considered in the classification 71. Tauopathies are distinguished based on the cellular distribution of tau pathology, exemplified by Pick's disease (PiD) and NFT‐dementia or primary age‐related tauopathy (PART), progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), argyrophilic grain disease (AGD), and globular glial tauopathies (GGT), showing variable involvement of neurons and glial cells 67. Indeed, the morphology of astrocytic pathology is crucial to distinguish PSP and CBD 67, furthermore, its importance is highlighted in aging‐related tau astrogliopathy (ARTAG) 72. Neuropathological classification conforms with different bands and isoforms demonstrated in Western blots of insoluble tau 87. α‐Synuclein pathology characterizes dementia with Lewy bodies (DLB) and Parkinson disease (PD) and MSA. DLB and PD are distinguished upon clinical symptoms and show predominance of intraneuronal cytoplasmic and neuritic deposits (cortical and brainstem type Lewy bodies and Lewy neurites) showing stages or different anatomical patterns 11, 16, 109. MSA is dominated by oligodendroglial cytoplasmic inclusions (GCIs) 160. All forms show various morphologies of astrocytic α‐syn deposition, however, considered as irrelevant for disease‐classification 68. TDP‐43 is a major component of the ubiquitin‐positive inclusions that characterize amyotrophic lateral sclerosis (ALS) and a common form of frontotemporal lobar degeneration (FTLD) 122. The spectrum of TDP‐43 immunoreactive structures includes neuronal cytoplasmic and intranuclear inclusions, dystrophic neurites, and oligodendroglial cytoplasmic inclusions 85; therefore, astrocytic TDP‐43 protein is not included in classification systems 101, 102. This is true for FUS (FET)‐proteinopathies, including basophilic inclusion body disease, atypical FTLD‐U, and neuronal intermediate filament inclusion disease (NIFID), where mostly neuronal and less frequently oligodendroglial inclusions are detected 114, 118, 120, 121.
In summary, astrocytic protein pathology is not unprecedented or unusual in NDD; however, morphological characterization of these is considered only for the neuropathological diagnosis and classification of tauopathies. Thus, there is a need for a closer look at the spectrum (Figures 1 and 2) and relationships of astrocytic protein deposition to neuronal protein or extracellular pathological aggregates so here we provide a careful reconsideration of the importance of PAG.
Figure 1.
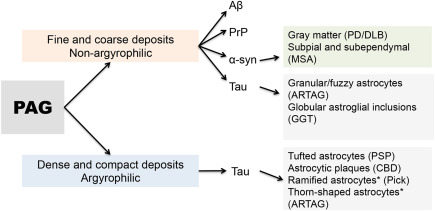
Stratification of PAG in neurodegenerative diseases. * Indicates that these are variably argyrophilic.
Figure 2.
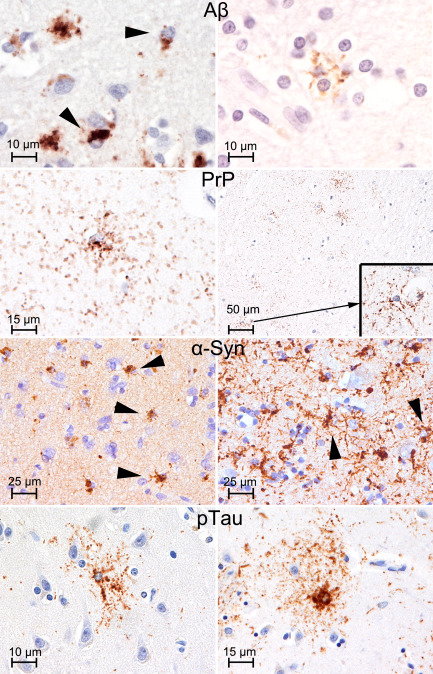
Various type of PAG. Amyloid‐β (Aβ; left: frontal cortex; right: frontal white matter; antibody used: anti‐Aβ, clone 6F/3D directed against amino acids 8–17 of the peptide, Dako, Glostrup, Denmark, 1:100), prion protein (PrP: temporal cortex; antibody used: anti‐PrP 12F10, Cayman Chemical, Ann Arbor, MI, USA, 1:2000), and α‐synuclein (left: striatum; right: temporal cortex; antibody used: anti‐α‐synuclein 5G4, Roboscreen, Leipzig, Germany, 1:4000) PAG showing coarse and fine granules of immunoreactivity: compare with granular/fuzzy astrocytes of ARTAG (temporal cortex; antibody used: anti‐tau AT8 pS202/pT205, Pierce Biotechnology, Rockford, IL, USA, 1:200).
PAG in Non‐Tauopathy Neurodegenerative Diseases
Aβ‐PAG
Aβ is produced by not only neurons but also non‐neural cells; indeed human astrocytes produce high levels of intact Aβ 19. Using different anti‐Aβ antibodies, Funato et al reported dot‐like Aβ deposits, associated mostly with Aβ40 immunoreactive diffuse plaques in the aging human brain 41. These deposits colocalized with GFAP positive astrocytes and further ultrastructural studies revealed their association with lipofuscin granules. The authors theorized that astrocytes may take up and degrade Aβ in lysosomes in the aging brain 41. An ultrastructural study of AD brain biopsies also found Aβ immunoreactivity in astrocytes 84. Thal et al showed that large numbers of GFAP‐positive astrocytes containing N‐terminal‐truncated Aβ fragments appear in the vicinity of N‐terminal‐truncated Aβ deposits, thus the authors concluded that N‐terminal‐truncated Aβ peptide may be cleared preferentially from the extracellular space by astrocytic uptake and processing 158. Another study examining the entorhinal cortex in AD affected brains showed that activated astrocytes are abundant near amyloid plaques and they showed Aβ42 immunoreactivity in astrocytes in the molecular layer 116. They discussed the possibility that subpial plaques are in fact astrocyte‐derived following the lysis of the astrocytes 116. Oide et al considered Aβ deposits of diffuse and neuritic plaques to be shredded by astrocytic processes from the marginal zone of plaques, and to gradually disintegrate into smaller compartments 126. In a further study on individuals with Down syndrome (under the age of 30 years) reported Aβ1–28 and Aβ40 immunostaining in astrocytic cell bodies and in processes that extended to the vicinity of blood vessels 46. These studies raise the possibility that astrocytes interact with Aβ deposition in the brain. However, more studies are needed for better interpretation, particularly that, similarly to the detection of intraneuronal Aβ deposits, most of the antibodies that show intracellular deposits are unable to distinguish Aβ from AβPP 2.
PrP‐PAG
Prion diseases are characterized by the deposition of disease‐associated PrP together with spongiform change of the neuropil associated with neuronal loss and reactive astrogliosis 71. Early IHC studies reported granular PrP deposits in astrocytes in Creutzfeldt‐Jakob disease 99; this has already been described in an experimental mouse model with scrapie infection 28. Indeed, it has been shown, that this PrP accumulates in astrocytes prior to the cardinal neuropathological changes in scrapie 28. Double immunolabeling confocal microscopy studies in human prion disease confirmed that astroglial cells harbor intracytoplasmic disease‐associated PrP granules and that there is additionally a periastrocytic accumulation of disease‐associated PrP deposits suggesting that astroglial cells may have a role in the processing, degradation, or removal of disease‐associated PrP 77. However, in the early stages of prion diseases in humans this cannot be ascertained with certainty; therefore astrocytic PrP is never seen as a pure neuropathological feature and also not considered in the diagnostic description of immunostaining patterns. Finally, the role of astrocytes in the formation of neuropil vacuolation has been addressed in a comprehensive ultrastructural study. This revealed that the ultrastructural correlates of spongiform change develop within neuronal processes and, most likely, not astrocytic processes, in contrast to the intra‐astrocytic vacuolation found in some rodent models 92.
TDP‐43‐PAG and FUS‐PAG
TDP‐43 proteinopathies are associated with the mislocalization of TDP‐43 from the neuronal nucleus into the cytoplasm; however, TDP‐43 pathology is consistently observed in the oligodendrocytes as well. In contrast, astrocytic TDP‐43 pathology is not a consistent feature of, or less examined in, FTLD‐TDP or ALS. Interestingly, astrocytic TDP‐43 immunoreactivity has been reported in non‐TDP‐43 disorders 119. For example, Uryu et al reported TDP‐43 in astrocytic plaques of the FTLD‐tau disease CBD 161. Furthermore, a study evaluating three cases with FTLD‐TDP due to mutations in the gene for progranulin and a case of familial LB disease due to the A53T mutation in the gene encoding α‐syn demonstrated astrocytic end‐feet with abnormal TDP‐43 fibrillary inclusions, while glial fibrils of reactive astrocytes were negative for TDP‐43 93. In addition, round phospho‐TDP‐43‐positive structures associated with astrocytes have been described in Cockayne syndrome 141. Lee et al reported TDP‐43 in astrocyte‐related Rosenthal fibers and eosinophilic granular bodies associated with low‐grade tumors and reactive brain tissue 88. A further study demonstrated widespread TDP‐43 pathology in astrocytes of Alexander disease, a primarily astrocytic neurodegenerative disease, which lacks neuronal TDP‐43 deposits 167. Interestingly, phosphorylation, and increased insolubility of TDP‐43 is similar in Alexander disease to what has been reported in other neurodegenerative diseases, however, insoluble C‐terminal fragments of TDP‐43 were absent or barely detectable in immunoblots contrasting neurodegenerative TDP‐43 proteinopathies. One of the most frequent genetic causes of FTLD‐TDP is related to C9orf72 mutation; this leads to prominent TDP‐43 pathology and also shows TDP‐43 negative ubiquitinated neuronal cytoplasmic inclusions, which are immunoreactive for antibodies generated against putative GGGGCC repeat RAN‐translated peptides (anti‐C9RANT); however, astroglia do not show these peptide deposits 10.
Similarly to TDP‐43 proteinopathies, astrocytic FUS‐inclusions are not considered to be major pathological hallmarks of FTLD‐FUS. Astrocytic FUS inclusions have been described in the white matter in NIFID, but relatively infrequently when compared with neuronal inclusions 9. A comparative analysis of six cases of ALS‐FUS revealed glial inclusions; however, in spite of a ramified morphology of some of these, an astrocytic origin could not be confirmed by double‐labeling experiments using sensitive and specific markers of these cell types, therefore, these inclusions were not considered to be astroglial 100. Finally, the glial intranuclear inclusions in intranuclear inclusion body disease show FUS immunoreactivity 111.
α‐Synuclein PAG
It is generally accepted that PD is a neuron‐predominant, while MSA is a glia‐predominant α‐synucleinopathy. However, α‐syn inclusions in oligodendrocytes can be seen in PD; this has been reported first in the substantia nigra 6, 164 and later it has been shown that oligodendroglial inclusions, with overlapping features to that seen in MSA, can be seen in the pallidothalamic tract 137. Astrocytic α‐syn pathology has received less attention. Wakabayashi et al mentioned already that some astrocytes in the brainstem show crescent‐shaped inclusions 164. Terada et al reported astrocytic star‐like inclusions detectable also by Gallyas–Braak staining mostly in the temporal lobe in DLB cases; however, the anti‐α‐syn antibody used in that study did not label these 156. Braak and colleagues performed a comprehensive study using different anti‐α‐syn antibodies and found immunoreactive astrocytes in Braak PD stage 4 or higher preferentially in prosencephalic regions (amygdala, thalamus, septum, striatum, claustrum, and cerebral cortex) 17. Importantly, these were detected mostly by antibodies covering the NAC (non‐amyloid component) region of the α‐syn molecule, but they were ubiquitin and p62 negative and were also undetectable using silver methods, including the Gallyas stain 17; thus, contrasting with the findings of Terada et al 156. Braak et al concluded that labeling of astrocytes appears to accompany the formation of neuronal inclusion bodies and the presence of Aβ protein or neurofibrillary changes of the Alzheimer type, do not appear to influence the development of these 17. A further study, focusing on neostriatal phosphorylated‐α‐syn pathology, revealed coiled‐like glial inclusions correlating with Braak PD stage 113. The morphology of these glial inclusions was different from that reported by Braak et al 17 suggesting that phosphorylated‐α‐syn antibodies do not reveal the whole spectrum of astrocytic α‐syn pathology as seen by selected non‐phospho antibodies 17, 70. Indeed, an antibody (5G4) against the epitope containing amino acids 47–53 in α‐syn, which detects exclusively disease‐associated α‐syn but not the monomer form 81, revealed abundant non‐ubiquitinated α‐syn astroglial pathology correlating with dots and thin neurites but not with the thick neurites and neuronal immunoreactivity 70. This study showed that in the cortex and amygdala the morphology is more star‐like, while in the striatum fine granular astrocytic deposits were seen; ultrastructural examinations revealed a relation to endocytosis 70. These observations can be better interpreted in the context of a comprehensive study on experimental models and human brains 90. This showed an endocytosis‐dependent transfer of α‐syn to astrocytes and suggested that α‐syn proteins may be released from neurons and therefore could be a source of astroglial α‐syn pathology and an important mediator of astroglial inflammatory responses 90.
It also should be mentioned that based on a double‐labeling immunofluorescence study α‐syn‐positive doughnut‐shaped often ubiquitin‐positive structures were located in the GFAP‐positive, swollen processes of Bergmann glia of the cerebellum in DLB/PD and less in MSA 129. Furthermore, several types of oxidized‐γ‐syn positive astrocytes with different morphologies were reported in PD/DLB and also in controls 153. An interesting aspect of glial α‐syn immunoreactivity was highlighted by a study comparing different epitope‐retrieval methods in distinctly processed tissue: this revealed that in the normal human brain both oligodendroglia and astroglia show prominent α‐syn immunoreactivity detectable in vibratome sections using proteinase K and formic acid pretreatment 112. This study emphasizes the pathogenic role of posttranslationally modified α‐syn in the disease‐processes of α‐synucleinopathies 112.
In contrast to PD/DLB, another morphological type of α‐syn inclusion has been described in MSA: phosphorylated‐α‐syn and 5G4 81 immunoreactive processes appeared in the subpial surface of the spinal cord and brainstem, as well as the subependymal region of the lateral ventricles 117. They were not visualized by Gallyas–Braak staining and were ubiquitin and p62 negative. While the morphology of these α‐syn inclusions is reminiscent of subpial and subependymal ARTAG 72, however, the anatomical distribution in the brainstem is different; mostly dorsolateral for tau in ARTAG 79 and ventrolateral for α‐syn in MSA 117.
TAU‐PAG in Primary FTLD‐Tauopathies and Aging
In spite of the application of silver‐staining methods many decades prior to IHC, only the introduction and development of tau IHC methods enable the recognition that astroglial tau pathology is an important component of FTLD‐tau. Many studies described these as glial fibrillary tangles, by analogy to neuronal NFTs, however, this does not reflect the fact that tau pathology involves different astroglial populations and associates with dramatically different morphological appearances. It has also been noted that the descriptions of morphologies varied between disorders and also between publications, the latter possibly complicating the comparison of studies. The recognition that the nomenclature and definitions of astrocytic tau morphologies in FTLD tauopathies and aging can vary between experts signifies that this topic still requires further consensus studies 82. Recent experimental studies show that glial tau pathology can be reproduced in animal models of tauopathies through injections of diseased brain homogenates from AD and FTLD‐Tau patients 12, 22, thereby suggesting the existence of pathological tau strains involving different astrocytic populations or inducing distinct astrocytic responses. Currently, there is emerging consensus that at least six types of astrocytic tau pathologies should be distinguished (Figure 3): tufted astrocytes, astrocytic plaques, ramified astrocytes, globular astroglial inclusions seen mostly in primary FTLD‐tauopathies, while thorn‐shaped astrocytes (TSA) and granular/fuzzy astrocytes (GFA) are discussed in the context of ARTAG 72.
Figure 3.
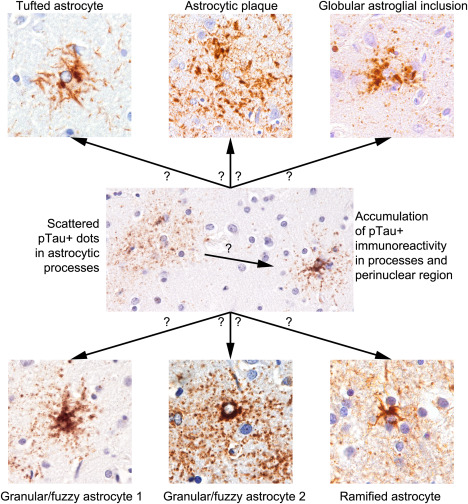
Conceptual summary of the development of astroglial tau pathologies.
Astrocytic tau pathology in progressive supranuclear palsy
In 1988, Probst et al described a surprisingly high number of stellate neurons showing Gallyas positive material 133; re‐evaluation of the images (see Figure 1A in that paper) suggest that these would be now interpreted as tufted astrocytes. Hauw and colleagues mentioned tufts of abnormal fibers in PSP cases using the Bodian silver method and tau IHC 48, soon confirmed to be related to astrocytes 168, 170. Tufted astrocytes are characterized by the accumulation of phospho‐tau immunoreactivity in the proximal part of astrocytic processes that shows argyrophilia particularly with Gallyas staining 67. They are found mostly in the striatum, frontal and motor cortices. Ultrastructural observations described tubular profiles and suggested that tufted astrocytes are protoplasmic astrocytes 7, 124. Importantly, tufted astrocytes do not represent a reactive process but rather astrocytic degeneration, which does not seem to contribute to gliosis or neuronal loss, therefore its clinical significance has yet to be clarified 159. The interesting finding that tufted astrocytes are usually GFAP negative 65, 149 raises questions about whether they are really protoplasmic astrocytes, however, they are negative for another type of astrocyte, NG2, as well 149. It has been speculated that oxidative damage leads to GFAP fragmentation, explaining its lost, all together indicating dysfunction of pathological tau‐harboring protoplasmic astrocytes associated with neuronal dysfunction 142, 149. Contrasting NFTs with tufted astrocytes shows that the latter remain more localized in their distribution and may retain their original regular pattern of clustering in subcortical areas, and this is associated with a lack of spatial correlation that has be observed between neuronal and glial cell pathologies in specific regions 8. A further aspect on the origin of tufted astrocytes was highlighted by a study showing that these, together with astrocytic plaques of CBD, are positioned in close proximity to blood vessels 147. Tufted astrocytes were found to be closer to blood vessels than were astrocytic plaques 147. In the original paper by Nishimura et al on glial pathology in PSP, in addition to tufted astrocytes, argyrophilic masses with flame‐ or thorn‐like shape were described 124. TSA are GFAP positive 56 and they have received more focus in the context of aging‐related changes 72. Since PSP pathology can be seen associated with different clinical presentations 27, the distribution and amount of astrocytic pathology might vary between these forms, making the development of widely applicable staging system more difficult.
Astrocytic tau pathology in corticobasal degeneration
In 1994, a study by Wakabayashi et al reported that flame‐shaped glial inclusions revealed by Bielschowsky silver staining also showed IHC positivity for tau, but they remained undetectable with the Bodian silver method while the Gallyas silver staining was not used 165. Ultrastructurally, these inclusions were demonstrated to contain straight tubules and the affected cells were interpreted to be in oligodendroglia 165. Similar observations were reported by others at the same time 83. The observation of tau positive pathology resembling neuritic plaques, but without Aβ amyloid cores in CBD 105 was a first step toward the recognition that, apart from oligodendrocytes, tau appears in astroglia in CBD, mainly in the distal segments of astrocytic processes 32. Accordingly astrocytic plaques in CBD are defined as focal and densely tau‐immunoreactive stubby dilatations of distal processes of astrocytes giving an Aβ senile‐plaque‐like appearance without Aβ amyloid cores 72. Gallyas staining demonstrates astrocytic plaques clearly, and ultrastructurally, these are characterized by randomly arranged bundles of straight and twisted tubules with diameters of 15–20 nm 172. It has been noted that the number of astrocytic plaques may vary between cases; furthermore, description of so‐called incidental CBD cases suggested that indeed astrocytic tau pathology in the cortex might be one of the first pathological steps in the onset of CBD 95. This has been supported by observations in CBD cases with prominent neuronal tau pathology where astrocytes harboring tau immunoreactive pathology appear in areas without neuronal tau pathology 79. Indeed, considerable CBD pathology can exist in individuals without significant clinical symptoms suggesting that astrocytic plaques are not the major tau pathologies that are responsible for clinical symptomatology in CBD 104, 110. Komori et al reported that astrocytic plaques and tufted astrocytes, the two morphologically different types of astroglial inclusions found in CBD and PSP brains, respectively, do not coexist 66. Notably, CBD‐like and PSP‐like disorders are also caused by MAPT mutations that are pathogenic for hereditary tauopathies initially referred as frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP‐17). Interestingly, although protoplasmic astrocytes seem to be abnormal and reactive in CBD as in PSP, abnormal tau proteins accumulate in these astrocytes in PSP but not CBD 149.
Astrocytic tau pathology in argyrophilic grain disease
AGD has been described as a disorder showing accumulation of argyrophilic grains 14. In spite of the presence of argyrophilic oligodendroglial coiled bodies, astrocytic tau pathology does not show striking argyrophilia or dense inclusions as in PSP. The tau immunoreactive morphology has also been described as bush‐like astrocytes without glial fibrillary tangles 13. Furthermore, thin astrocytic plaques, periventricular and subpial astrocytes as well as small clusters of astrocytic processes have also been mentioned in reports on some AGD cases 38. Tau immunoreactive astrocytes vary between cases 38 and thought to become more abundant in later stages of AGD 139. Moreover, the significant morphological and anatomical overlap of these astrocytes with those seen in elderly individuals without argyrophilic grains supports the concept that these astrocytes are better interpreted as GFA representing gray matter ARTAG 72, 75, 79. Finally, one comprehensive study on AGD and PSP cases indicated that at least some of these GFA‐like morphologies (termed tufted astrocyte‐like astrocytic lesions in that study) can potentially evolve into Gallyas‐positive tufted astrocytes in AGD brains 58.
Astrocytic tau pathology in globular glial tauopathies
The concept of GGT unified glial‐predominant tauopathy disorders either with globular inclusions mainly in oligodendroglia or in astrocytes 1. Globular astroglial inclusions are defined as tau immunoreactive distinct globules (up to the size of the astroglial nucleus; 1–5 μm) and dots (1–2 μm) in the perikarya and proximal parts of astrocytic processes, found in the gray matter 72. These are somewhat reminiscent of tufted astrocytes, but they are non‐argyrophilic thereby contrasting not only with tufted astrocytes but also with the globular oligodendroglial inclusions of GGT 1, 67. GGT types are distinguished based on the anatomical involvement and the predominance of astroglial or oligodendroglial inclusions 1. In cases with predominantly white matter and oligodendroglial involvement, astrocytic globular inclusions are seen mostly in the frontal and temporal cortical areas, amygdala, striatum and medial thalamus 74. A subset of GGT cases involving frontotemporal, motor cortex and corticospinal tract show abundant astroglial inclusions 40.
Astrocytic tau pathology in Pick's disease
PiD is considered to show small amounts of astrocytic tau pathology, termed ramified astrocytes, when compared with neuronal tau pathology. However, there is considerable variability among reports on this in the literature. This can be due to the fact that before the introduction of isoform specific tau antibodies (3R and 4R) the definition of Pick's disease was not uniform 26, 80. Feany et al provided a precise description of the morphology of ramified astrocytes in Pick's disease, indicating that these astrocytic inclusions “tended to occupy more of the cell body and to ramify into the astrocytic cell processes,” and that they were “often localized to one side of the cell soma” 33. Ultrastructurally, the tau aggregates in ramified astrocytes are formed by straight tubules 65, and may show 3R immunoreactivity 36, 80, which is clearly different from PSP and CBD, where the astrocytic tau pathology is 4R isoform immunoreactive. Although ramified astrocytes appear in severely damaged cortical areas 59, 65, it has been suggested that astroglial tau pathology that is distinct from ramified astrocytes can appear in regions without neuronal inclusions 59, 79. For the sake of completeness it must be mentioned that glial inclusions in PiD with globular morphology occurring in the white matter are partly in oligodendrocytes 169 (usually smaller than the cell nucleus in contrast to the globular oligodendroglial inclusions of GGT with larger globules) 67, however, astrocytes in the subcortical white matter can also harbor globular tau inclusions 171. Ultrastructurally, these show juxtanuclear aggregates of abnormal filaments with straight tubules practically indistinguishable from those seen in neuronal Pick bodies 171.
Astrocytic tau pathology in MAPT mutations
FTDP‐17 or hereditary frontotemporal dementia associated with mutations in the MAPT gene shows a wide variety of tau pathologies 42. In addition to different tau positive neuronal inclusions, astroglial and oligodendroglial tau positive inclusions are described 43. Since some of these are reminiscent of the tau pathologies seen in primary FTLD‐tauopathies, papers on FTLDP‐17 often refer to these tau pathologies as MAPT mutation associated inclusions with disease phenotypes that resemble sporadic PSP, CBD, Pick's disease or GGT Although oligodendroglial inclusions in MAPT mutation cases are usually termed coiled bodies 69, meticulous descriptions of astrocytic tau inclusions are lacking in many papers while a lack of consensus terminology on astroglial tau pathologies also makes comparison of these publications difficult. Ghetti et al noted that the cell types involved vary according to the location of the MAPT gene mutation; usually mutations in exons 1, 10, 11, and 12 as well as introns following exons 9 and 10 show glial (i.e., oligo‐ and astroglial) inclusions 43. For example rare cortical tuft‐shaped astrocytes have been reported in exon 1 R5H mutation 49, numerous tufted astrocytes in the caudate, putamen, and thalamus in exon 1 R5L mutation 132. The L266V mutation is one of the rare mutations in exon 9 where astrocytic tau pathology has been described. Kobayashi et al reported unique features of these frequent tau positive astrocytic inclusions in the cerebral cortex, some which were stained positively with the Bodian silver method, but they were rarely stained with Gallyas–Braak method, and other inclusions showing fibrillary morphologies were strongly stained with Bodian and Gallyas–Braak methods 63. Hogg et al noted coarse astrocytic tau immunoreactivity in the cortex in another case with this mutation 52. Tufted astrocytes and tau‐immunoreactive inclusions have been reported in introns 9 and 10 MAPT mutations 43, 103. Exon 10 mutations are not infrequently associated with astrocytic tau pathologies; however, the details of the descriptions vary. In the N279K MAPT mutation cases mostly oligodendroglial tau‐inclusions are seen, and only rare astrocytic tau positive inclusions have been noted but without showing a clear similarity to tufted astrocytes or astrocytic plaques 25, 43, 134. Glial tau pathology was mentioned but not specified whether being astrocytic in L284L 29 and N296N 150 mutations, while prominent granular astrocytic tau inclusions (but not like tufted astrocytes) have been mentioned in association with cortical neuronal loss in a N296H MAPT mutation case 60. Furthermore, a wide range of morphologies of astrocytic tau inclusions, referred to as tufted forms, have been linked to the deln296 MAPT mutation 37. The P301L, P301S, and P301T MAPT mutations are associated with a wide range of tau positive glial inclusion morphologies described as tufted astrocytes and astrocytic plaques 43 while in P301L globular astrocytic inclusions (as in GGT) and GFA (as in ARTAG) have been emphasized 155. Unspecified intense tau immunostaining in affected astrocytes has been mentioned in a G303V MAPT mutation case 138 and also in S305N MAPT mutation cases 54, 62. The S305S mutation shows a spectrum of different astrocytic tau immunoreactivities, termed glial tangles, tufted astrocytes 152 and astrocytic plaques 47 by the authors. Unspecified extensive astrocytic tau deposits have been reported in the exon 11 L315V MAPT mutation 162. Prominent astroglial tau immunoreactive inclusions have been reported to be associated with the K317M MAPT mutation 174; a later study on the K317N MAPT mutation, which described globular astroglial inclusions similar to GGT, pointed out the resemblance of the astrocytic and oligodendroglial tau inclusions in the K317M MAPT mutation cases with that of GGT 154. In the exon 12 G335S MAPT mutation cases, astrocytes containing granular and/or punctate tau‐immunoreactive deposits in cell bodies and the proximal portion of processes (reminiscent of tufted astrocytes) or in the distal part only (reminiscent of astrocytic plaques) have been described 151. These resemble GFA of gray matter ARTAG supporting the concept that some of these might be pre‐mature forms of astrocytic tau pathologies 79. Interestingly, further ARTAG types (subpial TSA), together with cortical astrocytic plaques, have been observed in a 49‐year‐old demented individual with MAPT gene duplication 3, 91. This case indicates that an imbalance of tau homeostasis contributes to the development of a clearly age‐related pathology as observed earlier with mutations that alter the 3R:4R tau isoform ratio 53. Further descriptions of ARTAG type pathology in other MAPT mutations are lacking. Finally, astrocytic tau immunoreactive inclusions without specification of the morphology, have been reported in the cerebral cortex and white matter in the Q336R MAPT mutation 130, in the frontal white matter in E342V MAPT mutation 96, and astrocytes interpreted as tufted astrocytes by the authors in the K369I MAPT mutation 123. Notably, the concomitant presence of astrocytic plaques and tufted astrocytes, a phenomenon not recognized in primary FTLD‐tauopathies 66, has also been reported in MAPT mutation cases. In summary, much is known about the spectrum of MAPT mutation‐associated astroglial tau pathologies but little is known how the astrocytic tau pathology relates to primary FTLD‐tauopathies, and there is a lack of harmonized terminology in the reports on these pathologies.
Astrocytic tau pathology in aging: ARTAG
In the aging brain astrocytic tau pathology has been described in the subpial and subependymal regions of the gray and white matter and frequently in the depths of gyri, as well as in the basal forebrain and brainstem, morphologically in the form of TSA 55, 56, 57. Similarity to some astrocytes described in PSP 124 has also been noted. Later Schultz et al reported a high prevalence of TSA in aged human, particularly at the level of the amygdala; interestingly similar astroglial tau pathology has also been observed in baboons 144, 146. The study by the MRC‐CFAS group supported these findings and mentioned additionally that less commonly TSA can be observed also in the vicinity of neuronal cell bodies in gray matter areas such as amygdala and dentate gyrus 86. TSA have been considered as non‐specific but are relatively underreported in studies since neuron‐related tau pathology, such as NFTs and AGD seemed to be more relevant age‐related tau pathologies 23, 38. Moreover, the descriptions focused on the medial temporal lobe and did not provide details on further tau astroglial tau pathologies such as those in the gray matter. Importantly, Munoz et al described “argyrophilic thorny astrocyte clusters (ATACs)” in the frontal, temporal, and parietal cortices and in subcortical white matter in a cohort of patients with nonfluent variant of primary progressive aphasia associated with AD pathology and discussed the possibility that these might have clinical relevance 115. The question whether gray matter astroglial tau pathologies have any relevance for clinical symptoms was further highlighted in a study reporting a peculiar constellation of tau pathology in cases wherein diffuse granular immunopositivity of astrocytic processes and patchy accumulation of thin threads were observed in a distinctive distribution (frontal and temporal cortices, hippocampus, amygdala, basal ganglia, locus coeruleus, and substantia nigra) 76. In a follow‐up of this study on a community based‐cohort gray matter tau astrogliopathy was grouped into four groups based on the anatomical involvement and morphological appearance: Group I (medial temporal lobe with few astrocytes with granular tau); Group II (amygdala with granular, thorny and tufted‐like astrogliopathy type); Group III (limbic regions‐basal ganglia‐substantia nigra with granular and thorny astrogliopathy); and Group IV (hippocampal‐dentate gyrus‐amygdala predominant with granular and thorny astrogliopathy). These tau pathologies were either not (e.g., Group I) or were associated with various constellations of clinical symptoms 75. Together with a paper on focal glial tau pathology associated with progressive aphasia 166, these studies argued that tau astrogliopathies in the gray matter might reflect neuronal dysfunction leading to clinical symptoms. In a series of studies Ferrer and colleagues demonstrated that these tau astrogliopathies in the white and gray matter show a distinct biochemical signature such as inconsistent detectability using phospho‐specific anti‐tau antibody Ser262 or conformational tau modifications at amino acids 312 to 322 (MC1), or tau truncated at aspartic acid 421 (tau‐C3) 36, 98, 142. Further reports in elderly patients exemplified by the description of isolated tufted astrocytes in the occipitotemporal gyrus in a population‐representative cohort 86, or prominent subcortical white matter astrocytic tau pathology in brains from two elderly patients in whom CBD was considered 140, emphasized the need for the harmonization of terminology and evaluation strategies. Therefore, the umbrella term ARTAG has been introduced to encompass all of these aging‐related astroglial tau pathologies, with or without accompanying morphological features of other NDD, including primary FTLD‐tauopathies 72. Accordingly, ARTAG should be considered when detecting either TSA or GFA (Figure 4A–F). TSA represents tau immunoreactivity in astrocytic perikarya with extension into the proximal parts of the astrocytic processes, as well as tau positive inclusions in the astrocytic endfeet at the glia limitans around blood vessels and at the pial surface; GFA represents fine granular immunoreactivity of branching processes of gray matter astrocytes where the perinuclear soma is densely immunoreactive in most of these 72. A four‐step characterization algorithm called TReSS to signify Type, Regional involvement, Severity, Subregional involvement has been proposed: 1 identification of five types based on the location of either morphologies of tau astrogliopathy: subpial, subependymal, perivascular, white matter, gray matter; 2 documentation of the regional involvement: medial temporal lobe, lobar, subcortical, and brainstem; 3 documentation of the severity of tau astrogliopathy; and 4 description of subregional involvement 72. A recent multisite study proposed more specified description of the severity 82. Furthermore, the latter study suggested that the spectrum of coexisting pathological astrocytic tau immunoreactivities might be wider than generally assumed in primary FTLD‐Tau disorders if more care is taken to describe these lesions 82. ARTAG (except for the gray matter type) shows a predilection for the basal brain regions. A study focusing on the anterior basal forebrain showed that ARTAG is most prevalent in the subpial location (39.13%) followed by a perivascular distribution (30.43%) and the authors of this study emphasized that ARTAG might share common pathways with chronic traumatic encephalopathy (CTE) 97. A comprehensive study on ARTAG reported the systematic mapping of ARTAG types in different anatomical regions in more than six hundred cases and correlated these with clinicopathological and genetic variables 79. This study further highlighted the considerable overlap of ARTAG with CTE and primary FTLD‐tauopathy‐related tau pathologies presenting a concept that some of the gray matter ARTAG may represent early forms of primary FTLD‐tauopathies 79. A notable observation was the strong association of AD‐related variables with the presence of lobar white matter ARTAG. Furthermore, five constellations were recognized: (i) Subpial ± white matter ± perivascular ARTAG in basal brain areas and medial temporal lobe; (ii) Subpial ± white matter ± perivascular ARTAG in the brainstem; (iii) Subpial ± perivascular ± gray matter ARTAG in lobar areas; (iv) White matter ± perivascular ARTAG in lobar areas; (v) Gray matter ARTAG in limbic ± neocortical ± subcortical ± brainstem areas with or without features of primary FTLD‐tauopathies. These constellations might reflect different etiologies or eventually different entities 79. Finally, a remarkable mix of neuronal and astroglial tauopathy compatible with ARTAG has been described in the mammillary bodies in AD patients 131 distinct from the sex‐dependent cytoskeletal changes consisting of NFTs, a network of dystrophic neurites, and terminal‐like vessel‐associated processes seen in the infundibular hypothalamic nucleus in elderly men 143, 145.
Figure 4.
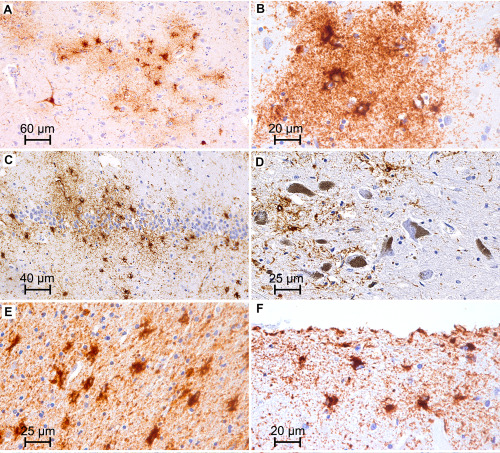
Examples of astroglial tau immunoreactivities in the aging brain. A,B. Clusters of granular/fuzzy astrocytes in the gray matter (accumbens nucleus). C. Thorny astrocytes in the dentate gyrus. D. Granular/fuzzy astrocytes in the substantia nigra. E. Thorn‐shaped astrocytes in the temporal white matter. F. Subpial thorn‐shaped astrocytes.
Further astrocytic tau pathologies
As previously mentioned, astrocytic tau pathology is an important component of the morphological alterations reported in CTE, a disorder associated with repetitive brain trauma and progressive neurological deterioration 106, 107, 108. Indeed, there are many overlaps with ARTAG, such as accumulation of subpial astrocytes in basal brain regions, and also in dorsolateral lobar areas, and additionally an overrepresentation of males in both CTE and ARTAG, or association of ARTAG with ventricular enlargement, an alteration seen in CTE as well 79, 97, 106, 107, 108. Therefore, for the definition of CTE‐associated lesions the presence of neuronal tau pathology is important to emphasize 106 while the presence of pure subpial of cortical clusters of astrocytic tau immunoreactivities should not be at once interpreted as CTE. Using strict criteria and standardized approaches can help to distinguish the rare CTE‐like pathology from the frequent ARTAG‐related tau pathology in the diagnostic practice 125 or research studies 64.
Tufted astrocytes in PSP or other astrocytic tau pathologies usually are less prominent or spare the cornu ammonis subregions of the hippocampus. In this context it is important to mention recent reports on cases with or without PSP pathology and peculiar 4R isoform tau immunoreactive spherical neuronal inclusions in the hippocampus, which show prominent argyrophilic astrocytic tau pathology in the hippocampus 73. Unexpectedly, a genetic prion disease associated with the V203I mutation in the prion protein gene (PRNP) also showed hippocampal astrocytic tau pathology reminiscent of these but without the neuronal inclusions 78. Finally, a recent study on a familial behavioral variant frontotemporal in two sisters born from consanguineous parents described prominent astrocytic tau pathology 35. The tau‐positive astrocytes were reminiscent of reactive astrocytes but in addition to the cytoplasm, perivascular foot processes around the majority of cortical blood vessels were decorated with heavy deposits of hyper‐phosphorylated tau, massively involving cortical, hippocampal and subcortical regions 35. In addition, cerebellar Bergmann glia were also heavily stained with the AT8 antibody 35. These studies indicate also that we are far from understanding the variability of tau accumulations in different populations of astrocytes in different anatomical regions.
Conceptual summary of astrocytic tau pathology
Tau mRNA is found in neurons in the human brain 44 suggesting that astrocytic tau immunoreactivity could be reflecting purely uptake of tau from neurons. However, astrocytic tau pathology is seen in regions without other types of tau pathology 79, 95. Observations of preclinical and incidental forms of primary FTLD‐tauopathies 31, 75, 95, 104, 110, 173 can help to elucidate the pathogenic role of astrocytes. Recent studies suggest that the first step of astrocytic pathology might be the fine granular accumulation in astrocytic processes; these tau deposits are then transported to distal or proximal segment of the astrocytic cytoskeleton and eventually aggregate, become argyrophilic and/or ubiquitinated 58, 79. Therefore, pure detection of single astrocytes with fine granular (dot‐like) phospho‐tau immunoreactivity in the human brain might be just a less likely and potentially transient expression of a phospho‐tau epitope as a reaction to a yet to be identified pathogenic event, however, it can represent an early preclinical form of primary FTLD‐tauopathy or ARTAG (Figure 3) 79. In particular, that the regional appearance of these single astrocytes usually overlap with the anatomical regions involved in primary FTLD tauopathies and ARTAG, moreover, gray matter ARTAG also overlaps with the involvement patterns of primary FTLD‐tauopathies and potentially could represent early subtypes of those 79.
Concluding Remarks
Immunohistochemical and molecular biological methods highlighted the wide spectrum and involvement of astrocytes in the disease protein pathology characteristic of NDD. Moreover, there are further protein deposits associated with astrocytes, such as those seen in the hyaline protoplasmic astrocytopathy of neocortex usually detected in the clinical setting of epilepsy and/or psychomotor retardation in younger individuals 50. Apart from clearly distinct PAG morphologies and staining patterns such as argyrophilia, there are peculiar similarities to be noted. For example, subpial accumulation of PAG is seen in MSA (α‐syn), PSP, CBD, ARTAG, and CTE (tau) and AD (Aβ). Subependymal PAG in seen in MSA (α‐syn) and ARTAG (tau). In the gray matter GFA (tau) or morphologies reminiscent of GFA are seen in PD/DLB (α‐syn) and rarely in prion disease.
What can be the role and consequence of astrocytic protein pathology? The fine granular (non‐argyrophilic) morphology of protein immunoreactivity in astrocytes seen in prion diseases (prion protein), Lewy body disorders (α‐syn) and tauopathies (tau) raises the hypothesis that common pathogenic steps might underlie these astroglial pathologies (Figures 2 and 5), such as phagocytosis of pathologically altered proteins. Indeed, astrocytes have been found to highly express an array of phagocytic receptors exemplified by their contribution to phagocytizing synapses 21 or axonal mitochondria 24 in the brain. Experimental studies in tau transgenic mouse model of astrocytic tau pathologies suggest that this pathology contributes to glial degeneration 51; furthermore as functional consequence of astrocytic tau pathology neuronal degeneration can be detected in the absence of neuronal tau inclusions 39. Moreover, astrocytes play an important role in the clearance of toxic α‐syn species from the extracellular space 94 and uptake and spreading of α‐syn from astrocytes to neurons can lead to neuronal death 20. Glial cells are discussed as mediators of Aβ removal from the brain, and many of the proteins involved in the enzymatic degradation of Aβ are produced by glial cells 135 so they should be able to endocytose monomeric and oligomeric Aβ and degrade them 89. Thus, astroglial NDD proteinopathies might reflect their contribution to disease spreading or clearance of disease‐associate proteins, and, depending on the disease protein (i.e., tau), it might lead to astroglial dysfunction and eventually contribute to the degenerations of neurons.
Figure 5.
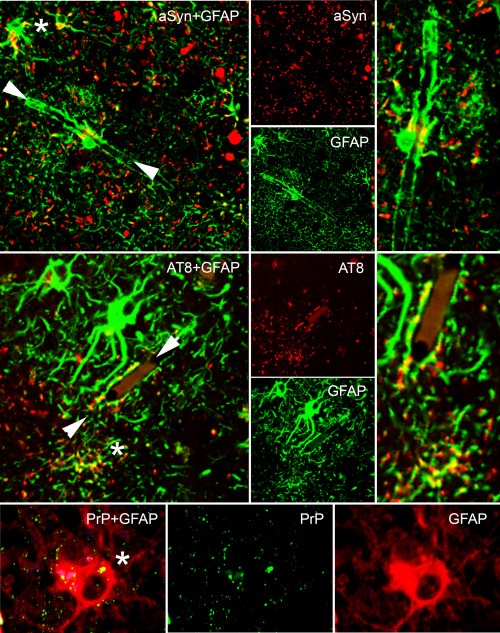
Overlapping immunoreactive patterns in PAG. Astroglial granular immunoreactivity (indicated by asterisk) can be seen for example for α‐synuclein, tau, and PrP, moreover astroglial (see enlarged image in the right indicated by white arrowheads in the left images) end‐feets show immunoreactivity in α‐synucleinopathies or tauopathies.
In conclusion, the studies presented here and in other review articles 34, 163 show that astrocytes and oligodendroglial cells 136, 137 may have an underappreciated spectrum of roles to play in mechanisms NDD. Thus, the time may now have arrived for astrocytes and oligodendroglia to receive more attention and become the focus of studies of NDD mechanisms as well for investigating potential NDD therapies.
Conflict of Interest
Authors report no conflict of interest.
Acknowledgments
Support for this work was provided by grants from the National Institute on Aging of the National Institutes of Health (P30‐AG10124, PO1‐AG17586, NS088341, and NS094003).
References
- 1. Ahmed Z, Bigio EH, Budka H, Dickson DW, Ferrer I, Ghetti B et al (2013) Globular glial tauopathies (GGT): consensus recommendations. Acta Neuropathol 126:537–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Aho L, Pikkarainen M, Hiltunen M, Leinonen V, Alafuzoff I (2010) Immunohistochemical visualization of amyloid‐beta protein precursor and amyloid‐beta in extra‐ and intracellular compartments in the human brain. J Alzheimer's Dis 20:1015–1028. [DOI] [PubMed] [Google Scholar]
- 3. Alexander J, Kalev O, Mehrabian S, Traykov L, Raycheva M, Kanakis D et al (2016) Familial early‐onset dementia with complex neuropathologic phenotype and genomic background. Neurobiol Aging 42:199–204. [DOI] [PubMed] [Google Scholar]
- 4. Alzheimer A (1911) Über eigenartige Krankheitsfälle des späten Alters. Zeitschr Gesamte Neurol Psychiat 1911:365–385. [Google Scholar]
- 5. Arai N, Oda M (1996) A variety of glial pathological structures by the modified Gallyas–Braak method. Neuropathology 16:133–138. [Google Scholar]
- 6. Arai T, Ueda K, Ikeda K, Akiyama H, Haga C, Kondo H et al (1999) Argyrophilic glial inclusions in the midbrain of patients with Parkinson's disease and diffuse Lewy body disease are immunopositive for NACP/alpha‐synuclein. Neurosci Lett 259:83–86. [DOI] [PubMed] [Google Scholar]
- 7. Arima K (2006) Ultrastructural characteristics of tau filaments in tauopathies: immuno‐electron microscopic demonstration of tau filaments in tauopathies. Neuropathology 26:475–483. [DOI] [PubMed] [Google Scholar]
- 8. Armstrong RA, Cairns NJ (2013) Spatial patterns of the tau pathology in progressive supranuclear palsy. Neurol Sci 34:337–344. [DOI] [PubMed] [Google Scholar]
- 9. Armstrong RA, Gearing M, Bigio EH, Cruz‐Sanchez FF, Duyckaerts C, Mackenzie IR et al (2011) The spectrum and severity of FUS‐immunoreactive inclusions in the frontal and temporal lobes of ten cases of neuronal intermediate filament inclusion disease. Acta Neuropathol 121:219–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ash PE, Bieniek KF, Gendron TF, Caulfield T, Lin WL, Dejesus‐Hernandez M et al (2013) Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 77:639–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Beach TG, Adler CH, Lue L, Sue LI, Bachalakuri J, Henry‐Watson J et al (2009) Unified staging system for Lewy body disorders: correlation with nigrostriatal degeneration, cognitive impairment and motor dysfunction. Acta Neuropathol 117:613–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Boluda S, Iba M, Zhang B, Raible KM, Lee VM, Trojanowski JQ (2015) Differential induction and spread of tau pathology in young PS19 tau transgenic mice following intracerebral injections of pathological tau from Alzheimer's disease or corticobasal degeneration brains. Acta Neuropathol 129:221–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Botez G, Probst A, Ipsen S, Tolnay M (1999) Astrocytes expressing hyperphosphorylated tau protein without glial fibrillary tangles in argyrophilic grain disease. Acta Neuropathol 98:251–256. [DOI] [PubMed] [Google Scholar]
- 14. Braak H, Braak E (1987) Argyrophilic grains: characteristic pathology of cerebral cortex in cases of adult onset dementia without Alzheimer changes. Neurosci Lett 76:124–127. [DOI] [PubMed] [Google Scholar]
- 15. Braak H, Braak E (1991) Neuropathological stageing of Alzheimer‐related changes. Acta Neuropathol 82:239–259. [DOI] [PubMed] [Google Scholar]
- 16. Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E (2003) Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging 24:197–211. [DOI] [PubMed] [Google Scholar]
- 17. Braak H, Sastre M, Del Tredici K (2007) Development of alpha‐synuclein immunoreactive astrocytes in the forebrain parallels stages of intraneuronal pathology in sporadic Parkinson's disease. Acta Neuropathol 114:231–241. [DOI] [PubMed] [Google Scholar]
- 18. Brettschneider J, Del Tredici K, Lee VM, Trojanowski JQ (2015) Spreading of pathology in neurodegenerative diseases: a focus on human studies. Nat Rev Neurosci 16:109–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Busciglio J, Gabuzda DH, Matsudaira P, Yankner BA (1993) Generation of beta‐amyloid in the secretory pathway in neuronal and nonneuronal cells. Proc Natl Acad Sci USA 90:2092–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cavaliere F, Cerf L, Dehay B, Ramos‐Gonzalez P, De Giorgi F, Bourdenx M et al (2017) In vitro alpha‐synuclein neurotoxicity and spreading among neurons and astrocytes using Lewy body extracts from Parkinson disease brains. Neurobiol Dis 103:101–112. [DOI] [PubMed] [Google Scholar]
- 21. Chung WS, Clarke LE, Wang GX, Stafford BK, Sher A, Chakraborty C et al (2013) Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature 504:394–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Clavaguera F, Akatsu H, Fraser G, Crowther RA, Frank S, Hench J et al (2013) Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc Natl Acad Sci USA 110:9535–9540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Crary JF, Trojanowski JQ, Schneider JA, Abisambra JF, Abner EL, Alafuzoff I et al (2014) Primary age‐related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol 128:755–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Davis CH, Kim KY, Bushong EA, Mills EA, Boassa D, Shih T et al (2014) Transcellular degradation of axonal mitochondria. Proc Natl Acad Sci USA 111:9633–9638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Delisle MB, Murrell JR, Richardson R, Trofatter JA, Rascol O, Soulages X et al (1999) A mutation at codon 279 (N279K) in exon 10 of the Tau gene causes a tauopathy with dementia and supranuclear palsy. Acta Neuropathol 98:62–77. [DOI] [PubMed] [Google Scholar]
- 26. Dickson DW (1998) Pick's disease: a modern approach. Brain Pathol 8:339–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dickson DW, Ahmed Z, Algom AA, Tsuboi Y, Josephs KA (2010) Neuropathology of variants of progressive supranuclear palsy. Curr Opin Neurol 23:394–400. [DOI] [PubMed] [Google Scholar]
- 28. Diedrich JF, Bendheim PE, Kim YS, Carp RI, Haase AT (1991) Scrapie‐associated prion protein accumulates in astrocytes during scrapie infection. Proc Natl Acad Sci USA 88:375–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. D'Souza I, Poorkaj P, Hong M, Nochlin D, Lee VM, Bird TD, Schellenberg GD (1999) Missense and silent tau gene mutations cause frontotemporal dementia with parkinsonism‐chromosome 17 type, by affecting multiple alternative RNA splicing regulatory elements. Proc Natl Acad Sci USA 96:5598–5603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Duyckaerts C, Delatour B, Potier MC (2009) Classification and basic pathology of Alzheimer disease. Acta Neuropathol 118:5–36. [DOI] [PubMed] [Google Scholar]
- 31. Evidente VG, Adler CH, Sabbagh MN, Connor DJ, Hentz JG, Caviness JN et al (2011) Neuropathological findings of PSP in the elderly without clinical PSP: possible incidental PSP? Parkinsonism Relat Disord 17:365–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Feany MB, Dickson DW (1995) Widespread cytoskeletal pathology characterizes corticobasal degeneration. Am J Pathol 146:1388–1396. [PMC free article] [PubMed] [Google Scholar]
- 33. Feany MB, Mattiace LA, Dickson DW (1996) Neuropathologic overlap of progressive supranuclear palsy, Pick's disease and corticobasal degeneration. J Neuropathol Exp Neurol 55:53–67. [DOI] [PubMed] [Google Scholar]
- 34. Ferrer I (2017) Diversity of astroglial responses across human neurodegenerative disorders and brain aging. Brain Pathol. doi: 10.1111/bpa.12538. [DOI] [PMC free article] [PubMed]
- 35. Ferrer I, Legati A, Garcia‐Monco JC, Gomez‐Beldarrain M, Carmona M, Blanco R et al (2015) Familial behavioral variant frontotemporal dementia associated with astrocyte‐predominant tauopathy. J Neuropathol Exp Neurol 74:370–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ferrer I, Lopez‐Gonzalez I, Carmona M, Arregui L, Dalfo E, Torrejon‐Escribano B et al (2014) Glial and neuronal tau pathology in tauopathies: characterization of disease‐specific phenotypes and tau pathology progression. J Neuropathol Exp Neurol 73:81–97. [DOI] [PubMed] [Google Scholar]
- 37. Ferrer I, Pastor P, Rey MJ, Munoz E, Puig B, Pastor E et al (2003) Tau phosphorylation and kinase activation in familial tauopathy linked to deln296 mutation. Neuropathol Appl Neurobiol 29:23–34. [DOI] [PubMed] [Google Scholar]
- 38. Ferrer I, Santpere G, van Leeuwen FW (2008) Argyrophilic grain disease. Brain 131:1416–1432. [DOI] [PubMed] [Google Scholar]
- 39. Forman MS, Lal D, Zhang B, Dabir DV, Swanson E, Lee VM, Trojanowski JQ (2005) Transgenic mouse model of tau pathology in astrocytes leading to nervous system degeneration. J Neurosci 25:3539–3550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Fu YJ, Nishihira Y, Kuroda S, Toyoshima Y, Ishihara T, Shinozaki M et al (2010) Sporadic four‐repeat tauopathy with frontotemporal lobar degeneration, Parkinsonism, and motor neuron disease: a distinct clinicopathological and biochemical disease entity. Acta Neuropathol 120:21–32. [DOI] [PubMed] [Google Scholar]
- 41. Funato H, Yoshimura M, Yamazaki T, Saido TC, Ito Y, Yokofujita J et al (1998) Astrocytes containing amyloid beta‐protein (Abeta)‐positive granules are associated with Abeta40‐positive diffuse plaques in the aged human brain. Am J Pathol 152:983–992. [PMC free article] [PubMed] [Google Scholar]
- 42. Ghetti B, Oblak AL, Boeve BF, Johnson KA, Dickerson BC, Goedert M (2015) Invited review: frontotemporal dementia caused by microtubule‐associated protein tau gene (MAPT) mutations: a chameleon for neuropathology and neuroimaging. Neuropathol Appl Neurobiol 41:24–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ghetti B, Wszolek Z, Boeve B, Spina S, Goedert M (2011) Frontotemporal dementia and parkinsonism linked to chromosome 17. In: Neurodegeneration: The Molecular Pathology of Dementia and Movement Disorders, Dickson DW, Weller RO (eds), pp. 110–134. Blackwell: Chichester. [Google Scholar]
- 44. Goedert M, Spillantini MG, Potier MC, Ulrich J, Crowther RA (1989) Cloning and sequencing of the cDNA encoding an isoform of microtubule‐associated protein tau containing four tandem repeats: differential expression of tau protein mRNAs in human brain. EMBO J 8:393–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Guo JL, Lee VM (2014) Cell‐to‐cell transmission of pathogenic proteins in neurodegenerative diseases. Nat Med 20:130–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gyure KA, Durham R, Stewart WF, Smialek JE, Troncoso JC (2001) Intraneuronal Abeta‐amyloid precedes development of amyloid plaques in Down syndrome. Arch Pathol Lab Med 125:489–492. [DOI] [PubMed] [Google Scholar]
- 47. Halliday GM, Song YJ, Creasey H, Morris JG, Brooks WS, Kril JJ (2006) Neuropathology in the S305S tau gene mutation. Brain 129:E40. [DOI] [PubMed] [Google Scholar]
- 48. Hauw JJ, Verny M, Delaere P, Cervera P, He Y, Duyckaerts C (1990) Constant neurofibrillary changes in the neocortex in progressive supranuclear palsy. Basic differences with Alzheimer's disease and aging. Neurosci Lett 119:182–186. [DOI] [PubMed] [Google Scholar]
- 49. Hayashi S, Toyoshima Y, Hasegawa M, Umeda Y, Wakabayashi K, Tokiguchi S et al (2002) Late‐onset frontotemporal dementia with a novel exon 1 (Arg5His) tau gene mutation. Ann Neurol 51:525–530. [DOI] [PubMed] [Google Scholar]
- 50. Hedley‐Whyte ET, Goldman JE, Nedergaard M, Friedman A, Han X, Schmidt RE, Powers JM (2009) Hyaline protoplasmic astrocytopathy of neocortex. J Neuropathol Exp Neurol 68:136–147. [DOI] [PubMed] [Google Scholar]
- 51. Higuchi M, Ishihara T, Zhang B, Hong M, Andreadis A, Trojanowski J, Lee VM (2002) Transgenic mouse model of tauopathies with glial pathology and nervous system degeneration. Neuron 35:433–446. [DOI] [PubMed] [Google Scholar]
- 52. Hogg M, Grujic ZM, Baker M, Demirci S, Guillozet AL, Sweet AP et al (2003) The L266V tau mutation is associated with frontotemporal dementia and Pick‐like 3R and 4R tauopathy. Acta Neuropathol 106:323–336. [DOI] [PubMed] [Google Scholar]
- 53. Hong M, Zhukareva V, Vogelsberg‐Ragaglia V, Wszolek Z, Reed L, Miller BI et al (1998) Mutation‐specific functional impairments in distinct tau isoforms of hereditary FTDP‐17. Science 282:1914–1917. [DOI] [PubMed] [Google Scholar]
- 54. Iijima M, Tabira T, Poorkaj P, Schellenberg GD, Trojanowski JQ, Lee VM et al (1999) A distinct familial presenile dementia with a novel missense mutation in the tau gene. NeuroReport 10:497–501. [DOI] [PubMed] [Google Scholar]
- 55. Ikeda K (1996) Glial fibrillary tangles and argyrophilic threads: classification and disease specificity. Neuropathology 16:71–77. [Google Scholar]
- 56. Ikeda K, Akiyama H, Arai T, Nishimura T (1998) Glial tau pathology in neurodegenerative diseases: their nature and comparison with neuronal tangles. Neurobiol Aging 19:S85–S91. [DOI] [PubMed] [Google Scholar]
- 57. Ikeda K, Akiyama H, Kondo H, Haga C, Tanno E, Tokuda T, Ikeda S (1995) Thorn‐shaped astrocytes: possibly secondarily induced tau‐positive glial fibrillary tangles. Acta Neuropathol 90:620–625. [DOI] [PubMed] [Google Scholar]
- 58. Ikeda C, Yokota O, Nagao S, Ishizu H, Oshima E, Hasegawa M et al (2016) The relationship between development of neuronal and astrocytic tau pathologies in subcortical nuclei and progression of argyrophilic grain disease. Brain Pathol 26:488–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Irwin DJ, Brettschneider J, McMillan CT, Cooper F, Olm C, Arnold SE et al (2016) Deep clinical and neuropathological phenotyping of Pick disease. Ann Neurol 79:272–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Iseki E, Matsumura T, Marui W, Hino H, Odawara T, Sugiyama N et al (2001) Familial frontotemporal dementia and parkinsonism with a novel N296H mutation in exon 10 of the tau gene and a widespread tau accumulation in the glial cells. Acta Neuropathol 102:285–292. [DOI] [PubMed] [Google Scholar]
- 61. Jucker M, Walker LC (2013) Self‐propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 501:45–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kobayashi K, Kidani T, Ujike H, Hayashi M, Ishihara T, Miyazu K et al (2003) Another phenotype of frontotemporal dementia and parkinsonism linked to chromosome‐17 (FTDP‐17) with a missense mutation of S305N closely resembling Pick's disease. J Neurol 250:990–992. [DOI] [PubMed] [Google Scholar]
- 63. Kobayashi T, Ota S, Tanaka K, Ito Y, Hasegawa M, Umeda Y et al (2003) A novel L266V mutation of the tau gene causes frontotemporal dementia with a unique tau pathology. Ann Neurol 53:133–137. [DOI] [PubMed] [Google Scholar]
- 64. Koga S, Dickson DW, Bieniek KF (2016) Chronic traumatic encephalopathy pathology in multiple system atrophy. J Neuropathol Exp Neurol 75:963–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Komori T (1999) Tau‐positive glial inclusions in progressive supranuclear palsy, corticobasal degeneration and Pick's disease. Brain Pathol 9:663–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Komori T, Arai N, Oda M, Nakayama H, Mori H, Yagishita S et al (1998) Astrocytic plaques and tufts of abnormal fibers do not coexist in corticobasal degeneration and progressive supranuclear palsy. Acta Neuropathol 96:401–408. [DOI] [PubMed] [Google Scholar]
- 67. Kovacs GG (2015) Invited review: neuropathology of tauopathies: principles and practice. Neuropathol Appl Neurobiol 41:3–23. [DOI] [PubMed] [Google Scholar]
- 68. Kovacs GG (2016) Molecular pathological classification of neurodegenerative diseases: turning towards precision medicine. Int J Mol Sci 17. pii: E189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kovacs GG (2017) Globular glial inclusions unveil enigmas of MAPT mutations. Neuropathol Appl Neurobiol 43:191–193. [DOI] [PubMed] [Google Scholar]
- 70. Kovacs GG, Breydo L, Green R, Kis V, Puska G, Lorincz P et al (2014) Intracellular processing of disease‐associated alpha‐synuclein in the human brain suggests prion‐like cell‐to‐cell spread. Neurobiol Dis 69:76–92. [DOI] [PubMed] [Google Scholar]
- 71. Kovacs GG, Budka H (2009) Molecular pathology of human prion diseases. Int J Mol Sci 10:976–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kovacs GG, Ferrer I, Grinberg LT, Alafuzoff I, Attems J, Budka H et al (2016) Aging‐related tau astrogliopathy (ARTAG): harmonized evaluation strategy. Acta Neuropathol 131:87–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kovacs GG, Kwong LK, Grossman M, Irwin DJ, Lee EB, Robinson JL et al (2016) Tauopathy with hippocampal 4‐repeat tau immunoreactive spherical inclusions: a report of three cases. Brain Pathol. doi: 10.1111/bpa.12482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kovacs GG, Majtenyi K, Spina S, Murrell JR, Gelpi E, Hoftberger R et al (2008) White matter tauopathy with globular glial inclusions: a distinct sporadic frontotemporal lobar degeneration. J Neuropathol Exp Neurol 67:963–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Kovacs GG, Milenkovic I, Wohrer A, Hoftberger R, Gelpi E, Haberler C et al (2013) Non‐Alzheimer neurodegenerative pathologies and their combinations are more frequent than commonly believed in the elderly brain: a community‐based autopsy series. Acta Neuropathol 126:365–384. [DOI] [PubMed] [Google Scholar]
- 76. Kovacs GG, Molnar K, Laszlo L, Strobel T, Botond G, Honigschnabl S et al (2011) A peculiar constellation of tau pathology defines a subset of dementia in the elderly. Acta Neuropathol 122:205–222. [DOI] [PubMed] [Google Scholar]
- 77. Kovacs GG, Preusser M, Strohschneider M, Budka H (2005) Subcellular localization of disease‐associated prion protein in the human brain. Am J Pathol 166:287–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Kovacs GG, Rahimi J, Strobel T, Lutz MI, Regelsberger G, Streichenberger N et al (2017) Tau pathology in Creutzfeldt‐Jakob disease revisited. Brain Pathol 27:332–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Kovacs GG, Robinson JL, Xie SX, Lee EB, Grossman M, Wolk DA et al (2017) Evaluating the patterns of aging‐related tau astrogliopathy unravels novel insights into brain aging and neurodegenerative diseases. J Neuropathol Exp Neurol 76:270–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Kovacs GG, Rozemuller AJ, van Swieten JC, Gelpi E, Majtenyi K, Al‐Sarraj S et al (2013) Neuropathology of the hippocampus in FTLD‐Tau with Pick bodies: a study of the BrainNet Europe Consortium. Neuropathol Appl Neurobiol 39:166–178. [DOI] [PubMed] [Google Scholar]
- 81. Kovacs GG, Wagner U, Dumont B, Pikkarainen M, Osman AA, Streichenberger N et al (2012) An antibody with high reactivity for disease‐associated alpha‐synuclein reveals extensive brain pathology. Acta Neuropathol 124:37–50. [DOI] [PubMed] [Google Scholar]
- 82. Kovacs GG, Xie SX, Lee EB, Robinson JL, Caswell C, Irwin DJ et al (2017) Multisite assessment of aging‐related tau astrogliopathy (ARTAG). J Neuropathol Exp Neurol. doi: 10.1093/jnen/nlx041. [DOI] [PMC free article] [PubMed]
- 83. Ksiezak‐Reding H, Morgan K, Mattiace LA, Davies P, Liu WK, Yen SH et al (1994) Ultrastructure and biochemical composition of paired helical filaments in corticobasal degeneration. Am J Pathol 145:1496–1508. [PMC free article] [PubMed] [Google Scholar]
- 84. Kurt MA, Davies DC, Kidd M (1999) beta‐Amyloid immunoreactivity in astrocytes in Alzheimer's disease brain biopsies: an electron microscope study. Exp Neurol 158:221–228. [DOI] [PubMed] [Google Scholar]
- 85. Kwong LK, Uryu K, Trojanowski JQ, Lee VM (2008) TDP‐43 proteinopathies: neurodegenerative protein misfolding diseases without amyloidosis. Neurosignals 16:41–51. [DOI] [PubMed] [Google Scholar]
- 86. Lace G, Ince PG, Brayne C, Savva GM, Matthews FE, de Silva R et al (2012) Mesial temporal astrocyte tau pathology in the MRC‐CFAS ageing brain cohort. Dement Geriatr Cogn Disord 34:15–24. [DOI] [PubMed] [Google Scholar]
- 87. Lee VM, Goedert M, Trojanowski JQ (2001) Neurodegenerative tauopathies. Annu Rev Neurosci 24:1121–1159. [DOI] [PubMed] [Google Scholar]
- 88. Lee EB, Lee VM, Trojanowski JQ, Neumann M (2008) TDP‐43 immunoreactivity in anoxic, ischemic and neoplastic lesions of the central nervous system. Acta Neuropathol 115:305–311. [DOI] [PubMed] [Google Scholar]
- 89. Lee SJ, Seo BR, Koh JY (2015) Metallothionein‐3 modulates the amyloid beta endocytosis of astrocytes through its effects on actin polymerization. Mol Brain 8:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Lee HJ, Suk JE, Patrick C, Bae EJ, Cho JH, Rho S et al (2010) Direct transfer of alpha‐synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. J Biol Chem 285:9262–9272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Le Guennec K, Quenez O, Nicolas G, Wallon D, Rousseau S, Richard AC et al (2016) 17q21.31 duplication causes prominent tau‐related dementia with increased MAPT expression. Mol Psychiatry [doi: 10.1038/mp.2016.226]. [DOI] [PubMed] [Google Scholar]
- 92. Liberski PP, Streichenberger N, Giraud P, Soutrenon M, Meyronnet D, Sikorska B, Kopp N (2005) Ultrastructural pathology of prion diseases revisited: brain biopsy studies. Neuropathol Appl Neurobiol 31:88–96. [DOI] [PubMed] [Google Scholar]
- 93. Lin WL, Castanedes‐Casey M, Dickson DW (2009) Transactivation response DNA‐binding protein 43 microvasculopathy in frontotemporal degeneration and familial Lewy body disease. J Neuropathol Exp Neurol 68:1167–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Lindstrom V, Gustafsson G, Sanders LH, Howlett EH, Sigvardson J, Kasrayan A et al (2017) Extensive uptake of alpha‐synuclein oligomers in astrocytes results in sustained intracellular deposits and mitochondrial damage. Mol Cell Neurosci 82:143–156. [DOI] [PubMed] [Google Scholar]
- 95. Ling H, Kovacs GG, Vonsattel JP, Davey K, Mok KY, Hardy J et al (2016) Astrogliopathy predominates the earliest stage of corticobasal degeneration pathology. Brain 139:3237–3252. [DOI] [PubMed] [Google Scholar]
- 96. Lippa CF, Zhukareva V, Kawarai T, Uryu K, Shafiq M, Nee LE et al (2000) Frontotemporal dementia with novel tau pathology and a Glu342Val tau mutation. Ann Neurol 48:850–858. [PubMed] [Google Scholar]
- 97. Liu AK, Goldfinger MH, Questari HE, Pearce RK, Gentleman SM (2016) ARTAG in the basal forebrain: widening the constellation of astrocytic tau pathology. Acta Neuropathol Commun 4:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Lopez‐Gonzalez I, Carmona M, Blanco R, Luna‐Munoz J, Martinez‐Mandonado A, Mena R, Ferrer I (2013) Characterization of thorn‐shaped astrocytes in white matter of temporal lobe in Alzheimer's disease brains. Brain Pathol 23:144–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. MacDonald ST, Sutherland K, Ironside JW (1996) A quantitative and qualitative analysis of prion protein immunohistochemical staining in Creutzfeldt‐Jakob disease using four anti prion protein antibodies. Neurodegeneration 5:87–94. [DOI] [PubMed] [Google Scholar]
- 100. Mackenzie IR, Ansorge O, Strong M, Bilbao J, Zinman L, Ang LC et al (2011) Pathological heterogeneity in amyotrophic lateral sclerosis with FUS mutations: two distinct patterns correlating with disease severity and mutation. Acta Neuropathol 122:87–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Mackenzie IR, Neumann M (2017) Reappraisal of TDP‐43 pathology in FTLD‐U subtypes. Acta Neuropathol 134:79–96. [DOI] [PubMed] [Google Scholar]
- 102. Mackenzie IR, Neumann M, Baborie A, Sampathu DM, Du Plessis D, Jaros E et al (2011) A harmonized classification system for FTLD‐TDP pathology. Acta Neuropathol 122:111–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Malkani R, D'Souza I, Gwinn‐Hardy K, Schellenberg GD, Hardy J, Momeni P (2006) A MAPT mutation in a regulatory element upstream of exon 10 causes frontotemporal dementia. Neurobiol Dis 22:401–403. [DOI] [PubMed] [Google Scholar]
- 104. Martinez‐Maldonado A, Luna‐Munoz J, Ferrer I (2016) Incidental corticobasal degeneration. Neuropathol Appl Neurobiol 42:659–663. [DOI] [PubMed] [Google Scholar]
- 105. Mattiace LA, Wu E, Aronson M, Dickson DW (1991) A new type of neuritic plaque without amyloid in corticonigral degeneration without achromasia. J Neuropathol Exp Neurol 50:310. (abstr). [Google Scholar]
- 106. McKee AC, Cairns NJ, Dickson DW, Folkerth RD, Keene CD, Litvan I et al (2016) The first NINDS/NIBIB consensus meeting to define neuropathological criteria for the diagnosis of chronic traumatic encephalopathy. Acta Neuropathol 131:75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. McKee AC, Stein TD, Kiernan PT, Alvarez VE (2015) The neuropathology of chronic traumatic encephalopathy. Brain Pathol 25:350–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. McKee AC, Stern RA, Nowinski CJ, Stein TD, Alvarez VE, Daneshvar DH et al (2013) The spectrum of disease in chronic traumatic encephalopathy. Brain 136:43–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. McKeith IG, Dickson DW, Lowe J, Emre M, O'Brien JT, Feldman H et al (2005) Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology 65:1863–1872. [DOI] [PubMed] [Google Scholar]
- 110. Milenkovic I, Kovacs GG (2013) Incidental corticobasal degeneration in a 76‐year‐old woman. Clin Neuropathol 32:69–72. [DOI] [PubMed] [Google Scholar]
- 111. Mori F, Tanji K, Kon T, Odagiri S, Hattori M, Hoshikawa Y et al (2012) FUS immunoreactivity of neuronal and glial intranuclear inclusions in intranuclear inclusion body disease. Neuropathol Appl Neurobiol 38:322–328. [DOI] [PubMed] [Google Scholar]
- 112. Mori F, Tanji K, Yoshimoto M, Takahashi H, Wakabayashi K (2002) Demonstration of alpha‐synuclein immunoreactivity in neuronal and glial cytoplasm in normal human brain tissue using proteinase K and formic acid pretreatment. Exp Neurol 176:98–104. [DOI] [PubMed] [Google Scholar]
- 113. Mori F, Tanji K, Zhang H, Kakita A, Takahashi H, Wakabayashi K (2008) alpha‐Synuclein pathology in the neostriatum in Parkinson's disease. Acta Neuropathol 115:453–459. [DOI] [PubMed] [Google Scholar]
- 114. Munoz DG, Neumann M, Kusaka H, Yokota O, Ishihara K, Terada S et al (2009) FUS pathology in basophilic inclusion body disease. Acta Neuropathol 118:617–627. [DOI] [PubMed] [Google Scholar]
- 115. Munoz DG, Woulfe J, Kertesz A (2007) Argyrophilic thorny astrocyte clusters in association with Alzheimer's disease pathology in possible primary progressive aphasia. Acta Neuropathol 114:347–357. [DOI] [PubMed] [Google Scholar]
- 116. Nagele RG, D'Andrea MR, Lee H, Venkataraman V, Wang HY (2003) Astrocytes accumulate Abeta 42 and give rise to astrocytic amyloid plaques in Alzheimer disease brains. Brain Res 971:197–209. [DOI] [PubMed] [Google Scholar]
- 117. Nakamura K, Mori F, Kon T, Tanji K, Miki Y, Tomiyama M et al (2016) Accumulation of phosphorylated alpha‐synuclein in subpial and periventricular astrocytes in multiple system atrophy of long duration. Neuropathology 36:157–167. [DOI] [PubMed] [Google Scholar]
- 118. Neumann M, Bentmann E, Dormann D, Jawaid A, DeJesus‐Hernandez M, Ansorge O et al (2011) FET proteins TAF15 and EWS are selective markers that distinguish FTLD with FUS pathology from amyotrophic lateral sclerosis with FUS mutations. Brain 134:2595–2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Neumann M, Kwong LK, Truax AC, Vanmassenhove B, Kretzschmar HA, Van Deerlin VM et al (2007) TDP‐43‐positive white matter pathology in frontotemporal lobar degeneration with ubiquitin‐positive inclusions. J Neuropathol Exp Neurol 66:177–183. [DOI] [PubMed] [Google Scholar]
- 120. Neumann M, Rademakers R, Roeber S, Baker M, Kretzschmar HA, Mackenzie IR (2009) A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain 132:2922–2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Neumann M, Roeber S, Kretzschmar HA, Rademakers R, Baker M, Mackenzie IR (2009) Abundant FUS‐immunoreactive pathology in neuronal intermediate filament inclusion disease. Acta Neuropathol 118:605–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT et al (2006) Ubiquitinated TDP‐43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314:130–133. [DOI] [PubMed] [Google Scholar]
- 123. Neumann M, Schulz‐Schaeffer W, Crowther RA, Smith MJ, Spillantini MG, Goedert M, Kretzschmar HA (2001) Pick's disease associated with the novel Tau gene mutation K369I. Ann Neurol 50:503–513. [DOI] [PubMed] [Google Scholar]
- 124. Nishimura M, Namba Y, Ikeda K, Oda M (1992) Glial fibrillary tangles with straight tubules in the brains of patients with progressive supranuclear palsy. Neurosci Lett 143:35–38. [DOI] [PubMed] [Google Scholar]
- 125. Noy S, Krawitz S, Del Bigio MR (2016) Chronic traumatic encephalopathy‐like abnormalities in a routine neuropathology service. J Neuropathol Exp Neurol 75:1145–1154. [DOI] [PubMed] [Google Scholar]
- 126. Oide T, Kinoshita T, Arima K (2006) Regression stage senile plaques in the natural course of Alzheimer's disease. Neuropathol Appl Neurobiol 32:539–556. [DOI] [PubMed] [Google Scholar]
- 127. Papp MI, Kahn JE, Lantos PL (1989) Glial cytoplasmic inclusions in the CNS of patients with multiple system atrophy (striatonigral degeneration, olivopontocerebellar atrophy and Shy‐Drager syndrome). J Neurol Sci 94:79–100. [DOI] [PubMed] [Google Scholar]
- 128. Pekny M, Pekna M, Messing A, Steinhauser C, Lee JM, Parpura V et al (2016) Astrocytes: a central element in neurological diseases. Acta Neuropathol 131:323–345. [DOI] [PubMed] [Google Scholar]
- 129. Piao YS, Mori F, Hayashi S, Tanji K, Yoshimoto M, Kakita A et al (2003) Alpha‐synuclein pathology affecting Bergmann glia of the cerebellum in patients with alpha‐synucleinopathies. Acta Neuropathol 105:403–409. [DOI] [PubMed] [Google Scholar]
- 130. Pickering‐Brown SM, Baker M, Nonaka T, Ikeda K, Sharma S, Mackenzie J et al (2004) Frontotemporal dementia with Pick‐type histology associated with Q336R mutation in the tau gene. Brain 127:1415–1426. [DOI] [PubMed] [Google Scholar]
- 131. Plowey ED, Ziskin JL (2016) Hippocampal phospho‐tau/MAPT neuropathology in the fornix in Alzheimer disease: an immunohistochemical autopsy study. Acta Neuropathol Commun 4:114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Poorkaj P, Muma NA, Zhukareva V, Cochran EJ, Shannon KM, Hurtig H et al (2002) An R5L tau mutation in a subject with a progressive supranuclear palsy phenotype. Ann Neurol 52:511–516. [DOI] [PubMed] [Google Scholar]
- 133. Probst A, Langui D, Lautenschlager C, Ulrich J, Brion JP, Anderton BH (1988) Progressive supranuclear palsy: extensive neuropil threads in addition to neurofibrillary tangles. Very similar antigenicity of subcortical neuronal pathology in progressive supranuclear palsy and Alzheimer's disease. Acta Neuropathol 77:61–68. [DOI] [PubMed] [Google Scholar]
- 134. Reed LA, Schmidt ML, Wszolek ZK, Balin BJ, Soontornniyomkij V, Lee VM et al (1998) The neuropathology of a chromosome 17‐linked autosomal dominant parkinsonism and dementia (“pallido‐ponto‐nigral degeneration”). J Neuropathol Exp Neurol 57:588–601. [DOI] [PubMed] [Google Scholar]
- 135. Ries M, Sastre M (2016) Mechanisms of Abeta clearance and degradation by glial cells. Front Aging Neurosci 8:160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Rohan Z, Matej R, Rusina R, Kovacs GG (2014) Oligodendroglial response in the spinal cord in TDP‐43 proteinopathy with motor neuron involvement. Neurodegener Dis 14:117–124. [DOI] [PubMed] [Google Scholar]
- 137. Rohan Z, Milenkovic I, Lutz MI, Matej R, Kovacs GG (2016) Shared and distinct patterns of oligodendroglial response in alpha‐synucleinopathies and tauopathies. J Neuropathol Exp Neurol 75:1100–1109. [DOI] [PubMed] [Google Scholar]
- 138. Ros R, Thobois S, Streichenberger N, Kopp N, Sanchez MP, Perez M et al (2005) A new mutation of the tau gene, G303V, in early‐onset familial progressive supranuclear palsy. Arch Neurol 62:1444–1450. [DOI] [PubMed] [Google Scholar]
- 139. Saito Y, Ruberu NN, Sawabe M, Arai T, Tanaka N, Kakuta Y et al (2004) Staging of argyrophilic grains: an age‐associated tauopathy. J Neuropathol Exp Neurol 63:911–918. [DOI] [PubMed] [Google Scholar]
- 140. Sakai K, Piao YS, Kikugawa K, Ohara S, Hasegawa M, Takano H et al (2006) Corticobasal degeneration with focal, massive tau accumulation in the subcortical white matter astrocytes. Acta Neuropathol 112:341–348. [DOI] [PubMed] [Google Scholar]
- 141. Sakurai A, Makioka K, Fukuda T, Takatama M, Okamoto K (2013) Accumulation of phosphorylated TDP‐43 in the CNS of a patient with Cockayne syndrome. Neuropathology 33:673–677. [DOI] [PubMed] [Google Scholar]
- 142. Santpere G, Ferrer I (2009) Delineation of early changes in cases with progressive supranuclear palsy‐like pathology. Astrocytes in striatum are primary targets of tau phosphorylation and GFAP oxidation. Brain Pathol 19:177–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Schultz C, Braak H, Braak E (1996) A sex difference in neurodegeneration of the human hypothalamus. Neurosci Lett 212:103–106. [DOI] [PubMed] [Google Scholar]
- 144. Schultz C, Dehghani F, Hubbard GB, Thal DR, Struckhoff G, Braak E, Braak H (2000) Filamentous tau pathology in nerve cells, astrocytes, and oligodendrocytes of aged baboons. J Neuropathol Exp Neurol 59:39–52. [DOI] [PubMed] [Google Scholar]
- 145. Schultz C, Ghebremedhin E, Braak E, Braak H (1999) Sex‐dependent cytoskeletal changes of the human hypothalamus develop independently of Alzheimer's disease. Exp Neurol 160:186–193. [DOI] [PubMed] [Google Scholar]
- 146. Schultz C, Ghebremedhin E, Del Tredici K, Rub U, Braak H (2004) High prevalence of thorn‐shaped astrocytes in the aged human medial temporal lobe. Neurobiol Aging 25:397–405. [DOI] [PubMed] [Google Scholar]
- 147. Shibuya K, Yagishita S, Nakamura A, Uchihara T (2011) Perivascular orientation of astrocytic plaques and tuft‐shaped astrocytes. Brain Res 1404:50–54. [DOI] [PubMed] [Google Scholar]
- 148. Sofroniew MV, Vinters HV (2010) Astrocytes: biology and pathology. Acta Neuropathol 119:7–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Song YJ, Halliday GM, Holton JL, Lashley T, O'Sullivan SS, McCann H et al (2009) Degeneration in different parkinsonian syndromes relates to astrocyte type and astrocyte protein expression. J Neuropathol Exp Neurol 68:1073–1083. [DOI] [PubMed] [Google Scholar]
- 150. Spillantini MG, Yoshida H, Rizzini C, Lantos PL, Khan N, Rossor MN et al (2000) A novel tau mutation (N296N) in familial dementia with swollen achromatic neurons and corticobasal inclusion bodies. Ann Neurol 48:939–943. [DOI] [PubMed] [Google Scholar]
- 151. Spina S, Murrell JR, Yoshida H, Ghetti B, Bermingham N, Sweeney B et al (2007) The novel Tau mutation G335S: clinical, neuropathological and molecular characterization. Acta Neuropathol 113:461–470. [DOI] [PubMed] [Google Scholar]
- 152. Stanford PM, Halliday GM, Brooks WS, Kwok JB, Storey CE, Creasey H et al (2000) Progressive supranuclear palsy pathology caused by a novel silent mutation in exon 10 of the tau gene: expansion of the disease phenotype caused by tau gene mutations. Brain 123: 880–893. [DOI] [PubMed] [Google Scholar]
- 153. Surgucheva I, Newell KL, Burns J, Surguchov A (2014) New alpha‐ and gamma‐synuclein immunopathological lesions in human brain. Acta Neuropathol Commun 2:132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Tacik P, DeTure M, Lin WL, Sanchez Contreras M, Wojtas A, Hinkle KM et al (2015) A novel tau mutation, p.K317N, causes globular glial tauopathy. Acta Neuropathol 130:199–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Tacik P, Sanchez‐Contreras M, DeTure M, Murray ME, Rademakers R, Ross OA et al (2017) Clinicopathologic heterogeneity in frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP‐17) due to microtubule‐associated protein tau (MAPT) p.P301L mutation, including a patient with globular glial tauopathy. Neuropathol Appl Neurobiol 43:200–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Terada S, Ishizu H, Haraguchi T, Takehisa Y, Tanabe Y, Kawai K, Kuroda S (2000) Tau‐negative astrocytic star‐like inclusions and coiled bodies in dementia with Lewy bodies. Acta Neuropathol 100:464–468. [DOI] [PubMed] [Google Scholar]
- 157. Thal DR, Rub U, Orantes M, Braak H (2002) Phases of Abeta‐deposition in the human brain and its relevance for the development of AD. Neurology 58:1791–1800. [DOI] [PubMed] [Google Scholar]
- 158. Thal DR, Schultz C, Dehghani F, Yamaguchi H, Braak H, Braak E (2000) Amyloid beta‐protein (Abeta)‐containing astrocytes are located preferentially near N‐terminal‐truncated Abeta deposits in the human entorhinal cortex. Acta Neuropathol 100:608–617. [DOI] [PubMed] [Google Scholar]
- 159. Togo T, Dickson DW (2002) Tau accumulation in astrocytes in progressive supranuclear palsy is a degenerative rather than a reactive process. Acta Neuropathol 104:398–402. [DOI] [PubMed] [Google Scholar]
- 160. Trojanowski JQ, Revesz T, Neuropathology Working Group on MSA (2007) Proposed neuropathological criteria for the post mortem diagnosis of multiple system atrophy. Neuropathol Appl Neurobiol 33:615–620. [DOI] [PubMed] [Google Scholar]
- 161. Uryu K, Nakashima‐Yasuda H, Forman MS, Kwong LK, Clark CM, Grossman M et al (2008) Concomitant TAR‐DNA‐binding protein 43 pathology is present in Alzheimer disease and corticobasal degeneration but not in other tauopathies. J Neuropathol Exp Neurol 67:555–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. van Herpen E, Rosso SM, Serverijnen LA, Yoshida H, Breedveld G, van de Graaf R et al (2003) Variable phenotypic expression and extensive tau pathology in two families with the novel tau mutation L315R. Ann Neurol 54:573–581. [DOI] [PubMed] [Google Scholar]
- 163. Verkhratsky A, Zorec R, Parpura V (2017) Stratification of astrocytes in healthy and diseased brain. Brain Pathol. doi: 10.1111/bpa.12537. [DOI] [PMC free article] [PubMed]
- 164. Wakabayashi K, Hayashi S, Yoshimoto M, Kudo H, Takahashi H (2000) NACP/alpha‐synuclein‐positive filamentous inclusions in astrocytes and oligodendrocytes of Parkinson's disease brains. Acta Neuropathol 99:14–20. [DOI] [PubMed] [Google Scholar]
- 165. Wakabayashi K, Oyanagi K, Makifuchi T, Ikuta F, Homma A, Homma Y et al (1994) Corticobasal degeneration: etiopathological significance of the cytoskeletal alterations. Acta Neuropathol 87:545–553. [DOI] [PubMed] [Google Scholar]
- 166. Wakabayashi K, Shibasaki Y, Hasegawa M, Horikawa Y, Soma Y, Hayashi S et al (2000) Primary progressive aphasia with focal glial tauopathy. Neuropathol Appl Neurobiol 26:477–481. [DOI] [PubMed] [Google Scholar]
- 167. Walker AK, Daniels CM, Goldman JE, Trojanowski JQ, Lee VM, Messing A (2014) Astrocytic TDP‐43 pathology in Alexander disease. J Neurosci 34:6448–6458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Yamada T, Calne DB, Akiyama H, McGeer EG, McGeer PL (1993) Further observations on Tau‐positive glia in the brains with progressive supranuclear palsy. Acta Neuropathol 85:308–315. [DOI] [PubMed] [Google Scholar]
- 169. Yamada T, McGeer PL (1990) Oligodendroglial microtubular masses: an abnormality observed in some human neurodegenerative diseases. Neurosci Lett 120:163–166. [DOI] [PubMed] [Google Scholar]
- 170. Yamada T, McGeer PL, McGeer EG (1992) Appearance of paired nucleated, Tau‐positive glia in patients with progressive supranuclear palsy brain tissue. Neurosci Lett 135:99–102. [DOI] [PubMed] [Google Scholar]
- 171. Yamazaki M, Nakano I, Imazu O, Kaieda R, Terashi A (1994) Astrocytic straight tubules in the brain of a patient with Pick's disease. Acta Neuropathol 88:587–591. [DOI] [PubMed] [Google Scholar]
- 172. Yoshida M (2014) Astrocytic inclusions in progressive supranuclear palsy and corticobasal degeneration. Neuropathology 34:555–570. [DOI] [PubMed] [Google Scholar]
- 173. Yoshida K, Hata Y, Kinoshita K, Takashima S, Tanaka K, Nishida N (2017) Incipient progressive supranuclear palsy is more common than expected and may comprise clinicopathological subtypes: a forensic autopsy series. Acta Neuropathol 133:809–823. [DOI] [PubMed] [Google Scholar]
- 174. Zarranz JJ, Ferrer I, Lezcano E, Forcadas MI, Eizaguirre B, Atares B et al (2005) A novel mutation (K317M) in the MAPT gene causes FTDP and motor neuron disease. Neurology 64:1578–1585. [DOI] [PubMed] [Google Scholar]