

Abstract
Staphylococcus pseudintermedius is an opportunistic pathogen in dogs and cats and occasionally causes infections in humans. S. pseudintermedius is often resistant to multiple classes of antimicrobials. It requires a reliable detection so that it is not misidentified as S. aureus. Phenotypic and currently-used molecular-based diagnostic assays lack specificity or are labour-intensive using multiplex PCR or nucleic acid sequencing. The aim of this study was to identify a specific target for real-time PCR by comparing whole genome sequences of S. pseudintermedius and non-pseudintermedius.Genome sequences were downloaded from public repositories and supplemented by isolates that were sequenced in this study. A Perl-script was written that analysed 300-nt fragments from a reference genome sequence of S. pseudintermedius and checked if this sequence was present in other S. pseudintermedius genomes (n = 74) and non-pseudintermedius genomes (n = 138). Six sequences specific for S. pseudintermedius were identified (sequence length between 300–500 nt). One sequence, which was located in the spsJ gene, was used to develop primers and a probe. The real-time PCR showed 100% specificity when testing for S. pseudintermedius isolates (n = 54), and eight other staphylococcal species (n = 43). In conclusion, a novel approach by comparing whole genome sequences identified a sequence that is specific for S. pseudintermedius and provided a real-time PCR target for rapid and reliable detection of S. pseudintermedius.
Introduction
Staphylococcus pseudintermedius is an opportunistic coagulase-positive pathogen in dogs and cats, which generally causes topical infections and otitis externa [1]. Infections in humans occur sporadically [2], and often have a zoonotic origin [3–5]. S. pseudintermedius is a normal inhabitant of the skin and mucosa and healthy dogs and cats can be carriers [6,7]. Its methicillin-resistant variant–MRSP–is often multi-resistant, and sometimes resistant against all common antimicrobials [8,9], which makes clinical infections with this organism difficult to treat. Multi Locus Sequence Typing (MLST) of MRPS showed a highly clonal population structure, with ST71 as the predominant type in Northern Europe, ST68 in Northern America [10,11] and ST45 being dominant in Asia [12], but the population of methicillin-susceptible S. speudintermedius has not been studied in detail.
S. pseudintermedius belongs together with two other species, S. intermedius and S. delphini, to the SIG group. These species were long considered to be the same species [10] and were traditionally distinguished by the different hosts they were isolated from. When coagulase-positive staphylococci are isolated from dogs, the conventional biochemical identification cannot reliably differentiate between S. intermedius and S. aureus, what resulted in S. intermedius based on the host species. A molecular diagnostic test using PCR-RFLP of the pta gene was not specific for identification of S. pseudintermedius [13,14]. Approaches for partial sequence analysis of the tuf [15] or 16S rRNA genes [16] and sodA and hsp60 genes [17] were not always conclusive. A conventional multiplex PCR has been described for identification of different coagulase-positive staphylococci, targeting 900 bp of the nuc gene, but this gene contains insufficient variation for species-specific detection of S. pseudintermedius by real-time PCR (RT-PCR) [18]. More recently it was shown that S. pseudintermedius isolates can reliably be distinguished from other members of the SIG group by MALDI-TOF MS [19,20]. This is an accurate technique, and the costs per sample are low, but the high cost of the mass spectrometer itself makes that it is not available for all laboratories. An easy-to-use PCR targeting a single sequence for species-specific detection of S. pseudintermedius is currently not available, but PCR equipment is available in many research and diagnostic laboratories, both in developed and developing countries.
As whole-genome sequences (WGS) of S. pseudintermedius are becoming increasingly available, and now the cost of sequencing is dropping can comparative genome analysis identify novel nucleic acid diagnostic targets, for use in the species-specific detection of S. pseudintermedius. Until now, a suitable nucleic acid target for PCR was usually selected manually and was limited to genes that are known to be conserved. The aim of this study was to apply a bioinformatics approach on WGS comparisons to automate the discovery of suitable nucleic acid sequences for species-specific identification of S. pseudintermedius by means of RT-PCR.
Materials and methods
Whole-genome sequencing
Fifteen genomes of S. pseudintermedius and 119 genomes of other staphylococci were downloaded from public repositories at NCBI and DNA-nexus (strain IDs and accession numbers are listed in S1 Table). The set of WGS was supplemented by sequencing 59 clinical isolates of S. pseudintermedius, five of S. intermedius, and 15 of S. delphini isolates from dogs that were isolated at the Veterinary Microbiology Diagnostic Centre, Utrecht University, the Netherlands. This resulted in 74 S. pseudintermedius and 138 non-pseudintermedius genomes from different hosts (Table 1).
Table 1. Number of staphylococcal genome sequences used for sliding-frame analysis and strains for PCR validation.
| Species | No. of genomes | Host | No. of strains for PCR validation |
|---|---|---|---|
| S. agentis | 1 | Ovine | |
| S. aureus | 94 | human (n = 84), ovine (n = 3), porcine (n = 1), bovine (n = 2), food (n = 4) | 5 |
| S. capitis | 1 | human | |
| S. carnosus | 1 | meat | |
| S. delphini | 15 | equine (n = 10, ovine (n = 1), rodent (n = 1), delphinidae (n = 1), unknown (n = 2) | 15 |
| S. epidermidis | 4 | human (n = 3), bovine (1) | |
| S. equorum | 1 | food | 5 |
| S. haemolyticus | 2 | human | |
| S. hyicus | 1 | porcine | 3 |
| S. intermedius | 5 | ovine (n = 4), canine (n = 1) | 5 |
| S. lugdunensis | 2 | human | |
| S. pasteuri | 1 | unknown | |
| S. pseudintermedius | 74 | canine (n = 71), feline (n = 3) | 54 |
| S. saprophyticus | 1 | human | 3 |
| S. schleiferi | 5 | canine | 4 |
| S. warneri | 1 | human | |
| S. xylosus | 3 | ursidae (n = 1), unknown (n = 2) | 3 |
| Total | 212 | 97 |
The species of the clinical isolates were confirmed by MALDI-TOF MS (Bruker Daltonik), and the strains were sequenced using Illumina-sequencing performed at the Utrecht Sequencing Facility (Hubrecht Institute, KNAW, the Netherlands). The quality of the WGS data was assessed with the Checkm tool [21]. Sequence reads were assembled de novo using the program SPAdes v3.1.1 [22]. The final number of genomes per bacterial species is summarised in Table 1, and a list with genotype and accession numbers is available in S1 Table. The sequences of household genes 16S rRNA, tuf, sodA, and nuc were extracted from the genomes and aligned to confirm the taxonomy of the genome sequences and their species assignment in GenBank. The MLST sequence types were extracted from the genome sequences of S. pseudintermedius.
Genome comparisons
For phylogenetic analysis of the species in the SIG group the core genomes were determined of at least one genome of each Staphylococcus species from the strain list. Using the program Roary v3.5.6 [23], a gene presence/absence analysis was determined. A gene was considered to be part of the Staphylococcus core genome if at least 99% of the genomes contained this gene with at least 50% identity and 50% overlap. For the core genome comparison a SNP-alignment was performed and a Neighbour-Joining (NJ) tree was constructed using FastTree [24]. Macrococcus caseolyticus (GenBank accession AP009484.1) was included as root.
Sliding-frame genome analysis
In order to identify a sequence that is conserved in S. pseudintermedius, but not present in other staphylococci an iterative script was written in Perl to compare all genome sequences with a novel sliding-frame approach The genome of S. pseudintermedius isolate 14S02884-1 was chosen as an a priori reference genome. The reference genome was converted to frames by taking the first 300 nt of the reference sequence. Then, the frame of 300 nt shifted 100 nt downstream and the sequence in the frame was also recorded (nucleotides 100–400); this new frame had an overlap of 200 nt with the first frame. This process was continued until the end of the genome was reached and the reference genome was available in 300 nt sequences with 200 nt overlap with the neighbouring frame. First, a BLASTn of all S. pseudintermedius frames against the database with 138 non-pseudintermedius genomes was performed. If a frame was present in non-pseudintermedius genomes it would be unsuitable for a pseudintermedius-specific target for PCR and was excluded from further analysis. Frames were first compared to non-pseudintermedius genomes because it was shown by the genome comparison described above that all staphylococci have a large part of their genome sequences in common and a large part of the frames could be excluded from further analyses, which saved time. The frames that met the pre-set criteria (Identity score<20 or Coverage score<20) were analysed with BLASTn to the first non-reference genome of S. pseudintermedius, and the remaining frames were iteratively compared to the other S. pseudintermedius genomes (BLASTn filter was set to 100% identity and 100% query coverage for S. pseudintermedius genomes). In this way, each iteration consisted of less BLASTn searches than the previous, because frames (sequences) that were not suitable as a PCR target were excluded immediately. See the diagram in S1 Fig for a schematic overview of the approach. The analysis included complete genomes and genome sequences in contigs. The script requires BioPerl and was run using Perl v5.18.2. The script is available on github (https://github.com/koenverst/slidingframe/).
PCR design and validation
Overlapping frames that were conserved in S. pseudintermedius genomes and absent in non-pseudintermedius genomes were combined. To confirm that these sequences were not present in other organisms of which the sequence was not included in the local sliding-frame analysis a BLAST search was performed in the online NCBI GenBank nr-database. Primers and a probe were developed using Primer3 software (http://primer3.ut.cc) and were ordered at Integrated DNA Technologies (Belgium).
Included for PCR validation were 97 isolates of S. pseudintermedius and non-pseudintermedius (Table 1 and S2 Table). All isolates were analysed by MALDI-TOF MS (Bruker Daltonik) to confirm their species before PCRs were performed. DNA was extracted for PCR analysis by suspending 2 colonies of a fresh overnight culture on Columbia Agar with sheep blood (Oxoid, the Netherlands) in 500 μL Tris-EDTA pH 8.0 (Sigma Aldrich, the Netherlands) and incubated at 95°C for 10 min followed by centrifugation at 20,000xg for 1 min. Five μL supernatant was used for conventional PCR with GoTaq Green G2 master mix (Promega, the Netherlands) and 500 nM of each primer, or for RT-PCR using the LightCycler 480 Probes Master (Roche, the Netherlands) on a LightCycler 480-II system, with an optimal oligonucleotide concentrations of 300 nM of each primer and 75 nM probe. The cycling program was 2 min enzyme activation at 95°C, followed by 45 cycles of 30 s. denaturation at 95°C, 30 sec annealing at 58°C, and 1 min elongation at 72°C. For conventional PCR 10 min elongation at 72°C before cooling to 4°C, was added.
Results
From the genomes that were downloaded from GenBank (S1 Table) were the 16S rRNA, tuf, sodA, and nuc sequences aligned to check for the species identification. All the species matched with the species names present in the online database (data not shown). Of the clinical Staphylococcal isolates that were sequenced for this study the initial MALDI-TOF MS identification matched with the species identification based on the WGS data (S1 Table).
Genome comparison
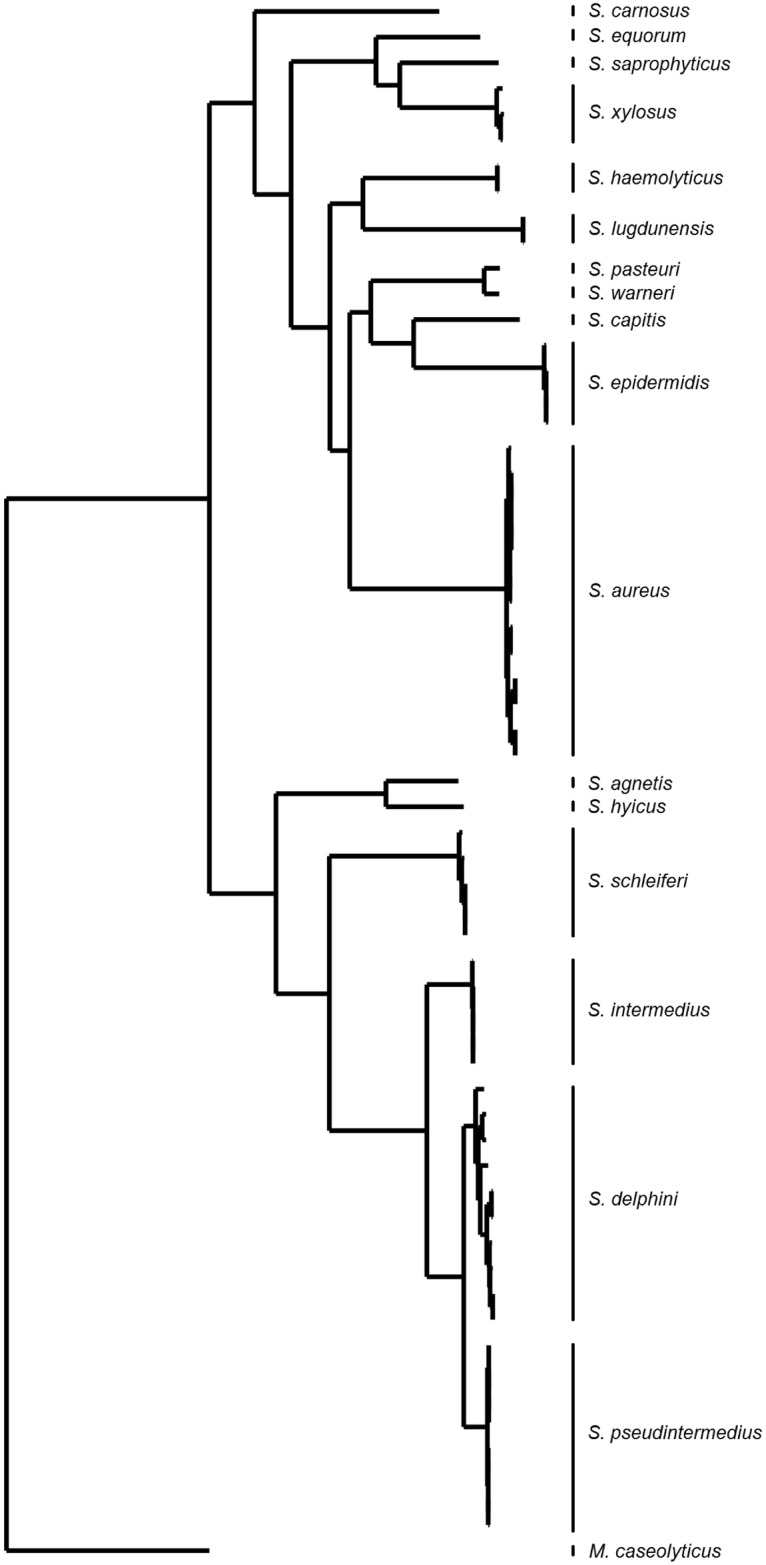
A phylogenetic genome comparison of the species of the SIG group and 115 other Staphylococcus species confirmed the previously observed genomic relatedness of the SIG species (Fig 1). The staphylococcal core genome consisted of 873 genes. Phylogenetic analysis showed that S. pseudintermedius is more closely related to S. delphini than to S. intermedius. Also, there is only little variation between genomes of S. pseudintermedius, while S. delphini genomes are more diverse (Fig 1). The genomes of S. pseudintermedius belonged to 24 different MLST sequence types (S1 Table).
Fig 1. Phylogenetic reconstruction based on comparison of the SNPs in the core-genomes of different staphylococcal species.
PCR target selection by sliding-frame genome analysis
The reference S. pseudintermedius genome contained a total of 30,391 frames to be analysed when a frame size of 300 and a frame shift of 100 nucleotides were chosen. After the first iteration–comparing the frames (300 nt sequences) of the reference genome to the non-pseudintermedius genomes– 24,992/30,391(80.2%) frames of the S. pseudintermedius reference genome were found to be present in the genomes of other Staphylococcus species (with more than 20% coverage or more than 20% identity) and were not considered in further BLASTn analysis because they would be unsuitable for a S. pseudintermedius-specific PCR. The remaining 5,399 frames were analysed against the other S. pseudintermedius genomes to find which frames were conserved in all genome sequences of S. pseudintermedius. When the script finished, 10 frames were left that met the pre-set filtering criteria. These sequences were identical in all S. pseudintermedius genomes and not present in genomes of other staphylococci. The number of remaining frames after each iteration is displayed in Fig 2. Overlapping frames could be combined into six sequences ranging from 300–500 nt, and an online BLAST search in the ED99 genome identified the function of the genes in which these sequences were present (Table 2). An online BLAST against all non-pseudintermedius sequences confirmed that the sequences were not present in any other GenBank entry (July 2016).
Fig 2. Iteration plot.
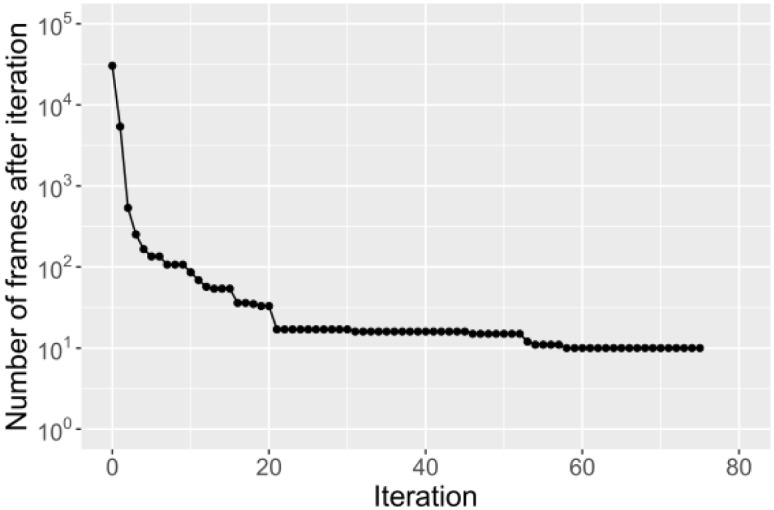
Number of frames that met the filter criteria after each iteration. At the 0th iteration all frames of the reference genome are present. The 1st iteration is the comparison to the non-pseudintermedius genomes. All following iterations are comparisons to S. pseudintermedius genomes.
Table 2. S. pseudintermedius-specific sequences and annotations in strain ED99 that resulted from the sliding-frame analysis.
| Seq. | Size (nt) | Annotated proteins in S. pseudintermedius ED99 (NC_017568.1) |
|---|---|---|
| 1 | 400 | Teichoic acid translocation permease protein (SPSE_1752) |
| 2 | 300 | Ribonuclease-diphosphate reductase, alpha subunit (nrdE) (SPSE_2002) |
| 3 | 300 | Accessory Sec System Translocase (secY2) (SPSE_0165) and (asp1) (SPSE_0166) |
| 4 | 500 | Hypothetical protein with conserved SDSD and STS motifs (spsJ)(SPSE_0164) |
| 5 | 300 | Molybdate ABC transporter, periplasmic molybdate-binding protein (modA) (SPSE_0543) |
| 6 | 400 | Multidrug-efflux transporter (SPSE_0785) |
The sequences of the six frames were analysed with Primer3 to identify oligonucleotides suitable for conventional and RT-PCR. These oligonucleotides were tested with conventional PCR. This identified that sequence 4 (500 nt) was best suited for PCR amplification. These oligonucleotides target a 198 bp sequence located in position 192532–192684 in the genome of S. pseudintermedius strain ED99 (GenBank accession NC_017568.1) in the 3’end of the spsJ gene. This gene is annotated as a putative cell wall anchor protein in strain ED99, or as hypothetical proteins in other S. speudintermedius genomes. The following oligonucleotides were used: stapse-Fw: 5'-ACC AAG GCC TGT AAG TAA AGC ACC-3'; stapse-Rev: 5'-TCT CTT TCA ACA TCG GCA TCA ACG C-3', and stapse-P: 5'-6FAM-ACT GTC GCT GAA TCG CTT GAT GAC G-BHQ1-3' as a probe. An online BLAST confirmed that the sequences were specifically detected all S. pseudintermedius sequences present in GenBank in July 2017.
PCR validation
The PCR was validated with 54 clinical isolates of S. pseudintermedius and 43 strains comprising eight staphylococcal species (Table 1). Of these isolates, 16 were reference isolates. The characteristics of the tested isolates are shown in S2 Table. For S. pseudintermedius, 21 methicillin-susceptible isolates and 33 methicillin-resistant isolates were tested, that were all positive in the RT-PCR. All non-pseudintermedius isolates (n = 43) were negative.
Discussion
The availability of WGS enabled us to compare genome sequences of S. pseudintermedius to find a conserved region in the genomes that is not present in other staphylococcal species, for the development of a species-specific PCR.
The core genome comparison showed that S. pseudintermedius genomes are highly related and are phylogenetically more related to S. delphini than to S. intermedius. This correlates with a previous genome comparison based on one isolate per species [10,25,26], and indicates that the close relatedness of strains from the SIG species is sustained after phylogenetic analysis of multiple genome sequences. Genome comparisons with other staphylococcal species confirmed that SIG species belong to a separated phylogenetic branch [27].
Identification of species-specific nucleotides for diagnostic PCR is limited by the availability of publically available genomes of S. pseudintermedius and related species. Therefore we generated additional genomes in this study, to reduce the risk that the sliding-frame script would identify conserved frames that are not conserved in other S. pseudintermedius genomes. As S. pseudintermedius is highly clonal we included the genomes of isolates belonging to 24 different MLST types, that were isolated in different geographical regions (the Netherlands, Switzerland, Japan and Sri Lanka), to cover genomic diversity. With the core genome analysis, no gene was identified that was specific for S. pseudintermedius and not present in the non-pseudintermedius genomes. The sliding-frame script, identified a six sequences that were species-specific and highly conserved in S. pseudintermedius, as this approach analysed the nucleotides of the whole genome. From the limited number of nucleic acid sequences that were retrieved from the S. peudintermedius genomes (i.e. six) only one sequences was suitable for RT-PCR development.
The nucleic acid sequence that was used in this study as PCR target, is located in the spsJ gene that encodes one of the 18 putative cell wall-anchored surface proteins in the genome of S. pseudintermedius strain ED99 [28]. The spsJ protein has homology with the sdrI gene in S. epidermidis, and is characterized by conserved motifs SDSD and STS [28]. However, a putative surface location of this protein could not be identified with proteome analysis and further characterization of the function is needed. Therefore is the spsJ gene annotated in other S. pseudintermedius genomes as hypothetical protein. The nucleic acid sequence that is targeted by the PCR is located in the 3’ end of the spsJ gene and is conserved and specific for S. pseudintermedius, and is most likely not under diversifying selection, in contrast to other parts of the encoding region that contain sequence variation (alignment is not shown). A GenBank BLASTn confirmed that the amplified fragment of the spsJ gene specifically detects S. pseudintermedius and is highly conserved in 74 S. pseudintermedius genomes. This putative diagnostic target was therefore further evaluated for the ability to accurately identify S. pseudintermedius and to distinguish it from other closely related Staphylococcal species. The RT-PCR was 100% specific for the tested clinical S. pseudintermedius isolates (n = 54) obtained from dogs (S2 Table). The PCR was not validated for detection of S. pseudintermedius in clinical samples as diagnostic is not based on a simple-presence of S. speudintermedius. Dogs can carry S. pseudintermedius asymptomatically [1], and the in this study developed RT-PCR can quantify the bacteria, enabling to study the association between clinical symptoms and bacterial load, especially when this RT-PCR is applied on samples from potential colonization sites.
Other approaches for genome comparison to identify potential diagnostic targets have been described. In a study on Haemophilus influenzae three genome sequences were compared for selection of putative diagnostic PCR targets, and subsequent sequence alignment and development of primers and probes [29]. Although this was a very successful approach, it required a lot of manual work and laboratory resources. The genome comparison approach we described in this study is scalable to large numbers of genome sequences and requires limited manual work once genome sequences are publically available. On the other hand, the approach for comparison of a few genomes, described by Coughlan et al. [29], is still very suitable for organisms for which only a few genomes are available in public databases. M-GCAT [30], and MAUVE [31] are also computer programs that are able to compare large numbers of whole genome sequences in a short time, but they can only show sequence similarities and dissimilarities. Our approach automatically filtered sequences that were present in the S. pseudintermedius genomes and absent in non-pseudintermedius genomes.
In conclusion, we successfully applied a sliding-frame approach for genome comparisons and an automated approach for PCR target selection. Using this approach, we developed a novel RT-PCR assay for species-specific detection of S. pseudintermedius. This is a promising approach to identify putative diagnostic PCR targets for other organisms.
Supporting information
All the frames from the reference genome are analysed with BLAST against the database with non-pseudintermedius staphylococci. The frames that meet the pre-set criteria (i.e. not present in non-pseudintermedius genomes) are iteratively compared to all remaining genomes of S. pseudintermedius, so that with each iteration less frames need to be analysed.
(PDF)
(PDF)
(PDF)
Acknowledgments
The authors are grateful to Dr. Aldert Zomer for helpful discussions and to the colleagues of the VMDC for collecting the isolates for validation of the PCR.
Data Availability
All whole-genome sequences that were obtained for this study are available from the NCBI repository. Accession numbers are available in S1 Table.
Funding Statement
The authors received no specific funding for this work.
References
- 1.Weese JS, van Duijkeren E (2010) Methicillin-resistant Staphylococcus aureus and Staphylococcus pseudintermedius in veterinary medicine. Vet Microbiol 140: 418–429. doi: 10.1016/j.vetmic.2009.01.039 [DOI] [PubMed] [Google Scholar]
- 2.Starlander G, Börjesson S, Grönlund-Andersson U, Tellgren-Roth C, Melhus Å (2014) Cluster of infections caused by methicillin-resistant Staphylococcus pseudintermedius in humans in a tertiary hospital. J Clin Microbiol 52: 3118–3120. doi: 10.1128/JCM.00703-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Riegel P, Jesel-Morel L, Laventie B, Boisset S, Vandenesch F, Prévost G. (2011) Coagulase-positive Staphylococcus pseudintermedius from animals causing human endocarditis. Int J Med Microbiol 301: 237–239. doi: 10.1016/j.ijmm.2010.09.001 [DOI] [PubMed] [Google Scholar]
- 4.Savini V, Barbarini D, Polakowska K, Gherardi G, Białecka A, Carretto E. (2013) Methicillin-resistant Staphylococcus pseudintermedius infection in a bone marrow transplant recipient. J Clin Microbiol 51: 1636–1638. doi: 10.1128/JCM.03310-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Somayaji R, Priyantha MAR, Rubin JE, Church D (2016) Human infections due to Staphylococcus pseudintermedius, an emerging zoonosis of canine origin: report of 24 cases. Diagn Microbiol Infect Dis 85: 471–476. doi: 10.1016/j.diagmicrobio.2016.05.008 [DOI] [PubMed] [Google Scholar]
- 6.Griffeth GC, Morris DO, Abraham JL, Shofer FS, Rankin SC (2008) Screening for skin carriage of methicillin-resistant coagulase-positive staphylococci and Staphylococcus schleiferi in dogs with healthy and inflamed skin. Vet Dermatol 19: 142–149. doi: 10.1111/j.1365-3164.2008.00663.x [DOI] [PubMed] [Google Scholar]
- 7.Vengust M, Anderson MEC, Rousseau J, Weese JS (2006) Methicillin-resistant staphylococcal colonization in clinically normal dogs and horses in the community. Lett Appl Microbiol 43: 602–606. doi: 10.1111/j.1472-765X.2006.02018.x [DOI] [PubMed] [Google Scholar]
- 8.McCarthy AJ, Harrison EM, Stanczak-Mrozek K, Leggett B, Waller A, Holmes M, et al. (2015) Genomic insights into the rapid emergence and evolution of MDR in Staphylococcus pseudintermedius. J Antimicrob Chemother 70: 997–1007. doi: 10.1093/jac/dku496 [DOI] [PubMed] [Google Scholar]
- 9.Kadlec K, Schwarz S (2012) Antimicrobial resistance of Staphylococcus pseudintermedius. Vet Dermatol 23: 276–282, e55 doi: 10.1111/j.1365-3164.2012.01056.x [DOI] [PubMed] [Google Scholar]
- 10.Perreten V, Kadlec K, Schwarz S, Grönlund Andersson U, Finn M, Greko C, et al. (2010) Clonal spread of methicillin-resistant Staphylococcus pseudintermedius in Europe and North America: an international multicentre study. J Antimicrob Chemother 65: 1145–1154. Available: http://www.ncbi.nlm.nih.gov/pubmed/20348087. [DOI] [PubMed] [Google Scholar]
- 11.Duim B, Verstappen KM, Broens EM, Laarhoven LM, van Duijkeren E, Hordijk J, et al. (2015) Changes in the population of methicillin-resistant Staphylococcus pseudintermedius and the dissemination of antimicrobial resistant phenotypes in the Netherlands. J Clin Microbiol. doi: 10.1128/JCM.01288-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perreten V, Chanchaithong P, Prapasarakul N, Rossano A, Blum SE, Elad D, et al. (2013) Novel pseudo-staphylococcal cassette chromosome mec element (ψSCCmec57395) in methicillin-resistant Staphylococcus pseudintermedius CC45. Antimicrob Agents Chemother 57: 5509–5515. doi: 10.1128/AAC.00738-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bannoehr J, Franco A, Iurescia M, Battisti A, Fitzgerald JR (2009) Molecular diagnostic identification of Staphylococcus pseudintermedius. J Clin Microbiol 47: 469–471. doi: 10.1128/JCM.01915-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Slettemeås JS, Mikalsen J, Sunde M (2010) Further diversity of the Staphylococcus intermedius group and heterogeneity in the MboI restriction site used for Staphylococcus pseudintermedius species identification. J Vet Diagn Invest 22: 756–759. http://www.ncbi.nlm.nih.gov/pubmed/20807936. [DOI] [PubMed] [Google Scholar]
- 15.Heikens E, Fleer A, Paauw A, Florijn A, Fluit AC (2005) Comparison of genotypic and phenotypic methods for species-level identification of clinical isolates of coagulase-negative staphylococci. J Clin Microbiol 43: 2286–2290. http://jcm.asm.org/cgi/doi/10.1128/JCM.43.5.2286-2290.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maiwald M (2004) Broad-range PCR for detection and identification of bacteria Molecular Microbiology: Diagnostic Principles and Practice. ASM Press; pp. 379–390. [Google Scholar]
- 17.Sasaki T, Kikuchi K, Tanaka Y, Takahashi N, Kamata S, Hiramatsu K. (2007) Reclassification of phenotypically identified Staphylococcus intermedius strains. J Clin Microbiol 45: 2770–2778. doi: 10.1128/JCM.00360-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sasaki T, Tsubakishita S, Tanaka Y, Sakusabe A, Ohtsuka M, Hirotaki S, et al. (2010) Multiplex-PCR method for species identification of coagulase-positive staphylococci. J Clin Microbiol 48: 765–769. doi: 10.1128/JCM.01232-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Decristophoris P, Fasola A, Benagli C, Tonolla M, Petrini O (2011) Identification of Staphylococcus intermedius Group by MALDI-TOF MS. Syst Appl Microbiol 34: 45–51. doi: 10.1016/j.syapm.2010.11.004 [DOI] [PubMed] [Google Scholar]
- 20.Murugaiyan J, Walther B, Stamm I, Abou-Elnaga Y, Brueggemann-Schwarze S, Vincze S, et al. (2014) Species differentiation within the Staphylococcus intermedius group using a refined MALDI-TOF MS database. Clin Microbiol Infect 20: 1007–1015. doi: 10.1111/1469-0691.12662 [DOI] [PubMed] [Google Scholar]
- 21.Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW (2015) CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res 25: 1043–1055. doi: 10.1101/gr.186072.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov A, et al. (2012) SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19: 455–477. doi: 10.1089/cmb.2012.0021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Page AJ, Cummins CA, Hunt M, Wong VK, Reuter S, Holden MT, et al. (2015) Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31: 3691–3693. doi: 10.1093/bioinformatics/btv421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Price MN, Dehal PS, Arkin AP (2009) FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol Biol Evol 26: 1641–1650. doi: 10.1093/molbev/msp077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ben Zakour NL, Beatson SA, van den Broek AHM, Thoday KL, Fitzgerald JR (2012) Comparative Genomics of the Staphylococcus intermedius Group of Animal Pathogens. Front Cell Infect Microbiol 2: 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bannoehr J, Ben Zakour NL, Waller AS, Guardabassi L, Thoday KL, Van Den Broek A, et al. (2007) Population genetic structure of the Staphylococcus intermedius group: insights into agr diversification and the emergence of methicillin-resistant strains. J Bacteriol 189: 8685–8692. doi: 10.1128/JB.01150-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Suzuki H, Lefébure T, Bitar PP, Stanhope MJ (2012) Comparative genomic analysis of the genus Staphylococcus including Staphylococcus aureus and its newly described sister species Staphylococcus simiae. BMC Genomics 13: 38 doi: 10.1186/1471-2164-13-38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bannoehr J, Ben Zakour NL, Reglinski M, Inglis NF, Prabhakaran S, Fossum E, et al. (2011) Genomic and surface proteomic analysis of the canine pathogen Staphylococcus pseudintermedius reveals proteins that mediate adherence to the extracellular matrix. Infect Immun 79: 3074–3086. doi: 10.1128/IAI.00137-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Coughlan H, Reddington K, Tuite N, Boo TW, Cormican M, Barrett L, et al. (2015) Comparative genome analysis identifies novel nucleic acid diagnostic targets for use in the specific detection of Haemophilus} influenzae. Diagn Microbiol Infect Dis 83: 112–116. doi: 10.1016/j.diagmicrobio.2015.06.013 [DOI] [PubMed] [Google Scholar]
- 30.Treangen TJ, Messeguer X (2006) M-GCAT: interactively and efficiently constructing large-scale multiple genome comparison frameworks in closely related species. BMC Bioinformatics 7: 433 doi: 10.1186/1471-2105-7-433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Treangen TJ, Ondov BD, Koren S, Phillippy AM (2014) The Harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol 15: 524 doi: 10.1186/s13059-014-0524-x [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
All the frames from the reference genome are analysed with BLAST against the database with non-pseudintermedius staphylococci. The frames that meet the pre-set criteria (i.e. not present in non-pseudintermedius genomes) are iteratively compared to all remaining genomes of S. pseudintermedius, so that with each iteration less frames need to be analysed.
(PDF)
(PDF)
(PDF)
Data Availability Statement
All whole-genome sequences that were obtained for this study are available from the NCBI repository. Accession numbers are available in S1 Table.