

Abstract
Traumatic brain injury (TBI) is the leading cause of trauma related morbidity in the developed world. TBI has been shown to trigger secondary injury cascades including endoplasmic reticulum (ER) stress, oxidative stress, and neuroinflammation. The link between secondary injury cascades and behavioral outcome following TBI is poorly understood warranting further investigation. Using our validated rodent blast TBI model, we examined the interaction of secondary injury cascades following single injury and how these interactions may contribute to impulsive-like behavior after a clinically relevant repetitive TBI paradigm. We targeted these secondary pathways acutely following single injury with the cellular stress modulator, salubrinal (SAL). We examined the neuroprotective effects of SAL administration on significantly reducing ER stress: janus-N-terminal kinase (JNK) phosphorylation and C/EBP homology protein (CHOP), oxidative stress: superoxide and carbonyls, and neuroinflammation: nuclear factor kappa beta (NFκB) activity, inducible nitric oxide synthase (iNOS) protein expression, and pro-inflammatory cytokines at 24 h post-TBI. We then used the more clinically relevant repeat injury paradigm and observed elevated NFκB and iNOS activity. These injury cascades were associated with impulsive-like behavior measured on the elevated plus maze. SAL administration attenuated secondary iNOS activity at 72 h following repetitive TBI, and most importantly prevented impulsive-like behavior. Overall, these results suggest a link between secondary injury cascades and impulsive-like behavior that can be modulated by SAL administration.
Keywords: Traumatic brain injury, Oxidative stress, Endoplasmic reticulum stress, Neuroinflammation, Impulsive-like behavior
1. Introduction
3.2 million Americans are currently living with disabilities from traumatic brain injury (TBI) (Zaloshnja et al., 2008). Impulsivity is one of the most common and potentially dangerous symptoms associated with brain injury (Schwarzbold et al., 2010; Adhikari et al., 2011; Logsdon et al., 2014; Michael et al., 2015). Impulsivity is a key finding in patients diagnosed with chronic traumatic encephalopathy (CTE) and presents early in disease progression (Banks et al., 2014; Rebetez et al., 2015). Currently, no therapies for impulsivity are available for patients diagnosed with CTE. The underlying mechanisms linking neurotrauma to subacute neuropsychiatric symptoms are still poorly understood (Lucke-Wold et al., 2014a).
The concept of an interrelationship between cellular stress and lasting degenerative changes remains to be elucidated. Endoplasmic reticulum (ER) stress and oxidative stress have emerged as contributors to neurodegeneration and behavioral dysfunction. ER stress has been shown to play a significant role in acute and chronic disease pathology following TBI (Zhang et al., 2012; Abdul-Muneer et al., 2014; Begum et al., 2014; Lucke-Wold et al., 2015a). We recently showed that markers of ER stress were increased in the brains of athletes diagnosed with CTE, and rodents exposed to repetitive blast injury (Lucke-Wold et al., 2016).
Janus-N-terminal kinase (JNK) is a common downstream component of ER stress (Urano et al., 2000), which is activated following TBI (Otani et al., 2002; Szmydynger-Chodobska et al., 2010). JNK activity can influence nuclear factor kappa beta (NFκB) translocation to the nucleus, which upregulates pro-inflammatory mediators (Ruan et al., 2015). It is well known that neural injury accelerates the release of pro-inflammatory cytokines which can signal lasting neuronal cell stress (Hong et al., 2016). If the cellular stress response is severe, or sustained, the neuron will undergo apoptosis (Nakagawa and Yuan, 2000), causing extensive gliosis and neuroinflammation (Harvey et al., 2015). We propose that TBI induces NOX4-mediated oxidative stress and JNK-mediated ER stress, which subsequently contributes to neuroinflammation through NFκB activation.
Simultaneous to ER stress activation, oxidative stress occurs and generates free radicals, which play a role in cell death and disease pathology following TBI (Toklu and Tumer, 2015). Free radicals damage cellular membranes, increase carbonyl formation, and can contribute to cell death and neurobehavioral dysfunction (Ferguson et al., 2010). We previously showed NOX4-mediated oxidative stress increased neuronal apoptosis following traumatic brain injury (Lucke-Wold et al., 2015b). In addition, Wu and colleagues used an In vitro model of endothelial injury to causally link NADPH-oxidase (Nox4)-mediated oxidative stress to Janus-N-terminal kinase (JNK)-mediated ER stress (Wu et al., 2014b). The group also silenced JNK-mediated ER stress and observed an attenuation of nuclear translocation NFκB (Wu et al., 2014b). These findings suggest NOX-mediated oxidative stress and JNK-mediated ER stress to be linked to NFκB activation.
In conjunction with these cell stress responses, chronic neuroinflammation has emerged as a possible contributory factor to behavior change (Faden et al., 2015). Preclinical models have shown that TBI is associated with a significant inflammatory burden (Kumar et al., 2014). Furthermore, it has been shown that neuroinflammation can persist years after injury in the brains of retired athletes (Coughlin et al., 2014). Recent clinical evidence ties neuroinflammation to neurobehavioral symptoms (Cho et al., 2013; Wu et al., 2014a). We propose that acute modulation of cellular stress after TBI will positively influence the extracellular inflammatory milieu leading to improved behavioral outcomes.
Salubrinal (SAL) is a modulator of cellular stress known to inhibit protein phosphatase 1, and attenuate global translation (Boyce et al., 2005). Reducing the ER workload promotes proteostasis and cell survival (Hotamisligil, 2010; Tsaytler et al., 2011; Walter and Ron, 2011) SAL has been shown to be neuroprotective in models of protein toxicity (Colla et al., 2012; Huang et al., 2012), stroke (Nakka et al., 2010), excitotoxicity (Sokka et al., 2007), and TBI (Rubovitch et al., 2015). In our model of TBI, have previously shown SAL to reduce ER-mediated apoptosis and to ameliorate impulsive-like behavior (Logsdon et al., 2014). In the present study, we investigated the effects of SAL on reducing neuroinflammation, and impulsive-like behavior following a more clinically relevant repetitive TBI paradigm (Fig. 1).
Fig. 1.
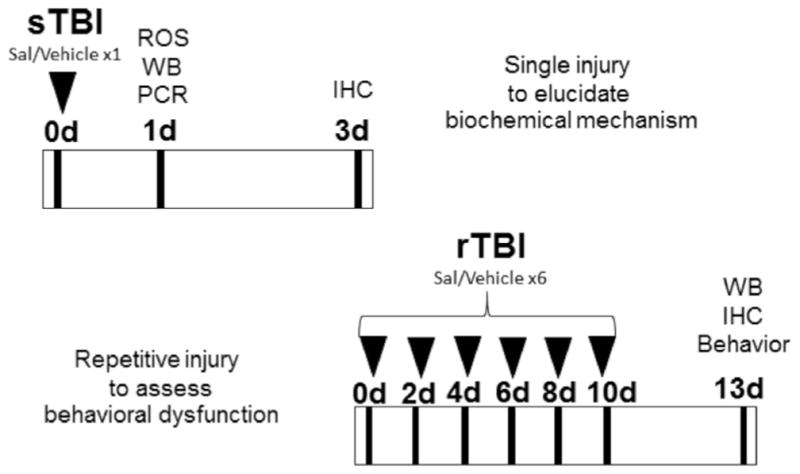
Experimental design. Detailed experimental timeline showing: sTBI (single blast) animals at top; rTBI (repetitive blast) animals at bottom. SAL (Salubrinal); ROS (Reactive oxygen species); WB (Western blot); PCR (Polymerase chain reaction); IHC (Immunohistochemistry).
2. Results
2.1. SAL attenuated markers of ER stress after single blast
ER stress is a common secondary cascade implicated in subacute injury expansion following TBI (Farook et al., 2013; Begum et al., 2014). Our previous study showed that markers of the acute phase ER stress were upregulated following sTBI (Logsdon et al., 2014). In this study, we investigated additional markers of ER stress, JNK phosphorylation and CHOP activation, which have been associated with NFκB activity (Deng et al., 2004; Tsai et al., 2012).
Fig. 2(A) indicates a significant difference between experimental groups in JNK activity after sTBI (F(2,9) =9.04; P<0.01). A significant increase in the ratio between JNK phosphorylation and total JNK expression was observed at 24 h after sTBI compared to control rats (q=5.79; P<0.01). SAL administration significantly attenuated JNK phosphorylation when compared to vehicle-treated sTBI rats (q=4.31; P<0.05).
Fig. 2.

SAL reduced ER stress markers after single blast. We measured a significant increase in JNK activity (pJNK/tJNK) at 24 h post-sTBI (**P<0.01 vs Vehicle); SAL significantly mitigated JNK activity (#P<0.05 vs sTBI+Vehicle). (A). We measured a significant increase in CHOP protein expression at 24 h post-sTBI (**P<0.01 vs Vehicle); SAL significantly mitigated CHOP expression (#P<0.05 vs sTBI+Vehicle) (B). One-way ANOVA, Newman-Keul’s post hoc. Mean±S.E.M. n=4.
Fig. 2(B) indicates a significant difference between experimental groups in CHOP activation after sTBI (F(2,9) =8.769; P<0.01). A significant increase in CHOP expression at 24 h was observed in sTBI rats as compared to control rats (q=5.86; P<0.01). SAL administration significantly attenuated CHOP activation when compared to vehicle-treated sTBI rats (q=3.673; P<0.05). SAL successfully reduced markers of ER stress when administered acutely after injury.
2.2. SAL reduced markers of oxidative stress after single blast
ER stress activation has been proposed to directly increase oxidative stress particularly in the striatum (Malhotra and Kaufman, 2007). ROS generation is a consequence of TBI mainly through membrane damage and subsequent NOX4 system activation (Zhang et al., 2012; Loane et al., 2013). Activation of the NOX4 system predominantly creates superoxide (Brennan et al., 2009; Lucke-Wold et al., 2015b). A previous report indicated that both NOX4 and superoxide were acutely elevated after TBI (Ansari et al., 2014).
Fig. 3(A) shows a significant difference in carbonyl levels between groups after sTBI (F(2,9) =10.21; P<0.01). Carbonyl formation is an end product of oxidative stress damage. A significant increase in carbonyl levels was measured in sTBI rats as compared to control rats (q=6.26; P<0.01). SAL administration significantly reduced carbonyl levels (q=4.23; P<0.05) when compared to vehicle-treated sTBI rats.
Fig. 3.

SAL reduced markers of oxidative stress after single blast. A significant increase in protein carbonyl levels was measured at 24 h post-sTBI (**P<0.01 vs Vehicle); SAL treatment significantly attenuated carbonyl levels (#P<0.01 vs sTBI) (A). A significant increase in superoxide levels was measured at 24 h post-sTBI (*P<0.05 vs Vehicle); SAL treatment significantly attenuated superoxide levels (##P<0.01 vs sTBI) (B). No significant differences were observed in total ROS levels (P<0.05) (C). We observed a significant increase in NOX4 (green) fluorescence at 24 h post-sTBI (***P<0.001 vs Vehicle); SAL significantly reduced NOX4 fluorescence (##P<0.01) (n=4). NOX4 images include nuclear counterstain DAPI (blue) (D). Images are from the striatum. Images are displayed at 20 ×; insets at 63 ×. (Scale bars =30 μm). One-way ANOVA, Newman-Keul’s post hoc. Mean±S.E.M. n=4.
Fig. 3(B) shows a significant difference in superoxide levels between experimental groups after sTBI (F(2,9) =7.68; P<0.05). A significant increase in superoxide levels was measured in sTBI rats when compared to control rats (q=4.92, P<0.05). SAL administration significantly reduced superoxide levels as compared to vehicle-treated sTBI rats (q=4.68, P<0.01). Fig. 3(C) shows no difference in total ROS levels (F(2,9) =0.16; P>0.05).
Fig. 3(D) shows a significant difference in corrected total cell fluorescence for NOX4 in the striatum at 24 h post-sTBI (F (2,27) =3.76; P<0.05). A significant increase in NOX4 fluorescence was observed in sTBI rats compared to control rats (q=5.46, P<0.001). SAL administration significantly reduced NOX4 fluorescence when compared to vehicle-treated sTBI rats (q=5.61, P<0.01). These results suggest that SAL attenuates NOX4-mediated oxidative stress by specifically reducing super-oxide in the striatum.
2.3. SAL attenuated markers of neuroinflammation after single blast
ER stress and oxidative stress have both been associated with increased neuroinflammation after TBI (Deslauriers et al., 2011; Bellezza et al., 2014; Hayashi, 2015). Integral to the process of neuroinflammation is activation and nuclear translocation of nuclear factor kappa B (NFκB) (Bracchi-Ricard et al., 2013). NFκB and inducible nitric oxide synthase (iNOS) are known to promote pro-inflammatory cytokines, such as, tumor necrosis factor alpha (TNFα) and interleukin 1 beta (IL-1β) (Hu et al., 2014). These inflammatory cytokines can signal changes that damage neuronal cell membranes and intracellular organelles (Abdullah and Bayraktutan, 2014).
Fig. 4(A) shows a significant difference in NFκB p65 expression between experimental groups at 24 h after sTBI (F(2,9) =4.67; P<0.05). NFκB p65 expression was significantly increased in sTBI rats compared to control rats (q=4.101; P<0.05). SAL administration significantly reduced NFκB p65 expression when compared to vehicle-treated sTBI rats (q=3.24; P<0.05).
Fig. 4.

SAL mitigated markers of neuroinflammation after single blast. We measured a significant increase in NFκB p65 expression at 24 h post-sTBI (*P<0.05 vs Vehicle); SAL significantly mitigated NFκB p65 expression (#P<0.05 vs sTBI+Vehicle) (A). We revealed a significant increase in iNOS protein expression at 24 h post-sTBI (*P <0.05 vs vehicle); SAL significantly mitigated iNOS expression (#P<0.05 vs sTBI+Vehicle) (B). We measured a significant increase in IL-1β mRNA abundance at 24 h post-sTBI (*P<0.05 vs Vehicle); SAL significantly mitigated IL-1β abundance (#P<0.05 vs sTBI +Vehicle) (C). We measured a significant increase in TNFα mRNA abundance at 24 h post-sTBI (*P<0.05 vs Vehicle); SAL significantly mitigated TNFα abundance (#P<0.05) (D). One-way ANOVA, Newman-Keul’s post hoc. Mean±S.E.M. n=4.
Fig. 4(B) shows a significant difference in iNOS expression at 24 h after sTBI (F(2,9) =6.81; P<0.05). A significant increase in iNOS expression was measured in sTBI rats compared to control rats (q=4.92; P<0.05). SAL administration significantly reduced iNOS expression when compared to vehicle-treated sTBI rats (q=3.97; P<0.05).
Fig. 4(C) shows a significant difference in IL-1β mRNA abundance at 24 h post-sTBI amongst the treatment groups (F(2,9) =5.62; P<0.05). A significant increase in IL-1β mRNA abundance was measured in sTBI rats compared to control rats (q=4.13; P<0.05). SAL administration significantly reduced IL-1β mRNA abundance when compared to vehicle-treated sTBI rats (q=4.08; P<0.05).
Fig. 4(D) shows a significant difference amongst the treatment groups in TNFα mRNA abundance at 24 h post-sTBI (F(2,9) =5.54; P<0.05). A significant increase in TNFα mRNA abundance was measured in sTBI rats compared to control rats (q=4.49; P<0.05). SAL administration significantly reduced TNFα mRNA abundance when compared to vehicle-treated sTBI rats (q=3.47; P<0.05). These results show that SAL significantly reduced neuroinflammatory markers following blast injury.
2.4. SAL attenuated markers of neurodegeneration after single blast
Gliosis and degenerative changes are initial signs of persistent neurodegeneration (Damjanac et al., 2007). We previously showed that ER stress is closely tied to neurodegenerative disease following neurotrauma (Lucke-Wold et al., 2016), and that TBI can produce gliosis and degenerative changes acutely post-injury (Turner et al., 2012).
Fig. 5(A) shows a significant difference in corrected total cell fluorescence for GFAP in the lateral orbitofrontal cortex at 72 h post-sTBI (F(2,29) =35.23; P<0.001). A significant increase in GFAP fluorescence was observed in sTBI rats compared to control rats (q=10.07, P<0.001). SAL administration significantly reduced GFAP fluorescence when compared to vehicle-treated sTBI rats (q=7.58, P<0.001).
Fig. 5.

SAL attenuated markers of neurodegeneration after single blast. We observed a significant increase in GFAP (green) fluorescence at 72 h post-sTBI (***P<0.001 vs Vehicle); SAL significantly reduced GFAP fluorescence (###P<0.001) (n=4). GFAP images include nuclear counterstain DAPI (blue) (A). We observed a significant increase in FJB (green) fluorescence at 72 h post-sTBI (*P<0.05 vs Vehicle); SAL significantly reduced FJB fluorescence (#P<0.05) (B). One-way ANOVA, Newman-Keul’s post hoc. Mean±S.E.M. n=4. Images are from the lateral orbitofrontal cortex. Images are displayed at 20 ×; insets at 63 ×. (Scale bars =30 μm).
Fig. 5(B) shows a significant difference in corrected total cell fluorescence for FJB in the lateral orbitofrontal cortex at 72 h post-sTBI (F(2,29) =7.43; P<0.01). A significant increase in FJB fluorescence was observed in sTBI rats compared to control rats (q=5.18, P<0.05). SAL administration significantly reduced FJB fluorescence when compared to vehicle-treated sTBI rats (q=3.32; P<0.05). These results demonstrate that SAL significantly reduces gliosis and neurodegeneration.
2.5. SAL reduces neuroinflammation after repetitive blast
Under persistent conditions, iNOS, can contribute to neurode-generation and behavioral symptoms (Jayakumar et al., 2014). Hsieh and colleagues discovered that iNOS activation is the mechanism by which ER stress causes the formation of ROS and is a direct tie to neuroinflammation (Hsieh et al., 2007).
Fig. 6(A) shows a significant difference in NFκB p65 expression between experimental groups at two weeks after rTBI (F (2,12) =14.04; P<0.05). NFκB p65 expression was significantly increased in rTBI rats compared to control rats (q=6.05; P<0.05). NFκB p65 expression was also increased in rTBI+SAL rats when compared to control rats (q=6.86; P<0.05).
Fig. 6.

SAL reduces neuroinflammation after repetitive blast. We measured a significant increase in NFκB p65 expression at two weeks post-rTBI (*P<0.05 vs Vehicle); and when SAL was administered post-rTBI (*P<0.05 vs Vehicle) (A). We revealed a significant increase in iNOS protein expression at two weeks post-rTBI (*P <0.05 vs vehicle); SAL significantly mitigated iNOS expression (#P<0.05 vs sTBI+Vehicle) (B). One-way ANOVA, Newman-Keul’s post hoc. Mean±S.E.M. n=4. Colocalization of iNOS (red) merged with CHOP (green) was determined by levels of yellow in each image (Overlap coefficient; r values). All panels display nuclear counterstain DAPI (blue). Arrows demarcate iNOS fluorescence (C). Images are from the lateral orbitofrontal cortex. Images are displayed at 20 ×; Insets displayed at 63 ×. (Scale bars =30 μm).
Fig. 6(B) shows a significant difference in iNOS expression between experimental groups at two weeks after rTBI (F(2,12) =6.25; P<0.05). A significant increase in iNOS expression was measured in rTBI rats compared to control rats (q=4.84; P<0.05). SAL administration significantly reduced iNOS expression when compared to vehicle-treated rTBI rats (q=3.49; P<0.05).
Fig. 6(C) shows IHC colocalization (yellow) of ER stress marker CHOP (green) with neuroinflammation marker iNOS (red) in the lateral orbitofrontal cortex. Overlap Pearson’s coefficient revealed a small correlation in vehicle controls (r=0.17), a large correlation in rTBI+Vehicle (r=0.65), and a small correlation in rTBI+SAL (r=0.22). These results suggest neuroinflammation and ER stress are increased in the same cells after repetitive blast exposure.
2.6. SAL ameliorated impulsive-like behavior after repetitive blast
Neuropsychiatric symptoms, such as impulsive-like behavior, commonly burden people who have suffered from multiple mild concussions, such as athletes and soldiers (Omalu et al., 2011; Bailes et al., 2013). Impulsive-like behavior becomes evident in rodents with damage to the lateral orbitofrontal cortex (Mar et al., 2011; Bidzan et al., 2012; Johnson et al., 2013).
We have previously observed impulsive-like behavior among rats exposed to a single blast from our TBI model (Logsdon et al., 2014). Using this more clinically relevant repetitive injury paradigm, we looked at impulsive-like behavior by measuring the time spent in the open arms of the EPM after repeat blast exposure.
Fig. 7(A) shows a significant difference in open arm time at two weeks post-rTBI amongst the treatment groups (F(2,24) =4.72; P<0.05). A significant increase was measured in the time that the rTBI rats spent in the open arms of the EPM compared to control rats (q=4.09; P<0.05). Interestingly, SAL administration significantly attenuated the open arm time when compared to vehicle-treated rTBI rats (q=3.31; P<0.05).
Fig. 7.

SAL ameliorated impulsive-like behavior after repetitive blast A significant increase was observed in the time that the rats spent in the open arms of the EPM at 72 h post-rTBI (*P<0.05 vs Vehicle); SAL significantly reduced the time spent (#P<0.05 vs rTBI+vehicle) (A). No significant differences were observed in the total distance moved among rats in each group (P<0.05) (B). Track plots from Anymaze™ were overlaid for each subject to provide a clear visual representation of the rats’ behavior during the EPM trials (C–E). One-way ANOVA, Newman-Keul’s post hoc. Mean±S.E.M. n=9.
Fig. 7(B) shows no difference in the total distance the rats travelled between the experimental groups (F(2,24) =2.54; P<0.05). Track plots from Anymaze™ were overlaid for each subject to provide a clear visual representation of the rats’ behavior during the EPM trials (Fig. 7(C)–(E)). SAL had a protective effect on reducing impulsive-like behavior.
2.7. Proposed mechanism of blast injury
Fig. 8 shows a proposed detailed schematic of the injury cascade following blast injury. We propose blast TBI causes vascular disruption and blood extravasation into the brain parenchyma that can damage plasma membranes and trigger secondary injury cascades (Uranga et al., 2009; Liu et al., 2013; Nisenbaum et al., 2014). The secondary injury cascades contribute to neurodegeneration and ultimately lead to neurobehavioral dysfunction.
Fig. 8.
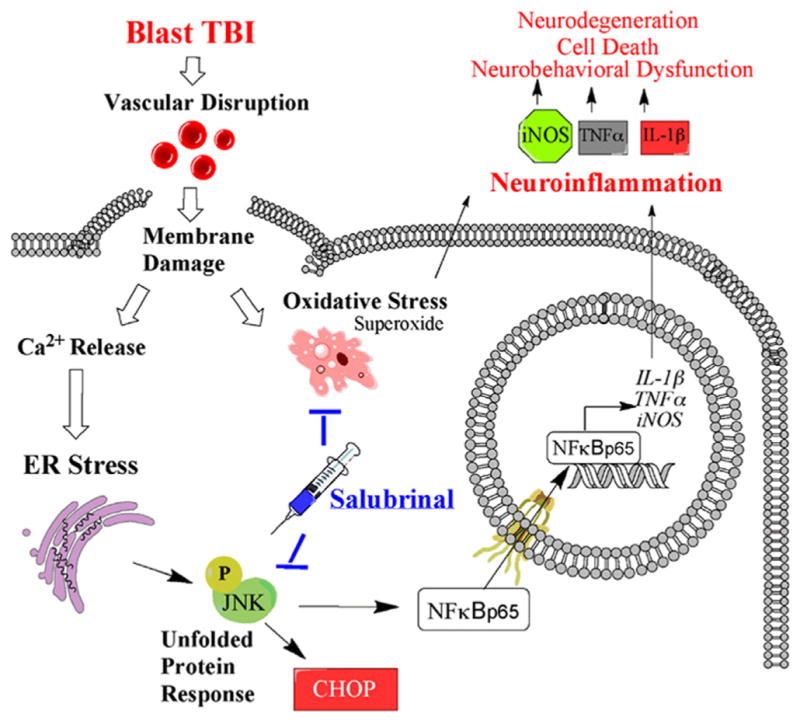
Blast injury cascade with SAL mechanism of action. Schematic of blast injury cascade and salubrinal mechanism of action. TBI=Traumatic Brain Injury; ER=Endoplasmic Reticulum; CHOP=C/EBP homology protein; NFκB=Nuclear factor kappa beta; JNK=Janus n-terminal kinase; iNOS=inducible Nitric oxide synthase; TNFα=Tumor necrosis factor alpha; IL-1β=Interleukin 1 beta.
3. Discussion
Blast TBI cause primary injury due, in part, to acceleration-deceleration forces to the rat’s head (Goldstein et al., 2012). The current study shows an initiation of secondary injury cascades including ER stress, oxidative stress, and neuroinflammation following primary injury. These acute injury cascades lead to additional glial reactivity and neurodegenerative changes. We also show that the cellular stress responses occurred concurrent with neuroinflammation after blast TBI. Neuroinflammation is a common secondary effect of brain injury demonstrated in a variety of rodent TBI models (Abdul-Muneer et al., 2013, 2014; Cho et al., 2013; Hu et al., 2014; Roth et al., 2014), and has been linked clinically to single and repetitive head injuries (Aungst et al., 2014; Webster et al., 2015). While previous studies have suggested an association between ER stress and neuroinflammation, what remains unknown is whether regulation of these responses would affect neurodegeneration and improve neurobehavioral outcome (Fenn et al., 2015). Additionally, ER stress activation has been proposed to directly increase oxidative stress through activation of iNOS particularly in the striatum (Malhotra and Kaufman, 2007). Using the ER stress modulator SAL, we show a potential link between these pathways. We also offer compelling evidence that targeting acute cellular stress cascades can prevent impulsive-like behavior after repeat injury as measured by time spent in the open arms of the EPM.
An important secondary injury cascade often increased following TBI and intimately linked to ER stress is oxidative stress (Cho et al., 2013). We report an increase in oxidative stress damage in the striatum following blast exposure, as evidenced by increased carbonyl and NOX4-mediated superoxide levels. Super-oxide production is a consequence of TBI mainly through activation of the NOX system (Brennan et al., 2009; Zhang et al., 2012; Loane et al., 2013). A previous report indicated that NOX4 and superoxide levels were elevated at 6 h after blast TBI and persisted for 72 h (Ansari et al., 2014). Additionally, NOX4 mediated oxidative stress perpetuates damage to neuronal membranes leading to cell death (Zhang et al., 2012). A late-onset symptom of CTE is motor dysfunction similar to Parkinson’s disease that indicates cell death and damage to the striatum (Stern et al., 2011). In a future study we will investigate late-onset motor disturbance associated with the striatal damage reported herein.
Persistent damage to the cell from oxidative stress can trigger a measurable increase in ER stress associated JNK phosphorylation (Quan et al., 2015), and subsequent NFkB activity, which affects neuroinflammation. An increase in both pJNK and NFkB p65 expression were demonstrated in this study. JNK can signal NFkB translocation to the nucleus, which may upregulate pro-in-flammatory mediators such as iNOS, TNFα, and IL-1β (Hu et al., 2014; Ruan et al., 2015). In line with this ensuing injury cascade following blast, we found increased levels of pro-inflammatory markers: NFκB, iNOS, TNFα, and IL-1β. These pro-inflammatory markers are thought to cause a sudden oxidative burst that overwhelms antioxidant defense cascades leading to further damage of surrounding brain tissue (Liao et al., 2013). We show repetitive blast TBI causes a subacute increase in iNOS fluorescence, suggesting a persistent inflammatory response. Consequently, while neuroinflammation is a necessary response to brain injury (Finnie, 2013), unabated, persistent neuroinflammation can lead to irreversible neurodegeneration and poor injury outcome.
Furthermore, we recently showed that ER stress is an important secondary injury response activated after blast TBI (Logsdon et al., 2014), and in human CTE (Lucke-Wold et al., 2016). ER stress can also lead to cognitive dysfunction following TBI as we previously demonstrated (Dash et al., 2015; Lucke-Wold et al., 2015a). In this paper, we show ER stress markers CHOP and pJNK fluorescence to be increased following single injury, and co-localized with iNOS following repeat injury. Similar to oxidative stress, the ER stress response has significant cross talk with neuroin-flammatory pathways through JNK signaling (Prell et al., 2014). A recent study suggests crosstalk between JNK phosphorylation and NFκB activity after inflammatory challenge with an ER stressor (Ruan et al., 2015).
Importantly, we show, for the first time, that SAL administration post-injury attenuates iNOS protein expression and JNK phosphorylation. This is in agreement with a recent study where SAL administration reduced NFκB and microglia activation in a model of Alzheimer’s disease (Huang et al., 2012). SAL also improved chronic motor performance in a model of spinal cord injury (Ohri et al., 2013). In a future study, we will investigate if SAL improves motor performance long-term following TBI. Further investigation is needed to see if acute modulation of cellular stress pathways will lead to sustained behavioral improvements. Our group and others showed that docosahexaenoic acid, another less-specific ER stress modulator, improved cognitive performance following TBI (Begum et al., 2013; Lucke-Wold et al., 2015a). Based on the results of this current study and the supporting background information from prior studies, we propose a novel mechanism of action for the beneficial properties of SAL through downstream regulation of NFκB activity. In support of this claim, we demonstrated that SAL administration after blast attenuated pJNK, NFκB, iNOS, TNFα, and IL-1β expression. SAL also decreased markers of glial reactivity after single blast exposure, suggesting a protective effect at the blood-brain barrier as we previously reported (Logsdon et al., 2014). Following repetitive blast, SAL furthermore was shown to reduce iNOS and ER stress markers.
Abnormal behavioral symptoms can be a manifestation of persistent neuroinflammation, and are of particular clinical relevance for patients suffering multiple mild-head injuries (Sominsky et al., 2015). We showed that in the more clinically relevant repetitive injury scenario, rats exposed to injury displayed impulsive-like behavior as measured on the EPM 72 h following the last blast. Interestingly, SAL administration reduced impulsive-like behavior when administered after each blast exposure. The data suggest that SAL may mitigate subacute markers of neuroinflammation and thereby reduce impulsive-like behavior after repetitive brain trauma.
These findings have broad reaching implications regarding the importance of targeting cellular stress acutely post-injury in order to reduce neurodegenerative changes and ameliorate neurobehavioral dysfunction. We showed that ER stress modulation after blast injury decreases acute superoxide formation in the striatum, attenuates neuroinflammatory markers in the frontal cortex, and ameliorates impulsive-like behavior in rats. In doing so, we map a unique interconnection between intracellular stress cascades and neuroinflammation. Further pre-clinical studies are warranted to determine the role of ER stress, oxidative stress, and neuroinflammation at subacute time points following brain injury and how these pathways contribute to neurobehavioral dysfunction.
4. Conclusions
In summary, cellular stress and neuroinflammation are intrinsically interconnected and play an important role in injury progression following TBI. We show that blast exposure induced markers of oxidative stress, which is known to contribute to the exacerbation of ER stress and neuroinflammation. Furthermore, we show that ER stress may be linked to neuroinflammation through the JNK-mediated NFκB pathway. Surprisingly, SAL reduced markers of ER stress and oxidative stress; thereby, reducing markers of neuroinflammation post-blast. The likely mechanism is reduction of JNK phosphorylation and iNOS activity. Most importantly, SAL ameliorated impulsive-like behavior when administered after each repetitive blast exposure. Secondary injury modulation with key multi-target pharmaceutics offers a promising approach to reduce the long-term neuropsychiatric symptoms associated with head injuries.
5. Experimental procedure
5.1. Animals
Fifty-one (51) male Sprague-Dawley rats (Hilltop Lab Animals) at 2–3 months of age were used in this study. The West Virginia University Animal Care and Use Committee approved all procedures involving rats. Rats were acclimated for 1 week prior to use and housed under 12 h light/dark conditions with food and water available ad libitum. Animal experiments were performed according to the principles of the Guide for the Care and Use of Laboratory Animals.
5.2. Salubrinal
SAL (Tocris Biosciences) was dissolved in 0.5% DMSO and delivered via intraperitoneal (i.p.) injection at a dose of 1 mg/kg (Sokka et al., 2007; Liu et al., 2014).
5.3. Blast overpressure exposure
Blast exposure was induced as previously described (Turner et al., 2013). Blast pressure with a 0.005″ membrane (15 psi incident wave; 50 psi reflected wave) was determined, from our previous work, to exhibit neural injury markers with no mortality (Turner et al., 2013; Logsdon et al., 2014; Lucke-Wold et al., 2014b). Following blast exposure, rats were returned to a holding cage equipped with a homeothermic heating pad. Once basic reflexes were restored, rats were returned to their home cage.
5.4. Experimental groups
A detailed timeline shows the course of injury and treatment for each group of rats (Fig. 1). Rats were randomly assigned to one of three treatment groups. Group 1 rats served as sham controls (anesthetized with 4% isoflurane). Following anesthesia, rats were injected with vehicle (0.5% DMSO; i.p.) 5 min after being placed on blast table. Group 2 rats served as our experimental control group, in which anesthetized rats were oriented similarly to sham rats and subsequently exposed to blast. Rats were injected with vehicle 5 min after each blast exposure. Group 3 served as our experimental group. Rats received blast exposure as group 2, but SAL (1 mg/kg; i.p.) was administered 5 min after each blast exposure (Rubovitch et al., 2015).
There was two blast TBI subgroupings: a single blast TBI (sTBI) and a repeated blast TBI (rTBI). Rats in the rTBI group received a blast every other day over two weeks, for a total of six blast injuries. This repetitive schedule was determined by our previous work that showed markers of ER stress activation (Lucke-Wold et al., 2015a), and from other TBI models that showed markers of neuroinflammation and neurodegeneration (Mouzon et al., 2012; Mouzon et al., 2014).
Twenty-four (24) sTBI rats were randomly divided into the three treatment groups and euthanized for biochemical analysis at 24 h post-blast (n=4 rats per group), and immunohistochemistry (IHC) at 72 h post-blast (n=4 rats per group). Twenty-seven (27) rTBI rats were randomly divided into the three treatment groups and were subject to behavioral testing 72 h after the repetitive blast schedule (n=9 rats per group). Four rTBI rats from each experimental group were euthanized for IHC after behavioral testing was complete.
5.5. Tissue preparation
Rats used for biochemical analysis were euthanized and their brains were rapidly removed in an ice-cold Halt™ protease/phosphatase inhibitor cocktail mix (Thermo Scientific). The striatum was dissected out from each hemisphere, flash frozen in liquid nitrogen, and stored at −80 °C for later measurement of carbonyl and reactive oxygen species (ROS) levels. The frontal cortex was dissected out from each hemisphere, flash frozen in liquid nitrogen and stored at −80 °C for later measurement of protein expression and mRNA abundance. Rats used for IHC were anesthetized with 4% isoflurane and perfused transcardially with ice-cold saline. Following perfusion, brains were removed, flash frozen in isopentane (−60 °C) and stored at −80 °C for IHC preparation.
5.6. Carbonyl measurement
For carbonyl measurement, striatal tissue samples from sTBI rats (n=4 rats per group) were assayed using an OxyBlot Protein Oxidation Detection kit (Millipore). Striatal tissues from each treatment group were sonicated in 6% SDS. Control samples were mixed with derivation control solution and experimental samples were mixed with 2,4-dinitrophenylhydrazine. Samples were incubated for 15 min followed by addition of neutralization solution. Samples were loaded (20 μl per well) and electrophoresed on a 10% acrylamide gel and immunoblotted as outlined in the Western blot methods below.
5.7. ROS measurement
For ROS detection, striatal tissue from sTBI rats were homogenized and cells were isolated by incubation in collagenase at 2 mg/ml for 30 min. Cells were separated by enzyme digestion and manual disruption by repeated pipetting. Cells were strained through a 70 μm nylon cell strainer followed by centrifugation at 400g for 5 min. Pellets were resuspended to a concentration of 1.0 × 106 cells/ml in DMEM.
Total ROS and superoxide levels were detected using a Total ROS/Superoxide Detection kit (Enzo Life Sciences) according to manufacturer’s instructions. In brief, 100 μl of suspended cells were added to each well of a dark-sided 96 well plate with a clear bottom. Cells were incubated overnight at 37 °C in DMEM. The following day, media was removed and 100 μl of ROS/Superoxide Detection Solution was added to each well and incubated in the dark for 1 h. Detection of total ROS (green) and superoxide (red/yellow) fluorescence was detected at an excitation wavelength of 488/520 nm and an emission wavelength of 550/610 nm, respectively. Data were collected using Gen5 software (BioTek). Concentrations were determined based on a known standard curve.
5.8. Western blot
Protein samples from sTBI and rTBI rats were prepared in 1% SDS and Western blot analysis was performed as previously described (Lucke-Wold et al., 2014b). Primary antibodies (and dilution factors) were rabbit anti-pJNK monoclonal antibody (mAB) (Thr183/Tyr185) (1:500), rabbit anti-binding immunoglobulin protein (BiP) mAB (1:1000), rabbit anti-JNK mAB (1:1000) (Cell Signaling); mouse anti-NFκB p65 polyclonal antibody (pAB) (1:200), rabbit anti-iNOS pAB (1:200) (Santa Cruz). A rabbit anti-β-actin mAB (1:10,000) (Cell Signaling) was used as an endogenous control to normalize protein loading. Secondary antibodies were IRDye® 800CW (goat anti-rabbit) and IRDye® 680RD (goat anti-mouse) (LI-COR Biosciences). Images were collected using the Odyssey Classic Infrared Imaging System (LI-COR Biosciences). Images were converted to gray scale, and detected bands were quantified using Image Studio Lite Software (LI-COR Biosciences). Bands were normalized to β-actin values to measure relative intensity.
5.9. Quantitative real-time polymerase chain reaction
RNA samples from sTBI and rTBI rats were prepared in TRIzol® reagent (Life Technologies) and confirmed for quality (1.8–2.1 absorbance ratio). Reverse transcription was conducted and real-time PCR analyses were performed on cDNA using the following oligonucleotide primer sets: TNFα (Rn01525859_g1), IL-1β (Rn00580432_m1), and 18s rRNA (Hs99999901_s1; endogenous control) (Life Technologies). Changes in mRNA abundance were determined by the ΔΔCt method with a threshold cycle value of 0.2 normalized to 18s rRNA.
5.10. Histology
Whole brains from sTBI and rTBI rats were mounted on a Leica CM3050S cryostat (Leica Microsystems) set to −20 °C. Coronal sections of the frontal cortex were sliced at a thickness of 20 μm and mounted onto glass slides for IHC staining as previously described (Lucke-Wold et al., 2015a). Briefly, brain slices were circumscribed, and incubated overnight with primary antibodies: iNOS (Santa Cruz), glial fibrillary acidic protein (GFAP), C/EBP homology protein (CHOP), BiP, and pJNK (Cell Signaling). The next day, an Alexa Flour® secondary antibody (Invitrogen) was applied to slides for 3 h, and coverslip mounted with Vectashield® 4′,6-diamidino-2-phenylindole (DAPI) nuclear counterstain (Vector). If staining for colocalization, a second set of primary and secondary antibodies were applied prior to fixing the coverslip. Fluorojade B (FJB) (Millipore), and ROS (Enzo Life Sciences) staining was performed in accordance with manufacturer’s instructions.
All immunohistochemistry was performed as described previously (Lucke-Wold et al., 2014b, 2015b). Briefly, images were acquired from the lateral orbitofrontal cortex (15 slides per animal). Fluorescent imaging was performed using a Zeiss Axio Observer Z1. For fluorescent staining, 10 distinct cells with clear morphology were randomly selected per slide, outlined, and measured with ImageJ software (NIH) by an observer blinded to experimental group. Density was adjusted per mean area to give corrected total cell fluorescence normalized to background. Co-localization quantification with the Just Another Co-localization plugin for ImageJ was used to determine overlap coefficient or Pearson’s coefficient (Bolte and Cordelieres, 2006). Overlap coefficient was calculated using k2= k1*k2 with values adjusted to threshold (Lucke-Wold et al., 2015a).
5.11. Elevated plus maze
Impulsive-like behavior was assessed in rTBI rats using the Elevated plus maze (EPM). Increased time spent in the open arms was considered a sign of impulsive-like behavior in the rodent (Mosienko et al., 2012; Johnson et al., 2013). The EPM was set at a height of 60 cm above the floor. The two open arms intersected perpendicular to the two closed arms. Each arm was 50 cm × 10 cm. The closed arms were encased by black siding 30 cm tall. Each rat was placed in the middle of the EPM facing an open arm and tracking was performed for 5 min using AnyMaze software (Stoelting), which pinpointed the location of the rat’s head and body continuously throughout the testing trial.
5.12. Statistical analysis
Data were analyzed using a one-way analysis of variance (AN-OVA) followed by Newman-Keul’s post hoc tests for between groups comparison. For colocalization studies, Pearson’s coefficient was obtained for control sections. Overlap coefficient was obtained for experimental groups to determine the extent of same-cell protein expression. Sample sizes were determined using a power analysis with an α of 0.05, a β of 0.2, and a sample effect of 0.4 for behavioral data and 0.3 for all other data (DSS Research Power Analysis). P<0.05 was considered significant.
Acknowledgments
We thank Dr. James P. O’Callaghan and Dr. Diane B. Miller of the Center for Disease Control (CDC) and the National Institute for Occupational Safety and Health (NIOSH). We appreciate guidance of Dr. James W. Simpkins during manuscript preparation (Director of Stroke Center). The authors acknowledge the assistance with animal work done by Xinlan Li (Dept. of Neurosurgery). The authors acknowledge the technical assistance performed by Kelly E. Smith. The authors are grateful for the assistance of Dr. Robert T.T. Gettens and Nicholas St. John of Western New England University for their help with design of the blast model and Mr. Peter Bennett and Mr. James Edward Robson for model construction. Imaging experiments and image analysis were performed in the West Virginia University Microscope Imaging Facility, which has been supported by the National Institutes of Health (NIH) Grants P20 RR016550, P30 RR032138/GM103488 and P20 RR016477. This work was supported by a Research Funding and Development (RFDG) grant from the West Virginia University Health Sciences Center Office of Research and Graduate Education (to JDH and CLR), a training grant from the NIH (to RCT) (5T32GM08174), and an American Foundation of Pharmaceutical Education pre-doctoral fellowship (to AFL and BPL). An American Medical Association Foundation Seed Grant and Neurosurgery Research & Education Foundation Medical Student Summer Research Fellowship was awarded (to BPL).
Abbreviations
- TBI
traumatic brain injury
- sTBI
single blast-induced traumatic brain injury
- rTBI
repeated blast-induced traumatic brain injury
- ER
endoplasmic reticulum
- SAL
salubrinal
- GFAP
Glial fibrillary acidic protein
- FJB
Fluorojade B
- JNK
c-jun N-terminal kinase
- NFκB
nuclear factor kappa beta
- iNOS
intrinsic nitric oxide synthase
- BiP
binding immunoglobulin protein
- CHOP
C/EBP homology protein
- TNFα
tumor necrosis factor alpha
- IL-1β
interleukin 1 beta
- NOX
NADPH-oxidase
Footnotes
Conflict of interest
The authors have no confiicts of interest to disclose.
Authors’ contributions
AFL conceived the study, designed the experiments, performed blast/injections, dissected out brain regions, executed biochemical analyses, analyzed the data, and wrote the manuscript. BPL conceived the study, designed the experiments, performed blast/injections, helped execute biochemical analyses, analyzed the data, and wrote the manuscript. LN performed the behavioral experiments and helped write the manuscript. RRM provided invaluable biochemical and behavioral knowledge, in addition to helping conceive the study. RCT assisted with blast exposure and helped write the manuscript. CLR provided invaluable clinical knowledge, in addition to helping conceive the study, design experiments, and manuscript preparation. JDH provided invaluable scientific knowledge, in addition to helping conceive the study, design experiments, and manuscript preparation.
References
- Abdul-Muneer PM, Chandra N, Haorah J. Interactions of oxidative stress and neurovascular inflammation in the pathogenesis of traumatic brain injury. Mol Neurobiol. 2014 doi: 10.1007/s12035-014-8752-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdul-Muneer PM, Schuetz H, Wang F, Skotak M, Jones J, Gorantla S, Zimmerman MC, Chandra N, Haorah J. Induction of oxidative and nitrosative damage leads to cerebrovascular inflammation in an animal model of mild traumatic brain injury induced by primary blast. Free Radic Biol Med. 2013;60:282–291. doi: 10.1016/j.freeradbiomed.2013.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdullah Z, Bayraktutan U. NADPH oxidase mediates TNF-alpha-evoked in vitro brain barrier dysfunction: roles of apoptosis and time. Mol Cell Neurosci. 2014;61:72–84. doi: 10.1016/j.mcn.2014.06.002. [DOI] [PubMed] [Google Scholar]
- Adhikari A, Topiwala MA, Gordon JA. Single units in the medial prefrontal cortex with anxiety-related firing patterns are preferentially influenced by ventral hippocampal activity. Neuron. 2011;71:898–910. doi: 10.1016/j.neuron.2011.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansari MA, Roberts KN, Scheff SW. A time course of NADPH-oxidase up-regulation and endothelial nitric oxide synthase activation in the hippocampus following neurotrauma. Free Radic Biol Med. 2014;77:21–29. doi: 10.1016/j.freeradbiomed.2014.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aungst SL, Kabadi SV, Thompson SM, Stoica BA, Faden AI. Repeated mild traumatic brain injury causes chronic neuroinflammation, changes in hippocampal synaptic plasticity, and associated cognitive deficits. J Cereb Blood Flow Metab. 2014;34:1223–1232. doi: 10.1038/jcbfm.2014.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailes JE, Petraglia AL, Omalu BI, Nauman E, Talavage T. Role of sub-concussion in repetitive mild traumatic brain injury. J Neurosurg. 2013;119:1235–1245. doi: 10.3171/2013.7.JNS121822. [DOI] [PubMed] [Google Scholar]
- Banks SJ, Mayer B, Obuchowski N, Shin W, Lowe M, Phillips M, Modic M, Bernick C. Impulsiveness in professional fighters. J Neuropsychiatry Clin Neurosci. 2014;26:44–50. doi: 10.1176/appi.neuropsych.12070185. [DOI] [PubMed] [Google Scholar]
- Begum G, Harvey L, Dixon CE, Sun D. ER stress and effects of DHA as an ER stress inhibitor. Transl Stroke Res. 2013;4:635–642. doi: 10.1007/s12975-013-0282-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begum G, Yan HQ, Li L, Singh A, Dixon CE, Sun D. Docosahexaenoic acid reduces ER stress and abnormal protein accumulation and improves neuronal function following traumatic brain injury. J Neurosci. 2014;34:3743–3755. doi: 10.1523/JNEUROSCI.2872-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellezza I, Grottelli S, Mierla AL, Cacciatore I, Fornasari E, Roscini L, Cardinali G, Minelli A. Neuroinflammation and endoplasmic reticulum stress are coregulated by cyclo(His-Pro) to prevent LPS neurotoxicity. Int J Biochem Cell Biol. 2014;51:159–169. doi: 10.1016/j.biocel.2014.03.023. [DOI] [PubMed] [Google Scholar]
- Bidzan L, Bidzan M, Pachalska M. Aggressive and impulsive behavior in Alzheimer’s disease and progression of dementia. Med Sci Monit. 2012;18:CR182–CR189. doi: 10.12659/MSM.882523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolte S, Cordelieres FP. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc. 2006;224:213–232. doi: 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]
- Boyce M, Bryant KF, Jousse C, Long K, Harding HP, Scheuner D, Kaufman RJ, Ma D, Coen DM, Ron D, Yuan J. A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science. 2005;307:935–939. doi: 10.1126/science.1101902. [DOI] [PubMed] [Google Scholar]
- Bracchi-Ricard V, Lambertsen KL, Ricard J, Nathanson L, Karmally S, Johnstone J, Ellman DG, Frydel B, McTigue DM, Bethea JR. Inhibition of astroglial NF-kappaB enhances oligodendrogenesis following spinal cord injury. J Neuroinflamm. 2013;10:92. doi: 10.1186/1742-2094-10-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan AM, Suh SW, Won SJ, Narasimhan P, Kauppinen TM, Lee H, Edling Y, Chan PH, Swanson RA. NADPH oxidase is the primary source of superoxide induced by NMDA receptor activation. Nat Neurosci. 2009;12:857–863. doi: 10.1038/nn.2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho HJ, Sajja VS, Vandevord PJ, Lee YW. Blast induces oxidative stress, inflammation, neuronal loss and subsequent short-term memory impairment in rats. Neuroscience. 2013;253:9–20. doi: 10.1016/j.neuroscience.2013.08.037. [DOI] [PubMed] [Google Scholar]
- Colla E, Coune P, Liu Y, Pletnikova O, Troncoso JC, Iwatsubo T, Schneider BL, Lee MK. Endoplasmic reticulum stress is important for the manifestations of alpha-synucleinopathy in vivo. J Neurosci. 2012;32:3306–3320. doi: 10.1523/JNEUROSCI.5367-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coughlin JM, Wang Y, Munro CA, Ma S, Yue C, Chen S, Airan R, Kim PK, Adams AV, Garcia C, Higgs C, Sair HI, Sawa A, Smith G, Lyketsos CG, Caffo B, Kassiou M, Guilarte TR, Pomper MG. Neuroinflammation and brain atrophy in former NFL players: an in vivo multimodal imaging pilot study. Neurobiol Dis. 2014;74C:58–65. doi: 10.1016/j.nbd.2014.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damjanac M, Rioux Bilan A, Barrier L, Pontcharraud R, Anne C, Hugon J, Page G. Fluoro-Jade B staining as useful tool to identify activated microglia and astrocytes in a mouse transgenic model of Alzheimer’s disease. Brain Res. 2007;1128:40–49. doi: 10.1016/j.brainres.2006.05.050. [DOI] [PubMed] [Google Scholar]
- Dash PK, Hylin MJ, Hood KN, Orsi SA, Zhao J, Redell TB, Tsvetkov AS, Moore AN. Inhibition of eIF2alpha phosphatase reduces tissue damage and improves learning and memory following experimental traumatic brain injury. J Neurotrauma. 2015 doi: 10.1089/neu.2014.3772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng J, Lu PD, Zhang Y, Scheuner D, Kaufman RJ, Sonenberg N, Harding HP, Ron D. Translational repression mediates activation of nuclear factor kappa B by phosphorylated translation initiation factor 2. Mol Cell Biol. 2004;24:10161–10168. doi: 10.1128/MCB.24.23.10161-10168.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deslauriers AM, Afkhami-Goli A, Paul AM, Bhat RK, Acharjee S, Ellestad KK, Noorbakhsh F, Michalak M, Power C. Neuroinflammation and endoplasmic reticulum stress are coregulated by crocin to prevent demyelination and neurodegeneration. J Immunol. 2011;187:4788–4799. doi: 10.4049/jimmunol.1004111. [DOI] [PubMed] [Google Scholar]
- Faden AI, Wu J, Stoica BA, Loane DJ. Progressive inflammatory-mediated neurodegeneration after traumatic brain or spinal cord injury. Br J Pharmacol. 2015 doi: 10.1111/bph.13179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farook JM, Shields J, Tawfik A, Markand S, Sen T, Smith SB, Brann D, Dhandapani KM, Sen N. GADD34 induces cell death through inactivation of Akt following traumatic brain injury. Cell Death Dis. 2013;4:e754. doi: 10.1038/cddis.2013.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenn AM, Skendelas JP, Moussa DN, Muccigrosso MM, Popovich PG, Lifshitz J, Eiferman DS, Godbout JP. Methylene blue attenuates traumatic brain injury-associated neuroinflammation and acute depressive-like behavior in mice. J Neurotrauma. 2015;32:127–138. doi: 10.1089/neu.2014.3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson S, Mouzon B, Kayihan G, Wood M, Poon F, Doore S, Mathura V, Humphrey J, O’Steen B, Hayes R, Roses A, Mullan M, Crawford F. Apolipoprotein E genotype and oxidative stress response to traumatic brain injury. Neuroscience. 2010;168:811–819. doi: 10.1016/j.neuroscience.2010.01.031. [DOI] [PubMed] [Google Scholar]
- Finnie JW. Neuroinflammation: beneficial and detrimental effects after traumatic brain injury. Inflammopharmacology. 2013;21:309–320. doi: 10.1007/s10787-012-0164-2. [DOI] [PubMed] [Google Scholar]
- Goldstein LE, Fisher AM, Tagge CA, Zhang XL, Velisek L, Sullivan JA, Upreti C, Kracht JM, Ericsson M, Wojnarowicz MW, Goletiani CJ, Maglakelidze GM, Casey N, Moncaster JA, Minaeva O, Moir RD, Nowinski CJ, Stern RA, Cantu RC, Geiling J, Blusztajn JK, Wolozin BL, Ikezu T, Stein TD, Budson AE, Kowall NW, Chargin D, Sharon A, Saman S, Hall GF, Moss WC, Cleveland RO, Tanzi RE, Stanton PK, McKee AC. Chronic traumatic encephalopathy in blast-exposed military veterans and a blast neurotrauma mouse model. Sci Transl Med. 2012;4:134ra160. doi: 10.1126/scitranslmed.3003716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey LD, Yin Y, Attarwala IY, Begum G, Deng J, Yan HQ, Dixon CE, Sun D. Administration of DHA reduces endoplasmic reticulum stress-associated inflammation and alters microglial or macrophage activation in traumatic brain injury. ASN Neuro. 2015:7. doi: 10.1177/1759091415618969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T. Conversion of psychological stress into cellular stress response: roles of the sigma-1 receptor in the process. Psychiatry Clin Neurosci. 2015;69:179–191. doi: 10.1111/pcn.12262. [DOI] [PubMed] [Google Scholar]
- Hong Y, Wang X, Sun S, Xue G, Li J, Hou Y. Progesterone exerts neuroprotective effects against Abeta-induced neuroinflammation by attenuating ER stress in astrocytes. Int Immunopharmacol. 2016;33:83–89. doi: 10.1016/j.intimp.2016.02.002. [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010;140:900–917. doi: 10.1016/j.cell.2010.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh YH, Su IJ, Lei HY, Lai MD, Chang WW, Huang W. Differential endoplasmic reticulum stress signaling pathways mediated by iNOS. Biochem Biophys Res Commun. 2007;359:643–648. doi: 10.1016/j.bbrc.2007.05.154. [DOI] [PubMed] [Google Scholar]
- Hu YC, Sun Q, Li W, Zhang DD, Ma B, Li S, Li WD, Zhou ML, Hang CH. Biphasic activation of nuclear factor kappa B and expression of p65 and c-Rel after traumatic brain injury in rats. Inflamm Res. 2014;63:109–115. doi: 10.1007/s00011-013-0677-1. [DOI] [PubMed] [Google Scholar]
- Huang X, Chen Y, Zhang H, Ma Q, Zhang YW, Xu H. Salubrinal attenuates beta-amyloid-induced neuronal death and microglial activation by inhibition of the NF-kappaB pathway. Neurobiol Aging. 2012;33(1007):e1009–e1017. doi: 10.1016/j.neurobiolaging.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayakumar AR, Tong XY, Ruiz-Cordero R, Bregy A, Bethea JR, Bramlett HM, Norenberg MD. Activation of NF-kappaB mediates astrocyte swelling and brain edema in traumatic brain injury. J Neurotrauma. 2014;31:1249–1257. doi: 10.1089/neu.2013.3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson EM, Traver KL, Hoffman SW, Harrison CR, Herman JP. Environmental enrichment protects against functional deficits caused by traumatic brain injury. Front Behav Neurosci. 2013;7:44. doi: 10.3389/fnbeh.2013.00044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar RG, Diamond ML, Boles JA, Berger RP, Tisherman S, Kochanek PM, Wagner AK. Acute CSF Interleukin-6 trajectories after TBI: associations with neuroinflammation, polytrauma, and outcome. Brain Behav Immun. 2014 doi: 10.1016/j.bbi.2014.12.021. [DOI] [PubMed] [Google Scholar]
- Liao Y, Liu P, Guo F, Zhang ZY, Zhang Z. Oxidative burst of circulating neutrophils following traumatic brain injury in human. PLoS One. 2013;8:e68963. doi: 10.1371/journal.pone.0068963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu HD, Li W, Chen ZR, Zhou ML, Zhuang Z, Zhang DD, Zhu L, Hang CH. Increased expression of ferritin in cerebral cortex after human traumatic brain injury. Neurol Sci. 2013;34:1173–1180. doi: 10.1007/s10072-012-1214-7. [DOI] [PubMed] [Google Scholar]
- Liu Y, Wang J, Qi SY, Ru LS, Ding C, Wang HJ, Zhao JS, Li JJ, Li AY, Wang DM. Reduced endoplasmic reticulum stress might alter the course of heart failure via caspase-12 and JNK pathways. Can J Cardiol. 2014;30:368–375. doi: 10.1016/j.cjca.2013.11.001. [DOI] [PubMed] [Google Scholar]
- Loane DJ, Stoica BA, Byrnes KR, Jeong W, Faden AI. Activation of mGluR5 and inhibition of NADPH oxidase improves functional recovery after traumatic brain injury. J Neurotrauma. 2013;30:403–412. doi: 10.1089/neu.2012.2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logsdon AF, Turner RC, Lucke-Wold BP, Robson MJ, Naser ZJ, Smith KE, Matsumoto RR, Huber JD, Rosen CL. Altering endoplasmic reticulum stress in a model of blast-induced traumatic brain injury controls cellular fate and ameliorates neuropsychiatric symptoms. Front Cell Neurosci. 2014;8:421. doi: 10.3389/fncel.2014.00421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucke-Wold BP, Turner RC, Logsdon AF, Bailes JE, Huber JD, Rosen CL. Linking traumatic brain injury to chronic traumatic encephalopathy: identification of potential mechanisms leading to neurofibrillary tangle development. J Neurotrauma. 2014a doi: 10.1089/neu.2013.3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucke-Wold BP, Logsdon AF, Smith KE, Turner RC, Alkon DL, Tan Z, Naser ZJ, Knotts CM, Huber JD, Rosen CL. Bryostatin-1 restores blood brain barrier integrity following blast-induced traumatic brain injury. Mol Neurobiol. 2014b doi: 10.1007/s12035-014-8902-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucke-Wold BP, Turner RC, Logsdon AF, Nguyen L, Bailes JE, Lee JM, Robson MJ, Omalu BI, Huber JD, Rosen CL. Endoplasmic reticulum stress implicated in chronic traumatic encephalopathy. J Neurosurg. 2015a:1–16. doi: 10.3171/2015.3.JNS141802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucke-Wold BP, Naser ZJ, Logsdon AF, Turner RC, Smith KE, Robson MJ, Bailes JE, Lee JM, Rosen CL, Huber JD. Amelioration of nicotinamide adenine dinucleotide phosphate-oxidase mediated stress reduces cell death after blast-induced traumatic brain injury. Transl Res. 2015b;166(509–528):e501. doi: 10.1016/j.trsl.2015.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucke-Wold BP, Turner RC, Logsdon AF, Nguyen L, Bailes JE, Lee JM, Robson MJ, Omalu BI, Huber JD, Rosen CL. Endoplasmic reticulum stress implicated in chronic traumatic encephalopathy. J Neurosurg. 2016;124(3):687–702. doi: 10.3171/2015.3.JNS141802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra JD, Kaufman RJ. Endoplasmic reticulum stress and oxidative stress: a vicious cycle or a double-edged sword? Antioxid Redox Signal. 2007;9:2277–2293. doi: 10.1089/ars.2007.1782. [DOI] [PubMed] [Google Scholar]
- Mar AC, Walker AL, Theobald DE, Eagle DM, Robbins TW. Dissociable effects of lesions to orbitofrontal cortex subregions on impulsive choice in the rat. J Neurosci. 2011;31:6398–6404. doi: 10.1523/JNEUROSCI.6620-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael AP, Stout J, Roskos PT, Bolzenius J, Mogul D, Gfeller J, Bucholz R. Evaluation of cortical thickness following traumatic brain injury in military veterans. J Neurotrauma. 2015 doi: 10.1089/neu.2015.3918. [DOI] [PubMed] [Google Scholar]
- Mosienko V, Bert B, Beis D, Matthes S, Fink H, Bader M, Alenina N. Exaggerated aggression and decreased anxiety in mice deficient in brain serotonin. Transl Psychiatry. 2012;2:e122. doi: 10.1038/tp.2012.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouzon B, Chaytow H, Crynen G, Bachmeier C, Stewart J, Mullan M, Stewart W, Crawford F. Repetitive mild traumatic brain injury in a mouse model produces learning and memory deficits accompanied by histological changes. J Neurotrauma. 2012;29:2761–2773. doi: 10.1089/neu.2012.2498. [DOI] [PubMed] [Google Scholar]
- Mouzon BC, Bachmeier C, Ferro A, Ojo JO, Crynen G, Acker CM, Davies P, Mullan M, Stewart W, Crawford F. Chronic neuropathological and neurobehavioral changes in a repetitive mild traumatic brain injury model. Ann Neurol. 2014;75:241–254. doi: 10.1002/ana.24064. [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Yuan J. Cross-talk between two cysteine protease families. Activation of caspase-12 by calpain in apoptosis. J Cell Biol. 2000;150:887–894. doi: 10.1083/jcb.150.4.887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakka VP, Gusain A, Raghubir R. Endoplasmic reticulum stress plays critical role in brain damage after cerebral ischemia/reperfusion in rats. Neurotox Res. 2010;17:189–202. doi: 10.1007/s12640-009-9110-5. [DOI] [PubMed] [Google Scholar]
- Nisenbaum EJ, Novikov DS, Lui YW. The presence and role of iron in mild traumatic brain injury: an imaging perspective. J Neurotrauma. 2014;31:301–307. doi: 10.1089/neu.2013.3102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohri SS, Hetman M, Whittemore SR. Restoring endoplasmic reticulum homeostasis improves functional recovery after spinal cord injury. Neurobiol Dis. 2013;58:29–37. doi: 10.1016/j.nbd.2013.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omalu B, Bailes J, Hamilton RL, Kamboh MI, Hammers J, Case M, Fitzsimmons R. Emerging histomorphologic phenotypes of chronic traumatic encephalopathy in American athletes. Neurosurgery. 2011;69:173–183. doi: 10.1227/NEU.0b013e318212bc7b. Discussion 183. [DOI] [PubMed] [Google Scholar]
- Otani N, Nawashiro H, Fukui S, Nomura N, Yano A, Miyazawa T, Shima K. Differential activation of mitogen-activated protein kinase pathways after traumatic brain injury in the rat hippocampus. J Cereb Blood Flow Metab. 2002;22:327–334. doi: 10.1097/00004647-200203000-00010. [DOI] [PubMed] [Google Scholar]
- Prell T, Lautenschlager J, Weidemann L, Ruhmer J, Witte OW, Grosskreutz J. Endoplasmic reticulum stress is accompanied by activation of NF-kappaB in amyotrophic lateral sclerosis. J Neuroimmunol. 2014;270:29–36. doi: 10.1016/j.jneuroim.2014.03.005. [DOI] [PubMed] [Google Scholar]
- Quan X, Wang J, Liang C, Zheng H, Zhang L. Melatonin inhibits tunicamycin-induced endoplasmic reticulum stress and insulin resistance in skeletal muscle cells. Biochem Biophys Res Commun. 2015 doi: 10.1016/j.bbrc.2015.06.065. [DOI] [PubMed] [Google Scholar]
- Rebetez MM, Rochat L, Ghisletta P, Walder B, Van der Linden M. Association between impulsivity, emotional/behavioural hyperactivation and functional outcome one year after severe traumatic brain injury. Brain Inj. 2015:1–7. doi: 10.3109/02699052.2015.1035326. [DOI] [PubMed] [Google Scholar]
- Roth TL, Nayak D, Atanasijevic T, Koretsky AP, Latour LL, McGavern DB. Transcranial amelioration of inflammation and cell death after brain injury. Nature. 2014;505:223–228. doi: 10.1038/nature12808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan J, Qi Z, Shen L, Jiang Y, Xu Y, Lan L, Luo L, Yin Z. Crosstalk between JNK and NF-kappaB signaling pathways via HSP27 phosphorylation in HepG2 cells. Biochem Biophys Res Commun. 2015;456:122–128. doi: 10.1016/j.bbrc.2014.11.045. [DOI] [PubMed] [Google Scholar]
- Rubovitch V, Barak S, Rachmany L, Goldstein RB, Zilberstein Y, Pick CG. The neuroprotective effect of salubrinal in a mouse model of traumatic brain injury. Neuromol Med. 2015;17:58–70. doi: 10.1007/s12017-015-8340-3. [DOI] [PubMed] [Google Scholar]
- Schwarzbold ML, Rial D, De Bem T, Machado DG, Cunha MP, dos Santos AA, dos Santos DB, Figueiredo CP, Farina M, Goldfeder EM, Rodrigues AL, Prediger RD, Walz R. Effects of traumatic brain injury of different severities on emotional, cognitive, and oxidative stress-related parameters in mice. J Neurotrauma. 2010;27:1883–1893. doi: 10.1089/neu.2010.1318. [DOI] [PubMed] [Google Scholar]
- Sokka AL, Putkonen N, Mudo G, Pryazhnikov E, Reijonen S, Khiroug L, Belluardo N, Lindholm D, Korhonen L. Endoplasmic reticulum stress inhibition protects against excitotoxic neuronal injury in the rat brain. J Neurosci. 2007;27:901–908. doi: 10.1523/JNEUROSCI.4289-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sominsky L, Walker AK, Hodgson DM. Editorial: Neuroinflammation and behavior. Front Neurosci. 2015;9:201. doi: 10.3389/fnins.2015.00201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern RA, Riley DO, Daneshvar DH, Nowinski CJ, Cantu RC, McKee AC. Long-term consequences of repetitive brain trauma: chronic traumatic encephalopathy. PM R. 2011;3:S460–S467. doi: 10.1016/j.pmrj.2011.08.008. [DOI] [PubMed] [Google Scholar]
- Szmydynger-Chodobska J, Fox LM, Lynch KM, Zink BJ, Chodobski A. Vasopressin amplifies the production of proinflammatory mediators in traumatic brain injury. J Neurotrauma. 2010;27:1449–1461. doi: 10.1089/neu.2010.1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toklu HZ, Tumer N. Oxidative Stress, brain edema, blood-brain barrier permeability, and autonomic dysfunction from traumatic brain injury. In: Kobeissy FHP, editor. Brain Neurotrauma: Molecular, Neuropsychological, and Rehabilitation Aspects. Boca Raton, FL: 2015. [Google Scholar]
- Tsai KH, Wang WJ, Lin CW, Pai P, Lai TY, Tsai CY, Kuo WW. NADPH oxidase-derived superoxide anion-induced apoptosis is mediated via the JNK-dependent activation of NF-kappaB in cardiomyocytes exposed to high glucose. J Cell Physiol. 2012;227:1347–1357. doi: 10.1002/jcp.22847. [DOI] [PubMed] [Google Scholar]
- Tsaytler P, Harding HP, Ron D, Bertolotti A. Selective inhibition of a regulatory subunit of protein phosphatase 1 restores proteostasis. Science. 2011;332:91–94. doi: 10.1126/science.1201396. [DOI] [PubMed] [Google Scholar]
- Turner RC, Naser ZJ, Bailes JE, Smith DW, Fisher JA, Rosen CL. Effect of slosh mitigation on histologic markers of traumatic brain injury: laboratory investigation. J Neurosurg. 2012;117:1110–1118. doi: 10.3171/2012.8.JNS12358. [DOI] [PubMed] [Google Scholar]
- Turner RC, Naser ZJ, Logsdon AF, DiPasquale KH, Jackson GJ, Robson MJ, Gettens RT, Matsumoto RR, Huber JD, Rosen CL. Modeling clinically relevant blast parameters based on scaling principles produces functional and histological deficits in rats. Exp Neurol. 2013;248:520–529. doi: 10.1016/j.expneurol.2013.07.008. [DOI] [PubMed] [Google Scholar]
- Uranga RM, Giusto NM, Salvador GA. Iron-induced oxidative injury differentially regulates PI3K/Akt/GSK3beta pathway in synaptic endings from adult and aged rats. Toxicol Sci. 2009;111:331–344. doi: 10.1093/toxsci/kfp152. [DOI] [PubMed] [Google Scholar]
- Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, Ron D. Coupling of stress in the ER to activation of JNK protein kinases by trans-membrane protein kinase IRE1. Science. 2000;287:664–666. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- Webster SJ, Van Eldik LJ, Watterson DM, Bachstetter AD. Closed head injury in an age-related Alzheimer mouse model leads to an altered neuroinflammatory response and persistent cognitive impairment. J Neurosci. 2015;35:6554–6569. doi: 10.1523/JNEUROSCI.0291-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Zhao Z, Sabirzhanov B, Stoica BA, Kumar A, Luo T, Skovira J, Faden AI. Spinal cord injury causes brain inflammation associated with cognitive and affective changes: role of cell cycle pathways. J Neurosci. 2014a;34:10989–11006. doi: 10.1523/JNEUROSCI.5110-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Wang D, Xiao Y, Zhou X, Wang L, Chen B, Li Q, Guo X, Huang Q. Endoplasmic reticulum stress plays a role in the advanced glycation end product-induced inflammatory response in endothelial cells. Life Sci. 2014b;110:44–51. doi: 10.1016/j.lfs.2014.06.020. [DOI] [PubMed] [Google Scholar]
- Zaloshnja E, Miller T, Langlois JA, Selassie AW. Prevalence of long-term disability from traumatic brain injury in the civilian population of the United States, 2005. J Head Trauma Rehabil. 2008;23:394–400. doi: 10.1097/01.HTR.0000341435.52004.ac. [DOI] [PubMed] [Google Scholar]
- Zhang QG, Laird MD, Han D, Nguyen K, Scott E, Dong Y, Dhandapani KM, Brann DW. Critical role of NADPH oxidase in neuronal oxidative damage and microglia activation following traumatic brain injury. PLoS One. 2012;7:e34504. doi: 10.1371/journal.pone.0034504. [DOI] [PMC free article] [PubMed] [Google Scholar]