

Abstract
The main structural component of connective tissues is fibrillar, cross-linked collagen whose fibrillogenesis can be modulated by Small Leucine-Rich Proteins/Proteoglycans (SLRPs). Not all SLRPs’ effects on collagen and extracellular matrix in vivo have been elucidated; one of the less investigated SLRPs is asporin. Here we describe the successful generation of an Aspn-/- mouse model and the investigation of the Aspn-/- skin phenotype. Functionally, Aspn-/- mice had an increased skin mechanical toughness, although there were no structural changes present on histology or immunohistochemistry. Electron microscopy analyses showed 7% thinner collagen fibrils in Aspn-/- mice (not statistically significant). Several matrix genes were upregulated, including collagens (Col1a1, Col1a2, Col3a1), matrix metalloproteinases (Mmp2, Mmp3) and lysyl oxidases (Lox, Loxl2), while lysyl hydroxylase (Plod2) was downregulated. Intriguingly no differences were observed in collagen protein content or in collagen cross-linking-related lysine oxidation or hydroxylation. The glycosaminoglycan content and structure in Aspn-/- skin was profoundly altered: chondroitin/dermatan sulfate was more than doubled and had an altered composition, while heparan sulfate was halved and had a decreased sulfation. Also, decorin and biglycan were doubled in Aspn-/- skin. Overall, asporin deficiency changes skin glycosaminoglycan composition, and decorin and biglycan content, which may explain the changes in skin mechanical properties.
Introduction
The bulk of the extracellular matrix (ECM) is composed of collagens woven into a fibrillar network that accommodates water-retaining proteoglycans and provides a cellular niche. Collagen fibrillogenesis and cross-linking can be modulated by Small Leucine-Rich Proteins/Proteoglycans (SLRPs); this constitutes a mechanism that contributes to the mechanical and physiological tissue properties [1–3]. The effects of SLRPs on collagen fibrillogenesis are evident in the organ-specific phenotypes of the various SLRP knockout mice: lumican-deficient mice have fragile skin and opaque cornea [4–6], fibromodulin-deficient mice have abnormally cross-linked and ill-fused collagen fibrils in tendons [3, 7], decorin-deficient mice have fragile skin, weak tendons and lower lung airway resistance [8–10], keratocan-deficient mice have flattened cornea (mimicking human cornea plana disorder caused by KERA mutations) [11, 12], and biglycan-deficient mice have osteoporotic bones [13]. Compound SLRP knockout mice have even graver multi-organ abnormities with more pronounced collagen fibril phenotypes [14, 15].
Not all SLRP knockout mouse phenotypes have been reported, but the collective knowledge of SLRPs’ tissue-specific effects and collagen interaction redundancy [16–18] will aid in understanding the complex mechanisms underlying the shaping of extracellular matrices. In this paper, we present an initial report on the phenotype of asporin-deficient mice. Compared with other SLRPs, asporin (ASPN or PLAP1) lacks glycosaminoglycan (GAG) chains [19] and carries a polymorphic calcium-binding polyaspartate sequence [17, 20]. Asporin expression has been detected in dermis, perichondrium and periosteum, tendon, and eye sclera [21, 22]. Concluded from several in vitro studies, asporin function has been related to collagen fibrillogenesis and collagen mineralization [17, 23–25]. Asporin has also been suggested to modulate cellular response to FGF-2 [26], BMP-2 [24], and TGF-β [20]. Asporin expression is induced by TGF-β [21, 27] and suppressed by IL-1β and TNF-α [27]. It can interact and inhibit TGF-β [28] and consequently act as a tumor suppressor [29], but other studies propose asporin to be an invasion-promoting protein [30–32].
In this paper we hypothesized that asporin functions in formation of a structurally coherent extracellular matrix in vivo. To test this hypothesis we generated an asporin knockout mouse model (Aspn-/-). We focused on analyzing the effect of asporin deficiency on skin extracellular matrix, where asporin has been detected [21, 22] and where we, in the current study, observed a biomechanical phenotype. To elucidate the underlying cause of this phenotype we also analyzed skin collagen, proteoglycans and GAGs. This report contributes to the overall understanding of the unique functions of SLRPs in connective tissues.
Materials and methods
Targeted disruption of the mouse Aspn gene
All animal experiments were conducted under approval of the regional animal ethical board committee (Malmö-Lunds djurförsöksetiska nämnd, Jordbruksverket, Sweden). Mice were euthanized by carbon dioxide followed by cervical dislocation. The BAC clone bMQ103e19 containing the 129/Sv mouse Aspn genomic material was obtained from Geneservice (UK). To construct the targeting vector a recombineering system described in [33] was used. The resulting targeting vector contained exons 2–5 (out of 8 present in the gene), where exons 2–3 were flanked with loxP sites, and the neo cassette, flanked with FRT sites, was placed upstreams of the second loxP site. Targeting vector also contained MCI-TK cassette for positive selection of the transformed 129/Sv ES cells that were screened for genomic integration of the vector with Southern blotting and PCR. The positive clone was injected into C57BL/6J blastocysts to generate chimeric mice that were later intercrossed to generate Aspnfneo/+ heterozygous animals. They were then crossed with FLPeR mice (Jackson Laboratory, Bar Harbor, ME) to remove the neo cassette and give Aspnf/+ mice. Finally, they were crossed with EIIa-Cre transgenic mice (Jackson Laboratory, Bar Harbor, ME) to remove exons 2–3 of the targeted Aspn allele to create Aspn-/+ mice (Fig 1A). Primer sequences in Fig 1 are: P1 5’-ACTTCATTTTAACTTCCTTTACTGAGA-3’, P2 5’-AAACAGCTGGGTCTGTCCAT-3’, P3 5’-CCGGCTGCATGTTTATTTTC-3’.
Fig 1. Aspn-/- mouse generation.

(A) Strategy for the creation of Aspn-null allele. The diagram shows wild-type, targeted, floxed, and excised alleles. Exons 1–4 (out of 8 present in the gene) are shown as rectangles. Primers used for genotyping are targeted to P1, P2, P3 sites and the expected PCR products’ sizes are denoted. (B) Genotyping PCR of Aspn-/- mice using primers targeted to sites P1, P2, P3. (C) Confirmation of Aspn protein expression loss in Aspn-/- mice by immunobloting tail SDS extracts for asporin.
Skin histology and immunohistochemistry
Mouse skins were fixed in HistoChoice (Sigma) for 24 h, dehydrated, embedded in paraffin and sectioned. Sections were stained with Masson Trichrome (Sigma) or processed for immunohistochemistry: rehydrated sections were washed 2 x 5 min in Tris-buffered saline (TBS) plus 0.025% Triton X-100 and blocked with 10% goat serum and 1% bovine serum albumin (BSA) for 2 h. Sections were then incubated for 16 h at 4°C with rat anti-mouse CD31 (Optistain, clone SZ31) or rat anti-mouse F4/80 (BioRad, clone CI:A3-1) diluted to 10 μg/mL in 1% BSA in TBS. Sections were rinsed 2 x 5 min with TBS plus 0.025% Triton X-100 and quenched with 0.3% hydrogen peroxide in TBS for 15 min. After rinsing with TBS, sections were stained with ultrasensitive ABC staining kit (ThermoFisher). Sections were washed 3 x 5 min with TBS and developed using diaminobenzidine peroxidase substrate (Vector Labs).
Immunoblots
Dorsal skin samples were boiled in SDS-PAGE loading buffer for 10 min. The proteins were run on 4–12% Bis-Tris reducing SDS-PAGE (ThermoFisher) and transferred to a nitrocellulose membrane. The membrane was blocked with 5% BSA in TBS, rinsed in wash buffer (TBS with 0.1% Tween-20) and immunoblotted in the incubation buffer (TBS with 0.1% Tween-20 and 1% BSA) using the following primary antibodies diluted to 1 μg/mL: anti-asporin (Abcam, ab31303), anti-decorin (kind gift from Professor Åke Oldberg, Lund University), anti-biglycan (LF106, kind gift from Professor Larry Fisher, NIH), anti-beta-actin (ab6276, Abcam). Membranes were washed 3 x 5 min and incubated with the corresponding secondary HRP-conjugated antibodies (all from GE Healthcare) diluted to 0.1 μg/mL for 1 h. Membranes were washed 3 x 5 min and developed with Luminata Forte (Millipore) using a CCD camera.
Electron microscopy
Dorsal skin samples were shaved and processed for transmission electron microscopy as described [34]. Collagen fibril thickness was quantified using images from three reticular dermis regions per mouse, at 50,000x magnification, using Adobe Photoshop.
Skin tensile strength determination
Mice were euthanized and kept on ice before shaving and gently removing the dorsal skin samples. Two parallel strips of skin (3 mm x 30 mm) were punched out in the craniocaudal direction, symmetrically on the left and right side of the spine. The samples were washed briefly in PBS, the central 5 mm were wrapped in PBS soaked gauze and the ends were allowed to dry for 90 min while keeping the central part moist with PBS. The dry ends were glued onto clamps using cyanoacrylate glue and after curing for 40 min the sample was placed in a PBS bath and mechanically tested (200-N tensile stage, petri dish version, Deben, Suffolk, UK). The test consisted of 4 preconditioning cycles to 15% strain, followed finally by a stretch to failure. All tests were at a rate of 6 mm/min with data recorded at 10 Hz. Sample width and length were measured by microscopy images. To avoid mechanically damaging the strips, thickness was measured with a constant-pressure caliper (model 293-334-30, Mitutoyo, Kawasaki, Japan) on a piece of skin adjacent to the tested strip. The failure test was analyzed for strain, stress and energy at both the yield point and at failure. The peak modulus was determined by a moving fit over a 10% strain range. Yield was defined as the first point where the modulus dropped below 50% of the peak value. For each animal, results for the two strips were averaged and groups were compared by unpaired two-tailed t-test with unequal variance.
Collagen molecular phenotype (mass spectrometry)
Dorsal skin collagen was defatted with chloroform/methanol (3:1 v/v). Collagen was solubilized by 3% acetic acid extraction at 4°C for 24 hrs or heat denaturation in SDS-PAGE sample buffer. The extracts were run on 6% SDS-PAGE and stained with Coomassie Blue. Bands corresponding to collagen α1(I) and α2(I) chains were cut out and processed for mass spectrometry using trypsin in-gel digestion, without reduction or alkylation. Collagen peptides were analyzed with LC-MS using an LTQ XL ion trap mass spectrometer (ThermoFisher) equipped with in-line liquid chromatography using a C4 5um capillary column (300um x 150mm; Higgins Analytical RS-15M3-W045) eluted at 4.5ul min. The LC mobile phase consisted of buffer A (0.1% formic acid in MilliQ water) and buffer B (0.1% formic acid in 3:1 acetonitrile:n-propanol v/v). The LC sample stream was introduced into the mass spectrometer by electrospray ionization (ESI) with a spray voltage of 4kV. Proteome Discoverer search software (Thermo Scientific) was used for peptide identification using the NCBI protein database. Proline and lysine modifications were examined manually by scrolling or averaging the full scan over several minutes so that all of the post-translational variations of a given peptide appeared together in the full scan.
Hydroxyproline determination
Dorsal skin samples were weighed and hydrolyzed in 6 M HCl for 4 hours at 120°C at 2 atm. Hydroxyproline was quantified as described earlier [35] and normalized to total tissue weight.
Reverse transcriptase- and real-time qPCR
Total RNA was isolated from skin mice using TRIzol reagent (ThermoFisher). 500 ng RNA was used for reverse transcription reaction using Superscript VILO (ThermoFisher). Real-time qPCR was conducted using Taqman probes and Taqman Gene Expression Master Mix (ThermoFisher) in an Applied Biosystems 7300 qPCR equipment. Gene expression was normalized to Actb transcripts. The Taqman probe list is presented in S1 Table.
Glycosaminoglycan analysis
Skins were lyophilized, weighed, and processed for GAG analyses as described in [34]. The proteins were degraded for 16 h at 55°C with 200 μg/ml pronase in 50 mm Tris/HCl, pH 8, 1 mM CaCl2, and 1% Triton X-100. After heat inactivation of the protease, MgCl2 was added to 2 mM and benzonase was added to a final 20 sigma units/ml and incubated for 2 h at 37°C. DNAase was heat-inactivated, NaCl was added to a final 0.1 M, and GAGs were purified by batch incubation with a DE52 anion exchange gel, followed by the transfer of the gel to disposable spin centrifuge tubes (Costar 8163). The gel was washed with buffer 1 (50 mM Tris/HCl, pH 8, 0.1 M NaCl, and 0.1% Triton X-100), buffer 2 (50 mM NaAc, pH 4, 0.1 M NaCl, and 0.1% Triton X-100), water, and 0.1 M NH4HCO3. Elution was achieved by 2 M NH4HCO3. After lyophilization, GAGs were provisionally and roughly estimated by carbazole [36]. Then, 500 ng GAGs were subjected to chondroitinase ABC (Sigma) overnight at 37°C in 20 μl 50 mM NH4OAc and 0.1 mg/ml BSA, containing 10 mIU enzyme, or chondroitinase B (R&D Systems) overnight at 37°C in 20 μl 50 mM Tris pH 7.5, 50 mM NaCl, 4 mM CaCl2, and 0.1 mg/ml BSA, containing 5 mIU enzyme, or with a mixture of heparinases (in-house preparation, purified from E.coli, stably singularly transfected with the pET-15b vector containing heparinase I, or vector pET-19b containing heparinase II or III, as provided by Professor Jian Liu (University of North Carolina; incubation overnight at 37°C in 20 μl 40 mM NaAc pH 7.0, 2 mM CaAc, 0.1 mg/ml BSA, containing 2 mIU of each enzyme). The samples were boiled and the supernatant was dried. Fluorophore-labelling of the resulting disaccharides was performed by adding 10 μl of 20 mM re-purified 2-aminoacridone (AMAC, Sigma), followed by a 20 min incubation at room temperature before the addition of 10 μl of 1 M NaBH3CN and incubation at 45°C for 16 h [37]. Pre-column AMAC-labeled disaccharides were analyzed with HPLC-fluorescence as described previously, with slight modifications [37]. Briefly, 20 μl samples were diluted to 100 μl in running buffer (98% A: NH4OAc, 60 mM, pH 5.6, and 2% B: MeCN) and injected (2 μl) onto an XBridge BEH Shield RP18 (2.1x100 mm, 2.5 μm). Disaccharides were separated using a 39 min gradient run at 0.35 ml/min (0–1 min: 98% A, 1–3 min: 98–96% A, 3–26 min: 96–85% A, 26–28 min: 85–10% A, 28–32 min: 10% A, 32–34 min: 10–98% A, 34–39 min: 98% A) on a Thermo Scientific UltiMate 3000 Quaternary Analytical system with an FLD-3400RS fluorescence detector (excitation λ = 428 and emission λ = 525). The column was kept at 30°C to improve performance and reproducibility. Quantification was done by comparison to known weight of standard disaccharides (Iduron, UK), mock-treated in the same buffers and enzymes as the samples in each series of runs.
Semi-quantitative analysis of decorin and biglycan protein cores
Proteins were extracted from two-month old mice skin (n = 4 per genotype) in a dissociating buffer (4 M guanidine chloride, 50 mM NaAc pH 5.5, 0.1% Triton X-100, complete protease inhibitor cocktail (Roche)) by consecutive use of a Polytron- and Potter-type homogenizers. After overnight incubation the extracts were spun down and the supernatant exchanged against a denaturating buffer (8 M urea, 50 mM NaAc pH 5.5, 0,2 M NaCl, 0.1% Triton X-100). Proteins were quantified by Bradford method (BioRad) and equal amount of proteins were used in anion-exchange purification of proteoglycans on DE-52 (Whatman), eluted by of 2M ammonium bicarbonate, 0.1% Triton X-100. The recovery from the DE-52 purification was assessed by co-incubation of 35S-labeled GAGs, which were protease-treated, with the skin extracts. The recovery of the labeled internal control was very similar among the eight samples (coefficient of variation = 11%). SDS-PAGE electrophoresis was done using stain-free gels (BioRad) and total proteins were visualized directly using the Chemi Doc Touch Imaging System (BioRad). For immunoblotting the samples were digested with chondroitinase ABC (as described in Glycosaminoglycan Analysis). Immunoblots were performed as described above.
Results
Generation of Aspn-/- mice
Our gene-targeting construct was designed to remove exons 2–3 in the Aspn gene where exon 2 contains the translation start site and 23 bp of upstream promoter sequence. First, we flanked exons 2–3 with two direct repeats of loxP sites, where the second loxP site was preceded by a neo cassette flanked with FRT sites. The gene-targeted mice were then bred with FLPeR mice in order to remove the neo cassette, and then with EIIa-Cre mice to remove exons 2–3 (Fig 1A). The resulting Aspn+/- mice were backcrossed with C57BL/6 mice for ten generations. When intercrossed, Aspn+/- mice generated Aspn+/+, Aspn+/- and Aspn-/- pups at normal Mendelian ratio. Excision of exons 2 and 3 was verified by genotyping (Fig 1B). No full size or fragments of asporin protein were present in Aspn-/- mice, confirming the functional ablation of the asporin gene (Fig 1C). All mice had normal fertility and external appearance. Five Aspn-/- mice were kept until 15 months of age and showed normal behavior. Histological and necroscopical analyses of 2 month-old Aspn-/- mice showed no gross abnormities in aorta, esophagus, intestine, kidney, liver, skeletal muscle, spleen and trachea.
Skin histology and electron microscopy
Asporin protein expression in dorsal skin was confirmed by immunoblotting SDS extracts. Also, absence of asporin protein in Aspn-/- mice skin was confirmed (Fig 2A). In 2 month-old female Aspn-/- mice Masson Trichrome staining did not reveal any apparent abnormities in the structure of dermis or hypodermis (n = 6 per genotype) (Fig 2B), and there were no apparent differences in the density of blood vessels (CD31-positive cells) or macrophages (F4/80-positive cells) (Fig 2C). We did not observe any histological abnormities in male mice skin either (n = 3 per genotype, S1 File). Observed in transmission electron microscopy, Aspn-/- reticular dermis had regularly contoured collagen fibrils (Fig 2D) with typical collagen banding pattern (Fig 2E). Collagen fibril diameter was 7% thinner in Aspn-/- skin (Fig 2F) with the average diameter of 88 ± 5 nm in wild-type (n = 6) and 82 ± 10 nm in Aspn-/- skin (n = 6; p = 0.23, Welch unpaired two-tailed t-test). Assessed by hydroxyproline determination, collagen quantity per wet tissue weight was not significantly changed (wild-type 67 ± 13 μg/mg, n = 5; Aspn-/- 58 ± 5 μg/mg, n = 5; p = 0.15, Welch unpaired two-tailed t-test).
Fig 2. Skin phenotype of Aspn-/- mice.
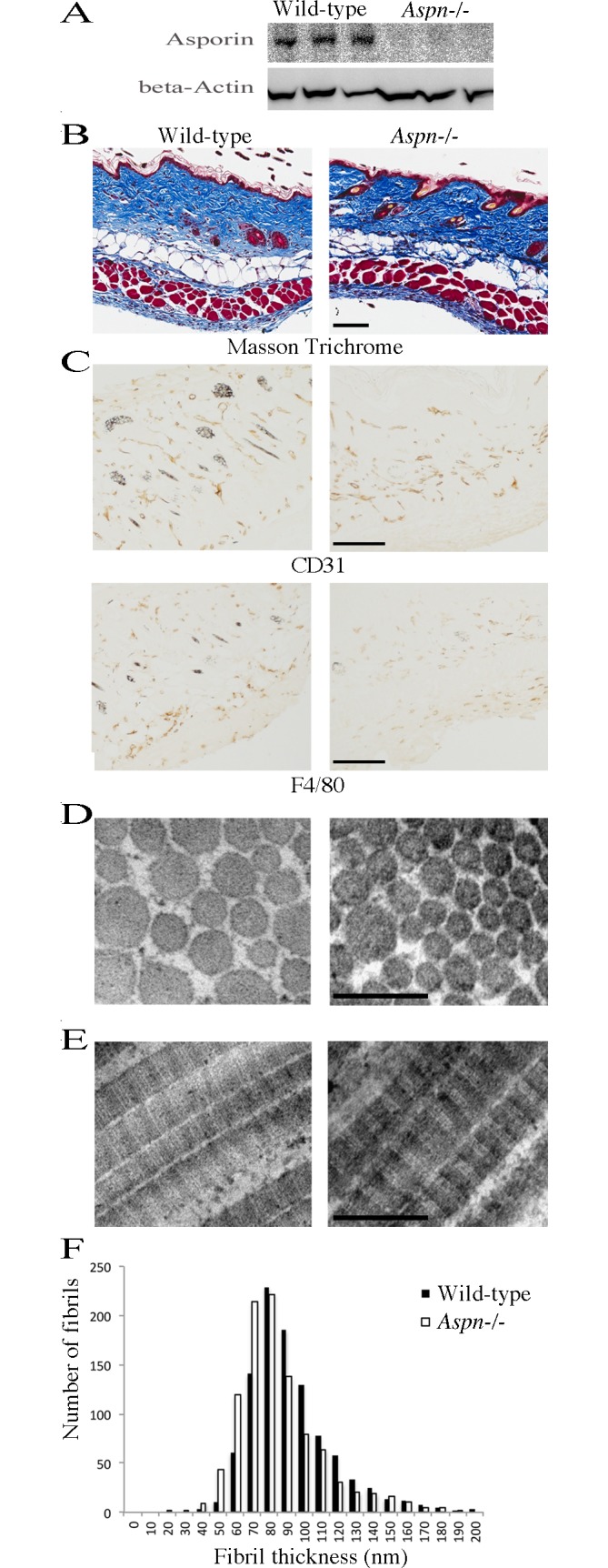
(A) Asporin immunoblotting of dorsal skin extracts from three wild-type and three Aspn-/- mice. (B) 2 month-old female dorsal skins were analyzed histologically by staining with Massson Trichrome, or (C) by immunohistochemistry by staining for the presence of CD31-positive blood vessels and F4/80-positive macrophages; bars in A and B are 100 μm. Similar results were obtained from analyses of six mice from each genotype. (D) Transmission electron microscopy on cross-sectioned collagen fibrils in reticular dermis. (E) Transmission electron microscopy on longitudinally oriented collagen fibrils in reticular dermis. Bars in C and D are 200 nm. (F) Quantification of collagen fibril diameter in reticular dermis. 1,000 fibrils were measured from electron microscopy images collected from six wild-type and six Aspn-/- mice.
Skin mechanical testing
Next, the general mechanical properties of Aspn-/- skin were assessed. In four-month old female Aspn-/- mice the skin toughness, or energy required to achieve failure, was almost 75% greater than in wild-type mice (Table 1). Other mechanical parameters, including failure stress and strain, were not significantly different but also tended toward superior strength of the Aspn-/- skin. Note that the parameters at yield are almost identical in both genotypes, indicating that most of the difference occurred after the yield point.
Table 1. Skin mechanical testing.
| Wild-type | Aspn-/- | ||||
|---|---|---|---|---|---|
| avg | sd | avg | sd | p value | |
| Sample Thickness (mm) | 0.48 | 0.11 | 0.39 | 0.03 | 0.13 |
| Modulus (MPa) | 13.8 | 7.3 | 16.1 | 6.4 | 0.59 |
| Yield Strain (%) | 47 | 7.2 | 46 | 9.2 | 0.88 |
| Yield Stress (MPa) | 4.0 | 1.8 | 4.4 | 1.2 | 0.67 |
| Yield Energy (MJ/m3) | 0.75 | 0.32 | 0.85 | 0.39 | 0.63 |
| Failure Strain (%) | 50 | 7 | 62 | 15 | 0.11 |
| Failure Stress (MPa) | 4.2 | 1.9 | 5.3 | 0.8 | 0.22 |
| Failure Energy (MJ/m3) | 0.89 | 0.38 | 1.55 | 0.46 | 0.022 |
Uniaxial tensile material properties in the craniocaudal direction, from duplicate measures on four-month old wild-type and Aspn-/- dorsal skin samples (n = 6 per genotype; avg = average, sd = standard deviation).
Skin collagen molecular phenotype
Because asporin is a collagen-binding protein we investigated if asporin deficiency could result in a changed molecular phenotype of collagen. Two-month old skin collagen was extracted with SDS or acetic acid and the collagen band pattern on Coomassie-stained SDS-PAGE was analyzed. We did not observe any differences between Aspn-/- and wild-type collagen (S1 Fig). We also used mass spectrometry to assess lysyl oxidase-mediated and lysyl hydroxylase-mediated modifications of collagen lysines involved in cross-linking. We could not detect any differences in the oxidation status of α1(I) chain C-telopeptidal lysine, and no differences in the hydroxylation status of α1(I) helical Lys-87, or in α2(I) helical Lys-90 (S1 Fig).
Skin gene expression
To assess the general extracellular matrix gene expression qPCR on selected gene transcripts was performed (Table 2). Aspn-/- skin had an increased expression of collagens (Col1a1, Col1a2, Col3a1), lysyl oxidases (Lox, Loxl2), collagenases (Mmp2, Mmp3), the collagen secretion-related Serpinh1 and Mia3, and the collagen fibril-modulating Lum. The expression of Plod2, the collagen telopeptide-targeted lysine hydroxylase, was decreased. The expression of Fmod and Fbn1 was unaltered.
Table 2. Quantification of gene transcripts using qPCR.
| Relative transcript amount in Aspn-/- (fold of wild-type) |
Wild-type | Aspn-/- | ||||
|---|---|---|---|---|---|---|
| p value | dCt | sd dCt | dCt | sd dCt | ||
| Col1a1 | 6.86 | 0.00004 | -0.21 | 0.71 | -2.99 | 0.27 |
| Col1a2 | 3.96 | 0.00004 | 2.63 | 0.28 | 0.64 | 0.46 |
| Col3a1 | 3.52 | 0.00008 | 1.85 | 0.37 | 0.03 | 0.41 |
| Fmod | 1.57 | 0.3 | 6.82 | 1.03 | 6.17 | 0.87 |
| Lox | 3.52 | 0.0002 | 5.73 | 0.34 | 3.91 | 0.54 |
| Loxl2 | 1.41 | 0.07 | 5.28 | 0.45 | 4.78 | 0.28 |
| Plod2 | 0.23 | 0.004 | 5.32 | 0.18 | 7.43 | 1.19 |
| Lum | 2.21 | 0.03 | 7.68 | 0.87 | 6.54 | 0.47 |
| Fbn1 | 1.02 | 0.9 | 3.91 | 0.57 | 3.88 | 0.16 |
| Mmp2 | 2.41 | 0.002 | 4.15 | 0.47 | 2.88 | 0.38 |
| Mmp3 | 3.19 | 0.0005 | 7.87 | 0.50 | 6.19 | 0.45 |
| Serpinh1 | 2.68 | 0.0001 | 5.36 | 0.31 | 3.94 | 0.35 |
| Mia3 | 1.91 | 0.001 | 8.81 | 0.31 | 7.88 | 0.31 |
Dorsal skin RNA from two-month old female mice (n = 5 per genotype) was used to determine mRNA levels of selected extracellular matrix-related genes. Aspn-/- mRNA levels are presented as percentage of wild-type mRNA levels, all normalized to Actb. P values were calculated using Student’s unpaired two-tailed t-test. Also shown are delta Ct (dCt) and standard deviation (sd dCt).
Skin glycosaminoglycan analysis
To characterize GAG content and structure in two-month old wild-type (n = 5) and Aspn-/- skin (n = 5), we utilized HPLC quantification and identification of disaccharides obtained after chondroitinase ABC- or B- or heparinases-digestion (representative chromatograms are presented in S2 Fig). Aspn-/- mice had higher chondroitin/dermatan sulfate (CS/DS) (wild-type 302 ± 65 ng/mg dry tissue; Aspn-/- 685 ± 105 ng/mg) but lower heparan sulfate (HS) content (wild-type 96 ± 25 ng/mg; Aspn-/- 49 ± 12 ng/mg). Hyaluronic acid quantification showed no differences (Fig 3A). Compositional analysis of CS/DS chains revealed a decrease in non-sulfated disaccharide structures UA-GalNAc (wild-type 8.7 ± 1.7%; Aspn-/- 4.7 ± 0.5%) compensated by an increase in the 4-O-sulfated disaccharide structures UA-GalNAc-4S (wild-type 82.2 ± 1.6%; Aspn-/- 87.7 ± 1.4%). The 2- and 4- disulfated structures UA-2S-GalNAc-4S were slightly reduced from 9.1 ± 0.4% in wild-type to 8.2 ± 0.5% in Aspn-/- (Fig 3B). In order to measure the content of iduronic acid in CS/DS we performed digestions with chondroitinase B, which cleaves only N-acetyl galactosamine-iduronic acid linkages [38]. The analyzed disaccharides must come from at least two adjacent iduronic acid-containing repeats in the native chain, most likely reflecting the long iduronic acid blocks present in the skin decorin and biglycan CS/DS chain. These iduronic acid-containing structures were increased in Aspn-/- skin (wild-type 200 ± 40 ng/mg dry tissue; Aspn-/- 470 ± 59 ng/mg; Fig 3A), showing that they constitute the majority of CS/DS chains in both genotypes (66% in wild-type and 69% in Aspn-/-), in agreement with a previous report [39]. As expected, these iduronic acid blocks contain mostly 4-sulfated iduronic-N-acetyl galactosamine (87 and 88% in wild-type and in Aspn-/-, respectively) and the remaining moieties are 2- and 4- disulfated.HS chains in Aspn-/- skin were less sulfated in all three positions, i.e. glucosamine N-sulfated and 6-O-sulfated, and hexuronic 2-O-sulfated (Fig 3C). In summary, Aspn-/- skin contained more CS/DS and less HS, and HS was less sulfated.
Fig 3. Skin glycosaminoglycan (GAG) analysis.
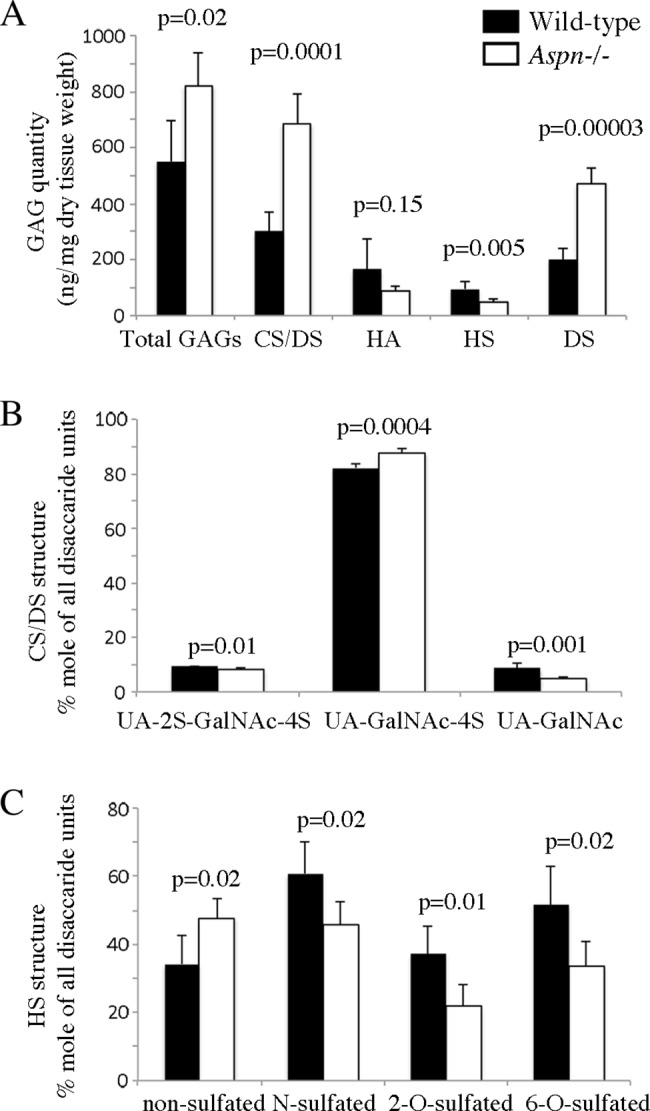
GAGs were purified from two-month old dorsal skin (n = 5 per genotype) and digested with chondroitinase ABC or chondroitinase B or a mixture of heparinases. The resulting disaccharides were fluorescently labeled, separated by HPLC and quantified. (A) Total GAGs are the sum of chondroitin/dermatan sulfate (CS/DS), hyaluronic acid (HA), and heparan sulfate (HS). (B) Compositional analysis of CS/DS chains. UA = unsaturated hexuronic acid obtained after chondroitinases digestion, either unsulfated or 2-O-sulfated (UA-2S); GalNAc = N-acetyl-galactosamine either unsulfated or 4-O-sulfated (GalNAc-4S). (C) Compositional analysis of HS chains. The measured HS disaccharides were grouped according to the sulfation position: non-sulfated = UA-GlcNAc; N-sulfated = UA-GlcNS + UA-2S-GlcNS + UA-GlcNS-6S + UA-2S-GlcNS-6S; 2-O-sulfated = UA-2S-GlcNAc + UA-2S-GlcNAc-6S + UA-2S-GlcNS + UA-2S-GlcNS-6S; 6-O-sulfated = UA-GlcNAc-6S + UA-2S-GlcNAc-6S + UA-GlcNS-6S + UA-2S-GlcNS-6S. UA = unsaturated hexuronic acid obtained after heparinases digestion, either unsulfated or 2-O-sulfated (UA-2S); Glc = glucosamine either N-acetylated (GlcNAc) or N-sulfated (GlcNS) which can be both O-unsulfated or 6-O-sulfated (GlcNAc-6S and GlcNS-6S). Error bars show standard deviation.
Semi-quantitative assessment of decorin and biglycan
To assess the quantities of skin proteoglycans we extracted proteins from dorsal skin samples (n = 4 per genotype) and applied similar total protein quantities (Fig 4A) to anion-exchange. The eluted total proteoglycans (Fig 4B) appeared twofold more abundant in Aspn-/- skin. To verify our findings and to identify the different proteoglycans we digested the samples with chondroitinase ABC and immunoblotted for decorin and biglycan. Aspn-/- skin contained approximately twofold more decorin (Fig 4C) and twofold more biglycan (Fig 4D).
Fig 4. Semi-quantitative assessment of decorin and biglycan.
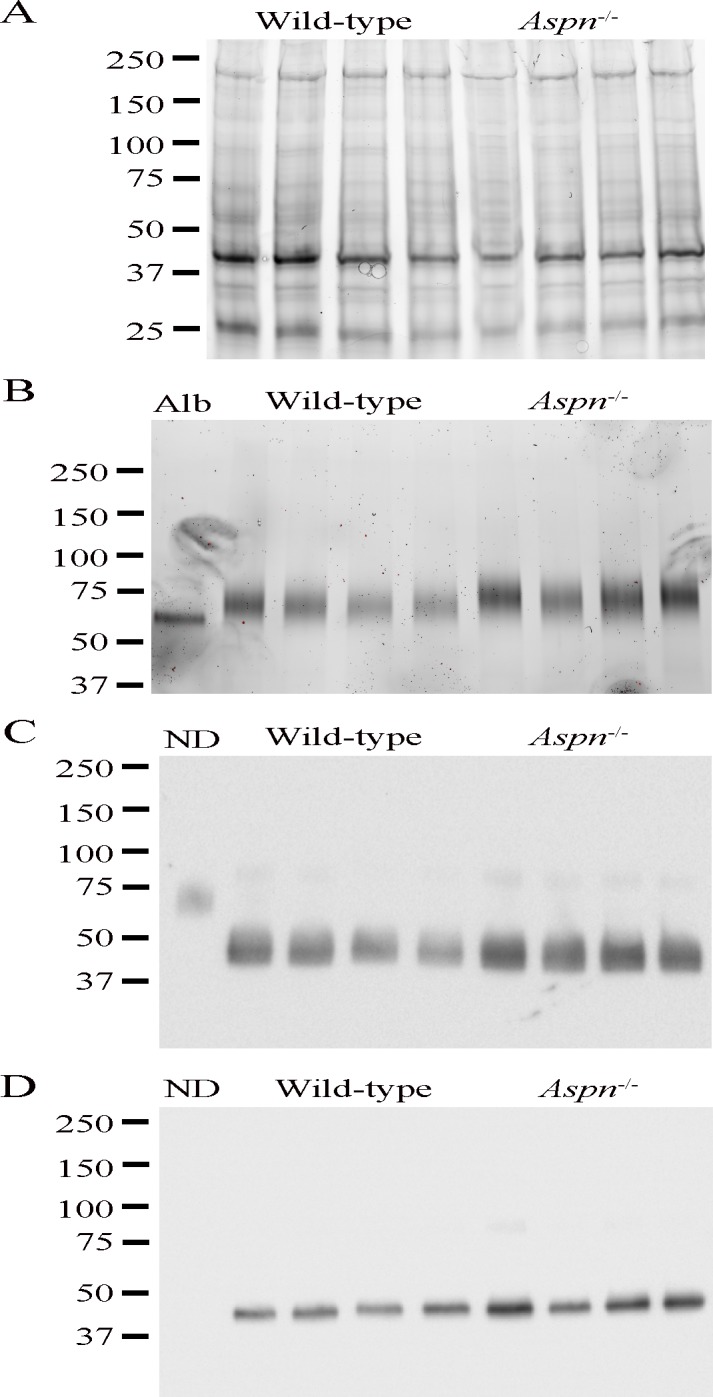
Dorsal skin proteins were extracted with guanidine- and dialyzed against urea-containing buffers. Total protein quantity was assessed by Bradford, and by gel imaging (A). Equal protein quantities were applied on anion-exchange beads and proteoglycans were eluted and imaged on gel (B); Alb is albumin control. Next, proteoglycan samples were digested with chondroitinase ABC and immunoblotted for decorin (C) and biglycan (D) to assess the quantities of the respective protein cores; ND is non-chondroitinase ABC-digested sample control (not visible in biglycan samples probably due to low quantity and smeary distribution).
Discussion
Collagens can be arranged and cross-linked in different ways, which is apparent from the variety of collagen fibril phenotypes in different tissues and organs. The question of mechanisms behind these processes remains intriguing, but several seemingly tissue-specific modulators of collagen fibrillogenesis have been identified in vitro and in vivo, especially among the SLRP family of proteins [1, 2]. In order to examine the role of the less investigated SLRPs we have characterized the in vivo extracellular matrix phenotype in a novel, SLRP-deficient, Aspn-/- mouse. In earlier studies asporin gene polymorphism has been associated with human knee osteoarthritis and lumbar disc degeneration [20, 40]; we were therefore intrigued that Aspn-/- mice did not show any apparent skeletal abnormities, even at histological examination. We suggest that the polymorphisms described in the previous papers [20, 40] have a different effect than total gene deletion. More studies are needed to elucidate the phenotypic impact of the different ASPN gene mutations.
In this paper we focused on skin phenotype–this is mainly because we, in this study, and other groups earlier [21, 22], detected asporin in dermis, and because we observed no abnormities in other major organs in Aspn-/- mice. In addition, we observed a changed biomechanical phenotype in Aspn-/- skin, which prompted the skin ECM analyses conducted in this study.
The skin extracellular matrix in Aspn-/- and wild-type mice was comparable in histological examinations, i.e. the relative amount of dermis and hypodermis, and the amount of blood vessels appeared similar in both genotypes. We observed no signs of inflammation, judging from the unaltered macrophage amounts.
Since asporin interacts with collagen and inhibits its fibrillogenesis in vitro [17] we conducted a detailed investigation of the molecular collagen phenotype in Aspn-/- skin. Collagen fibrils examined in an electron microscope were 7% thinner in Aspn-/- skin; this change, however, was not statistically significant and the fibrils were morphologically regular, in contrast to the fibril phenotypes reported in other SLRP-deficient mice, e.g. Dcn-/- and Lum-/- [6, 9]. Furthermore, the collagen band pattern on Coomassie-stained SDS-PAGE appeared normal, suggesting no defects in collagen post-translational modifications, e.g. proline hydroxylation. Despite a decrease in the amount of lysyl oxidase, the collagen α1(I)- and α2(I)-chain lysines involved in cross-linking were not modified differently than in wild-type collagen, in contrast to the phenotype reported in Fmod-/- mice [3]. Summarizing the collagen phenotype studies, we could detect only a small, statistically non-significant, difference in the fibril diameter in the Aspn-/- skin collagen. It is possible that the homologue decorin, which we found approximately doubled in Aspn-/- skin, binding to the same site on collagen as asporin [17] can compensate for the asporin deficiency, thus ensuring an essentially normal collagen fibril phenotype.
Despite the almost normal Aspn-/- skin collagen fibril phenotype we observed a robust transcript increase of collagens (Col1a1, Col1a2, Col3a1), collagen secretion-related Serpinh1 and Mia3, collagen cross-linking-related lysyl oxidases (Lox, Loxl2), the collagen fibrillogenesis-modulating SLRP Lum, and the collagenases Mmp2 and Mmp3. Altogether, the expression data could be explained by an overall augmented ECM turnover, whose effect, however, on the overall ECM structure observed by histology and electron microscopy appears minute.
While we did not detect any major changes in the overall skin and ECM structure or in collagen molecular phenotype we observed a major change in GAG content and structure. In the ECM a major component are chondroitin/dermatan (CS/DS) proteoglycans versican, decorin and biglycan. In Aspn-/- skin, CS/DS was more than doubled and the overall CS/DS sulfation was increased. The doubling of CS/DS corresponds well with the increased amount of decorin and biglycan protein cores in Aspn-/- skin. This could reflect a compensatory mechanism triggered by deficiency of one SLRP, as was the case in e.g. fibromodulin knockout mice [7]. Interestingly, HS amount decreased in Aspn-/- skin, and was considerably less sulfated, which could impact the cellular response to different growth factors. Overall, these changes in GAG content and structure could reflect a general cellular response to a changed extracellular matrix microenvironment. Our attempts at finding the mechanisms behind this response have so far not been fruitful.
The above-described changes in the ECM phenotype translate into a mechanically tougher skin in Aspn-/- mice. The strain-stress curves obtained from biomechanical analyses suggest that the difference between Aspn-/- and wild-type skin did not manifest in the initial elastic region where the deformation is reversible, but rather in the plastic region between the yield and the failure point, where sliding and possibly rupture of collagen fibrils lead to irreversible damage. We suggest that a compound effect of slightly thinner collagen fibrils and the large difference in GAG composition could both contribute to this biomechanical phenotype. While perhaps counterintuitive, the correlation of thinner collagen fibrils and higher mechanical strength has been described before, e.g. where decorin inhibited fibril assembly but increased fibril strength [41], and where thinner fibrils in 3D gels conferred increased tensile strength [42], possibly through promoting more interconnected fibrillar network (increased surface-to-volume ratio) [43]. Regarding GAGs’ influence on the mechanical properties, several papers argue against that possibility in a number of in vitro or ex vivo studies [41, 44, 45]. These studies, however, were performed on ligaments, where collagen fibers are largely aligned in parallel and have therefore limited compressive force components. Skin, unlike ligaments, will likely have more compression between fibers even under tensile load. This compression is likely to be influenced by the water-retaining GAGs, and in particular CS/DS. We suggest that the changed GAG composition and structure, as well as the increased amounts of decorin and biglycan, in Aspn-/- skin provide an explanation for the altered biomechanical properties.
Supporting information
(A) Two-month old dorsal skin collagen was extracted with either SDS-PAGE loading buffer or 3% acetic acid, run on 6% SDS-PAGE and stained with Coomassie Blue. The major collagen bands are denoted on the right. Similar results were obtained by analyzing nine mice of each genotype. (B) Mass spectrometry analysis of modifications of prolines and lysines around collagen cross-linking sites. The analyzed peptides’ sequences are shown at the top of each panel. Mass spectra show similar oxidation status of both prolines and lysines in both wild-type (WT) and Aspn-/- (KO) mice.
(TIF)
HPLC disaccharide chromatograms of CS/DS (A), DS (B), HS (C). GAGs were purified and digested with chondroitinase ABC (A), chondroitinase B (B), or heparinases I+II+III (C), then AMAC-labeled and HPLC-separated. In (A) and (B) the disaccharide identity is: peak 1: UA-2S-GalNAc-4S (B structure); peak 2: UA-GalNAc-4S-6S (E structure); peak 3: UA-2S-GalNAc-6S (D structure); peak 4: UA-GalNAc-4S (A structure); peak 5: UA-GalNAc-6S (C structure); peak 6: HA-derived UA-GlcNAc; peak 7: UA-GalNAc (0 structure). In (C) the disaccharide identity is: peak 1: UA-2S-GlcNS-6S; peak 2: UA-GlcNS-6S; peak 3: UA-2S-GlcNS; peak 4: UA-2S-GlcNAc; peak 5: UA-GlcNS; peak 6: UA-2S-GlcNAc-6S; peak 7: UA-GlcNAc-6S; peak 8: UA-GlcNAc. Asterisks indicate unidentified peaks. Similar chromatograms were obtained with other samples from the respective genotype.
(TIF)
(DOCX)
Images from dorsal skin Masson Trichrome stainings. Three male and three female mice per genotype were used. ASPN WT are wild-type, ASPN KO are Aspn-/-.
(ZIP)
The data were collected as described in Materials and Methods, Skin tensile strength determination.
(XLSX)
The data were collected as described in Materials and Methods, Electron microscopy.
(XLSX)
The data were collected as described in Materials and Methods, Reverse transcriptase- and real-time qPCR.
(XLSX)
Data Availability
All relevant data are within the paper and its Supporting Information files. All original data may be obtained by contacting the corresponding author. Primary data from mass spectrometry and GAG chromatography analyses are provided as Supplemental Figures.
Funding Statement
This study was supported by the Swedish Cancer Foundation, http://www.cancerfonden.se, to MM, SK; the Alfred Österlund Foundation, http://www.alfredosterlundsstiftelse.se/, to MM, SK; the Danish Council for Independent Research, http://ufm.dk/forskning-og-innovation/rad-og-udvalg/det-frie-forskningsrad, to RBS, grant DFF-1333-00052A; the NIH(NIAMS), http://www.nih.gov, to DE, grant AR037318; the Swedish Research Council, http://www.vr.se, to SK, grant 2008-6946; The Crafoord Foundation, http://en.crafoord.se/, to SK; The Åke Wiberg Foundation, http://ake-wiberg.se/, to SK; and The Magnus Bergvall Foundation, http://www.magnbergvallsstiftelse.nu/, to SK. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Kalamajski S, Oldberg A. The role of small leucine-rich proteoglycans in collagen fibrillogenesis. Matrix Biol. 2010;29(4):248–53. doi: 10.1016/j.matbio.2010.01.001 . [DOI] [PubMed] [Google Scholar]
- 2.Chen S, Birk DE. The regulatory roles of small leucine-rich proteoglycans in extracellular matrix assembly. FEBS J. 2013;280(10):2120–37. doi: 10.1111/febs.12136 ; PubMed Central PMCID: PMCPMC3651807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kalamajski S, Liu C, Tillgren V, Rubin K, Oldberg A, Rai J, et al. Increased C-telopeptide cross-linking of tendon type I collagen in fibromodulin-deficient mice. J Biol Chem. 2014;289(27):18873–9. doi: 10.1074/jbc.M114.572941 ; PubMed Central PMCID: PMCPMC4081928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chakravarti S, Petroll WM, Hassell JR, Jester JV, Lass JH, Paul J, et al. Corneal opacity in lumican-null mice: defects in collagen fibril structure and packing in the posterior stroma. Invest Ophthalmol Vis Sci. 2000;41(11):3365–73. ; PubMed Central PMCID: PMCPMC4318236. [PMC free article] [PubMed] [Google Scholar]
- 5.Yeh JT, Yeh LK, Jung SM, Chang TJ, Wu HH, Shiu TF, et al. Impaired skin wound healing in lumican-null mice. Br J Dermatol. 2010;163(6):1174–80. doi: 10.1111/j.1365-2133.2010.10008.x . [DOI] [PubMed] [Google Scholar]
- 6.Chakravarti S, Magnuson T, Lass JH, Jepsen KJ, LaMantia C, Carroll H. Lumican regulates collagen fibril assembly: skin fragility and corneal opacity in the absence of lumican. J Cell Biol. 1998;141(5):1277–86. ; PubMed Central PMCID: PMCPMC2137175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Svensson L, Aszodi A, Reinholt FP, Fassler R, Heinegard D, Oldberg A. Fibromodulin-null mice have abnormal collagen fibrils, tissue organization, and altered lumican deposition in tendon. J Biol Chem. 1999;274(14):9636–47. . [DOI] [PubMed] [Google Scholar]
- 8.Zhang G, Ezura Y, Chervoneva I, Robinson PS, Beason DP, Carine ET, et al. Decorin regulates assembly of collagen fibrils and acquisition of biomechanical properties during tendon development. J Cell Biochem. 2006;98(6):1436–49. doi: 10.1002/jcb.20776 . [DOI] [PubMed] [Google Scholar]
- 9.Danielson KG, Baribault H, Holmes DF, Graham H, Kadler KE, Iozzo RV. Targeted disruption of decorin leads to abnormal collagen fibril morphology and skin fragility. J Cell Biol. 1997;136(3):729–43. ; PubMed Central PMCID: PMCPMC2134287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fust A, LeBellego F, Iozzo RV, Roughley PJ, Ludwig MS. Alterations in lung mechanics in decorin-deficient mice. Am J Physiol Lung Cell Mol Physiol. 2005;288(1):L159–66. doi: 10.1152/ajplung.00089.2004 . [DOI] [PubMed] [Google Scholar]
- 11.Liu CY, Birk DE, Hassell JR, Kane B, Kao WW. Keratocan-deficient mice display alterations in corneal structure. J Biol Chem. 2003;278(24):21672–7. doi: 10.1074/jbc.M301169200 . [DOI] [PubMed] [Google Scholar]
- 12.Pellegata NS, Dieguez-Lucena JL, Joensuu T, Lau S, Montgomery KT, Krahe R, et al. Mutations in KERA, encoding keratocan, cause cornea plana. Nat Genet. 2000;25(1):91–5. doi: 10.1038/75664 . [DOI] [PubMed] [Google Scholar]
- 13.Xu T, Bianco P, Fisher LW, Longenecker G, Smith E, Goldstein S, et al. Targeted disruption of the biglycan gene leads to an osteoporosis-like phenotype in mice. Nat Genet. 1998;20(1):78–82. doi: 10.1038/1746 . [DOI] [PubMed] [Google Scholar]
- 14.Jepsen KJ, Wu F, Peragallo JH, Paul J, Roberts L, Ezura Y, et al. A syndrome of joint laxity and impaired tendon integrity in lumican- and fibromodulin-deficient mice. J Biol Chem. 2002;277(38):35532–40. doi: 10.1074/jbc.M205398200 . [DOI] [PubMed] [Google Scholar]
- 15.Zhang G, Chen S, Goldoni S, Calder BW, Simpson HC, Owens RT, et al. Genetic evidence for the coordinated regulation of collagen fibrillogenesis in the cornea by decorin and biglycan. J Biol Chem. 2009;284(13):8888–97. doi: 10.1074/jbc.M806590200 ; PubMed Central PMCID: PMCPMC2659246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Svensson L, Narlid I, Oldberg A. Fibromodulin and lumican bind to the same region on collagen type I fibrils. FEBS Lett. 2000;470(2):178–82. . [DOI] [PubMed] [Google Scholar]
- 17.Kalamajski S, Aspberg A, Lindblom K, Heinegard D, Oldberg A. Asporin competes with decorin for collagen binding, binds calcium and promotes osteoblast collagen mineralization. Biochem J. 2009;423(1):53–9. doi: 10.1042/BJ20090542 . [DOI] [PubMed] [Google Scholar]
- 18.Kalamajski S, Oldberg A. Homologous sequence in lumican and fibromodulin leucine-rich repeat 5–7 competes for collagen binding. J Biol Chem. 2009;284(1):534–9. doi: 10.1074/jbc.M805721200 . [DOI] [PubMed] [Google Scholar]
- 19.Lorenzo P, Aspberg A, Onnerfjord P, Bayliss MT, Neame PJ, Heinegard D. Identification and characterization of asporin. a novel member of the leucine-rich repeat protein family closely related to decorin and biglycan. J Biol Chem. 2001;276(15):12201–11. doi: 10.1074/jbc.M010932200 . [DOI] [PubMed] [Google Scholar]
- 20.Kizawa H, Kou I, Iida A, Sudo A, Miyamoto Y, Fukuda A, et al. An aspartic acid repeat polymorphism in asporin inhibits chondrogenesis and increases susceptibility to osteoarthritis. Nat Genet. 2005;37(2):138–44. doi: 10.1038/ng1496 . [DOI] [PubMed] [Google Scholar]
- 21.Kou I, Nakajima M, Ikegawa S. Expression and regulation of the osteoarthritis-associated protein asporin. J Biol Chem. 2007;282(44):32193–9. doi: 10.1074/jbc.M706262200 . [DOI] [PubMed] [Google Scholar]
- 22.Henry SP, Takanosu M, Boyd TC, Mayne PM, Eberspaecher H, Zhou W, et al. Expression pattern and gene characterization of asporin. a newly discovered member of the leucine-rich repeat protein family. J Biol Chem. 2001;276(15):12212–21. doi: 10.1074/jbc.M011290200 . [DOI] [PubMed] [Google Scholar]
- 23.Ueda M, Goto T, Kuroishi KN, Gunjigake KK, Ikeda E, Kataoka S, et al. Asporin in compressed periodontal ligament cells inhibits bone formation. Arch Oral Biol. 2016;62:86–92. doi: 10.1016/j.archoralbio.2015.11.010 . [DOI] [PubMed] [Google Scholar]
- 24.Yamada S, Tomoeda M, Ozawa Y, Yoneda S, Terashima Y, Ikezawa K, et al. PLAP-1/asporin, a novel negative regulator of periodontal ligament mineralization. J Biol Chem. 2007;282(32):23070–80. doi: 10.1074/jbc.M611181200 . [DOI] [PubMed] [Google Scholar]
- 25.Zhu F, Friedman MS, Luo W, Woolf P, Hankenson KD. The transcription factor osterix (SP7) regulates BMP6-induced human osteoblast differentiation. J Cell Physiol. 2012;227(6):2677–85. doi: 10.1002/jcp.23010 ; PubMed Central PMCID: PMCPMC3241898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Awata T, Yamada S, Tsushima K, Sakashita H, Yamaba S, Kajikawa T, et al. PLAP-1/Asporin Positively Regulates FGF-2 Activity. J Dent Res. 2015;94(10):1417–24. doi: 10.1177/0022034515598507 . [DOI] [PubMed] [Google Scholar]
- 27.Duval E, Bigot N, Hervieu M, Kou I, Leclercq S, Galera P, et al. Asporin expression is highly regulated in human chondrocytes. Mol Med. 2011;17(7–8):816–23. doi: 10.2119/molmed.2011.00052 ; PubMed Central PMCID: PMCPMC3146605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nakajima M, Kizawa H, Saitoh M, Kou I, Miyazono K, Ikegawa S. Mechanisms for asporin function and regulation in articular cartilage. J Biol Chem. 2007;282(44):32185–92. doi: 10.1074/jbc.M700522200 . [DOI] [PubMed] [Google Scholar]
- 29.Maris P, Blomme A, Palacios AP, Costanza B, Bellahcene A, Bianchi E, et al. Asporin Is a Fibroblast-Derived TGF-beta1 Inhibitor and a Tumor Suppressor Associated with Good Prognosis in Breast Cancer. PLoS Med. 2015;12(9):e1001871 doi: 10.1371/journal.pmed.1001871 ; PubMed Central PMCID: PMCPMC4556693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Simkova D, Kharaishvili G, Korinkova G, Ozdian T, Suchankova-Kleplova T, Soukup T, et al. The dual role of asporin in breast cancer progression. Oncotarget. 2016. doi: 10.18632/oncotarget.10471 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ding Q, Zhang M, Liu C. Asporin participates in gastric cancer cell growth and migration by influencing EGF receptor signaling. Oncol Rep. 2015;33(4):1783–90. doi: 10.3892/or.2015.3791 . [DOI] [PubMed] [Google Scholar]
- 32.Satoyoshi R, Kuriyama S, Aiba N, Yashiro M, Tanaka M. Asporin activates coordinated invasion of scirrhous gastric cancer and cancer-associated fibroblasts. Oncogene. 2015;34(5):650–60. doi: 10.1038/onc.2013.584 . [DOI] [PubMed] [Google Scholar]
- 33.Liu P, Jenkins NA, Copeland NG. A highly efficient recombineering-based method for generating conditional knockout mutations. Genome Res. 2003;13(3):476–84. doi: 10.1101/gr.749203 ; PubMed Central PMCID: PMCPMC430283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Olsson PO, Gustafsson R, In 't Zandt R, Friman T, Maccarana M, Tykesson E, et al. The Tyrosine Kinase Inhibitor Imatinib Augments Extracellular Fluid Exchange and Reduces Average Collagen Fibril Diameter in Experimental Carcinoma. Mol Cancer Ther. 2016;15(10):2455–64. doi: 10.1158/1535-7163.MCT-16-0026 . [DOI] [PubMed] [Google Scholar]
- 35.Berg RA. Determination of 3- and 4-hydroxyproline. Methods Enzymol. 1982;82 Pt A:372–98. . [DOI] [PubMed] [Google Scholar]
- 36.Bitter T, Muir HM. A modified uronic acid carbazole reaction. Anal Biochem. 1962;4:330–4. . [DOI] [PubMed] [Google Scholar]
- 37.Volpi N, Galeotti F, Yang B, Linhardt RJ. Analysis of glycosaminoglycan-derived, precolumn, 2-aminoacridone-labeled disaccharides with LC-fluorescence and LC-MS detection. Nat Protoc. 2014;9(3):541–58. doi: 10.1038/nprot.2014.026 . [DOI] [PubMed] [Google Scholar]
- 38.Michel G, Pojasek K, Li Y, Sulea T, Linhardt RJ, Raman R, et al. The structure of chondroitin B lyase complexed with glycosaminoglycan oligosaccharides unravels a calcium-dependent catalytic machinery. J Biol Chem. 2004;279(31):32882–96. doi: 10.1074/jbc.M403421200 ; PubMed Central PMCID: PMCPMC4135467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maccarana M, Kalamajski S, Kongsgaard M, Magnusson SP, Oldberg A, Malmstrom A. Dermatan sulfate epimerase 1-deficient mice have reduced content and changed distribution of iduronic acids in dermatan sulfate and an altered collagen structure in skin. Mol Cell Biol. 2009;29(20):5517–28. doi: 10.1128/MCB.00430-09 ; PubMed Central PMCID: PMCPMC2756890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Song YQ, Cheung KM, Ho DW, Poon SC, Chiba K, Kawaguchi Y, et al. Association of the asporin D14 allele with lumbar-disc degeneration in Asians. Am J Hum Genet. 2008;82(3):744–7. doi: 10.1016/j.ajhg.2007.12.017 ; PubMed Central PMCID: PMCPMC2427233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reese SP, Underwood CJ, Weiss JA. Effects of decorin proteoglycan on fibrillogenesis, ultrastructure, and mechanics of type I collagen gels. Matrix Biol. 2013;32(7–8):414–23. doi: 10.1016/j.matbio.2013.04.004 ; PubMed Central PMCID: PMCPMC3795835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Roeder BA, Kokini K, Sturgis JE, Robinson JP, Voytik-Harbin SL. Tensile mechanical properties of three-dimensional type I collagen extracellular matrices with varied microstructure. J Biomech Eng. 2002;124(2):214–22. . [DOI] [PubMed] [Google Scholar]
- 43.Roeder BA, Kokini K, Voytik-Harbin SL. Fibril microstructure affects strain transmission within collagen extracellular matrices. J Biomech Eng. 2009;131(3):031004 doi: 10.1115/1.3005331 . [DOI] [PubMed] [Google Scholar]
- 44.Lujan TJ, Underwood CJ, Henninger HB, Thompson BM, Weiss JA. Effect of dermatan sulfate glycosaminoglycans on the quasi-static material properties of the human medial collateral ligament. J Orthop Res. 2007;25(7):894–903. doi: 10.1002/jor.20351 . [DOI] [PubMed] [Google Scholar]
- 45.Lujan TJ, Underwood CJ, Jacobs NT, Weiss JA. Contribution of glycosaminoglycans to viscoelastic tensile behavior of human ligament. J Appl Physiol (1985). 2009;106(2):423–31. doi: 10.1152/japplphysiol.90748.2008 ; PubMed Central PMCID: PMCPMC2644241. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Two-month old dorsal skin collagen was extracted with either SDS-PAGE loading buffer or 3% acetic acid, run on 6% SDS-PAGE and stained with Coomassie Blue. The major collagen bands are denoted on the right. Similar results were obtained by analyzing nine mice of each genotype. (B) Mass spectrometry analysis of modifications of prolines and lysines around collagen cross-linking sites. The analyzed peptides’ sequences are shown at the top of each panel. Mass spectra show similar oxidation status of both prolines and lysines in both wild-type (WT) and Aspn-/- (KO) mice.
(TIF)
HPLC disaccharide chromatograms of CS/DS (A), DS (B), HS (C). GAGs were purified and digested with chondroitinase ABC (A), chondroitinase B (B), or heparinases I+II+III (C), then AMAC-labeled and HPLC-separated. In (A) and (B) the disaccharide identity is: peak 1: UA-2S-GalNAc-4S (B structure); peak 2: UA-GalNAc-4S-6S (E structure); peak 3: UA-2S-GalNAc-6S (D structure); peak 4: UA-GalNAc-4S (A structure); peak 5: UA-GalNAc-6S (C structure); peak 6: HA-derived UA-GlcNAc; peak 7: UA-GalNAc (0 structure). In (C) the disaccharide identity is: peak 1: UA-2S-GlcNS-6S; peak 2: UA-GlcNS-6S; peak 3: UA-2S-GlcNS; peak 4: UA-2S-GlcNAc; peak 5: UA-GlcNS; peak 6: UA-2S-GlcNAc-6S; peak 7: UA-GlcNAc-6S; peak 8: UA-GlcNAc. Asterisks indicate unidentified peaks. Similar chromatograms were obtained with other samples from the respective genotype.
(TIF)
(DOCX)
Images from dorsal skin Masson Trichrome stainings. Three male and three female mice per genotype were used. ASPN WT are wild-type, ASPN KO are Aspn-/-.
(ZIP)
The data were collected as described in Materials and Methods, Skin tensile strength determination.
(XLSX)
The data were collected as described in Materials and Methods, Electron microscopy.
(XLSX)
The data were collected as described in Materials and Methods, Reverse transcriptase- and real-time qPCR.
(XLSX)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files. All original data may be obtained by contacting the corresponding author. Primary data from mass spectrometry and GAG chromatography analyses are provided as Supplemental Figures.