

Abstract
Nanomedicine is a rapidly evolving form of therapy that holds a great promise in superior drug delivery efficiency and therapeutic efficacy than conventional cancer treatment. In this review, we attempt to cover the benefits and the limitations of current nanomedicines with special attention to covalent nanoconjugates for imaging and drug delivery in brain. The improvement in brain tumor treatment remains dismal despite decades of efforts in drug development and patient care. One of the major obstacles in brain cancer treatment is the poor drug delivery efficiency owing to the unique blood-brain-barrier (BBB) in the CNS. Although various anti-cancer agents are available to treat tumors outside of the CNS, the majority fails to cross the BBB. In this regard, nanomedicines have increasingly drawn attention due to their multi-functionality and versatility. Nano-drugs can penetrate BBB and other biological barriers, and selectively accumulate in tumor cells, while concurrently decreasing systemic toxicity.
Keywords: Covalent nanoconjugates, Blood-Brain-Barrier (BBB), Nanomedicine for brain imaging, Drug delivery, Brain cancer
Graphical abstract
Introduction
Medicines have been traditionally given to patients in the forms of pills (“small ball or round mass of medicine”, from Middle Dutch or Middle Low German “pille” [1] corresponding to modern encapsulated nanodrugs. Or in the form of a drug as in French “drogue”[2], a natural or synthetic soluble chemical, corresponding to covalent nanodrug. The fundamental difference between pills and drugs is that the active reagent in the pill or capsule is not immobilized by chemical bonds and thus free to evade the carrier material (micelle, liposome, suspended water insoluble precipitate), whereas the drug given e.g., in the form of a soluble nanodrug contains an active natural or synthetic compound, which is covalently bound to a macromolecular platform and often resembles a prodrug.
The main features of a nanodrug are precise targeting and delivery, which are equally important for successful treatment. Addressing safety is of great importance. Unsecured delivery could cause adverse reactions / side effects that occur when a toxic drug and targeted carrier are disconnected during delivery and the drug becomes available elsewhere. To achieve a rapid transport through the body’s vascular system, minimize clearance through kidneys and facilitate high penetration through tissue and membrane barriers, imaging or therapeutic delivery vehicles have been developed that cover a range of nanoscale sizes (5-400 nm) [3, 4].
Transported cargo may be both chemically (covalently) bound to the vehicle, and cleaved from the vehicle platform to become the pharmacologically active drug. The bound drugs are not free to diffuse from the carrier, whereas in contrast, encapsulating vehicles transport the drugs in their free unbound form. Encapsulating vehicles are liposomes, micelles, or one of several nanoparticles fabricated by dispersion or precipitation methods. Encapsulating devices release their cargo by spontaneous drug diffusion or after nanoparticle dissolution or capsule erosion. The nano capsules can be designed to “open” at the targeted delivery site in response to the typical environment such as local pH or enzyme cleavage activity. However, because of spontaneous diffusion and capsule-destabilizing environment, release can occur in an uncontrolled fashion and cause harmful damage to healthy tissue. Micelles have a structure, which is in dynamic equilibrium with their parts forming free constituents. They are self-assembled only when the concentration of the free constituents exceeds the so-called critical micelle concentration (CMC). Below CMC the micelles are instable and dissociate into the free constituents [5, 6], which may occur with injected micelles when they circulate in the vascular system. Concomitantly with the dissociation, the drug located in the micelle core will be released into plasma. Despite this possibility and uncontrolled drug diffusion out of capsules, micelles and other encapsulating devices are frequently used for targeted drug delivery. In this review, we analyze the best possibility for the drug delivery through the multiple bio-barriers with the special attention to the delivery to the brain.
1. Covalent Drug Delivery Systems (CDS)
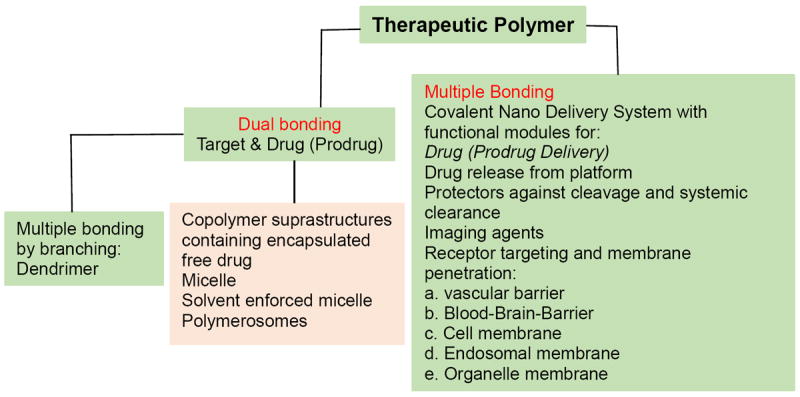
1.1 Ringsdorf’s vision of therapeutic polymers
Ringsdorf proposed the first version of Covalent Delivery Systems (CDS) (Table 1). They became known as “Therapeutic Polymers” because drugs and targeting molecules were attached covalently to a polymer platform [4, 5, 7]. Ringdorf’s view was of a linear polymeric nano drug with a central polymer platform: a module with conjugated prodrug binding covalently to the platform by a cleavable linker, and in addition, a module for targeting and a module providing solubility for the therapeutic polymer [8, 9].
Table 1.
Properties of therapeutic polymers
|
1.2 The multi modular architecture of nanoconjugates
This first concept has in the following years been refined to develop polymers that simultaneously carry multiple variable functional groups (modules) in particular, an additional moiety for releasing the therapeutic polymer from endosome vesicle, and aimed at rescuing prodrugs from lysosomal cleavage (Table 1 & Fig. 1). In fact, the only known type of escape until then had been based on the proton sponge effected osmotic disruption of membranes by polycationic devices such as polyethyleneimine, polyhistidine and polylysine for the endosome release of nucleic acids [10]. A decade ago, the tripeptide-based lipophilic endosome escape modules were introduced that could be applied to a large variety of payloads other than nucleic acid [11-14]. Another important module was the antibody for tumor targeting. This novelty allowed the CDS to cross multiple biobarriers including the blood-brain barrier (BBB) [15-22]. Recently, the concept of Mini Nano drugs was introduced that uses receptor affine peptides instead of specific antibodies for targeted drug delivery [23, 24]. In principle, the covalent concept foresees a plethora of modules depending on the number of anchorage sites on the polymer platform. The attached chemically or biologically reactive groups can function synchronously or independently allowing a diversity of reactions in a time and space dependent fashion [4, 5, 7]. Multiple attachments of cell targeting modules offer the possibility to physically connect CD20 receptors to unleash apoptosis in human non-Hodgkin’s lymphoma Raji B cells [25] or, as it may be proposed in a scenario with an analogy to special bispecific antibodies (BiTEs), to physically connect epidermal growth factor (EGFR) mutant receptors (EGFRvIII) on tumor cells with CD3 receptor on cytotoxic T-lymphocytes to initiate apoptosis of tumor cells [26, 27].
Figure 1.
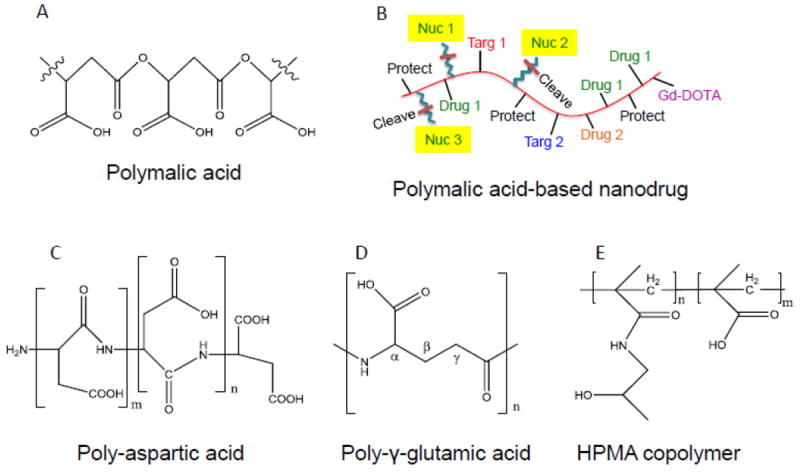
Common therapeutic polymers for nano imaging and therapeutics.
A, β-Poly(L-malic acid). B, β-Poly(L-malic acid)-based covalent delivery system (CDS). C, α-Poly(L-aspartic acid). D, γ-Poly(L-glutamic acid). E, Poly(N-(2-hydroxypropyl)methacrylamid copolymer). Functional modules are substituted at carboxylic groups. B, an example is shown for the variability of functional modules on a polymeric nanodrug: “Drug 1, Drug 2”, multiple copies of different chemotherapeutic drugs; “Nuc 1, Nuc 2, Nuc 3”, multiple different AONs; “Targ 1, Targ 2”, multiple different targeting antibodies or affine peptides; “Gd-DOTA”, Gadolinium-DOTA, an MRI tracer for imaging; “Protect”, polyethylene glycol PEG2000 or PEG5000; “Cleave”, linker cleavage site.
1.3 Structural and functional diversity of CDS
Because of the macromolecular structure, CDS are versatile molecules. The macromolecular diversity unfolds further when including branched polymers such as dendrimers, molecular rods, and in addition, spontaneous self-association (Table 1) [4, 5, 7]. In the case of a covalently bound drug, these delivery systems should not be confused with nanoparticles encapsulating free drugs.
2. Intramolecular dynamics and group mobility
Depending on their rigidity and status of branching, CDS differ by their ability to exercise intramolecular dynamics. Unbranched nano conjugates differ in rigidity due to the nature of their polymeric units and interconnecting covalent bonds. For example, nano conjugates with polysaccharides, nucleic acids and polypeptides have a stiffer and thus more rod-like structure because of hindered rotation around their backbone. In another example, nano conjugates with polyesters such as poly(acrylic acid), poly(hydroxy alkanoic acid) and poly(malic acid) have backbones interconnected by carbon-carbon and carbon-oxygen bonds. Segments of nanoconjugates are freely rotatable around these bonds. Consequently, spatial crowding inferred by neighboring positions of bulky substituents can be relieved by rotations around the bonds. Furthermore, the rotations contribute to structural flexibility within the platform and could allow spatial clustering of e.g., lipophilic residues [28, 29].
2.1 Antibodies and their replacement by affine peptides
In the absence of antibodies or other large proteins, CDS with free rotatable bonds and extended structures can exercise internal bending motions and coil formation. Bending motions can propel them at higher rates of diffusion than nano conjugates of similar molecular weight but with a spherical shape. Thus, they could achieve faster permeation through membrane barriers and are more effective for deep tissue penetration [30, 31]. Bulky ligands such as antibodies increase size, shape and motion. The rate of interaction of CDS with cell surface receptors is governed by the slow dissociation of the receptor complexes with high affinity binding antibody modules. Their replacement by peptides decreases the size and also binding affinity and accelerates dissociation [32, 33], (Table 2). In our recent preclinical study, the replacement resulted in the reduction of hydrodynamic diameter from 22 nm to 7.8 nm while retaining excellent therapeutic efficacy [23] (Fig. 2).
Table 2.
Replacement of antibody by affinity peptide: pro and contra
| Pro | Contra |
|---|---|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Figure 2.
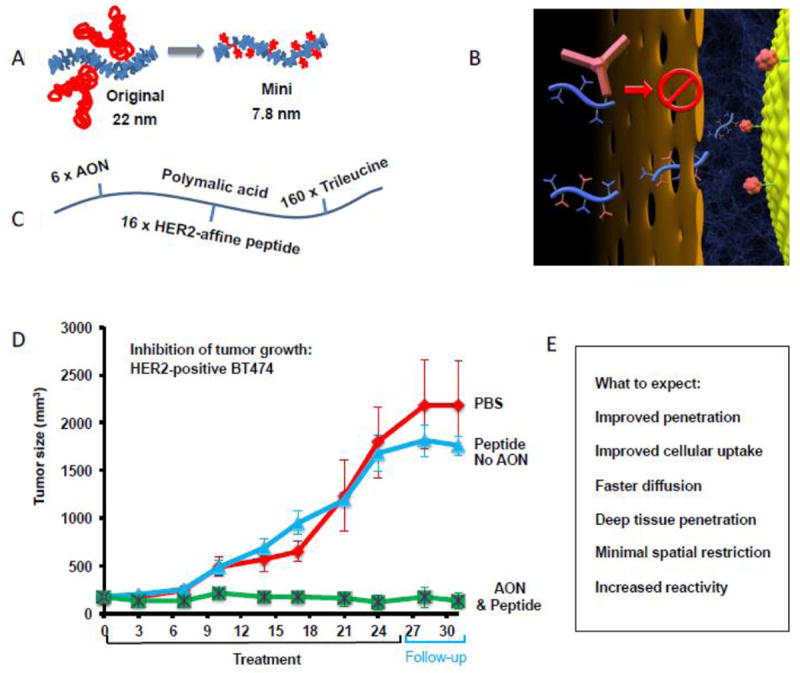
Concept of a mini nano drug. A, comparison of an antibody-targeted covalent drug delivery system with a peptide-targeted mini nano drug. B, cartoon displaying advantages of the mini nano drug over the parent nano drug in gaining access through size limiting passages and crowded extracellular matrix to reach a receptor for binding. C, example of modules built in a mini nano drug carrying AONs for blocking mRNA for HER2 synthesis. Platform (curved line): PMLA, molecular weight 50 kDa. Loading: 6 molecules AON; 16 molecules HER2 affine peptide; 160 molecules trileucine for mini nano drug release from endosome into cytoplasm. D, growth inhibition of HER2-positive human breast tumor in nude mice by the mini nano drug treatment. E, benefits expected from the mini nano drug vs. antibody containing nano drug. Modified from [23].
2.2 Prodrugs and cleavable linkers
CDS are macromolecular entities, which deliver prodrugs conjugated through appropriate linkers. Linkers have been developed that are cleaved in response to exposure to chemistries or enzymatic activities typically found at the site of delivery [34]. Examples include pH-responsive hydrolytic cleavage of activated ester or hydrazides, and the reductive cleavage of disulfide bonds by glutathione in the cytoplasm of recipient cells [4, 35]. Consensus peptide linkers have been introduced that are susceptible to cleavage by specific peptidases such as metalloproteinases MMP-2 or MMP-9 [36], and cathepsins in lysosomes [37, 38].
3. Modular organization of CDS with diverse functions
A CDS is a nano sized macromolecule built from modules specialized in specific functions. One category of modules provides platforms for the chemical assembly of the other modules. Center and “assembly” module is a polymer that provides chemically reactive sites, for example, −OH, -NH2, -COOH, -SH2, -CH=CH-, to attach functional modules. In a wider sense, the “assembly” module includes linkers that could be cleaved for e.g., drug release. Linkers connect the platform with functional modules including drugs, targeting antibodies or peptides, membrane destabilizing molecules or protecting molecules [11-23, 28, 39, 40]. Linkers such as peptides or polyethylene glycol (PEG) can vary in size and flexibility to allow a functional module approach, and interact optimally with its biological target such as a receptor or an enzyme active site, or anneal specifically with a nucleic acid of a given sequence.
Among functioning modules are targeting modules, drug molecules, imaging dyes, contrast agents (MRI), radioactive 125I-labeled tyrosine as tracer molecules, and protective polyethylene glycol (PEG). Targeting modules can be specific antibodies, peptides, oligosaccharides, oligonucleotides or ligands of diverse structures that bind to specific receptors. The targeting ligand must often adopt a certain geometry and orientation to access the specific binding site e.g., pass through a bottleneck at the entrance of a binding pocket or present itself in a conformation that binds a receptor with the highest affinity. An optimal linker should not present geometrical constraints nor unfavorable electrical charges or hydration. A drug conjugated with a linker could be active, or inactive (prodrug). The prodrug can be activated by cleavage of the linker. Converting a prodrug into an active drug is a powerful method for generating high site specificity.
In case a platform offers multiple sites, modules with different specificities can be assigned that function in programmed delivery through several bio barriers to the site of drug action. An example is targeted growth inhibition of lung and breast metastatic tumors in the brain [14]. The designed polymalic acid-based nanodrugs bind with their attached specific antibody to endothelial transferrin receptor (TfR), then pass through the blood-brain barrier (BBB) endothelial cell layer by transferrin receptor-mediated transcytosis. After release from the TfR specific antibody the nanodrugs attach with their Cetuximab or Herceptin antibody to tumor-overexpressed EGFR or HER2 receptors and enter the clathrin-mediated endosome pathway. The nanodrugs remain in the maturing endosomes until the increasing acidification activates the pH-sensitive membranolytic action of the tri-Leucine module [12, 28], and then they finally find their way into the cytoplasm. The pH-responsive membrane disruption is managed by a module consisting of tri-Leucine (LLL) residues conjugated with > 50% of the malyl units of the polymer. After the release of the nano conjugate, cytoplasmic glutathione (GSH) present in > 3 mM concentration reductively cleaves the disulfide containing linker to release EGFR- and HER2-antisense oligonucleotides (AONs) to block EGFR and HER2 specific mRNA. The delivery route and the cleavage of AONs from the polymer platform could be followed by fluorescence of the individually with AlexaFluor 680 and Rhodamine tagged AONs and polymalic acid platform by confocal microscopy. Ex vivo western blots showed that the synthesis of EGFR or HER2 and the downstream phosphorylation of Akt were significantly inhibited. PARP was cleaved in agreement with tumor cell apoptosis [14, 21, 22]. The delivery pathway from nanodrug injection to nanodrug uptake in targeted tumor cells parallels that of imaging enhancement through targeted contrast agents such as fluorescent dyes or MRI contrast agents. The application of brain tumor targeted MRI contrast agents for diagnostic purpose has been demonstrated in tumor bearing mouse models [14].
Covalent nano delivery devices with novel configurations of modules with specific features can open a variety of new avenues: (1) Multiple peptide or chemotherapeutic containing modules in proximal sites could be designed for multivalent binding in the nano and pico concentration range thereby surpassing the binding affinities of antibodies; (2) Linear polymers are polar regarding their termini and could be specifically conjugated at one of them with a (fluorescent) reporter agent. Such devices that could contain a fluorescent terminus could be used in mechanistic studies [41]. Using chemically identical polymer fragments for conjugation at the terminal ends could results in polymer elongation with or without change in direction of polarity, thus opening a new direction of synthetic approaches.
Due to the multiplicity and spatially precise attachment points, covalent delivery devices offer a variety of underexplored possibilities not only in controlled drug delivery but also in controlled interconnections between receptors on single cells and those on different cells or on arrays of cells, especially on immune system cells.
4. CDS structural coherence
Covalent nano delivery systems are macromolecular entities displaying coherence of all residues. This “covalent coherence” distinguishes CDS from “association driven coherence” of non-covalent nano carriers (NCDS).
Covalent coherence is controlled by a precise composition and structure. Although a biological CDS preparation is not monodisperse, the principle of coherence is maintained. This does not hold for NCDS. Solvent precipitation based nanoparticles slowly dissolve, and in the case of micelles nanoparticles follow self-association ruled by the CMC [6]. In these cases, the polydispersity reflects different polymer contents, and the particle surface constantly reforms due to the particle-internal mobility of the constituents. The structural interface to the solvent may depend on internal mobility, composition and chemistry of the constituents, solvent composition, the temperature and the time of circulation in the blood. Then, a serious consequence is the uncontrolled release of drug from the delivering nanoparticle e.g., by diffusion. Diffusion depends on chemical composition and physics as well as on size, shape and chemistry of the drug. Release could be extreme for hydrophilic and low molecular chemotherapeutics and could give rise to toxic side effects especially during prolonged treatment. Attempts to minimize the diffusion have involved crosslinking between the nano capsule forming components. However, diffusion can remain substantial especially during long-term circulation of nanoparticle. A typical example for uncontrolled in vitro release into plasma from nanoparticles manufactured from poly(D,L-lactide-co-glycolide) (PLGA)-poly(ethylene glycol)(PEG) has been reported for docetaxel (DTX) indicating a 50% release over circulation time [42].
5. Examples of Covalent Nanodelivery Systems based on polymer platform
5.1 Poly(β-l-malic acid)
Polymalic acid of high molecular weight was discovered by its ability to mimic nucleic acids and competitively inhibit DNA polymerase α of Physarum polycephalum, and, to a lesser extent, the activities of DNA polymerases from other organisms [43, 44] (Fig. 3). Later on, poly(β-l-malic acid) (PMLA) was shown to have a functionality as a highly suitable platform for successful covalent delivery system due to an easy chemical substitution chemistry, biodegradability, absence of toxicity, and absence of immunogenicity. In preclinical studies, it was proven as an optimal platform to deliver chemotherapeutics such as doxorubicin and temozolomide, as well as antisense oligonucleotides of the Morpholino type to solid human breast cancer, human brain tumor, and metastases [11-23, 28, 39, 40]. The maximum tolerated dose of poly (β-l-malic acid) is 1.0 g/Kg [19]. Polymalic acid is available by synthetic chemistry in its D,L racemic form and in structural variants involving the carboxylic group in α- or β-position of the malic acid building unit and in unbranched/branched variants (summarized in [45]). Nature-made PMLA is an unbranched polymer of the β-L-isomer [11, 43, 46, 47]. Molecular weights are 5-10 kDa when isolated from Aureobasidium and other fungal strains [46, 48, 49] and 30-300 kDa when isolated from the plasmodium, the vegetative cell form of Physarum polycephalum, and from other members of the myxomycetes clad [46]. Polymalic acid has pendant carboxylic acid groups, which are ionized at physiological pH and render the polymer highly soluble [45]. Approximately 50% of the carboxylate groups may be derivatized with leucine ethyl ester or tri-Leucine without notably affecting the solubility in serum [50]. Polymalic acid-based drug delivery systems called “Polycefins” [11], have been synthesized with highly purified pharmaceutical quality PMLA from the Physarum polycephalum isolate [13-15, 17-22, 39, 40] (Fig. 1A), whereas synthetic PMLA was also used for the synthesis of other conjugates [51]. Intravenously injected PMLA-based nanodrugs are delivered through the tumor vascular endothelium by conjugation to antibodies that recognize TfR overexpressed in tumor capillary system [11-14, 17-22, 39, 40]. Receptor binding is followed by endothelial transcytosis through BBB into brain tumors and subsequently into tumor cells receptor mediated endocytosis. Tumor cell uptake is targeted by antibodies against TfR, EGFR and HER2 in the case of glioblastoma and brain metastases of human triple negative breast tumor, and metastases of HER2-positive human breast cancer, as well as other metastatic tumors in the brain [12, 14]. The highly specific recognition and delivery to brain was demonstrated for animals containing simultaneously different brain tumors by applying MRI contrast agents equipped with the tumor specific targeting antibodies and which were otherwise the same as the nanodrugs used for treatment [14]. The successfully delivered antisense oligonucleotides specifically blocked mRNA synthesis of overexpressed HER2 and EGFR/EGFRvIII in the tumor cells achieving significant prolongation of tumor bearing animal survival [11-14, 17-22, 39, 40].
Figure 3.
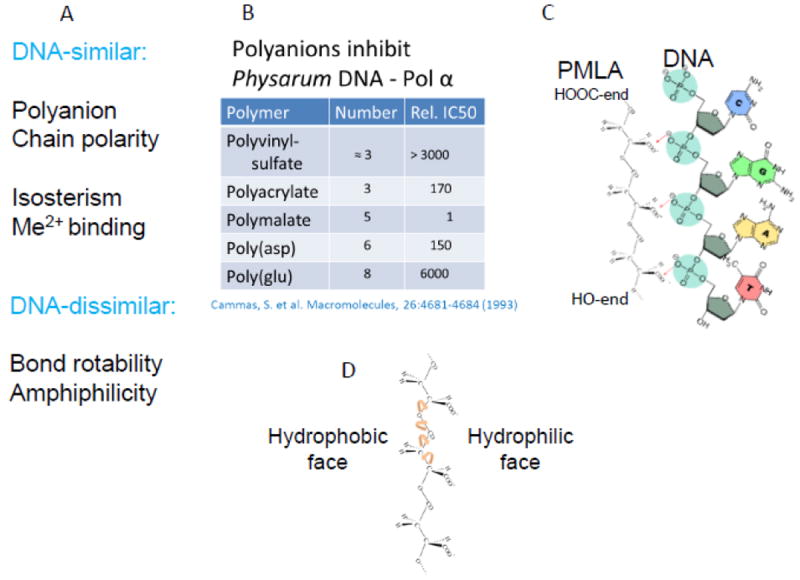
Diversity of macromolecular delivery systems distinguished by number, structures and functions of modules. Summary of structural and functional properties of PMLA from Physarum polycephalum and comparison with properties of nucleic acids: A, properties of PMLA and nucleic acids as similar and dissimilar. B, competitive inhibition of Physarum DNA polymerase-α comparing PMLA and other polyanions. The degree of inhibition correlates with the distance between negative charges. C, comparison of charge distances in DNA and PMLA. D, evidence of PMLA amphiphilic structure revealed by positioning of hydrophobic aliphatic groups and hydrophilic carboxylates.
5.2 Poly(L-amino acid)s
Anionic polymers structurally related to PMLA are homo polymers of aspartic (PAA) and glutamic (PGA) acid. Peptide bonds unlike ester bonds typically impose stiffness of the polymer backbone in contrast with high rotational flexibility of the ester backbone in polymalic acid. This restricts spatial orientations of covalently attached bulky groups such as proteins or oligonucleotides, and impairs intra and inter polymer interactions between intrinsic and extrinsic ligands.
The pendant carboxylic groups of these homopolymers are fully ionized at neutral pH giving rise to polyanions with slightly lower charge densities as for PMLA. Similarly, these polymers can occur in two structurally linear variants, one involving the carboxyl group in α-position (e.g., α-PGA), and the other one involving the carboxyl group in γ-position (γ-PGA) next to the amino group. Branched variants are possible when both types are verified in a single polymer molecule. α-PGA and γ-PGA are naturally synthesized. Low molecular weight α-PGA has been obtained by recombinant technology; however, the production is problematic, whereas chemical synthesis by nucleophile-initiated polymerization of the γ-protected N-carboxyanhydride of l-glutamic acid is feasible [52].
In comparison with PMLA, the peptide bonds of polyamino acids are relatively stable against spontaneous hydrolysis at neutral and moderately acidic pH. In human, α-PGA is systemically degraded by specific peptidases. Efficient degradation of γ-PGA after injection into humans is disputed, and it may not be cleaved by specific peptidases. When tested on cell culture level, maximum tolerated concentrations varied between 10 mg/kg and 100 mg/kg. At a dose of 1 mg/kg injected into mice intravenously, γ-PGA was well tolerated. The γ-PGA amide bonds are cleaved by extracellular peptidases of Bacillus subtilis and of other Bacillus strains [52-54]. Although mammals lack appropriate peptidases, γ-PGA was found to be edible. Efficient γ-PGA uptake into blood from the intestines or clearance through feces remain to be conclusively demonstrated. Cleavage by stomach hydrochloric acid and by the intestinal microbes [55] can occur during food digestion and may involve specific γ-glutamyl transferases [56] in mammals [57].
5.3 Poly(α-l-glutamic acid)
Poly(α-l-glutamic acid) (α-PGA) has a respectable history as a covalent drug delivery system. α-PGA platforms have been reported for the delivery of paclitaxel (TXL), camptothecin, and of other chemotherapeutics [58-60]. “Passive” delivery by the Enhanced Permeability and Retention (EPR) effect (tumor-compromised enhanced permeability of vasculature and lack of lymphatic drainage) has been highly effective in therapy with paclitaxel and camptothecin [58-60] without the need to involve specific receptors for active tumor targeting. The high tumor specificity has been suggested to depend on M2 macrophage uptake of α-PGA nano drugs before infiltrating tumors (reviewed in [58]). The chemotherapeutics were conjugated forming pH-labile responsive ester bonds with the γ-COOH of glutamic acid units [58, 59]. Release catalyzed through peptide bond cleavage by lysosomal cathepsin B ([61] and reviewed in [58]) has been considered as another pathway of paclitaxel activation and exit from endosomal compartments into the cytoplasm. Active targeting of TXL- α-PGA conjugate was achieved using cyclic RGD, c(RGDfK), or the dimer glutamate-c(RGDfK)2 targeting modules which specifically bind αvβ3 integrin receptor overexpressed on tumor endothelial and epithelial cells. Because the integrin targeted TXL- α-PGA conjugates have hydrodynamic diameters of 7-8 nm. They can be considered as “mini nano drugs” and reminiscent of the PMLA-based mini nano drug [23] but distinguished from most manufactured anti-tumor nano drugs of sizes 30-200 nm. Mini nano drugs are considered as providing improved anti-tumor activity through high diffusion rate and permeability. Delivery of paclitaxel, camptothecin, doxorubicin and other anti-tumor chemotherapeutics by conjugation with α-PGA and their use in clinical trials Phase I-III has been reviewed [58]. Paclitaxel α-PGA conjugate (Polyglumex) has entered clinical trials as radiation sensitizer in temozolomide radiation therapy of glioblastoma [62].
Polyglutamic acid is successfully used as conjugation platform for covalent attachment of gadolinium DTPA- and DO3A-complexes and of NIR813 fluorescent dye functioning in single and dual MRI combined fluorescence imaging (reviewed in [58]). As suggested for treatment, high uptake efficacy by certain tumors was noted, which supported the M2 macrophage uptake mechanism in favor of α-PGA specificity to target tumors. In the study of delivery efficacy, it was noticed that overly sustained blood circulation of α-PGA gadolinium contrast agents raised safety concerns. To accelerate clearance, α-PGA-cystamine-Gd(III)-DOTA conjugate was introduced, which had a short residing time [63]. However, because of the strong EPR effect, α-PGA-gadolinium contrast agents might not be suitable for tumor type diagnosis by clinical MRI.
5.4 Poly(γ-(DL)-glutamic acid)
γ-PGA is synthesized by a variety of microorganisms including cultured Bacillus subtilis (reviewed in [53, 54, 64]. The capsules of virulent strains of Bacillus anthracis contain solely γ-D-PGA [65] protecting the bacterial cells against phage infections and also preventing antibody recognition and uptake by macrophages [64, 66]. Naturally occurring γ-D-PGA is nontoxic, however, in combination with two other secreted factors the polymer becomes the dangerous Bacillus anthracis exotoxin [67, 68]. Although the B. anthrax capsule is not immunogenic, the poly(D-γ-glutamic acid) component or fragments could become immunogenic when conjugated to peptides or proteins. The highest levels of IgG anti-γ-D-PGA were elicited by decamers of γ-D-PGA at 10–20 mol per unspecified protein bound to the N- or C-terminal end [69]. Given these data, immunogenicity of γ-D-PGA oligomers or polymers could be of concern if any peptides and protein were bound to γ-D-PGA-nano conjugates for drug delivery.
In contrast to ribosomal synthesis of poly(α-l-glutamic acid), poly(γ-l(d)glutamic acid) is synthesized by a membrane-bound protein complex in bacteria [52, 64]. Polymers of high molecular weight ~105 - 8 × 106 Da can be isolated. Natural γ-PGA contains a mixture of l-glutamic acid and d-glutamic acid [64]. d-glutamic acid from B. anthracis and similar γ-(DL)PGA from other microorganisms take a helical conformation when in the unionized form, and a varying random coil conformation with increased ionization [53, 64, 70]. In contrast, several conformations are reported for α-PGA including α-helix, β-sheet and random coil at varying pH and salt concentrations [53, 64]. Unlike α-PGA, the γ-PGA backbone characteristically contains periodically repeated hydrophobic - (CH2)2 - alkyl segments that provoke hydrophobic interaction and aggregation, especially when pendant carboxylic groups of the polymer chain are neutralized.
γ-PGA shares many of the pharmacological properties of α-PGA, such as unusually effective passive (EPR) tumor targeting and long blood circulation after intravenous administration. Although γ-PGA has multiple pendant carboxylates for conjugation of chemotherapeutic and oligomeric nucleic acid drugs, it has not been used much for drug delivery in contrast to the multiple applications of PMLA, which also has a multiplicity of chemically reactive pendant carboxylic acid groups (see above). One of the reasons is that the hydrophobic nature of γ-PGA renders nano conjugates less water soluble with a high tendency for self-aggregation. Anti-breast tumor activity has been demonstrated for cis-platinum(II) compounds coordinated with inter chain and intra chain γ-PGA carboxyl groups [71]. Both PGA α- and γ-isomers may be applied for nucleic acid delivery because of their stabilization of nucleic acid complexes with polycations through hydrophobic interactions, or as nucleic acid binding conjugates with cationic ligands. The PGA is supposed to facilitate uptake and reduce cytotoxicity [72, 73]; however, nucleic acid delivering nanoparticles on the PGA basis that could target tissue or cells have not been forwarded to clinics. The reason for this could be the difficulty of chemical ligation of PGA and nucleic acids in overcoming the electrostatic repulsion between the negatively charged molecules. If this is the problem, it could be overcome by conjugating of PGA with neutral polynucleotide versions such as phosphodiamidate morpholino oligomers as demonstrated for conjugates with PMLA.
5.5 Poly(aspartic acid)
Similarly to poly(malic acid) and poly(glutamic acid), poly(aspartic acid) exists in D,L stereoisomers, structural α- and β- isomers and branched α,β variants. Synthesis and biodegradability of PAA have been reviewed [74]. Structurally, poly(α,β-(D,L)-aspartic acid) and poly(α,β-(D,L)-malic acid) are similar except the replacement of the amido group in PAA by the ester group in PMLA. Remarkably, the spatial distances of polymer pendant carboxylates are similar, which was recognized in experiments measuring the inhibition of DNA polymerases by polyanions of variable structures [44]. In this study, a similarity of the distance between the negatively charged carboxylates in PMLA and PAA with phosphate groups in nucleic acids has been noted. In another study where the degree of membrane destabilization by copolymers of poly(β-l-malic acid), poly(α-aspartic acid), poly(α-glutamic acid) and polyacrylic acid was measured, it was found that poly(β-l-malic acid) and poly(α-aspartic acid) had the highest membrane leakage activities [50]. These observations could inspire the designs of new PAA-based nano conjugates for drug delivery. To date, the following interesting results have been reported. (1) Hydrogels for drug delivery: an injectable hydrogel was obtained by hydrazone formation of aldehyde-modified PAA, which released cargo in response to late endosome and lysosomal pH [75]. In another version, poly(ethylene glycol) monomethyl ether (mPEG) and doxorubicin were conjugated onto polyasparihyazide (PAHy), prepared by hydrazinolysis of polysuccinimide, and formed effective anti-tumor nanoparticles of approximately 200 nm size suitable for pH-responsive delivery of doxorubicin [76]. (2) Poly(aspartic acid) segments have been shown to bind to hydroxyapatite (HA) and could be applied for the targeting of nanoparticles to bone tissue [77]. (3) Because of its biocompatibility including biodegradability and negligible immunogenicity, the potency of PAA to form different covalent combinations with polycations and cyclodextrin to form nucleic acid complexes for gene delivery have been recognized and developed into biodegradable nucleic acid delivery assemblies [78, 79]. In these studies, PAA was employed as the platform for covalent attachment of several kinds of nucleic acid interactive molecules, such as ethylamine derivatives to form polycation-like structures, benzyl alcohol esterification of pendant carboxyl groups to introduce hydrophobicity, and cyclodextrin for engaging host-guest nucleic acid interactions. Despite the structural similarity with PMLA, macromolecular (all covalent) conjugates of PAA have not been used as delivery vehicles. In contrast, the manufacture of micelles or of composites in combinations with nucleic acids and chemotherapeutics is preferred, which would allow stabilization of nucleic acid delivery vehicles through the addition of electrostatic interactions between polyanionic polyaspartate, various polycations, and hydrophobic polymers [80, 81].
5.6 Polyacrylic acid and its derivative N-(2-hydroxypropyl)methacrylamide copolymer (HPMA)
Polyacrylic acid (PAA) as a pH-responsive polymer, poly(propylacrylic acid) (PPAA) as a pH-responsive membrane destabilizing polymer [82], and especially N-(2-hydroxypropyl)methacryl amide copolymer (Fig. 1) combining pH responsivity, membrane destabilization activity, and ability for covalent drug delivery [83], are a group of polymers evolving from the favorable polymerization chemistry of acrylic acid. HPMA was introduced by the Kopecek laboratory in Prague during the early 1970s after appreciating its favorable relationship between hydrophilicity and biocompatibility, in particular the stable chemistry of the N-substituted methacrylamide group [84]. The PAA is not biodegradable due to the lack of enzymes that could cleave the C-C bonds of the polymer backbone. Moreover, because of their high molecular weights, HPMA and conjugates lack renal clearance. Longevity of such conjugates could be harmful with a risk of storage diseases. By interspacing short PHMA copolymer stretches permissive to renal clearance with peptides that are substrates of peptidolytic enzymes, such as papain or lysosomal cathepsin B, HPMA copolymers have been redesigned to be “biodegradable” [85-87]. HPMA copolymer has been explored into various applications to covalently attach or deliver a broad variety of chemotherapeutics, saccharides, peptides, and antibodies for specific targeting of cells and tissues [88], including chemotherapeutics that were membrane permeable but poorly soluble in aqueous media. Such chemotherapeutics are conjugated via a cathepsin B cleavable tetrapeptide linker GFLG or by a pH-sensitive hydrazone linker [89]. These drugs could escape the lysosomal compartment by their autonomous membrane permeation activity when cleaved from the HPMA conjugate. Several of these HPMA drug conjugates have entered clinical trials [90].
Among the variety of conjugates, an interesting variant was constructed, which colligated single cell surface CD20 antigens of human non-Hodgkin’s lymphoma (NHL) Raji B cells by binding to multivalent HPMA copolymer-Fab′ conjugates resulting in the induction of apoptosis [25]. In a further study, crosslinking was initiated by annealing of complementary morpholino antisense oligonucleotides (AON) located on independent Fab’ conjugates (a novel drug-free nanotherapeutic treatment of B-cell malignancies) [91]. However, this approach of complementary annealing of morpholino AON on HPMA to trigger cell death was not translated into using AON to block mRNA unlike the AON-PMLA conjugates that demonstrated impressive antitumor activity [13, 14, 17-22, 39, 40]. In an alternative approach, HPMA carrying triplex forming oligonucleotides (TFOs) were used for treating liver fibrosis by inhibiting the transcription of α1(I) collagen gene [92]. The 25-mer fully phosphorothioated oligomer TFO-3’-NH2 was conjugated with nitrophenylester-activated Poly (HPMA-co-GFLG-ONP) at the C-terminus of the cathepsin B specific peptide [93]. The α-D-mannopyranoside containing nano conjugate was targeted to hepatic stellate cells (HSCs). Type I collagen gene expression was significantly inhibited when HSC-T6 cells were transfected with this conjugate following tail vein injection into rats. In another approach, HPMA copolymer contained N-(2-(2-pyridyldithio)ethyl)methacryl amide. In a thioldisulfide exchange reaction miRNA was covalently attached to PHMA via the formed reducible disulfide linker [94, 95]. Whereas the delivery of covalent HPMA nucleic acid conjugates parallels the delivery of morpholino AON in PMLA-based nano drugs, the more classical approach of the delivery of nucleic acids involving noncovalent attachment through electrostatic anchorage has been reported on a large scale [96-98]. The HPMA conjugated cationic stretches were oligolysine (12-mers or lower) and oligolysine stretches attached to oligonucleotides [96-103]. These constructs bind and compact DNA with variable length tailored to the size of HPMA-oligolysine copolymers [102]. To reduce toxicity, the constructs contained either cathepsin-cleavable oligopeptides or reducible disulfide linkers that were cleaved by lysosomal peptidase or reductively in cytoplasm. To manage the escape from endosomes, the constructs carried histidine residues to induce proton sponge osmotic rupture of the endosomal membrane or contained lipophilic compounds for lytic membrane destabilization [103]. Novel syntheses based on covalent attachments to HPMA offer great advantage over the application of other polyanions due to their easy and controllable reversible-addition fragmentation chain-transfer (RAFT) polymerization, which has been well advanced [7, 100, 103, 104]. Amino acids and peptide derivatives containing a terminal vinyl group are readily copolymerized into strategically opportune positions in these constructs [60]. Beside the development of excellent nano drugs, HPMA copolymers have been designed for in vivo tumor tracking and imaging using various imaging agents [105-107] or radioactive isotopes [87], a recent example being using the IR-783 dye for uMUC-1-targeted near infrared colonoscopy[107].
6. Coherence of polymeric nano drug design
6.1 Hydrodynamic shape and size
The shape of a CDS in solution, whether rod, coil or sphere, depends on chemical design. Elongated and rod-like CDS have been built by chemical conjugation of modules with sites of functional groups distributed along unbranched polymers such as N-(2-hydroxypropyl)-methacrylamide copolymer (HPMA) or PMLA, PGA, dextran, and chitosan. The effect of substitution on CDS shape has hitherto not received much attention. Polymalic acid-based CDS will be considered here as an example.
The biologically produced PMLA is a polyester between the hydroxyl group and the β-carboxylic group of L-malic acid leaving the α-carboxylic group pendant position and reactive for substitutions activity [13, 14, 17-22, 39, 40]. Activated as N-hydroxy succinimide (NHS) ester the carboxyl group reacts with free amino groups as part of simple linkers, peptides and proteins. Linkers containing mercapto groups serve to form thioethers and disulfide substitutions including morpholino AON. Nearest neighbor effects on the substitution by hydrophobic residues such as leucine ethyl ester result in hydrophobic clusters that are responsible for membrane destabilization and permeation [28]. Studies of unsubstituted PMLA in distilled water by cryo-transmission electron microscopy indicated open, coiled forms and double stranded structures between non-ionized stretches of the polymer (unpublished). Calculation based on the chemical structure indicates the length of the polymer strand (50 kDa) of approximately 80 nm that is reconcilable with the length of the observed double stranded structures. Consistent with coiling is the hydrodynamic diameter of 4-7 nm measured by dynamic light scattering for PMLA of 50-100 kDa. After various degrees of substitutions with PEG (5%, percent denotes the fraction of malyl units conjugated with the ligand), AONs (1-4%), antibodies (1-5 molecules per polymer), and peptides as leucine ethyl ester or trileucine (40%), the nanoconjugates remain highly soluble and are biologically active. Hydrodynamic diameter measured as a function of molecular weight increased in a biphasic fashion with a break at the transition from free to substituted PMLA [19]. This biphasic behavior is interpreted by the assumption of a polymer coil in phosphate-buffered saline (PBS, pH 7.4) with the size indicated at the transition. When the polymer is conjugated with increasing number of modules, the particle size expands indicated by the second phase of hydrodynamic diameter as a function of molecular weight [19].
6.2 High axial ratio in support of CDS environmental interactions and directional mobility
CDS based on unbranched polymer platforms display their targeting modules and cargo (drug) along the platform polymer. By the interaction with solvents and functional ligands the coil can unfold to an extended configuration. In this state the nano conjugate has a high length to diameter ratio with a geometry that favors a high number of environmental contacts, e.g., of bound ligands with receptors, access to reagents and enzymes for linker cleavage and to receptors in control of degradation and clearance. A high axial ratio supports directionality of movement in the direction of the long axis, and also suggests that thermally induced bending motions propel the polymer through densely packed environment. The successful delivery of AON by a “streamlined” PMLA-based mini nanodrug is in agreement with the proposed mechanism [23].
6.3 Coherence and biodegradability
Another result of coherence is propensity for biodegradation. Biodegradation into fragments is usually carried out by enzymes starting cleavage from one of the termini (exo enzymes) or after recognizing signature sequences located within a polymer (endo enzymes). Biologically derived CDS are biodegradable. Well known examples are proteins, nucleic acids, carbohydrates. Whereas there is no problem with recognizing endolytic cleavage sites in CDS, such sites buried in micelles or solvent precipitation/evaporation manufactured particles are accessible only after nanoparticle dissolution. Biodegradable polymeric platforms are highly desirable in drug delivery systems precluding toxic storage diseases, immunological responses or toxic side effects due to unresolved degradation and systemic clearance. In case of HPMA, which contains exclusively non-biodegradable aliphatic C-C interchain bonds, an elegant solution has been invented by periodically placing short non-immunogenic peptides into HPMA using click chemistry [85, 86]. The short HPMA fragments remaining after cleavage by cathepsin B can be exported from the organism through renal clearance. Targeted peptidolytic cleavage has found application in tumor MR imaging and tumor targeting by cleavage at peptidase signature sites [108, 109].
Nature-made polymers are usually biodegradable in mammals. Examples used in nanomedicine are PMLA, α-PGA, γ-PGA, and chitosan. Unsubstituted PMLA is also degraded by spontaneous hydrolytic ester cleavage via oligomeric intermediates to L-malic acid (half-life related to molecular weight is10 hrs at pH 7.4, 37°C [110, 111]). The rate of spontaneous hydrolytic cleavage is considerably decreased after substitution at the pendant α-carboxylic groups of the polymer. A polymalate depolymerizing enzyme is secreted by Physarum polycephalum [112, 113], and enzymes have been identified in bacteria [114], fungi [46], and as serum lipases in human [115]. The malic acid generating activity of the Physarum PMLA hydrolase is stalled at branching points and pendant substitutions. Poly(L-glutamic acid) is cleaved by cathepsin B highly expressed in tumor lysosomes, but with no measurable activity in serum, allowing long-lived covalent conjugates of paclitaxel with exclusive activation of paclitaxel in tumor lysosomes [116]. Enzymes that cleave y-glutamyl linkages have been identified and characterized in mammals [117, 118], fungi [119], bacteria, and phages[120, 121]. Chitosan is degraded by chitosanases (EC 3.2.1.132), and products have been well characterized [122].
During treatment, injected CDS is spontaneously or enzymatically cleaved exolytically from one of the polymer ends by depolymerases yielding free polymer building units, or in most other cases endolytically forming fragments that may contain both targeting and pharmacologically active modules which are still prospective nano drugs. Fragments devoid of prodrugs may target receptors, acting as competitive inhibitors of biological or regulatory pathway(s). The kind of fragments containing only prodrugs will not develop side effects due to lack of cell uptake and drug activation. With time, degradation continues towards clearance through kidneys, liver, spleen and macrophages. In contrast, cleavage sites of NCDS are often buried inside the nanoparticles. But without cleavage, active drugs can be released as a result of spontaneous carrier dissolution due to change in ionic strength, pH, temperature, or by detergents and other solubilizing agents. Free drugs, acidification and carrier degradation products can give rise to systemic toxicity.
6.4 Examples of modules with specific functions
The central module of CDS is the polymer platform, which outlines the particle shape and dynamic flexibility. The platform carries all modules with functions to achieve optimal delivery (Fig. 1B): receptor targeting, cleavage from the delivery system and destabilizing of membranes. Linkers combine functional modules with the platform. They can be responsive to enzyme activity, pH or redox reactions outside or inside recipient cells. Other modules such as protective PEG, fluorescent dyes and MRI contrast agents (e.g., Gadolinium-DOTA) can be attached to the platform with the purpose of inhibiting enzymatic degradation, systemic clearance by macrophages, liver, spleen, or as means for local tracing of CDS, and MRI diagnostics.
6.5 Platforms, typical chemical/structural outfit
Covalent nano delivery systems are typically unbranched linear polymers, and branched typically spherical polymers (e.g., dendrimers). The polymer is built from repeating units, which in the case of linear CDS can be designed to carry functional groups along the polymer and in the case of dendrimers, functional groups in terminal positions of the branches. Examples of repeating units are amino acids such as glutamic acid and aspartic acid, or hydroxycarboxylic acids such as malic acid and citric acid, or saccharides such as glucose and a glucose amine. In principle, each unit can be chemically conjugated with specific functional ligands resulting in a plethora of CDS that cannot be matched by NCDS.
6.6 Platforms for Precision Molecular Therapy and Imaging
Using gene sequencing, molecular cancer markers can be identified within a short period of time after diagnosis. Using the antisense technology, gene expression can be efficiently silenced by clinical treatment with antisense modalities such as siRNA or AON. When using CDS with multiple attachments, one or several genes can be simultaneously targeted by the same platform delivering at the same time different AON or other drugs. The delivery and simultaneous multiple knock down has been demonstrated with a set of AON against EGFR, HER2, CK2, laminin α4 and laminin β1 genes [13, 14, 17-22, 39, 40].
7. Prodrugs
When drugs are covalently bound to CDS, they become inactive, i.e. “prodrugs”. Conjugation requires a chemical “handle” on the drug, which is removed from the drug during the release reaction or remains part of the cleavage product without interference with its pharmaceutical action. An example is the 3’-thiopropylamido group of AON that does not adversely affect annealing with the target mRNA. Examples of complete chemotherapeutic drug reconstitution are pH-responsive hydrolysis of 2’-carboxylic acid esters of paclitaxel and docetaxel or of hydrazone derivatives of doxorubicin, summarized in [35]. Examples of incomplete removal of linker components include the cleavage of linker peptides by lysosomal cathepsin B in the case of doxorubicin [38, 123] or reductive cleavage of disulfide linker by the cytoplasmic glutathione in the case of AON [11, 12]. Cleavable linkers have been recently reviewed [35].
8. Nucleic acids delivery by covalent nanodrugs
Nucleic acids have been permanently conjugated through peptides or reversibly through cleavable disulfide linkers. Two principle antisense variants are available: nucleic acids such as siRNA carrying negatively charged phosphodiester bonds, and neutrally charged synthetic variants replacing natural ribose phosphate backbone by neutrally charge amido phosphate morpholino residues. Whereas both variants anneal perfectly with template nucleic acids, the neutral version is invulnerable to nucleolytic activity and is synthetically easy to conjugate with polyanions such as PMLA. In addition, the morpholino variant behaves invariantly to most peptides and proteins when assembled on delivery platforms [33, 124]. Routinely used 25-mer morpholino AON are below renal threshold and have been reported as nontoxic. Because of morpholino AON, targeted nanomedicines with PMLA conjugates have been very successful in inhibiting tumor synthesis of molecular cancer markers such as laminin α4 and β1 chains, EGFR/EGFRvIII, and HER2 [11-14, 17-22, 39, 40].
9. Covalent Nanodelivery Systems for Imaging and treatment of Brain Tumors
9.1 Nanomedicine Overview
Based on their structural properties, nanodrugs can be divided into the following categories: liposomes, dendrimers, polymer conjugates, micelles, carbon nanotubes, gold nanoparticles and magnetic nanoparticles [125]. Due to their unparalleled payload capacity, versatile targeting, and multi-functional properties, nanomedicines can be equipped with almost all types of moieties such as chemotherapy, radioactive agents, nucleotide oligomers, antibodies, proteins or peptides as well as imaging agents for therapeutic or diagnostic purposes. More importantly, nanodrugs can significantly increase the drug bioavailability while decreasing its side effects via specific biological barrier penetration mechanisms as well as drug targeting mechanisms [126, 127]. Here, we briefly summarize the major nanostructures listed in Table 3 with an emphasis on their application to the brain diseases. A human brain is structurally unique and complex relative to other organs in the existence of multiple biological barriers both inside and outside the brain tissue [128]. Therefore, drug delivery to the brain has always been the most challenging task in drug development. In the context of brain delivery, each category of the nanostructure has its own strength and limitation as a drug carrier, and these structural features impose both challenges and opportunities in their application in drug development, especially in CNS diseases. Covalent nano conjugates with their chemical stability in plasma and superior tissue targetability are attractive drug candidates for the penetration of multiple biobarriers, delivery of the active moieties into the brain cells, and release without intermediate neurotoxic subproducts.
Table 3.
Major structures of nanodrugs: advantages and disadvantages for brain delivery
| Nanostructure | Advantages for Brain | Disadvantages for Brain |
|---|---|---|
| Comparison with nano covalent conjugates | ||
Liposome
|
|
|
Dendrimers
|
||
Polymer-conjugates
|
|
|
Micelles
|
|
|
Carbon Nanotubes
|
|
|
| Gold Nanoparticles |
|
|
Magnetic Nanoparticles
|
|
|
9.2 Brain Barriers: Blood-Brain-Barrier
A human brain is highly vascularized but is also selectively isolated from other parts of the body by the blood-brain-barrier (BBB). Described as the “bottleneck in brain drug development,” the BBB consists of brain endothelium held together by tight junctions that forms a barrier around the brain with highly selective permeability [135]. In addition to the endothelial cells, other participating cells lend even more regulation to this barrier, including astrocytes, pericytes, microglia, and the vascular basement membrane [132]. For example, astrocytes surround the BBB with their end-feet, secreting factors that upregulate tight junction proteins and express key enzymes and transporters that contribute to the proper functioning of the barrier [132]. Microglial cells are an important part of brain’s immune defense system, and they are promptly activated and able to swallow pathogens and present antigens for T cell activation when the foreign entities pass through BBB and invade brain environment [136]. Pericytes have been shown to play a critical role in regulating BBB permeability as well as the integration of other cell types [137].
Importantly, the BBB functions to maintain a constant environment inside the brain by regulating cerebral extracellular fluid and protecting the brain from environmental noise; but when pharmacological treatment is needed, the same barrier prevents the extravasation of most drugs useful for treatment of CNS diseases such as brain tumors [14, 134, 138]. The BBB excludes most polar molecules, but certain nutrients and ions can gain access through highly regulated receptors [139]. Although the BBB has been observed to be altered at the tumor core (termed the “brain-tumor barrier”, or BTB) with somewhat disrupted selectivity (Fig. 4), it is intact adjacent to the BTB, allowing peripheral malignant cells to evade treatment [140]. Some possible mechanisms for BBB penetration include passive diffusion, active transport by transcytosis/endocytosis, or by inhibiting efflux pumps (Fig 5 [140]). Furthermore, other physiological obstacles limit progress in brain cancer treatment, as will be described later in this review. We are going to use the term “BBB” instead the “BTB” formally used in the literature when speaking about brain tumors. We would like to emphasize the fact that despite the known differences between normal and tumor endothelial/vascular systems, they still represent similar obstacles for the drug delivery and treatment of brain cancer.
Figure 4.
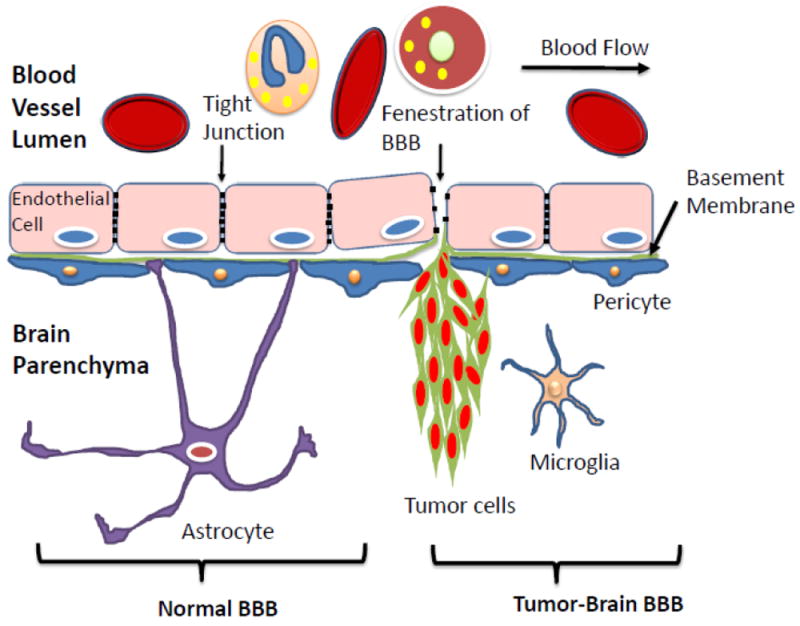
Blood-Brain-Barrier (BBB) architecture in normal and tumor brain.
Figure 5.
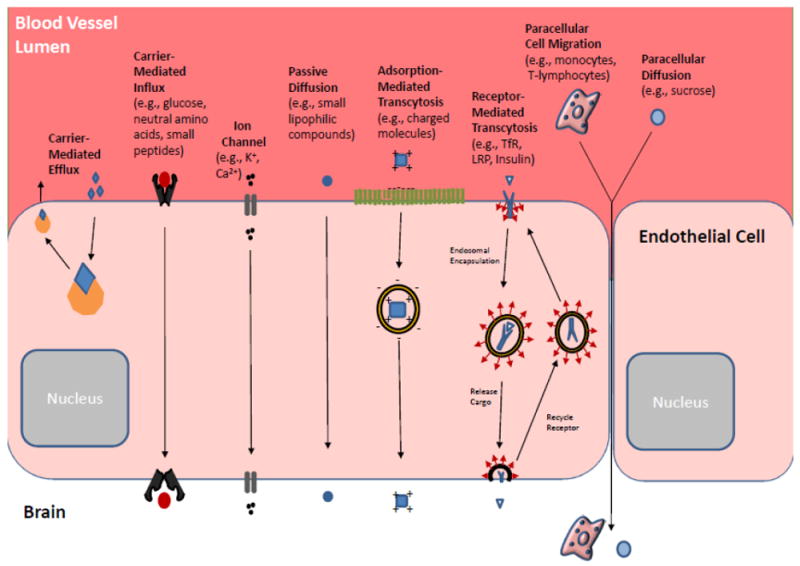
Molecular transport across normal BBB. In the healthy BBB, there are many mechanisms by which endogenous substances are able to migrate from blood to brain. In addition to simple diffusion, entry mechanisms involve channels, carriers, and receptors.
9.3 Brain Barriers: Blood-CSF Barrier (BCSFB)
The Blood-CSF Barrier (BCSFB) is formed by the epithelial cells of the choroid plexi and other circumventricular organs (CVO) [141]. Different from BBB, the capillary cells of the BCSFB do not have tight junctions, and they are fenestrated allowing for free permeation of various substances [142]. Circumscribing these capillaries are polarized epithelial cells with tight junctions, preventing free diffusion between blood and surrounding CSF. The choroid plexus produces CSF and regulates the movement of solutes between blood and CSF [141, 143]. Reservoirs of CSF exist in the subarachnoid space and the ventricles, exchanging fluid and solutes with surrounding tissue [143]. CSF cycles bidirectionally through brain parenchyma by mixing with the interstitial fluid and then reentering the reservoirs mentioned above [143]. The exchange of substances through this cycling is essential for maintaining homeostasis in the brain, and the integral membrane proteins aquaporins are critical for this movement of fluid. Three pathways have been outlined for drug delivery involving CSF, including blood borne (drug moves from CSF to blood and then to brain through BBB), diffusion (drug in CSF diffuses to brain through BCSFB), and convection (drug moves from CSF to brain through bulk flow of perivascular spaces), but these mechanisms have shown very limited effects with current methods [128]. Future research into the BCSFB may provide novel insight into delivery mechanisms. However, in exploring these mechanisms one must be aware that drug penetration into CSF cannot be used as a measure for BBB permeability, as they are completely distinct barriers with different characteristics. It is also essential to understand that drugs injected into the CSF are unlikely to reach deep brain parenchyma; novel methods of delivery are needed in order to exploit this pathway for therapeutic purposes [128].
9.4. Brain Cancers
Brain cancers, both primary and metastatic, are the leading cause of cancer-related deaths in the United States in patients below the age of 35 [144]. It is widely accepted that gliomas are among the most hostile tumors – with the most dangerous being Glioblastoma Multiforme (GBM) – and result in significant morbidity and mortality [138, 145, 146]. GBM is a Grade IV astrocytoma per WHO classification system for CNS tumors [147]. At the genetic level, GBM tumors can be divided into four molecular subtypes with distinct genetic signatures, termed Neuro, Proneuro, Mesenchymal and Classical subtypes [148]. Due to the nature of GBM, tumor recurrence is extremely common even with the most aggressive treatment regimens including surgery, radiotherapy, chemotherapy and even hemispherectomies [138, 144]. Physicians and scientists have attempted to tackle this aggressive cancer from all angles, employing innovative surgical and therapeutic techniques to curb the progression of this disease. However, improvement in the outcome of GBM patients has remained dismal in the past 25 years. Today, the standard of care in brain tumor treatment consists of cytoreductive surgery, followed by chemotherapy [140, 144, 149, 150]. Along with radiation, Temozolomide (TMZ) continues to be the first line chemotherapy since its first approval for GBM treatment by FDA. Whereas new treatments in other fields of medicine continue to improve remarkably, the little progress made in brain cancer treatment and diagnosis is due to the highly aggressive nature of brain tumors as well as to the inability of many therapeutic agents to overcome the biological barriers associated with the brain.
In 2016, a four-year initiative, National “Cancer Moonshot”, was launched with a $1 billion funding to jump start this program [151]. For reasons such as this, the near future holds great promise for innovation in the field of brain cancer treatment. This review will discuss the obstacles in brain cancer diagnosis and treatment, in addition to the potential application of nanomedicine. We will cover important considerations for the design of an ideal nanotherapeutic or imaging agent, and outline promising future directions of nanomedicine in the context of brain tumor diagnosis and treatment. Specific attention will be given to covalent nanomedicines (covalent nano conjugates) as stable in plasma and promising for the BBB delivery.
10 Application of Nanotechnology for Brain Cancer Imaging
When treating a brain tumor, the first step is its proper diagnosis, e.g., through imaging. Ideally, before a suitable course of treatment is prescribed, a physician needs to properly identify the grade, histological type, genomic markers, location, and origin of the tumor. Historically, tumor biopsies have been the major source of this information. However, unlike the situation in most organs, brain biopsies carry a certain danger for the patient with 6-12% complications, and are in some cases impossible to perform [Malone et al. (2015), World Neurosurgery]. For this reason, imaging, with its many different modalities, holds potential as a non-invasive technique to assess these parameters in the brain. Furthermore, imaging helps physicians determine whether surgical intervention is necessary [152].
With the rapid technological advancement, imaging has grown significantly from a once solely anatomical tool to the one that now assesses a range of different biological and morphological signals, making for a highly powerful diagnostic approach. Anatomy-based imaging of the past allowed for the evaluation of structural abnormality and tumor-related complications [153]. With the implementation of modern imaging techniques, it is now possible to read functional, hemodynamic, metabolic, cellular, and cytoarchitectural abnormalities [153, 154]. This continuing innovation now allows imaging to be used to diagnose and grade tumors, and to accurately monitor patient prognosis, all without physically breaching the brain.
Below, we will briefly cover the major modalities commonly used in brain imaging, which are summarized in Table 4.
Table 4.
Commonly used imaging technologies for brain cancer management
| Imaging Technology | Magnetic Resonance Imaging (MRI) | Computed Tomography (CT) | Positron Emission Tomography (PET) | Ultrasound (US) |
|---|---|---|---|---|
| Mechanism | Main diagnostic approach for brain cancers. MRI employs a strong magnetic field to force protons of water molecules in the body to align with the field, and the scanner can detect the changes in the direction and rotational axis of protons and the energy released. | CT uses X-ray to generate detailed scans of the areas in the body. | PET uses radiotracer to assess the functions of the tissue or organ, such as blood flow, oxygen or glucose consumption. | Ultrasound utilizes high frequency (> 20 kHz) sound waves to produce biological images and the image is produced based on the reflection of the waves off the body structures. |
| Strength | Fast, noninvasive, nonradioactive and good spatial resolution. | Fast (minutes ~ half an hour); noninvasive; images can be visualized in 2-D (slice) or 3-D fashion. | Often be used in combination with CT to provide accurate anatomic information. | Non-radioactive and quite versatile; for instance, focused ultrasound (FUS) has been adapted for BBB disruption for drug delivery, treatment of neurodegenerative and cerebrovascular diseases, and neurosurgical small volume tumor ablation [32, 141]. |
| Limitation | Contrast enhancing agent, such as gadolinium has high renal toxicity. | Radiation from X-ray; lack of anatomic information. | Radioactive tracers. | Limited spatial resolution. |
Nanotechnology offers many advantages in medical applications and has received tremendous focus in both the therapeutic and diagnostic settings. The goal has been to employ nanotechnology to create clearer images for more accurate diagnosis and treatment monitoring, while keeping non-specific toxicity to a minimum. The following sections will describe the impact nanotechnology has had on brain tumor imaging, and its applications in precision medicine.
Currently, many imaging modalities are used either alone or in combination to improve diagnostic capabilities. In nano imaging, the two main approaches are MRI and fluorescent imaging [156]. Nonetheless, other traditional imaging modalities such as CT, PET, and ultrasound are commonly engaged for the imaging of brain tumors.
10.1 Magnetic Resonance Imaging (MRI)
Currently, Gadolinium (Gd)-enhanced MRI is the leading imaging modality for brain tumor diagnosis, owing to Gd’s inherently large magnetic moment [157]. Although Gd is a highly toxic substance to humans, its chelated form is both safe and able to maintain its important paramagnetic properties when it is acting as a contrast-enhancing agent [158]. In the context of brain tumors, Gd-enhanced contrast increases the differentiation between tumor and brain tissue, allowing for visualization of a more defined tumor border [152]. However, there are limitations in traditional Gd-contrast agents: they have poor accumulation and retention within the brain. Furthermore, there are severe limitations in their imaging power: they are unable to determine the etiology of brain tumors; they are not able to distinguish between primary brain tumors and metastasized brain tumors; and they are unable to identify tiny early-phase metastatic lesions [159]. As a result, Gd-based nanoparticles have been formulated to achieve higher targeted Gd accumulation, longer retention within the brain, and stronger T1 contrast (due to higher accumulation), potentially allowing for the identification of tiny lesions [160]. Multiple Gd3+-based contrast nano formulations have been clinically accepted [161]. Recently, MR-guided focused ultrasound (MRgFUS), which is a non-invasive procedure that reversibly opens the BBB without damage to the surrounding neurons, has been employed for targeted drug delivery to the brain involving non-human primates. Results of a clinical trial designed to establish the feasibility, safety and preliminary efficacy of MRgFUS to open the BBB for the delivery of chemotherapeutic agents in brain tumors has been approved by Health Canada. Preliminary data have shown an increased Gd uptake in the sonicated area of the brain [162].
Recently, a landmark paper was published by Patil et al. in which the “MRI Virtual Biopsy” method was introduced [14]. This innovative method allows for the differential diagnosis of lung and breast brain metastatic tumors, the two most common primary cancer sites for brain metastasis. HER2 and EGFR antibodies were covalently conjugated to a CDS with PMLA backbone that also contained one or two imaging agents: chelated Gd for MR imaging, and/or Alexa Fluor 680 for optical imaging. This multi-modal imaging concept allowed for the confirmation of the MRI results by means of fluorescent imaging. The used nano imaging agents were able to preferentially accumulate in different tumors in the same brain tissue based on differences in molecular markers, allowing for non-invasive tumor typing helpful for subsequent therapy (Fig. 6).
Figure 6.
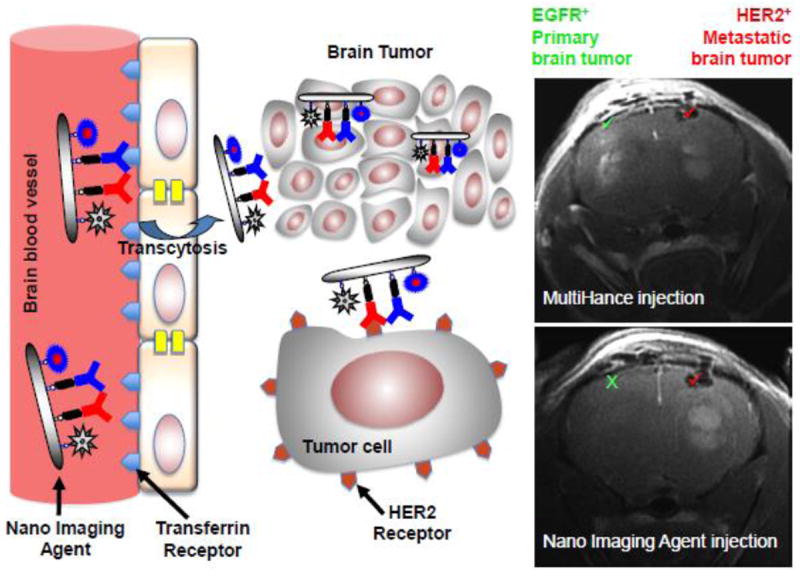
Schematic illustration of differential diagnosis of brain tumors by MRI using systemically administered Gd-conjugated polymeric nano imaging agents. Mice had different tumors grown in each hemisphere, an EGFR+ glioblastoma and HER2+ breast cancer. Using BBB-permeating and HER2-targeted PMLA-based nano imaging agent it is possible to noninvasively diagnose by MRI the HER2+ tumor (it is the only one visible), which can be subsequently treated with a similar nano agent (see Fig. 8). This is not possible with a standard MRI agent MultiHance that highlights both tumors. Reproduced from [14].
Though Gd is generally safe in its chelated form, Gd-induced nephrogenic systemic fibrosis (NSF) has been reported. It is believed that a small amount of Gd may de-chelate while in circulation inducing NSF [158]. Therefore, alternatives to Gd-based nano contrast agents have been investigated. The most common non-Gd-based MR contrast agents are based around iron-oxide. In 1978, Ohgushi et al. discovered that iron-oxide possessed powerful inherent contrast properties for T2 imaging [163]. As a matter of fact, iron-oxide-based nanoparticles were the first to be used for imaging ([164, 165]). In the context of brain tumor imaging, current clinical trials are focusing on Super-Paramagnetic Iron Oxide Nanoparticles (SPIONs) for the detection of microscopic metastatic lesions and enhancing the contrast of malignant brain tumors [164]. Today, multiple SPIONs are used in clinic: for example, Feridex/Endorem, and Resovist.
10.2 Fluorescent Imaging
Due to its non-invasive, highly sensitive, real-time imaging capabilities, optical imaging has become one of the most popular imaging modalities in nanomedicine. However, when applying it to the in vivo systems, problems such as autofluorescence and peripheral absorption arise [166]. These problems prevent deep-tissue imaging, as many biological entities act as fluorophores of particular wavelengths. Consequently, the poor penetration depth of many optical agents is a hindrance to their use. In response to the autofluorescence of endogenous biological structures, scientists have discovered that minimal interference occurs when exciting fluorophores with near-infrared (NIR) wavelengths [166]. Due to their potential cytotoxic effects, NIR-emitting dyes are still under investigation. They come in two major forms, but the most commonly employed in vivo forms are various synthetic fluorophores. A recent study by Kim et al. used glioma-bearing rats that were administered mesenchymal stem cells (MSCs) tagged with synthetic fluorophore-labeled nanoparticles. This group was the first to reveal that in vivo NIR imaging could be used to track the distribution of injected MSCs [167]. Importantly, no significant differences (e.g. cytotoxicity) were seen between labeled and non-labeled MSCs.
The second major type of NIR fluorophore is based on Quantum Dots (QDs). Due to their easily modifiable surfaces and high resistance to photobleaching, diverse QD fluorescent nanoparticles have been constructed. QDs usually contain elements from metal groups II, III, and V of the periodic table, as their electrons are loosely bound to the atom, allowing for their excitation and subsequent photon emission [168]. QDs are ideal agents for crossing the BBB, as they are small (<10 nm in diameter) and modifiable with homing moieties [166, 169]. Currently, QDs are still under investigation for their potential cytotoxic profiles. To minimize toxicity, QDs can be incorporated into nontoxic carriers such as dendrimers and liposomes [170, 171]. Despite their potential hazardous side effects, QDs hold a great promise for the future of brain cancer treatment. They may be very helpful in demarcating tumor boundaries for intraoperative detection, as fluorescent imaging is capable of highlighting clearly defined tumor borders [172, 173].
10.3 Computed Tomography (CT)
Advancements in nanotechnology have also facilitated improvements within the realm of CT. Due to its low cost and ability to quickly generate images, CT scans are commonly used today. However, even with the use of traditional iodine-based contrast agents, CT scans have severe drawbacks, including rapid renal clearance coupled with strong renal toxicity. Iodine-based contrast nano agents have been developed, improving both the retention and toxicity issues [166]. These contrast agents, which can be used for effective tumor detection, are based on high atomic number elements such as iodine, gold, and Gd [174, 175]. In particular, gold-based CT contrast nano agents have garnered attention recently due to their high X-ray absorption coefficient, easily modifiable surface, and biocompatibility [166]. Though nano-related CT imaging for brain tumor visualization is not explored well, some important findings were published recently. For example, Hainfeld et al. created a gold-based nanoparticle for brain tumor radiotherapy and CT imaging [176]. Due to high electron density of gold, these nanoparticles were visualized by high-resolution CT, which showed a 19:1 increased uptake between brain tumor tissue and normal tissue, respectively.
10.4 Positron Emission Tomography (PET)
In addition to MRI and CT, PET also serves as an important tool for imaging. Distinct from traditional MRI and CT, PET scans are functional, allowing for metabolic assessments on a molecular level. PET has multiple useful cancer applications, including important drug-delivery measures such as biodistribution and pharmacokinetics [177], defining the “actual” tumor volume [178], and differentiating between metabolically active and post-treatment necrotic tumor masses [179]. However, due to its lack of structural anatomical information, PET alone is not a preferred imaging technique for cancer diagnosis. Instead, PET coupled with CT or MRI is often used, as these dual-imaging techniques reveal both anatomical and functional molecular information. Though PET/CT is the traditional form of dual-modal imaging, PET/MRI has recently gained popularity in cancer imaging, as certain types of cancers (e.g., brain, head, neck) are better visualized by MRI than by CT [180]. For example, a study demonstrated that radioisotope-bearing nano micelles can be constructed and used for the treatment and imaging of rat GBMs. To assess certain parameters such as anatomy, accumulation, biodistribution, and local blood volume, PET/MRI dual-modal imaging was applied [181]. However, few studies have been published so far involving nano agents in PET-based imaging for brain tumors.
10.5 Ultrasound (US)
US scans are commonly employed in imaging because of their high safety, real-time imaging capabilities, and low cost. Currently, a drawback of US is its poor contrast resolution; even with the use of microbubble contrast agents, the resolution is not as high as with other imaging modalities [164]. Moreover, microbubbles are too large to readily extravasate into tumors, thus limiting the application of US for tumor imaging. Due to these limitations, perfluorocarbon-based nanodroplets have been developed, which are capable of entering tumors [164]. However, these nanodroplets have poorer contrast than the more traditional microbubbles, which is a major deterrent for use in tumor imaging [182]. Luke et al. recently reported “super-resolution ultrasound imaging” of a new generation of nano-sized contrast agents, laser-activated nanodroplets (LANDs), which seem to hold future promise for deep tissue molecular imaging [183]. Further investigations need to be conducted to explore the application of LANDs to brain tumor targeting.
Unlike other imaging modalities, US also serves a vital functional role outside of imaging. In particular, focused ultrasound (FUS) has become an important technique for drug delivery across the BBB (Fig. 7), with reported sub-millimeter precision [184]. In a recent study, Chen et al. synthesized TGF-β1-conjugated microbubbles to facilitate the extravasation of nanoparticles into mouse glioma cells with the help of FUS [185]. When FUS was applied, the microbubbles physically disturbed the BBB, rendering it more permeable. Furthermore, application of FUS allowed the microbubbles to release TGF-β1, which further disturbed the BBB by reducing pericyte wrapping of the endothelial cells. It was determined that, in the presence of microbubbles, FUS facilitated nanoparticle extravasation into the glioma cells [185].
Figure 7.
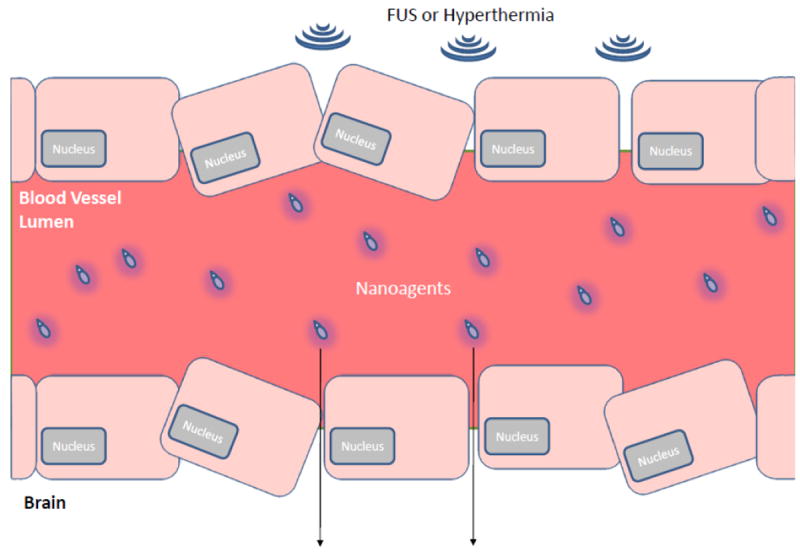
BBB disruption by physical forces. The two most common physical manipulations to promote CNS drug delivery are shown: Focused Ultrasound (FUS) and Hyperthermia.
11. Brain Cancer Treatment
11.1 Primary Brain Cancer Treatment
Gliomas are the most common and aggressive primary brain tumors [145, 186]. They can be divided into four malignancy levels according to WHO grading: low grade tumors, grades I (pilocytic astrocytoma) and II (diffuse astrocytoma), are characterized by faster-than-normal proliferation and a possibility of spreading to nearby tissues [147]; high-grade tumors, grades III (anaplastic astrocytoma) and IV (glioblastoma multiforme), are characterized by increased cell and blood vessel density, atypical cells, necrosis, and high mitotic and metastatic activity [147, 186]. Statistically, 10% of glioblastomas are secondary neoplasms; the remaining 90% are de novo GBMs, which have a rapid onset of only 3 months (compared to 4-5 years with secondary glioblastomas) [145]. Very limited options have been made available for treating primary brain cancer in recent history: from the 1960s until 1998, only two drugs were approved for the treatment of brain tumors [187]. In 1999, Temozolomide (TMZ), a DNA alkylator, was approved for treating primary brain tumors and remains the standard of care today ([150, 188]. Chemotherapy for recurrent GBM is very limited and usually results in little if any benefit. For instance, Carmustine (BCNU) wafers are widely used for treating glioblastoma, however, many studies showed that BCNU treatment did not increase patient survival or the quality of patients’ life [189, 190]. A small fraction of GBM patients showed promising response to tyrosine kinase inhibition, such as to EGFR inhibitors. However, the elevated PI3K/AKT activity in GBM tumors often diminishes the therapeutic advantage of EGFR inhibition [191]. Despite collective effort of physicians and the scientific community, the median post-diagnostic survival of patients with GBM remains 13-16 months on average [145, 147].
11.2 Secondary Brain Tumor Treatment
Brain metastases (BM) are a common and severe complication of other cancers, and they present even stronger challenges to treatment due to their significant morbidity. The most common origin of BM includes cancers arising from the lung, breast, colon, kidney, and skin [192]. BM are present in up to 40%-50% of metastatic lung cancers and 25% of metastatic breast cancers, with autopsy revealing about twice as many cases [193, 194]. Progress in primary breast cancer treatment has led to an increase in patient longevity but has also increased the chance of residual cells metastasizing, in particular to the brain [153]. Even lower than that of primary brain tumors, the median survival of patients with BM is only under 6 months [194].
Pharmacological treatments in the clinic for primary breast and lung cancers commonly include their respective therapeutic monoclonal antibodies, such as trastuzumab and cetuximab. Trastuzumab, which targets a receptor tyrosine kinase HER2, an EGFR family member, and cetuximab, which targets EGFR, are effective for treatment of certain primary breast and lung cancers, respectively (Fig. 8, [14]). However, like most drugs, they cannot penetrate the BBB to reach brain tumors on their own, and thus fail to treat BM [153].
Figure 8.
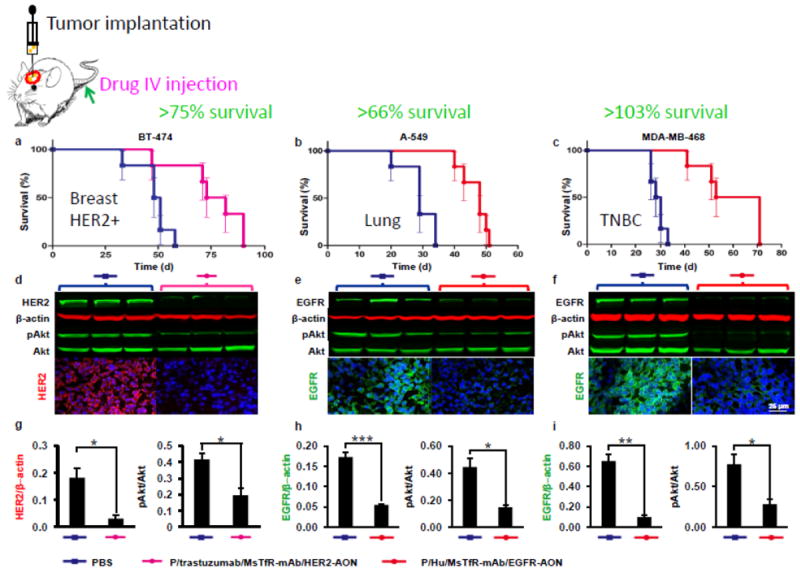
Survival of mice with brain metastases from primary HER2+ breast, triple negative breast cancer (TNBC) and lung cancers, which are positive for EGFR+, after treatment with tumor-targeted therapeutic polymers inhibiting the expression of HER2 or EGFR. Treatment was performed after MRI-based differential diagnoses of brain lesions made with nanoimaging agents (see Fig. 6). Reproduced from [14].
12. Designing a Nanodrug for Treating Brain Tumors
The primary objective of a chemotherapeutic drug is to be able to treat cancerous tissue specifically. However, most traditional drugs produce non-specific toxicity, causing unwanted damage to healthy tissue. As will be described in this section, one of the major advantages of nanomedicine is that it allows for homing modifications and for selective targeting. Thus, nanotherapeutic agents (NTs) are viable candidates for the treatment of many diseases, including brain tumors, as the sites of interest can be specifically targeted to reduce general toxicity [195]. However, to produce an effective NT, many factors must be taken into consideration.
12.1 Choosing an appropriate nanocarrier
Depending on the purpose of the NT, and the region in which it will act, different nanocarriers are chosen. There are pros and cons to using any type of nanocarrier, and in Table 3, we summarized commonly used BBB-crossing nanostructures with their advantages and disadvantages, respectively. The size, shape, surface properties and mechanical stiffness (termed as 4S Parameters) of the nanodrugs are particularly critical for their transportation and stability in the circulation, tumor-specific accumulation as well as clearance [196]. In all these areas, certain preferences do exist. For example, in the treatment of CNS diseases, lipid-based formulations are preferred due to their hydrophobic nature, which may allow for passive diffusion across cell membranes [197]. However, it is important not to limit designs according to these preferences. Promising in vivo results have been obtained in brain tumor research by using non-lipid-based nano formulations. In one of such examples, our work successfully demonstrated BBB permeation in mice using a non-toxic, biodegradable nanobiopolymer, PMLA, as the nanocarrier for delivering GBM therapeutics (Fig. 9, [14, 19, 198]).
Figure 9.
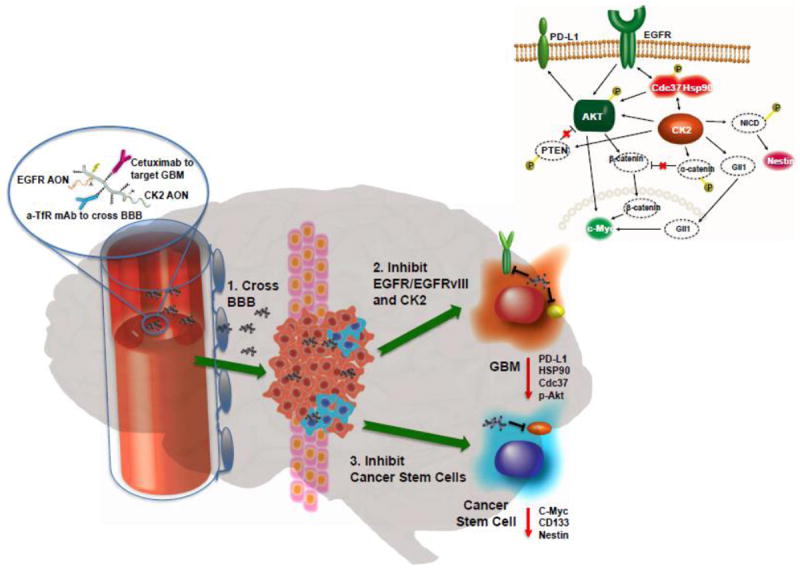
Schematic illustration of the inhibition of brain tumor growth and glioma stem cells by PMLA-based nano drugs. A novel mechanism of cross-talk between protein kinase CK2, an ubiquitous serine/threonine protein kinase, and wild and mutated variant vIII (EGFR/EGFRvIII) EGFR pathways is depicted. Reproduced from [198].
12.2 Optimizing Circulation Time
For a therapy through systemic delivery to work, the drug has to stay in the circulation for an optimal period of time. One of the advantages of nanomedicines is the increased circulation time relative to traditional treatments [199]. Yet, even with NTs, circulation can be cut short by reticulo-endothelial system (RES) clearance [200]. When highly hydrophobic molecules form aggregates with hydrophobic plasma proteins, blood clots form. The formation of these clots triggers phagocytic RES macrophages to remove the source of the problem, which is the hydrophobic molecule [200, 201]. Thus, to evade macrophage clearance, NTs are commonly modified with hydrophilic conjugates, such as PEG [199]. By adding a hydrophilic stabilizer, protein aggregation is prevented, thus removing the trigger that initiates macrophage-dependent clearance [202].
For many years, PEG has been the go-to for macrophage evasion. Currently, there are over 10 FDA-approved PEGylated nanomedicines, and more than 20 are in clinical trials [203]. In the context of CNS delivery, it has been widely reported that PEG does not prevent BBB crossing. However, PEG influence on BBB-crossing mechanisms has not been extensively researched [204]. Furthermore, PEG may cause other side effects; for example, PEG can induce an immune response, initiating the production of anti-PEG IgM antibodies [205]. These anti-PEG antibodies allow macrophages to quickly recognize PEGylated molecules upon re-exposure, thus facilitating RES clearance [205]. Nonetheless, PEGylated nanomedicines remain the golden standard today.
In addition to RES clearance, NTs can also be removed by the kidneys, though this is not nearly as significant of an obstacle. Renal clearance is far easier to overcome than RES clearance, due to its strict size dependence: if the particle’s hydrodynamic diameter is less than 6 nm, it will undergo rapid renal clearance [206]. Thus, renal clearance could easily be avoided by increasing the size of NT [206].
It is important to note that there is an optimal circulation time: an NT should remain in the body long enough to produce an effect, but not so long that it starts to cause problems. Prolonged drug accumulation could potentially cause toxicity. Importantly, it has been demonstrated that the biodistribution of drug conjugates could be tuned based on their physicochemical properties. For example, Hamilton et al. demonstrated that larger particles had higher accumulation across virtually every tissue in the body, whereas the smaller particles were more readily excreted [207].
12.3 Targeting and Crossing the Blood-Brain Barrier
One of the biggest obstacles in the treatment of brain tumors and other CNS diseases is bypassing the BBB [197]. The BBB is exceptionally particular in the material it allows to cross from blood to brain. Studies have shown that the BBB prevents 100% of large molecules and 98% of small molecules from diffusing freely across the barrier [208]. However, nanomedicine often exploits two phenomena that allow for NTs to selectively accumulate in the tumor mass: the EPR effect and BBB-receptor targeting.
As previously mentioned, one of the most exciting advantages of nanomedicine is its ability to limit general toxicity through specific tissue targeting. Typically, the development of a brain tumor triggers abnormal angiogenesis, as tumors have an imbalance of proangiogenic and antiangiogenic factors [202, 209]. As a result of the disordered angiogenesis, “leaky” blood vessels with compromised structural integrity are formed, increasing BBB permeability in certain areas of the tumor vasculature [202]. This well-known phenomenon, the EPR effect, is the passive mechanism by which small, polar NTs can selectively enter brain tumors [210]. However, the effect is minimal compared to a more active form of targeting, as the EPR effect is limited to the small areas of the BBB that are impaired (Fig. 5).
Distinct from the passive targeting mechanism, NTs are capable of actively targeting tumors, further facilitating their accumulation in the tumor cells [211]. This ability is conferred to NTs through the conjugation of specific targeting moieties. Importantly, this ability can only be exploited because different physiological structures express distinctive biomarkers [199]: for instance, malignant endothelial cells overexpress the TfR, as they require a larger amount of iron [211, 212]. This overexpression allows NTs to actively bypass the strict constraints imposed by the BBB: by carrying transferrin-targeting moieties, such as transferrin peptides, NTs can target and bind to endogenous BBB TfRs [213]. Once the NTs engage their targeted receptors, they are shuttled into the tumor mass via receptor-mediated endothelial transcytosis [214]. In addition to receptor overexpression by malignant cells, certain receptors are highly concentrated in specific areas of the BBB, allowing for selective drug delivery [215]. For example, Tumor Necrosis Factor (TNF) transporters are highly expressed near the hypothalamus - this locally increased receptor expression would allow TNF-targeting NTs to specifically target hypothalamic gliomas [216].
The two mechanisms described above – EPR and receptor targeting – are the most common ones seen in NT BBB crossing. Other less common mechanisms worth mentioning include adsorption-mediated endocytosis and carrier-mediated transport [217]. Furthermore, physical manipulations such as FUS and hyperthermia have been also employed for BBB penetration (Fig. 7) [208, 218, 219].
12.4 Penetrating the Brain Tumor
As described in the previous section, various mechanisms may be employed to direct NTs into brain tumors. Yet, there is still an important hurdle to navigate once the NTs enter the tumor: the tumor vasculature. The abnormal angiogenesis responsible for the EPR effect also results in the formation of irregular blood vessel networks. Due to this vascular abnormality, blood vessels are not as uniformly dispersed throughout tumors as they are throughout healthy tissue. An extreme example of this irregularity is displayed throughout GBM vasculature [209, 220]. Therefore, the perfusion of drugs from blood to tumor is heterogeneous, resulting in large pockets within the tumor mass that cannot be easily accessed by the NTs [221]. One promising strategy to mitigate this issue can be an anti-angiogenic treatment, potentially normalizing the vasculature and permitting the drug to better penetrate the tumor [209, 222].
However, fixing the irregular blood vessel network would not completely solve the diffusion problem: once inside the tumor mass, the NT must also be able to travel through the extracellular matrix (ECM). Diffusion through the tumor ECM is regulated by collagen grids, which are highly upregulated in glioma ECM [223]. Low-collagen tumors facilitate interstitial diffusion more effectively than high-collagen tumors [224]. Thus, the upregulation of collagen in the stroma of gliomas makes it more difficult for NT diffusion. In addition, the heterogeneous nature of the collagen networks (and their associated interstitial molecules) further prevents NTs from homogeneously penetrating tumors, leaving untreated pockets within the tumor mass [225]. A potential strategy to improve NT interstitial diffusion is to degrade the tumor ECM using enzymes such as collagenases [225].
Aside from collagen content, particle-related factors such as electric charge, size and shape also determine the ease with which the NT can traverse the tumor interstitial matrix [226-228]. NTs, which could be hundreds of nanometers in size, have difficulties spreading through the interstitial matrix of the tumor due to limited space [229]. Thus, NT-matrix interactions significantly reduce the amount of nanodrug reaching tumor cells [222]. Nance et al. conducted in vivo experiments in which they determined that nanoparticles up to 100 nm in size effectively diffused through normal brain tissue, whereas 200-nm nanoparticles did not [230]. However, it is important to note that the optimal size for deep brain tumor penetration varies across the literature.
12.5 Internalization
A nanodrug might need to traverse hundreds of micrometers of tumor stroma before it reaches malignant cells [231]. Moreover, it is possible that the nanodrug simply diffuses past the cell without recognizing it. Thus, to improve the recognition process, targeting moieties are often conjugated to the NT [15]. For example, we have engineered a dual monoclonal antibody NT for improved drug targeting: the anti-mouse TfR antibody allows the NT to bind the BBB TfR and get transcytosed, whereas the anti-nucleosome antibody 2C5 allows the NT to bind to nucleosome receptors on the cancer cells [232]. It is important to note that the presence of a targeting ligand allows the NT to interact with its respective receptor when within a few nanometers from it; the targeting ligand does not, however, home the NT directly to the receptor from across the tumor mass by using antibodies or peptides [23, 233].
In addition to facilitating cell recognition, the targeting conjugates often promote cellular uptake [234]. Upon binding to their targeted receptors, the NTs are generally shuttled into the cell via endocytosis – the exact endocytic pathway is dependent on factors such as the tumor microenvironment and physicochemical properties of the NT [208]. Yet, receptor-mediated interactions do not always promote internalization [234]. To overcome these non-permitting interactions, other entry mechanisms could be exploited. For example, NTs could be conjugated with cell-penetrating peptides (CPPs) [235]. CPPs are short peptides that are readily internalized by cells, and have been shown to effectively cross the BBB for CNS drug delivery [236]. However, the mechanisms by which CPPs enter cells are poorly understood – they are thought to be taken up through direct translocation, tertiary endocytosis, or other possible mechanisms [208]. An important drawback of CPPs is that they act in a non-specific manner [237], although this problem could be resolved through nanocarriers: in order to overcome their lack of specificity, CPPs can be covalently coupled to target-specific NTs. This covalent coupling allows CPPs to act specifically on targeted cancer cells and shuttle the covalently linked cargo into the target cell [235].
12.6 Endosomal Escape
Currently, endocytosis is the most common route for cellular uptake of NTs: this holds true for both CPP-mediated and receptor-mediated internalization [238, 239]. Regardless of the precise mechanism of endocytosis, the NT enters the cell encapsulated in an endosome. Aside from caveolae-mediated endocytosis, the downstream fate for all other endosomes is degradation through lysosomal digestion [238]. Thus, to evade lysosomal fusion and digestion, the NT must be able to escape the endosome in a timely manner. Within the literature, there are a variety of mechanisms by which endosomolysis has been achieved. One of the most common mechanisms is via the action of polyethyleneimine (PEI), a polymer with many unsaturated amino groups [233]. PEI acts by binding endosomal protons, causing counter ions and water to be pumped into the endosome. The increased osmotic pressure induces swelling, causing the endosome to burst and release its cargo directly into the cytosol [233].
Yet, PEI has been reported to have undesirable cytotoxic effects [240]. In light of this, another common technique for endosomal escape is via the action of CPPs, which are able to induce endosomolysis under sub-toxic concentrations [241]. However, the mechanisms by which they function vary significantly and require extensive mechanistic studies [208, 242]. Nonetheless, pH-sensitive peptide residues possess great potential for overcoming endosomal entrapment, as endosomes become more acidic as they proceed from their early to late stages (Fig. 10, [12]). Thus, other peptide-based formulations have been composed. For example, we have achieved successful endosomolysis within GBM cells through a pH-sensitive tripeptide, trileucine (LLL). LLL is activated by the acidic environment of the late endosome causing disruption of the endosomal membrane to facilitate cargo release [12]. Furthermore, physical manipulations for endosomal escape such as temperature- and photo-induced disruptions have been reported [243].
Figure 10.
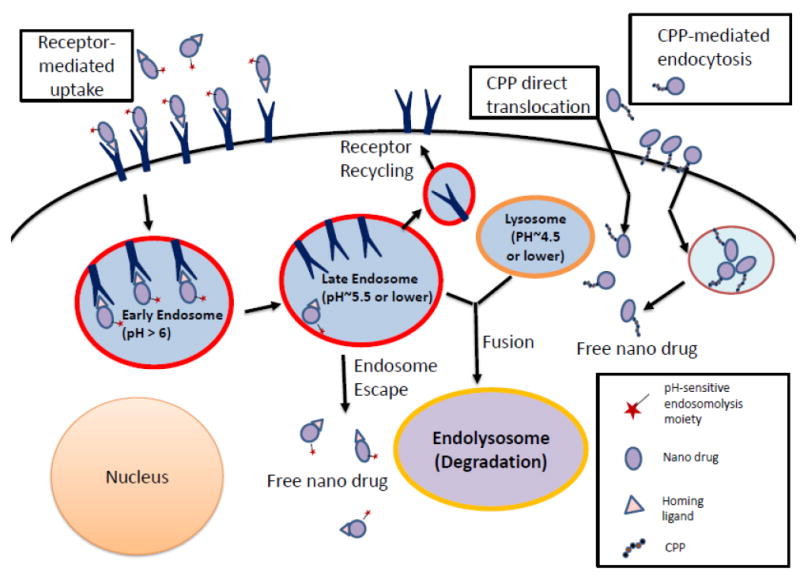
Receptor-mediated uptake and cytoplasmic trafficking of polymeric nano drugs. Modifications of nano drugs with homing ligand (antibody or peptide) or with cell penetrating peptide (CPP) are shown.
12.7 Tumor-Suppressing Activity
At the point that NTs escape the endosome, they enter the cytoplasm. In virtue of their size, NTs are able to act within cells on a molecular level [244] and are programmed to act within the cytoplasm. Depending on their action, the NTs can either act on structures within the cytoplasm or on specific organelles.
One preferred mechanism of action in the cytoplasm involves the use of RNA-interfering agents, primarily AON and siRNA. We have synthesized a glioma-targeting nanobioconjugate that suppressed tumor activity through silencing by AON the expression of two chains (α4 and β1) of vascular laminin-411 overexpressed in GBM [12, 39]. Similarly, siRNA can be used to interfere with the translation of tumor-promoting gene product. For example, Kozielski et al. synthesized siRNA-loaded polymeric nanoparticles that targeted GBM cells. When the nanoparticles entered the reducing cytoplasmic environment, the siRNA was cleaved off their platforms and could bind to their respective mRNAs [245].
Certain tumor-suppressing mechanisms require access to particular organelles. The current standard of care for gliomas, TMZ, acts through such a mechanism. For example, we synthesized a PMLA-TMZ nanoconjugate, which released TMZ into the cytoplasm of glioma cells upon endosomal escape. TMZ, a DNA-alkylating agent, then had to enter the nucleus by diffusion to exert its effects on DNA [13]. However, certain complexes could be too large to enter the nucleus by diffusion, and must interact with the nuclear pore complex to gain access to the interior of the nucleus. To accomplish this, larger complexes could be conjugated with nuclear localization sequences, which allow them to specifically bind to the nuclear membrane [157]. Similarly, if a NT target is the mitochondria, it could be actively transported across the mitochondrial membrane via a mitochondrial localization sequence.
13 Future Directions
Although enormous challenges exist in brain tumor treatment, we are confident that nanomedicine is able to confer great advantages to brain drug delivery. One very interesting application for nanomedicine in the treatment of brain cancer is the effective delivery of gene editing tools like CRISPR/Cas9. Although the “off-target” effect is still a big concern, a variety of applications of CRISPR/Cas9 system as therapeutics are currently under investigation [246, 247]. In cancer treatment, one strategy to use this technology is to attack oncogenes or modify the epigenome of the cancer cells by CRISPR/Cas9 [248]. After its first adaption for genome editing in eukaryotic system [249, 250], CRISPR/Cas9 system has been rapidly developed into a powerful research toolset as well as a promising therapeutic approach [251]. However, no effective method for intravenous, targeted delivery of CRISPR/Cas9 and its penetration through the BBB is readily available. Thus, nanomedicine holds a great potential to solve this problem and revolutionize both the fields of gene editing and the treatment of brain cancer [252].
Another area of potential innovation in brain cancer treatment is the focus on multimodal drug combinations. Nanomedicines have the ability to carry different therapeutic agents simultaneously and this ability can confer great advantages in cancer treatment and tumor suppression, if given appropriate targets. Defining the most effective use of covalently conjugated and encapsulated (or both) drugs could take advantage of synergistic, additive, and potentiative models of chemotherapeutic drug delivery [253]. In the meantime, the impressive treatment efficacy of cancer immunotherapies has gained attention from the field of nanomedicine. The simultaneous delivery of molecular inhibitors to modulate tumor microenvironment and IL-2 cytokine as part of anti-HER2 fusion monoclonal antibody to boost tumor immune system at the same time was introduced by engineering a nano conjugate to successfully treat HER2+ breast cancer in an animal model [20]. The development of nano immunotherapy is well underway. A novel nano vaccine recently reported to treat tumors, is composed of a mixture of an antigen and a synthetic polymeric nanoparticle, PC7A NP, which generates a strong cytotoxic T-cell response. The PC7A NP activated type I interferon-stimulated genes (STING), which led to tumor growth inhibition in melanoma, colon cancer, and human papilloma virus-E6/E7 tumor models. The combination of the PC7A nano vaccine and an anti-PD-1 antibody showed great synergy in mice [162, 254]. Additionally, nanoparticles have been used to engineer human stem cells to express an apoptotic inducer, such as TNF-related apoptosis-inducing ligand (TRAIL), for treating GBM in a mouse model [255].
In the field of brain imaging, we clearly need not only an improvement in diagnostic applications, but also an advanced imaging technique to guide treatment and this may be extremely helpful in achieving real precision medicine. The ability to predict recurrence of aggressive brain tumors like GBM would improve physicians’ ability to design effective treatment regimens, and provide new knowledge about cancer cell migration and penetration patterns. For example, a mutation in IDH1/2 in GBM was found to correlate with better survival as compared with tumors with WT-IDH genes, and the mutated IDH genes can be detected by MR imaging [256, 257]. Another study found a correlation between metabolic ratios of certain factors (choline-containing compounds (Cho: N-acetyl aspartate and creatinine) in the recurrence pathway of GBM using H-MRS imaging techniques. Problems with this method still exist, including low spatial resolution and long acquisition time [194, 258]. Nanoimaging agents may therefore be able to provide better resolution. Furthermore, we recently showed that differential diagnosis of brain tumors of different origins (primary vs. metastatic tumors) can be effectively made using MRI [14]. In this regard, using imaging methods to understand and detect cancer invasion, distribution of drugs in vivo, and tumorigenesis in real-time may bring new mechanistic discoveries in target identification and management, and lead to a more effective treatment [134, 186]. In the light of modern scientific achievements in cancer immunology and a comprehensive individual genomic/proteomic/metabolomic tumor characterization, the versatile covalent nano drug delivery systems can provide unlimited imaging and treatment possibilities for cancers that are difficult to treat and for many other pathological conditions.
Acknowledgments
This work was supported by the following grants: NIH R01 CA123495, U01 CA151815, R01 CA188743, R01 CA206220, R01 EY013431, and R01 CA 209921.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.http://www.etymonline.com/index.php?allowed_in_frame=0&search=pill
- 2.http://www.etymonline.com/index.php?allowed_in_frame=0&search=drug
- 3.Longmire M, Choyke PL, Kobayashi H. Clearance properties of nano-sized particles and molecules as imaging agents: considerations and caveats. Nanomedicine (Lond) 2008;3:703–717. doi: 10.2217/17435889.3.5.703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Haag R, Kratz F. Polymer therapeutics: concepts and applications. Angewandte Chemie International Edition. 2006;45:1198–1215. doi: 10.1002/anie.200502113. [DOI] [PubMed] [Google Scholar]
- 5.Pelegri-O’Day EM, Lin E-W, Maynard HD. Therapeutic protein–polymer conjugates: advancing beyond PEGylation. Journal of the American Chemical Society. 2014;136:14323–14332. doi: 10.1021/ja504390x. [DOI] [PubMed] [Google Scholar]
- 6.Owen SC, Chan DP, Shoichet MS. Polymeric micelle stability. Nano Today. 2012;7:53–65. [Google Scholar]
- 7.Huang Y, Wang D, Zhu X, Yan D, Chen R. Synthesis and therapeutic applications of biocompatible or biodegradable hyperbranched polymers. Polymer Chemistry. 2015;6:2794–2812. [Google Scholar]
- 8.Gros L, Ringsdorf H, Schupp H. Polymeric antitumor agents on a molecular and on a cellular level? Angewandte Chemie International Edition in English. 1981;20:305–325. [Google Scholar]
- 9.Ringsdorf H. Journal of Polymer Science: Polymer Symposia. Wiley Online Library; 1975. Structure and properties of pharmacologically active polymers; pp. 135–153. [Google Scholar]
- 10.Boussif O, Lezoualc’h F, Zanta MA, Mergny MD, Scherman D, Demeneix B, Behr J-P. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. Proceedings of the National Academy of Sciences. 1995;92:7297–7301. doi: 10.1073/pnas.92.16.7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ljubimova JY, Fujita M, Ljubimov AV, Torchilin VP, Black KL, Holler E. Poly(malic acid) nanoconjugates containing various antibodies and oligonucleotides for multitargeting drug delivery. Nanomedicine. 2008;3:247–265. doi: 10.2217/17435889.3.2.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ding H, Inoue S, Ljubimov AV, Patil R, Portilla-Arias J, Hu J, Konda B, Wawrowsky KA, Fujita M, Karabalin N, Sasaki T, Black KL, Holler E, Ljubimova JY. Inhibition of brain tumor growth by intravenous poly(β-l-malic acid) nanobioconjugate with pH-dependent drug release. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:18143–18148. doi: 10.1073/pnas.1003919107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Patil R, Portilla-Arias J, Ding H, Inoue S, Konda B, Hu J, Wawrowsky KA, Shin PK, Black KL, Holler E, Ljubimova JY. Temozolomide delivery to tumor cells by a multifunctional nano vehicle based on poly(β-L-malic acid) Pharmaceutical Research. 2010;27:2317–2329. doi: 10.1007/s11095-010-0091-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Patil R, Ljubimov AV, Gangalum PR, Ding H, Portilla-Arias J, Wagner S, Inoue S, Konda B, Rekechenetskiy A, Chesnokova A, Markman JL, Ljubimov VA, Li D, Prasad RS, Black KL, Holler E, Ljubimova JY. MRI virtual biopsy and treatment of brain metastatic tumors with targeted nanobioconjugates: nanoclinic in the brain. ACS Nano. 2015;9:5594–5608. doi: 10.1021/acsnano.5b01872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee B-S, Fujita M, Khazenzon NM, Wawrowsky KA, Wachsmann-Hogiu S, Farkas DL, Black KL, Ljubimova JY, Holler E. Polycefin, a new prototype of a multifunctional nanoconjugate based on poly(β-l-malic acid) for drug delivery. Bioconjugate Chemistry. 2006;17:317–326. doi: 10.1021/bc0502457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ljubimova J, Fujita M, Lee B, Khazenzon N, Wachsmann-Hogiu S, Farkas D, Black K, Holler E. Nanoconjugates of poly (malic acid) with functional modules for drug delivery. NSTI-Nanotech. 2006:354–357. [Google Scholar]
- 17.Ljubimova JY, Fujita M, Khazenzon NM, Lee B-S, Wachsmann-Hogiu S, Farkas DL, Black KL, Holler E. Nanoconjugate based on polymalic acid for tumor targeting. Chemico-biological interactions. 2008;171:195–203. doi: 10.1016/j.cbi.2007.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ljubimova JY, Ding H, Portilla-Arias J, Patil R, Gangalum PR, Chesnokova A, Inoue S, Rekechenetskiy A, Nassoura T, Black KL. Polymalic acid-based nano biopolymers for targeting of multiple tumor markers: an opportunity for personalized medicine? JoVE (Journal of Visualized Experiments) 2014:e50668–e50668. doi: 10.3791/50668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ljubimova JY, Portilla-Arias J, Patil R, Ding H, Inoue S, Markman JL, Rekechenetskiy A, Konda B, Gangalum PR, Chesnokova A, Ljubimov AV, Black KL, Holler E. Toxicity and efficacy evaluation of multiple targeted polymalic acid conjugates for triple-negative breast cancer treatment. Journal of Drug Targeting. 2013;21:956–967. doi: 10.3109/1061186X.2013.837470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ding H, Helguera G, Rodríguez JA, Markman J, Luria-Pérez R, Gangalum P, Portilla-Arias J, Inoue S, Daniels-Wells TR, Black K. Polymalic acid nanobioconjugate for simultaneous immunostimulation and inhibition of tumor growth in HER2/neu-positive breast cancer. Journal of Controlled Release. 2013;171:322–329. doi: 10.1016/j.jconrel.2013.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Inoue S, Ding H, Portilla-Arias J, Hu J, Konda B, Fujita M, Espinoza A, Suhane S, Riley M, Gates M. Polymalic acid based nanobiopolymer provides efficient systemic breast cancer treatment by inhibiting both HER2/neu receptor synthesis and activity. Cancer research. 2011;71:1454–1464. doi: 10.1158/0008-5472.CAN-10-3093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Inoue S, Patil R, Portilla-Arias J, Ding H, Konda B, Espinoza A, Mongayt D, Markman JL, Elramsisy A, Phillips HW. Nanobiopolymer for direct targeting and inhibition of EGFR expression in triple negative breast cancer. PLoS One. 2012;7:e31070. doi: 10.1371/journal.pone.0031070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ding H, Gangalum PR, Galstyan A, Fox I, Patil R, Hubbard P, Murali R, Ljubimova JY, Holler E. HER2-positive breast cancer targeting and treatment by a peptide-conjugated mini nanodrug, Nanomedicine: Nanotechnology. Biology and Medicine. 2017;13:631–639. doi: 10.1016/j.nano.2016.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ding H, Fox I, Patil R, Galstyan A, Black KL, Ljubimova JY, Holler E. Polymalic acid tritryptophan copolymer interacts with lipid membrane resulting in membrane solubilization. Journal of Nanomaterials. 2017;2017 doi: 10.1155/2017/4238697. Article ID 4238697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chu T-W, Yang J, Kopeček J. Anti-CD20 multivalent HPMA copolymer-Fab′ conjugates for the direct induction of apoptosis. Biomaterials. 2012;33:7174–7181. doi: 10.1016/j.biomaterials.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Choi BD, Kuan C-T, Cai M, Archer GE, Mitchell DA, Gedeon PC, Sanchez-Perez L, Pastan I, Bigner DD, Sampson JH. Systemic administration of a bispecific antibody targeting EGFRvIII successfully treats intracerebral glioma. Proceedings of the National Academy of Sciences. 2013;110:270–275. doi: 10.1073/pnas.1219817110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Choi BD, Pastan I, Bigner DD, Sampson JH. A novel bispecific antibody recruits T cells to eradicate tumors in the “immunologically privileged” central nervous system. Oncoimmunology. 2013;2:e23639. doi: 10.4161/onci.23639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ding H, Portilla-Arias J, Patil R, Black KL, Ljubimova JY, Holler E. Distinct mechanisms of membrane permeation induced by two polymalic acid copolymers. Biomaterials. 2013;34:217–225. doi: 10.1016/j.biomaterials.2012.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Drake AW, Myszka DG, Klakamp SL. Characterizing high-affinity antigen/antibody complexes by kinetic-and equilibrium-based methods. Analytical biochemistry. 2004;328:35–43. doi: 10.1016/j.ab.2003.12.025. [DOI] [PubMed] [Google Scholar]
- 30.Salami S, Rondeau-Mouro C, Barhoum M, van Duynhoven J, Mariette F. Translational and rotational diffusion of flexible PEG and rigid dendrimer probes in sodium caseinate dispersions and acid gels. Biopolymers. 2014;101:959–965. doi: 10.1002/bip.22492. [DOI] [PubMed] [Google Scholar]
- 31.Lee KL, Hubbard LC, Hern S, Yildiz I, Gratzl M, Steinmetz NF. Shape matters: the diffusion rates of TMV rods and CPMV icosahedrons in a spheroid model of extracellular matrix are distinct. Biomaterials science. 2013;1:581–588. doi: 10.1039/C3BM00191A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Read T, Olkhov RV, Williamson ED, Shaw AM. Label-free Fab and Fc affinity/avidity profiling of the antibody complex half-life for polyclonal and monoclonal efficacy screening. Analytical and bioanalytical chemistry. 2015;407:7349–7357. doi: 10.1007/s00216-015-8897-6. [DOI] [PubMed] [Google Scholar]
- 33.Summerton J. Morpholino antisense oligomers: the case for an RNase H-independent structural type. Biochimica et Biophysica Acta (BBA)-Gene Structure and Expression. 1999;1489:141–158. doi: 10.1016/s0167-4781(99)00150-5. [DOI] [PubMed] [Google Scholar]
- 34.Ge Z, Liu S. Functional block copolymer assemblies responsive to tumor and intracellular microenvironments for site-specific drug delivery and enhanced imaging performance. Chemical Society Reviews. 2013;42:7289–7325. doi: 10.1039/c3cs60048c. [DOI] [PubMed] [Google Scholar]
- 35.Böhme D, Beck-Sickinger AG. Drug delivery and release systems for targeted tumor therapy. Journal of Peptide Science. 2015;21:186–200. doi: 10.1002/psc.2753. [DOI] [PubMed] [Google Scholar]
- 36.Cathcart J, Pulkoski-Gross A, Cao J. Targeting matrix metalloproteinases in cancer: bringing new life to old ideas. Genes & Diseases. 2015;2:26–34. doi: 10.1016/j.gendis.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Duncan R, Cable HC, Lloyd JB, Rejmanova P, Kopecek J. Degradation of side-chains of N-(2-hydroxypropyl)methacrylamide copolymers by lysosomal thiol-proteinases. Bioscience Report. 1982;2:1041–1046. doi: 10.1007/BF01122173. [DOI] [PubMed] [Google Scholar]
- 38.Dubowchik GM, Firestone RA. Cathepsin B-sensitive dipeptide prodrugs. 1. A model study of structural requirements for efficient release of doxorubicin. Bioorganic & medicinal chemistry letters. 1998;8:3341–3346. doi: 10.1016/s0960-894x(98)00609-x. [DOI] [PubMed] [Google Scholar]
- 39.Fujita M, Khazenzon NM, Ljubimov AV, Lee B-S, Virtanen I, Holler E, Black KL, Ljubimova JY. Inhibition of laminin-8 in vivo using a novel poly (malic acid)-based carrier reduces glioma angiogenesis. Angiogenesis. 2006;9:183–191. doi: 10.1007/s10456-006-9046-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Patil R, Portilla-Arias J, Ding H, Konda B, Rekechenetskiy A, Inoue S, Black KL, Holler E, Ljubimova JY. Cellular delivery of doxorubicin via pH-controlled hydrazone linkage using multifunctional nano vehicle based on poly (β-L-malic acid) International journal of molecular sciences. 2012;13:11681–11693. doi: 10.3390/ijms130911681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Karl M, Gasselmaier B, Krieg RC, Holler E. Localization of fluorescence-labeled poly (malic acid) to the nuclei of the plasmodium of Physarum polycephalum. European Journal of Biochemistry. 2003;270:1536–1542. doi: 10.1046/j.1432-1033.2003.03521.x. [DOI] [PubMed] [Google Scholar]
- 42.Hrkach J, Von Hoff D, Ali MM, Andrianova E, Auer J, Campbell T, De Witt D, Figa M, Figueiredo M, Horhota A. Preclinical development and clinical translation of a PSMA-targeted docetaxel nanoparticle with a differentiated pharmacological profile. Science translational medicine. 2012;4:128ra139–128ra139. doi: 10.1126/scitranslmed.3003651. [DOI] [PubMed] [Google Scholar]
- 43.Fischer H, Erdmann S, Holler E. An unusual polyanion from Physarum polycephalum that inhibits homologous DNA-polymerase. alpha. in vitro. Biochemistry. 1989;28:5219–5226. doi: 10.1021/bi00438a045. [DOI] [PubMed] [Google Scholar]
- 44.Holler E, Achhammer G, Angerer B, Gantz B, Hambach C, Reisner H, Seidel B, Weber C, Windisch C, Braud C. Specific inhibition of Physarum polycephalum DNA-polymerase-α-primase by poly (l-malate) and related polyanions. European Journal of Biochemistry. 1992;206:1–6. doi: 10.1111/j.1432-1033.1992.tb16894.x. [DOI] [PubMed] [Google Scholar]
- 45.Lee BS, Vert M, Holler E. Water-soluble aliphatic polyesters: poly (malic acid)s. Biopolymers Online. 2002 [Google Scholar]
- 46.Rathberger K, Reisner H, Willibald B, Molitoris H-P, Holler E. Comparative synthesis and hydrolytic degradation of poly (L-malate) by myxomycetes and fungi. Mycological Research. 1999;103:513–520. [Google Scholar]
- 47.Cammas S, Guerin P, Girault J, Holler E, Gache Y, Vert M. Natural poly (L-malic acid): NMR shows a poly (3-hydroxy acid)-type structure. Macromolecules. 1993;26:4681–4684. [Google Scholar]
- 48.Cheng C, Zhou Y, Lin M, Wei P, Yang S-T. Polymalic acid fermentation by Aureobasidium pullulans for malic acid production from soybean hull and soy molasses: Fermentation kinetics and economic analysis. Bioresource Technology. 2017;223:166–174. doi: 10.1016/j.biortech.2016.10.042. [DOI] [PubMed] [Google Scholar]
- 49.Manitchotpisit P, Skory CD, Peterson SW, Price NP, Vermillion KE, Leathers TD. Poly (β-L-malic acid) production by diverse phylogenetic clades of Aureobasidium pullulans. Journal of industrial microbiology & biotechnology. 2012;39:125–132. doi: 10.1007/s10295-011-1007-7. [DOI] [PubMed] [Google Scholar]
- 50.Ding H, Portilla-Arias J, Patil R, Black KL, Ljubimova JY, Holler E. The optimization of polymalic acid peptide copolymers for endosomolytic drug delivery. Biomaterials. 2011;32:5269–5278. doi: 10.1016/j.biomaterials.2011.03.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Loyer P, Cammas-Marion S. Natural and synthetic poly (malic acid)-based derivates: a family of versatile biopolymers for the design of drug nanocarriers. Journal of drug targeting. 2014;22:556–575. doi: 10.3109/1061186X.2014.936871. [DOI] [PubMed] [Google Scholar]
- 52.Buescher JM, Margaritis A. Microbial biosynthesis of polyglutamic acid biopolymer and applications in the biopharmaceutical, biomedical and food industries. Critical reviews in biotechnology. 2007;27:1–19. doi: 10.1080/07388550601166458. [DOI] [PubMed] [Google Scholar]
- 53.Shih I-L, Van Y-T. The production of poly-(γ-glutamic acid) from microorganisms and its various applications. Bioresource Technology. 2001;79:207–225. doi: 10.1016/s0960-8524(01)00074-8. [DOI] [PubMed] [Google Scholar]
- 54.Luo Z, Guo Y, Liu J, Qiu H, Zhao M, Zou W, Li S. Microbial synthesis of poly-γ-glutamic acid: current progress, challenges, and future perspectives. Biotechnology for Biofuels. 2016;9:134. doi: 10.1186/s13068-016-0537-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tam NK, Uyen NQ, Hong HA, Duc LH, Hoa TT, Serra CR, Henriques AO, Cutting SM. The intestinal life cycle of Bacillus subtilis and close relatives. Journal of bacteriology. 2006;188:2692–2700. doi: 10.1128/JB.188.7.2692-2700.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Morelli CF, Calvio C, Biagiotti M, Speranza G. pH-Dependent hydrolase, glutaminase, transpeptidase and autotranspeptidase activities of Bacillus subtilis γ-glutamyltransferase. FEBS Journal. 2014;281:232–245. doi: 10.1111/febs.12591. [DOI] [PubMed] [Google Scholar]
- 57.Liao Z-X, Peng S-F, Ho Y-C, Mi F-L, Maiti B, Sung H-W. Mechanistic study of transfection of chitosan/DNA complexes coated by anionic poly (γ-glutamic acid) Biomaterials. 2012;33:3306–3315. doi: 10.1016/j.biomaterials.2012.01.013. [DOI] [PubMed] [Google Scholar]
- 58.Melancon MP, Li C. Multifunctional synthetic poly (L-glutamic Acid)–based cancer therapeutic and imaging agents. Molecular imaging. 2011;10 7290.2011. 00007. [PMC free article] [PubMed] [Google Scholar]
- 59.Zou Y, Wu Q, Tansey W, Chow D, Hung M, Charnsangavej C, Wallace S, Li C. Effectiveness of water soluble poly (L-glutamic acid)-camptothecin conjugate against resistant human lung cancer xenografted in nude mice. International journal of oncology. 2001;18:331–336. doi: 10.3892/ijo.18.2.331. [DOI] [PubMed] [Google Scholar]
- 60.Singer JW, Vries P, Bhatt R, Tulinsky J, Klein P, Li C, Milas L, Lewis RA, Wallace S. Conjugation of camptothecins to poly-(L-glutamic acid) Annals of the New York Academy of Sciences. 2000;922:136–150. doi: 10.1111/j.1749-6632.2000.tb07032.x. [DOI] [PubMed] [Google Scholar]
- 61.Kanamala M, Wilson WR, Yang M, Palmer BD, Wu Z. Mechanisms and biomaterials in pH-responsive tumour targeted drug delivery: a review. Biomaterials. 2016;85:152–167. doi: 10.1016/j.biomaterials.2016.01.061. [DOI] [PubMed] [Google Scholar]
- 62.Jeyapalan S, Boxerman J, Donahue J, Goldman M, Kinsella T, Dipetrillo T, Evans D, Elinzano H, Constantinou M, Stopa E. Paclitaxel poliglumex, temozolomide, and radiation for newly diagnosed high-grade glioma: a Brown University Oncology Group Study. American journal of clinical oncology. 2014;37:444–449. doi: 10.1097/COC.0b013e31827de92b. [DOI] [PubMed] [Google Scholar]
- 63.Lu Z-R, Wang X, Parker DL, Goodrich KC, Buswell HR. Poly (l-glutamic acid) Gd (III)-DOTA conjugate with a degradable spacer for magnetic resonance imaging. Bioconjugate chemistry. 2003;14:715–719. doi: 10.1021/bc0340464. [DOI] [PubMed] [Google Scholar]
- 64.Ogunleye A, Bhat A, Irorere VU, Hill D, Williams C, Radecka I. Poly-γ-glutamic acid: production, properties and applications. Microbiology. 2015;161:1–17. doi: 10.1099/mic.0.081448-0. [DOI] [PubMed] [Google Scholar]
- 65.Tomcsik J, Szongott H. Ǘber ein spezifisches Protein der kapsel des milzbrandbazillus. Zeitschrift Immunitaet-Forschung. 1933;78:86–99. [Google Scholar]
- 66.Zwartouw H, Smith H. Polyglutamic acid from Bacillus anthracis grown in vivo: structure and aggressin activity. Biochemical Journal. 1956;63:437. doi: 10.1042/bj0630437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Little SF, Ivins BE. Molecular pathogenesis of Bacillus anthracis infection. Microbes and Infection. 1999;1:131–139. doi: 10.1016/s1286-4579(99)80004-5. [DOI] [PubMed] [Google Scholar]
- 68.Jang J, Cho M, Lee H-R, Cha K, Chun J-H, Hong K-J, Park J, Rhie G-e. Monoclonal antibody against the poly-γ-D-glutamic acid capsule of Bacillus anthracis protects mice from enhanced lethal toxin activity due to capsule and anthrax spore challenge. Biochimica et Biophysica Acta (BBA)-General Subjects. 2013;1830:2804–2812. doi: 10.1016/j.bbagen.2012.11.006. [DOI] [PubMed] [Google Scholar]
- 69.Schneerson R, Kubler-Kielb J, Liu T-Y, Dai Z-D, Leppla SH, Yergey A, Backlund P, Shiloach J, Majadly F, Robbins JB. Poly (γ-D-glutamic acid) protein conjugates induce IgG antibodies in mice to the capsule of Bacillus anthracis: a potential addition to the anthrax vaccine. Proceedings of the National Academy of Sciences. 2003;100:8945–8950. doi: 10.1073/pnas.1633512100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Muroga Y, Nakaya A, Inoue A, Itoh D, Abiru M, Wada K, Takada M, Ikake H, Shimizu S. Conformation of poly (γ-glutamic acid) in aqueous solution. Biopolymers. 2016;105:191–198. doi: 10.1002/bip.22776. [DOI] [PubMed] [Google Scholar]
- 71.Feng Z, Lai Y, Ye H, Huang J, Xi XG, Wu Z. Poly (γ, L-glutamic acid)-cisplatin bioconjugate exhibits potent antitumor activity with low toxicity: A comparative study with clinically used platinum derivatives. Cancer science. 2010;101:2476–2482. doi: 10.1111/j.1349-7006.2010.01708.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kurosaki T, Kitahara T, Kawakami S, Higuchi Y, Yamaguchi A, Nakagawa H, Kodama Y, Hamamoto T, Hashida M, Sasaki H. γ-Polyglutamic acid-coated vectors for effective and safe gene therapy. Journal of controlled release. 2010;142:404–410. doi: 10.1016/j.jconrel.2009.11.010. [DOI] [PubMed] [Google Scholar]
- 73.Kodama Y, Yatsugi Y, Kitahara T, Kurosaki T, Egashira K, Nakashima M, Muro T, Nakagawa H, Higuchi N, Nakamura T. Quaternary complexes modified from pDNA and poly-L-lysine complexes to enhance pH-buffering effect and suppress cytotoxicity. Journal of pharmaceutical sciences. 2015;104:1470–1477. doi: 10.1002/jps.24364. [DOI] [PubMed] [Google Scholar]
- 74.Thombre SM, Sarwade BD. Synthesis and biodegradability of polyaspartic acid: a critical review. Journal of Macromolecular Science, Part A. 2005;42:1299–1315. [Google Scholar]
- 75.Lu C, Wang X, Wu G, Wang J, Wang Y, Gao H, Ma J. An injectable and biodegradable hydrogel based on poly (α, β-aspartic acid) derivatives for localized drug delivery. Journal of Biomedical Materials Research Part A. 2014;102:628–638. doi: 10.1002/jbm.a.34725. [DOI] [PubMed] [Google Scholar]
- 76.Wang X, Wu G, Lu C, Zhao W, Wang Y, Fan Y, Gao H, Ma J. A novel delivery system of doxorubicin with high load and pH-responsive release from the nanoparticles of poly (α, β-aspartic acid) derivative. European Journal of Pharmaceutical Sciences. 2012;47:256–264. doi: 10.1016/j.ejps.2012.04.007. [DOI] [PubMed] [Google Scholar]
- 77.Jiang T, Yu X, Carbone EJ, Nelson C, Kan HM, Lo KW-H. Poly aspartic acid peptide-linked PLGA based nanoscale particles: potential for bone-targeting drug delivery applications. International journal of pharmaceutics. 2014;475:547–557. doi: 10.1016/j.ijpharm.2014.08.067. [DOI] [PubMed] [Google Scholar]
- 78.Song H-Q, Dou X-B, Li R-Q, Yu B-R, Zhao N-N, Xu F-J. A general strategy to prepare different types of polysaccharide-graft-poly aspartic acid as degradable gene carriers. Acta biomaterialia. 2015;12:156–165. doi: 10.1016/j.actbio.2014.10.041. [DOI] [PubMed] [Google Scholar]
- 79.Nie J-J, Dou X-B, Hu H, Yu B, Chen D-F, Wang R-X, Xu F-J. Poly (aspartic acid)-based degradable assemblies for highly efficient gene delivery. ACS applied materials & interfaces. 2015;7:553–562. doi: 10.1021/am506730t. [DOI] [PubMed] [Google Scholar]
- 80.Feng S-T, Li J, Luo Y, Yin T, Cai H, Wang Y, Dong Z, Shuai X, Li Z-P. pH-sensitive nanomicelles for controlled and efficient drug delivery to human colorectal carcinoma LoVo cells. PloS one. 2014;9:e100732. doi: 10.1371/journal.pone.0100732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Huang H, Li Y, Sa Z, Sun Y, Wang Y, Wang J. A smart drug delivery system from charge-conversion polymer–drug conjugate for enhancing tumor therapy and tunable drug release. Macromolecular bioscience. 2014;14:485–490. doi: 10.1002/mabi.201300337. [DOI] [PubMed] [Google Scholar]
- 82.Stayton P, El-Sayed ME, Murthy N, Bulmus V, Lackey C, Cheung C, Hoffman AS. ‘Smart’ delivery systems for biomolecular therapeutics. Orthodontics & craniofacial research. 2005;8:219–225. doi: 10.1111/j.1601-6343.2005.00336.x. [DOI] [PubMed] [Google Scholar]
- 83.Yang J, Kopeček J. Macromolecular therapeutics. Journal of Controlled Release. 2014;190:288–303. doi: 10.1016/j.jconrel.2014.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Duncan R, Kopeček J. Soluble synthetic polymers as potential drug carriers. Polymers in medicine. 1984:51–101. [Google Scholar]
- 85.Luo K, Yang J, Kopečková P, Kopecek J. Biodegradable multiblock poly [N-(2-hydroxypropyl) methacrylamide] via reversible addition – fragmentation chain transfer polymerization and click chemistry. Macromolecules. 2011;44:2481–2488. doi: 10.1021/ma102574e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lomkova EA, Chytil P, Janoušková O, Mueller T, Lucas H, Filippov SK, Trhlíková O, Aleshunin PA, Skorik YA, Ulbrich K. Biodegradable micellar HPMA-based polymer–drug conjugates with betulinic acid for passive tumor targeting. Biomacromolecules. 2016;17:3493–3507. doi: 10.1021/acs.biomac.6b00947. [DOI] [PubMed] [Google Scholar]
- 87.Zhang L, Zhang R, Yang J, Wang J, Kopeček J. Indium-based and iodine-based labeling of HPMA copolymer–epirubicin conjugates: Impact of structure on the in vivo fate. Journal of Controlled Release. 2016;235:306–318. doi: 10.1016/j.jconrel.2016.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yang J, Kopeček J. Design of smart HPMA copolymer-based nanomedicines. Journal of Controlled Release. 2016;240:9–23. doi: 10.1016/j.jconrel.2015.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chytil P, Etrych T, Koňák Č, Šírová M, Mrkvan T, Říhová B, Ulbrich K. Properties of HPMA copolymer doxorubicin conjugates with pH-controlled activation: effect of polymer chain modification. Journal of Controlled Release. 2006;115:26–36. doi: 10.1016/j.jconrel.2006.06.028. [DOI] [PubMed] [Google Scholar]
- 90.Duncan R, Vicent MJ. Do HPMA copolymer conjugates have a future as clinically useful nanomedicines? A critical overview of current status and future opportunities. Advanced Drug Delivery Reviews. 2010;62:272–282. doi: 10.1016/j.addr.2009.12.005. [DOI] [PubMed] [Google Scholar]
- 91.Chu T-W, Kosak KM, Shami PJ, Kopeček J. Drug-free macromolecular therapeutics induce apoptosis of patient chronic lymphocytic leukemia cells. Drug Delivery And Translational Research. 2014;4:389–394. doi: 10.1007/s13346-014-0209-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ye Z, Houssein HSH, Mahato RI. Bioconjugation of oligonucleotides for treating liver fibrosis. Oligonucleotides. 2007;17:349–404. doi: 10.1089/oli.2007.0097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yang N, Ye Z, Li F, Mahato RI. HPMA polymer-based site-specific delivery of oligonucleotides to hepatic stellate cells. Bioconjugate chemistry. 2009;20:213–221. doi: 10.1021/bc800237t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yang J, Zhang R, Radford DC, Kopeček J. FRET-trackable biodegradable HPMA copolymer-epirubicin conjugates for ovarian carcinoma therapy. Journal of Controlled Release. 2015;218:36–44. doi: 10.1016/j.jconrel.2015.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lundy BB, Convertine A, Miteva M, Stayton PS. Neutral polymeric micelles for RNA delivery. Bioconjugate chemistry. 2013;24:398–407. doi: 10.1021/bc300486k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Carlson PM, Schellinger JG, Pahang JA, Johnson RN, Pun SH. Comparative study of guanidine-based and lysine-based brush copolymers for plasmid delivery. Biomaterials science. 2013;1:736–744. doi: 10.1039/C3BM60079C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Novo L, Takeda KM, Petteta T, Dakwar GR, van den Dikkenberg JB, Remaut K, Braeckmans K, van Nostrum CF, Mastrobattista E, Hennink WE. Targeted decationized polyplexes for siRNA delivery. Molecular Pharmaceutics. 2015;12:150–161. doi: 10.1021/mp500499x. [DOI] [PubMed] [Google Scholar]
- 98.Shi J, Schellinger JG, Pun SH. Engineering biodegradable and multifunctional peptide-based polymers for gene delivery. Journal of biological engineering. 2013;7:25. doi: 10.1186/1754-1611-7-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Oupický D, Koňák Č, Ulbrich K, Wolfert M, Seymour L. DNA delivery systems based on complexes of DNA with synthetic polycations and their copolymers. Journal of Controlled Release. 2000;65:149–171. doi: 10.1016/s0168-3659(99)00249-7. [DOI] [PubMed] [Google Scholar]
- 100.Tappertzhofen K, Weiser F, Montermann E, Reske-Kunz A, Bros M, Zentel R. poly-L-lysine–poly [HPMA] Block copolymers obtained by RAFT polymerization as polyplex-transfection reagents with minimal toxicity. Macromolecular bioscience. 2015;15:1159–1173. doi: 10.1002/mabi.201500022. [DOI] [PubMed] [Google Scholar]
- 101.Burke RS, Pun SH. Synthesis and characterization of biodegradable HPMA-oligolysine copolymers for improved gene delivery. Bioconjugate Chemistry. 2010;21:140–150. doi: 10.1021/bc9003662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Johnson RN, Chu DS, Shi J, Schellinger JG, Carlson PM, Pun SH. HPMA-oligolysine copolymers for gene delivery: optimization of peptide length and polymer molecular weight. Journal of controlled release. 2011;155:303–311. doi: 10.1016/j.jconrel.2011.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tappertzhofen K, Beck S, Montermann E, Huesmann D, Barz M, Koynov K, Bros M, Zentel R. Bioreducible poly-L-lysine–poly [HPMA] block copolymers obtained by RAFT polymerization as efficient polyplex-transfection reagents. Macromolecular bioscience. 2016;16:106–120. doi: 10.1002/mabi.201500212. [DOI] [PubMed] [Google Scholar]
- 104.Zhang Y, Guo C, Li S, Luo K, Hu J, Gu Z. The potential of poly [N-2-hydroxypropyl methacrylamide] via reversible addition-fragmentation chain transfer polymerization as safe nanocarrier. Journal of Nanoscience and Nanotechnology. 2016;16:5746–5754. doi: 10.1166/jnn.2016.11749. [DOI] [PubMed] [Google Scholar]
- 105.Lu Z-R. Molecular imaging of HPMA copolymers: visualizing drug delivery in cell, mouse and man. Advanced Drug Delivery Reviews. 2010;62:246–257. doi: 10.1016/j.addr.2009.12.007. [DOI] [PubMed] [Google Scholar]
- 106.Fan W, Shi W, Zhang W, Jia Y, Zhou Z, Brusnahan SK, Garrison JC. Cathepsin S-cleavable, multi-block HPMA copolymers for improved SPECT/CT imaging of pancreatic cancer. Biomaterials. 2016;103:101–115. doi: 10.1016/j.biomaterials.2016.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kogan-Zviagin I, Shamay Y, Nissan A, Sella-Tavor O, Golan M, David A. Intra-colonic administration of a polymer-bound NIRF probe for improved colorectal cancer detection during colonoscopy. Journal of Controlled Release. 2014;192:182–191. doi: 10.1016/j.jconrel.2014.06.058. [DOI] [PubMed] [Google Scholar]
- 108.Olson ES, Jiang T, Aguilera TA, Nguyen QT, Ellies LG, Scadeng M, Tsien RY. Activatable cell penetrating peptides linked to nanoparticles as dual probes for in vivo fluorescence and MR imaging of proteases. Proceedings of the National Academy of Sciences. 2010;107:4311–4316. doi: 10.1073/pnas.0910283107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Nguyen QT, Olson ES, Aguilera TA, Jiang T, Scadeng M, Ellies LG, Tsien RY. Surgery with molecular fluorescence imaging using activatable cell-penetrating peptides decreases residual cancer and improves survival. Proceedings of the National Academy of Sciences. 2010;107:4317–4322. doi: 10.1073/pnas.0910261107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Braud C, Vert M. Degradation of poly (β-malic acid)-monitoring of oligomers formation by aqueous SEC and HPCE. Polymer Bulletin. 1992;29:177–183. [Google Scholar]
- 111.Braud C, Vert M. Poly(β-malic acid) based biodegradable polyesters aimed at pharmacological uses. Trend Polym Sci. 1993;3:57–65. [Google Scholar]
- 112.Gasslmaier B, Holler E. Specificity and direction of depolymerization of β-poly (L-malate) catalysed by polymalatase from Physarum polycephalum. The FEBS Journal. 1997;250:308–314. doi: 10.1111/j.1432-1033.1997.0308a.x. [DOI] [PubMed] [Google Scholar]
- 113.Gasslmaier B, Krell CM, Seebach D, Holler E. Synthetic substrates and inhibitors of β-poly (L-malate)-hydrolase (polymalatase) The FEBS Journal. 2000;267:5101–5105. doi: 10.1046/j.1432-1327.2000.01573.x. [DOI] [PubMed] [Google Scholar]
- 114.Gödde C, Liebergesell M, Steinbüchel A. Isolation of poly (β-l-malic acid)-degrading bacteria and purification and characterization of the PMA hydrolase from Comamonas acidovorans strain 7789. FEMS Microbiology Letters. 1999;173:365–372. doi: 10.1111/j.1574-6968.1999.tb13527.x. [DOI] [PubMed] [Google Scholar]
- 115.Portilla-Arias JA, García-Alvarez M, de Ilarduya AM, Holler E, Galbis JA, Muñoz-Guerra S. Synthesis, degradability, and drug releasing properties of methyl esters of fungal poly(β, L-malic acid) Macromolecular Bioscience. 2008;8:540–550. doi: 10.1002/mabi.200700248. [DOI] [PubMed] [Google Scholar]
- 116.Melancon MP, Wang W, Wang Y, Shao R, Ji X, Gelovani JG, Li C. A novel method for imaging in vivo degradation of poly (L-glutamic acid), a biodegradable drug carrier. Pharmaceutical Research. 2007;24:1217–1224. doi: 10.1007/s11095-007-9253-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Galivan J, Ryan TJ, Chave K, Rhee M, Yao R, Yin D. Glutamyl hydrolase: pharmacological role and enzymatic characterization. Pharmacology & Therapeutics. 2000;85:207–215. doi: 10.1016/s0163-7258(99)00063-7. [DOI] [PubMed] [Google Scholar]
- 118.Yao R, Schneider E, Ryan TJ, Galivan J. Human gamma-glutamyl hydrolase: cloning and characterization of the enzyme expressed in vitro. Proceedings of the National Academy of Sciences. 1996;93:10134–10138. doi: 10.1073/pnas.93.19.10134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Tanaka T, Hiruta O, Futamura T, Uotani K, Satoh A, Taniguchi M, Susumu O. Purification and characterization of poly (γ-glutamic acid) hydrolase from a filamentous fungus, Myrothecium sp. TM-4222. Bioscience, Biotechnology, And Biochemistry. 1993;57:2148–2153. [Google Scholar]
- 120.Kimura K, Itoh Y. Characterization of poly-γ-glutamate hydrolase encoded by a bacteriophage genome: possible role in phage infection of Bacillus subtilis encapsulated with poly-γ-glutamate. Applied and Environmental Microbiology. 2003;69:2491–2497. doi: 10.1128/AEM.69.5.2491-2497.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Uchida I, Makino S, Sasakawa C, Yoshikawa M, Sugimoto C, Terakado N. Identification of a novel gene, dep, associated with depolymerization of the capsular polymer in Bacillus anthracis. Molecular Microbiology. 1993;9:487–496. doi: 10.1111/j.1365-2958.1993.tb01710.x. [DOI] [PubMed] [Google Scholar]
- 122.Mekasha S, Toupalová H, Linggadjaja E, Tolani HA, Anděra L, Arntzen MØ, Vaaje-Kolstad G, Eijsink VG, Agger JW. A novel analytical method for d-glucosamine quantification and its application in the analysis of chitosan degradation by a minimal enzyme cocktail. Carbohydrate Research. 2016;433:18–24. doi: 10.1016/j.carres.2016.07.003. [DOI] [PubMed] [Google Scholar]
- 123.Kopeček J, Kopečková P, Minko T, Lu Z-R. HPMA copolymer–anticancer drug conjugates: design, activity, and mechanism of action. European Journal of Pharmaceutics and Biopharmaceutics. 2000;50:61–81. doi: 10.1016/s0939-6411(00)00075-8. [DOI] [PubMed] [Google Scholar]
- 124.Moulton J. Guide for Morpholino Users: Toward therapeutics. Journal of Drug Discovery, Development and Delivery. 2016;3:270–282. [Google Scholar]
- 125.Shi J, Kantoff PW, Wooster R, Farokhzad OC. Cancer nanomedicine: progress, challenges and opportunities. Nature Reviews on Cancer. 2017;17:20–37. doi: 10.1038/nrc.2016.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lajoie JM, Shusta EV. Targeting receptor-mediated transport for delivery of biologics across the blood-brain barrier. Annual Review of Pharmacology and Toxicology. 2015;55:613–631. doi: 10.1146/annurev-pharmtox-010814-124852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Lai SK, Wang Y-Y, Hanes J. Mucus-penetrating nanoparticles for drug and gene delivery to mucosal tissues. Advanced Drug Delivery Reviews. 2009;61:158–171. doi: 10.1016/j.addr.2008.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Pardridge WM. CSF, blood-brain barrier, and brain drug delivery. Expert Opinion on Drug Delivery. 2016;13:963–975. doi: 10.1517/17425247.2016.1171315. [DOI] [PubMed] [Google Scholar]
- 129.Karim R, Palazzo C, Evrard B, Piel G. Nanocarriers for the treatment of glioblastoma multiforme: Current state-of-the-art. Journal of Controlled Release. 2016;227:23–37. doi: 10.1016/j.jconrel.2016.02.026. [DOI] [PubMed] [Google Scholar]
- 130.Meng J, Agrahari V, Youm I. Advances in targeted drug delivery approaches for the central nervous system tumors: the inspiration of nanobiotechnology. Journal of Neuroimmune Pharmacology. 2017;12:84–98. doi: 10.1007/s11481-016-9698-1. [DOI] [PubMed] [Google Scholar]
- 131.Webster DM, Sundaram P, Byrne ME. Injectable nanomaterials for drug delivery: Carriers, targeting moieties, and therapeutics. European Journal of Pharmaceutics and Biopharmaceutics. 2013;84:1–20. doi: 10.1016/j.ejpb.2012.12.009. [DOI] [PubMed] [Google Scholar]
- 132.Aparicio-Blanco J, Martín-Sabroso C, Torres-Suárez A-I. In vitro screening of nanomedicines through the blood brain barrier: A critical review. Biomaterials. 2016;103:229–255. doi: 10.1016/j.biomaterials.2016.06.051. [DOI] [PubMed] [Google Scholar]
- 133.Canal F, Sanchis J, Vicent MJ. Polymer–drug conjugates as nano-sized medicines. Current Opinion in Biotechnology. 2011;22:894–900. doi: 10.1016/j.copbio.2011.06.003. [DOI] [PubMed] [Google Scholar]
- 134.Caffo M, Raudino G, Caruso G. Nanotechnology and brain tumors drug delivery. Recent Patents on Nanomedicine. 2013;3:26–36. [Google Scholar]
- 135.Pardridge WM. The Blood-Brain Barrier: Bottleneck in Brain Drug Development. NeuroRx. 2005;2:3–14. doi: 10.1602/neurorx.2.1.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Lécuyer M-A, Kebir H, Prat A. Glial influences on BBB functions and molecular players in immune cell trafficking. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 2016;1862:472–482. doi: 10.1016/j.bbadis.2015.10.004. [DOI] [PubMed] [Google Scholar]
- 137.Armulik A, Genove G, Mae M, Nisancioglu MH, Wallgard E, Niaudet C, He L, Norlin J, Lindblom P, Strittmatter K, Johansson BR, Betsholtz C. Pericytes regulate the blood-brain barrier. Nature. 2010;468:557–561. doi: 10.1038/nature09522. [DOI] [PubMed] [Google Scholar]
- 138.Kim S-S, Harford JB, Pirollo KF, Chang EH. Effective treatment of glioblastoma requires crossing the blood brain barrier and targeting tumors including cancer stem cells: The promise of nanomedicine. Biochemical and Biophysical Research Communications. 2015;468:485–489. doi: 10.1016/j.bbrc.2015.06.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Ramanathan A, Nelson AR, Sagare AP, Zlokovic BV. Impaired vascular-mediated clearance of brain amyloid beta in Alzheimer’s disease: the role, regulation and restoration of LRP1. Frontiers in Aging Neuroscience. 2015;7:136. doi: 10.3389/fnagi.2015.00136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Dréan A, Goldwirt L, Verreault M, Canney M, Schmitt C, Guehennec J, Delattre J-Y, Carpentier A, Idbaih A. Blood-brain barrier, cytotoxic chemotherapies and glioblastoma. Expert Review of Neurotherapeutics. 2016;16:1285–1300. doi: 10.1080/14737175.2016.1202761. [DOI] [PubMed] [Google Scholar]
- 141.Begley DJ. Delivery of therapeutic agents to the central nervous system: the problems and the possibilities. Pharmacology & Therapeutics. 2004;104:29–45. doi: 10.1016/j.pharmthera.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 142.Liddelow SA. Development of the choroid plexus and blood-CSF barrier. Frontiers in Neuroscience. 2015;9:32. doi: 10.3389/fnins.2015.00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Matsumae M, Sato O, Hirayama A, Hayashi N, Takizawa K, Atsumi H, Sorimachi T. Research into the physiology of cerebrospinal fluid reaches a new horizon: intimate exchange between cerebrospinal fluid and interstitial fluid may contribute to maintenance of homeostasis in the central nervous system. Neurologia medicochirurgica. 2016;56:416–441. doi: 10.2176/nmc.ra.2016-0020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Woodworth GF, Dunn GP, Nance EA, Hanes J, Brem H. Emerging insights into barriers to effective brain tumor therapeutics. Frontiers in Oncology. 2014;4:126. doi: 10.3389/fonc.2014.00126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Popescu AM, Purcaru SO, Alexandru O, Dricu A. New perspectives in glioblastoma antiangiogenic therapy. Contemporary Oncology/Współczesna Onkologia. 2015;20:109–118. doi: 10.5114/wo.2015.56122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kamran N, Calinescu A, Candolfi M, Chandran M, Mineharu Y, Asad AS, Koschmann C, Nunez FJ, Lowenstein PR, Castro MG. Recent advances and future of immunotherapy for glioblastoma. Expert Opinion on Biological Therapy. 2016;16:1245–1264. doi: 10.1080/14712598.2016.1212012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, Scheithauer BW, Kleihues P. The 2007 WHO classification of tumours of the central nervous system. Acta neuropathologica. 2007;114:97–109. doi: 10.1007/s00401-007-0243-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Verhaak RGW, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, Alexe G, Lawrence M, O’Kelly M, Tamayo P, Weir BA, Gabriel S, Winckler W, Gupta S, Jakkula L, Feiler HS, Hodgson JG, James CD, Sarkaria JN, Brennan C, Kahn A, Spellman PT, Wilson RK, Speed TP, Gray JW, Meyerson M, Getz G, Perou CM, Hayes DN C.G.A.R. Network. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nanocarriers as an emerging platform for cancer therapy. Nat Nano. 2007;2:751–760. doi: 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]
- 150.Becker KP, Yu J. Status quo—standard-of-care medical and radiation therapy for glioblastoma. The Cancer Journal. 2012;18:12–19. doi: 10.1097/PPO.0b013e318244d7eb. [DOI] [PubMed] [Google Scholar]
- 151.House TW. FACT SHEET: Investing in the cancer Moonshot. 2016 [Google Scholar]
- 152.Anzalone N, Gerevini S, Scotti R, Vezzulli P, Picozzi P. Detection of cerebral metastases on magnetic resonance imaging: intraindividual comparison of gadobutrol with gadopentetate dimeglumine. Acta Radiologica. 2009;50:933–940. doi: 10.1080/02841850903095385. [DOI] [PubMed] [Google Scholar]
- 153.Cha S. Update on Brain Tumor Imaging: From Anatomy to Physiology. American Journal of Neuroradiology. 2006;27:475. [PMC free article] [PubMed] [Google Scholar]
- 154.Li X, Zhang X-N, Li X-D, Chang J. Multimodality imaging in nanomedicine and nanotheranostics. Cancer Biology & Medicine. 2016;13:339–348. doi: 10.20892/j.issn.2095-3941.2016.0055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Leinenga G, Langton C, Nisbet R, Gotz J. Ultrasound treatment of neurological diseases — current and emerging applications. Nat Rev Neurol. 2016;12:161–174. doi: 10.1038/nrneurol.2016.13. [DOI] [PubMed] [Google Scholar]
- 156.Gobbo OL, Sjaastad K, Radomski MW, Volkov Y, Prina-Mello A. Magnetic nanoparticles in cancer theranostics. Theranostics. 2015;5:1249–1263. doi: 10.7150/thno.11544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Hild WA, Breunig M, Goepferich A. Quantum dots – nano-sized probes for the exploration of cellular and intracellular targeting. European Journal of Pharmaceutics and Biopharmaceutics. 2008;68:153–168. doi: 10.1016/j.ejpb.2007.06.009. [DOI] [PubMed] [Google Scholar]
- 158.Montagne A, Toga AW, Zlokovic BV. Blood-brain barrier permeability and gadolinium: Benefits and potential pitfalls in research. JAMA Neurology. 2016;73:13–14. doi: 10.1001/jamaneurol.2015.2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Runge VM, Aoki S, Bradley WG, Jr, Chang K-H, Essig M, Ma L, Ross JS, Valavanis A. Magnetic resonance imaging and computed tomography of the brain—50 years of innovation, with a focus on the future. Investigative radiology. 2015;50:551–556. doi: 10.1097/RLI.0000000000000170. [DOI] [PubMed] [Google Scholar]
- 160.Hadjipanayis CG, Bouras A, Chang S. Applications of multifunctional nanoparticles in malignant brain tumours. European Association of NeuroOncology Magazine. 2013;4:9–15. [Google Scholar]
- 161.Mura S, Couvreur P. Nanotheranostics for personalized medicine. Advanced Drug Delivery Reviews. 2012;64:1394–1416. doi: 10.1016/j.addr.2012.06.006. [DOI] [PubMed] [Google Scholar]
- 162.Luo M, Wang H, Wang Z, Cai H, Lu Z, Li Y, Du M, Huang G, Wang C, Chen X. A STING-activating nanovaccine for cancer immunotherapy. Nature Nanotechnology. 2017 doi: 10.1038/nnano.2017.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Ohgushi M, Nagayama K, Wada A. Dextran-magnetite: A new relaxation reagent and its application to T2 measurements in gel systems. Journal of Magnetic Resonance. 1969;29(1978):599–601. [Google Scholar]
- 164.Spence T, De Souza R, Dou Y, Stapleton S, Reilly RM, Allen C. Integration of imaging into clinical practice to assess the delivery and performance of macromolecular and nanotechnology-based oncology therapies. Journal of Controlled Release. 2015;219:295–312. doi: 10.1016/j.jconrel.2015.09.036. [DOI] [PubMed] [Google Scholar]
- 165.Remsen LG, McCormick CI, Roman-Goldstein S, Nilaver G, Weissleder R, Bogdanov A, Hellström I, Kroll RA, Neuwelt EA. MR of carcinoma-specific monoclonal antibody conjugated to monocrystalline iron oxide nanoparticles: the potential for noninvasive diagnosis. American Journal of Neuroradiology. 1996;17:411. [PMC free article] [PubMed] [Google Scholar]
- 166.Key J, Leary JF. Nanoparticles for multimodal in vivo imaging in nanomedicine. International journal of nanomedicine. 2014 doi: 10.2147/IJN.S53717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Kim SM, Jeong CH, Woo JS, Ryu CH, Lee J-H, Jeun S-S. In vivo near-infrared imaging for the tracking of systemically delivered mesenchymal stem cells: tropism for brain tumors and biodistribution. International Journal of Nanomedicine. 2016;11:13–23. doi: 10.2147/IJN.S97073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Posadas I, Monteagudo S, Ceña V. Nanoparticles for brain-specific drug and genetic material delivery, imaging and diagnosis. Nanomedicine. 2016;11:833–849. doi: 10.2217/nnm.16.15. [DOI] [PubMed] [Google Scholar]
- 169.Cabral Filho PE, Cardoso ALC, Pereira MIA, Ramos APM, Hallwass F, Castro MMCA, Geraldes CFGC, Santos BS, Pedroso de Lima MC, Pereira GAL, Fontes A. CdTe quantum dots as fluorescent probes to study transferrin receptors in glioblastoma cells. Biochimica et Biophysica Acta (BBA) - General Subjects. 2016;1860:28–35. doi: 10.1016/j.bbagen.2015.09.021. [DOI] [PubMed] [Google Scholar]
- 170.Zhang L, Habib AA, Zhao D. Phosphatidylserine-targeted liposome for enhanced glioma-selective imaging. Oncotarget. 2016;7:38693–38706. doi: 10.18632/oncotarget.9584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Bai Q, Zhao Z, Sui H, Chen J, Xie X, Wen F. The preparation and application of dendrimer modified cdte/cds near infrared quantum dots for brain cancer cells imaging. Applied Sciences. 2015;5 [Google Scholar]
- 172.Suganami A, Iwadate Y, Shibata S, Yamashita M, Tanaka T, Shinozaki N, Aoki I, Saeki N, Shirasawa H, Okamoto Y, Tamura Y. Liposomally formulated phospholipid-conjugated indocyanine green for intra-operative brain tumor detection and resection. International Journal of Pharmaceutics. 2015;496:401–406. doi: 10.1016/j.ijpharm.2015.10.001. [DOI] [PubMed] [Google Scholar]
- 173.Qiu L, Zhang F, Shi Y, Bai Z, Wang J, Li Y, Lee D, Ingraham C, Feng X, Yang X. Gliomas: motexafin gadolinium-enhanced molecular MR imaging and optical imaging for potential intraoperative delineation of tumor margins. Radiology. 2015;279:400–409. doi: 10.1148/radiol.2015150895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.van Schooneveld MM, Cormode DP, Koole R, van Wijngaarden JT, Calcagno C, Skajaa T, Hilhorst J, ’t Hart DC, Fayad ZA, Mulder WJM, Meijerink A. A fluorescent, paramagnetic and PEGylated gold/silica nanoparticle for MRI, CT and fluorescence imaging. Contrast media & molecular imaging. 2010;5:231–236. doi: 10.1002/cmmi.376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Regino CAS, Walbridge S, Bernardo M, Wong KJ, Johnson D, Lonser R, Oldfield EH, Choyke PL, Brechbiel MW. A dual CT-MR dendrimer contrast agent as a surrogate marker for convection-enhanced delivery of intracerebral macromolecular therapeutic agents. Contrast Media & Molecular Imaging. 2008;3:2–8. doi: 10.1002/cmmi.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Hainfeld JF, Smilowitz HM, O’Connor MJ, Dilmanian FA, Slatkin DN. Gold nanoparticle imaging and radiotherapy of brain tumors in mice. Nanomedicine. 2012;8:1601–1609. doi: 10.2217/nnm.12.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Ding H, Wu F. Image guided biodistribution and pharmacokinetic studies of theranostics. Theranostics. 2012;2:1040–1053. doi: 10.7150/thno.4652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Plotkin M, Gneveckow U, Meier-Hauff K, Amthauer H, Feußner A, Denecke T, Gutberlet M, Jordan A, Felix R, Wust P. 18 F-FET PET for planning of thermotherapy using magnetic nanoparticles in recurrent glioblastoma. International Journal of Hyperthermia. 2006;22:319–325. doi: 10.1080/02656730600734128. [DOI] [PubMed] [Google Scholar]
- 179.Malek E, Sendilnathan A, Yellu M, Petersen A, Fernandez-Ulloa M, Driscoll JJ. Metabolic tumor volume on interim PET is a better predictor of outcome in diffuse large B-cell lymphoma than semiquantitative methods. Blood Cancer Journal. 2015;5:e326. doi: 10.1038/bcj.2015.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Yoo HJ, Lee JS, Lee JM. Integrated whole body MR/PET: where are we? Korean Journal of Radiology. 2015;16:32–49. doi: 10.3348/kjr.2015.16.1.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Seo JW, Ang J, Mahakian LM, Tam S, Fite B, Ingham ES, Beyer J, Forsayeth J, Bankiewicz KS, Xu T, Ferrara KW. Self-assembled 20-nm 64Cu-micelles enhance accumulation in rat glioblastoma. Journal of Controlled Release. 2015;220(Part A):51–60. doi: 10.1016/j.jconrel.2015.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Yildirim A, Chattaraj R, Blum NT, Goldscheitter GM, Goodwin AP. Stable Encapsulation of air in mesoporous silica nanoparticles: fluorocarbon-free nanoscale ultrasound contrast agents. Advanced Healthcare Materials. 2016;5:1290–1298. doi: 10.1002/adhm.201600030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Luke GP, Hannah AS, Emelianov SY. Super-resolution ultrasound imaging in vivo with transient laser-activated nanodroplets. Nano Letters. 2016;16:2556–2559. doi: 10.1021/acs.nanolett.6b00108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Timbie KF, Mead BP, Price RJ. Drug and gene delivery across the blood brain barrier with focused ultrasound. Journal of Controlled Release. 2015;219:61–75. doi: 10.1016/j.jconrel.2015.08.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Chen Y-C, Chiang C-F, Wu S-K, Chen L-F, Hsieh W-Y, Lin W-L. Targeting microbubbles-carrying TGFβ1 inhibitor combined with ultrasound sonication induce BBB/BTB disruption to enhance nanomedicine treatment for brain tumors. Journal of Controlled Release. 2015;211:53–62. doi: 10.1016/j.jconrel.2015.05.288. [DOI] [PubMed] [Google Scholar]
- 186.Chaichana KL, Pinheiro L, Brem H. Delivery of local therapeutics to the brain: working toward advancing treatment for malignant gliomas. Therapeutic delivery. 2015;6:353–369. doi: 10.4155/tde.14.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Sengupta S, Marrinan J, Frishman C, Sampath P. Impact of temozolomide on immune response during malignant glioma chemotherapy. Clinical and Developmental Immunology. 2012;2012:831090. doi: 10.1155/2012/831090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Donato V, Papaleo A, Castrichino A, Banelli E, Giangaspero F, Salvati M, Delfini R. Prognostic implication of clinical and pathologic features in patients with glioblastoma multiforme treated with concomitant radiation plus temozolomide. Tumori. 2007;93:248. doi: 10.1177/030089160709300304. [DOI] [PubMed] [Google Scholar]
- 189.Attenello FJ, Mukherjee D, Datoo G, McGirt MJ, Bohan E, Weingart JD, Olivi A, Quinones-Hinojosa A, Brem H. Use of gliadel (BCNU) wafer in the surgical treatment of malignant glioma: a 10-year institutional experience. Annals of Surgical Oncology. 2008;15:2887. doi: 10.1245/s10434-008-0048-2. [DOI] [PubMed] [Google Scholar]
- 190.Souhami L, Seiferheld W, Brachman D, Podgorsak EB, Werner-Wasik M, Lustig R, Schultz CJ, Sause W, Okunieff P, Buckner J, Zamorano L, Mehta MP, Curran WJ., Jr Randomized comparison of stereotactic radiosurgery followed by conventional radiotherapy with carmustine to conventional radiotherapy with carmustine for patients with glioblastoma multiforme: Report of Radiation Therapy Oncology Group 93-05 protocol. International Journal of Radiation Oncology*Biology*Physics. 2004;60:853–860. doi: 10.1016/j.ijrobp.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 191.Mellinghoff IK, Wang MY, Vivanco I, Haas-Kogan DA, Zhu S, Dia EQ, Lu KV, Yoshimoto K, Huang JHY, Chute DJ, Riggs BL, Horvath S, Liau LM, Cavenee WK, Rao PN, Beroukhim R, Peck TC, Lee JC, Sellers WR, Stokoe D, Prados M, Cloughesy TF, Sawyers CL, Mischel PS. Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. New England Journal of Medicine. 2005;353:2012–2024. doi: 10.1056/NEJMoa051918. [DOI] [PubMed] [Google Scholar]
- 192.Pekmezci M, Perry A. Neuropathology of brain metastases. Surgical Neurology International. 2013;4:S245–S255. doi: 10.4103/2152-7806.111302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Chamberlain MC, Baik CS, Gadi VK, Bhatia S, Chow LQM. Systemic therapy of brain metastases: non–small cell lung cancer, breast cancer, and melanoma. Neuro-Oncology. 2017;19:i1–i24. doi: 10.1093/neuonc/now197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Owonikoko TK, Arbiser J, Zelnak A, Shu H-KG, Shim H, Robin AM, Kalkanis SN, Whitsett TG, Salhia B, Tran NL, Ryken T, Moore MK, Egan KM, Olson JJ. Current approaches to the treatment of metastatic brain tumours. Nat Rev Clin Oncol. 2014;11:203–222. doi: 10.1038/nrclinonc.2014.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Parveen S, Misra R, Sahoo SK. Nanoparticles: a boon to drug delivery, therapeutics, diagnostics and imaging. Nanomedicine: Nanotechnology, Biology and Medicine. 2012;8:147–166. doi: 10.1016/j.nano.2011.05.016. [DOI] [PubMed] [Google Scholar]
- 196.Anchordoquy TJ, Barenholz Y, Boraschi D, Chorny M, Decuzzi P, Dobrovolskaia MA, Farhangrazi ZS, Farrell D, Gabizon A, Ghandehari H, Godin B, La-Beck NM, Ljubimova J, Moghimi SM, Pagliaro L, Park J-H, Peer D, Ruoslahti E, Serkova NJ, Simberg D. Mechanisms and barriers in cancer nanomedicine: addressing challenges, looking for solutions. ACS Nano. 2017;11:12–18. doi: 10.1021/acsnano.6b08244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Wong HL, Wu XY, Bendayan R. Nanotechnological advances for the delivery of CNS therapeutics. Advanced Drug Delivery Reviews. 2012;64:686–700. doi: 10.1016/j.addr.2011.10.007. [DOI] [PubMed] [Google Scholar]
- 198.Chou ST, Patil R, Galstyan A, Gangalum PR, Cavenee WK, Furnari FB, Ljubimov VA, Chesnokova A, Kramerov AA, Ding H, Falahatian V, Mashouf L, Fox I, Black KL, Holler E, Ljubimov AV, Ljubimova JY. Simultaneous blockade of interacting CK2 and EGFR pathways by tumor-targeting nanobioconjugates increases therapeutic efficacy against glioblastoma multiforme. J Control Release. 2016;244:14–23. doi: 10.1016/j.jconrel.2016.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Xu X, Ho W, Zhang X, Bertrand N, Farokhzad O. Cancer nanomedicine: from targeted delivery to combination therapy. Trends in Molecular Medicine. 2015;21:223–232. doi: 10.1016/j.molmed.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Cole AJ, David AE, Wang J, Galbán CJ, Hill HL, Yang VC. Polyethylene glycol modified, cross-linked starch-coated iron oxide nanoparticles for enhanced magnetic tumor targeting. Biomaterials. 2011;32:2183–2193. doi: 10.1016/j.biomaterials.2010.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Gupta AK, Gupta M. Synthesis and surface engineering of iron oxide nanoparticles for biomedical applications. Biomaterials. 2005;26:3995–4021. doi: 10.1016/j.biomaterials.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 202.Sengupta S, Kulkarni A. Design principles for clinical efficacy of cancer nanomedicine: a look into the basics. ACS Nano. 2013;7:2878–2882. doi: 10.1021/nn4015399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Jokerst JV, Lobovkina T, Zare RN, Gambhir SS. Nanoparticle PEGylation for imaging and therapy. Nanomedicine. 2011;6:715–728. doi: 10.2217/nnm.11.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Rabanel J-M, Faivre J, Paka GD, Ramassamy C, Hildgen P, Banquy X. Effect of polymer architecture on curcumin encapsulation and release from PEGylated polymer nanoparticles: Toward a drug delivery nano-platform to the CNS. European Journal of Pharmaceutics and Biopharmaceutics. 2015;96:409–420. doi: 10.1016/j.ejpb.2015.09.004. [DOI] [PubMed] [Google Scholar]
- 205.Ernsting MJ, Murakami M, Roy A, Li S-D. Factors controlling the pharmacokinetics, biodistribution and intratumoral penetration of nanoparticles. Journal of Controlled Release. 2013;172:782–794. doi: 10.1016/j.jconrel.2013.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Soo Choi H, Liu W, Misra P, Tanaka E, Zimmer JP, Itty Ipe B, Bawendi MG, Frangioni JV. Renal clearance of quantum dots. Nat Biotech. 2007;25:1165–1170. doi: 10.1038/nbt1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Hamilton JL, Imran ul-haq M, Abbina S, Kalathottukaren MT, Lai BFL, Hatef A, Unniappan S, Kizhakkedathu JN. In vivo efficacy, toxicity and biodistribution of ultra-long circulating desferrioxamine based polymeric iron chelator. Biomaterials. 2016;102:58–71. doi: 10.1016/j.biomaterials.2016.06.019. [DOI] [PubMed] [Google Scholar]
- 208.Zhang F, Xu C-L, Liu C-M. Drug delivery strategies to enhance the permeability of the blood brain barrier for treatment of glioma. Drug Design, Development and Therapy. 2015;9:2089–2100. doi: 10.2147/DDDT.S79592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307:58–62. doi: 10.1126/science.1104819. [DOI] [PubMed] [Google Scholar]
- 210.Maeda H, Bharate GY, Daruwalla J. Polymeric drugs for efficient tumor-targeted drug delivery based on EPR-effect. European Journal of Pharmaceutics and Biopharmaceutics. 2009;71:409–419. doi: 10.1016/j.ejpb.2008.11.010. [DOI] [PubMed] [Google Scholar]
- 211.Cheng Y, Morshed RA, Auffinger B, Tobias AL, Lesniak MS. Multifunctional nanoparticles for brain tumor imaging and therapy. Advanced Drug Delivery Reviews. 2014;66:42–57. doi: 10.1016/j.addr.2013.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Daniels TR, Delgado T, Rodriguez JA, Helguera G, Penichet ML. The transferrin receptor part I: Biology and targeting with cytotoxic antibodies for the treatment of cancer. Clinical Immunology. 2006;121:144–158. doi: 10.1016/j.clim.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 213.Kang T, Jiang M, Jiang D, Feng X, Yao J, Song Q, Chen H, Gao X, Chen J. Enhancing glioblastoma-specific penetration by functionalization of nanoparticles with an iron-mimic peptide targeting transferrin/transferrin receptor complex. Molecular Pharmaceutics. 2015;12:2947–2961. doi: 10.1021/acs.molpharmaceut.5b00222. [DOI] [PubMed] [Google Scholar]
- 214.Pardridge WM. Blood-brain barrier drug targeting: the future of brain drug development. Molecular interventions. 2003;3:90. doi: 10.1124/mi.3.2.90. [DOI] [PubMed] [Google Scholar]
- 215.Banks WA. From blood-brain barrier to blood-brain interface: new opportunities for CNS drug delivery. Nat Rev Drug Discov. 2016;15:275–292. doi: 10.1038/nrd.2015.21. [DOI] [PubMed] [Google Scholar]
- 216.Moinuddin A, Morley JE, Banks WA. Regional variations in the transport of interleukin-1α across the blood-brain barrier in icr and aging SAMP8 mice. Neuroimmunomodulation. 2000;8:165–170. doi: 10.1159/000054814. [DOI] [PubMed] [Google Scholar]
- 217.Ren J, Shen S, Wang D, Xi Z, Guo L, Pang Z, Qian Y, Sun X, Jiang X. The targeted delivery of anticancer drugs to brain glioma by PEGylated oxidized multi-walled carbon nanotubes modified with angiopep-2. Biomaterials. 2012;33:3324–3333. doi: 10.1016/j.biomaterials.2012.01.025. [DOI] [PubMed] [Google Scholar]
- 218.Sabel M, Rommel F, Kondakci M, Gorol M, Willers R, Bilzer T. Laser induced thermotherapy and blood-brain barrier changes: a review. medical laser application. 2002;17:164–169. [Google Scholar]
- 219.Yang F-Y, Wang H-E, Lin G-L, Lin H-H, Wong T-T. Evaluation of the increase in permeability of the blood brain barrier during tumor progression after pulsed focused ultrasound. International Journal of Nanomedicine. 2012;7:723–730. doi: 10.2147/IJN.S28503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Winkler F, Kozin SV, Tong RT, Chae S-S, Booth MF, Garkavtsev I, Xu L, Hicklin DJ, Fukumura D, di Tomaso E, Munn LL, Jain RK. Kinetics of vascular normalization by VEGFR2 blockade governs brain tumor response to radiation: Role of oxygenation, angiopoietin-1, and matrix metalloproteinases. Cancer Cell. 2004;6:553–563. doi: 10.1016/j.ccr.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 221.Endrich B, Reinhold HS, Gross JF, Intaglietta M. Tissue perfusion inhomogeneity during early tumor growth in rats. Journal of the National Cancer Institute. 1979;62:387–395. [PubMed] [Google Scholar]
- 222.Jain RK, Stylianopoulos T. Delivering nanomedicine to solid tumors. Nat Rev Clin Oncol. 2010;7:653–664. doi: 10.1038/nrclinonc.2010.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Shimizu T, Kurozumi K, Ishida J, Ichikawa T, Date I. Adhesion molecules and the extracellular matrix as drug targets for glioma. Brain Tumor Pathology. 2016;33:97–106. doi: 10.1007/s10014-016-0261-9. [DOI] [PubMed] [Google Scholar]
- 224.Netti PA, Berk DA, Swartz MA, Grodzinsky AJ, Jain RK. Role of extracellular matrix assembly in interstitial transport in solid tumors. Cancer Research. 2000;60:2497. [PubMed] [Google Scholar]
- 225.McKee TD, Grandi P, Mok W, Alexandrakis G, Insin N, Zimmer JP, Bawendi MG, Boucher Y, Breakefield XO, Jain RK. Degradation of fibrillar collagen in a human melanoma xenograft improves the efficacy of an oncolytic herpes simplex virus vector. Cancer Research. 2006;66:2509. doi: 10.1158/0008-5472.CAN-05-2242. [DOI] [PubMed] [Google Scholar]
- 226.Krol A, Maresca J, Dewhirst MW, Yuan F. Available volume fraction of macromolecules in the extravascular space of a fibrosarcoma: implications for drug delivery. Cancer Research. 1999;59:4136. [PubMed] [Google Scholar]
- 227.Nugent LJ, Jain RK. Extravascular diffusion in normal and neoplastic tissues. Cancer Research. 1984;44:238. [PubMed] [Google Scholar]
- 228.Stylianopoulos T, Poh M-Z, Insin N, Bawendi MG, Fukumura D, Munn Lance L, Jain RK. Diffusion of particles in the extracellular matrix: the effect of repulsive electrostatic interactions. Biophysical Journal. 2010;99:1342–1349. doi: 10.1016/j.bpj.2010.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Pluen A, Boucher Y, Ramanujan S, McKee TD, Gohongi T, di Tomaso E, Brown EB, Izumi Y, Campbell RB, Berk DA, Jain RK. Role of tumor-host interactions in interstitial diffusion of macromolecules: cranial vs. subcutaneous tumors. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:4628–4633. doi: 10.1073/pnas.081626898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Nance EA, Woodworth GF, Sailor KA, Shih T-Y, Xu Q, Swaminathan G, Xiang D, Eberhart C, Hanes J. A dense poly(ethylene glycol) coating improves penetration of large polymeric nanoparticles within brain tissue. Science Translational Medicine. 2012;4:149ra119. doi: 10.1126/scitranslmed.3003594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Tong R, Langer R. Nanomedicines targeting the tumor microenvironment. The Cancer Journal. 2015;21:314–321. doi: 10.1097/PPO.0000000000000123. [DOI] [PubMed] [Google Scholar]
- 232.Fujita M, Lee B-S, Khazenzon NM, Penichet ML, Wawrowsky KA, Patil R, Ding H, Holler E, Black KL, Ljubimova JY. Brain tumor tandem targeting using a combination of monoclonal antibodies attached to biopoly(β-l-malic acid) Journal of Controlled Release. 2007;122:356–363. doi: 10.1016/j.jconrel.2007.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Nichols JW, Bae YH. Odyssey of a cancer nanoparticle: From injection site to site of action. Nano Today. 2012;7:606–618. doi: 10.1016/j.nantod.2012.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Núñez-Lozano R, Cano M, Pimentel B, de la Cueva-Méndez G. ‘Smartening’ anticancer therapeutic nanosystems using biomolecules. Current Opinion in Biotechnology. 2015;35:135–140. doi: 10.1016/j.copbio.2015.07.005. [DOI] [PubMed] [Google Scholar]
- 235.Wang F, Wang Y, Zhang X, Zhang W, Guo S, Jin F. Recent progress of cell-penetrating peptides as new carriers for intracellular cargo delivery. Journal of Controlled Release. 2014;174:126–136. doi: 10.1016/j.jconrel.2013.11.020. [DOI] [PubMed] [Google Scholar]
- 236.Gu G, Xia H, Hu Q, Liu Z, Jiang M, Kang T, Miao D, Tu Y, Pang Z, Song Q, Yao L, Chen H, Gao X, Chen J. PEG-co-PCL nanoparticles modified with MMP-2/9 activatable low molecular weight protamine for enhanced targeted glioblastoma therapy. Biomaterials. 2013;34:196–208. doi: 10.1016/j.biomaterials.2012.09.044. [DOI] [PubMed] [Google Scholar]
- 237.Torchilin V. Intracellular delivery of protein and peptide therapeutics. Drug Discovery Today: Technologies. 2008;5:e95–e103. doi: 10.1016/j.ddtec.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 238.Kou L, Sun J, Zhai Y, He Z. The endocytosis and intracellular fate of nanomedicines: Implication for rational design. Asian Journal of Pharmaceutical Sciences. 2013;8:1–10. [Google Scholar]
- 239.Koren E, Torchilin VP. Cell-penetrating peptides: breaking through to the other side. Trends in Molecular Medicine. 2012;18:385–393. doi: 10.1016/j.molmed.2012.04.012. [DOI] [PubMed] [Google Scholar]
- 240.Kleemann E, Neu M, Jekel N, Fink L, Schmehl T, Gessler T, Seeger W, Kissel T. Nano-carriers for DNA delivery to the lung based upon a TAT-derived peptide covalently coupled to PEG-PEI. Journal of Controlled Release. 2005;109:299–316. doi: 10.1016/j.jconrel.2005.09.036. [DOI] [PubMed] [Google Scholar]
- 241.Hou KK, Pan H, Schlesinger PH, Wickline SA. A role for peptides in overcoming endosomal entrapment in siRNA delivery — A focus on melittin. Biotechnology Advances. 2015;33:931–940. doi: 10.1016/j.biotechadv.2015.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Rozema DB, Ekena K, Lewis DL, Loomis AG, Wolff JA. Endosomolysis by masking of a membrane-active agent (EMMA) for cytoplasmic release of macromolecules. Bioconjugate Chemistry. 2003;14:51–57. doi: 10.1021/bc0255945. [DOI] [PubMed] [Google Scholar]
- 243.Shete HK, Prabhu RH, Patravale VB. Endosomal escape: a bottleneck in intracellular delivery. Journal of nanoscience and nanotechnology. 2014;14:460–474. doi: 10.1166/jnn.2014.9082. [DOI] [PubMed] [Google Scholar]
- 244.Sahoo SK, Parveen S, Panda JJ. The present and future of nanotechnology in human health care, Nanomedicine: Nanotechnology. Biology and Medicine. 2007;3:20–31. doi: 10.1016/j.nano.2006.11.008. [DOI] [PubMed] [Google Scholar]
- 245.Kozielski KL, Tzeng SY, Hurtado De Mendoza BA, Green JJ. Bioreducible Cationic Polymer-Based Nanoparticles for Efficient and Environmentally Triggered Cytoplasmic siRNA Delivery to Primary Human Brain Cancer Cells. ACS Nano. 2014;8:3232–3241. doi: 10.1021/nn500704t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Yi L, Li J. CRISPR-Cas9 therapeutics in cancer: promising strategies and present challenges. Biochimica et Biophysica Acta (BBA) - Reviews on Cancer. 2016;1866:197–207. doi: 10.1016/j.bbcan.2016.09.002. [DOI] [PubMed] [Google Scholar]
- 247.Sachdeva M, Sachdeva N, Pal M, Gupta N, Khan IA, Majumdar M, Tiwari A. CRISPR/Cas9: molecular tool for gene therapy to target genome and epigenome in the treatment of lung cancer. Cancer Gene Ther. 2015;22:509–517. doi: 10.1038/cgt.2015.54. [DOI] [PubMed] [Google Scholar]
- 248.Savić N, Schwank G. Advances in therapeutic CRISPR/Cas9 genome editing. Translational Research. 2016;168:15–21. doi: 10.1016/j.trsl.2015.09.008. [DOI] [PubMed] [Google Scholar]
- 249.Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science. 2013;339:819. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. RNA-Guided Human Genome Engineering via Cas9. Science. 2013;339:823. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Fellmann C, Gowen BG, Lin P-C, Doudna JA, Corn JE. Cornerstones of CRISPR-Cas in drug discovery and therapy. Nat Rev Drug Discov. 2017;16:89–100. doi: 10.1038/nrd.2016.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Nair M, Jayant RD, Kaushik A, Sagar V. Getting into the brain: Potential of nanotechnology in the management of NeuroAIDS. Advanced Drug Delivery Reviews. 2016;103:202–217. doi: 10.1016/j.addr.2016.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Hu Q, Sun W, Wang C, Gu Z. Recent advances of cocktail chemotherapy by combination drug delivery systems. Advanced Drug Delivery Reviews. 2016;98:19–34. doi: 10.1016/j.addr.2015.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Shao L, Yang Z-J, Andrén D, Johansson P, Käll M. Gold Nanorod Rotary Motors Driven by Resonant Light Scattering. ACS Nano. 2015;9:12542–12551. doi: 10.1021/acsnano.5b06311. [DOI] [PubMed] [Google Scholar]
- 255.Jiang X, Fitch S, Wang C, Wilson C, Li J, Grant GA, Yang F. Nanoparticle engineered TRAIL-overexpressing adipose-derived stem cells target and eradicate glioblastoma via intracranial delivery. Proc Natl Acad Sci U S A. 2016;113:13857–13862. doi: 10.1073/pnas.1615396113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Chen J-R, Yao Y, Xu H-Z, Qin Z-Y. Isocitrate Dehydrogenase (IDH)1/2 Mutations as Prognostic Markers in Patients With Glioblastomas. Medicine. 2016;95:e2583. doi: 10.1097/MD.0000000000002583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Zhang B, Chang K, Ramkissoon S, Tanguturi S, Bi WL, Reardon DA, Ligon KL, Alexander BM, Wen PY, Huang RY. Multimodal MRI features predict isocitrate dehydrogenase genotype in high-grade gliomas. Neuro-Oncology. 2017;19:109–117. doi: 10.1093/neuonc/now121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Shim H, Wei L, Holder CA, Guo Y, Hu XP, Miller AH, Olson JJ. Use of High-Resolution Volumetric MR Spectroscopic Imaging in Assessing Treatment Response of Glioblastoma to an HDAC Inhibitor. American Journal of Roentgenology. 2014;203:W158–W165. doi: 10.2214/AJR.14.12518. [DOI] [PMC free article] [PubMed] [Google Scholar]