


Abstract
Bisulfite genomic sequencing is the method of choice for the generation of methylation maps with single-base resolution. The method is based on the selective deamination of cytosine to uracil by treatment with bisulfite and the sequencing of subsequently generated PCR products. In contrast to cytosine, 5-methylcytosine does not react with bisulfite and can therefore be distinguished. In order to investigate the potential for optimization of the method and to determine the critical experimental parameters, we determined the influence of incubation time and incubation temperature on the deamination efficiency and measured the degree of DNA degradation during the bisulfite treatment. We found that maximum conversion rates of cytosine occurred at 55°C (4–18 h) and 95°C (1 h). Under these conditions at least 84–96% of the DNA is degraded. To study the impact of primer selection, homologous DNA templates were constructed possessing cytosine-containing and cytosine-free primer binding sites, respectively. The recognition rates for cytosine (≥97%) and 5-methylcytosine (≥94%) were found to be identical for both templates.
INTRODUCTION
In vertebrates and higher plants methylation of cytosine plays an important role in the organization of gene expression. Elucidation of the DNA methylation patterns will be of great benefit to our understanding of the structure of complex genomes. The dideoxy sequencing approach (1) is the method of choice for sequencing of cloned or PCR amplified DNA. It does not, however, distinguish between cytosine and 5-methylcytosine (5mC). For this reason Frommer et al. (2) developed a selective chemical derivation method using bisulfite which is known as ‘bisulfite genomic sequencing’. Essentially, the method is based on the complete deamination of cytosine to uracil by modification with bisulfite followed by PCR of the modified genomic DNA, direct sequencing of the PCR products or subcloning and sequencing of the subclones. 5mC does not react with bisulfite (3). In the final sequence pattern all original cytosines appear as thymines while 5mC residues are displayed as cytosines.
The bisulfite method was successfully used for methylation analysis of genomic DNA from different sources (4–9). All investigators followed the modification protocol originally developed by Frommer et al. (2): genomic DNA is prepared, fragmented by shearing or digestion with restriction enzymes, denatured with sodium hydroxide, treated with a concentrated bisulfite–hydroquinone solution at pH 5, desalted and desulfonated with sodium hydroxide. Finally, the DNA is neutralized, desalted and resolved in water or storage buffer. Over the last few years, several groups have studied the original reaction conditions and suggested technical improvements (8,10–12). However, some of these observations are controversial and no comprehensive investigation of all the parameters has been published so far. Since the method is based on the complete conversion of cytosine and the complete non-conversion of 5mC, we decided to quantify and to compare the sensitivity and specificity at different time/temperature combinations for the incubation with bisulfite. DNA degradation is an undesired side-effect of the bisulfite treatment and has an impact on the detection limit of the method. Currently, it is not known how much of the DNA is actually lost during the treatment. We decided therefore to determine the degree of DNA degradation by two independent methods: HPLC and quantitative PCR (qPCR).
The only way to determine DNA methylation patterns with single-molecule and single-base resolution is to subclone the PCR products into appropriate vectors and to sequence the inserts of individual clones. Since our laboratory has an interest in methylation patterns of individual molecules we have focussed our investigation on this method. The comparison with other procedures [e.g. direct sequencing of the PCR products (13), Ms-SNuPE (14), MSP (15)] would be beyond the scale of the presented study and deserves a dedicated investigation on its own. We present in this work for the first time a comprehensive investigation of the influence of time and temperature of the bisulfite reaction on the sensitivity and specificity of the bisulfite sequencing method and deliver an estimation of the degree of DNA degradation during the treatment.
MATERIALS AND METHODS
Bisulfite genomic sequencing
As experimental target, we designed an artificial double- stranded DNA molecule of 193 bp with a G+C content of 49.2% and named it ARTα (GenBank accession no. AF316370). This DNA was inserted into pGEM-T (Promega). It possesses primer binding sites for the primers art-1 (5′-GTG ATT AGT GTT TTG AGG TAT TT) and art-2 (5′-CTT TCA AAA CTA AAC AAA CAA A) that allow amplification of the DNA only if the cytosines within the primer binding sites are converted to uracil (Fig. 1). For this type of DNA we introduce the term ‘selective template’. In addition, a second template molecule ARTβ was generated (GenBank accession no. AF316371). ARTβ is nearly identical to ARTα but has primer binding sites that contain T instead of C. This type of DNA, which allows for amplification with or without bisulfite treatment, we call ‘non-selective template’.
Figure 1.

Alignment of model DNA used in this study. Primers art-1 and art-2 are given in lower case. ARTα and ARTβ are identical with the exception of the primer binding sites and six more mutations, one of them an A to G transition in position 92 which allows identification of the templates after bisulfite treatment.
Alkaline denaturation. The initial denaturation step as well as the final desulfonation reaction were already optimized (16). To obtain single-stranded DNA, the DNA was incubated in 0.3 M NaOH at 37°C for 20 min (16). The plasmid DNA pGEM#ARTα (0.5 fg to 50 ng representing 150 to 1.5 × 1010 target molecules) was supplemented with 10 µg salmon sperm DNA (Boehringer), yeast RNA (Boehringer) or tRNA (Sigma R-8508) as carrier. The reaction volume was adjusted to 100 µl with sterile water and 11 µl of 3 M NaOH were added. In our hands these conditions resulted in complete strand separation. However, it was shown that an increase of the incubation temperature to 42°C was necessary to obtain full denaturation of a G+C rich DNA region (9).
Deamination. For the deamination step, sodium bisulfite solutions of two different concentrations were used. Initially, solution I (3.87–4.26 M HSO3–) was applied to deliver results that are comparable to earlier studies (11). Since the reaction kinetics of the cytosine deamination are dependent on bisulfite concentration (17) a saturated bisulfite solution (solution II) was used in the subsequent experiments. Solution I: 4.05 g sodium bisulfite (ACS reagent grade Sigma S-8890) were dissolved without vigorous shaking in 8 ml water and the pH was adjusted to a final pH of 5.0 with 400 µl of 10 M NaOH. Hydroquinone (0.22 g; Sigma H-9003) was dissolved in 10 ml water and 500 µl of this solution were added to the bisulfite solution resulting in a final concentration of 10 mM which has been previously determined to be optimal (18). After complete dissolution of the sodium bisulfite the volume was adjusted to 10 ml and the solution was passed through a 0.45 µm filter membrane. Since commercially available sodium bisulfite is a mixture of sodium bisulfite and metabisulfite the final concentration of HSO3– can only be estimated to be between 3.87 and 4.26 M. Solution II was prepared like solution I but 5.41 g sodium bisulfite were used resulting in a saturated solution of 5.20–5.69 M HSO3–. Sodium bisulfite and hydroquinone powders were stored under vacuum. Freshly prepared bisulfite solution (1200 µl) was added directly to the denatured DNA. The reaction was overlaid with 200 µl mineral oil (Sigma M-3516) and incubated in the dark for 1, 4, 18 and 24 h, respectively, at several temperatures between 0 and 95°C. Each bisulfite treatment was carried out independently between two and five times.
Desulfonation. The DNA was desalted using either QIAex II (Qiagen) or Wizard DNA Clean-Up systems (Promega) (19) according to the protocol provided by the manufacturer and resolved in 110 µl of 1 mM Tris–Cl pH 8. 100 µl of this solution were combined with 11 µl of a 3 M NaOH solution and incubated at 37°C for 20 min. Incubation times up to 45 min did not harm the DNA. Sodium hydroxide concentrations in the final solution below 0.3 M result in lower PCR yields indicating incomplete desulfonation (data not shown).
Neutralization and desalting. After desulfonation the DNA was neutralized with 47 µl of 10 M ammonium acetate, precipitated with 500 µl of 96% ethanol under addition of 1 µg carrier tRNA at –20°C overnight, washed with 500 µl of 70% ethanol and dried. The addition of carrier nucleic acid in the denaturation, deamination and precipitation steps is essential for the complete recovery of the bisulfite treated DNA. In our hands, it did not matter which sort of nucleic acid was applied as a carrier; however, the use of either RNA resulted in a clearer PCR product and certainly eliminates the risk of undesired co-amplification of carrier DNA.
Storage conditions of bisulfite-treated DNA. The DNA was resolved in 20–100 µl sterile 1 mM Tris–Cl pH 8 and stored at –20°C. Under these conditions it can be stored for at least 6 months without affecting the PCR yield. Storage of the bisulfite-treated DNA in unbuffered water resulted in considerable hydrolyzation within 3 days and should be avoided.
Amplification, cloning and sequencing. The bisulfite treated DNA was PCR amplified in 50 µl reaction volume containing reaction buffer (Perkin Elmer), 70–250 µM each dNTP, 2.5 U AmpliTaq (Perkin Elmer), 50 pmol primers art-1 and art-2 overlaid with mineral oil (Sigma M-3516). Cycling conditions were: 2 min at 95°C, 30 cycles of 30 s at 95°C, 1 min at 54°C, 1 min at 72°C subsequently followed by 10 min at 72°C. PCR products were precipitated with PEG mix (26.2% PEG 8000, 6.6 mM MgCl2, 0.6 M sodium acetate) (20), subcloned into pGEM-T (Promega) or pCR 2.1 (Invitrogen TA cloning kit), respectively. Twelve clones of each single bisulfite treatment were sequenced using a Ready Reaction DyeDeoxy Terminator Cycle Sequencing Kit (Perkin Elmer). Sequencing reactions were separated and analyzed on an ABI Prism DNA sequencer 377 (Perkin Elmer). To calculate the conversion rate the resulting clones from each bisulfite treatment were aligned and the converted cytosines were counted. The ratio of converted cytosines to the total of cytosines before the treatment was calculated in percent. For direct sequencing, the PCR products were purified by gel filtration with Sepharose CL-6B (Pharmacia), and sequenced using cycle sequencing, [α-33P]ddNTP and primer art-1. Reaction products were separated on a (bis)acrylamide gel and exposed for autoradiography. Autoradiograms were manually inspected.
Methylation at the HpaII site of ARTα and ARTβ
Plasmid DNA (500 ng) was incubated with 80 µM S-adenosylmethionine and 4 U HpaII methyltransferase (NEB) in HpaII methylase buffer (NEB) for 2 h at 37°C in a total volume of 20 µl, followed by deactivation at 70°C for 10 min. DNA was precipitated, dried and resolved in 20 µl NEbuffer 1 (NEB) and incubated with 2 U HpaII (NEB) for 2 h at 37°C in parallel with unmethylated pGEM#ARTα and pGEM#ARTβ, respectively, as a control. The reaction mixtures were applied on a 1% agarose (Serva) TAE gel (21) and the undigested and therefore methylated band was cut out and purified using the QIAEX II gel extraction kit (Qiagen).
Quantification of DNA loss by HPLC and competitive qPCR
Single-stranded M13mp18 (5 µg) was treated with bisulfite for 5, 15 and 60 min at 55°C as indicated above with the difference that no other nucleic acids were added as carrier. In the control, the bisulfite solution was replaced by 1 mM Tris pH 8. After the neutralization step the DNA was desalted, digested with nuclease P1 to 5′ monophosphate nucleotides and quantified on a Smart HPLC system (Pharmacia) with a MiniQ PC 3.2/3 column using 20 mM Tris–Cl pH 8.22 and a 0–1 M NaCl gradient as buffer system. The recovery (mol found/mol expected × 100) for dCMP (96.5 ± 6.6%), dTMP (100.5 ± 5.6%), dAMP (113.8 ± 4.7%) and dGMP (86.7 ± 21.5%) was quantitative. Competitive quantitative PCR (22) was chosen as an independent proof of the HPLC result. pGEM#ARTβ (50, 5 and 0.5 ng; Fig. 1) was bisulfite-treated as described above. This DNA was PCR-amplified in the presence of a constant amount of 0.1 ng pGEM#ARTγ SmaI+ (GenBank accession no. AF316373) competitor DNA. The PCR product was purified and cut with SmaI. Ten microliters of the restriction digest was applied on a 2% TAE agarose gel, stained and photographed after electrophoresis. Mass standards from 5 to 100 ng (GibcoBRL) were applied on every gel and used to estimate the amount of undigested wild-type DNA band and of the 140 bp fragment band of the competitor DNA. A standard curve was created using a dilution series of 0.7–0.02 ng wild-type pGEM#ARTγ and 0.1 ng pGEM#ARTγ SmaI+ as competitor. ARTγ (GenBank accession no. AF316372) is identical to successfully bisulfite-treated ARTβ. This standard curve (r2 = 0.96) was used to determine the concentration of wild-type DNA pGEM#ARTβ after the bisulfite treatment.
RESULTS
The optimal bisulfite genomic sequencing method must deliver complete conversion of cytosine residues to uracil while the minor base 5mC should remain intact. In addition, the loss of DNA during the modification reaction due to non-specific degradation should be kept as small as possible. Finally, the result should be independent of the primer sequences. These four points were investigated one by one based on the general procedure outlined above.
Under which incubation time/temperature combinations is cytosine completely converted to uracil? The results of the experiments with subcloned PCR products are summarized in Table 1. Full cytosine deamination can be achieved under several time/temperature combinations. The most robust temperature is 55°C with complete conversion between 4 and 18 h (4 h, 99.48 ± 0.39%; 18 h, 99.65 ± 0.52%). At 95°C >98% cytosines were converted after 1 h (98.05 ± 2.2%) but not yet after 30 min (95%). When incubated >2 h at 95°C, no PCR product could be amplified anymore. Our data indicate that both deamination as well as DNA degradation proceeds faster at higher temperatures giving a time window of 14 h for 55°C but only 1 h for 95°C. Incubation at 15 or 0°C seemed to deliver an alternative for the incubation at high temperatures. Incubation for 4 h at 15°C gave 98.35 ± 0.5% deamination rate; however, the PCR yield was very low. In order to make enough PCR product for direct sequencing we increased the concentration of Taq polymerase. This improved the yield but the percentage of converted cytosines in the PCR products was lower than estimated from the subcloned products. Incubation at 0°C for 1 h resulted in 98% conversion but only in one out of three experiments could a PCR product be amplified. Also in this case, PCR yield was very low and addition of extra Taq polymerase resulted only in a weak improvement. Taken together, we considered these reaction conditions not to be robust enough for further investigation. All other time/temperature combinations delivered conversion rates <98% and were rejected. To exclude that our findings are severly biased by the subcloning procedure, PCR products were sequenced directly. An example for 4 h treatment is shown in Figure 2. Clearly, complete conversion could only be achieved at 55°C. The results for this series of experiments were obtained with bisulfite solution I. For all following experiments saturated bisulfite solution II was used.
Table 1. Summary of the relationship of cytosine deamination rate with incubation temperature and incubation time with the bisulphite–hydroquinone solution.
| Temperature (°C) |
1 h |
n |
4 h |
n |
18 h |
n |
| 0 | 98.00 | 1 (10) | 78.30 ± 25.03 | 2 (20) | 82.80 | 1 (11) |
| 15 | 88.60 ± 3.68 | 2 (24) | 98.35 ± 0.49 | 2 (22) | 54.25 ± 11.24 | 2 (18) |
| 35 | 30.05 ± 0.07 | 2 (19) | 69.75 ± 1.34 | 2 (21) | 94.10 ± 5.09 | 2 (21) |
| 55 | 77.30 ± 2.40 | 2 (23) | 99.48 ± 0.39 | 4 (41) | 99.65 ± 0.52 | 4 (45) |
| 80 | 92.69 ± 9.22 | 2 (23) | – | – | – | – |
| 85 | 94.65 ± 0.49 | 2 (24) | 93.20 ± 9.62 | 2 (19) | 93.40 ± 0.32 | 2 (24) |
| 95 | 98.05 ± 2.20 | 5 (53) | – | – | – | – |
The ratio of deaminated cytosine is given as a percentage of total cytosine content in between the primer binding sites of the upper strand of ARTα. n indicates the number of independent experiments that yielded a PCR product. The total number of sequenced clones is given in parentheses.
Figure 2.
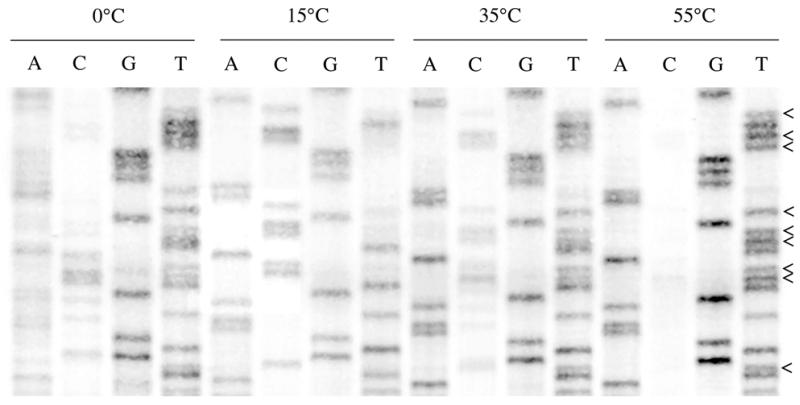
Part of an autoradiogram for four sequencing reactions. The unconverted native sequence is GAC TCC GGG AAC GCC TAC CTG ATA AGT GCT A (positions 48–64 of ARTα). Arrowheads mark the cytosine positions. Plasmid DNA was treated with sodium bisulfite solution for 4 h at 0, 15, 35 and 55°C, PCR amplified and sequenced. Only at 55°C did complete cytosine conversion occur. The low PCR yield for DNA treated at 0°C thwarts accurate sequencing of the PCR product.
Does the PCR selectively amplify the deaminated strands? In the selective template approach outlined in the Materials and Methods, deaminated DNA strands are selectively amplified because its primer binding sites contain cytosines and the primers can only bind if these are converted to uracil. It could be argued that not all cytosines are actually deaminated but simply escape the PCR amplification because the primers do not hybridize to these templates. To test this hypothesis, a new model DNA named ARTβ was created by PCR mutagenesis of ARTα. ARTα and ARTβ are identical with exception of the primer binding sites and six more mutations, one of them an A to G transition in position 92, allowing tracing of both homologs after bisulfite treatment (Fig. 1). The primer binding sites of ARTβ contain thymines instead of cytosines. The primers art-1 and art-2 amplify this template regardless of whether the bisulfite treatment was successful or not. The use of ARTβ allows estimation of the conversion efficiency independently from a possible amplification bias towards deaminated templates.
PCR products were subcloned and sequenced as described above. Experiments with non-selective template ARTβ and 1 h incubation at 0°C showed only marginal overall conversion of cytosine to uracil (0.95 ± 1.34%) and this experimental line was not followed anymore, leaving 55°C for 4 or 18 h and 95°C for 1 h as possible incubation conditions. The results for these time/temperature combinations are summarized in Table 2. The data show that both the selective and the non-selective approaches result in identical recognition rates for cytosine.
Table 2. The recognition ratio as a mol percentage of unmethylated and methylated cytosine as a function of bisulfite incubation time and temperature.
| |
|
Recognition of cytosine (%) |
|
Recognition of 5mC (%) |
|
| Selective | Non-selective | Selective | Non-selective | ||
| 4 h, 55°C | Sens. | 99.50 ± 0.45 | 98.11 ± 2.78 | 100.00 ± 0.00 | 88.89 ± 19.25 |
| Spec. | 100.00 ± 0.00 | 99.68 ± 0.55 | 86.25 ± 12.16 | 71.69 ± 30.19 | |
| 18 h, 55°C | Sens. | 99.76 ± 0.21 | 99.89 ± 0.20 | 92.80 ± 6.46 | 100.00 ± 0.00 |
| Spec. | 99.78 ± 0.21 | 100.00 ± 0.00 | 92.50 ± 6.61 | 96.67 ± 5.77 | |
| 1 h, 95°C | Sens. | 94.08 ± 7.48 | 93.73 ± 7.31 | 94.57 ± 8.57 | 96.00 ± 8.94 |
| Spec. | 99.84 ± 0.25 | 99.89 ± 0.26 | 55.24 ± 35.05 | 45.66 ± 24.32 |
Three different parameter combinations were used (first column). In the following columns sensitivity [sens. = true/(true + false negative)] and specificity [spec. = true/(true + false positive)] are shown. Values of 100% indicate that either complete deamination of unmethylated cytosine (columns 2 and 3) or no deamination at all for 5mC (columns 4 and 5) was achieved. The values for the selective and non-selective approach are given in parallel. A selective template would only be amplified when at least the cytosines in the primer binding sites are deaminated to uracil. The primer binding sites of the non-selective template contain no cytosine and it will be amplified irrespective of the deamination efficiency. In consequence the non-selective values are free of selection impact by primers and provide a good estimate of the actual conversion rate. At least three independent bisulfite treatments were carried out for each data point and a minimum of 22 sequencing reactions was analyzed per data entry. The recognition rate for 5mC at 55°C for 4 h (non-selective) was calculated as the average from three experiments one giving 67% and two giving 100%. The very low recognition rate of 67% is probably due to incomplete enzymatic methylation and digest and not a result of unspecific deamination since longer incubation gives better results. Specificity values are usually higher for the recognition of unmethylated cytosine resulting in a weak overestimation of 5mC content.
To investigate whether 5mC remains intact under reaction parameters that cause deamination of virtually all cytosines, ARTα and ARTβ DNA was methylated with HpaII methylase (NEB) at position 53 (Fig. 1) and treated with bisulfite.
Results are shown in Table 2. Even under the harshest conditions (95°C for 1 h in saturated bisulfite solution) at least 94% of the 5mCs are detected. All three time/temperature conditions delivered comparable results.
How much DNA is lost during the bisulfite treatment? It was shown that degradation of the template DNA during the bisulfite treatment is a major side reaction (18) but so far it was not known how much of the DNA is lost. A rapid and sufficiently accurate method for the quantification of DNA is the visual inspection or densitometry of DNA bands in electrophoresis gels. However, this method requires distinct bands and therefore cannot be applied to the DNA smears that are present after the bisulfite treatment. In order to quantify the DNA loss, two alternative approaches were therefore developed: (i) HPLC of single-strand M13 DNA digested with endonuclease P1 after treatment with bisulfite and (ii) quantitative PCR of model DNA ARTβ after the treatment.
Figure 3 displays the time course of DNA degradation measured with the HPLC approach. After 5 min incubation the intact DNA is already reduced to 9.0 ± 2.1 mass % and after 1 h incubation time only 4.2 ± 0.1 mass % of the total DNA is present. It should be noted that after this time virtually no cytosine is detectable but uracil is now present. Its exact quantification is difficult because no base line separation of dAMP and dUMP could be achieved.
Figure 3.

Degradation of single-stranded M13mp18 DNA during bisulfite treatment as a function of time: M13mp18 DNA was treated with sodium bisulfite for 5, 15 and 60 min at 55°C. After the treatment the DNA was incubated with nuclease P1 to generate 5′ monophosphate nucleotides. The amount of these dNMPs was quantified on a Smart HPLC system with MiniQ PC 3.2/3 column (Pharmacia). DNA degradation proceeds very fast and after 60 min only 4.2 ± 0.1 mass % of the initial DNA amount is detectable. At this time the dCMP concentration is below the detection limit. dUMP can be found after 15 min.
By quantitative PCR of pGEM#ARTβ we found that 16 ± 11 mass % of the DNA is present after 4 h of bisulfite treatment at 55°C. This rate is ∼10% higher than the one determined by HPLC after 1 h treatment. It is not clear whether this is due to the addition of carrier tRNA, an effect of the higher standard error of the quantitative PCR method or a result of the different quantities of starting material (5 µg DNA for HPLC and 50, 5 and 0.5 ng for qPCR).
Our data indicate that between 84 and 96% of the DNA is degraded during the bisulfite treatment.
DISCUSSION
Our findings show that at a given bisulfite concentration, the reaction kinetics of the conversion of cytosine to uracil in DNA molecules depends on the reaction temperature and the reaction time in a non-linear fashion (Table 1). Complete selective conversion of cytosine to uracil can be achieved by incubation of alkaline denatured DNA with saturated bisulfite solution at several temperature/time combinations. Three temperature/time combinations were investigated in detail: 55°C/4 h, 55°C/18 h and 95°C/1 h. Under these conditions, the deamination of cytosine is complete whereas 5mC remains unaltered. Currently, most experiments are carried out at 55°C (9,16,18,23,24). Raising the temperature up to 95°C can indeed only be recommended if sufficient amounts of DNA are available because the DNA degrades much faster under these conditions. Bisulfite incubation times >1 h lead to complete degradation of the modified DNA. However, 5mC is not deaminated at 95°C and as far as this fact is concerned every temperature above 55°C can be used. Our data show that also at 55°C after 4 h most of the DNA (84–96%) is degraded during the modification reaction. However, the remaining DNA is usually sufficient for the subsequent PCR amplification. To reduce the problems associated with DNA degradation when very few DNA is available, the embedding of DNA or whole cells in agarose was introduced by Olek et al. (23). They reported a successful nested PCR with 100 cells representing ∼500 pg template DNA.
Interestingly, our findings indicate that the bisulfite conversion has a minimum at around 25°C (Table 1). Although the sulfonation step itself is exothermic (24) the overall deamination rate increases with raising temperature (17). On the other hand double-stranded DNA is unreactive to bisulfite. We conclude from our results that <25°C the renaturation rate of DNA is higher than the deamination rate, a relation that is reversed beyond this temperature. To our knowledge our results provide the first direct evidence for this hypothesis. Renaturation is inhibited at low temperatures. It has been shown that lowering the reaction temperature to 0°C could indeed help to resolve a blockage in cytosine conversion (11). But in the hands of other authors no PCR product could be obtained after bisulfite treatment at this temperature (12). We found indeed higher cytosine conversion rates at 0°C than between 10 and 35°C indicating that the renaturation was inhibited at this temperature. However, in many cases no PCR product could be amplified and in general the standard errors of the experiments were very high. In addition, amplification under non-selective conditions showed that only a minority of DNA strands is converted at 0°C and that the cytosine conversion rates observed with the selective template were due to a selective PCR amplification of the converted molecules. Taken together, we do not think that incubation at low temperatures provides a good alternative to other means of strand separation. Incubation with bisulfite for 24 h did not improve the average conversion rate at any of the investigated temperatures (data not shown). Since DNA degradation increases with time it is evident to shorten the incubation time as much as possible, and the reaction time of 24 h was not further investigated.
With rare exceptions (6) investigators relied on primers that only amplify the bisulfite treated DNA when the deamination was successful in the primer binding sites. It was not clear whether the conversion rates observed in these experiments were at least in part a consequence of this selection effect. However, it could be argued that heterogeneity in the deamination along the DNA strand could lead to amplification of DNA that is only partially deaminated in between the fully converted primer binding sites, and unconverted cytosines would erroneously be assigned as 5mC. In order to investigate the impact of primer selection a neutral template was created that could be amplified with or without successful deamination. Its non-selective amplification allows measurement of the actual overall cytosine conversion rate without possible distortion by primer selection. The comparison presented in Table 2 shows that both the selective and non-selective approaches give identical results for incubation at 55°C (4 and 18 h) and 95°C (1 h). We conclude that under these conditions indeed all cytosines have been converted. As a consequence, our data indicate that it is legitimate to use selective primers, i.e. primers containing thymine residues instead of the original cytosine or adenine instead of guanine, respectively. This is a necessity in almost all cases because DNA simply does not contain enough cytosine free primer binding sites. While we cannot rule out that with other templates and other primers a selection of converted DNA strands takes place, the occurrence of selection in our idealized model system would have thrown serious doubt on the reliability of the method.
Bisulfite treatment of genomic DNA combined with the sequencing of subcloned PCR products delivers the methylation patterns of individual molecules. Frequently, the method is also used to estimate the distribution of average cytosine methylation along a DNA strand, or for the general quantification of the 5mC content of a DNA fragment. Quantification can, however, be jeopardized by biased PCR amplification and cloning bias. The problem of amplification bias has been addressed before (25) and it was shown that the same primer pair can preferentially amplify either the methylated or the unmethylated molecules. The question of whether the base composition of the PCR products also influences the cloning efficiency has not yet been studied. Anecdotal evidence suggests that PCR products that are free of cytosines in the top-strand are difficult to ligate into a vector or to propagate in Escherichia coli. It will be interesting to further investigate this detail also with respect to E.coli hosts and cultivation conditions. In any case from the proportion of cytosines in individual clones, conclusions should be drawn with care and under consideration of the limits of the method.
For most routine PCR reactions 50 ng human genomic DNA as template delivers a sufficient amount of PCR product. Our findings indicate that during the bisulfite treatment ∼90% of the template DNA is lost. That would imply that 500 ng genomic DNA has to be deployed for each genomic sequencing approach. In many applications this is not feasible. Therefore, it is more appropriate to use a nested PCR approach, i.e. two subsequent rounds of PCR using nested or semi-nested primer pairs (4,7,8,26). Primers will in general be longer than usual PCR primers and the primer binding sites should not contain methylatable sites. As a rule, several primers have to be tested for each PCR until a set of successful primers will be established. A guide to primer design has been published (16).
Recently, our laboratory described the methylation patterns of several genes in human (27) and Arabidopsis thaliana (28). In these studies, the bisulfite treatment was carried out at 55°C for 4 h to minimize degradation. At least 500 pg but usually 50 ng genomic template DNA was used for each nested PCR (not considering the loss during the treatment). In contrast, in experimental classes with undergraduate students we routinely applied the 95°C/1 h combination, higher DNA concentrations and non-nested PCR to tighten the schedule.
Bisulfite genomic sequencing is a powerful technique for the analysis of DNA methylation patterns. However, it is usually not easy to introduce this technique as a new method in the laboratory and to get it work reliably. Our work delivers for the first time a comprehensive description of the influence of the most critical reaction parameters. This improves the scientific basis of this technique and will help to avoid potential error sources in the experimental setup.
References
- 1.Sanger F., Nicklein,S. and Coulson,A.R. (1977) DNA sequencing with chain-terminator inhibitors. Proc. Natl Acad. Sci. USA, 74, 5463–5467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Frommer M., McDonald,L.E., Millar,D.S., Collis,C.M., Watt,F., Grigg,G.W., Molloy,P.L. and Paul,C.L. (1992) A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc. Natl Acad. Sci. USA, 89, 1827–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang R.Y.-H., Gehrke,C.W. and Ehrlich,M. (1980) Comparison of bisulphite modification of 5-methyldeoxycytidine and deoxycytidine residues. Nucleic Acids Res., 8, 4777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ngô V., Gourdji,D. and Laverrière,J.N. (1996) Site-specific methylation of the rat prolactin and growth hormone promotors correlates with gene expression. Mol. Cell. Biol., 16, 3245–3254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clark S.J., Harrison,J. and Frommer,M. (1995) CpNpG methylation in mammalian cells. Nat. Genet., 11, 20–27. [DOI] [PubMed] [Google Scholar]
- 6.Selker E.U., Fritz,D.Y. and Singer,M.J. (1993) Dense nonsymmetrical DNA methylation resulting from repeat-induced point mutations in Neurospora. Science, 262, 1724–1728. [DOI] [PubMed] [Google Scholar]
- 7.Loebel D.A.F. and Johnston,P.G. (1996) Methylation analysis of a marsupial X-linked CpG island by bisulfite genomic sequencing. Genome Res., 6, 114–123. [DOI] [PubMed] [Google Scholar]
- 8.McDonald L.E. and Kay,G.F. (1997) Methylation analysis using bisulfite genomic sequencing: application to small numbers of intact cells. Biotechniques, 22, 272–274. [DOI] [PubMed] [Google Scholar]
- 9.Stöger R., Kajimura,T.M., Brown,W.T. and Laird,C.D. (1997) Epigenetic variation illustrated by DNA methylation patterns of the fragile-X gene FMR1. Hum. Mol. Gen., 6, 1791–1801. [DOI] [PubMed] [Google Scholar]
- 10.Olek A. and Walter,J. (1997) The pre-implantation ontogeny of the H19 methylation imprint. Nat. Genet., 17, 275–276. [DOI] [PubMed] [Google Scholar]
- 11.Feil R., Charlton,J., Bird,A.P., Walter,J. and Reik,W. (1994) Methylation analysis of individual chromosomes: improved protocol for bisulphite genomic sequencing. Nucleic Acids Res., 22, 695–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Paulin R., Grigg,G.W., Davey,M.W. and Piper,A.A. (1998) Urea improves efficiency of bisulphite-mediated sequencing of 5′-methylcytosine in genomic DNA. Nucleic Acids Res., 26, 5009–5010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Paul C.L. and Clark,S.J. (1996) Cytosine methylation: quantitation by automated genomic sequencing and GENESCAN™ analysis. Biotechniques, 21, 126–133. [DOI] [PubMed] [Google Scholar]
- 14.Gonzalgo M.L. and Jones,P.A. (1997) Rapid quantitation of methylation differences at specific sites using methylation-sensitive single nucleotide primer extension (Ms-SNuPE). Nucleic Acids Res., 25, 2529–2531. [DOI] [PMC free article] [PubMed]
- 15.Herman J.G., Graff,J.G., Myöhänen,S., Nelkin,B.D. and Baylin,S.B. (1996) Methylation-specific PCR: a novel PCR assay for methlation status of CpG islands. Proc. Natl Acad. Sci. USA, 93, 9821–9826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Clark S.J. and Frommer,M. (1997) Bisulphite genomic sequencing of methylated cytosines. In Taylor,G.R. (ed.), Laboratory Methods for the Detection of Mutations and Polymorphisms in DNA. CRC Press, Boca Raton, New York, London, Tokyo, pp. 151–161.
- 17.Sono M., Wataya,Y. and Hayatsu,H. (1972) Role of bisulfite in the deamination and the hydrogen isotope exchange of cytidylic acid. J. Am. Chem. Soc., 95, 4745–4749. [DOI] [PubMed] [Google Scholar]
- 18.Raizis A.M., Schmitt,F. and Jost,J.-P. (1995) A bisulfite method of 5-methylcytosine mapping that minimizes template degradation. Anal. Biochem., 226, 161–166. [DOI] [PubMed] [Google Scholar]
- 19.Reeben M. and Prydz,H. (1994) An improved method for detection of 5-methyl-cytosine by PCR-based genomic sequencing. Biotechniques, 16, 416–417. [PubMed] [Google Scholar]
- 20.Rosenthal A., Coutelle,O. and Craxton,M. (1992) Large-scale production of DNA sequencing templates by microtitre format PCR. Nucleic Acids Res., 21, 173–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning, A Labortatory Manual, 2nd Edn, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 22.Gilliland G., Perrin,S. and Bunn,H.F. (1990) Competitive PCR for quantification of mRNA. In Innis,M.A., Gelfand,D.H., Sninsky,J.J. and White,T.J. (eds), PCR Protocols: A Guide to Methods and Applications. Academic Press, San Diego, CA, pp. 60–69.
- 23.Olek A., Oswald,J. and Walter,J. (1996) A modified and improved method for bisulphite based cytosine methylation analysis. Nucleic Acids Res., 24, 5064–5066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Clark S.J. and Frommer,M. (1995) Deamination with NaHSO3 in DNA methylation studies. In Saluz,H.P. and Wiebauer,K. (eds), DNA and Nucleoprotein Structure In Vivo. Springer Verlag, Heidelberg, Germany, pp. 123–135.
- 25.Warnecke P.M., Stirzacker,C., Melki,J.R., Millar,D.S., Paul,C.L. and Clark,S.J. (1997) Detection and measurement of PCR bias in quantitative methylation analysis of bisulphite-treated DNA. Nucleic Acids Res., 25, 4422–4426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Clark S.J., Harrison,J., Paul,C.L. and Frommer,M. (1994) High sensitivity mapping of methylated cytosines. Nucleic Acids Res., 22, 2990–2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grunau C., Hindermann,W. and Rosenthal,A. (2000) Large-scale methylation analysis of human genomic DNA reveals tissue-specific differences between the methylation profiles of genes and pseudogenes. Hum. Mol. Genet., 9, 2651–2663. [DOI] [PubMed] [Google Scholar]
- 28.Rohde A., Grunau,C., De Beck,L., Van Montagu,M., Rosenthal,A. and Boerjan,W. (1999) Carpel, a new Arabidopsis epi-mutant of the SUPERMAN gene: phenotypic analysis and DNA methylation status. Plant Cell Physiol., 40, 961–972. [DOI] [PubMed] [Google Scholar]