

Abstract
Mutations of the phosphoinositide-3-kinase (PI3K) catalytic subunit alpha gene (PIK3CA) are frequent in endometrial cancer. We sequenced exon9 and exon20 of PIK3CA in 280 primary endometrial cancers to assess the relationship with clinicopathologic variables, patient survival and associations with PIK3CA mRNA and phospho-AKT1 by gene expression and protein data, respectively. While PIK3CA mutations generally had no impact on survival, and were not associated with clinicopathological variables, patients with exon9 charge-changing mutations, providing a positive charge at the substituted amino acid residue, were associated with poor survival (p = 0.018). Furthermore, we characterized PIK3CA mutations in the metastatic setting, including 32 patients with matched primary tumors and metastases, and found a high level of concordance (85.7%; 6 out of 7 patients), suggesting limited heterogeneity. PIK3CA mRNA levels were increased in metastases compared to the primary tumors (p = 0.031), independent of PIK3CA mutation status, which rather associated with reduced PIK3CA mRNA expression. PIK3CA mutated tumors expressed higher p-AKT/AKT protein levels, both within primary (p < 0.001) and metastatic lesion (p = 0.010). Our results support the notion that the PI3K signaling pathway might be activated, both dependent- and independently of PIK3CA mutations, an aspect that should be considered when designing PIK3 pathway targeting strategies in endometrial cancer.
Introduction
Endometrial cancer is the most frequent female pelvic malignancy in industrialized countries and the incidence is increasing1, 2. The most common basis for determining prognosis and risk for developing systemic disease is the distinction into type I and II, based on histologic type and grade3. Molecular studies attempt to improve this classification since up to 20% of the type I cancers will recur, while 50% of the type II cancers do not recur2. Such characterization has shown that type I cancers are more often mutated in Homo sapiens v-Ki-ras2 Kirsten rat sarcoma viral oncogene (KRAS), fibroblast growth factor receptor 2 (FGFR2) and phosphatase and tensin homology (PTEN), and tend to be microsatellite instable and estrogen- and progesterone receptor positive2, 4. Type II cancers, that often display hormone receptor loss, have altered expression of p53 and p16 and HER2 overexpression and amplification2, 4. In addition, recent comprehensive genetic profiling has refined the molecular classification of endometrial carcinomas, reflecting clinical phenotypes2, 5. While these differences have prognostic value, they have so far not been utilized therapeutically4, 6.
The phosphoinositide 3-kinase (PI3K) signaling pathway is critical in maintaining a normal balance between cell survival and apoptosis. Activating alterations in the PI3K pathway are frequently found in cancer, and deregulation of components of this pathway is associated with carcinogenesis7–10. Class IA PI3Ks are heterodimers consisting of one catalytic (p110α p110β and p110δ and one regulatory subunit (p85α p85β and p55γ. p110α is encoded by the PI3K catalytic subunit alpha gene PIK3CA and is the most frequently altered isoform in human cancer8, 11. Under normal conditions, the PI3K pathway is activated through transmembrane tyrosine kinase growth factor receptors (RTK) or by G-protein coupled receptors, relieving the inhibitory activity of the regulatory subunit, allowing the catalytic subunit to phosphorylate phosphatidylinositol(4,5)-bisphosphate (PIP2) to generate phos-phatidylinositol(3,4,5)-triphosphate (PIP3). The reverse process is regulated by PTEN that is able to dephosphorylate PIP3 to PIP2. PIP3 acts as a docking molecule for the serine/threonine kinases phosphoinositide-dependent kinase 1 (PDK1) and AKT/protein kinase-B (PKB), enabling phosphorylation and activation of AKT1 (p-AKT) at the T308 residue11, 12. Phosphorylated AKT (p-AKT) leads to activation of mammalian target of rapamycin (mTOR) complex and increased protein synthesis of its effector p70S6K and phosphorylation of ribosomal S6 protein11.
In primary endometrial cancer lesions, PIK3CA is the second most frequently significant mutated gene after PTEN, with a frequency of 53% according to comprehensive genomic profiling including whole exome sequencing performed by The Cancer Genome Atlas (TCGA)5, 13, 14. Mutations at exon9 hotspot residues, p.E542, p.E545 and p.Q546, may cause a charge-plus change; that is a change from negatively charged glutamic acid (E) (or glutamine [Q], uncharged) to positively charged Lysine (K), and lead to an abrogation of the interaction with the regulatory subunit p85α (encoding gene: PIK3R1) and hence, relieving the inhibitory effect on p110α15–20. In contrast to the characterization performed on primary tumors, PI3K/AKT alterations in metastases or recurrences are not well described.
Despite the fact that most PIK3CA mutations lead to an overactive enzyme with growth promoting properties16, 19, the literature is inconsistent when describing the prognostic impact of PIK3CA mutations and in particular regarding the different classes of PIK3CA mutations on patient survival in different cancer types21–23. There is conflicting evidence of the effect of PIK3CA mutations on clinical variables in endometrial cancer, although relations to depths of myometrial infiltration and differentiation grade have been reported24, 25.
The purpose of this study was to analyze the impact of PIK3CA mutations, on survival and clinical characteristics, and their relation with additional markers in the PI3K pathway, including assessment of AKT1 activation status (p-AKT_T308/AKT ratio), focusing on patients with systemic disease. Next, we wanted to assess the degree of concordance of PIK3CA hotspot mutations in a set of 32 matched pairs of primary endometrial cancers and metastases.
Results
The occurrence of PIK3CA helical and kinase mutations in primary endometrial cancer
To investigate the functional consequences of mutations located in the helical membrane association domain and/or in the kinase domain of PIK3CA, we sequenced exon9 and exon20, respectively. The mutation rate was 15.7% (Fig. 1 and Supplementary Table S1), in agreement with earlier and similar reports of PIK3CA mutations (range 12–39%; by Sanger sequencing)24, 25, 35–37. 25 different missense mutations and one nonsense mutation predicted to affect 24 different amino acids (AAs) in the PIK3CA encoded protein (p110α) were identified (Fig. 1b and Supplementary Table S2). Specifically, 21 of the mutations were found within four AAs located in the helical domain, while 26 mutations were focused at eight AAs in the kinase domain in the primary tumors. Of these, two cases had two mutations in exon9, and one case had mutations in both exons 9 and 20. In total, we identified 47 mutations, distributed over 44 cases with primary tumors.
Figure 1.

Occurrence of mutations within exon9 and exon20 of the PIK3CA gene in endometrial cancer. (a) Overview of PIK3CA gene, encoding the p110α protein, a catalytic subunit of PI3K class IA proteins. Protein regions corresponding to exon9; helical domain and exon20; kinase domain, are shown with known mutational hotspot positions indicated on top. Abbreviations: p85BD; p85α regulatory subunit binding domain, RBD; RAS-binding domain, C2D; protein-kinase-C-homology-2 domain, Helical D; helical domain, Kinase D; kinase domain. (b) Overview of primary tumor (PT) and metastasis (M) sample sets and overlapping matching PT and M paired sample set (left side). Number of patients is denoted in parenthesis. Twenty-four unique mutations were observed in primary and (top-side of the exons) or in a primary tumor (under-side of the metastatic lesions by Sanger sequencing. Mutations found in primary and metastatic lesions are indicated at each side of the exon. Each dot represents a mutation in either a metastatic lesion exons). The blue dots represent a mutation that have been previously reported in the Catalogue of Somatic Mutations in cancer (COSMIC, v79) or The Cancer Genome Atlas (TCGA) databases, while the pink dots represent novel mutations not previously found in any cancer types or not seen in endometrial cancer. Three primary tumors revealed double mutations: p.E545A/E545D, p.E542V/E542*(STOP), p.Q546R/G1049S.
Four positions on PIK3CA were more frequently mutated and considered hotspot sites in agreement with other studies; p.E542, p.E545, p.Q546 and p.H1047, with 6, 9, 5 and 15 mutations, respectively (Fig. 1 and Supplementary Table S2)24, 25, 38. Mutations within these AAs accounted for 75% of all the mutations we detected in primary tumors, comparable to the rate in TCGA and Catalogue of Somatic Mutations in cancer (COSMIC)13, 14, 39 (74.4% and 75%, respectively; Supplementary Table S1).
In addition to hotspot mutations, we detected p.E542*(STOP), a PIK3CA mutation never observed in any cancer and two mutations that had not previously been reported in endometrial cancer; p.L540V and p.G1050S (Fig. 1b and Supplementary Table S2)5, 13, 14, 39.
Relation of PIK3CA mutations to clinicopathological variables, survival and markers of the PI3K pathway
The PIK3CA mutations in primary tumors were not associated with any of the clinicopathologic variables or biomarkers investigated (Table 1 and Supplementary Table S3). PIK3CA mutations did not influence disease specific endometrial carcinoma survival (Fig. 2a). However, structural analyses of PIK3CA mutations have elucidated that helical and kinase mutations induce gain-of-function of p110α through different mechanisms7, 40. When stratifying according to localization of the PIK3CA mutations (Fig. 2b), patients with exon9 mutations in the helical domain, tended to have a worse outcome compared to patients with exon20 mutations of the kinase domain (p = 0.113) or patients with no PIK3CA mutations detected (p = 0.033).
Table 1.
Comparison of clinicopathologic variables and PIK3CA mutation status in 280 primary endometrial carcinomas assessed by Sanger sequencing.
| PIK3CA mutation | Total cases n | No | Yes | p-valuea |
|---|---|---|---|---|
| Variable | n (%) | n (%) | ||
| Age | 280 | 0.4 | ||
| <66 | 123 (86.6) | 19 (13.4) | ||
| ≥66 | 114 (82.6) | 24 (17.4) | ||
| BMI | 271 | 0.6 | ||
| <30 | 153 (83.6) | 30 (16.4) | ||
| ≥30 | 76 (86.4) | 12 (13.6) | ||
| FIGO stage | 280 | 0.8 | ||
| I-II | 191 (84.9) | 34 (15.1) | ||
| III-IV | 46 (83.6) | 9 (16.4) | ||
| Myometrial infiltration | 278 | 0.1 | ||
| <50% infiltration | 140 (88.1) | 19 (11.9) | ||
| ≥50% infiltration | 96 (80.7) | 23 (19.3) | ||
| Lymph node metastasis | 235 | 0.9 | ||
| No | 170 (84.2) | 32 (15.8) | ||
| Yes | 28 (84.8) | 5 (15.8) | ||
| Histologic type | 280 | 0.4 | ||
| Endometrioid | 195 (85.5) | 33 (14.5) | ||
| Non-Endometrioid | 42 (80.8) | 10 (19.2) | ||
| Grade | 277 | 0.6 | ||
| High-medium | 149 (85.6) | 25 (14.4) | ||
| Low | 85 (83.3) | 17 (16.7) | ||
| DNA ploidy | 231 | 0.4 | ||
| Diploid | 147 (86.0) | 24 (14.0) | ||
| Aneuploid | 49 (81.7) | 11 (18.3) | ||
| ERα | 273 | 0.8 | ||
| Positive | 175 (85.3) | 31 (15.0) | ||
| Negative | 56 (83.6) | 11 (16.4) | ||
| PR | 276 | 0.6 | ||
| Positive | 174 (84.1) | 33 (15.9) | ||
| Negative | 60 (87.0) | 9 (13.0) |
aThe p-value was estimated using the Chi-square test. n = number of cases analyzed. Abbreviations: BMI; body mass index, FIGO; International Federation of Gynecology and Obstetrics. ERα; Estrogen receptor alpha. PR; progesterone receptor.
Figure 2.
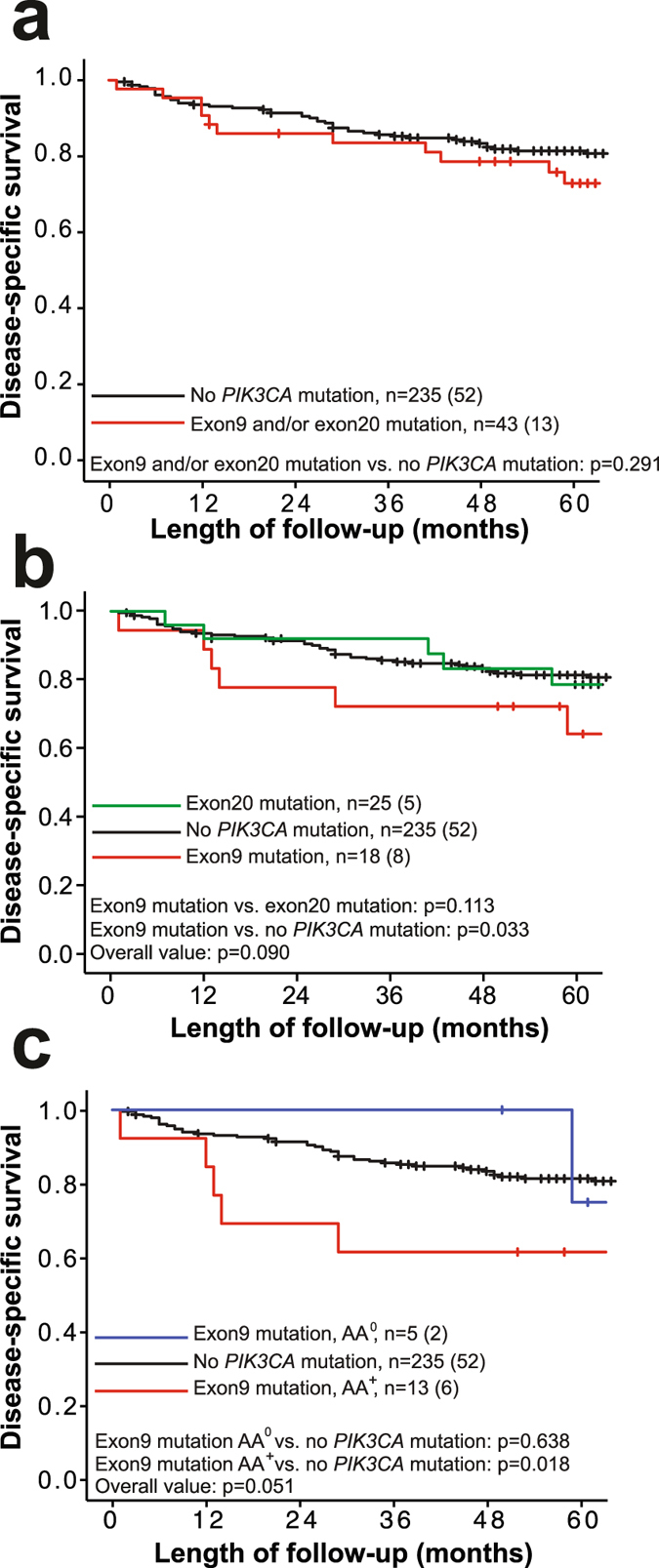
Disease-specific survival according to the PIK3CA mutation status in endometrial cancer. (a) PIK3CA mutation (exon9 and/or exon20) compared to no mutation detected. (b) PIK3CA exon9 mutations compared to PIK3CA exon20 mutations or non-mutated. (c) PIK3CA exon9 mutation leading to a change from negative to positive charge at affected amino acids (AA+), compared to mutation not leading to change of charge (AA°) and compared to no mutation. Curves are plotted by the Kaplan-Meier method; n = number of cases in each category followed by number of disease specific deaths, with comparison of survival between categories using log-rank (Mantel-Cox) test.
Mutations of the helical domain, encoded by exon9 were further stratified into two categories; those predicting a charge-plus alteration, (from negative to positive charged AA, herein denoted as AA+) and those mutations predicting not to cause such a change (AA°) (Supplementary Table S2). We added p.Q546R and p.Q546H to the group of AA+ previously described (p.E542K, p.E545K, p.Q546K)16, since these mutations also provide a change to a positively charged side chain of the substituted AA. Patients with AA+ mutations had poorer survival (p = 0.018) compared to patients with wild type PIK3CA or substitutions without charge-plus change (Fig. 2c). In multivariate survival analyses, the AA+ substitutions demonstrated an independent prognostic impact (p = 0.012, HR of 3.00, CI 1.27–7.07) when adjusting for important clinicopathologic variables in a multivariate Cox model (age, histologic type and grade; Supplementary Table S4). Further survival analysis based on these charge-positive substitutions showed reduced survival in non-endometrioid histologic type (p = 0.019; Supplementary Fig. S1a), but they did not in the endometrioid type tumors (Supplementary Fig. S1b). There was no correlation between exon9 mutations and histologic type when split into two groups (endometrioid and non-endometrioid; p = 0.749; Fisher’s Exact Test), nor with specific histologic subtypes listed in Supplementary Table S6 (p = 0.759; Pearson’s Chi-Square test). Similarly, exon9 AA+ cases were neither correlated to histologic types (endometrioid and non-endometrioid; p = 0.267, Fisher’s Exact Test, Supplementary Table S3) and nor to specific histologic subtypes (Supplementary Table S6, p = 0.499; Pearson’s Chi-Square test). Similar results were found when analyzing recurrence-free survival, where cases with initial metastatic disease were excluded. Also, in this setting, exon9 mutations and exon9 AA+ mutations also provided significantly lower survival compared to non-mutated (Supplementary Fig. S2; p = 0.007 and p = 0.036, respectively).
As PIK3CA mutations may affect different targets in the PI3K signaling pathway, we investigated these mutations in relation to several molecular markers known to be associated with PI3K pathway activation or to poor prognosis in endometrial cancer (Supplementary Table S5); including protein expression of Stathmin, p85α and PTEN (immunohistochemistry; IHC), KRAS mutations, PIK3CA amplification (assessed by Fluorescent in situ hybridization; FISH) or PIK3CA mRNA expression (Agilent microarray)7, 11, 24, 27, 30. The PIK3CA mutations did not correlate with any of the abovementioned factors. In contrast, a significant correlation (p = 0.007) was revealed between PIK3CA mutations and high p-AKT/AKT expression (ratio of RPPA-values), indicative of PI3K pathway activation (Supplementary Table S5).
Specific PIK3CA mutations are consistent from primary tumor to metastasis in paired tumor samples
PIK3CA mutation rates were similar for primary tumors that later metastasized (17.5%; n = 97) compared to those that did not metastasize (14.7%; n = 184). Within the unique set of primary tumors with corresponding metastases (n = 32), the mutation frequency in metastases (21.9%) was similar to the primary tumors (18.8%). Seven patients out of 32 with overlapping primary and metastatic lesions available for sequencing were found to harbor PIK3CA mutations (Fig. 3). The exact same mutation in the primary and corresponding metastatic lesion was detected in six out of the eight specific mutations detected in these cases. Two discordant cases presented a PIK3CA mutation only in one of the two types of lesions: Patient EC-1 presented the same mutation in two metastatic lesions that was not found in the primary tumor. Patient EC-3 had two PIK3CA mutations in the primary tumor, but only one of these mutations was detected in its corresponding metastasis (Fig. 3). Possible explanations of the mutation discordance between the primary tumor and the metastatic lesion for the two patients may be attributed to too low sensitivity of the applied sequencing method, tumor heterogeneity, or the content of tumor cells versus stromal contamination. High-throughput sequencing data has reported substantial intratumor heterogeneity as well as intertumor heterogeneity between primary tumor and metastasis41, 42. Interestingly, in an unrelated in depth whole exome sequencing study, we were able to detect the “missing” mutation also in the EC-1 primary tumor43. Thus, the final concordance of PIK3CA mutations is 87.5% in our sample set.
Figure 3.
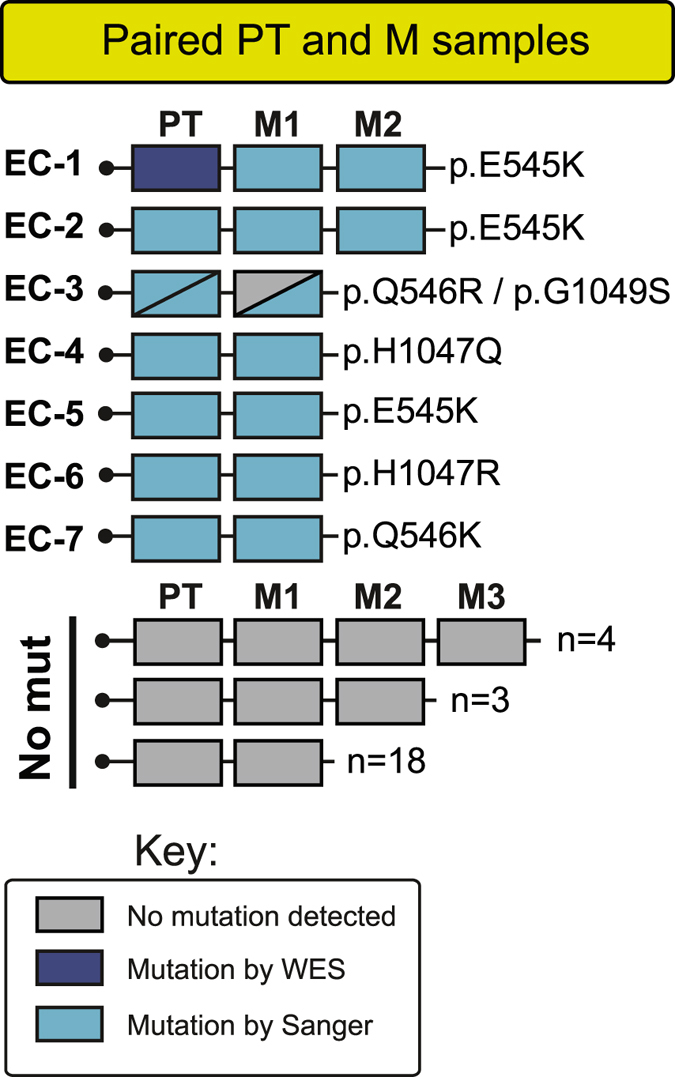
The specific PIK3CA mutations are concordant in the metastases compared with their paired corresponding primary endometrial cancer tumor. Most specific mutations in PIK3CA within primary tumors are detected in their corresponding metastasis counterpart within same patient. Key-box with color indications for mutation status and method used for detecting mutation. Abbreviations: PT; primary tumor, M1-3; unique individual metastases numbered for patients with multiple metastatic lesions, WES; whole exome sequencing.
RNF183 expression is significantly upregulated in PIK3CA mutated primary tumors
We exploited our array-derived mRNA expression data for primary tumors with accompanying mutational status (n = 135), to search for differentially expressed genes between tumors that were PIK3CA mutated or not, through the Significance Analysis of Microarray (SAM) method. Applying the cutoffs as described in the methods section, we only found one gene; the E3 ubiquitin ligase ring finger protein 183 (RNF183), to be significantly differentially expressed between the two groups with a fold change of 3.7 (log2-scale) in the PIK3CA mutated tumors compared to those with no mutation detected. Thus, global mRNA expression was not noticeably different in PIK3CA mutated compared to non-mutated tumors.
Metastatic lesions show increased PIK3CA mRNA expression independent of mutation status, and increased p-AKT/AKT protein levels dependent of PIK3CA mutations
We investigated the mRNA expression of PIK3CA, and found it increased from primary tumor to paired metastases within paired samples (p = 0.031; Fig. 4a). This increase was mostly attributable to cases without mutations, as half of the patients with mutations in this paired set showed reduced expression levels considering directional PIK3CA mRNA changes from primary to metastatic lesion within same case (Fig. 4b). To elucidate the effect of PIK3CA mutations on PIK3CA expression, we investigated the subset of patients with both mutation and expression data available (n = 169; Supplementary Fig. S3a), and found that mutations significantly reduced the PIK3CA expression, but only in the metastases (p = 0.017). High PIK3CA expression in primary tumors alone was associated with significant poorer survival compared to cases with low expression (p < 0.001; Supplementary Fig. S1c).
Figure 4.

PIK3CA expression level increases from corresponding primary to metastatic lesions. (a) The PIK3CA mRNA expression level increases significantly from the primary tumors to metastatic lesions measured within same patient. (b) Binary directional shift in PIK3CA expression level in the metastasis compared to the primary tumor. The absence of mutation is most likely to increase PIK3CA expression towards metastatic state, while half of the cases with mutations also display reduced PIK3CA expression in the metastatic lesions. Number of cases is indicated as affected cases/total cases in each group. (c) The PI3K mRNA signature score defined by Gustafson et al.29 and (d) expression of p-AKT/AKT comparing the paired samples of corresponding primary and metastatic tumors. For simplicity, first metastatic lesion (M1) was used as comparison when multiple metastases were available. Statistical test: Mann-Whitney U test.
We employed the PI3K signature score developed by Gustafson29, as a measure of PI3K pathway activation, on the set of paired primary and metastatic tumors, and found a tendency of increased PI3K activation in metastases (borderline significance of p = 0.055; Fig. 4c). The PI3K activation score seemed to be independent of the PIK3CA mutation status (Supplementary Fig. S3b). Measuring the p-AKT/AKT-ratio, with AKT acting downstream of PIK3CA, we found no significant differences between primary and metastatic lesions (Fig. 4d; RPPA data). However, a correlation between PIK3CA mutations and p-AKT/AKT was observed, with increased p-AKT/AKT in mutated compared to non-mutated PIK3CA tumors, both within primary tumors (p < 0.001) and within metastases (p = 0.010; Supplementary Fig. S3c).
We also investigated how the expression levels of these PI3K-pathway factors influenced cancer progression towards metastasis. The expression of PIK3CA mRNA increased concurrent with more invasive features within primary tumors (Supplementary Fig. S3d), comparing primary tumors that did not recur during follow-up to those that were diagnosed as metastatic at primary surgery (p = 0.005) or those that recurred later (p = 0.003). The PI3K signature score did not reach significance within these groups (Supplementary Fig. S3e). However, the p-AKT/AKT-ratio was higher in cases with no recurrence compared to those that were metastatic at primary surgery (p = 0.003; Supplementary Fig. S3f).
Discussion
PIK3CA mutations are known to activate the PI3K pathway. Despite extensive genetic characterization, the impact of PIK3CA mutations on clinical outcome and their relation to clinical variables remains unsettled in many cancer types, including endometrial cancer11, 16, 44. Considering the high prevalence of PIK3CA mutations in endometrial cancer, and the unresolved impact in clinical and metastatic settings, we focused our study on mutations of PIK3CA 5.
In endometrial cancer, findings related to patient survival are contradictory and have suggested that PIK3CA mutations are favorable45, unfavorable46 or have neutral effect on patient survival36, 47.
In our data, the presence of a PIK3CA mutation, considering all PIK3CA mutations, had no impact on survival, possibly because mutations in different exons may act through different mechanisms7, 40. However, we found an association between PIK3CA exon9 mutations and poor outcome in this study, contrasting exon20 mutations that were associated with improved survival. Data supporting our findings, relating exon9 mutations and survival have been reported for non-small cell lung cancer, soft-tissue sarcomas and breast cancer22, 48, 49, wheremutations were associated with reduced survival.
Interestingly, we found that patients with exon9 charge-plus changing substitutions in the helical domain showed even poorer survival (p = 0.018; Fig. 2c). Activating mutations in the helical domain (hotspots p.E542, p.E545 and p.Q546) are positioned at an exposed surface and believed to have the same effect as PI3K activation through RTK; abrogating the inhibitory effect of regulatory subunit p85α on the p110α catalytic subunit by interfering with the nSH2 binding to p110α15, 16, 20, 50, 51. These interactions are dominated by electrostatic interactions where the predominantly negatively charged loop of p110α (residues 542–546) interacts with the positively charged nSH2 domain of p85α, positioning nSH2 in an inhibitory structure15, 16, 18, 20, 51. Our finding supports earlier studies indicating that mutations that increase the positive surface charge of p110α could lead to gain of function and suggesting that exon9 mutations in the helical domain of p110α protein may have a functional and possibly clinically relevant effect16. We note with interest that when stratifying by histology, the effect of exon9 AA+ mutations on survival appeared prevalent in patients of non-endometrioid histologic type, as survival was impaired significantly in this group (p = 0.019; Supplementary Fig. S1a, mutations observed in 8.8% of cases) but not within the endometrioid types (p = 0.426; Supplementary Fig. S1b, mutations observed in 4.4% of cases). Neither exon9 nor exon9 AA+ mutations were associated with non-endometrioid histologic type that as such could have confounded the survival analysis. Further studies, including more patients with charge-plus changing PIK3CA substitutions, in particular those with non-endometrioid tumors, and a better understanding of the molecular mechanism of p110α (and interaction partners) is required to confirm and extend our observations.
We did not find any correlation between the PIK3CA mutation status and clinicopathological variables in our data. This is in line with previous observations where no relation between PIK3CA mutations and clinical variables were found24, 25, 35, 36, 45 and PIK3CA mutations appeared randomly distributed within the recently suggested molecular subgroups of endometrial cancer5. In the literature, there is no reported association of PIK3CA mutations with age, histology, grade, FIGO stage, ER and PR status24, 25, 36, 45, although PIK3CA mutations have been associated with myometrial invasion depth24 and poor differentiation grade25. It is possible that analysis of the PIK3CA mutations in larger cohorts will reveal subgroups that might be useful in disease stratification and future treatment. Such subgroups could include critical regulators of the PI3K pathway, such PIK3R1 and PTEN, again also found altered in endometrial cancer5, 11.
The finding of only one differentially expressed gene (RNF183) in our genome-wide array-based expression data, comparing tumors with and without PIK3CA mutations, is in line with the observed lack of associations with clinical and survival data we observe. Interestingly, the RNF183 gene has previously been identified as a potential biomarker for detection of endometrial cancer in uterine aspirates52 and was also found to be an independent factor for the prediction of lymph node metastasis in this disease53.
Our sequencing strategy has some limitations. First, our overall level of detection is lesser than that what can be achieved by high throughput sequencing approaches, although comparable at hotspot sites (Supplementary Table S1). Second, as we choose to target exon9 and exon20 (encoding helical and kinase domains of p110α, including hotspot sites p.E542, p.E545, p.Q546 and p.H1047), the larger part of PIK3CA was excluded from the mutation screen. Previous data has indicated recurrent mutations within the amino-terminal of p110α located in exons 1–7 of PIK3CA, suggesting additional hotspot mutations such as the p.R88Q within exon 15, 17, 54. Investigating N-terminal mutations (and other regions of p110α), and in particular their relation to clinical data, should be included in future studies; if not more high throughput approaches are selected that will cover this.
Metastases remains a key problem in oncology, as metastatic disease is responsible for up to 90% of cancer-related mortality, and yet one of the most poorly understood aspects of cancer55. PIK3CA mutations might predict response and resistance to targeted treatment with PI3K/AKT/mTOR pathway inhibitors as shown in clinical trials56. As a potential target, it is also relevant to explore the relationship of PIK3CA mutations between primary tissue and metastatic lesions, which is generally lesser explored due to lack of available tissue.
Our data show a concordance rate of 85.7% (6/7 cases) of specific mutations within our small sample set of matching primary tumor or metastases. The high consistency in our data seems to contradict the general heterogeneity issue revealed by high-throughput studies41, 42, but the homogeneity of PIK3CA mutations may particularly be attributed to two related observations. First, PIK3CA mutations do occur early in endometrial cancer progression, as shown earlier, and this may therefore contribute to the consistency of mutations we observe in our matched sample set28, 43. Second, PIK3CA mutations appear to be highly clonal in endometrial tumors as described in a recent high throughput sequencing study investigating spatial genetic relationships in the primary-metastasis setting43. A third explanation to the apparent “conserved state of PIK3CA mutations” is perhaps our focus on hotspot mutations in exon9 and −20, excluding mutation discoveries in other exons, and hence, possible masking the heterogeneity that might have been seen with other approaches. Bergström et al. 57 reported a concordance of 60% (6 of 10 cases) between paired samples of PIK3CA mutations in endometrial cancer. Focusing on their equivalent PIK3CA hotspot mutations (exons 9 and 20), they achieved a 57% concordance rate, which is somewhat lower compared to our study, although both studies display few paired-samples cases with PIK3CA mutations of either primary tumors, metastases or both. Thus, conclusions should be drawn with caution, and studies with larger set of paired primary tumors and metastases are warranted.
Our observation that PIK3CA mutations are present in the primary tumors and metastases with similar frequency, rules out the possibility that increased PIK3CA mutation rates in the metastases (rare mutations or a specific hotspot mutation) are “drivers” of the metastatic process itself. This emphasizes that increased knowledge about PIK3CA mutations, and their relation to other markers, is required to understand tumor initiation and progression, and for utilizing PIK3CA as a target in personalized therapeutic modalities
Investigation of the p-AKT/AKT-ratio in matched primary tumors and metastases showed no differences in expression levels. Thus, p-AKT levels may not be important in the metastatic progression, also underpinned by the association of increased tumor aggressiveness and decreased p-AKT/AKT-ratio (p = 0.003; Supplementary Fig. S3f), indicating that high p-AKT might be more important earlier during tumor development. On the other side, PIK3CA mutations seemed to activate AKT, as expression was higher among mutated primary and metastatic tumors (Supplementary Table S5 and Supplementary Fig. S3c), similar to observations made in colorectal cancer58. This suggests that PIK3CA mutations activate an AKT dependent pathway that, as suggested by Vasudevan et al.59, is most important in the formation of primary tumors.
PIK3CA mRNA expression increased from primary tumors to their corresponding metastasis (p = 0.031; Fig. 4a), also seen as a tendency of increased PI3K signature score (although only borderline significant p = 0.055; Fig. 4c). Previously, PIK3CA mRNA expression has been found to increase from normal control tissue to endometrial cancer tissues and from endometrioid to non-endometrioid histologic type47. Interestingly, in our study PIK3CA mutations seemed most likely to affect PIK3CA expression negatively (Fig. 4b and Supplementary Fig. S3a). We also found that high PIK3CA mRNA expression associated with poor prognosis and showed increased expression in metastases, suggesting independence of PIK3CA mutation status.
Conclusion
In summary, this study suggests that exon9 mutations of PIK3CA, in particular those leading to charge-plus changing substitutions, could affect structural properties of the encoded protein p110α, and this may provide a functional link to the reduced survival observed in affected endometrial cancer patients. Moreover, PIK3CA mutations seem to be present in primary tumors and metastases at relatively high consistency, suggesting early occurrence and “genetic conservation” during progression towards metastases. Finally, our data supports several studies demonstrating that AKT-signaling may be both dependent and independent of PIK3CA mutations in cancer.
Materials and Methods
Patient series
Since May 2001, fresh frozen tumor samples from paired primary and metastatic lesions have been prospectively sampled together with clinical data and formalin-fixed paraffin-embedded (FFPE) tissues from endometrial carcinoma patients at the Department of Gynecology and Obstetrics, Haukeland University Hospital, Bergen, Norway. Fresh frozen primary tumors from 280 patients were subjected to molecular profiling. From this group of patients, 32 cases also had additional corresponding fresh frozen tissue available for metastatic lesions. In nine of these patients, several recurrences were available adding up to the total number of 45 metastatic lesions. In total, 532 patients were included in this study, with histologic types as described in Supplementary Table S6.
Primary treatment consisted of abdominal hysterectomy with bilateral salpingo-oophorectomy, unless surgery was deemed contraindicated. For the majority of the patients, the primary treatment also consisted of pelvic lymphadenectomy as a surgical staging procedure. Adjuvant therapy was recommended for patients with FIGO stages ≥ II and high-risk FIGO I patients, defined as non-endometrioid tumors or deeply infiltrating endometrioid grade 3 tumors, according to national guidelines and as previously reported26.
Comprehensive clinicopathologic data were available for all patients. Clinical data included patient age at diagnosis, International Federation of Gynecology and Obstetrics (FIGO) stage according to the 2009 criteria, histologic type and grade, lymph node status, myometrial infiltration, adjuvant/further treatment and follow-up informationMedian follow-up time was 59 months, last time point February 2017.
The study was approved by the Norwegian Data Inspectorate, Norwegian Social Sciences Data Services (15501), and the Western Regional Committee for Medical and Health Research Ethics (REK_2009/2315). All patients gave written informed consent. All experiments were performed in accordance with relevant guidelines and regulations.
DNA sequencing
Mutations in PIK3CA exon9 and −20 from endometrial carcinomas were investigated by employing genomic DNA obtained from freshly frozen tissue samples. For metastatic samples, DNA was obtained by whole-genome amplified DNA (GenomePlex Complete WGA kit 2, Sigma-Aldrich, St. Louis, MO, USA) and used as template in PCR reactions to conserve the original material. Twenty-five ng genomic DNA was used as template in the PCR reactions together with 0.4 μM forward/reverse primers (Supplementary Table S7a,b) applying the Multiplex PCR kit or HotStarTaq Plus Master Mix kit (both Qiagen, Hilden, Germany). PCR products were run by agarose gel electrophoresis prior to treatment with ExoSAP-IT (USB, Cleveland, OH, USA) and direct Sanger sequencing (both directions) employing the BigDye Terminator Sequencing Kit version 3.1, and Applied Biosystems 3730XL Analyzer (Foster City, CA, USA). The sequencing chromatograms were visualized with eBioX version 1.5.1 (output examples of hotspot mutations shown in Supplementary Fig. S4). Previously published KRAS mutation data27 were compared with the new mutational data for 250 overlapping cases in the current study.
Oligonucleotide DNA microarray and analysis
The RNA was extracted, obtained and hybridized as previously reported28. Briefly, RNA was hybridized to Agilent whole human genome microarray 44k (Cat.no G4112F, Santa Clara, CA, USA), scanned with Agilent Microarray Scanner Bundle. Median spot signals were employed as expression values, which were subsequently quantile normalized and log2-transformed. Expression data were available for 272 cases of endometrial cancer.
The PI3K pathway activation signature was based on the expression of 160 genes derived from in vitro cell line experiments29. When calculating the signature score for each sample, we subtracted the expression values of the down-regulated genes included in the signature, from the up-regulated genes. The expression values were mean normalized and scaled to the same standard deviation30.
Significance analysis of microarray (SAM) was performed to evaluate genes differentially expressed between various PIK3CA mutated and non-mutated groups by using J-Express31. We applied cutoff levels of False Discovery Rate (FDR) < 0.001, q-value < 0.001 and fold change ≥1.5 for calling significantly mutated genes.
For stratification of PIK3CA mRNA into “high/low expression”, the 5-year disease-free survival plots (Kaplan-Meier method) according to quartiles of PIK3CA mRNA expression were explored. Quartiles 1–3 clustered together with favorable prognosis and were defined as “low expression”. The 4th quartile showed significant poorer survival compared to the rest, and was set to “high expression”.
Fluorescent in situ hybridization (FISH)
FFPE tumor tissues from primary tumors (hysterectomy specimens) were mounted on tissue micro arrays (TMA) as previously described27, 28. The PIK3CA gene copy number was evaluated by FISH in TMAs using the Histology FISH Accessory Kit (Dako Agilent, Santa Clara, CA, USA) according to the manufacturer recommendations with minor modifications28. Slides were heated over night at 58 °C and deparaffinized for 3 × 10 min in xylene. De- and rehydrations were carried out in three steps with 100%, 85% and 70% ethanol before 15 min of pepsin digestion. The TMAs were incubated with PIK3CA-/CEP3 Dual Color probe (Abnova, Taipei City, Taiwan) at 75 °C for 5 min and 37 °C for approximately 72 hours. All spots from one case were screened (1–9 spots per patient) looking for copy number increases. Then, taking the whole area of each spot into account, the area with optimal signal quality and quantity to detect possible copy number increases was selected for assessment of gene and centromere signals in 20 non-overlapping nuclei. Status of PIK3CA gene copy levels were evaluated as “polysomy” (CN ≥ 2.3) or as “focal amplification” (PIK3CA/CEP3 probe ratio ≥1.15). Amplified cases were classified with polysomy only if not focally amplified.
Immunohistochemical staining (IHC)
For detection of the p85α regulatory subunit of PI3K, TMAs were dewaxed in xylene, rehydrated in ethanol before microwave antigen retrieval at pH9 and incubation for 30 min with the mouse monoclonal antibody SC-1637 (Santa Cruz Biotechnology, Santa Cruz, CA), diluted 1:20. EnVision + System-HRP, anti-mouse was used as secondary antibody (Dako K4001). Evaluation of the staining index (index score 1–9), considering the product of staining intensity and area of maximal staining determined the level of staining32. Details, indexing and cut-off levels for IHC markers are summarized in Supplementary Table S8 60, 61.
Reverse Phase Protein Array (RPPA)
Protein levels of AKT (AKT1) and phosphorylated AKT (p-AKT_T308 and p-AKT_S473) were assessed by RPPA in 376 patients with endometrial cancer (p-AKT reported here as p-AKT_T308 only, as results were similar with p-AKT_S47333). The procedure was done as previously described5, 34. In brief, fresh frozen tumor samples were homogenized in lysis buffer followed by denaturation using SDS and subsequently serially diluted in lysis buffer. The lysates were printed on nitrocellulose coated slides before staining with RPPA-validated primary antibodies. The signal was captured by secondary antibodies, using a BioGenix autostainer and revealed by Dako Cytomation-catalyzed systems and DAB colorimetric reaction. Array-Pro analyzer (Media Cybernetics, Washington DC, USA) was used for quantifying spot signal intensities, before scanning of slides by CanoScan 9000F. Relative protein levels were determined by fitting each dilution curve with a logistic model (“Supercurve Fitting”; http://bioinformatics.mdanderson.org/OOMPA). Determination of cut-off in survival analysis for groups of high or low expression was performed as described for PIK3CA mRNA.
Statistical analysis
Statistics was performed using Statistical Package of Social Sciences (SPSS), version 20.0 (IBM Inc., Chicago, IL). The P-values represent two-sided tests considered to be significant when less than 0.05. For evaluating associations between categorical variables, the Pearson’s χ 2-test was used. Mann-Whitney U-test was used for analysis of continuous variables between categories. One case with mutations in both exons9 and exon20 was excluded from the analysis correlating specific exon mutations to phenotype. When comparing exons, this double mutated case was categorized as being exon9 mutated due to presence of the p.Q546R hotspot mutation. Chi squared test was used to compare the mutation frequency between primary tumors without recurrence, primary tumors with recurrence and metastatic lesions, and also within the paired set of primary and corresponding metastatic lesions. Univariate survival analyses were performed using the Kaplan-Meier method, with date of primary surgery as entry date, and death specifically due to endometrial carcinoma (disease-specific survival) or no recurrence of endometrial cancer (recurrence-free survival) as end points. For estimation of differences in survival between groups the two-sided log-rank (Mantel-Cox) tests was used. The Cox proportional hazard regression model was employed to evaluate the prognostic impact of PIK3CA exon9 charge-plus changing substitutions (AA+) adjusted for other prognostic parameters including age, histologic type and grade, which are factors with prognostic impact in endometrial cancer.
Electronic supplementary material
Acknowledgements
We thank Britt Edvardsen, Ellen Valen, Kadri Madissoo, Hua My Hoang and Inger Marie Løes for their expertise and support. This study was conducted with the support from the Research Council of Norway, the Norwegian Cancer Society, Helse-Vest, the University of Bergen and the Bergen Research Foundation.
Author Contributions
Conceived and designed the study: S.M., E.B., H.B.S., E.A.H. Collected samples and performed the experiments: S.M., H.M.J.W., E.B., F.H., M.K.H., I.L.T., K.K.M., A.M.O., K.-H.K., C.K., J.T., H.B.S., E.A.H. Data processing and analysis: S.M., H.M.J.W., E.B., F.H., A.B., M.K.H., I.L.T., K.K., K.K.M., A.M.O., K.-H.K., A.W.L., G.B.M., C.K., J.T., H.B.S., E.A.H. Wrote the manuscript: S.M., E.A.H. All authors critically read and approved the final manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Helga B. Salvesen is deceased.
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-10717-z
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Cancer Registry of Norway. Cancer in Norway 2015 - Cancer incidence, mortality, survival and prevalence in Norway. http://www.kreftregisteret.no (2016).
- 2.Salvesen HB, Haldorsen IS, Trovik J. Markers for individualised therapy in endometrial carcinoma. Lancet Oncol. 2012;13:e353–61. doi: 10.1016/S1470-2045(12)70213-9. [DOI] [PubMed] [Google Scholar]
- 3.Bokhman JV. Two pathogenetic types of endometrial carcinoma. Gynecol Oncol. 1998;15:10–7. doi: 10.1016/0090-8258(83)90111-7. [DOI] [PubMed] [Google Scholar]
- 4.Dedes KJ, Wetterskog D, Ashworth A, Kaye SB, Reis-Filho JS. Emerging therapeutic targets in endometrial cancer. Nat Rev Clin Oncol. 2011;8:261–71. doi: 10.1038/nrclinonc.2010.216. [DOI] [PubMed] [Google Scholar]
- 5.Cancer Genome Atlas Research Network. et al. Integrated genomic characterization of endometrial carcinoma. Nature. 497, 67–73 (2013). [DOI] [PMC free article] [PubMed]
- 6.Colombo N, et al. ESMO-ESGO-ESTRO Consensus Conference on Endometrial Cancer: Diagnosis. Treatment and Follow-up. Int J Gynecol Cancer. 2016;26:2–30. doi: 10.1097/IGC.0000000000000609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhao L, Vogt PK. Helical domain and kinase domain mutations in p110alpha of phosphatidylinositol 3-kinase induce gain of function by different mechanisms. Proc Natl Acad Sci USA. 2008;105:2652–7. doi: 10.1073/pnas.0712169105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kandoth C, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333–9. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Samuels Y, Ericson K. Oncogenic PI3K and its role in cancer. Curr Opin Oncol. 2006;18:77–82. doi: 10.1097/01.cco.0000198021.99347.b9. [DOI] [PubMed] [Google Scholar]
- 10.Thorpe LM, Yuzugullu H, Zhao JJ. PI3K in cancer: divergent roles of isoforms, modes of activation and therapeutic targeting. Nat Rev Cancer. 2015;15:7–24. doi: 10.1038/nrc3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Polivka J, Jr., Janku F. Molecular targets for cancer therapy in the PI3K/AKT/mTOR pathway. Pharmacol Ther. 2014;142:164–75. doi: 10.1016/j.pharmthera.2013.12.004. [DOI] [PubMed] [Google Scholar]
- 12.Alessi DR, et al. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol. 1997;7:261–9. doi: 10.1016/S0960-9822(06)00122-9. [DOI] [PubMed] [Google Scholar]
- 13.Cerami E, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–4. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gao J, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6 doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang CH, et al. The structure of a human p110alpha/p85alpha complex elucidates the effects of oncogenic PI3Kalpha mutations. Science. 2007;318:1744–8. doi: 10.1126/science.1150799. [DOI] [PubMed] [Google Scholar]
- 16.Gymnopoulos M, Elsliger MA, Vogt PK. Rare cancer-specific mutations in PIK3CA show gain of function. Proc Natl Acad Sci USA. 2007;104:5569–74. doi: 10.1073/pnas.0701005104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Millis SZ, Ikeda S, Reddy S, Gatalica Z, Kurzrock R. Landscape of Phosphatidylinositol-3-Kinase Pathway Alterations Across 19784 Diverse Solid Tumors. JAMA Oncol. 2016;2:1565–73. doi: 10.1001/jamaoncol.2016.0891. [DOI] [PubMed] [Google Scholar]
- 18.Gabelli SB, et al. Structural effects of oncogenic PI3Kalpha mutations. Curr Top Microbiol Immunol. 2010;347:43–53. doi: 10.1007/82_2010_53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vogt PK, et al. Phosphatidylinositol 3-kinase: the oncoprotein. Curr Top Microbiol Immunol. 2010;347:79–104. doi: 10.1007/82_2010_80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hon WC, Berndt A, Williams RL. Regulation of lipid binding underlies the activation mechanism of class IA PI3-kinases. Oncogene. 2012;31:3655–66. doi: 10.1038/onc.2011.532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mei ZB, Duan CY, Li CB, Cui L, Ogino S. Prognostic role of tumor PIK3CA mutation in colorectal cancer: a systematic review and meta-analysis. Ann Oncol. 2016;27:1836–48. doi: 10.1093/annonc/mdw264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang L, et al. PIK3CA mutations frequently coexist with EGFR/KRAS mutations in non-small cell lung cancer and suggest poor prognosis in EGFR/KRAS wildtype subgroup. PLoS One. 2016;9 doi: 10.1371/journal.pone.0088291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang SX, Polley E, Lipkowitz S. New insights on PI3K/AKT pathway alterations and clinical outcomes in breast cancer. Cancer Treat Rev. 2016;45:87–96. doi: 10.1016/j.ctrv.2016.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Catasus L, Gallardo A, Cuatrecasas M, Prat J. PIK3CA mutations in the kinase domain (exon 20) of uterine endometrial adenocarcinomas are associated with adverse prognostic parameters. Mod Pathol. 2008;21:131–9. doi: 10.1038/modpathol.3800992. [DOI] [PubMed] [Google Scholar]
- 25.Konopka B, et al. PIK3CA mutations and amplification in endometrioid endometrial carcinomas: relation to other genetic defects and clinicopathologic status of the tumors. Hum Pathol. 2011;42:1710–9. doi: 10.1016/j.humpath.2010.01.030. [DOI] [PubMed] [Google Scholar]
- 26.Trovik J, et al. Stathmin overexpression identifies high-risk patients and lymph node metastasis in endometrial cancer. Clin Cancer Res. 2011;17:3368–77. doi: 10.1158/1078-0432.CCR-10-2412. [DOI] [PubMed] [Google Scholar]
- 27.Birkeland E, et al. KRAS gene amplification and overexpression but not mutation associates with aggressive and metastatic endometrial cancer. Br J Cancer. 2012;107:1997–2004. doi: 10.1038/bjc.2012.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Berg A, et al. Molecular profiling of endometrial carcinoma precursor, primary and metastatic lesions suggests different targets for treatment in obese compared to non-obese patients. Oncotarget. 2012;6:1327–39. doi: 10.18632/oncotarget.2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gustafson AM, et al. Airway PI3K pathway activation is an early and reversible event in lung cancer development. Sci Transl Med. 2010;2 doi: 10.1126/scitranslmed.3000251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Salvesen HB, et al. Integrated genomic profiling of endometrial carcinoma associates aggressive tumors with indicators of PI3 kinase activation. Proc Natl Acad Sci USA. 2009;106:4834–9. doi: 10.1073/pnas.0806514106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stavrum, A.K., Petersen, K., Jonassen, I. & Dysvik, B. Analysis of gene-expression data using J-Express. Curr Protoc Bioinformatics. Chapter 7; doi:10.1002/0471250953.bi0703s21 (2008). [DOI] [PubMed]
- 32.Krakstad C, et al. Loss of GPER identifies new targets for therapy among a subgroup of ERalpha-positive endometrial cancer patients with poor outcome. Br J Cancer. 2012;106:1682–8. doi: 10.1038/bjc.2012.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Samuels Y, et al. Mutant PIK3CA promotes cell growth and invasion of human cancer cells. Cancer Cell. 2005;7:561–73. doi: 10.1016/j.ccr.2005.05.014. [DOI] [PubMed] [Google Scholar]
- 34.Hennessy BT, et al. A Technical Assessment of the Utility of Reverse Phase Protein Arrays for the Study of the Functional Proteome in Non-microdissected Human Breast Cancers. Clin Proteomics. 2010;6:129–51. doi: 10.1007/s12014-010-9055-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hayes MP, et al. PIK3CA and PTEN mutations in uterine endometrioid carcinoma and complex atypical hyperplasia. Clin Cancer Res. 2006;12:5932–5. doi: 10.1158/1078-0432.CCR-06-1375. [DOI] [PubMed] [Google Scholar]
- 36.Oda K, Stokoe D, Taketani Y, McCormick F. High frequency of coexistent mutations of PIK3CA and PTEN genes in endometrial carcinoma. Cancer Res. 2005;65:10669–73. doi: 10.1158/0008-5472.CAN-05-2620. [DOI] [PubMed] [Google Scholar]
- 37.Velasco A, et al. PIK3CA gene mutations in endometrial carcinoma: correlation with PTEN and K-RAS alterations. Hum Pathol. 2006;37:1465–72. doi: 10.1016/j.humpath.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 38.Samuels Y, Velculescu VE. Oncogenic mutations of PIK3CA in human cancers. Cell Cycle. 2004;3:1221–4. doi: 10.4161/cc.3.10.1164. [DOI] [PubMed] [Google Scholar]
- 39.Forbes SA, et al. COSMIC: exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015;43:D805–11. doi: 10.1093/nar/gku1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gabelli SB, et al. Activation of PI3Kalpha by physiological effectors and by oncogenic mutations: structural and dynamic effects. Biophys Rev. 2014;6:89–95. doi: 10.1007/s12551-013-0131-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gerlinger M, et al. Intratumour heterogeneity in urologic cancers: from molecular evidence to clinical implications. Eur Urol. 2015;67:729–37. doi: 10.1016/j.eururo.2014.04.014. [DOI] [PubMed] [Google Scholar]
- 42.Gerlinger M, et al. Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nat Genet. 2014;46:225–33. doi: 10.1038/ng.2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gibson WJ, et al. The genomic landscape and evolution of endometrial carcinoma progression and abdominopelvic metastasis. Nat Genet. 2016;48:848–55. doi: 10.1038/ng.3602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Westin SN, et al. PTEN loss is a context-dependent outcome determinant in obese and non-obese endometrioid endometrial cancer patients. Mol Oncol. 2015;9:1694–703. doi: 10.1016/j.molonc.2015.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lin DI. Improved survival associated with somatic PIK3CA mutations in copy-number low endometrioid endometrial adenocarcinoma. Oncol Lett. 2015;10:2743–8. doi: 10.3892/ol.2015.3702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McIntyre JB, et al. PIK3CA missense mutation is associated with unfavorable outcome in grade 3 endometrioid carcinoma but not in serous endometrial carcinoma. Gynecol Oncol. 2014;132:188–93. doi: 10.1016/j.ygyno.2013.11.015. [DOI] [PubMed] [Google Scholar]
- 47.Catasus L, Gallardo A, Cuatrecasas M, Prat J. Concomitant PI3K-AKT and p53 alterations in endometrial carcinomas are associated with poor prognosis. Mod Pathol. 2009;22:522–9. doi: 10.1038/modpathol.2009.5. [DOI] [PubMed] [Google Scholar]
- 48.Barretina J, et al. Subtype-specific genomic alterations define new targets for soft-tissue sarcoma therapy. Nat Genet. 2010;42:715–21. doi: 10.1038/ng.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Barbareschi M, et al. Different prognostic roles of mutations in the helical and kinase domains of the PIK3CA gene in breast carcinomas. Clin Cancer Res. 2007;13:6064–9. doi: 10.1158/1078-0432.CCR-07-0266. [DOI] [PubMed] [Google Scholar]
- 50.Gabelli SB, Mandelker D, Schmidt-Kittler O, Vogelstein B, Amzel LM. Somatic mutations in PI3Kalpha: structural basis for enzyme activation and drug design. Biochim Biophys Acta. 2009;1804:533–40. doi: 10.1016/j.bbapap.2009.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mandelker D, et al. A frequent kinase domain mutation that changes the interaction between PI3Kalpha and the membrane. Proc Natl Acad Sci USA. 2009;106:16996–7001. doi: 10.1073/pnas.0908444106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Colas E, et al. Molecular markers of endometrial carcinoma detected in uterine aspirates. Int J Cancer. 2011;129:2435–44. doi: 10.1002/ijc.25901. [DOI] [PubMed] [Google Scholar]
- 53.Bou Zgheib N, et al. Molecular determinants for lymph node metastasis in clinically early-stage endometrial cancer. Oncol Lett. 2015;11:323–9. doi: 10.3892/ol.2015.3883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rudd ML, et al. A unique spectrum of somatic PIK3CA (p110alpha) mutations within primary endometrial carcinomas. Clin Cancer Res. 2011;17:1331–40. doi: 10.1158/1078-0432.CCR-10-0540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science. 2011;331:1559–64. doi: 10.1126/science.1203543. [DOI] [PubMed] [Google Scholar]
- 56.Janku F, et al. PI3K/AKT/mTOR inhibitors in patients with breast and gynecologic malignancies harboring PIK3CA mutations. Journal of clinical oncology. 2012;30:777–82. doi: 10.1200/JCO.2011.36.1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bergstrom CP, Geest K, O’Gara R, Corless CL, Morgan TK. Discordant Mutations in Paired Primary and Metastatic Endometrial Adenocarcinomas Identified by Semiconductor-Based Sequencing for Rapid Cancer Genotyping. Reprod Sci. 2016;23:1575–9. doi: 10.1177/1933719116648213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Baba Y, et al. Phosphorylated AKT expression is associated with PIK3CA mutation, low stage, and favorable outcome in 717 colorectal cancers. Cancer. 2011;117:1399–408. doi: 10.1002/cncr.25630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vasudevan KM, et al. AKT-independent signaling downstream of oncogenic PIK3CA mutations in human cancer. Cancer Cell. 2009;16:21–32. doi: 10.1016/j.ccr.2009.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lotan TL, et al. PTEN protein loss by immunostaining: analytic validation and prognostic indicator for a high risk surgical cohort of prostate cancer patients. Clinical cancer research. 2011;17:6563–73. doi: 10.1158/1078-0432.CCR-11-1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tangen IL, et al. Loss of progesterone receptor links to high proliferation and increases from primary to metastatic endometrial cancer lesions. European journal of cancer. 2014;50:3003–10. doi: 10.1016/j.ejca.2014.09.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.