

Abstract
Sequencing of tumors is now routine and guides personalized cancer therapy. Mutant allele fractions (MAFs, or the ‘mutation dose’) of a driver gene may reveal the genomic structure of tumors and influence response to targeted therapies. We performed a comprehensive analysis of MAFs of driver alterations in unpaired primary and metastatic colorectal cancer (CRC) at our institution from 2010 to 2015 and studied their potential clinical relevance. Of 763 CRC samples, 622 had detailed annotation on overall survival in the metastatic setting (OSmet) and 89 received targeted agents matched to KRAS (MEK inhibitors), BRAF (BRAF inhibitors), or PIK3CA mutations (PI3K pathway inhibitors). MAFs of each variant were normalized for tumor purity in the sample (adjMAFs). We found lower adjMAFs for BRAFV 600E and PIK3CA than for KRAS,NRAS, and BRAF non‐V600 variants. TP53 and BRAFV 600E adjMAFs were higher in metastases as compared to primary tumors, and high KRAS adjMAFs were found in CRC metastases of patients with KRAS wild‐type primary tumors previously exposed to EGFR antibodies. Patients with RAS‐ or BRAFV 600E‐mutated tumors, irrespective of adjMAFs, had worse OSmet. There was no significant association between adjMAFs and time to progression on targeted therapies matched to KRAS,BRAF, or PIK3CA mutations, potentially related to the limited antitumor activity of the employed drugs (overall response rate of 4.5%). In conclusion, the lower BRAFV 600E and PIK3CA adjMAFs in subsets of primary CRC tumors indicate subclonality of these driver genes. Differences in adjMAFs between metastases and primary tumors suggest that approved therapies may result in selection of BRAFV 600E‐ and KRAS‐resistant clones and an increase in genomic heterogeneity with acquired TP53 alterations. Despite significant differences in prognosis according to mutations in driver oncogenes, adjMAFs levels did not impact on survival and did not help predict benefit with matched targeted agents in the metastatic setting.
Keywords: clonality, colorectal cancer, driver gene, mutant allele fraction
Abbreviations
- CRC
colorectal cancer
- MAFs
mutant allele fractions
1. Introduction
Tumor next‐generation sequencing (NGS) is routine part of prescreening programs to guide precision cancer therapy. NGS allows the identification of mutations in driver genes in a very sensitive and quantitative manner. The mutant allele fractions (MAFs), also called ‘mutation dose’, represent the number of mutant reads divided by the total number of reads – coverage – at a specific genomic position. In some scenarios, the MAFs of driver genes may have important clinical implications. Examples include the upfront resistance to anti‐EGFR therapies in metastatic colorectal cancer (CRC) with KRAS MAFs as low as 1% (Azuara et al., 2016; Laurent‐Puig et al., 2015), or the positive association between higher EGFR L858R MAFs in lung cancer specimens and longer duration of treatment benefit with gefitinib and erlotinib in the metastatic setting (Ono et al., 2014; Zhou et al., 2011). More recently, investigators have been tracking MAFs of driver genes to infer mutational timeline and depict dynamic clonal evolution in individual tumors exposed to targeted agents (McGranahan et al., 2015; Murtaza et al., 2015; Russo et al., 2016).
In tissue samples, the MAFs are largely influenced by tumor purity (fraction of neoplastic cells in the sample) and ploidy (either copy number gains or losses of wild‐type/mutant alleles). It is possible to partially adjust the MAF of a mutation by normalizing it to the neoplastic cell content of the sample, which can be named an ‘adjusted MAF’ (adjMAF). Interestingly, when examining NGS results, MAFs do not clearly correlate with tumor purity, and in samples with more than one mutation, adjMAFs are often different among the affected genes (Normanno et al., 2015). These findings suggest either coexisting gene mutations and copy number alterations or intratumor genomic heterogeneity, with clonal (truncal) and subclonal driver gene mutations in the same tumor sample.
In many cancer types, including CRC, a comprehensive analysis of driver genes adjMAFs remains to be performed, with particular attention to differences between primary and metastatic lesions, or after exposure to standard therapies. In addition, the potential prognostic effect of a driver gene ‘mutation dose’ in solid tumors has not been investigated in detail. In this manuscript, we present an in‐depth analysis of adjMAFs of driver genes in CRC and estimate their relative clonal and subclonal distribution. We also investigate their potential clinical impact in a large patient cohort with outcome annotation, focusing on survival in the metastatic setting and on the magnitude of response to matched targeted therapies, namely anti‐MEK, anti‐BRAF, and anti‐PI3K agents according to KRAS, BRAF V600E, and PIK3CA adjMAFs, respectively.
2. Materials and methods
2.1. Mutation analysis and populations of interest
From 2010 to 2015, 763 consecutive patients with metastatic CRC were eligible for targeted sequencing at our institution (Vall d'Hebron Institute of Oncology, VHIO) as part of a molecular prescreening program (MPP) for early drug development. From January 2010 to May 2014, mutation detection and quantification was performed using a multiplex mass spectrometry‐based technology (massARRAY Sequenom® platform, with a 24 oncogene panel of hotspot mutations, including most frequent variants in KRAS, NRAS, BRAF, and PIK3CA). Thereafter, we moved to an amplicon‐based NGS technology (MiSeq Illumina® platform, with a 61 oncogene plus tumor suppressor panel, covering most frequently mutated exons of KRAS, NRAS, BRAF, PIK3CA, APC, and TP53). Technical details of mutation analysis are described in Doc S1. Both tests were performed in‐house in our Cancer Genomics Lab under ISO accreditation (UNE‐EN ISO 15189:2013) including mutation detection and quantification. Average sequencing depth was 1000× allowing precise estimates for low MAFs (mutations were called at a minimum MAF of 3%). We used archived formalin‐fixed paraffin‐embedded tissues for sequencing after pathological assessment of neoplastic cell content in the sample by the Molecular Oncology Lab. Tumor purity was defined as the amount of sample occupied by cancer cells and not by surrounding stromal and immune/inflammatory cells, that is, percentage of transformed (neoplastic) cells. In order to mitigate variability, the quantification of neoplastic cells was performed by an experienced pathologist (P.N.) always in the same section used for sequencing, as recently recommended by other groups (Lhermitte et al., 2017). A minimum of 20% tumor purity was required for sample processing (resolution at 5% level). Heterogeneous tissue samples were macrodissected for tumor purity assessment and molecular analysis. The calculated adjMAFs (MAF/tumor purity) of driver genes of interest were used to infer clonality of the events. In summary, for oncogenes, the expected adjMAF is close to 0.5 if the event is clonal and < 0.5 if subclonal. For tumor suppressors, as deletion of the wild‐type allele (loss of heterozygosity) is a common genomic event, the expected adjMAFs is > 0.5 if the event is clonal.
Of 763 patients with targeted mutation profiling (molecular population), 622 received oncologic treatment at VHIO and had complete survival annotation (molecular + clinical population). Data curators from the Oncology Data Science (ODysSey) group prospectively extracted this information from medical records in structured clinical–molecular databases. The remaining patients were external referrals to MPP and did not have clinical interventions at our institution. From 622 molecularly and clinically eligible patients, 34 with KRAS‐mutated tumors were enrolled in clinical trials with MEK inhibitors, 20 with BRAF V600E‐mutated tumors received anti‐BRAF therapy, and 35 with PIK3CA‐mutated tumors were treated with an anti‐PI3K agent. Treatment on phase 1 studies continued until disease progression or unacceptable toxicity and was carried out according to the specific requirements of each protocol. Tumor responses were classified as complete, partial, stable disease, or progressive disease as per Response Evaluation Criteria in Solid Tumors (RECIST) v 1.1. Time to progression (TTP) was defined as the time interval from the start of a therapy to its discontinuation for disease progression or death, whichever occurred first (patients with permanent treatment discontinuation for toxicity without evidence of progressive disease were censored at the time of last dose). Overall survival in the metastatic setting (OSmet) was defined as time from first diagnosis of metastasis until death or last follow‐up.
2.2. Statistical and ethical considerations
Nonparametric tests (Mann–Whitney U and Kruskal–Wallis) were used to cross‐compare adjMAFs of different genes and correlate with variables of interest, such as tissue source (CRC primary vs. metastatic site). Survival analyses (TTP and OSmet) were conducted using the Kaplan–Meier method and compared with the log‐rank test. We constructed univariate and multivariable Cox proportional hazard models for OSmet. The association between TTP and adjMAFs was measured using Pearson's correlation. All tests were two‐sided, and a P value < 0.05 was considered statistically significant. Statistical analyses were conducted using r version 3.2.3 (survival and phenoTest packages). All patients that participated in our institutional MPP signed informed consent form giving investigators access to molecular and clinical data for research purposes. All clinical trials were conducted in accordance with the guidelines of the VHIO Institutional Review Board.
3. Results
3.1. Biological insights from analysis of driver genes adjMAFs in CRC
As shown in Table 1, sequencing was mostly performed on samples derived from CRC primary tissue. Patients whose metastatic sites were used for profiling had prior exposure to systemic therapies at the time of sample acquisition. This population had sequencing performed exclusively in the metastatic site – unpaired samples. Median tumor purity was 50% (IQR 35–70%), with no significant differences when comparing samples that harbored mutations in driver genes and those wild‐type for the respective genes (P > 0.05) or according to the tissue source used for profiling (P = 0.16).
Table 1.
Population characteristics
| Molecular population (n = 763) | ||
| Mutation analysis | Sequenom® | 460 (60.3%) |
| MiSeq® | 303 (39.7%) | |
| Tissue source | CRC primary | 586 (79.7%) |
| Metastasis | 149 (20.3%) | |
| Missing | 28 | |
| Driver mutation | KRAS | 365 (47.8%) |
| NRAS | 29 (3.8%) | |
| BRAF | 65 (8.5%) | |
| PIK3CA | 128 (16.7%) | |
| APC (MiSeq® only) | 154 (50.8%) | |
| TP53 (MiSeq® only) | 191 (63.0%) | |
| Molecular–clinical population (n = 622) | ||
| Age at diagnosis | Median (range) | 58 years (22–85) |
| Gender | Male | 386 (62%) |
| Female | 236 (38%) | |
| Stage at diagnosis | Early | 252 (41%) |
| Metastatic | 370 (59%) | |
| CRC primary site | Right | 178 (30%) |
| Left | 260 (45%) | |
| Rectum | 145 (25%) | |
| Number of metastatic sites at diagnosis of metastasis | One | 419 (67%) |
| Two | 163 (26%) | |
| Three or more | 40 (7%) | |
| Metastatic sites | Liver | 407 (65%) |
| Lung | 186 (30%) | |
| Node | 131 (21%) | |
| Peritoneal | 94 (15%) | |
| Other | 57 (9%) | |
| Surgical treatment for metastasis | Any | 285 (46%) |
| Liver | 195 (31%) | |
| Lung | 52 (8%) | |
| Other sites | 57 (9%) | |
| Pharmacological treatment | Oxaliplatin based | 599 (97%) |
| Irinotecan based | 549 (89%) | |
| Antiangiogenic therapy | 342 (55%) | |
| Anti‐EGFR therapy | 285 (46%) | |
| Anti‐MEK therapya | 52 (8%) | |
| Anti‐BRAF therapyb | 20 (3%) | |
| Anti‐PI3K therapyc | 70 (11%) | |
| Any other experimental therapy | 169 (28%) | |
aRAS mutated or wild‐type, single agent or combo; b BRAF mutated, single agent, or combo; c PIK3CA mutated or wild‐type, single agent, or combo.
The prevalence of oncogene mutations in our cohort is depicted in Fig. 1A. The lower prevalence of APC mutations (51%) as compared to published literature (around 70%; The Cancer Genome Atlas, 2012) is related to a limited APC exon coverage of our NGS panel. Inspection of adjMAFs distribution (Fig. 1B,C) revealed major differences across driver genes. Relative to a simulated normal distribution of adjMAF according to the ‘one‐hit hypothesis’ for oncogenes (median 0.5, IQR 0.25–0.75), we observed significantly lower adjMAFs for BRAF (median 0.31, IQR 0.23–0.50; P < 0.001) and PIK3CA (median 0.38, IQR 0.25–0.56; P < 0.001), suggesting a potential subclonality of these genomic alterations. TP53 adjMAFs (median 0.66, IQR 0.40–0.85) were significantly higher than simulated cohort (P < 0.001), indicating a clonal event plus deletion of wild‐type allele in most samples. There were no significant differences between adjMAFs of oncogenes KRAS (median 0.56, IQR 0.42–0.77; P = 0.66) and NRAS (median 0.49, IQR 0.33–0.66; P = 0.44) as compared with normal distribution, reinforcing clonality of these events. The same was true for APC adjMAFs (median 0.50, IQR 0.32–0.83; P = 0.87). Of note, we compared the distribution of oncogene adjMAF according to platform (Sequenom® or MiSeq Illumina®) and found no statistically significant differences (P > 0.05 for KRAS, NRAS, BRAF, and PIK3CA).
Figure 1.
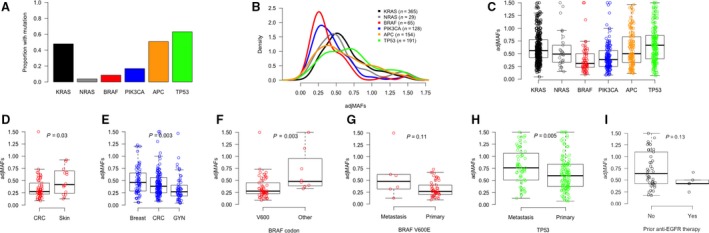
Biological insights into mutant allele fractions adjusted for tumor purity (adjMAFs) in CRC. Proportion of CRC samples with mutations in driver genes (A) and distribution of adjMAFs for the respective genes (B, C). BRAFV 600E adjMAFs are higher in melanomas as compared to CRC (D), and PIK3CA adjMAFs are higher in breast cancer and lower in gynecological malignancies as compared to CRC (E). BRAF adjMAFs are different according to codon affected (higher in non‐V600 mutations as compared to V600; F) and tissue source (trend for higher counts in metastases as compared to CRC primaries; G). TP53 adjMAFs are also higher in metastases as compared to CRC primaries (H). KRAS adjMAFs in metastases of patients with prior exposure to EGFR antibodies (originally KRAS wild‐type in the primary tissue) are not significantly different from those without prior targeted treatment (with constitutive KRAS mutations in primary tissue) (I).
Next, we investigated in more detail the potential subclonality of BRAF and PIK3CA mutations in CRC. Taking advantage of our institutional molecular database, we compared adjMAFs in CRC with those of other malignancies having frequent mutations in these genes. We selected samples from other tumors profiled during the same time period and using similar platforms. As shown in Fig. 1D, BRAF V600E adjMAFs were significantly higher in skin melanomas (median 0.42, IQR 0.25–0.68; n = 25; P = 0.03) than in CRC. With regard to PIK3CA mutations, depicted in Fig. 1E, an intermediate adjMAFs level was seen in CRC as compared to breast cancer (median 0.46, IQR 0.29–0.65; n = 64; P = 0.002) and gynecological malignancies (median 0.27, IQR 0.19–0.40; n = 56; P = 0.05). This suggests that PIK3CA mutations may be either clonal or subclonal events in CRC. Indeed, when comparing adjMAFs of PIK3CA and KRAS in CRC samples with co‐occurring mutations (Fig. S1A), we confirmed that potential subclonality of PIK3CA mutations is restricted to a subset of CRC tumors (32 of 86 samples [37.2%] with KRAS/PIK3CA adjMAFs ratio > 1.5).
We then explored differences in adjMAFs of driver oncogenes in CRC according to codon or domain affected and tissue source. As illustrated in Fig. S1B, only BRAF adjMAFs vary according to codon affected, being significantly higher in non‐V600 mutations (median 0.48, IQR 0.39–0.85; n = 9) as compared to V600 variants (median 0.28, IQR 0.22–0.44; n = 56; P = 0.003; Fig. 1F). A stratified analysis based on tissue source (Fig. S1C) showed TP53 adjMAFs significantly higher in metastases (median 0.76, IQR 0.51–1.00; n = 63) than CRC primaries (median 0.60, IQR 0.38–0.83; n = 126; P = 0.005; Fig. 1G), suggesting that TP53 copy number alterations are more frequent in CRC metastases. A trend for higher BRAF V600E adjMAFs in metastatic sites (median 0.48, IQR 0.32–0.62; n = 6) than CRC primaries (median 0.27, IQR 0.21–0.40; n = 48; P = 0.11; Fig. 1H) was also identified. Four of six BRAF V600E metastatic lesions had prior exposure to anti‐EGFR therapy. Finally, we identified a population with KRAS‐mutated metastatic tumors that received treatment with EGFR antibodies prior to biopsy of metastatic site, based on a diagnosis of KRAS wild‐type in CRC primary performed outside our institution. As shown in Fig. 1I, KRAS adjMAFs of these samples (median 0.43, IQR 0.43–0.50; n = 5) were not significantly different from those without prior anti‐EGFR therapy exposure (median 0.6, IQR 0.43–1.1; n = 41; P = 0.13).
3.2. Clinical impact of driver genes adjMAFs in CRC
First, we investigated whether the presence of driver oncogene mutations had an impact on prognosis of metastatic CRC patients. As shown in Table 1, this represents an unselected population treated at our institution in the last 6 years, including patients eligible to surgical resection of metastasis during the course of their disease (46%). Most patients had liver metastasis only at diagnosis and were exposed to oxaliplatin‐ and irinotecan‐based chemotherapy plus antiangiogenic agents or anti‐EGFR therapy (if RAS wild‐type). In addition, close to 40% of our patients received experimental agents in early clinical trials in the third‐ or fourth‐line settings. Median follow‐up of patients alive was 38 months (IQR 22–64 months). When aggregating patients in subgroups based on oncogene mutations, as illustrated in Kaplan–Meier curves of Fig. 2A, we observed major differences in median OSmet. In univariate Cox models, detailed in Table 2, we found that patients whose tumors harbored either RAS or BRAF mutations had significantly worse OSmet when compared to quadruple wild‐type (KRAS, NRAS, BRAF, and PIK3CA) tumors. PIK3CA mutations, when having either a more clonal or subclonal pattern of adjMAFs coexisting with RAS mutations, did not negatively impact on OSmet, as illustrated in Fig. S2A. Survival was not significantly different according to KRAS or NRAS codon affected, and only BRAF V600E‐mutated tumors had a statistical association with worse OSmet as compared with tumors harboring KRAS G12 events, as shown in Fig. S2B and detailed in Table 2. We also found no effect of KRAS mutation clonality on survival when considering KRAS adjMAFs as a continuous variable (P = 0.37), as illustrated in Fig. 2B. Similarly, in the BRAF V600E‐mutated population, BRAF adjMAFs did not impact on prognosis (P = 0.34; results not shown). Next, we constructed a multivariable Cox model with all clinicopathological and molecular covariates that demonstrated statistically significant (P < 0.05) association with OSmet in univariate models. The results of our model that included gender, location of CRC primary, the number of metastatic sites, surgical resection of metastasis, RAS mutations, and BRAF V600E mutations are detailed in Table 2. Surgical treatment of metastasis was the strongest determinant of survival in our population, followed by driver oncogene mutations. We also observed a statistically significant effect of CRC primary location on OSmet, with higher risk of death for patients with right‐sided tumors, irrespective of mutations in RAS and BRAF V600E.
Figure 2.
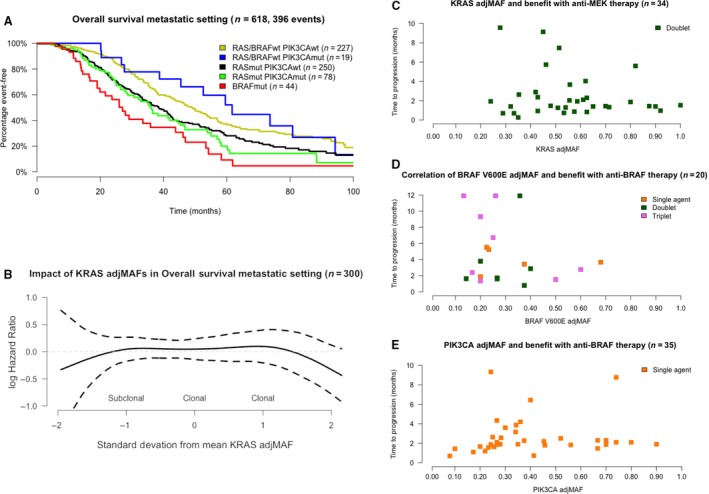
Clinical insights into mutant allele fractions adjusted for tumor purity (adjMAFs) in CRC. Overall survival in the metastatic setting is affected by mutation subgroup (A). The clonality of KRAS mutations (as continuous adjMAFs values) does not impact on survival models (B). There is no correlation between KRAS,BRAFV 600E, and PIK3CA adjMAFs and TTP on matched targeted therapies, as shown in C–E.
Table 2.
Univariate and multivariable Cox models
| Univariate models | |||||
|---|---|---|---|---|---|
| Subgroup | No. at risk | No. events | Median OSmet (months) [CI 95%] | Univariate HR [CI 95%] | P value (log‐rank) |
| RAS/BRAFwt PIK3CAwt | 227 | 143 | 48.7 [42.4–55.7] | Control | |
| RAS/BRAFwt PIK3CAmut | 19 | 12 | 59.2 [43.9–NR] | 0.84 [0.47–1.52] | 0.57 |
| RASmut PIK3CAwt | 250 | 157 | 37.0 [32.8–42.2] | 1.43 [1.14–1.79] | 0.002 |
| RASmut PIK3CAmut | 78 | 53 | 36.5 [28.8–46.8] | 1.60 [1.17–2.20] | 0.004 |
| BRAFmut | 44 | 31 | 27.2 [19.9–46.2] | 2.31 [1.57–3.43] | < 0.001 |
| RASmut PIK3CAwt | 250 | 157 | 37.0 [32.8–42.2] | Control | |
| RASmut PIK3CAmut clonal | 44 | 32 | 36.8 [27.8–51.4] | 1.20 [0.82–1.76] | 0.35 |
| RASmut PIK3CAmut subclonal | 28 | 19 | 32.8 [23.7–NR] | 1.15 [0.71–1.85] | 0.57 |
| KRASmut codon 12 | 228 | 156 | 39.3 [35.1–42.9] | Control | |
| KRASmut codon 13 | 46 | 29 | 29.5 [22.6–42.8] | 1.45 [0.97–2.18] | 0.06 |
| KRASmut codon other | 29 | 15 | 36.2 [29.7–NR] | 0.79 [0.46–1.34] | 0.38 |
| NRASmut any codon | 25 | 12 | 51.4 [35.3–NR] | 0.68 [0.38–1.23] | 0.21 |
| BRAFmut V600E | 38 | 25 | 23.7 [19.0–52.5] | 1.70 [1.11–2.60] | 0.01 |
| BRA mut other | 6 | 6 | 41.0 [29.1–NR] | 1.31 [0.46–1.34] | 0.38 |
| Multivariable model | ||
|---|---|---|
| Variable; n = 582, number of events = 371 | Multivariable HR [CI 95%] | P value (log‐rank) |
| Male (vs. female) | 1.16 [0.93–1.45] | 0.18 |
| Number of metastatic sites (2+ vs. 1) | 1.29 [1.04–1.62] | 0.02 |
| Rectum (vs. left) | 1.13 [0.87–1.46] | 0.34 |
| Right colon (vs. left) | 1.28 [1.01–1.64] | 0.05 |
| Surgical resection of metastasis (vs. no) | 0.35 [0.28–0.45] | < 0.001 |
| BRAF V600E mutation (vs. wt) | 1.56 [0.98–2.49] | 0.06 |
| RAS mutation (vs. wt) | 1.42 [1.15–1.77] | 0.001 |
As prognosis was not affected by clonality of driver oncogenes, we investigated their potential impact on duration of treatment benefit with matched targeted agents. Detailed description of the population and regimens under investigation can be seen in Table 3. Complete or partial responses were only observed in five patients (20%) with BRAF V600E‐mutated tumors treated with combination regimens. Median TTP was 1.8 months (CI 95% 1.4–2.4) with MEK inhibitors (given as doublets with another targeted agent) in patients with KRAS‐mutated tumors, 3.15 months (CI 95% 1.9–6.7) with BRAF inhibitors (given as single agents, doublets, or triplets) in patients with BRAF V600E‐mutated tumors and 2.10 months (CI 95% 1.9–2.6) with PI3K inhibitors (given as single agents) in patients with PIK3CA‐mutated tumors. As illustrated in Fig. 2C–E, we found no association between adjMAFs and TTP on matched therapy: Pearson's correlations of −0.12 (CI 95% −0.44 to 0.22; P = 0.48), 0.02 (CI 95% −0.46 to 0.43; P = 0.94), and 0.05 (CI 95% −0.29 to 0.37; P = 0.79) for MEK, BRAF, and PI3K inhibitors, respectively.
Table 3.
Matched targeted agent population
| Anti‐MEK (n = 34) | Anti‐BRAF (n = 20) | Anti‐PI3K (n = 35) | |
|---|---|---|---|
| Gene | KRAS mutated | BRAF mutated | PIK3CA mutated |
| Variant | G12 = 24 (71%) | V600E = 20 (100%) | Helical = 28 (80%) |
| G13 = 6 (17%) | Kinase = 6 (17%) | ||
| Other = 4 (12%) | Other = 1 (3%) | ||
| Coexisting mutation | PIK3CA mutation = 12 (35%) | PIK3CA mutation = 4 (20%) | KRAS mutation = 13 (63%) |
| adjMAF (median, IQR) | 0.55 (0.43–0.64) | 0.25 (0.20–0.37) | 0.34 (0.25–0.54) |
| Profiling | |||
| Sequenom® | 31 (91%) | 12 (60%) | 27 (77%) |
| MiSeq® | 3 (9%) | 8 (40%) | 8 (23%) |
| CRC primary | 28 (82%) | 19 (95%) | 27 (77%) |
| Metastasis | 6 (18%) | 1 (5%) | 8 (23%) |
| Regimen | |||
| Single‐agent inhibitor | 0 | 5 (25%) | 35 (100%) |
| Doublet inhibitor | 34 (100%) | 7 (35%) | 0 |
| MEK + PI3K = 22 (65%) | BRAF + MEK = 4 (20%) | ||
| MEK + IGFR1 = 8 (23%) | BRAF + EGFR = 5 (25%) | ||
| MEK + HER = 4 (12%) | |||
| Triplet inhibitor | 0 | 8 (40%) | 0 |
| BRAF + EGFR + PI3K = 4 (20%) | |||
| BRAF + EGFR + WNT = 2 (10%) | |||
| BRAF + EGFR + CDK = 1 (5%) | |||
| BRAF + EGFR + MEK = 1 (5%) | |||
| Response | |||
| Complete | 0 | 1 (5%) | 0 |
| Partial | 0 | 4 (20%) | 0 |
| Stable disease | 7 (44%) | 9 (45%) | 11 (31%) |
| Progressive | 19 (56%) | 6 (30%) | 22 (63%) |
| NA | 0 | 0 | 2 (6%) |
| Discontinuation | |||
| Ongoing | 0 | 1 (5%) | 1 (3%) |
| Progression | 31 (91%) | 19 (95%) | 30 (87%) |
| Toxicity | 3 (9%) | 0 | 2 (5%) |
| Other | 0 | 0 | 2 (5%) |
4. Discussion
NGS of patient tumors has been rapidly incorporated into both prescreening programs and clinical trials over the last years, with the goal of identifying gene alterations that can guide individualized decisions. MAFs of driver genes reflect the genomic complexity of tumors, which may influence prognosis and response to targeted therapies. However, MAFs are frequently underreported or overlooked, which prompted us to investigate whether they have an impact in CRC evolution in the metastatic setting. Our results indicate clonality of RAS mutations and potential subclonality of BRAF V600E mutations and a subset of PIK3CA mutations in primary CRC tumors. Normanno et al. (2015) also found that in most CRC, the majority of neoplastic cells carry mutant KRAS or NRAS, while in BRAF‐ and PIK3CA‐mutant cases, only a fraction of neoplastic cells harbor the mutant allele. Alternatively, repetitive copy number alterations co‐occurring with mutations in driver genes could reduce BRAF and PIK3CA adjMAFs counts. However, the published literature does not support this hypothesis: BRAF and PIK3CA mutations rarely co‐occur with copy number gains in wild‐type alleles (The Cancer Genome Atlas, 2012; Zack et al., 2013). Arm‐level 7q gains (BRAF locus) have been described in up to 40% of CRC samples, mainly chromosomally instable tumors lacking BRAF mutations (The Cancer Genome Atlas, 2012). In fact, from 20 BRAF V600E‐mutated samples in TCGA cohort, only three cases (15%) had coexisting low‐level BRAF copy number gains that could possibly explain a lower adjMAF count (The Cancer Genome Atlas, 2012). Similarly, repetitive chromosome 3q gains (PIK3CA locus) have not been reported in non‐Asian CRC samples (He et al., 2003). Indeed, from 33 PIK3CA‐mutated samples in TCGA cohort, only two cases (6%) had coexisting low‐level PIK3CA copy number gains (The Cancer Genome Atlas, 2012). Therefore, we believe that BRAF V600E and PIK3CA mutations are real subclonal events in subsets of primary CRC tumors.
Other studies have also described clonal–subclonal frequencies of driver alterations in cancer. In a comprehensive analysis of TCGA data in nine solid tumors, McGranahan et al. (2015) found a clear tendency for mutations in driver genes to be clonal compared to mutations in noncancer genes. Interestingly, genes involved in the PI3K‐AKT‐mTOR pathway, such as PIK3CA, had a higher proportion of subclonal events compared to genes associated with RAS‐MAPK pathway, including KRAS, NRAS, and BRAF. There were clear differences in clonality of PIK3CA and BRAF V600E events according to tumor type, and we also identified a more clonal distribution in breast cancer and melanoma, respectively, as compared with CRC. On the other hand, our results suggest distinctive genomic structures according to BRAF codon affected, with non‐V600‐mutated tumors harboring a clear clonal pattern. Importantly, none of the studies reported above, including ours, have analyzed MAFs in light of microsatellite instability (MSI), which is associated with hypermutation rates and low copy number alterations (The Cancer Genome Atlas, 2012).
Previous studies have found that the presence of a low fraction of KRAS‐mutated cells within primary tumors may provide a reservoir for acquired resistance to EGFR antibodies (Azuara et al., 2016; Laurent‐Puig et al., 2015). It is surprising that the measurement of MAFs in primary samples correlates with the effects of therapy in the metastatic setting, which suggests that in addition to concordance of mutation events in primary and metastatic samples, the relapsed lesions most likely retain a similar genomic structure, with the same distribution of MAFs. However, we found high KRAS adjMAFs in metastatic sites of patients with a previous diagnosis of KRAS wild‐type CRC and exposed to anti‐EGFR therapy, suggesting clonal selection of KRAS‐mutant cells. Furthermore, differences in MAFs of primary and unmatched metastatic sites for BRAF V600E and TP53 mutations potentially reflect clonal selection and/or acquired copy number events after therapy. Indeed, the heterogeneity in copy number alterations between matched primary tumors and different metastatic lesions may explain some of the differences in adjMAFs across sites (Sveen et al., 2016).
With regard to the clinical implications of driver gene mutations, we observed that RAS and BRAF V600E mutations, irrespective of adjMAFs, have a negative effect on survival in the metastatic setting. These results indicate that even if subclonal, a driver event is biologically relevant. We acknowledge the fact that MSI, a poor prognostic factor in metastatic CRC, has not been taken into consideration in prognostic models (Kim et al., 2016). Additionally, we found that the clonality of KRAS, BRAF V600E, and PIK3CA mutations did not predict benefit with matched targeted agents. The negative findings, different from other reports in EGFR‐mutated lung cancer with EGFR tyrosine kinase inhibitors (Ono et al., 2014; Zhou et al., 2011), may be explained by the limited benefit with the targeted therapies explored in our cohort of chemotherapy‐refractory CRC, particularly with MEK and PI3K inhibitors in the KRAS‐ and PIK3CA‐mutant populations, respectively – the latter harboring coexisting KRAS mutations known to confer primary resistance to PI3K inhibitors as single agents (Dienstmann et al., 2012). For patients treated with BRAF inhibitors, while the poor response to targeted therapy can be explained by constitutive activation of alternative signaling pathways (Prahallad et al., 2012), the lack of correlation between BRAF V600E adjMAFs measured in CRC primary tissues and TTP in the metastatic setting may be related to a potential shift in clonality status of BRAF V600E events from primaries to metastases. The association between MAFs of driver gene events identified through circulating tumor DNA (ctDNA) NGS and clinical outcome under targeted therapy should be investigated.
5. Conclusion
To conclude, our results suggest that driver gene mutations can be subclonal and even ‘low MAF’ events in NGS tests should be reported. As of today, the absolute MAFs numbers cannot be used to optimize prediction of prognosis or response to matched targeted therapy in CRC. Major limitations of our study include the relatively small and targeted gene panel investigated, lack of copy number data to more precisely define clonality, and the absence of MSI status annotation. From a research perspective, more work needs to be conducted to increase biology understanding before clinical translation. Finally, we believe that analysis of paired primary and metastatic samples from the same patient (longitudinal sampling), with detailed treatment annotation, is crucial to further assess clonality and subclonality patterns of genomic events in cancer. Our work represents a foundation for future efforts assessing the clinical significance of a tumor's genomic structure to guide precision cancer therapy, opening the door for additional investigations on the dynamics of clonal evolution after chemotherapies and targeted drugs.
Author contribution
RD, IM, ES‐G, CO, MV, FRP, CV, and AG collected, analyzed, and interpreted the data. SL, HGP, PN, and AV involved in molecular data generation and interpretation. EE, GA, IM, ES‐G, TM, JC, MA, TS, HV, JR, and JT involved in clinical data generation and interpretation.
Disclaimers
Partial results of this study were previously presented at American Society of Clinical Oncology 2016 Annual Meeting as an oral communication in the Colorectal Cancer Clinical Science Symposium, The Clone Wars.
Supporting information
Fig. S1. A subset of samples with co‐occurring KRAS and PIK3CA mutations has a ‘subclonal’ pattern of PIK3CA adjMAFs (defined as KRAS/PIK3CA adjMAFs ratio > 1.5; A). Driver genes adjMAFs according to codon or domain affected (B) and tissue source (C).
Fig. S2. Overall survival in the metastatic setting in KRAS mutated colorectal cancer, stratified by co‐occurring PIK3CA mutations, either clonal or subclonal events (A). Overall survival in the metastatic setting in patients with tumor mutations in driver genes of the MAPK pathway, stratified by codon affected (B).
Doc. S1. Supplementary methods.
Acknowledgements
The authors acknowledge the FERO Foundation for supporting research in gastrointestinal malignancies and the Cellex Foundation for providing research facilities and equipment.
References
- Azuara D, Santos C, Lopez‐Doriga A, Grasselli J, Nadal M, Sanjuan X, Marin F, Vidal J, Montal R, Moreno V et al (2016) Nanofluidic digital PCR and extended genotyping of RAS and BRAF for improved selection of metastatic colorectal cancer patients for anti‐EGFR therapies. Mol Cancer Ther 15, 1106–1112. [DOI] [PubMed] [Google Scholar]
- Dienstmann R, Serpico D, Rodon J, Saura C, Macarulla T, Elez E, Alsina M, Capdevila J, Perez‐Garcia J, Sánchez‐Ollé G et al (2012) Molecular profiling of patients with colorectal cancer and matched targeted therapy in phase I clinical trials. Mol Cancer Ther 11, 2062–2071. [DOI] [PubMed] [Google Scholar]
- He QJ, Zeng WF, Sham JS, Xie D, Yang XW, Lin HL, Zhan WH, Lin F, Zeng SD, Nie D et al (2003) Recurrent genetic alterations in 26 colorectal carcinomas and 21 adenomas from Chinese patients. Cancer Genet Cytogenet 144, 112–118. [DOI] [PubMed] [Google Scholar]
- Kim CG, Ahn JB, Jung M, Beom SH, Kim C, Kim JH, Heo SJ, Park HS, Kim JH, Kim NK et al (2016) Effects of microsatellite instability on recurrence patterns and outcomes in colorectal cancers. Br J Cancer 115, 25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurent‐Puig P, Pekin D, Normand C, Kotsopoulos SK, Nizard P, Perez‐Toralla K, Rowell R, Olson J, Srinivasan P, Le Corre D et al (2015) Clinical relevance of KRAS‐mutated subclones detected with picodroplet digital PCR in advanced colorectal cancer treated with anti‐EGFR therapy. Clin Cancer Res 21, 1087–1097. [DOI] [PubMed] [Google Scholar]
- Lhermitte B, Egele C, Weingertner N, Ambrosetti D, Dadone B, Kubiniek V, Burel‐Vandenbos F, Coyne J, Michiels JF, Chenard MP et al (2017) Adequately defining tumor cell proportion in tissue samples for molecular testing improves interobserver reproducibility of its assessment. Virchows Arch 470, 21–27. [DOI] [PubMed] [Google Scholar]
- McGranahan N, Favero F, de Bruin EC, Birkbak NJ, Szallasi Z and Swanton C (2015) Clonal status of actionable driver events and the timing of mutational processes in cancer evolution. Sci Transl Med 7, 283ra254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murtaza M, Dawson SJ, Pogrebniak K, Rueda OM, Provenzano E, Grant J, Chin SF, Tsui DW, Marass F, Gale D et al (2015) Multifocal clonal evolution characterized using circulating tumour DNA in a case of metastatic breast cancer. Nat Commun 6, 8760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Normanno N, Rachiglio AM, Lambiase M, Martinelli E, Fenizia F, Esposito C, Roma C, Troiani T, Rizzi D, Tatangelo F et al (2015) Heterogeneity of KRAS, NRAS, BRAF and PIK3CA mutations in metastatic colorectal cancer and potential effects on therapy in the CAPRI GOIM trial. Annals Oncol 26, 1710–1714. [DOI] [PubMed] [Google Scholar]
- Ono A, Kenmotsu H, Watanabe M, Serizawa M, Mori K, Imai H, Taira T, Naito T, Murakami H, Nakajima T et al (2014) Mutant allele frequency predicts the efficacy of EGFR‐TKIs in lung adenocarcinoma harboring the L858R mutation. Annals Oncol 25, 1948–1953. [DOI] [PubMed] [Google Scholar]
- Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, Zecchin D, Beijersbergen RL, Bardelli A, Bernards R (2012) Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 483, 100–103. [DOI] [PubMed] [Google Scholar]
- Russo M, Siravegna G, Blaszkowsky LS, Corti G, Crisafulli G, Ahronian LG, Mussolin B, Kwak EL, Buscarino M, Lazzari L et al (2016) Tumor heterogeneity and lesion‐specific response to targeted therapy in colorectal cancer. Cancer Discov 6, 147–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sveen A, Løes IM, Alagaratnam S, Nilsen G, Høland M, Lingjærde OC, Sorbye H, Berg KC, Horn A, Angelsen JH et al (2016) Intra‐patient inter‐metastatic genetic heterogeneity in colorectal cancer as a key determinant of survival after curative liver resection. PLoS Genet 12, e1006225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas (2012) Comprehensive molecular characterization of human colon and rectal cancer. Nature 487, 330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zack TI, Schumacher SE, Carter SL, Cherniack AD, Saksena G, Tabak B, Lawrence MS, Zhsng CZ, Wala J, Mermel CH et al (2013) Pan‐cancer patterns of somatic copy number alteration. Nat Genet 45, 1134–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Zhang XC, Chen ZH, Yin XL, Yang JJ, Xu CR, Yan HH, Chen HJ, Su J, Zhong WZ et al (2011) Relative abundance of EGFR mutations predicts benefit from gefitinib treatment for advanced non‐small‐cell lung cancer. J Clin Oncol 29, 3316–3321. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. A subset of samples with co‐occurring KRAS and PIK3CA mutations has a ‘subclonal’ pattern of PIK3CA adjMAFs (defined as KRAS/PIK3CA adjMAFs ratio > 1.5; A). Driver genes adjMAFs according to codon or domain affected (B) and tissue source (C).
Fig. S2. Overall survival in the metastatic setting in KRAS mutated colorectal cancer, stratified by co‐occurring PIK3CA mutations, either clonal or subclonal events (A). Overall survival in the metastatic setting in patients with tumor mutations in driver genes of the MAPK pathway, stratified by codon affected (B).
Doc. S1. Supplementary methods.