

Abstract
Ewing sarcomas (ES) are highly malignant, osteolytic bone or soft tissue tumors, which are characterized by EWS–ETS translocations and early metastasis to lung and bone. In this study, we investigated the role of the BRICHOS chaperone domain‐containing endochondral bone protein chondromodulin I (CHM1) in ES pathogenesis. CHM1 is significantly overexpressed in ES, and chromosome immunoprecipitation (ChIP) data demonstrate CHM1 to be directly bound by an EWS–ETS translocation, EWS‐FLI1. Using RNA interference, we observed that CHM1 promoted chondrogenic differentiation capacity of ES cells but decreased the expression of osteolytic genes such as HIF1A,IL6,JAG1, and VEGF. This was in line with the induction of the number of tartrate‐resistant acid phosphatase (TRAP +)‐stained osteoclasts in an orthotopic model of local tumor growth after CHM1 knockdown, indicating that CHM1‐mediated inhibition of osteomimicry might play a role in homing, colonization, and invasion into bone tissues. We further demonstrate that CHM1 enhanced the invasive potential of ES cells in vitro. This invasiveness was in part mediated via CHM1‐regulated matrix metallopeptidase 9 expression and correlated with the observation that, in an xenograft mouse model, CHM1 was essential for the establishment of lung metastases. This finding is in line with the observed increase in CHM1 expression in patient specimens with ES lung metastases. Our results suggest that CHM1 seems to have pleiotropic functions in ES, which need to be further investigated, but appears to be essential for the invasive and metastatic capacities of ES.
Keywords: CHM1, endochondral bone, Ewing sarcoma, invasion, metastasis
Abbreviations
- ABCG2
ATP‐binding cassette, subfamily G (WHITE), member 2
- CHM1
leukocyte cell‐derived chemotaxin 1 (chondromodulin 1, CNMD)
- DKK2
dickkopf WNT signaling pathway inhibitor 2
- ES
Ewing sarcoma
- HIF1A
hypoxia‐inducible factor 1, alpha subunit
- IL6
interleukin 6
- JAG1
jagged 1
- MMP
matrix metallopeptidase
- NANOG
nanog homeobox
- OPN
secreted phosphoprotein 1 (SPP1)
- PROM1
prominin 1
- RANKL
tumor necrosis factor superfamily member 11 (TNFSF11)
- TGFB1
transforming growth factor beta
- TRAP
tartrate‐resistant acid phosphatase
- TSS
transcription start site
- VEGF
vascular endothelial growth factor receptor 1
1. Introduction
Ewing sarcomas (ES) are the second most common malignancy of bone and soft tissues in children and adolescents, which accounts for 10–15% of all primary bone tumors (Burchill, 2003). Genetically, ES are defined by EWS–ETS translocations encoding aberrant transcription factors presumed to induce the highly malignant phenotype of this disease (Delattre et al., 1994; Lessnick and Ladanyi, 2012; Mackintosh et al., 2010; Sorensen et al., 1994). Other contributing somatic mutations involved in disease development have only been observed at low frequency (Agelopoulos et al., 2015; Brohl et al., 2014; Crompton et al., 2014; Tirode et al., 2014). ES are characterized by early metastasis into lung and bone tissues. Metastasis is commonly hematogenous and related to stemness (Burdach et al., 2009; Richter et al., 2009; Schmidt et al., 1985). Even though prognosis for patients with ES has markedly improved during the development of multimodal therapeutic approaches, the survival rate of patients with advanced, multifocal disease is still associated with fatal outcome (Burdach et al., 1993, 2010; Thiel et al., 2011); especially, multifocal bone or bone marrow disease and the development of metastases in bones are catastrophic events in the clinical course of patients with ES (Burdach and Jurgens, 2002; Coleman, 2006; Thiel et al., 2016).
Based on our previous microarray analysis, we identified the dickkopf WNT signaling pathway inhibitor 2 (DKK2) critical for terminal bone development (Li et al., 2005) and two BRICHOS domain‐containing genes important for chondrogenic differentiation (Deleersnijder et al., 1996; Klinger et al., 2011), to be overexpressed in ES (Hauer et al., 2013; Staege et al., 2004). We demonstrated DKK2 to be an agonist of the canonical WNT/β‐catenin pathway and to be a key player in ES metastasis, bone invasiveness, and osteolysis (Hauer et al., 2013).
Here, we analyzed one of the BRICHOS domain‐containing genes, leukocyte cell‐derived chemotaxin 1 (also known as chondromodulin 1; CHM1; CNMD), for its function in chondro‐osseous tumor growth and invasiveness. Sanchez‐Pulido et al. (2002) observed that the BRICHOS domain itself seems to be involved in post‐translational processing of the corresponding pro‐proteins and/or to have a chaperone‐like activity. CHM1 expression has been previously associated with chondrosarcoma and BRICHOS domain mutations in the surfactant protein C precursor have been linked to endoplasmic reticulum stress, proteasome dysfunction, and caspase 3 activation, suggesting a role for the BRICHOS chaperone domain in microenvironmental regulation (Hedlund et al., 2009; Sanchez‐Pulido et al., 2002). Under normal conditions, CHM1 is almost exclusively expressed in the cartilage and has a strong antiangiogenic function (Hiraki and Shukunami, 2000; Hiraki et al., 1997; Yoshioka et al., 2006). The secreted, mature form of the glycoprotein is a key factor in chondrocyte proliferation and development and simultaneously inhibits terminal chondrocyte hypertrophy and endochondral ossification (Klinger et al., 2011; Shukunami and Hiraki, 2001). These characteristics indicated that CHM1 might be important in ES malignancy, as ES progenitor cells seem to be of premature chondrogenic origin arrested at early osteo‐chondrogenic differentiation (Hauer et al., 2013; von Heyking et al., 2016; Tanaka et al., 2014).
In the present study, we observed that CHM1 reduced the endothelial but enhanced the chondrocytic differentiation ability of ES. CHM1 simultaneously increased the expression of several stem cell genes such as PROM1. Furthermore, CHM1 overexpression promoted in vitro invasiveness, as well as lung metastasis of ES cells in a xenograft mouse model. In line with these findings, expression of CHM1 is significantly higher in lung metastases samples of patients with ES than in samples derived from different bone localizations. This indicates CHM1 to be important for ES malignancy, especially for maintaining an undifferentiated, metastatic phenotype in ES.
2. Materials and methods
2.1. Cell lines
ES lines (MHH‐ES1, RD‐ES, SK‐ES1, SK‐N‐MC, and TC‐71), neuroblastoma lines (CHP126, MHH‐NB11, SHSY5Y, and SIMA), and pediatric human B‐cell precursor leukemic lines (cALL2, NALM6, and 697) were obtained from the German Collection of Microorganisms and Cell Cultures (DSMZ, Braunschweig, Germany). ES line VH64 was kindly provided by Marc Hotfilder (Münster University, Münster, Germany); osteosarcoma lines (HOS, HOS‐58, MG‐63, MNNG, SaOS, SJSA01, U2OS, and ZK‐58) by Jan Smida and Michaela Nathrath, Institute of Pathology and Radiation Biology (HMGU, Neuherberg, Germany). A673 was purchased from ATCC (LGC Standards, Teddington, UK). SB‐KMS‐KS1 and SB‐KMS‐MJ1 are ES cell lines that were established in our laboratory (Grunewald et al., 2012; Richter et al., 2009). Retrovirus packaging cell line PT67 was obtained from Takara Bio Europe/Clontech (Saint‐Germain‐en‐Laye, France). Cells were maintained in a humidified incubator at 37 °C in 5–8% CO2 atmosphere in RPMI 1640 or DMEM (both Life Technologies, Carlsbad, CA, USA) containing 10% heat‐inactivated fetal bovine serum (Biochrom, Berlin, Germany) and 100 μg·mL−1 gentamicin (Life Technologies). Cell lines were checked routinely for purity (e.g., EWS‐FLI1 translocation product, surface antigen or HLA phenotype) and mycoplasma contamination.
2.2. RNA interference (RNAi)
For transient RNA interference, cells were transfected with small interfering RNA (siRNA) as described previously (Richter et al., 2009). To test transfection efficiency and gene silencing, RNA was extracted and gene expression assessed by quantitative real‐time PCR. All siRNA sequences are provided in the supplementary data.
2.3. Constructs and retroviral gene transfer
For stable silencing of CHM1 expression, oligonucleotides were designed corresponding to the most efficient siRNA used for transient RNA interference and retroviral gene transfer was performed as described previously (Richter et al., 2009). The used oligonucleotides are provided in the Supporting Information (Doc. S1).
2.4. Quantitative Real‐time PCR (qRT‐PCR)
Total RNA was isolated and reverse‐transcribed using the High Capacity cDNA Reverse Transcription Kit (Life Technologies) according to the manufacturer's instructions. Differential gene expression was then analyzed by qRT‐PCR using TaqMan Universal PCR Master Mix and fluorescence detection with an AB 7300 Real‐Time PCR System (both Life Technologies) as described previously (Richter et al., 2009, 2013). Gene expression was normalized to glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH). A list of used assays is provided in the Supporting Information. NTC: nontemplate control.
2.5. ChIP and quantitative real‐time PCR
ChIP was performed using ChIP‐IT® Express Kit (Active Motif, Carlsbad, CA, USA) according to the manufacturer's instructions. In brief, 2 × 107 SK‐N‐MC and TC‐71 cells, respectively, were fixed with methanol‐free formaldehyde (Life Technologies, Darmstadt, Germany) at a final concentration of 1% for 10 min. After neutralization with glycine, cells were lysed in RIPA buffer with protease inhibitors. Samples were sonicated to an average DNA length of 200–400 bp using a M220 Focused‐ultrasonicator™ (Covaris, Woburn, MA, USA). ChIP was carried out using 5 μg of anti‐FLI1 antibody (C‐19, sc‐356X; Santa Cruz) or anti‐rabbit IgG (sc‐2027X; Santa Cruz), respectively. DNA was cleaned up using IPure kit (Diagenode, Seraing, Belgium). Quantitative real‐time PCR (qPCR) using SYBR Green (Bio‐Rad, München, Germany) was performed for different loci of the CHM1 promoter and one positive control loci at −1081 bp upstream of the transcription start site (TSS) of the EZH2 promoter. FLI1 binding was normalized to IgG control antibody using the ΔΔCT method (Livak and Schmittgen, 2001).
2.6. Proliferation assay
Cell proliferation was determined with an impedance‐based instrument system (xCELLigence, Roche/ACEA Biosciences, Basel, Switzerland) enabling label‐free real‐time cell analysis. Briefly, 1–3 × 104 cells were seeded into 96‐well plate with 200 μL media containing 10% FBS and allowed to grow up to 60 h. Cellular impedance was measured periodically every four hours and gene knockdown was monitored by qRT‐PCR.
2.7. Colony forming assay
Cells were seeded in duplicate into a 35‐mm plate at a density of 5 × 103 cells per 1.5 mL methylcellulose‐based media (R&D Systems, Minneapolis, MN, USA) according to the manufacturer's instructions and cultured for 10–14 days at 37 °C/5% CO2 in a humidified atmosphere.
2.8. In vitro invasion assay
To study cell invasion, the BioCoat™ Angiogenesis System: Endothelial Cell invasion was used (BD Biosciences, San Jose, CA, USA) according to the manufacturer's instructions as described previously (Grunewald et al., 2012).
2.9. Differentiation assay
Cellular tube formation was tested by the use of a commercial Matrigel matrix assay (Biocoat; BD Biosciences) according to the manufacturer's instruction. Briefly, cells were seeded at 5 × 104 cells per well in a 96‐well plate and grown at 37 °C (5% CO2) in a humidified atmosphere. After 16–18 h, cells were stained with 1 μg·mL−1 Calcein AM Fluorescent Dye (BD Biosciences) for 30 min in the dark. Cells were imaged by fluorescence microscopy by using a Nikon Eclipse TS 100 with an attached Nikon Coolpix 5400 camera (Nikon, Tokyo, Japan).
2.10. Elisa
An ELISA with 48 strip wells from MyBioSource (San Diego, CA, USA) to detect CHM1 levels (MBS937594) was performed according to the manufacturer's instructions.
2.11. Microarray analysis
Patient material was obtained from clinical studies of the Cooperative Ewing Sarcoma Study Group in Europe. All patients provided informed consent. Biotinylated target cRNA was prepared as previously described (Richter et al., 2009). A detailed protocol is available at www.affymetrix.com. Samples were hybridized to Affymetrix Human Gene 1.0 ST microarrays and analyzed by affymetrix software expression console (Affymetrix, High Wycombe, UK), version 1.1. For the data analysis, robust multichip average normalization was performed, including background correlation, quantile normalization, and median polish summary method. Array data were submitted at GEO (GSE45544).
2.12. Animal model
Immunodeficient Rag2−/−γc−/− mice on a BALB/c background were obtained from the Central Institute for Experimental Animals (Kawasaki, Japan) and maintained in our animal facility under pathogen‐free conditions in accordance with the institutional guidelines and approval by local authorities (Regierung von Oberbayern). Experiments were performed in 6‐ to 20‐week‐old mice.
2.13. In vivo experiments
To examine in vivo tumorigenicity, 2 × 106 ES cells and derivatives were injected subcutaneously into the inguinal region of immunodeficient Rag2−/−γc−/− mice, and when the tumor reached 1 cm3, mice were sacrificed and tumor samples were analyzed.
For the analysis of in vivo metastatic potential, 1.5–2 × 106 ES cells and derivatives were injected in a volume of 0.2 mL into the tail vein of immunodeficient Rag2−/−γc−/− mice as described previously (Grunewald et al., 2012; Richter et al., 2009). Mice were sacrificed after five weeks, and metastatic spread was examined in individual organs.
To investigate bone invasiveness and osteolysis, mice were anesthetized with 500 mg·mL−1 novaminsulfon (Ratiopharm, Ulm, Germany) and isoflurane (Abbott, Abbott Park, IL, USA) and A673 or TC‐71 derivatives were injected as described previously (Hauer et al., 2013). Briefly, a 30‐gauge needle was introduced through the proximal tibia plateau and 2 × 105 ES cells in a volume of 20 μL were injected into the medullary cavity. In all experiments, tumors and affected tissues were recovered and processed for histological analyses. Intratibial tumor formation was monitored by X‐ray radiography.
2.14. Histology
Murine organs were fixed in phosphate‐buffered 4% formaldehyde and embedded in paraffin; 3‐ to 5‐μm‐thick sections were stained with hematoxylin and eosin (H&E). Hind limb bones were decalcified and paraffin‐embedded; the histological analysis with H&E was complemented by quantification of tartrate‐resistant acid phosphatase (TRAP+)‐stained osteoclasts. All sections were reviewed and interpreted by two pathologists (J. C‐W.; F. N. or I.E.).
2.15. Statistical analyses
Data are mean ± SEM as indicated. Differences were analyzed by unpaired two‐tailed Student's t‐test as indicated using Excel (Microsoft, Redmond, WA, USA) or Prism 5 (GraphPad Software, San Diego, CA, USA); P values < 0.05 were considered statistically significant (*P < 0.05; **P < 0.005; ***P < 0.0005).
3. Results
3.1. CHM1 is highly expressed in Ewing sarcomas
Previously, we identified CHM1 to be highly expressed in ES (Staege et al., 2004). As shown in Fig. 1A,B, we observed high levels of CHM1 expression exclusively in ES, compared to different normal and fetal tissues (Fig. 1A), or various other pediatric or adult cancer types such as neuroblastoma, medulloblastoma, leukemia, and various carcinomas (Fig. 1B). To further validate overexpression of CHM1 in ES, we tested nine common ES cell lines against a series of different osteosarcoma, neuroblastoma, and ALL cell lines using qRT‐PCR. As expected, CHM1 was strongly up‐regulated in ES cell lines, but not in neuroblastoma and ALL cell lines (Fig. 1C). Furthermore, analysis of mRNA levels revealed no expression of CHM1 in osteosarcoma cell lines (Fig. 1C), while CHM1 was previously associated with inhibition of endochondral ossification (Deleersnijder et al., 1996; Klinger et al., 2011).
Figure 1.
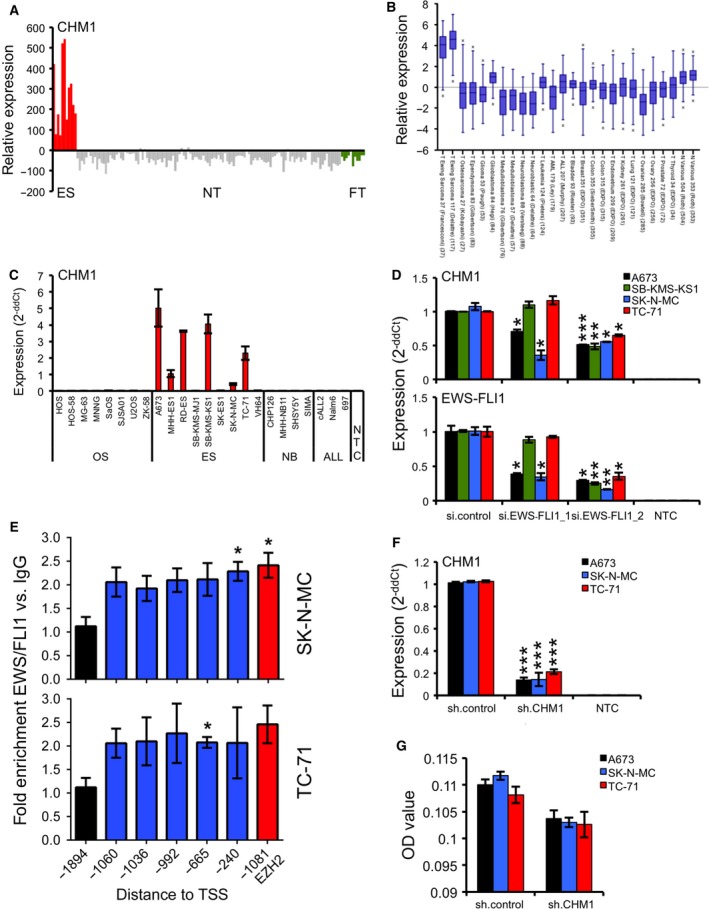
CHM1 is highly overexpressed in ES. (A) Expression profile of CHM1 in primary ES in comparison with normal tissue (NT) and fetal tissue (FT). ES, NT, and FT samples were analyzed using EOS‐Hu01 microarrays (Staege et al., 2004). (B) Expression levels of CHM1 in different pediatric small, round, blue cell tumors, carcinomas, and normal tissues by box plot presentation using a comparative study of the amc onco‐genomics software tool (www.amc.com). Results are 2‐log‐centered for better representation of results. The number of samples in each cohort is given in brackets. (C) CHM1 expression in different tumor cell lines analyzed by qRT‐PCR. Data are mean ± SEM. (D) RNA interference of EWS‐FLI1 expression (bottom) does reduce CHM1 expression (top). si.EWS‐FLI1_1 (less efficient) and si.EWS‐FLI1_2 represent the specific siRNAs (si.control: nonsilencing siRNA). Results of qRT‐PCR 48 h after transfection are shown. Data are mean ± SEM of two independent experiments; t‐test. (E) EWS‐FLI1 enrichment at the CHM1 promoter in SK‐N‐MC and TC‐71 cells. ChIP analysis was performed with FLI1 and control IgG antibodies, respectively, and analyzed by quantitative PCR for binding to different regions of the CHM1 promoter. FLI1 enrichment was detected at different ETS recognition sites −1060, −1036, −992, −665, and −240 bp upstream of the TSS of CHM1. The −1894‐bp region, which is devoid of ETS recognition sequences, served as negative control. The ETS consensus site at −1081 bp of the EZH2 promoter (Richter et al., 2009) was used as positive control for FLI1 binding. Data represent the mean of two independent experiments, and error bars represent standard deviations. (F) Constitutive suppression of CHM1 expression after infection of ES cells with CHM1‐specific shRNA constructs as measured by qRT‐PCR (sh.CHM1 and sh.control). qRT‐PCR data are mean ± SEM of 10 independent experiments; t‐test. (G) ELISA detection of CHM1 levels in the supernatant of ES cells stably transfected with CHM1 shRNA or control. Data are mean ± SEM; t‐test. *P < 0.05; **P < 0.005; ***P < 0.0005 (see 2.15. Statistical analyses).
Subsequently, we analyzed whether the oncogenic fusion protein EWS‐FLI1 can influence CHM1 expression in four different ES cell lines. As shown in Fig. 1D, RNA interference‐mediated EWS‐FLI1 silencing led to a significant, efficiency‐dependent suppression of CHM1 levels, which indicates CHM1 expression to be associated with EWS‐FLI1. We next performed ChIP analysis with FLI1 and IgG antibodies to analyze binding of FLI1 to the CHM1 promoter. FLI1 enrichment was detected at different ETS recognition sites −1060, −1036, −992, −665, and −240 bp upstream of the TSS of CHM1 (Fig. 1E). These data suggest CHM1 to be directly regulated by the ES chimeric transcription factor, EWS‐FLI1. For subsequent analysis, we constitutively down‐regulated CHM1 in different ES cell lines (A673, SK‐N‐MC, and TC‐71) to further elucidate the influence of this gene on ES pathogenesis (Fig. 1F,G).
3.2. CHM1 influences the endothelial as well as chondrocytic differentiation potential of ES
Due to the well‐known antiangiogenic function of CHM1 (Hiraki et al., 1997; Yoshioka et al., 2006), we first tested the endothelial differentiation capacity of A673 and MHH‐ES1 cells either stable‐transfected with sh.CHM1 or sh.control or transiently with CHM1 or control siRNA, respectively (Fig. S1A), in a Matrigel matrix assay. As shown in Fig. 2A, CHM1 expression clearly inhibited the potential to form cellular tubes in ES cell lines irrespective of whether we investigated constitutive or transient knockdown of CHM1. Furthermore, CHM1 seems to be a key factor in chondrocyte development and proliferation inhibiting terminal chondrocyte differentiation to a hypertrophic phenotype during the process of endochondral ossification (Klinger et al., 2011; Shukunami and Hiraki, 2001). Thus, we incubated three ES cell lines stably transfected with sh.CHM1 and sh.control with specific differentiation media to induce chondrogenic or osteogenic differentiation. The differentiation potential was determined by qRT‐PCR using specific marker genes (Vater et al., 2011). As shown in Fig. S1B,C, the chondrogenic, and to a lesser extent the osteogenic, differentiation ability was significantly impaired after CHM1 knockdown. Based on these findings, we asked whether CHM1 might be important for the maintenance of an immature, chondrocytic phenotype of this tumor. Therefore, we analyzed the expression of different stem cell genes, namely ATP‐binding cassette, subfamily G (WHITE), member 2 (ABCG2; Szepesi et al., 2015; Zhou et al., 2001), nanog homeobox (NANOG; Mitsui et al., 2003), and prominin 1 (PROM1; Katoh and Katoh, 2007), in ES cell lines with CHM1 knockdown and respective controls. As shown in Fig. S1D, suppression of CHM1 decreased the expression of ABCG2 and PROM1 compared to sh.control‐transfected cells, of which only PROM1, important for maintaining stemness and pluripotency, was down‐regulated down to 13.7% (32.9%), especially in A673 cells, after CHM1 knockdown at the protein level (Fig. S1E).
Figure 2.
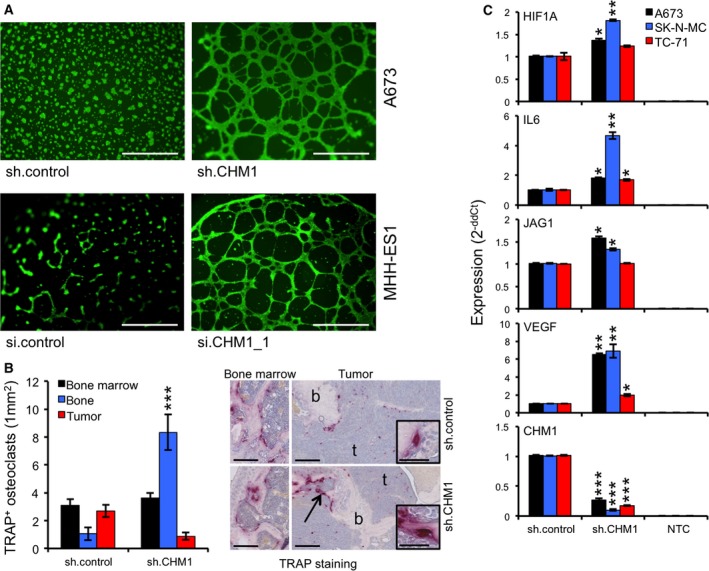
CHM1 inhibits tube formation and influences osteomimicry. (A) Tube formation assay with constitutively transfected A673 (sh.control and sh.CHM1) and transiently transfected MHH‐ES1 (si.control and si.CHM1_1) cells demonstrated CHM1 to clearly inhibit endothelial differentiation potential (scale bar 0.5 mm). (B) Analysis of osteolysis of A673 sh.CHM1 and negative controls (sh.control) in an orthotopic bone xenotransplantation model (five to eight mice per group). Affected bones were assessed by histology (TRAP staining, scale bar 0.25 mm or 0.05 mm). Left panel: quantitative summary of the average number of osteoclasts (mm2) in unaffected bone marrow, tumor samples, and attached to the bone in tumor tissues (bone). Data are mean ± SEM of at least two independent samples (at least 40 segments counted); t‐test. Right panel: Representative pictures are shown. CHM1 knockdown significantly enhanced the amount of TRAP‐positive osteoclasts attached to the bone (b) in the area of tumor (arrow) and thus increased the osteolytic phenotype. (C) Different ES cell lines with constitutive CHM1 knockdown and respective controls were analyzed by qRT‐PCR for expression of osteolytic genes such as HIF1A,IL6,JAG1, and VEGF. Data are mean ± SEM of two independent experiments; t‐test. *P < 0.05; **P < 0.005; ***P < 0.0005 (see 2.15. Statistical analyses).
3.3. CHM1 represses osteomimicry of ES
Due to the particular effect of CHM1 especially on the chondrogenic differentiation potential of ES cells, we asked whether CHM1 may influence bone‐associated tumor growth of ES in vivo, as well. We injected constitutive sh.CHM1‐ or sh.control‐infected A673 cells (see Materials and methods 2.13) into the tibiae of immunodeficient Rag2−/−γc−/− mice and analyzed bone infiltration and destruction by X‐ray radiography and histology. Many mice developed severe osteolytic lesions (both around 80%), regardless of whether mice were injected with A673 sh.control or sh.CHM1 cells (data not shown). However, the number of TRAP+ osteoclasts was significantly increased within bone tissue, but decreased within the tumor tissue in sh.CHM1 samples as compared to negative controls (Fig. 2B). A similar experiment with TC‐71 sh.CHM1 and sh.control cells could confirm these findings, even though only few mice (40%) developed a tumor regardless of whether injecting TC‐71 sh.CHM1 or sh.control cells (Fig. S2).
The increased osteolytic phenotype in bone tissue after CHM1 knockdown might result in better localization to bone in combination with a change in the expression pattern of cancer cells, also known as osteomimicry. We determined the mRNA levels of different genes known to be associated with osteolysis. As shown in Fig. 2C, CHM1 knockdown significantly increased the expression levels of osteolytic genes such as hypoxia‐inducible factor 1, alpha subunit (HIF1A), interleukin 6 (IL6), jagged 1 (JAG1), and vascular endothelial growth factor receptor 1 (VEGF) (Weilbaecher et al., 2011), which may further increase the osteolytic and malignant activity within bone observed here (Fig. 2B).
3.4. CHM1 enhances proliferation in ES
To further analyze the impact of CHM1 overexpression on the pathogenesis and malignancy of ES, we next examined the effect of CHM1 on in vitro proliferation using an xCELLigence‐based proliferation assay. As shown in Fig. 3A, constitutive down‐regulation of CHM1 significantly decreased contact‐dependent growth of all three ES cell lines investigated without affecting the cell cycle (Fig. S3). Interestingly, CHM1 similarly enhanced colony formation on methylcellulose matrices in A673, SK‐N‐MC, and TC‐71 cells in vitro (Fig. 3B). Subsequently, we analyzed whether CHM1 affects in vivo tumorigenicity of ES, too. We injected stably transfected A673 and TC‐71 cells with sh.CHM1 and sh.control subcutaneously into the inguinal region of immunodeficient Rag2−/−γc−/− mice and analyzed local tumor growth. However, in contrast to in vitro proliferation, suppression of CHM1 only marginally delayed local tumor growth in vivo (Fig. 3C).
Figure 3.
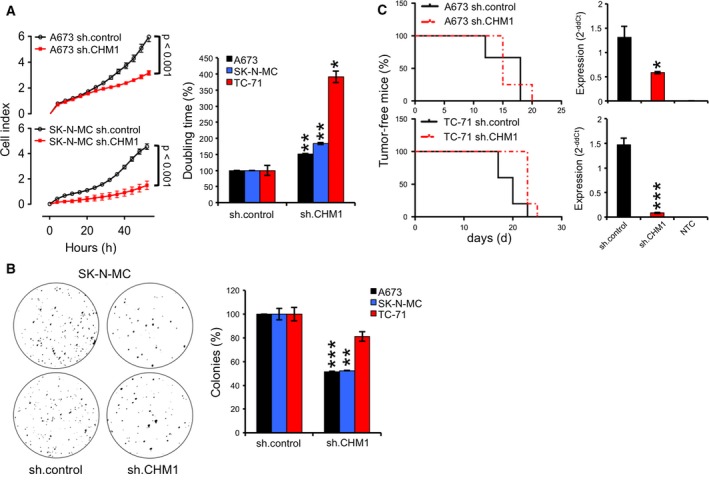
CHM1 delayed proliferation in ES in vitro. (A) Analysis of contact‐dependent growth of constitutively sh.CHM1‐ and sh.control‐infected ES cell lines with xCELLigence. Left panel: Cellular impedance was measured every four hours (relative cell index). Data are mean ± SEM (hexaplicate/group); t‐test. Right panel: doubling time of constitutive A673, SK‐N‐MC, and TC‐71 CHM1 shRNA infectants. Data are mean ± SEM of two independent experiments/cell line (hexaplicate/group); t‐test. B. Effect of CHM1 knockdown on anchorage‐independent growth in A673, SK‐N‐MC, and TC‐71 cells using methylcellulose matrices. Left panel: A representative experiment with SK‐N‐MC cells was shown as macrograph. Right panel: The average number of colonies of at least two different experiments with three different ES cell lines was shown after stable CHM1 suppression. (C) Left panel: evaluation of tumorigenicity of constitutive A673 and TC‐71 CHM1 shRNA infectants in immunodeficient Rag2−/−γc−/− mice (3–5 mice per group). Right panel: post ex vivo CHM1 expression using qRT‐PCR. Data are mean ± SEM, t‐test. *P < 0.05; **P < 0.005; ***P < 0.0005 (see 2.15. Statistical analyses).
3.5. CHM1 enhances invasiveness and metastasis in ES
Invasiveness and metastasis are important hallmarks of cancer (Hanahan and Weinberg, 2011). Therefore, we tested three ES cell lines with constitutive CHM1 knockdown and respective controls in a Matrigel invasion assay. Stably silenced CHM1 ES cell lines showed a clear reduction in invasion down to 4% in SK‐N‐MC cells compared to control cells (Fig. 4A). As previously reported by our group, matrix metallopeptidases (MMPs) appear to be important for ES invasiveness (Grunewald et al., 2012; Hauer et al., 2013; Richter et al., 2013). Thus, we next examined the mRNA expression of MMP1, MMP7, and MMP9 after CHM1 knockdown. As shown in Fig. 4B, suppression of CHM1 clearly reduced mRNA levels of MMP9, in contrast to MMP1 and MMP7 (Fig. S4A). Simultaneously transient MMP9 knockdown significantly decreased the amount of cells crossing the Matrigel, albeit not as strong as observed after CHM1 suppression (Fig. 4A,B bottom), indicating additional factors involved.
Figure 4.
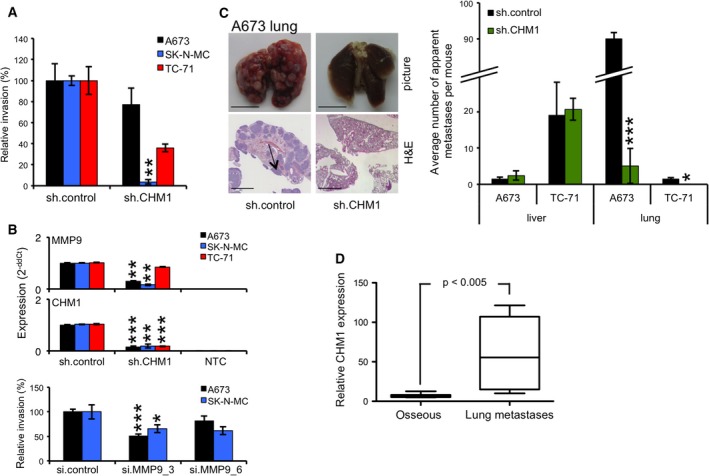
CHM1 enhances metastasis in ES in vivo. (A) Analysis of invasiveness of ES cell lines through Matrigel after transfection with specific CHM1 shRNA constructs. Data are mean ± SEM of two independent experiments; t‐test. (B) Upper panel: qRT‐PCR of MMP9 expression after stable CHM1 knockdown. Data are mean ± SEM of two independent experiments; t‐test. Lower panel: analysis of the invasive potential of A673 and SK‐N‐MC cells after transient transfection with two specific MMP9 siRNAs 48 h before seeding. Data are mean ± SEM; t‐test. (C) Analysis of metastasis using A673 and TC‐71 cells with stable CHM1 suppression and respective controls (four to five mice per group). Left panel: Representative lungs with corresponding H&E staining of A673‐injected mice are shown (scale bar 5 or 2 mm). Right panel: Average number of apparent metastases per mouse in lung and liver tissues is illustrated; t‐test. (D) DotBlot of relative CHM1 expression in ES osseous tumor samples compared to ES lung metastases samples using microarray analysis of 14 patient tumor samples. *P < 0.05; **P < 0.005; ***P < 0.0005 (see 2.15. Statistical analyses).
Finally, we investigated the metastatic potential of ES cells constitutively transfected with sh.CHM1 and sh.control in immunodeficient Rag2−/−γc−/− mice. Even though there is no difference in cell size and there is only a minimal increase in granularity between sh.CHM1 and sh.control cells (Fig. S4B), suppression of CHM1 significantly reduced the number of lung metastases after inoculation with A673 cells (Fig. 4C). However, no clear differences were observed for liver metastases for these cells (Fig. 4C, right). These results were confirmed with TC‐71 cells; while no lung metastases were observed after CHM1 suppression, the number of liver metastases was not affected (Fig. 4C). Interestingly, modulation of angiogenesis did not seem to contribute to ES metastasis in our in vivo mouse model. Although CHM1 clearly inhibited the endothelial differentiation potential in vitro (Fig. 2A) and in vivo, no differences in angiogenesis were observed as demonstrated by CD31 and Mac‐3 staining of different lung and liver tumor samples (Fig. S4C,D).
To further determine the relevance of these results in the clinical setting, we analyzed samples from 14 patients with ES. Interestingly, microarray analysis revealed a significantly higher expression of CHM1 (P‐value < 0.005) in tumor samples derived from lung metastases than from different local relapses in bone localizations (Fig. 4D).
4. Discussion
The current study investigated the role of CHM1 for the biology and pathology of ES. We observed that EWS‐FLI1 specifically induced CHM1 expression.
CHM1 is a known antiangiogenic factor, which has been demonstrated to play a role in bone development and to be expressed in growth plate cartilage of hypertrophic and calcified zones (Hiraki et al., 1997; Miura et al., 2014; Yoshioka et al., 2006). Previously, we have shown that reduced tumor perfusion is associated with resistance and poor prognosis in ES (Dunst et al., 2001). CHM1 influences endochondral ossification as well as chondrocyte development and proliferation (Klinger et al., 2011; Shukunami and Hiraki, 2001). Its function may be mediated by its secreted form or by its intracellular effect on different pathways, respectively (Mera et al., 2009). ES cells secrete CHM1 as demonstrated via ELISA, but we have no direct information on potential membrane‐bound forms as available antibodies so far do not work reproducibly in western blot analysis (data not shown). However, following RNA interference, we observed CHM1 to affect endothelial, as well as chondrocytic, differentiation potential of ES, presumably via its intracellular activity. Because CHM1 maintains a more undifferentiated chondrocytic phenotype and represses endothelial differentiation of ES, we further investigated the expression of several stem cell genes. Although we did not observe a distinct phenotype, we could show that CHM1 enhanced the expression of ABCG2 and PROM1. ABCG2 is expressed in a wide variety of stem cells (Zhou et al., 2001), while PROM1 is so in embryonic and adult as well as cancer stem cells and maintains stem cell properties by suppressing differentiation (Katoh and Katoh, 2007). ABCG2, in addition, seems to be a good marker for stem cells with enhanced osteogenic and chondrogenic differentiation potential (Szepesi et al., 2015). Remarkably, Tanaka et al. (2014) recently demonstrated that cells present in the embryonic superficial zone of long bones and of osteo‐chondrogenic origin are possible ES progenitor cells. Furthermore, epigenetic suppression of CHM1 in malignant tumor of bone such as osteosarcoma (Aoyama et al., 2004) is supportive for its presumed role maintaining an immature chondrocytic phenotype in ES.
While investigating how CHM1 influences tumor growth in our orthotopic xenograft mouse model (Hauer et al., 2013), we observed that overall tumor growth was relatively unaffected although an increase in TRAP+ osteoclasts in bone tissue following CHM1 suppression was detected. In line with this observation, we noticed an increased expression of malignancy‐promoting/osteolytic genes after CHM1 knockdown in ES cells, which might enhance aggressiveness and result in better localization to bone in combination with a change in the expression pattern of cancer cells, also known as osteomimicry. Expression of the transcription factor HIF1A by tumor cells inhibits osteoblast differentiation and enhances the differentiation and maturation of osteoclasts, in part via VEGF induction (Dunn et al., 2009; Hiraga et al., 2007; Weilbaecher et al., 2011). Furthermore, Guan et al. demonstrated that VEGF increases RANKL promoter activity in ES, leading to induced bone lysis (Guan et al., 2009), which may explain the increased osteolytic phenotype of ES after CHM1 knockdown in our osteotropic tumor model as observed here. In addition, JAG1, a potent downstream mediator of TGFB1, which promotes osteolysis in breast cancer cells by activating the NOTCH signaling pathway, leads to increased IL6 expression (Sethi et al., 2011; Tao et al., 2011). However, in ES, NOTCH signaling is switched off via EWS‐FLI1‐mediated repression (Ban et al., 2008; Bennani‐Baiti et al., 2011). IL6 is a pro‐proliferative cytokine, which promotes tumor growth (Ara et al., 2009) and is enhanced after CHM1 suppression in ES cells (Fig. 2C). Another prominent example with regard to osteomimicry observed here was the expression of OPN, which also increased after CHM1 knockdown especially in A673 cells (Fig. S1C). OPN is normally expressed by osteoclasts and facilitates attachment of osteoclasts to the bone matrix (Reinholt et al., 1990). Moreover, OPN is known to be secreted by tumor cells and promotes bone marrow cell recruitment and tumor formation in bones (Anborgh et al., 2010; Weilbaecher et al., 2011). Overall, these results may provide hints that CHM1 may balance a certain level of chondro‐osseous differentiation capability and supports stronger CHM1 expression in lung metastases compared to bone samples of patients with ES, as observed here.
Further analysis of ES malignancy revealed that CHM1 significantly enhances contact‐dependent as well as contact‐independent growth of different ES cell lines in vitro, but only marginally influences local tumor growth in xenograft mice after subcutaneous injection. Presumably, the CHM1‐mediated growth advantage in vitro may be reduced by a poorer supplement/support of tumor growth in vivo due to the known antiangiogenic function of this glycoprotein (Hiraki et al., 1997; Yoshioka et al., 2006).
Additionally, we clearly observed that CHM1 enhances in vitro invasiveness and significantly increased the mRNA expression of MMP9 in different ES cell lines. In previous studies (Grunewald et al., 2012; Hauer et al., 2013; Richter et al., 2013), we demonstrated MMP1 to be the most important factor influencing ES invasiveness in vitro and in vivo. However, these results were not confirmed after knockdown of CHM1. Transient suppression of MMP9 clearly reduced the invasive potential of ES cells, as well, introducing MMP9 as another important factor in ES invasiveness. This observation is confirmed by different publications identifying MMP9 as a crucial factor associated with invasion in other tumor entities, such as breast and prostate cancer (Bin Hafeez et al., 2009; Wang et al., 2011). In line with these findings, in vivo knockdown of CHM1 mainly suppressed the development of lung metastases of different ES cells investigated in our mouse model, indicating CHM1 to be important for the development of lung but not for liver or bone metastases. These results were complemented by clinical data reinforcing a role of CHM1 for ES invasiveness and metastasis especially to lung tissues (Fig. 4D).
In summary, our results indicate that CHM1 preserves the immature chondrocytic phenotype of this disease and enhances clonality as well as invasiveness and the metastatic potential especially for lung metastasis in vivo, thereby promoting the malignant potential of this disease.
Author contributions
KvH, SG, OS, DS, AF, and GHSR performed experiments. KvH, CMT, TH, and GHSR analyzed data. JCW, FN, and IE carried out pathology assessments and IHC analyses. SB and GHSR initiated the project. PS provided key insights into data interpretation. KvH and GHSR wrote the manuscript.
Supporting information
Fig. S1. CHM1 maintains an undifferentiated phenotype of ES.
Fig. S2. In vivo bone invasion and osteolysis.
Fig. S3. Cell cycle distribution analyses.
Fig. S4. CHM1 knock down does not influence in vivo angiogenesis.
Doc S1. Materials and methods.
Acknowledgements
The authors would like to thank Laura Roth, Melanie Thiede, and Eleonore Samson for their support. This work was supported by grants from the Else‐Kröner‐Fresenius Stiftung (2013_A49) and the Wilhelm‐Sander Stiftung (2009.901.3). It is part of the Translational Sarcoma Research Network (TransSaRNet; 01GM1104B), ‘Rare Diseases’ and Prospective Validation of Biomarkers in Ewing Sarcoma for Personalised Translational Medicine (PROVABES; 01KT1311), Funding Programs of the Federal Ministry of Education and Research (BMBF), Germany. Work in Irene Esposito's laboratory is supported by the National Genome Research Network (NGFNplus, 01GS0850) and in Frauke Neff's laboratory by Infrafrontier (01KX1012). Work in Carsten Müller‐Tidow's laboratory was supported by DFG Mu1328/14‐1.
References
- Agelopoulos K, Richter GH, Schmidt E, Dirksen U, von Heyking K, Moser B, Klein HU, Kontny U, Dugas M, Poos K et al (2015) Deep sequencing in conjunction with expression and functional analyses reveals activation of FGFR1 in Ewing sarcoma. Clin Cancer Res 21, 4935–4946. [DOI] [PubMed] [Google Scholar]
- Anborgh PH, Mutrie JC, Tuck AB and Chambers AF (2010) Role of the metastasis‐promoting protein osteopontin in the tumour microenvironment. J Cell Mol Med 14, 2037–2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoyama T, Okamoto T, Nagayama S, Nishijo K, Ishibe T, Yasura K, Nakayama T, Nakamura T and Toguchida J (2004) Methylation in the core‐promoter region of the chondromodulin‐I gene determines the cell‐specific expression by regulating the binding of transcriptional activator Sp3. J Biol Chem 279, 28789–28797. [DOI] [PubMed] [Google Scholar]
- Ara T, Song L, Shimada H, Keshelava N, Russell HV, Metelitsa LS, Groshen SG, Seeger RC and DeClerck YA (2009) Interleukin‐6 in the bone marrow microenvironment promotes the growth and survival of neuroblastoma cells. Cancer Res 69, 329–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ban J, Bennani‐Baiti IM, Kauer M, Schaefer KL, Poremba C, Jug G, Schwentner R, Smrzka O, Muehlbacher K, Aryee DN et al (2008) EWS‐FLI1 suppresses NOTCH‐activated p53 in Ewing's sarcoma. Cancer Res 68, 7100–7109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennani‐Baiti IM, Aryee DN, Ban J, Machado I, Kauer M, Muhlbacher K, Amann G, Llombart‐Bosch A and Kovar H (2011) Notch signalling is off and is uncoupled from HES1 expression in Ewing's sarcoma. J Pathol 225, 353–363. [DOI] [PubMed] [Google Scholar]
- Bin Hafeez B, Adhami VM, Asim M, Siddiqui IA, Bhat KM, Zhong W, Saleem M, Din M, Setaluri V and Mukhtar H (2009) Targeted knockdown of Notch1 inhibits invasion of human prostate cancer cells concomitant with inhibition of matrix metalloproteinase‐9 and urokinase plasminogen activator. Clin Cancer Res 15, 452–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brohl AS, Solomon DA, Chang W, Wang J, Song Y, Sindiri S, Patidar R, Hurd L, Chen L, Shern JF et al (2014) The genomic landscape of the Ewing Sarcoma family of tumors reveals recurrent STAG2 mutation. PLoS Genet 10, e1004475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burchill SA (2003) Ewing's sarcoma: diagnostic, prognostic, and therapeutic implications of molecular abnormalities. J Clin Pathol 56, 96–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdach S and Jurgens H (2002) High‐dose chemoradiotherapy (HDC) in the Ewing family of tumors (EFT). Crit Rev Oncol Hematol 41, 169–189. [DOI] [PubMed] [Google Scholar]
- Burdach S, Jurgens H, Peters C, Nurnberger W, Mauz‐Korholz C, Korholz D, Paulussen M, Pape H, Dilloo D, Koscielniak E et al (1993) Myeloablative radiochemotherapy and hematopoietic stem‐cell rescue in poor‐prognosis Ewing's sarcoma. J Clin Oncol 11, 1482–1488. [DOI] [PubMed] [Google Scholar]
- Burdach S, Plehm S, Unland R, Dirksen U, Borkhardt A, Staege MS, Muller‐Tidow C and Richter GH (2009) Epigenetic maintenance of stemness and malignancy in peripheral neuroectodermal tumors by EZH2. Cell Cycle 8, 1991–1996. [DOI] [PubMed] [Google Scholar]
- Burdach S, Thiel U, Schoniger M, Haase R, Wawer A, Nathrath M, Kabisch H, Urban C, Laws HJ, Dirksen U et al (2010) Total body MRI‐governed involved compartment irradiation combined with high‐dose chemotherapy and stem cell rescue improves long‐term survival in Ewing tumor patients with multiple primary bone metastases. Bone Marrow Transplant 45, 483–489. [DOI] [PubMed] [Google Scholar]
- Coleman RE (2006) Clinical features of metastatic bone disease and risk of skeletal morbidity. Clin Cancer Res 12, 6243s–6249s. [DOI] [PubMed] [Google Scholar]
- Crompton BD, Stewart C, Taylor‐Weiner A, Alexe G, Kurek KC, Calicchio ML, Kiezun A, Carter SL, Shukla SA, Mehta SS et al (2014) The genomic landscape of pediatric Ewing sarcoma. Cancer Discov 4, 1326–1341. [DOI] [PubMed] [Google Scholar]
- Delattre O, Zucman J, Melot T, Garau XS, Zucker JM, Lenoir GM, Ambros PF, Sheer D, Turc‐Carel C, Triche TJ et al (1994) The Ewing family of tumors – a subgroup of small‐round‐cell tumors defined by specific chimeric transcripts. N Engl J Med 331, 294–299. [DOI] [PubMed] [Google Scholar]
- Deleersnijder W, Hong G, Cortvrindt R, Poirier C, Tylzanowski P, Pittois K, Van Marck E and Merregaert J (1996) Isolation of markers for chondro‐osteogenic differentiation using cDNA library subtraction. Molecular cloning and characterization of a gene belonging to a novel multigene family of integral membrane proteins. J Biol Chem 271, 19475–19482. [DOI] [PubMed] [Google Scholar]
- Dunn LK, Mohammad KS, Fournier PG, McKenna CR, Davis HW, Niewolna M, Peng XH, Chirgwin JM and Guise TA (2009) Hypoxia and TGF‐beta drive breast cancer bone metastases through parallel signaling pathways in tumor cells and the bone microenvironment. PLoS One 4, e6896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunst J, Ahrens S, Paulussen M, Burdach S and Jurgens H (2001) Prognostic impact of tumor perfusion in MR‐imaging studies in Ewing tumors. Strahlenther Onkol 177, 153–159. [DOI] [PubMed] [Google Scholar]
- Grunewald TG, Diebold I, Esposito I, Plehm S, Hauer K, Thiel U, da Silva‐Buttkus P, Neff F, Unland R, Muller‐Tidow C et al (2012) STEAP1 is associated with the invasive and oxidative stress phenotype of Ewing tumors. Mol Cancer Res 10, 52–65. [DOI] [PubMed] [Google Scholar]
- Guan H, Zhou Z, Cao Y, Duan X and Kleinerman ES (2009) VEGF165 promotes the osteolytic bone destruction of Ewing's sarcoma tumors by upregulating RANKL. Oncol Res 18, 117–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D and Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144, 646–674. [DOI] [PubMed] [Google Scholar]
- Hauer K, Calzada‐Wack J, Steiger K, Grunewald TG, Baumhoer D, Plehm S, Buch T, Prazeres da Costa O, Esposito I, Burdach S et al (2013) DKK2 mediates osteolysis, invasiveness, and metastatic spread in Ewing sarcoma. Cancer Res 73, 967–977. [DOI] [PubMed] [Google Scholar]
- Hedlund J, Johansson J and Persson B (2009) BRICHOS – a superfamily of multidomain proteins with diverse functions. BMC Res Notes 2, 180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Heyking K, Roth L, Ertl M, Schmidt O, Calzada‐Wack J, Neff F, Lawlor ER, Burdach S and Richter GH (2016) The posterior HOXD locus: its contribution to phenotype and malignancy of Ewing sarcoma. Oncotarget 7, 41767–41780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiraga T, Kizaka‐Kondoh S, Hirota K, Hiraoka M and Yoneda T (2007) Hypoxia and hypoxia‐inducible factor‐1 expression enhance osteolytic bone metastases of breast cancer. Cancer Res 67, 4157–4163. [DOI] [PubMed] [Google Scholar]
- Hiraki Y, Inoue H, Iyama K, Kamizono A, Ochiai M, Shukunami C, Iijima S, Suzuki F and Kondo J (1997) Identification of chondromodulin I as a novel endothelial cell growth inhibitor. Purification and its localization in the avascular zone of epiphyseal cartilage. J Biol Chem 272, 32419–32426. [DOI] [PubMed] [Google Scholar]
- Hiraki Y, Shukunami C (2000) Chondromodulin‐I as a novel cartilage‐specific growth‐modulating factor. Pediatr Nephrol 14, 602–605. [DOI] [PubMed] [Google Scholar]
- Katoh Y and Katoh M (2007) Comparative genomics on PROM1 gene encoding stem cell marker CD133. Int J Mol Med 19, 967–970. [PubMed] [Google Scholar]
- Klinger P, Surmann‐Schmitt C, Brem M, Swoboda B, Distler JH, Carl HD, von der Mark K, Hennig FF and Gelse K (2011) Chondromodulin 1 stabilizes the chondrocyte phenotype and inhibits endochondral ossification of porcine cartilage repair tissue. Arthritis Rheum 63, 2721–2731. [DOI] [PubMed] [Google Scholar]
- Lessnick SL and Ladanyi M (2012) Molecular pathogenesis of Ewing sarcoma: new therapeutic and transcriptional targets. Annu Rev Pathol 7, 145–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Liu P, Liu W, Maye P, Zhang J, Zhang Y, Hurley M, Guo C, Boskey A, Sun L et al (2005) Dkk2 has a role in terminal osteoblast differentiation and mineralized matrix formation. Nat Genet 37, 945–952. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐Delta Delta C(T)) Method. Methods 25, 402–408. [DOI] [PubMed] [Google Scholar]
- Mackintosh C, Madoz‐Gurpide J, Ordonez JL, Osuna D and Herrero‐Martin D (2010) The molecular pathogenesis of Ewing's sarcoma. Cancer Biol Ther 9, 655–667. [DOI] [PubMed] [Google Scholar]
- Mera H, Kawashima H, Yoshizawa T, Ishibashi O, Ali MM, Hayami T, Kitahara H, Yamagiwa H, Kondo N, Ogose A et al (2009) Chondromodulin‐1 directly suppresses growth of human cancer cells. BMC Cancer 9, 166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsui K, Tokuzawa Y, Itoh H, Segawa K, Murakami M, Takahashi K, Maruyama M, Maeda M and Yamanaka S (2003) The homeoprotein Nanog is required for maintenance of pluripotency in mouse epiblast and ES cells. Cell 113, 631–642. [DOI] [PubMed] [Google Scholar]
- Miura S, Kondo J, Takimoto A, Sano‐Takai H, Guo L, Shukunami C, Tanaka H and Hiraki Y (2014) The N‐terminal cleavage of chondromodulin‐I in growth‐plate cartilage at the hypertrophic and calcified zones during bone development. PLoS One 9, e94239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinholt FP, Hultenby K, Oldberg A and Heinegard D (1990) Osteopontin – a possible anchor of osteoclasts to bone. Proc Natl Acad Sci U S A 87, 4473–4475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter GH, Fasan A, Hauer K, Grunewald TG, Berns C, Rossler S, Naumann I, Staege MS, Fulda S, Esposito I et al (2013) G‐Protein coupled receptor 64 promotes invasiveness and metastasis in Ewing sarcomas through PGF and MMP1. J Pathol 230, 70–81. [DOI] [PubMed] [Google Scholar]
- Richter GH, Plehm S, Fasan A, Rossler S, Unland R, Bennani‐Baiti IM, Hotfilder M, Lowel D, von Luettichau I, Mossbrugger I et al (2009) EZH2 is a mediator of EWS/FLI1 driven tumor growth and metastasis blocking endothelial and neuro‐ectodermal differentiation. Proc Natl Acad Sci U S A 106, 5324–5329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez‐Pulido L, Devos D and Valencia A (2002) BRICHOS: a conserved domain in proteins associated with dementia, respiratory distress and cancer. Trends Biochem Sci 27, 329–332. [DOI] [PubMed] [Google Scholar]
- Schmidt D, Harms D and Burdach S (1985) Malignant peripheral neuroectodermal tumours of childhood and adolescence. Virchows Arch A Pathol Anat Histopathol 406, 351–365. [DOI] [PubMed] [Google Scholar]
- Sethi N, Dai X, Winter CG and Kang Y (2011) Tumor‐derived JAGGED1 promotes osteolytic bone metastasis of breast cancer by engaging notch signaling in bone cells. Cancer Cell 19, 192–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukunami C, Hiraki Y (2001) Role of cartilage‐derived anti‐angiogenic factor, chondromodulin‐I, during endochondral bone formation. Osteoarthritis Cartilage 9(Suppl A), S91–S101. [DOI] [PubMed] [Google Scholar]
- Sorensen PH, Lessnick SL, Lopez‐Terrada D, Liu XF, Triche TJ and Denny CT (1994) A second Ewing's sarcoma translocation, t(21;22), fuses the EWS gene to another ETS‐family transcription factor, ERG. Nat Genet 6, 146–151. [DOI] [PubMed] [Google Scholar]
- Staege MS, Hutter C, Neumann I, Foja S, Hattenhorst UE, Hansen G, Afar D and Burdach SE (2004) DNA microarrays reveal relationship of Ewing family tumors to both endothelial and fetal neural crest‐derived cells and define novel targets. Cancer Res 64, 8213–8221. [DOI] [PubMed] [Google Scholar]
- Szepesi A, Matula Z, Szigeti A, Varady G, Szabo G, Uher F, Sarkadi B and Nemet K (2015) ABCG2 is a selectable marker for enhanced multilineage differentiation potential in periodontal ligament stem cells. Stem Cells Dev 24, 244–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka M, Yamazaki Y, Kanno Y, Igarashi K, Aisaki K, Kanno J and Nakamura T (2014) Ewing's sarcoma precursors are highly enriched in embryonic osteochondrogenic progenitors. J Clin Invest 124, 3061–3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao J, Erez A and Lee B (2011) One NOTCH further: Jagged 1 in bone metastasis. Cancer Cell 19, 159–161. [DOI] [PubMed] [Google Scholar]
- Thiel U, Wawer A, von Luettichau I, Bender HU, Blaeschke F, Grunewald TG, Steinborn M, Roper B, Bonig H, Klingebiel T et al (2016) Bone marrow involvement identifies a subgroup of advanced Ewing sarcoma patients with fatal outcome irrespective of therapy in contrast to curable patients with multiple bone metastases but unaffected marrow. Oncotarget 7, 70959–70968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiel U, Wawer A, Wolf P, Badoglio M, Santucci A, Klingebiel T, Basu O, Borkhardt A, Laws HJ, Kodera Y et al (2011) No improvement of survival with reduced‐ versus high‐intensity conditioning for allogeneic stem cell transplants in Ewing tumor patients. Ann Oncol 22, 1614–1621. [DOI] [PubMed] [Google Scholar]
- Tirode F, Surdez D, Ma X, Parker M, Le Deley MC, Bahrami A, Zhang Z, Lapouble E, Grossetete‐Lalami S, Rusch M et al (2014) Genomic landscape of Ewing sarcoma defines an aggressive subtype with co‐association of STAG2 and TP53 mutations. Cancer Discov 4, 1342–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vater C, Kasten P and Stiehler M (2011) Culture media for the differentiation of mesenchymal stromal cells. Acta Biomater 7, 463–477. [DOI] [PubMed] [Google Scholar]
- Wang X, Lu H, Urvalek AM, Li T, Yu L, Lamar J, DiPersio CM, Feustel PJ and Zhao J (2011) KLF8 promotes human breast cancer cell invasion and metastasis by transcriptional activation of MMP9. Oncogene 30, 1901–1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weilbaecher KN, Guise TA and McCauley LK (2011) Cancer to bone: a fatal attraction. Nat Rev Cancer 11, 411–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshioka M, Yuasa S, Matsumura K, Kimura K, Shiomi T, Kimura N, Shukunami C, Okada Y, Mukai M, Shin H et al (2006) Chondromodulin‐I maintains cardiac valvular function by preventing angiogenesis. Nat Med 12, 1151–1159. [DOI] [PubMed] [Google Scholar]
- Zhou S, Schuetz JD, Bunting KD, Colapietro AM, Sampath J, Morris JJ, Lagutina I, Grosveld GC, Osawa M, Nakauchi H et al (2001) The ABC transporter Bcrp1/ABCG2 is expressed in a wide variety of stem cells and is a molecular determinant of the side‐population phenotype. Nat Med 7, 1028–1034. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. CHM1 maintains an undifferentiated phenotype of ES.
Fig. S2. In vivo bone invasion and osteolysis.
Fig. S3. Cell cycle distribution analyses.
Fig. S4. CHM1 knock down does not influence in vivo angiogenesis.
Doc S1. Materials and methods.