

Abstract
Ubiquitylation is one of the cardinal post‐translational modifications in the cell, balancing several distinct biological processes and acting as a pathogen recognition receptor during bacterial pathogen invasion. A dense layer of polyubiquitin chains marks invading bacteria that gain access to the host cytosol for their selective clearance via xenophagy. However, the enzymes that mediate recognition of cytosolic bacteria and generate this ubiquitin (Ub) coat remain largely elusive. To address this, we employed an image‐based RNAi screening approach to monitor the loss of Ub on Salmonella upon depletion of human Ub E3 ligases in cells. Using this approach, we identified ARIH1 as one of the ligases involved in the formation of Ub coat on cytosolic bacteria. In addition, we provide evidence that the RING‐between‐RING ligase ARIH1, together with LRSAM1 and HOIP, forms part of a network of ligases that orchestrates recognition of intracellular Salmonella and participates in the activation of the host cell immune response.
Keywords: ARIH1, HHARI, RBR E3 ligase, Salmonella, ubiquitin
Subject Categories: Microbiology, Virology & Host Pathogen Interaction; Post-translational Modifications, Proteolysis & Proteomics
Introduction
Salmonella enterica are gram‐negative facultative anaerobic bacteria that can be divided into several subspecies and thousands of serovars based on the lipopolysaccharide and flagellar antigens. These serovars can be roughly categorized as typhoidal and non‐typhoidal. Salmonella enterica ssp. enterica ser. Typhimurium (S. Typhimurium) belongs to the latter type that generally leads to food poisoning and gastrointestinal disease in a wide range of hosts. In mice, the bacteria are capable of breaching the epithelial barrier of the gastrointestinal tract by phagocytic engulfment or through endocytosis using M cells 1 and cross the gut vascular barrier to disseminate into the liver and the spleen 2. Invasion and intracellular proliferation are facilitated by various effector proteins delivered into host cells by two distinct type III secretion systems (T3SSs) encoded on the Salmonella pathogenicity islands 1 and 2 (SPI‐1 and SPI‐2) 3. In general, the SPI‐1 T3SS enables invasion and stimulates the initial inflammatory response while the SPI‐2 T3SS contributes to the intracellular proliferation within the Salmonella‐containing vacuole (SCV) 4.
Macroautophagy (hereafter referred to as autophagy) is a fundamental, evolutionarily conserved, cellular process that enables cells to engulf and digest portions of their cytoplasm in a regulated manner 5. Anti‐bacterial autophagy (xenophagy) serves as a cell‐autonomous immune mechanism against invasive intracellular bacteria 6. This defensive response includes the formation of a dense coat of polyubiquitin chains that serves as pathogen recognition receptor and directs intracellular bacteria for autophagic degradation 7, 8. Specifically, S. Typhimurium was shown to be targeted and subsequently degraded by the host's ubiquitin (Ub) and autophagy systems, respectively 9. Ubiquitylated bacteria are recruited to autophagosomes by concerted action of the mammalian Atg8 homologues of the LC3 subfamily, which are anchored in the membrane of the forming autophagosome, and several autophagy cargo receptors, namely NDP52, OPTN, and p62 that bind ubiquitylated proteins via their respective Ub binding domains and LC3 through their LC3‐interacting regions 10.
Ubiquitin is a small protein modifier that labels proteins in a highly specific manner. Conjugation of Ub to a targeted lysine of a protein is regulated by sequential activity of Ub activating (E1), conjugating (E2), and ligating (E3) enzymes. The presence of seven lysines in Ub allows formation of seven homotypic linkage types and multiple possible heterotypic chains 11. Moreover, Ub can form linear (M1) chains in which Ub moieties are connected in a head‐to‐tail orientation involving the N‐terminal methionine 12. The proteins directly involved in detecting bacteria in the host cytosol and generating the ubiquitylation signal required for their subsequent autophagosomal degradation remain loosely defined. For example, RNF166 was recently shown to localize to S. Typhimurium and recruit autophagy receptors and LC3 13. However, ubiquitylation of S. Typhimurium by RNF166 was not tested in this study. To date, the E3 ligases LRSAM1 14 and HOIP 15, which is the catalytic subunit of the linear ubiquitin chain assembly complex (LUBAC), have been demonstrated to be involved in bacteria‐associated ubiquitylation during infection with S. Typhimurium. Intriguingly, ubiquitylation of bacteria is only reduced but not abolished in LRSAM1‐depleted or LRSAM1‐deficient cells 14, and the recruitment of LUBAC requires a pre‐existing ubiquitin signal 15. Moreover, in addition to the M1‐linked polyUb layer that is regulated by the deubiquitinase OTULIN 16, lysine 63 (K63)‐linked polyUb chains have also been detected in the Ub coat surrounding S. Typhimurium 17, while on the other hand LRSAM1 was shown to mediate K6‐ and K27‐linked polyubiquitylation in vitro 14. Thus, it seems highly plausible that other E3 ligases than LRSAM1 and HOIP are involved in this process. The observation that Parkin mediates ubiquitylation of Mycobacterium tuberculosis 18 led us to speculate whether other members of this RING‐between‐RING (RBR) E3 ligase family besides HOIP play a role in antagonizing S. Typhimurium infections.
Results
ARIH1 is required to ubiquitylate cytosolic bacteria during S. Typhimurium infection
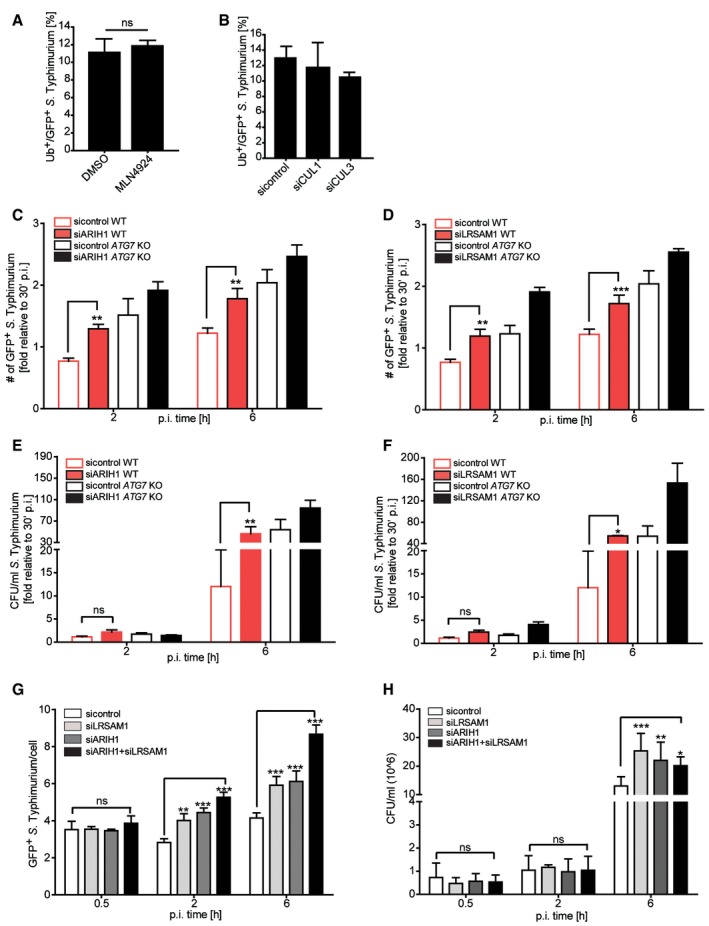
To systematically identify host machineries involved in the ubiquitylation events directed to eliminate S. Typhimurium, we employed an image‐based RNAi screening approach using siRNA pools individually targeting all 14 members of the human RBR E3 ligase family 19. Reverse transfection of HeLa cells with validated siRNAs (Fig EV1A) was followed by infection with a GFP‐expressing wild‐type S. Typhimurium or a strain lacking the bacterial effector SifA (ΔsifA). The latter bacteria undergo more frequent escape from SCVs 20, leading to increased numbers of ubiquitylated cytosolic S. Typhimurium. Importantly, in both strains the expression of GFP was under the control of the glucose 6‐phosphate‐responsive promoter 21 (hereafter referred to as cytoGFP), which allowed exclusive detection of S. Typhimurium that escaped their SCVs. Immunostaining of fixed cells with an Ub‐specific antibody (FK2) was employed to monitor cytosolic bacteria for the loss of their Ub coat upon depletion of RBR family members 2 hours (h) post‐infection (p.i.). High‐content image acquisition with an automated high‐throughput laser‐spinning confocal microscope and an algorithm‐based image analysis suite was employed to unbiasedly determine the number of GFP‐positive S. Typhimurium in cells and the percentage of these bacteria to which Ub colocalized. Importantly, RNAi transfection per se did not affect bacterial invasion or SCV integrity, since transfection with a non‐targeting control siRNA did not alter the number of cytosolic S. Typhimurium compared to mock transfection (Fig EV1E). Our screening effort revealed a substantial increase in the number of GFP‐positive ΔsifA S. Typhimurium with a marked concurrent decrease in the Ub‐positive fraction of these bacteria upon depletion of ARIH1 (also known as HHARI) (Fig 1A). When determining z‐scores for the number of GFP‐positive as well as Ub‐ and GFP‐positive bacteria in cells depleted for RBR family members, only knockdown of ARIH1 resulted in values that passed the significance threshold of +2 and ‐2, respectively (Fig 1B and C). Notably, we were unable to detect significant changes upon depletion of any RBR ligase using wild‐type S. Typhimurium (Fig EV1B–D). For deconvolution of the pooled ARIH1 siRNAs, we employed four individual siRNAs, which all efficiently depleted ARIH1 from cells (Fig EV1F). Upon infection with cytoGFP‐expressing ∆sifA S. Typhimurium, three out of four siRNAs resulted in significantly increased levels of GFP‐positive bacteria with a concomitant decrease in bacterial Ub colocalization. This phenotype was comparable to the effect of LRSAM1 depletion (Figs 1D–F and EV1G). Importantly, a similar accumulation of GFP‐positive S. Typhimurium was also observed upon knockdown of the core autophagy genes ATG5 and ATG7 (Fig EV1H–J). Together, our findings indicate that ARIH1 is involved in ubiquitylating cytosolic S. Typhimurium and restricting the bacterial load in the cytosol.
Figure EV1. Infection, knockdown, and wild‐type S. Typhimurium controls (related to Fig 1).
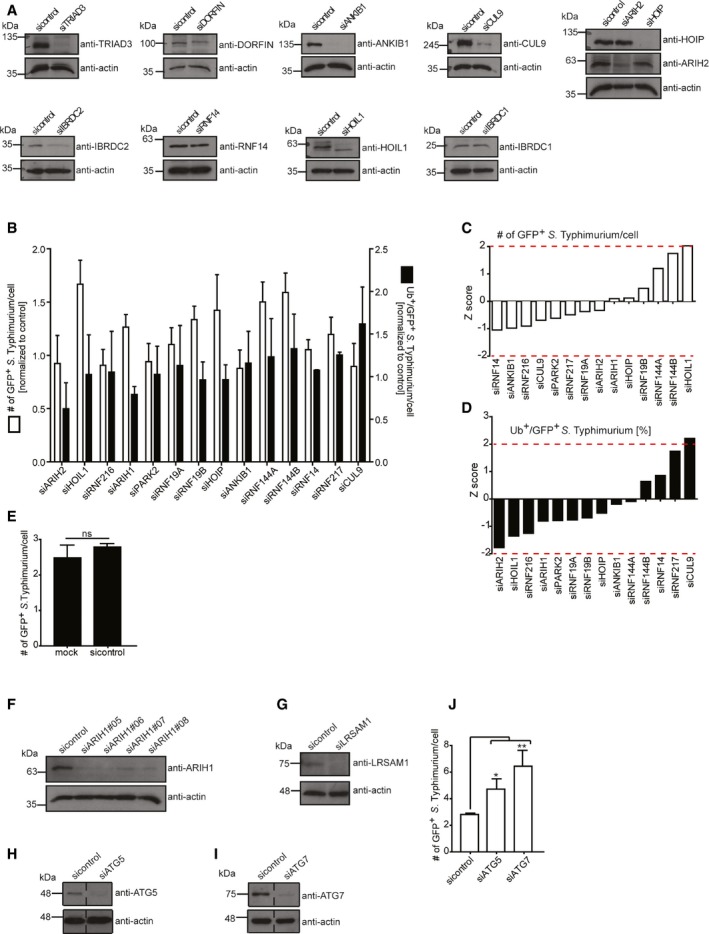
-
ALysates from HeLa cells transfected with indicated pooled siRNAs for 72 h were analyzed by SDS–PAGE and immunoblotting. Due to low specificity of commercially available antibodies, the efficiency of RNAi‐mediated depletion of NKLAM and RNF144A could not be assessed by immunoblotting. Notably, Parkin is not expressed in HeLa cells.
-
BHeLa cells reversely transfected with sicontrol or pooled siRNAs targeting all 14 known RBR Ub E3 ligases for 72 h were infected with wild‐type cytoGFP‐expressing S. Typhimurium for 2 h prior to fixation and immunolabeling with anti‐polyUb antibody (FK2). Number of GFP‐positive (GFP+) and Ub‐positive and GFP‐positive (Ub+/GFP+) bacteria was determined using an automated quantification software and normalized to sicontrol counting on average 800 cells/sample (GFP‐positive S. Typhimurium/cell = 3.90 ± 0.42, ubiquitylated S. Typhimurium [%] = 11.76 ± 2.10). Data represent mean ± SD. n = 2 biological replicates.
-
C, Dz‐scores of GFP+ (C) and Ub+/GFP+ (D) bacteria from (B).
-
EHeLa cells transfected with sicontrol or left untreated (mock) for 72 h were infected with ΔsifA cytoGFP‐expressing S. Typhimurium for 2 h prior to fixation. Number of GFP+ bacteria in at least 250 cells/sample was determined using automated quantification. Data represent mean ± SD. Significance was determined using unpaired Student's t‐test. ns = not significant. n = 3 biological replicates.
-
F–ILysates from HeLa cells transfected with indicated single siRNAs for 72 h were analyzed by SDS–PAGE and immunoblotting.
-
JHeLa cells transfected with indicated single siRNAs for 72 h were infected as in (B) followed by fixation and confocal microscopy. Number of GFP+ bacteria was determined by automated quantification in 250 cells/sample on average. Data represent mean ± SD. Significance was determined using one‐way ANOVA. *P < 0.05, **P < 0.01. n = 3 biological replicates.
Figure 1. Image‐based RNAi screening of RBR E3 ligases involved in bacterial Ub coat formation upon S. Typhimurium infection.
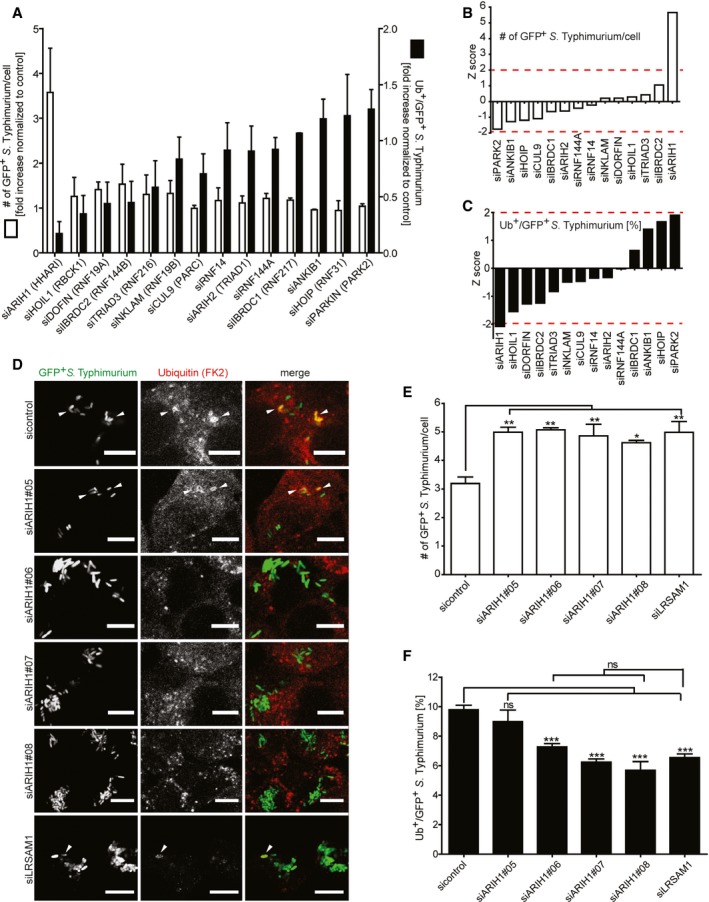
-
AHeLa cells reversely transfected for 72 h with non‐targeting control (sicontrol) or pooled siRNAs targeting all 14 known RBR Ub E3 ligases were infected with cytoGFP‐expressing ΔsifA S. Typhimurium for 2 h prior to fixation and immunolabeling with anti‐Ub antibody (FK2). Number of GFP‐positive (GFP+) and Ub‐ and GFP‐positive (Ub+/GFP+) bacteria was determined using automated quantification software and normalized to sicontrol counting on average 800 cells/sample (GFP‐positive S. Typhimurium/cell = 3.50 ± 0.13, ubiquitylated S. Typhimurium [%] = 8.32 ± 1.10). Data represent mean ± SD. n = 2 biological replicates.
-
B, Cz‐scores of GFP+ (B) and Ub+/GFP+ (C) bacteria from (A).
-
DHeLa cells were transfected with indicated single siRNAs, infected, fixed, and immunolabeled as in (A). Scale bar: 10 μm. Arrowheads indicate colocalization events.
-
E, FAutomated quantification of GFP+ (E) and Ub+/GFP+ (F) bacteria in on average 250 cells/sample. Data represent mean ± SD. Significance was determined using one‐way ANOVA. *P < 0.05, **P < 0.01, ***P < 0.001, ns = not significant. n = 3 biological replicates.
ARIH1 precedes LRSAM1 in the recruitment to cytosolic S. Typhimurium where both ligases colocalize
To examine whether ARIH1 is recruited to cytosolic bacteria during infection, we performed immunofluorescent labeling of cells infected with cytoGFP‐expressing ∆sifA S. Typhimurium. Consistent with its ability to ubiquitylate bacteria, we indeed detected endogenous ARIH1 on GFP‐positive S. Typhimurium that were also decorated with Ub (Fig 2A), while a reduced colocalization was observed upon ARIH1 knockdown (Fig EV2A). Importantly, a similar recruitment was also observed for the wild‐type cytoGFP strain (Fig EV2B). Single‐molecule localization microscopy revealed a nanoscale patchlike localization pattern of ARIH1 on these bacteria (Fig 2B). Intriguingly, LRSAM1 showed a similar clustered colocalization on the surface of cytosolic bacteria (Fig 2C). By examining the recruitment kinetics of both E3 ligases to cytosolic bacteria at endogenous levels, ARIH1 was found to localize to S. Typhimurium within the first 30 min p.i. while LRSAM1 was recruited later (2 h p.i.) and to a lesser extent than ARIH1 (Fig 2D and E). However, both ligases were found to colocalize on some of the SCV‐escaped bacteria (Fig 2D). At 6 h p.i., bacterial colocalization of ARIH1 and LRSAM1 was heavily diminished, but some LRSAM1‐positive S. Typhimurium could still be observed (Fig 2D and E). These results show that ARIH1 is recruited faster to cytosolic S. Typhimurium than LRSAM1 while LRSAM1 persists longer on these bacteria.
Figure 2. ARIH1 is recruited to the cytosolic S. Typhimurium, localizes with LRSAM1, and contributes K48‐linked polyUb chains to their Ub coat.
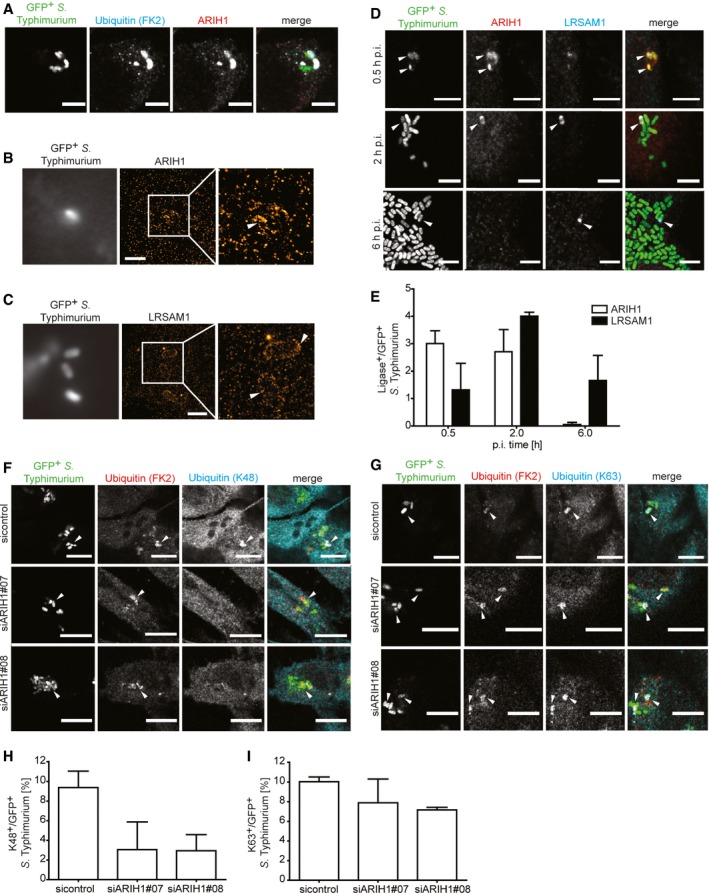
-
AHeLa cells were infected with cytoGFP ΔsifA S. Typhimurium for 2 h followed by fixation, immunolabeling with anti‐Ub (FK2) and anti‐ARIH1 antibodies, and confocal microscopy. Scale bar: 5 μm.
-
B, CHeLa cells were infected and fixed as in (A) and immunolabeled with anti‐ARIH1 (B) or anti‐LRSAM1 (C) antibodies followed by additional fixation and super‐resolution dSTORM imaging. Scale bar: 2 μm. Arrowheads indicate nanoscale protein patches.
-
DHeLa cells were infected for indicated p.i. time points and fixed as in (A) prior to immunolabeling with anti‐ARIH1 and anti‐LRSAM1 antibodies and confocal microscopy. Scale bar: 5 μm. Arrowheads indicate colocalization events.
-
EAutomated quantification of LRSAM1‐positive and ARIH1‐positive S. Typhimurium from (D) in at least 100 cells/sample. Data represent mean ± SD. n = 2 biological replicates.
-
F, GHeLa cells transfected with indicated single siRNAs for 72 h and infected as in (A) prior to fixation and immunolabeling with anti‐Ub (FK2) and anti‐K48 (F) or ‐K63 polyUb (G) antibodies. Scale bar: 10 μm. Arrowheads indicate colocalization events.
-
H, IAutomated quantification of K48+/GFP+ (F) or K63+/GFP+ (G) bacteria in at least 100 cells/sample from (F and G). Data represent mean ± SD. n = 2 biological replicates.
Figure EV2. Neddylated CRLs are not required for ubiquitylation of cytosolic S. Typhimurium (related to Figs 2, 3, 4).
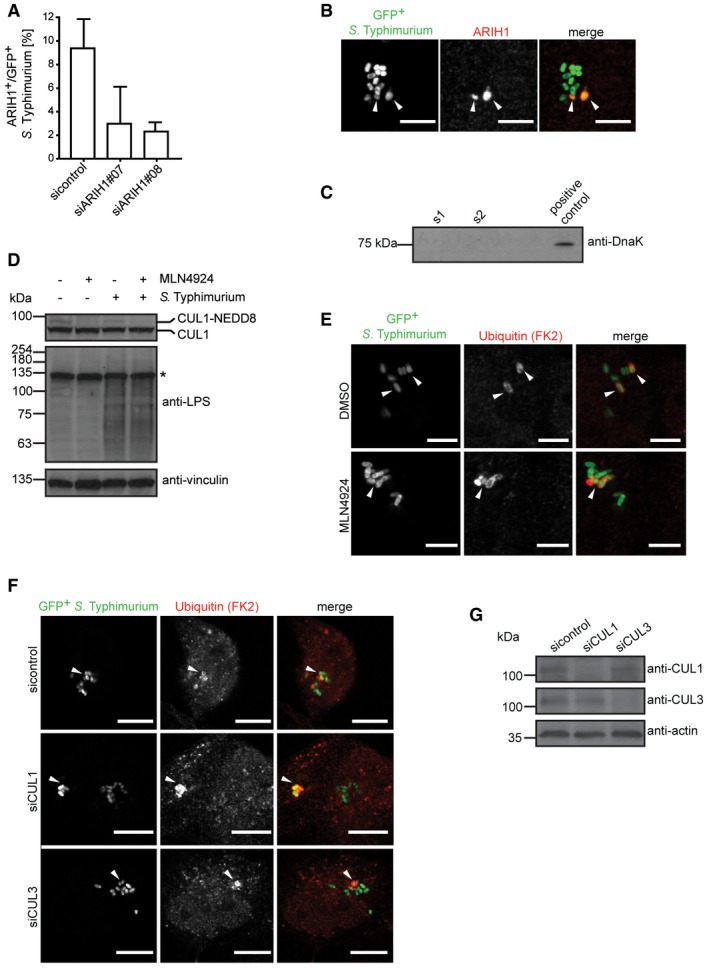
- HeLa cells transfected with indicated single siRNAs for 72 h were infected with cytoGFP‐expressing ΔsifA S. Typhimurium for 2 h followed by fixation and anti‐ARIH1 immunolabeling. Number of ARIH1+/GFP+ bacteria in at least 250 cells/sample was determined using automated quantification. Data represent mean ± SD. n = 2 biological replicates.
- HeLa cells were infected with cytoGFP wild‐type S. Typhimurium for 2 h followed by fixation, immunolabeling with anti‐ARIH1 antibody, and confocal microscopy. Arrowheads indicate colocalization events. Scale bar: 5 μm.
- Integrity control of bacteria. While performing the in vitro ubiquitylation reaction, bacterial supernatants were sampled right before (s1) and immediately after (s2) the reaction. Intact bacteria were used as a positive control.
- HeLa cells infected as in (A) or left uninfected were treated with 2 μM MLN4924 or DMSO during the course of the infection prior to lysis. Lysates were analyzed by SDS–PAGE and immunoblotting.
- HeLa cells infected as in (A) were treated with MLN4924 or DMSO as in (D) prior to fixation and immunolabeling with anti‐Ub antibody (FK2). Arrowheads indicate colocalization events. Scale bar: 5 μm.
- HeLa cells were transfected with indicated pooled siRNAs and infected as in (A) prior to fixation and anti‐Ub (FK2) immunolabeling. Arrowheads indicate colocalization events. Scale bar: 5 μm.
- HeLa cells were reversely transfected with indicated pooled siRNAs for 72 h and lysed. Lysates were analyzed by SDS–PAGE and immunoblotting.
ARIH1 contributes K48‐linked chains to the Ub coat surrounding cytosolic S. Typhimurium
M1‐ and K63‐linked polyUb chains were reported to localize to S. Typhimurium 17. Since linear ubiquitylation is exclusively conferred by LUBAC 12, 15, we sought to address whether ARIH1 modifies cytosolic bacteria with K63‐linked polyUb using a chain‐specific antibody. As K48‐linked polyUb was found to surround M. tuberculosis 18 and cytosolic Mycobacterium marinum in macrophages 22, we also included a K48‐specific antibody in our immunofluorescence analysis. To our surprise, we observed that cytosolic S. Typhimurium were positive for both chain types (Fig 2F and G). Unexpectedly, only K48‐linked Ub chains were substantially decreased on cytosolic bacteria when ARIH1 was depleted in cells (Fig 2H and I). Thus, these results indicate that the Ub coat surrounding S. Typhimurium in the cytosol is composed of at least three different Ub chain types of which ARIH1 regulates the levels of K48‐linked Ub.
ARIH1 ubiquitylates S. Typhimurium in vitro
To examine whether ARIH1 alone is sufficient to ubiquitylate S. Typhimurium, we set up an in vitro ubiquitylation assay with purified components (Fig 3A). Consistent with previous reports 23 incubation of an ARIH1 variant lacking the autoinhibitory C‐terminal Ariadne domain (∆Ariadne) with HA‐tagged Ub, UBA1 as E1, and UBCH7 (alias UBE2L3) as E2 enzymes for 1 h at 37°C resulted in robust autoubiquitylation of ARIH1 in an ATP‐dependent manner while full‐length ARIH1 failed to ubiquitylate itself (Fig 3B). Next, we repeated the above reaction in the presence of S. Typhimurium (cytoGFP ΔsifA). Once ubiquitylation was stopped by EDTA, bacteria were separated from ARIH1 and other ubiquitylation reaction components by repeated centrifugation and washing. Subsequent immunoblot analysis revealed that these bacteria were modified by Ub in a manner dependent on active ARIH1 and ATP (Fig 3C). Notably, the buffer conditions of the in vitro ubiquitylation reaction did not affect the integrity of bacteria (Fig EV2C). To assess the specificity of this in vitro reaction, we examined whether ARIH1 also ubiquitylates purified mitochondria from the filamentous, ascomycete fungus Podospora anserina as an alternative substrate. Although ARIH1 was active in these reactions, we failed to detect any Ub signal on these mitochondria (Fig 3D). Since recent ubiquitinome profiling revealed that outer membrane proteins (OMPs) of S. Typhimurium become ubiquitylated upon their escape to the cytosol 24, we assessed whether ARIH1‐mediated S. Typhimurium ubiquitylation requires the presence of OMPs. As expected, ARIH1 was unable to ubiquitylate S. Typhimurium that were stripped off their OMPs by proteinase K pretreatment (Fig 3E, two left lanes). As evident from ARIH1 autoubiquitylation, ubiquitylation reaction components were fully functional in these reactions due to the addition of a serine protease inhibitor (Fig 3E, two right lanes). Collectively, these results show that ARIH1 is sufficient to specifically ubiquitylate S. Typhimurium OMPs.
Figure 3. In vitro ubiquitylation of S. Typhimurium by ARIH1.
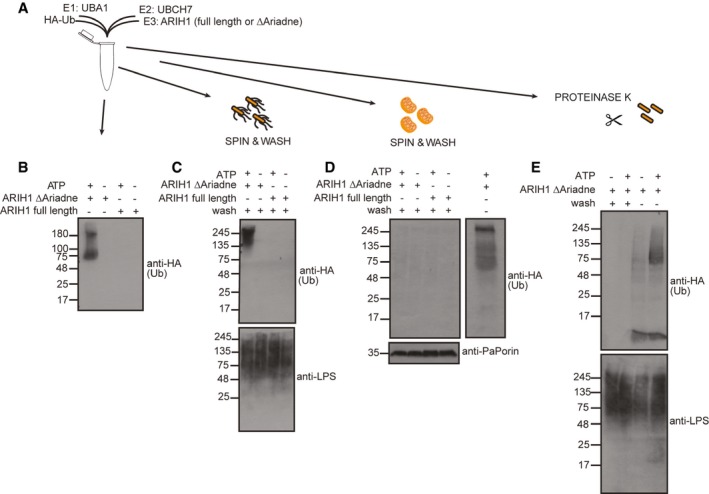
-
AIn vitro ubiquitylation reaction scheme.
-
BPurified inactive full‐length or C‐terminally truncated, active ARIH1 (Δariadne) were incubated with HA‐Ub, UBA1, and UBCH7 in the absence or presence of ATP at 37°C for 1 h. Reactions were stopped by addition of EDTA and subjected to SDS–PAGE and immunoblot analysis.
-
C–EReactions were carried out as in (B) in the presence of cytoGFP ΔsifA S. Typhimurium (C), purified mitochondria from P. anserina (D), or proteinase K‐pretreated S. Typhimurium (E). Once the reactions were stopped, bacteria or mitochondria were repeatedly pelleted by centrifugation and washed followed by separation on SDS–PAGE and immunoblotting. Notably, PMSF was added to the ubiquitylation buffer in (E) to inhibit proteinase K activity during the ubiquitylation reaction.
Ubiquitylation of cytosolic S. Typhimurium does not require cullin‐RING ligases
Since ARIH1 was shown to require activation by neddylated members of the cullin‐RING ligases (CRLs) family in vitro 25 and cooperatively binds the latter to monoubiquitylate CRL substrates 26, we assessed whether activated CRLs were involved in ubiquitylation of S. Typhimurium. To this end, we performed infection experiments in the absence and presence of the neddylation inhibitor MLN4924 27, which interferes with the NEDD8 conjugation cascade and leads to a rapid loss of cullin neddylation (Fig EV2D). Contrary to ARIH1 depletion, the number of Ub‐positive cytosolic bacteria was unchanged upon MLN4924 treatment (Figs 4A and EV2E). Consistently, ubiquitylation of cytoplasmic S. Typhimurium was similarly unaffected upon depletion of CUL1 or CUL3 (Figs 4B and EV2F and G), which were both shown to interact and cooperate with ARIH1 in the ubiquitylation of CRL substrates 26. Thus, these results suggest that in response to S. Typhimurium infection ARIH1 is activated by a mechanism that is independent of neddylated CRLs.
Figure 4. ARIH1 function during bacterial infection does not require activation by CRLs and encompasses both autophagy‐dependent and autophagy‐independent roles.
-
AHeLa cells infected with cytoGFP‐expressing ΔsifA S. Typhimurium for 2 h or left uninfected were treated with 2 μM MLN4924 or DMSO during the course of infection, fixed, and immunolabeled with anti‐Ub antibody (FK2). Number of Ub+/GFP+ bacteria in at least 250 cells/sample was determined by using automated quantification. Data represent mean ± SD. Significance was determined using unpaired Student's t‐test. ns = not significant. n = 3 biological replicates.
-
BHeLa cells were transfected with indicated pooled siRNAs for 72 h, infected, fixed, and immunolabeled as described in (A) prior to fixation. Number of Ub+/GFP+ bacteria was determined by automated quantification in at least 100 cells/sample. Data represent mean ± SD. n = 2 biological replicates.
-
C–HWild‐type or ATG7 CRISPR/Cas9 knockout HeLa cells transfected for 72 h with indicated siRNAs were infected during indicated p.i. time points as in (A) prior to fixation (C, D, and G) or lysis and serial dilution plating (E, F, and H). Number of GFP+ bacteria in at least 250 cells/sample was determined using automated quantification (C, D, and G). Results in (C–F) were normalized to the 30 min p.i. time point. Data represent mean ± SD. Significance was determined using two‐way ANOVA. *P < 0.05, **P < 0.01, ***P < 0.001, ns = not significant. n = 3 (wild‐type HeLa) or n = 2 (ATG7 knockout HeLa) biological replicates.
ARIH1 and LRSAM1 have xenophagy‐dependent and xenophagy‐independent functions
To examine whether ARIH1 exclusively functions in xenophagy during S. Typhimurium infection, we monitored the amount of intracellular bacteria in wild‐type and ATG7 CRISPR/Cas9 knockout HeLa cells at different p.i. time points upon ARIH1 depletion by quantifying GFP‐positive bacteria and colony formation units (CFUs). As a positive control, we also analyzed the effect of LRSAM1 knockdown in both conditions. The loss of ATG7 (Fig EV3A) increased the numbers of cytosolic S. Typhimurium at 2 and 6 h p.i. to a similar level than depletion of ARIH1 or LRSAM1 in wild‐type cells (Fig 4C and D). Intriguingly, depletion of LRSAM1 or ARIH1 in cells lacking ATG7 led to substantially further increased numbers of GFP‐positive bacteria at both p.i. time points (Fig 4C and D). Similar results were obtained for both ligases in wild‐type and ATG7‐deleted cells at 6 h p.i. by monitoring CFUs (Fig 4E and F). These additive effects of bacterial counts in ATG7 knockout cells upon depletion of either one of the ligases indicate that ARIH1 and LRSAM1 potentially protect the cell against pathogens by mechanisms both dependent and independent of autophagy.
Figure EV3. Colocalization of xenophagy components to cytosolic S. Typhimurium upon ARIH1 depletion (related to Fig 4).
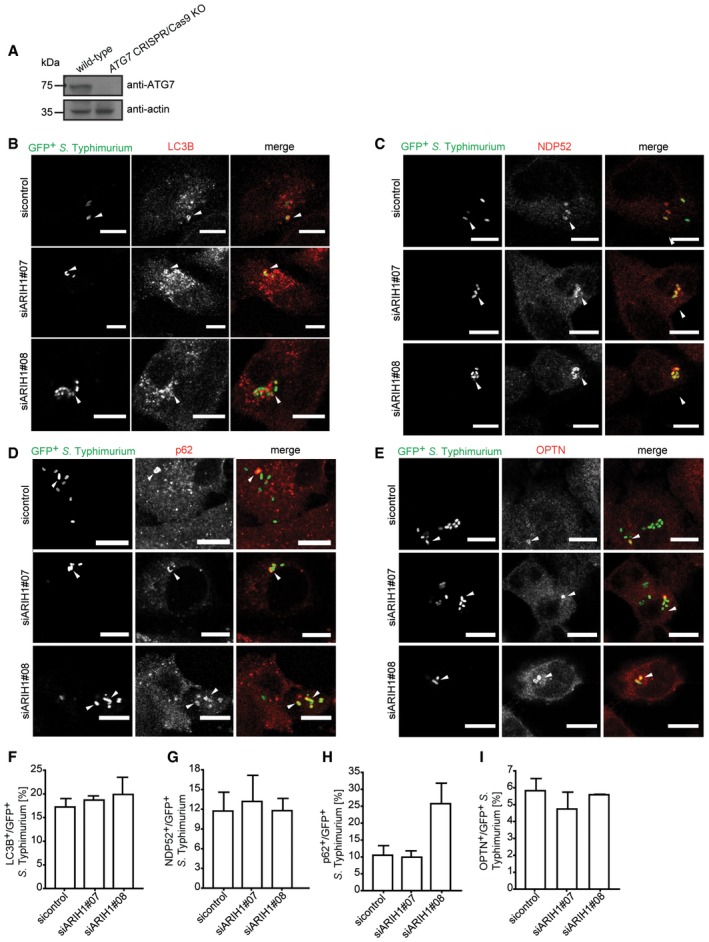
-
ALysates from wild‐type and ATG7 CRISPR/Cas9 knockout HeLa cells were analyzed by SDS–PAGE and immunoblotting.
-
B–IHeLa cells transfected with indicated siRNAs for 72 h were infected with ΔsifA cytoGFP‐expressing S. Typhimurium for 2 h, fixed, and immunolabeled with anti‐LC3B (B), anti‐NDP52 (C), anti‐p62 (D), or anti‐OPTN (E) antibodies. Number of GFP+ bacteria that colocalized with LC3B (F), NDP52 (G), p62 (H), or OPTN (I) was determined by automated quantification in at least 100 cells/sample. Data represent mean ± SD. n = 2 biological replicates.
ARIH1 and LRSAM1 play different roles but function in the same anti‐bacterial pathway
Given that ARIH1 and LRSAM1 both colocalize on bacteria, ubiquitylate cytosolic bacteria and limit the cytosolic pool of S. Typhimurium, we sought to address whether these two ligases share a genetic interaction. To this end, we compared the effect of single and double depletion of ARIH1 and LRSAM1 on the numbers of cytosolic bacteria and CFUs. Noteworthy, there were no significant changes observed in any of the experiments during the first 30 min p.i., indicating that neither single nor double knockdowns affect infection levels or SCV integrity (Fig 4G and H). Intriguingly, the effect of the combined knockdown of ARIH1 and LRSAM1 led to a significant but smaller increase in GFP‐positive bacteria than expected based on the phenotypes caused by individual depletion of either ligase (Fig 4G). Furthermore, when taking into account the whole intracellular population of S. Typhimurium, the double knockdown of ARIH1 and LRSAM1 did not further deteriorate the bacterial colony count phenotype compared to the single knockdowns (Fig 4H). This result indicates that ARIH1 and LRSAM1 have a positive, alleviating interaction and likely act in the same pathway. Since LRSAM1 was shown to be required for the colocalization of LC3B, p62, and NDP52 to cytosolic bacteria 14, we examined whether ARIH1 is likewise involved in recruiting xenophagy components to S. Typhimurium. However, in contrary to LRSAM1, we observed by and large no substantial differences in bacterial localization of LC3B (Fig EV3B and F), p62 (Fig EV3C and G), OPTN (Fig EV3D and H), or NDP52 (Fig EV3E and J) upon ARIH1 knockdown. Next, we analyzed the effect of a combined depletion of ARIH1 and LRSAM1 on the bacterial Ub coat. However, unlike individual depletions of ARIH1 or LRSAM1, combined knockdown of both ligases did surprisingly not affect ubiquitylation of cytosolic bacteria (Fig 5A and B). Since cytosolic bacteria were shown to be decorated with polyUb of different topology including linear Ub chains 15, 17 and the anti‐Ub antibody FK2 also recognized M1‐linked ubiquitin moieties (Fig EV4A and B), we speculated that depletion of ARIH1 and LRSAM1 may trigger linear ubiquitylation of S. Typhimurium. To test this hypothesis, we monitored colocalization of M1‐linked Ub to cytosolic bacteria in cells depleted of ARIH1, LRSAM1, or both. Intriguingly, we observed a significant increase in the number of linearly ubiquitylated bacteria irrespectively of whether ARHI1, LRSAM1, or both ligases were depleted from cells (Fig 5C and D). This suggests again that ARIH1 and LRSAM1 both play a role in the same pathway by localizing to cytosolic S. Typhimurium and that the loss of either or both ligases is buffered by induction of linear ubiquitylation of these bacteria. Interestingly, the linear Ub signal on bacteria was not sufficient to restrict the cytosolic pool of S. Typhimurium, indicating that linear ubiquitylation of bacteria might have other roles than xenophagic targeting. One possibility is that these M1‐linked Ub chains may actually serve to sense and/or signal the presence of persistent cytosolic bacteria. Consistent with this notion, cells depleted of ARIH1 or/and LRSAM1 showed reduced protein abundance of IκBα (Fig 5E and F) and a partially increased phosphorylation of p65 (Fig 5E and G), both of which are indicative of an active NF‐κB signaling.
Figure 5. Depletion of either ARIH1, LRSAM1, or both triggers linear ubiquitylation of S. Typhimurium and activation of the NF‐κB pathway.
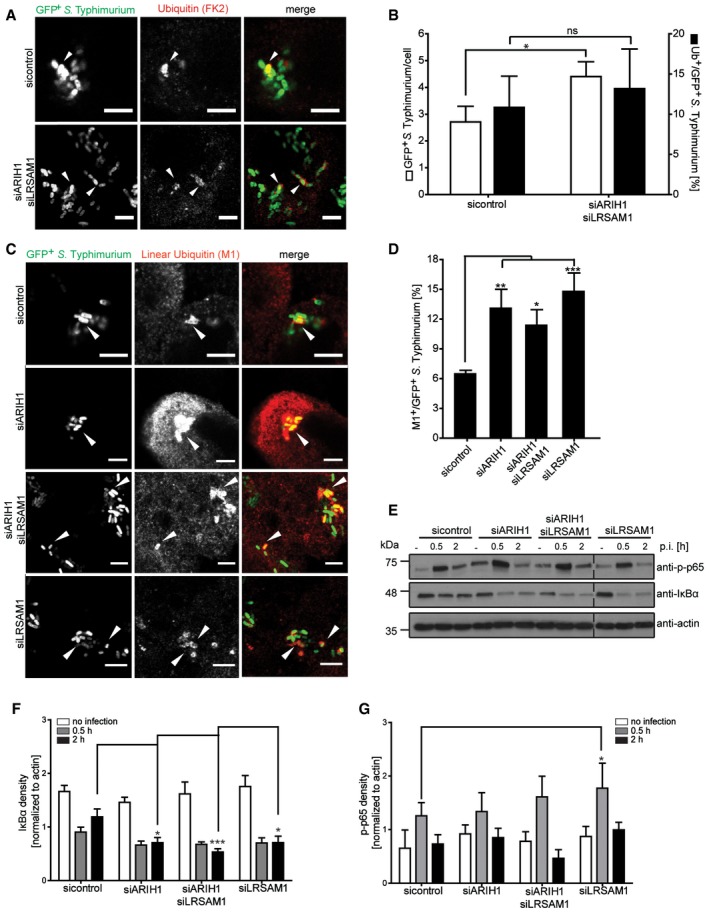
-
AWild‐type HeLa cells transfected with indicated single siRNAs for 72 h were left infected for 2 h with cytoGFP‐expressing ΔsifA S. Typhimurium prior to fixation and immunolabeling with anti‐Ub antibody (FK2). Scale bar: 5 μm. Arrowheads indicate colocalization events.
-
BAutomated quantification of GFP+ and Ub+/GFP+ bacteria in at least 250 cells/sample from (A). Data represent mean ± SD. Significance was determined using two‐way ANOVA. *P < 0.05, ns = not significant. n = 3 biological replicates.
-
CHeLa cells transfected with indicated siRNAs and infected as in (A) were fixed and immunolabeled with an antibody specific for linear Ub chains (M1‐Ub). Scale bar: 5 μm. Arrowheads indicate colocalization events.
-
DAutomated quantification of M1+/GFP+ bacteria in at least 250 cells/sample from (C). Data represent mean ± SD. Significance was determined using one‐way ANOVA. *P < 0.05, **P < 0.01, ***P < 0.001. n = 3 biological replicates.
-
ELysates from HeLa cells were transfected and infected as in (A) during indicated p.i. time points or left uninfected and analyzed by SDS–PAGE and immunoblotting.
-
F, GQuantification of IkBα protein abundance (F) and phosphorylation of p65 (G) from (E). All density values were normalized to actin. Data represent mean ± SD. Significance was determined using two‐way ANOVA. *P < 0.05, ***P < 0.001, n = 4 biological replicates.
Figure EV4. Immune detection of Ub chains topologies (related to Fig 5).
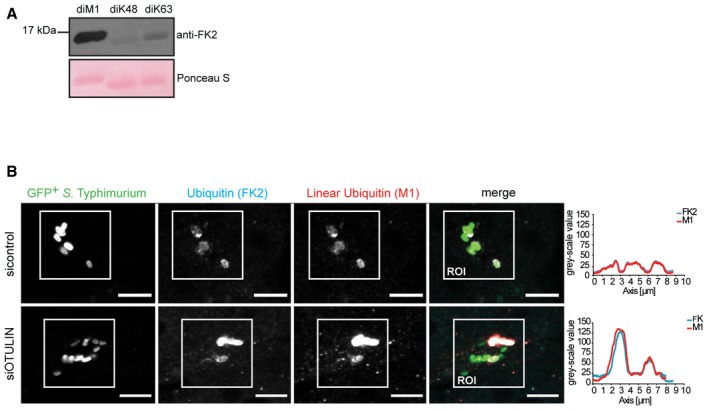
- Purified linear (M1), K48‐linked, and K63‐linked di‐Ub variants were separated by SDS–PAGE and analyzed by immunoblotting.
- HeLa cells were transfected with indicated siRNAs and infected as in (A) followed by fixation and immunolabeling with anti‐polyUb antibodies (FK2 and M1). Scale bar: 5 μm. Regions of interest (ROIs) show colocalization of FK2‐positive and M1‐positive bacteria and a concomitant increase in fluorescence intensities.
ARIH1 is linked with LRSAM1 and HOIP in a regulative anti‐bacterial network
Since our results indicated a positive genetic interaction of ARIH1 and LRSAM1, we asked whether both ligases are coordinately regulated in the course of S. Typhimurium infection. Intriguingly, knockdown of LRSAM1 led to a significant increase in the recruitment of endogenous ARIH1 to GFP‐positive bacteria (Fig 6A and B), suggesting that both ligases are indeed connected in a compensatory manner. A robust patchlike recruitment of ARIH1 upon LRSAM1 depletion was additionally confirmed by dSTORM imaging (Fig 6F). On the other hand, we observed no substantial differences in recruitment of endogenous LRSAM1 upon depletion of ARIH1 (Fig 6D, E and G). Since depletion of ARIH1 and/or LRSAM1 induced linear ubiquitylation of S. Typhimurium, we examined whether HOIP and ARIH1 are also linked in a similar way than ARIH1 and LRSAM1. HOIP knockdown resulted in a robust increase in colocalization of endogenous ARIH1 and cytosolic bacteria (Figs 6A and C, and EV5A). Notably, depletion of HOIP resulted in a moderate but not significant increase in ubiquitylated GFP‐positive bacteria compared to the control in our primary RBR screen (Fig 1A). Given these seemingly compensatory phenotypes, we assayed whether ARIH1 associates with LRSAM1 or HOIP. However, we did not detect any interaction between these ligases (Fig EV5B). Collectively, these results indicate that ARIH1, LRSAM1, and HOIP are part of an Ub E3 ligase network that functions to restrict the bacterial load in the course of S. Typhimurium infection.
Figure 6. ARIH1, LRSAM1, and HOIP mutually regulate localization of host ligases to cytosolic bacteria.
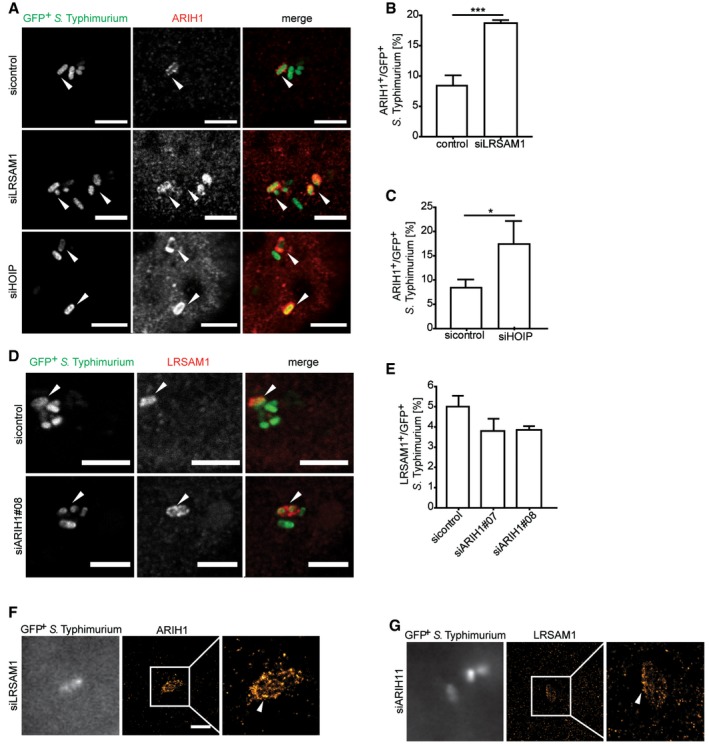
-
AHeLa cells transfected with indicated siRNAs were infected with cytoGFP ΔsifA S. Typhimurium for 2 h followed by fixation and immunolabeling with anti‐ARIH1 antibody. Scale bar: 5 μm. Arrowheads indicate colocalization events.
-
B, CAutomated quantification of ARIH1+ bacteria upon LRSAM1 (B) or HOIP (C) knockdown in on average 250 cells/sample from (A). Data represent mean ± SD. Significance was determined using unpaired Student's t‐test. *P < 0.05, ***P < 0.001. n = 3 biological replicates.
-
DHeLa cells transfected with indicated siRNA were infected as in (A), fixed and immunolabeled with anti‐LRSAM1 antibody. Scale bar: 5 μm. Arrowheads indicate colocalization events.
-
EAutomated quantification of LRSAM1+ S. Typhimurium upon knockdown of ARIH1 in at least 100 cells/sample. Data represent mean ± SD. n = 2 biological replicates.
-
F, GHeLa cells were transfected with indicated siRNA, fixed, immunolabeled with anti‐ARIH1 (F), or anti‐LRSAM1 (G) antibody, followed by additional post‐staining fixation and super‐resolution dSTORM imaging. Scale bar: 2 μm. Arrowheads indicate nanoscale protein patches.
Figure EV5. ARIH1 does not bind to LRSAM1 or HOIP (related to Fig 6).
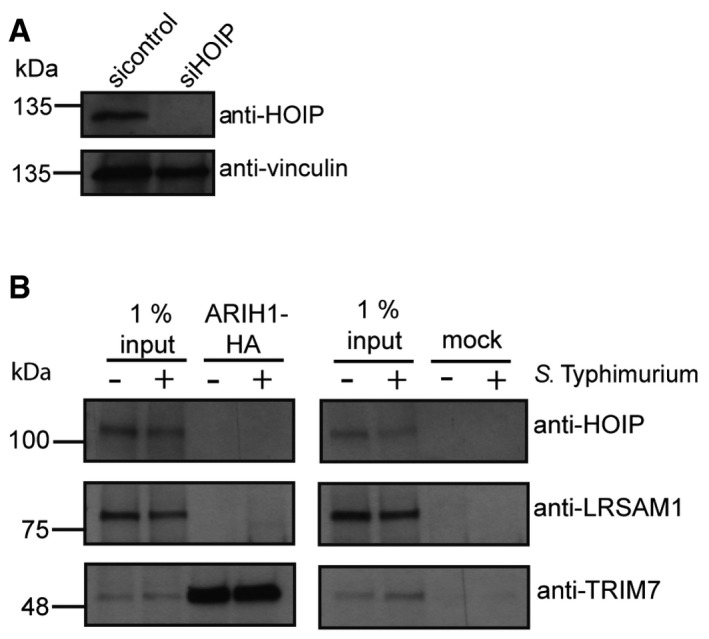
- HeLa cells were reversely transfected with indicated single siRNA for 72 h and lysed. Lysates were analyzed by SDS–PAGE and immunoblotting.
- Lysates from HeLa cells expressing C‐terminally HA‐tagged ARIH1 were subjected to HA immunoprecipitation and analyzed by SDS–PAGE and immunoblotting.
Discussion
In this report, we identified ARIH1 as an E3 ligase involved in recognition and ubiquitylation of invasive S. Typhimurium and provide evidence that ARIH1, together with LRSAM1 and HOIP, form a cooperative E3 ligase network that functions in restricting the bacterial load in the cytosol. Our findings are consistent with a recent report showing the recruitment of LUBAC to cytosol‐exposed S. Typhimurium and the formation of a M1‐linked polyUb layer surrounding these bacteria 15. However, the observation that a pre‐existing layer of polyUb is required for LUBAC localization on S. Typhimurium and that depletion or deletion of LRSAM1 does not completely abolish ubiquitylation of S. Typhimurium 14 points to the involvement of several host E3 ligases in the defense against intracellular pathogens. In line with the notion that ARIH1 and LRSAM1 operate in the same pathway, we observed that knockdown of ARIH1 only partially reduces the Ub coat formation on S. Typhimurium. Intriguingly, ARIH1 was responsible for the modification of cytosolic bacteria with K48 polyUb. Moreover, knockdown of both ligases in turn increased the number of bacteria tagged by linear Ub chains, suggesting a compensating activation of LUBAC in order to preserve the Ub coat surrounding the bacteria. Moreover, by promoting the generation of M1‐linked Ub during S. Typhimurium infection, the host cell may boost NF‐κB signaling to antagonize intracellular bacteria in an attempt to compensate for the loss of ARIH1. Consistent with this notion, the Dikic and Randow laboratories recently demonstrated that the incorporation of linear Ub moieties in the coat formed around S. Typhimurium triggers activation of the NF‐κB pathway 15, 16. Conversely, ARIH1 can compensate for the loss of LRSAM1 or HOIP, which is reflected in an increased localization of endogenous ARIH1 to cytosolic bacteria upon depletion of either of these two E3 ligases. These results imply the existence of a dynamic equilibrium of different Ub chain types that form the Ub coat surrounding the intracellular bacteria upon depletion of LRSAM1 and/or ARIH1. It remains to be addressed how ARIH1 is targeted to S. Typhimurium in the host cytosol and which bacterial OMPs are the direct substrates of ARIH1. In addition, further studies are required to understand how the three E3 ligases cooperate with each other and which signaling mechanisms are involved in this concerted pathogen defense action.
Recently, a new regulatory mechanism of substrate ubiquitylation was reported involving ARIH1 and CLRs. CLRs and ARIH1 associate in an E3‐E3 cascade where ARIH1 is first rendered active by NEDD8‐modified CLRs, which in turn enables ARIH1 to monoubiquitylate CLR client substrates and serves as a priming E3 ligase 26. In our experiments, however, we observed no significant changes in the ubiquitylation of S. Typhimurium upon treatment with the neddylation inhibitor MLN4924 or the depletion of two ARIH1‐associated CLRs, suggesting that in the course of bacterial evasion into the cytosol ARIH1 is activated by a CRL‐independent mechanism. Thus, further work is needed to elucidate the molecular basis of this activation.
Materials and Methods
Antibodies
The following antibodies were used: anti‐actin (A1978, Sigma), anti‐ATG5 (2630, Cell Signaling), anti‐ATG7 (8558, Cell Signaling), anti‐ANKIB1 (A301‐368A, Bethyl), anti‐ARIH1 (ab3891, Abcam), anti‐ARIH2 (ab17014, Abcam), anti‐CUL1 (A303‐373A‐M, Bethyl), anti‐CUL3 (A301‐109A, Bethyl), anti‐CUL9 (A300‐098A, Bethyl), anti‐DORFIN (A303‐105A, Bethyl), anti‐FK2 (PW8810, Enzo), anti‐GFP (11814460001, Roche), anti‐HA (MMS‐101P, Covance), anti‐HOIL1 (NBP2‐88301, Novus), anti‐HOIP (ab2135, Abcam), anti‐IBRDC1 (ab58041, Abcam), anti‐IBRDC2 (ab110458, Abcam), anti‐IκBα (4812, Cell Signaling), anti‐K48 polyubiquitin (UBIQ: 7044, Genentech), anti‐K63 polyubiquitin (UBIQ: 7045, Genentech), anti‐LC3B (2775, Cell Signaling), anti‐M1 polyubiquitin antibody (UBIQ: 8349, Genentech), anti‐LPS (ab8274, Abcam), anti‐LRSAM1 (ab73113, Abcam), anti‐NDP52 (9036, Cell Signaling), anti‐optineurin (ab23666, Abcam), anti‐p62 (M162‐3, MBL), anti‐p‐p65 (3031, Cell Signaling), anti‐PCNA (sc‐7907, Santa Cruz), anti‐RNF14 (ab134927, Abcam), anti‐TRIAD3 (A304‐113A), and anti‐TRIM7 (HPA039213, Sigma). Secondary antibodies coupled with a horseradish peroxidase or a fluorophore used in the experiments were the following: anti‐rabbit IgG (W4011, Promega), anti‐mouse IgG (W4021, Promega), anti‐goat IgG (V8051, Promega), anti‐human IgG Cy3 (ab97170, Abcam), anti‐human IgG Alexa Fluor 647 (A‐21445, ThermoFisher), anti‐mouse IgG Alexa Fluor 594 (A‐11005, ThermoFisher), anti‐goat IgG Cy5 (705‐175‐147, Jackson Immunolab), anti‐mouse IgG Cy3 (715‐585‐150, Jackson Immunolab), anti‐rabbit IgG Alexa Fluor 594 (A11012, ThermoFisher), anti‐goat IgG Alexa Fluor 594 (A11058, ThermoFisher), and anti‐mouse Cy5 (715‐175‐151, Jackson Immunolab).
Cell culture, transfections, and genome editing
HeLa cells were grown in DMEM (Gibco) supplemented with 10% FBS and 2 mM l‐glutamine (Gibco) and grown at 37°C and 5% CO2. RNAi pools targeting RBR E3 ligases were purchased from Dharmacon (GE Healthcare). HeLa cells were reversely transfected for 72 h with 30 or 5 nM (for siHOIP) siRNAs using Lipofectamine RNAiMax (Life Technologies) according to manufacturer's instructions. For control and deconvolution experiments, cells were transfected with individual siRNAs targeting ARIH1 (#05‐CGAGAUAUUUCCCAAGAUU, #06‐GGAUAUGCCUUGUCAGAUC, #07‐GAGAGUCGACGAAGGGUUU, #08‐CCAAAUGCCAUGUCACAAU), LRSAM1 (UGACGGAGUUAGAAGCCAA), ATG7 (GGAAUAUCCUGCAGAAGAA), ATG5 (GCCCACAGAUGGAGUAGCA), HOIP (UGAACAUCCUGGAGAAAUA), OTULIN (GACUGAAAUUUGAUGGGAA), or a non‐targeting siON‐TARGET control (GAUCCGCAGCGACAUCAACCUGAUU) (all GE Healthcare). Plasmids were transfected using Lipofectamine 2000 (Life Technologies) according to manufacturer's instructions or with 35 μM polyethylenimine. The ORF of ARIH1 (a kind gift from C. Johnson) was cloned into pDONR223 and recombined into a Gateway destination vector for the expression in mammalian cells. To generate ATG7 knockout HeLa cells, a sgRNA targeting the ATG7 gene was selected with the help of the algorithm created by Fheng Zhang (http://crispr.mit.edu:8079/) and cloned into the pSpCas9(BB)‐2A‐Puro (PX459) (Addgene #48139) vector according to the published protocol 28. Upon transient transfection, HeLa cells were cultured in 1 μg/ml puromycin and single clones were selected for further growth and immunoblot analysis.
Salmonella infection
Bacterial infections were performed with S. Typhimurium SL1344‐derived wild‐type strain SFH2 21 and ΔsifA strain SFH4 (obtained from Dirk Bumann), which both express GFP under the control of a glucose 6‐phosphate promoter. Glucose 6‐phosphate is exclusively present in the cytoplasm. SFH4 was constructed by lambda red‐mediated recombination 29 using the primers sifA_1 AAAAAGGGTCGATTTAATCAATTATGTAGTCATTTTTACTCCAGTATAAGTGAGATTAATGTGTAGGCTGGAGCTGCTTC and sifA_2 GCAAACAGGAAGTACGTGAGTAAACCCTGAACGTGACGTCTGAGAAAGCGTCGTCTGATTATTCCGGGGATCCGTCGACC followed by FLIP recombinase‐mediated excision of the aphT resistance marker using plasmid pCP20. Notably, complementation was not yet tested in this strain. Upon 1:33 dilution of a stationary culture in Luria‐Bertani (LB) broth containing carbenicillin (100 μg/ml) (SHF2) and kanamycin (30 μg/ml) (SFH4), respectively, the bacteria were grown at 37°C with aeration for 3 h to reach an OD600 of 1.7–2.4. Infection was performed at a multiplicity of infection (MOI) 200 for 35 min. Subsequently, gentamicin was added to the medium at a final concentration of 100 μg/ml and incubated for 30 min, 2 or 6 h p.i. Cells were washed twice with 1× PBS to remove dead bacteria prior to further experiments.
Colony formation assay
Cells were infected as described and lysed at different p.i. time points. Cell were washed twice with sterile 1× PBS and lysed in 1% Triton X‐100/PBS. Serial dilution series (102–105) of lysates were dropped in triplicates on LB plates and incubated overnight at 37°C.
Immunofluorescence
HeLa cells grown on glass coverslips in 12‐well plates or in 384‐well glass bottom plates (PerkinElmer) were fixed with 4% paraformaldehyde for 20 min at room temperature followed by three wash steps with 1× PBS, 10‐min permeabilization with 0.1% Triton X‐100 in 1× PBS and blocking with 3% BSA in 1× PBS for 45 min at 37°C. Primary antibodies were incubated in 3% BSA in 1× PBS for 2–3 h at 37°C or overnight at 4°C. After repeated washing with 1× PBS, fluorophore‐coupled secondary antibodies were added in 3% BSA and incubated for 15 min at 37°C followed by three times washing with 1× PBS. Coverslips were mounted on glass plates with ProlongGold Antifade (Life Technologies), and 384‐well plates were subjected to DRAQ5 (Life Technologies) staining with a 1:5,000 dilution for 10 min at room temperature and stored in 1× PBS at 4°C. For double immunostainings, antibodies were incubated sequentially. For dSTORM imaging, samples were additionally subjected to a 5 min post‐staining fixation with 2% PFA/PBS, followed by several 1× PBS washing steps and storage in 0.01% NaN3/PBS. Images of cells on coverslips were observed on a TCS SP8 point scanner microscope with an HC PL APO CS263×/1.40 oil immersion objective using excitation beam splitters (DD 488/552 and TD 488/552/638) and photomultiplier tube detectors (PMT1: 493–574 nm, PMT2: 574–643 nm, PMT3: 643–778 nm). Images were acquired in xyz space resolution with 400 Hz scan speed and in a 1,024 × 1,024 format (0.241 × 0.241 μm pixel/voxel size) with a uniform pinhole diameter of 95.5 μm and analyzed with the LAS AF Lite software (Leica). 384‐well plates were imaged by an Opera automated spinning disk confocal microscope with a UPLAPO 60×/1.20 water objective. Images were acquired with a High QE CCD camera and analyzed with the Acapella 2.6 Studio software (PerkinElmer).
Statistical analysis
All data show mean ± SD, and the statistical analysis was performed using GraphPad Prism. Assumption of Normality was applied to all data based on the sample size (n > 30). Statistical significance was determined by one‐way, two‐way ANOVA, or unpaired Student's t‐test. The standard scores or z‐scores were calculated by the formula: z = (x‐μ)/σ; where x is the raw score, μ is the mean of the population, and σ is the standard deviation of the population.
Single‐molecule localization microscopy
Super‐resolution imaging was performed using a custom‐built wide‐field microscope as described earlier 30. Briefly, a 647‐nm laser (iBeam smart, Toptica Photonics) was coupled into an inverted microscope (IX71, Olympus) equipped with a 100× oil immersion objective (NA 1.45). Fluorescence was recovered by a 405‐nm laser (CUBE 405‐50C, Coherent). Fluorescence emission was separated from excitation light with a dichroic mirror (HC Quad 410/504/582/669, AHF) and a bandpass filter (700/75, AHF) and detected with an EMCCD camera (iXon3, Andor). A buffer consisting of 100 mM β‐mercaptoethylamine (MEA), 10 (w/v) % glucose, and an oxygen scavenger system (50 U/ml glucose oxidase, 5,000 U/ml catalase, 2.5 mM KCl, 1.1 mM Tris–HCl pH 7.5, 2.5% glycerol, 0.2 mM Tris(2‐carboxylethyl)phosphine hydrochloride) in PBS (pH adjusted to 8 with 1 M NaOH) was added to the samples to enable photoswitching of the fluorophore Alexa Fluor 647, applying the concept of direct stochastic optical reconstruction microscopy (dSTORM) 31. Super‐resolution microscopy was performed using inclined illumination of the sample. Single‐molecule movies were recorded with 20 Hz and of 10,000 frames. Super‐resolution images were obtained by processing the movies with rapidSTORM 32. Channel alignment was performed with Fiji 33.
Immunoprecipitation
HA‐tagged ARIH1 was transiently expressed in HeLa cells for 24 h. Cells were washed and harvested with ice cold 1× PBS followed by immediate lysis in MCLB (50 mM Tris–HCl pH 7.4, 150 mM NaCl, 0.5% NP40). Supernatants were passed through 0.45‐μm spin filters (Millipore) and subjected to immunoprecipitation with anti‐HA agarose (Sigma) overnight rotating at 4°C. Samples were washed four times with MCLB, and bound material was eluted by boiling at 95°C for 5 min in Laemmli buffer.
In vitro ubiquitylation assay
In vitro reactions included UBA1 (5 nM; Boston Biochem), UBCH7 (100 nM; Boston Biochem), active (Δariadne) or autoinhibited (full‐length) ARIH1 (1 μM; kind gifts from B. Schulman), HA‐tagged ubiquitin (0.02 mg/ml; Boston Biochem), ubiquitylation buffer (5 mM Tris–HCl pH 7.4, 0.5 mM MgCl2, 0.1 mM DTT, 0.01% Tween) and were performed in the absence and presence of 2 mM Mg‐ATP and S. Typhimurium (1 × 108 bacteria/reaction) or purified mitochondria from P. anserina (50 μg/reaction) for 1 h at 37°C. Reactions were stopped by addition of EDTA to a final concentration of 100 μM, washed twice with 1× PBS and subjected to SDS–PAGE and immunoblotting. For ubiquitylation reactions with protease shed S. Typhimurium, bacteria were pre‐treated with 1.25 μg/ml proteinase K (Roth) for 15 min at 50°C in 30 mM Tris–HCl, pH 8.0, followed by buffer exchange to the ubiquitylation buffer containing the serine proteinase inhibitor PMSF (Sigma).
Author contributions
MSD and MH performed dSTORM imaging. MP performed all other experiments. MP and CB conceived the study and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Review Process File
Acknowledgements
We would like to thank C. Johnson for the ARIH1 ORF, B. Schulman for purified ARHI1 variants, H. Osiewacz for purified P. anserina mitochondria, K. Husnjak for purified di‐ubiquitin moieties, A. Kirsch for help with the Acapella script optimization, I. Dikic and members of his group as well as T. Bionda and A. Ernst for reagents and advice, D. Bumann and B. Claudi for bacterial strains, and V. Kempf, P. Kraiczy, and G. Devraj for technical support and advice with Salmonella infections. This work was supported by the Deutsche Forschungsgemeinschaft (German Research Foundation) within the framework of the Munich Cluster for Systems Neurology (EXC 1010 SyNergy) and of the Collaborative Research Centre on Autophagy (SFB1177), the LOEWE grant Ub‐Net and the European Research Council (282333‐XABA).
EMBO Reports (2017) 18: 1572–1585
References
- 1. Jones BD, Ghori N, Falkow S (1994) Salmonella Typhimurium initiates murine infection by penetrating and destroying the specialized epithelial M cells of the Peyer's patches. J Exp Med 180: 15–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Spadoni I, Zagato E, Bertocchi A, Paolinelli R, Hot E, Di Sabatino A, Caprioli F, Bottiglieri L, Oldani A, Viale G et al (2015) A gut‐vascular barrier controls the systemic dissemination of bacteria. Science 350: 830–834 [DOI] [PubMed] [Google Scholar]
- 3. Marcus SL, Brumell JH, Pfeifer CG, Finlay BB (2000) Salmonella pathogenicity islands: big virulence in small packages. Microbes Infect 2: 145–156 [DOI] [PubMed] [Google Scholar]
- 4. Ochman H, Groisman EA (1996) Distribution of pathogenicity islands in Salmonella spp. Infect Immun 64: 5410–5412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mizushima N, Levine B, Cuervo AM, Klionsky DJ (2008) Autophagy fights disease through cellular self‐digestion. Nature 451: 1069–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Levine B (2005) Eating oneself and uninvited guests: autophagy‐related pathways in cellular defense. Cell 120: 159–162 [DOI] [PubMed] [Google Scholar]
- 7. Benjamin JL, Sumpter R Jr, Levine B, Hooper LV (2013) Intestinal epithelial autophagy is essential for host defense against invasive bacteria. Cell Host Microbe 13: 723–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Perrin AJ, Jiang X, Birmingham CL, So NS, Brumell JH (2004) Recognition of bacteria in the cytosol of Mammalian cells by the ubiquitin system. Curr Biol 14: 806–811 [DOI] [PubMed] [Google Scholar]
- 9. Birmingham CL, Smith AC, Bakowski MA, Yoshimori T, Brumell JH (2006) Autophagy controls Salmonella infection in response to damage to the Salmonella‐containing vacuole. J Biol Chem 281: 11374–11383 [DOI] [PubMed] [Google Scholar]
- 10. Gomes LC, Dikic I (2014) Autophagy in antimicrobial immunity. Mol Cell 54: 224–233 [DOI] [PubMed] [Google Scholar]
- 11. Pickart CM, Fushman D (2004) Polyubiquitin chains: polymeric protein signals. Curr Opin Chem Biol 8: 610–616 [DOI] [PubMed] [Google Scholar]
- 12. Kirisako T, Kamei K, Murata S, Kato M, Fukumoto H, Kanie M, Sano S, Tokunaga F, Tanaka K, Iwai K (2006) A ubiquitin ligase complex assembles linear polyubiquitin chains. EMBO J 25: 4877–4887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Heath RJ, Goel G, Baxt LA, Rush JS, Mohanan V, Paulus GL, Jani V, Lassen KG, Xavier RJ (2016) RNF166 determines recruitment of adaptor proteins during antibacterial autophagy. Cell Rep 17: 2183–2194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Huett A, Heath RJ, Begun J, Sassi SO, Baxt LA, Vyas JM, Goldberg MB, Xavier RJ (2012) The LRR and RING domain protein LRSAM1 is an E3 ligase crucial for ubiquitin‐dependent autophagy of intracellular Salmonella Typhimurium. Cell Host Microbe 12: 778–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Noad J, von der Malsburg A, Pathe C, Michel MA, Komander D, Randow F (2017) LUBAC‐synthesized linear ubiquitin chains restrict cytosol‐invading bacteria by activating autophagy and NF‐kappaB. Nat Microbiol 2: 17063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. van Wijk SJL, Fricke F, Herhaus L, Gupta J, Hotte K, Pampaloni F, Grumati P, Kaulich M, Sou YS, Komatsu M et al (2017) Linear ubiquitination of cytosolic Salmonella Typhimurium activates NF‐kappaB and restricts bacterial proliferation. Nat Microbiol 2: 17066 [DOI] [PubMed] [Google Scholar]
- 17. van Wijk SJ, Fiskin E, Putyrski M, Pampaloni F, Hou J, Wild P, Kensche T, Grecco HE, Bastiaens P, Dikic I (2012) Fluorescence‐based sensors to monitor localization and functions of linear and K63‐linked ubiquitin chains in cells. Mol Cell 47: 797–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Manzanillo PS, Ayres JS, Watson RO, Collins AC, Souza G, Rae CS, Schneider DS, Nakamura K, Shiloh MU, Cox JS (2013) The ubiquitin ligase parkin mediates resistance to intracellular pathogens. Nature 501: 512–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Spratt DE, Walden H, Shaw GS (2014) RBR E3 ubiquitin ligases: new structures, new insights, new questions. Biochem J 458: 421–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Beuzon CR, Meresse S, Unsworth KE, Ruiz‐Albert J, Garvis S, Waterman SR, Ryder TA, Boucrot E, Holden DW (2000) Salmonella maintains the integrity of its intracellular vacuole through the action of SifA. EMBO J 19: 3235–3249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Spinnenhirn V, Farhan H, Basler M, Aichem A, Canaan A, Groettrup M (2014) The ubiquitin‐like modifier FAT10 decorates autophagy‐targeted Salmonella and contributes to Salmonella resistance in mice. J Cell Sci 127: 4883–4893 [DOI] [PubMed] [Google Scholar]
- 22. Collins CA, De Maziere A, van Dijk S, Carlsson F, Klumperman J, Brown EJ (2009) Atg5‐independent sequestration of ubiquitinated mycobacteria. PLoS Pathog 5: e1000430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Duda DM, Olszewski JL, Schuermann JP, Kurinov I, Miller DJ, Nourse A, Alpi AF, Schulman BA (2013) Structure of HHARI, a RING‐IBR‐RING ubiquitin ligase: autoinhibition of an Ariadne‐family E3 and insights into ligation mechanism. Structure 21: 1030–1041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fiskin E, Bionda T, Dikic I, Behrends C (2016) Global analysis of host and bacterial ubiquitinome in response to Salmonella Typhimurium Infection. Mol Cell 62: 967–981 [DOI] [PubMed] [Google Scholar]
- 25. Kelsall IR, Duda DM, Olszewski JL, Hofmann K, Knebel A, Langevin F, Wood N, Wightman M, Schulman BA, Alpi AF (2013) TRIAD1 and HHARI bind to and are activated by distinct neddylated Cullin‐RING ligase complexes. EMBO J 32: 2848–2860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Scott DC, Rhee DY, Duda DM, Kelsall IR, Olszewski JL, Paulo JA, de Jong A, Ovaa H, Alpi AF, Harper JW et al (2016) Two distinct types of E3 ligases work in unison to regulate substrate ubiquitylation. Cell 166: 1198–1214 e24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S, Brownell JE, Burke KE, Cardin DP, Critchley S et al (2009) An inhibitor of NEDD8‐activating enzyme as a new approach to treat cancer. Nature 458: 732–736 [DOI] [PubMed] [Google Scholar]
- 28. Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F (2013) Genome engineering using the CRISPR‐Cas9 system. Nat Protoc 8: 2281–2308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Datsenko KA, Wanner BL (2000) One‐step inactivation of chromosomal genes in Escherichia coli K‐12 using PCR products. Proc Natl Acad Sci USA 97: 6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dietz MS, Fricke F, Kruger CL, Niemann HH, Heilemann M (2014) Receptor‐ligand interactions: binding affinities studied by single‐molecule and super‐resolution microscopy on intact cells. ChemPhysChem 15: 671–676 [DOI] [PubMed] [Google Scholar]
- 31. Heilemann M, van de Linde S, Schuttpelz M, Kasper R, Seefeldt B, Mukherjee A, Tinnefeld P, Sauer M (2008) Subdiffraction‐resolution fluorescence imaging with conventional fluorescent probes. Angew Chem Int Ed Engl 47: 6172–6176 [DOI] [PubMed] [Google Scholar]
- 32. Wolter S, Schuttpelz M, Tscherepanow M, Van De Linde S, Heilemann M, Sauer M (2010) Real‐time computation of subdiffraction‐resolution fluorescence images. J Microsc 237: 12–22 [DOI] [PubMed] [Google Scholar]
- 33. Schindelin J, Arganda‐Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B et al (2012) Fiji: an open‐source platform for biological‐image analysis. Nat Methods 9: 676–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Review Process File