

Abstract
Precursor B lymphocytes expand upon expression of a pre‐B cell receptor (pre‐BCR), but then transit into a resting state in which immunoglobulin light chain gene recombination is initiated. This bi‐phasic sequence is orchestrated by the IL‐7 receptor (IL‐7R) and pre‐BCR signaling, respectively, but little is known about microRNAs fine‐tuning these events. Here, we show that pre‐B cells lacking miR‐15 family functions exhibit prolonged proliferation due to aberrant expression of the target genes cyclin E1 and D3. As a consequence, they fail to trigger the transcriptional reprogramming normally accompanying their differentiation, resulting in a developmental block at the pre‐B cell stage. Intriguingly, our data indicate that the miR‐15 family is suppressed by both IL‐7R and pre‐BCR signaling, suggesting it is actively integrated into the regulatory circuits of developing B cells. These findings identify the miR‐15 family as a novel element required to promote the switch from pre‐B cell proliferation to differentiation.
Keywords: B Lymphocytes, lymphopoiesis, microRNA, miR‐15
Subject Categories: Development & Differentiation, Immunology, RNA Biology
Introduction
B cell development in the bone marrow is tightly linked to the sequential rearrangement of immunoglobulin gene segments that constitute the mature B cell receptor (BCR). Initiated at the pre‐pro‐B stage, progenitors first undergo diversity (D) to joining (J) recombination, closely followed by variable (V) to DJ recombination in late pro‐B cells 1. If in‐frame, this gives rise to an immunoglobulin heavy chain (Igμ) that is expressed on the cell surface as part of the pre‐B cell receptor (pre‐BCR) 2. The pre‐BCR, composed of two identical copies of the Igμ chain and the germ line‐encoded surrogate light chain components lambda5 and VpreB, marks the onset of the pre‐B cell stage, which imposes an important checkpoint during early B cell development 3. Upon expression of a signaling‐competent pre‐BCR, pre‐B cells clonally expand to enrich for a population of cells with a successful Igμ rearrangement, but then transit into a resting state, also referred to as small pre‐B cell stage. Here, the recombination of the light chain gene loci is initiated, eventually resulting in the expression of the genuine BCR in a subset of cells that enables their further maturation 4. The described bi‐phasic setup of differentiation events is mainly established through two signaling circuits, driven by the IL‐7 receptor (IL‐7R) and the pre‐BCR, respectively 5. Together, they effectively separate pre‐B cell proliferation from DNA rearrangement and pre‐B‐to‐immature B cell differentiation, thereby ensuring genomic integrity throughout development, documented by the fact that errors in these processes can promote blood cancer 6, 7, 8, 9. However, despite recent advances, it is fair to say that the precise molecular processes that drive these cell fate decisions at the pre‐B cell stage are incompletely understood, in particular when considering the contribution of non‐coding genomic elements such as miRNAs.
MiRNAs are short RNAs that mediate post‐transcriptional silencing of the majority of protein‐coding genes in mammals 10. In consequence, they have emerged as an additional layer of gene regulation in almost all biological processes including the development and function of the immune system 11. Early B cell development, for example, is shaped by a diverse set of miRNAs including the miR‐17‐92 cluster, miR‐150, and miR‐34a, whereas terminal differentiation and function of B cells is fine‐tuned, for example, by miR‐155 and miR‐181b 12, 13, 14, 15, 16, 17, 18, 19, 20. However, with about 100 different miRNAs expressed throughout hematopoiesis, controlling a large number of protein‐coding genes both individually and synergistically, we are only beginning to decipher the complexity of their biological activities in this and other systems.
Here, we have screened a miRNA sponge library in a well‐defined pre‐B cell differentiation model, in an attempt to cover one of the last white spots in miRNA‐regulated B cell development, and observed that cells lacking miR‐15 function fail to induce the transcriptional reprogramming that normally accompanies differentiation, resulting in enhanced proliferation and a severe block at the pre‐B cell stage. To our surprise, miR‐15 family members mainly target genes involved in cell‐cycle regulation to modulate B cell differentiation. Intriguingly, ectopic expression of the cell‐cycle regulators cyclin E1 and D3 is sufficient to largely recapitulate the miR‐15 family loss‐of‐function phenotype. This indicates that miR‐15 family‐mediated suppression of these genes is indispensable for proper B cell maturation, providing a possible explanation how loss of miR‐15‐family function may contribute to blood cancer in humans, that is, by stabilizing an immature but highly proliferative developmental state. Consistently, we find that the miR‐15 family is suppressed by IL‐7R signaling, but becomes activated upon differentiation, suggesting its pivotal integration into the decisive regulatory circuits controlling pre‐B‐to‐immature B cell transition.
Results
A loss‐of‐function screen identifies miRNAs regulating early B cell development
To unravel the roles of individual miRNA families in early B cell development, we chose a “sponge”‐based loss‐of‐function approach in order to avoid the promiscuity associated with miRNA overexpression 21. MiRNA sponges or decoys are expression constructs consisting of a cDNA encoding a fluorescent marker, for example, dsRedExpress2 (hereafter abbreviated as dsRed), and concatemeric repeats of specific miRNA‐binding sites in the 3′‐UTR (Fig 1A). Once transcribed, the sponge mRNA functions as bait that competes with the endogenous mRNAs for specific miRNA‐of‐interest/RISC complexes, resulting in a derepression of endogenous target genes. At the same time, the fluorescent marker allows the discrimination of sponge‐positive versus sponge‐negative cells. Notably, miRNA sponges have been reported to effectively target not only single miRNA species, but the complete miRNA family as defined by their seed sequence 21.
Figure 1. A loss‐of‐function screen identifies miRNAs involved in pre‐B cell differentiation.
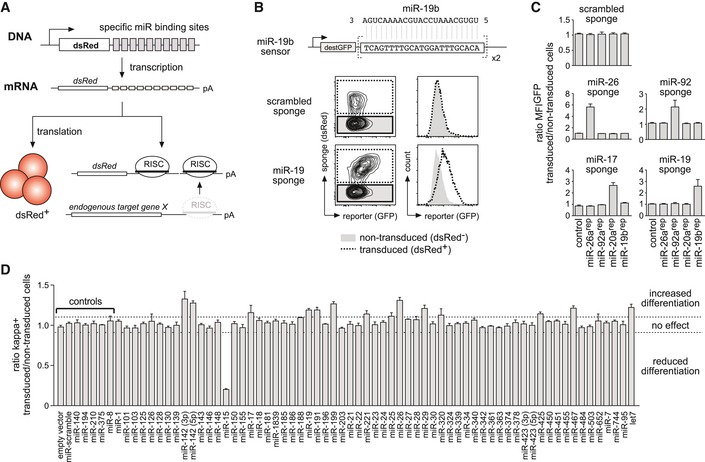
- Schematic illustration of the experimental setup to induce functional miRNA knockdown by miRNA sponges.
- Derepression of a fluorescent miRNA sensor by a miRNA sponge. Pre‐B cells expressing a GFP‐based sensor for miR‐19b (upper lane) were transduced with a scrambled sponge as a control or with a sponge targeting the miR‐19 family (both expressing dsRed as a marker). Contour plots as well as histograms depict the GFP fluorescence distribution of the dsRed‐negative, non‐transduced population, and the dsRed‐positive, sponge‐expressing cells.
- Bar graphs representing an experiment as in (B), comprising five miRNA reporter constructs and five different miRNA sponges. Depicted values represent the ratio of GFP MFI of transduced compared to non‐transduced cells within the same sample. Data show mean + SD and are representative of four independent experiments yielding similar results.
- Pre‐B cells retrovirally transduced with the respective sponge constructs were cultured without IL‐7 for 60 h to induce their differentiation into immature B cells. Differentiation was quantified by FACS analysis for surface expression of the mature B cell receptor (as measured by kappa light chain expression). An empty vector, a scrambled sponge, and sponges targeting miRNAs not expressed in early B cells were used as controls, respectively. Individual bars show the ratio of percentage of kappa‐positive cells comparing transduced and non‐transduced cells within one sample, as a mean (+ SD) of at least three independent experiments. Dashed lines mark the region in which miRNA sponges were defined as having no effect (ratio of 0.9–1.1). MFI, mean fluorescence intensity.
To validate the specificity of our approach, we initially tested a subset of sponge constructs on fluorescent sensors in which two specific miRNA target sites were cloned into the 3′‐UTR of a GFP cDNA (Fig 1B). When expressed in a cell, these sensors give rise to a certain GFP mean fluorescence intensity (MFI) based on the endogenous miRNA levels. Co‐expression of a control sponge encoding a scrambled sequence repeat did not alter GFP MFI in this setup (Fig 1B). In contrast, a specific sponge promoted a strong derepression of GFP, reflecting a sequestration of endogenous miRNAs by the sponge. By combining additional sponges and reporter cell lines, we furthermore confirmed the specificity of the individual miRNA sponge constructs (Fig 1C).
Next, we generated a retroviral miRNA sponge library encompassing 63 constructs (Appendix Table S1) targeting all miRNA families reported to be expressed throughout early B cell development, based on available small RNAseq data 22. This library was then applied for an in vitro pre‐B‐to‐immature B cell differentiation screen, using the pre‐B cell line wk3, lacking the adaptor protein SLP‐65, a crucial mediator of signaling downstream of the pre‐BCR. Notably, SLP‐65−/− pre‐B cells can be cultured indefinitely in the presence of IL‐7, but immediately start to differentiate into BCR+ immature B cells upon IL‐7 withdrawal 23. When individually expressed in wk3 cells, a subset of the sponge constructs tested provoked clear phenotypes, promoting or suppressing normal pre‐B cell differentiation compared to controls based on surface Igκ expression (Fig 1D). Of note, the sponge constructs that showed an activity in this assay mainly targeted miRNA families reported to be strongly expressed in B cell precursors 22, suggesting that miRNA expression has to exceed a certain threshold to be physiologically relevant (Appendix Fig S1).
Functional knockdown of the miR‐15 family interferes with pre‐B cell differentiation, apoptosis, and proliferation in vitro
The most striking effect in differentiating pre‐B cells was observed upon functional knockdown of the miR‐15 family, which comprises the miR‐15a/miR‐16‐1, miR‐15b/miR‐16‐2, and miR‐497/miR‐195 clusters. MiR‐15a/16‐1 is deleted or downregulated in the majority of cases in chronic lymphocytic leukemia (CLL), clearly demonstrating a tumor‐suppressive function of the miR‐15 family in B cells 24, 25. However, little is known about its role in normal B cell development. In pre‐B cells, reducing the activity of the miR‐15 family resulted in a severe block in differentiation upon IL‐7 withdrawal, as measured by a reduced percentage of cells that were able to recombine the kappa light chain loci and express the BCR on the cell surface (Fig 2A) as well as by diminished Rag1/2 recombinase activity quantified by a fluorescent reporter (Fig EV1A and B). Of note, this block in differentiation was also observed in a number of other pre‐B cell lines, all arrested at the pre‐B stage by loss of the adapter SLP‐65 (Fig EV1C). However, while reconstitution of SLP‐65 into these pre‐B cells promoted differentiation, as has been previously shown 23, the relative suppressive function of the miR‐15 sponge was retained even in this setup (Fig EV1D). This indicates that the observed effect does not depend on SLP‐65 deficiency.
Figure 2. Functional knockdown of the miR‐15 family interferes with pre‐B cell differentiation in vitro .
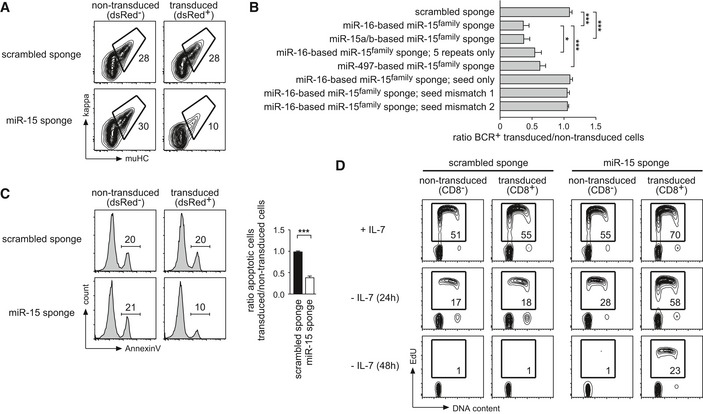
- Wk3 pre‐B cells transduced with constructs encoding a scrambled sponge as a control or a sponge targeting the miR‐15 family were cultured without IL‐7 for 72 h. Differentiation was quantified by FACS analysis of BCR expression as measured by mu heavy chain and kappa light chain. Contour plots compare non‐transduced and transduced cells within one sample. Numbers represent the percentage of cells within the respective region.
- Bar graphs describing an experiment as in (A), comparing different sponges targeting the miR‐15 family as well as several sponge mutants. Values show the ratio in the percentage of BCR‐positive cells in transduced compared to non‐transduced cells within the same sample. Bars represent means of at least 4 independent experiments ± SD. Groups were compared by a one‐way ANOVA and Bonferroni testing; ***P < 0.001, *P < 0.05.
- MiR‐15 family knockdown protects against apoptosis induced by growth factor withdrawal. Wk3 pre‐B cells transduced with the depicted constructs were cultured without IL‐7 for 48 h. Histograms show a representative experiment in which cells gated for intact membrane integrity (PI−) were analyzed for their apoptotic rate by flow cytometry, comparing non‐transduced and transduced cells. Numbers represent the percentage of cells within the respective gate. The bar graph depicts the ratio of apoptotic cells comparing the transduced and the non‐transduced population of each sample (mean ± SD of five independent experiments). Individual groups were analyzed by a paired t‐test; ***P < 0.001.
- Reduced miR‐15 family activity enables prolonged proliferation upon growth factor withdrawal. Wk3 pre‐B cells transduced with constructs as indicated were cultured with IL‐7 or without IL‐7 for 24 h and 48 h, respectively, before labeling with EdU for 45 min, staining, and FACS analysis. Contour plots compare the non‐transduced and the transduced population within one sample. Numbers represent the percentage of cells in EdU‐positive gate. Data are representative of at least three independent experiments yielding highly similar results. BCR, B cell receptor; PI, propidium iodide; EdU, 5‐ethynyl‐2′‐deoxyuridine.
Figure EV1. The miR‐15 sponge‐mediated suppression of pre‐B cell differentiation reduces Rag1/2 activity in a fluorescent reporter and can be observed in independent pre‐B cell lines.
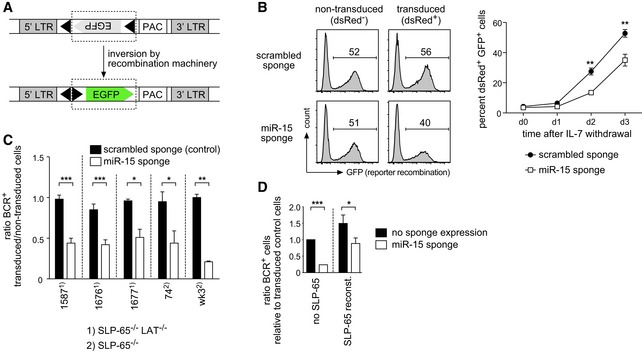
- Schematic overview of the fluorescent reporter for kappaLC recombination. An inverted EGFP cDNA flanked by kappaLC recombination signal sequences (black triangles) is expressed from a retroviral LTR. Upon Rag1/2‐mediated recombination, the GFP cassette is inverted, giving rise to GFP+ cells. PAC, puromycin resistance gene.
- Sequestering miR‐15 family members reduces the activity of the recombination machinery in pre‐B cells. Wk3 cells expressing the reporter as shown in (A) were transduced with the scrambled sponge as a control or the miR‐15 sponge and cultured without IL‐7 to induce light chain recombination. The histogram plots depict the GFP expression in the non‐transduced, dsRed− population and the transduced, dsRed+ population of a representative experiment on day 3. Numbers indicate the percentage of cells in the respective gate. The line graph shows the percentage of GFP+ cells in the dsRed+ population over the course of 3 days (mean ± SD of three independent experiments). Statistical significance was calculated by a paired t‐test; **P < 0.01.
- Different pre‐B cell lines (SLP‐65−/− or SLP‐65−/−LAT−/− as indicated) including the wk3 line used throughout the study transduced with vectors encoding the scrambled sponge or the sponge targeting the miR‐15 family were cultured without IL‐7 to induce differentiation. After 60–72 h, cells were analyzed for expression of the mature BCR (as measured by anti‐kappaLC and anti‐muHC antibodies). Individual bars depict the ratio in the percentage of BCR+ cells comparing transduced and non‐transduced cells. Groups were compared by a paired t‐test; ***P < 0.001, **P < 0.01, *P < 0.05. Data represent means ± SD of three independent experiments.
- Wk3 pre‐B cells were co‐transduced with vectors encoding the miR‐15 sponge (dsRed as a marker) and SLP‐65 (GFP as a marker) or the scrambled sponge and the empty vector as a control. After 72 h, non‐transduced, dsRed+, GFP+, and dsRed+GFP+ cells were analyzed for expression of the mature BCR. Individual bars depict the ratio in the percentage of BCR+ cells comparing the miR‐15 sponge and/or SLP‐65‐expressing cells with cells expressing their respective control constructs (means ± SD of three independent experiments). Groups were compared by a paired t‐test; ***P < 0.001, *P < 0.05.
To demonstrate that this reduced pre‐B cell differentiation was indeed provoked by miRNA/RISC sequestration, rather than a non‐specific effect due to sponge expression, we repeated the initial assay with two sponges based on other miR‐15 family members as well as with several sponge mutants (Appendix Table S2). In addition to the sponge based on miR‐16 that was used for the initial screen (hereafter referred to as miR‐15 sponge), a sponge based on the miR‐15a/b sequences also induced a strong block in differentiation (Fig 2B). As expected for a competitive decoy, a reduction in the number of miRNA‐binding site repeats from > 15 to 5 compromised sponge function, as shown by the comparison of the miR‐16‐based sponges. Likewise, a mutation from A to G in the sponge corresponding to position 1 in the miRNA, which has been shown to diminish miRNA function 26, lowered sponge activity (miR‐497‐based sponge). On the other hand, two individual seed mismatches completely abrogated sponge function, which was also observed for a sponge containing only the region corresponding to the miRNA seed, but lacking any non‐seed interactions (Fig 2B). Strikingly, the ability of the individual sponge constructs to suppress pre‐B cell differentiation correlated perfectly well with their activity on miR‐15a, miR‐15b, and miR‐16 reporters (Fig EV2A and B), strongly indicating that the block in pre‐B cell differentiation is indeed due to endogenous miRNA sequestration. Supporting this, we furthermore found that the reciprocal experiment, that is, ectopic expression of miR‐15a‐16‐1 and miR‐15b‐16‐1 clusters, promoted differentiation already 24 h after IL‐7 withdrawal (Fig EV2C).
Figure EV2. Effects of different sponge variants and miR‐15 family overexpression on differentiation and etoposide‐induced apoptosis.
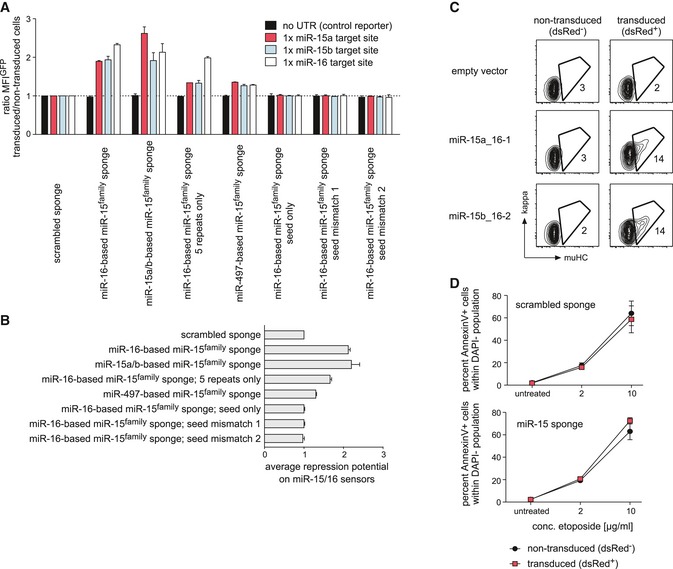
- Different sponge variants all targeting the miR‐15 family differ in their derepressive potential in reporter assays. Wk3 pre‐B cells selected for expression of GFP‐based reporter constructs for miR‐15a, miR‐15b, and miR‐16 as well as a control construct without any miR binding site in its 3′‐UTR were transduced with different sponge constructs as indicated (see Appendix Table S2 for details). After 48 h, cells were analyzed for the GFP MFI in transduced and non‐transduced cells, respectively. Bars display the ratio of GFP MFI comparing these population within one sample, and values were normalized to the scrambled sponge control (set as 1). Note that the three constructs on the right side contain exactly five repeats, whereas the other sponges consist of more than 12 repeats if not otherwise indicated. Data represent means ± SD of two independent experiments.
- Average derepressive potential of the individual sponges as shown in (A), calculated as the average of the measured GFP MFI ratio for miR‐15a/b and miR‐16: [(ratio MFI_miR‐15a + ratio MFI_miR‐15b)/2 + ratio MFI_miR‐16]/2.
- Ectopic expression of miR‐15a‐16‐1 and miR‐15b‐16‐2 clusters promotes pre‐B cell differentiation. Wk3 pre‐B cells transduced with an empty control plasmid or with plasmids encoding the miR‐15a‐16‐1 and miR‐15b‐16‐2 clusters, respectively, were cultured without IL‐7 for 24 h. Differentiation was quantified by FACS analysis of BCR expression. Contour plots compare non‐transduced (left column) and transduced cells (right column) within one sample. Numbers represent the percentage of cells within the respective region. Data are representative of three independent experiments.
- Sequestration of the miR‐15 family does not protect against etoposide‐induced apoptosis. Wk3 cells expressing the scrambled sponge (upper graph) or the miR‐15 sponge (lower graph) were left untreated or were treated with 2 or 10 μg/ml etoposide for 12 h before flow cytometric analysis for apoptosis. The line graphs depict the percentage of Annexin V+ cells within the DAPI− population, shown for the non‐transduced and for the transduced, dsRed+ fraction within the same sample. Data represent means ± SD of three independent experiments.
Beyond differentiation, we furthermore investigated whether loss of miR‐15 family function also compromises cell survival and proliferation, two additional processes that are tightly regulated in pre‐B cell precursors. Indeed, while we found no changes in survival under steady‐state conditions (data not shown), the rate of apoptotic cell death induced by IL‐7 withdrawal in miR‐15 sponge‐expressing cells was reduced to about 50% of the untransduced control population (Fig 2C). Notably, the treatment of scrambled sponge and miR‐15 sponge‐expressing cells with the topoisomerase II inhibitor etoposide, a potent inducer of DNA strand breaks and apoptosis, did not reveal any protective impact by the latter (Fig EV2D). This suggests that the miR‐15 sponge is not anti‐apoptotic per se, but only enhances survival in the context of growth factor withdrawal. Moreover, miR‐15 family knockdown increased the percentage of proliferating cells in the presence of IL‐7 and also enabled prolonged proliferation in the absence of growth factor (Fig 2D). Together, these data suggest that miR‐15 family members limit proliferation and promote pre‐B cell differentiation under physiologic conditions in vitro, which is consistent with its tumor‐suppressive capacity reported in CLL.
Loss of miR‐15 family function provides a competitive advantage and counteracts pre‐B cell differentiation in vivo
To investigate whether the inhibition of the miR‐15 family affects early B cell development also in an adoptive transfer setting mimicking the physiological situation more closely, we injected the same preparation of a wk3 mixture containing cells expressing either the scrambled sponge (dsRed+), a sponge targeting the miR‐15 family (GFP+), or no sponge at all (no fluorescence) into Rag1‐deficient recipients (Fig 3A). After 12 days, bone marrow cells were isolated and analyzed for two parameters: First, their GFP:dsRed ratio to unravel any positive or negative impact of the sponge constructs on the relative cellular fitness. Second, the percentage of B cell precursors in which light chain recombination had taken place, giving rise to BCR+ cells as measured by muHChi expression. With respect to the former, cells isolated after 12 days showed a massive shift toward the GFP+, miR‐15 sponge‐expressing population when compared to the ratio before injection (Fig 3B and C, upper panel), indicating that the loss of miR‐15 activity enhances the competitive cellular fitness also in vivo. Likewise, the inhibition of the miR‐15 family also severely compromised pre‐B cell differentiation (Fig 3B and C, lower panel), recapitulating the results of our in vitro screen and follow‐up experiments. Together, this suggests that miR‐15 family members control cellular fitness and developmental progression under physiological conditions, and that loss of this regulation impacts on pre‐B cell proliferation and survival, thereby preventing developmental progression into the immature B cell stage.
Figure 3. Knockdown of the miR‐15 family in pre‐B cells promotes their enrichment and counteracts their differentiation in vivo .
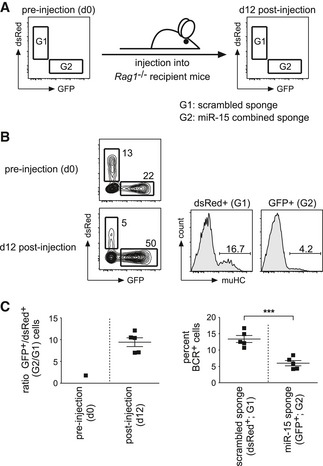
-
A, B(A) Schematic illustration of the in vivo experiment. Wk3 pre‐B cells individually transduced with a scrambled sponge as a control (dsRed+) or with a sponge targeting the miR‐15 family (combining binding site repeats corresponding to miR‐16 and miR‐15a; GFP+) were mixed, analyzed for their GFP:dsRed ratio, and injected into five Rag1‐deficient recipients. At d12, mice were sacrificed, and bone marrow cells were harvested and re‐analyzed for their GFP:dsRed ratio as well as for the percentage of differentiated, BCR+ cells that had undergone light chain recombination in the GFP+ versus dsRed+ population. A representative example is shown in (B). For the post‐injection analysis, bone marrow cells were gated for CD19+ckit− to exclude endogenous B cell progenitors from the recipients.
-
CPlots for the GFP:dsRed ratio before injection and at d12 as well as for the differentiation of pre‐B cells expressing a scrambled sponge or a miR‐15 sponge. Data show mean + SD. Groups were compared by an unpaired t‐test; ***P < 0.001.
MiR‐15 knockdown suppresses the transcriptional reprogramming in differentiating pre‐B cells
Proliferation and differentiation are processes that are regulated by complex gene regulatory networks. To gain insight into the molecular mechanism underlying the strong effect of miR‐15 family knockdown, we compared the transcriptome of sponge‐expressing and control pre‐B cells, both in the presence of IL‐7 and upon growth factor withdrawal for 36 h, by microarray analysis. As expected, control cells switching from a proliferative phase to differentiation upon IL‐7 withdrawal showed massive transcriptional changes, comprising almost 1,500 annotated coding genes that became differentially expressed (Fig 4A). In contrast, the transcriptional reprogramming in miR‐15 sponge cells was severely impaired, with < 200 genes being differentially expressed by at least twofold under these conditions. Notably, miR‐15 sponge cells and scrambled sponge control cells displayed only minor differences in the presence of IL‐7, which was reflected not only by the low number of 60 differentially expressed genes (DEGs), but also by cluster analysis of the individual microarray samples (Fig 4B). Here, cells cultured in the presence of IL‐7 formed a group that was clearly separated from the control samples cultured without IL‐7. Strikingly, miR‐15 sponge cells cultured without IL‐7 clustered with the former, indicating that inhibition of the miR‐15 family suppresses or delays the transcriptional reprogramming that accompanies differentiation. Illustrating this defective reprogramming, quantitative PCR analysis demonstrated that, for example, the expression levels of Rag1 and Rag2, two key genes essential for immunoglobulin heavy and light chain recombination and thus differentiation, were significantly repressed upon miR‐15 family knockdown already in the presence of IL‐7 and only became weakly induced upon growth factor deprivation (Fig 4C). Moreover, the induction of several genes encoding key transcription factors in early B cell development, such as Irf4, Irf8, and Ikzf3 (also referred to as Aiolos) 27, 28, 29, was compromised in the absence of miR‐15 function (Fig 4C). Taken together, this suggests that the functional knockdown of the miR‐15 family counteracts the transcriptional changes induced by growth factor withdrawal, thus establishing a status that efficiently prevents pre‐B cell differentiation.
Figure 4. Functional knockdown of miR‐15 family members counteracts the transcriptional reprogramming that accompanies pre‐B cell differentiation.
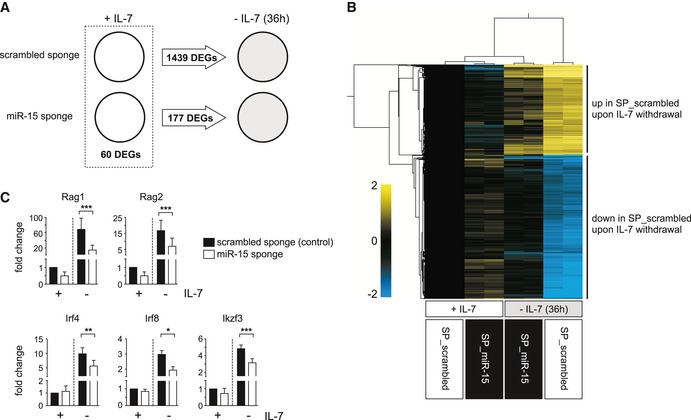
- Graphical illustration of the transcriptional changes in pre‐B cells expressing a scrambled sponge and a sponge directed against the miR‐15 family, both in the presence of IL‐7 and upon IL‐7 withdrawal for 36 h. Total RNA of pre‐B cells selected for expression of the indicated constructs was reverse transcribed into cDNA and subjected to microarray analysis. Differentially expressed genes (DEGs) were defined as annotated genes with a more than twofold change (P < 0.05) in expression levels between the respective populations.
- Heat map generated of microarray data reflecting changes in gene expression levels (restricted to annotated genes that become deregulated upon IL‐7 withdrawal in cells expressing the scrambled sponge) for biological replicates of each condition (expression of scrambled sponge and miR‐15 sponge; +IL‐7 and IL‐7 withdrawal for 36 h), normalized to the +IL‐7 scrambled sponge samples. Genes and samples were subjected to hierarchical clustering. The color code indicates log2‐fold changes.
- Bar graphs showing quantitative PCRs for Rag1, Rag2, Irf4, Irf8, and Ikzf3 gene expression in control and in miR‐15 sponge pre‐B cells, respectively, as used for the microarray analysis. Each experiment has been normalized to the scrambled sponge sample in the presence of IL‐7. Groups were compared by a paired t‐test; ***P < 0.001, **P < 0.01, *P < 0.05. Data show mean + SD and represent at least four independent experiments.
The miR‐15 family targets interphase cyclins in B cell precursors
Having revealed the functional relevance of the miR‐15 family in pre‐B cells, we were wondering about how the miR‐15 family directly affects the transcriptome. Thus, we again analyzed the microarray data, this time focusing on the identification of putative target genes. In particular, we compared the miR‐15 sponge and scrambled sponge samples and retrieved genes that were upregulated by at least 1.25‐fold in the former, both in the presence and in the absence of IL‐7 (Fig 5A). About 500 (+IL‐7) or 1,900 (−IL‐7) annotated genes, respectively, were further filtered for the presence of conserved miR‐15a, miR‐15b, or miR‐16 binding sites using the PicTar and miRanda algorithms 30, 31. This gave rise to 168 putative target genes in the presence of IL‐7, and 468 genes in the absence of IL‐7, with an overlap of 109 genes (Fig 5A; Appendix Table S3). A gene ontology analysis of the protein‐coding genes in this overlap region revealed a strong enrichment for processes involved in cell‐cycle regulation (Appendix Fig S2). This prompted us to manually screen this putative target list for candidates whose derepression might explain the observed phenotype. Indeed, we found several such genes, for example, cyclin E1, cyclin D3, Cdc6, and Cdc25a, all involved in interphase progression and DNA replication, whose derepression upon miR‐15 family knockdown was validated by quantitative PCR (Figs 5B and EV3A). Notably, the anti‐apoptotic gene Bcl2, a previously reported target of the miR‐15 family in CLL 32, was not retrieved from the microarray data using the abovementioned strategy. Supporting this, we failed to observe any differences in Bcl2 gene expression on transcript and protein level in miR‐15 sponge‐expressing cells compared to a control (Fig EV3B and C). Moreover, a functional analysis revealed that the miR‐15 loss‐of‐function phenotype could not be recapitulated by exogenous Bcl2 expression (Fig EV3D), arguing against Bcl2 as an important miR‐15 target gene in pre‐B cells.
Figure 5. The miR‐15 family targets cyclins E1 and D3 in pre‐B cells.
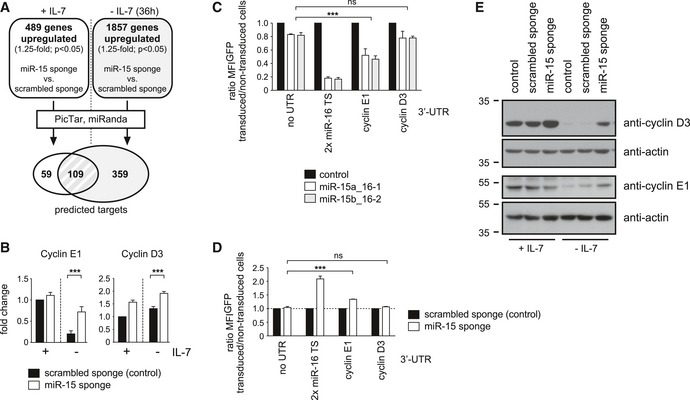
-
ASchematic illustration of the applied strategy for miR‐15 target identification. Genes upregulated (more than 1.25‐fold, P < 0.05) in the microarray data of miR‐15 sponge versus scrambled sponge samples, both in the presence and in the absence of IL‐7, were filtered for predicted, conserved miR‐15a, miR‐15b, and miR‐16 binding sites in their 3′‐UTR using the PicTar and miRanda algorithms.
-
BQuantitative PCR for cyclin E1 and cyclin D3 gene expression in control and in miR‐15 sponge pre‐B cells, respectively, in the presence of IL‐7 and upon IL‐7 withdrawal (36 h). Individual experiments were normalized to the scrambled sponge; +IL‐7 sample. Groups were compared by a paired t‐test; ***P < 0.001. Bars represent means of 10 independent experiments ± SD.
-
C, DDirect regulation of cyclin E1, but indirect regulation of cyclin D3, by the miR‐15 family. Pre‐B cells selected for expression of a control construct encoding GFP without a 3′‐UTR, a positive control with two perfect miR‐16 target sites (TS) as well as reporters with the 3′‐UTRs of cyclin E1 and cyclin D3 were transduced with an empty vector as well as with expression constructs for miR‐15a_16‐1 and miR‐15b_16‐2 clusters (C). In a reverse approach, the reporter cells were furthermore transduced with the scrambled sponge as a control or with the sponge construct targeting the miR‐15 family (D). Individual bars display the ratio in GFP MFI comparing transduced and non‐transduced cells measured by flow cytometry after 48 h (ratios were normalized to the empty vector control, respectively, which was set as 1). Values (combined for miR‐15a_16‐1 and miR‐15b_16‐2 in C) were compared by a one‐way ANOVA followed by Bonferroni testing; ***P < 0.001. Bars represent means ± SD of least three independent experiments.
-
EPre‐B cells selected for the scrambled sponge, the miR‐15 sponge, or the empty control vector were cultured with IL‐7 or without IL‐7 for 36 h before lysis, SDS–PAGE, and Western blotting with anti‐cyclin D3, anti‐cyclin E1, and anti‐actin antibodies. Shown data are representative for three independent experiments.
Source data are available online for this figure.
Figure EV3. Cdc6 and Cdc25a, but not Bcl2 as additional putative target genes of the miR‐15 family.
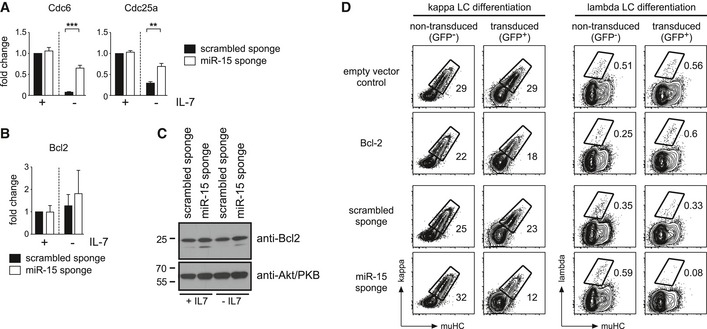
-
A, BPre‐B cells selected for expression of the scrambled sponge and the sponge targeting the miR‐15 family were cultured in the presence of IL‐7 or without IL‐7 for 36 h. Total RNA isolated from these cultures was converted into cDNA, and expression of Cdc6, Cdc25a (A), and Bcl2 (B) was determined by quantitative PCR. Bars display the fold change, normalized to the scrambled sponge +IL‐7 sample (set as 1). Groups were compared by a paired t‐test; ***P < 0.001, **P < 0.01. Bars represent means ± SD of at least three independent experiments.
-
CBcl‐2 protein levels are not altered by the miR‐15 sponge. Pre‐B cells selected for the scrambled sponge or the miR‐15 sponge were cultured with IL‐7 or without IL‐7 for 36 h before lysis, SDS–PAGE and Western blotting with anti‐Bcl‐2 and anti‐Akt/PKB antibodies as a loading control. Data show a representative example of three independent experiments.
-
DExogenous expression of Bcl‐2 does not resemble the miR‐15 loss‐of‐function phenotype in pre‐B cells. Wk3 cells were transduced with an empty vector control or a Bcl‐2 overexpression construct as well as with the scrambled sponge or the miR‐15 sponge. After 3 days of culture without IL‐7 to induce differentiation, cells were analyzed for muHC and kappaLC (left panels) or lambdaLC expression (right panel) by flow cytometry. Contour plots depict the non‐transduced (GFP−) and the transduced (GFP+) population within the same samples. Numbers indicate the percentage of cells within the respective gates. Note that Bcl‐2 induces a mild reduction of differentiation into kappa+ cells, but provokes an increase in the lambda+ population. In contrast, sequestration of the miR‐15 family interferes with differentiation toward both kappa+ and lambda+ cells, suggesting a different mode of action. Data are representative of three independent experiments.
Source data are available online for this figure.
Hence, we focused only on cell‐cycle regulatory genes, especially on cyclin E1 and cyclin D3, due to their reported key functions in cell‐cycle control. While cyclin E1 is a well‐known regulator of the G1/S phase transition transcriptionally induced downstream of cyclin D/Cdk4‐dependent Rb inhibition, cyclin D3 has been described as a key factor integrating cytokine and pre‐BCR‐dependent signals to expand the pool of pre‐B cells 33, 34, 35.
To investigate whether cyclin D3 and cyclin E1 are indeed direct target genes of the miR‐15 family in pre‐B cells, we performed 3′‐UTR assays in which the genomic region containing the putative binding site was expressed as the 3′‐UTR of a reporter gene. When expressed together with the miR‐15a/miR‐16‐1 or the miR‐15b/miR‐16‐2 clusters, the cyclin E1 UTR reporter showed a clear repression compared to the controls, indicating direct regulation by the miR‐15 family (Fig 5C). The 3′‐UTR of cyclin D3, however, was not repressed significantly by the miRNAs, pointing to an indirect, UTR‐independent mode of regulation. This was confirmed by the reciprocal experiment, in which the 3′‐UTR reporters were coexpressed together with a miR‐15 sponge. Again, miR‐15 family loss of function promoted a derepression of the cyclin E1 UTR reporter, but did affect GFP expression in case of the cyclin D3 3′‐UTR only mildly (Fig 5D). Nevertheless, both genes were clearly targeted by the miR‐15 family in pre‐B cells and this targeting was also confirmed on protein level by Western blot analysis (Fig 5E).
Enforced expression of target genes cyclin E1 and D3 mimics the miR‐15 loss‐of‐function phenotype
Having identified cyclin E1 and cyclin D3 as direct and indirect target genes of the miR‐15 family in pre‐B cells, respectively, we asked whether their aberrant expression would be sufficient to induce a similar phenotype as observed upon sponge‐mediated miR‐15 sequestration. To this end, we transduced pre‐B cells with either the miR‐15 sponge or with constructs encoding cyclin E1 or D3, both expressed from an exogenous promoter and devoid of their 3′‐UTR. Consistent with our hypothesis, enforced expression of cyclin E1 and D3 indeed suppressed pre‐B cell differentiation upon IL‐7 withdrawal (Fig 6A). Surprisingly, this also extended into the transcriptional reprogramming normally induced by growth factor deprivation in pre‐B cells, as exemplified by the impaired induction of Rag1 and Irf4 genes under these conditions (Fig 6B). This points to essential cell‐cycle regulators as the key target genes of the miR‐15 family in early B cell development. Supporting this, co‐expression of the Cdk inhibitor p27Kip1, a potent inducer of cell‐cycle arrest at the G1 phase (Appendix Fig S3) 36, almost completely rescued the miR‐15 sponge‐mediated block in pre‐B cell differentiation (Fig 6C and D). This suggests that the miR‐15 family loss‐of‐function phenotype can be mainly attributed to its pro‐proliferative effect.
Figure 6. Enforced expression of target genes cyclin E1 and cyclin D3 phenocopies the effect induced by miR‐15 inhibition.
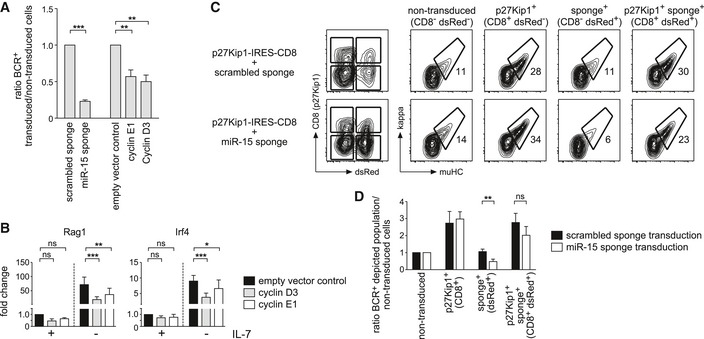
- Wk3 pre‐B cells transduced with expression vectors for the miR‐15 sponge and for cyclin E1 and D3 as well as control vectors were cultured without IL‐7 and analyzed by flow cytometry for BCR expression after 72 h. Bars depict the ratio in the percentage of BCR surface expression comparing transduced to non‐transduced cells within the same sample. Individual values were normalized to their respective controls (scrambled sponge and empty vector control, respectively; set as 1). Individual groups were compared by a one‐way ANOVA followed by Bonferroni testing; ***P < 0.001, **P < 0.01. Data represent mean ± SD of three independent experiments.
- Cells treated as in (A) were analyzed for transcriptional changes of Rag1 and Irf4 genes by quantitative PCR. The bar graphs display the fold changes in gene expression (normalized to the scrambled sponge, +IL‐7 value). Groups were compared by a one‐way ANOVA and Bonferroni testing; ***P < 0.001, **P < 0.01, *P < 0.05. Bars represent means of five independent experiments ± SD.
- Wk3 pre‐B cells expressing the scrambled or the miR‐15 sponge (dsRed as markers), respectively, were transduced with a construct encoding p27Kip1 (CD8 as a marker), expanded in the presence of IL‐7 for 24 h and then cultured without IL‐7 for 72 h before FACS analysis. Based on expression of CD8 and dsRed, cells were categorized as non‐transduced (NT), p27Kip1‐expressing (CD8+), sponge‐expressing (dsRed+) or double‐positive for both p27Kip1 and the sponge (CD8+ dsRed+; left panels). Contour plots show the differentiation of the subpopulations as measured by BCR surface expression for a representative experiment.
- The bar graph depicts the ratio in the percentage of BCR surface expression comparing the respective population to non‐transduced cells within the same sample for experiments as shown in (C). Groups were compared by a paired t‐test; **P < 0.01. Bars represent means of five independent experiments ± SD.
The miR‐15 family is regulated by IL‐7 receptor and pre‐BCR signals
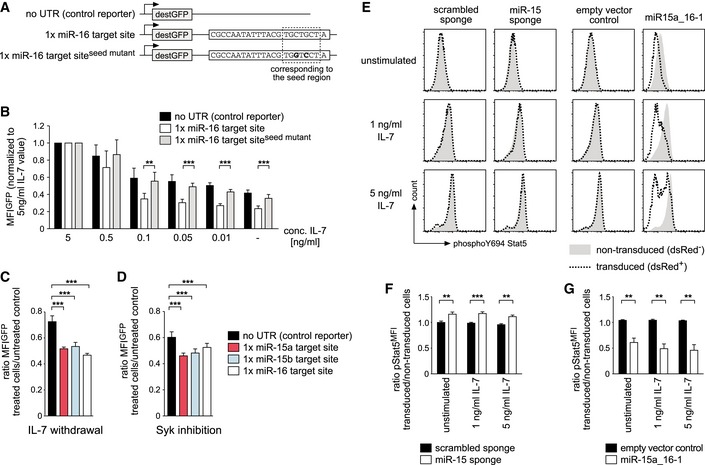
Together, our in vitro and in vivo results, the microarray data as well as the target analysis pointed toward a regulatory role of the miR‐15 family in the switch from proliferation to differentiation. Since this switch is mainly controlled by the interplay of IL‐7 receptor (IL‐7R) and pre‐BCR signaling, we were wondering whether one or both of them directly regulates the activity of the miR‐15 family. To investigate this, we expressed a fluorescent reporter for miR‐16 as a representative of the miR‐15 family, as well as two controls, in pre‐B cells and cultured them in graded concentrations of IL‐7 (Fig 7A and B). While reduced levels of growth factor decreased reporter intensity, most likely by reducing the activity of the promoter driving GFP expression, miR‐16 reporter cells displayed significantly lower GFP levels at IL‐7 concentrations ≤ 0.1 ng/ml. This indicated that miR‐16 activity significantly increases under limiting concentrations of IL‐7. In a corresponding setup, we confirmed that also miR‐15a and miR‐15b are regulated in the same manner (Fig 7C). To analyze whether pre‐BCR signaling also affects miR‐16 activity, we subjected the same set of reporter pre‐B cells to inhibition of Syk (Appendix Fig S4), the major kinase downstream of the pre‐BCR. In analogy to the abovementioned experiment, this resulted in a repression of miR‐15a, miR‐15b, and miR‐16 reporters compared to the control, suggesting that Syk‐mediated pre‐BCR signaling can suppress the activity of the miR‐15 family (Fig 7D). Of note, this increased relative activity of miR‐15 family members in the absence of IL‐7 and pre‐BCR signaling was not accompanied by an increase in miRNA levels as measured by qPCR (Fig EV4A).
Figure 7. The miR‐15 family is integrated into the IL‐7R and pre‐BCR signaling networks.
-
ASchematic illustration of the reporter constructs used for experiments shown in (B and C). The region in the miR‐16 target site that corresponds to the miRNA seed region as well as the nucleotides that are mutated to abolish reporter function are marked.
-
BReduced signaling via the IL‐7 receptor increases miR‐16 activity. Pre‐B cells sorted for expression of the indicated GFP reporters were cultured for 48 h in different concentrations of IL‐7 and analyzed by flow cytometry. Individual bars represent the GFP MFI for each IL‐7 concentration, normalized to the respective reporter cells cultured in 5 ng/ml IL‐7. Groups for the miR‐16 target site and the mutated control reporter were compared by a one‐way ANOVA and Bonferroni testing; ***P < 0.001, **P < 0.01. Data represent means ± SD of at least four independent experiments.
-
CLoss of IL‐7 signaling activates the whole miR‐15 family. Pre‐B cells expressing the reporters as indicated were cultured without IL‐7 for 48 h. Individual bars display the mean ± SD (four independent experiments) of the ratio in GFP MFI comparing cells without IL‐7 and cells under normal IL‐7 culture conditions. Statistical significance was calculated by ANOVA and a Bonferroni test; ***P < 0.001.
-
DPre‐BCR signals suppress the activity of the miR‐15 family. Pre‐B cells with reporters as in (C) were subjected to Syk inhibition for 24 h. Bars compare treated and non‐treated cells expressing the respective reporters and display the mean ratio in GFP MFI ± SD (four independent experiments). Statistical significance was calculated by ANOVA and a Bonferroni test; ***P < 0.001.
-
EMiR‐15 activity regulates signaling downstream of the IL‐7 receptor. Pre‐B cells expressing the miR‐15 sponge or miR‐15a‐16‐1 as indicated were starved without IL‐7 in medium containing 0.5% FCS at 37°C for 1 h, followed by stimulation with the indicated concentrations of IL‐7 for 30 min. Histogram overlays of a representative experiment show the Stat5 Y694 phosphorylation of non‐transduced and transduced cells within the same sample as measured by flow cytometry.
-
F, GAnalysis of four independent experiments comparing the scrambled sponge and the miR‐15 sponge (F) or an empty vector and miR‐15a_16‐1 (G) as shown in (E). Individual bars depict means ± SD of the ratio of pStat5 MFI values comparing transduced and the non‐transduced populations of each sample. Samples were compared by a paired t‐test; ***P < 0.001, **P < 0.01.
Figure EV4. Analysis of miRNA and IL‐7R expression in the absence of IL‐7 and pre‐BCR signaling as well as in a miR‐15 loss‐of‐function/gain‐of‐function setting.
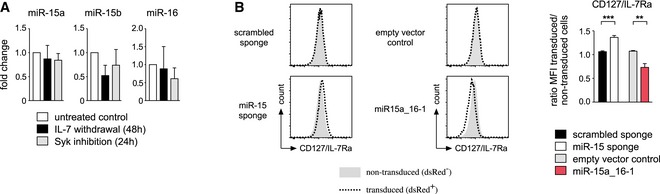
- Increased miRNA activity in differentiating pre‐B cells is not accompanied by increased miRNA expression. Total RNA isolated from wk3 cells cultured without IL‐7 for 48 h, with the Syk inhibitor R406 (2 μM) for 24 h or left untreated was converted into cDNA, and expression of miR‐15a, miR‐15b, miR‐16, and sno‐202 as a reference was determined by quantitative PCR. Bars display the fold change, normalized to the untreated control sample (set as 1). Values are expressed as mean ± SD and represent four independent experiments.
- IL‐7R surface expression is regulated by the activity of the miR‐15 family. Pre‐B cells expressing the miR‐15 sponge or miR‐15a‐16‐1 as indicated were analyzed for surface IL‐7R as measured by CD127/IL‐7Ra expression. Histograms display a representative experiment, comparing non‐transduced (gray graph) and transduced cells (dotted line) within the same sample. The bar graph shows the corresponding means ± SD of four independent experiments, in each case compared by a paired t‐test; ***P < 0.001, **P < 0.01.
The observed control of the miR‐15 family by IL‐7R and pre‐BCR signaling prompted us to investigate whether there is a regulatory loop also in reverse, that is, whether miR‐15 family members also affect how cells react to growth factor receptor signaling. To this end, we stimulated pre‐B cells expressing either the miR‐15 sponge or the miR‐15a‐16‐1 cluster and the respective controls with defined IL‐7 amounts and analyzed the phospho‐Stat5 (pStat5) levels as a readout for signaling downstream of the IL‐7R. Interestingly, cells in which the miR‐15 family members were sequestered by the sponge displayed slightly increased levels of basal pStat5 and retained this also upon stimulation with IL‐7 (Fig 7E, left panel and Fig 7F). Correspondingly, increasing the activity of the miRNAs by overexpression of the miR‐15a‐16‐1 cluster lowered basal pStat5 levels (Fig 7E, right panel and Fig 7G). Moreover, a significant fraction of the cells appeared refractory to stimulation with IL‐7, indicating that miR‐15 activity indeed affects signaling downstream of the IL‐7R. To investigate whether this effect could be contributed to altered receptor expression, we quantified steady‐state IL‐7R levels on pre‐B cells upon functional miR‐15 knockdown or overexpression, respectively. Strikingly, surface CD127/IL‐7R alpha expression in sponge‐expressing cells was increased by about 30% despite comparable RNA expression levels, whereas overexpression of miR‐15a‐16‐1 significantly reduced surface IL‐7R (Fig EV4B and data not shown). Thus, the activity of the miR‐15 family appears to control IL‐7R signaling, at least to some extent by regulating IL‐7R expression, thereby establishing a reinforcing feedback loop (Fig EV5). Together, this indicates that the miR‐15 family is an essential component of regulatory circuits controlled by pre‐BCR and IL‐7R signaling.
Figure EV5. Model for the functional role of the miR‐15 family at the pre‐B‐to‐immature B cell transition.
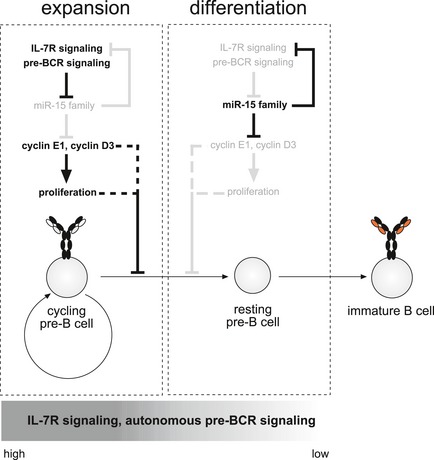
Upon entering the pre‐B cell stage, B cell precursors receive strong signals through the autonomously active pre‐BCR and are highly dependent on IL‐7 receptor signaling, which dampens the activity of the miR‐15 family. This enables strong expression of its direct and indirect targets genes such as cyclin E1 and cyclin D3, thereby promoting cell proliferation and expansion of the pre‐B cell pool. This already appears to be sufficient to counteract transcriptional reprogramming and further maturation, although we cannot exclude that this is a direct, proliferation‐independent effect of cyclin D3 and/or E1 (dotted line). However, once these cells begin to differentiate, a process that is induced by pre‐BCR signaling as well as by decreasing signals through the IL‐7R, this suppression of the miR‐15 family is reverted. Loss of pre‐BCR surface expression by downregulation of pre‐BCR components, reduced IL‐7R expression as well as decreasing IL‐7 concentrations in the microenvironment increase the activity of the miR‐15 family. This is accompanied by the miR‐15‐mediated suppression of IL‐7Ra expression, establishing an enforcing feedback loop. In consequence, the miR‐15 family becomes fully active, now being able to repress its target genes. Transcriptional downregulation of genes such as cyclin E1 and cyclin D3 further supports the transition from expansion to differentiation, ultimately resulting in the transcriptional reprogramming that is a prerequisite for proper B cell development.
Discussion
Although the miR‐15 family has been extensively studied in the context of cancer, its physiological function has been largely neglected. Here, an unbiased screen revealed a novel role for the miR‐15 family in early B cell development, defining it as a critical regulator of the bi‐phasic program that efficiently separates proliferation from potentially hazardous DNA rearrangements during differentiation of pre‐B cells. According to the current model, these processes are mainly established by the signaling networks downstream of the IL‐7R and the pre‐BCR, respectively, which can have both synergistic and opposing effects depending on the cellular state 5. Signals emanating from the IL‐7R primarily contribute to the expansion and survival of pro‐ and pre‐B cells by inducing the expression of cell‐cycle regulators such as cyclin D3 33, 37 and anti‐apoptotic modulators such as Mcl‐1 and Bcl‐2 38, 39, 40. At the same time, however, these signaling cascades also efficiently suppress recombination of the light chain loci by preventing both the expression of the recombination machinery as well as locus accessibility 41, 42. The pre‐BCR, on the other hand, initially provides proliferative signals, thus synergizing with the IL‐7R in enabling the expansion of cells with a successfully recombined mu heavy chain. However, it then induces a response that directly counteracts the IL‐7R signaling‐induced program. In response to transcription factors such as E2A, IRF4, IRF8, and Aiolos, which have been shown to become activated downstream of the pre‐BCR, cells undergo cell‐cycle arrest and open the light chain locus for recombination 27, 29, 37, 43, 44. This is complemented by the direct interference of pre‐BCR signaling with the IL‐7R‐induced PI3K‐AKT pathway, resulting in activation of FOXO transcription factors and subsequent expression of Rag genes as a prerequisite for pre‐B cell differentiation 45, 46.
Here, we show that the loss of miR‐15 function clearly opposes the reprogramming that accompanies normal pre‐B differentiation, that is, a significant number of genes are not appropriately induced, including key transcription factors such as Irf4, Irf8, and Aiolos as well as components of the recombination machinery. While the distinct contribution of these genes to the phenotype is difficult to access, it is nevertheless feasible that their collective flawed expression, even if individual changes appear small, has a profound impact on the cellular response.
We can only speculate about how exactly miR‐15 interferes with the transcriptional reprogramming on the molecular level. Due to enormous the number of genes whose induction is impaired upon IL‐7 withdrawal, it appears likely that the miR‐15 family represses key transcription factors or processes placed upstream of those. In this respect, our detailed analysis for putative target genes revealed that the miR‐15 family mainly represses interphase cell‐cycle regulators in B cell progenitors, such as cyclin D3, cyclin E1, Cdc6, and Cdc25a, which is in accordance with findings in CLL 25, 47, 48. Of those, we confirmed direct targeting of cyclin E1, a classic driver of the G1‐to‐S phase transition, by the miR‐15 family. G1 arrest, normally induced by attenuated IL‐7R signaling and by signals downstream of the pre‐BCR, is a prerequisite for stabilization of Rag2 protein and thus VDJ recombination 7. Increased levels of cyclin E1 in consequence of miR‐15 family knockdown, on the other hand, most likely force cells into S phase independent of the cellular status. Along the same line, we found cyclin D3 to be clearly targeted by the miR‐15 family as well, although in an indirect manner. The inefficient upregulation of Aiolos without miR‐15 family function, which has been shown to suppress cyclin D3 expression in differentiating pre‐B cells, may contribute to this indirect derepression 37. Of note, cyclin D3 has been described not only as a potent promoter of proliferation, but also counteracts the opening of the Ig light chain locus 33, 42. Thus, it is tempting to speculate that aberrant expression of cyclin D3 targets both processes in the absence of the miR‐15 family, thereby efficiently suppressing pre‐B cell differentiation.
In pre‐B cells, the proliferative effect of cyclins E1 and D3 is most likely aggravated by the impact of the miR‐15 family on IL‐7R signaling. Our data clearly indicate that miR‐15 loss‐of‐function cells retain higher levels of Stat5 phosphorylation after starvation and upon re‐stimulation. It is therefore feasible that the absence of miR‐15 activity promotes cell‐cycle progression also via increased signaling downstream of the IL‐7R. How miR‐15 determines the cellular response to the growth factor is unclear, but a contributing factor may be the impact of miR‐15 family members on IL‐7R expression.
Of note, the miR‐15 family not only regulates the cellular behavior to growth factors, but is also itself controlled by IL‐7R signals, thereby establishing a reinforcing feedback loop. This is in line with a previous report that demonstrated a repression of miR‐15 on the transcriptional level by Stat5 activity 49. Surprisingly, in our experiments, the loss of IL‐7R signaling clearly activated miR‐15a, miR‐15b, and miR‐16; however, this increase in activity was not accompanied by altered miRNA levels. While this may appear counterintuitive, studies have demonstrated that the relative activity of a particular miRNA is not only determined by its own expression level, but also by the ratio of the miRNA to its target genes. 50. Thus, it is possible that miR‐15 family activity may increase upon IL‐7 withdrawal due to a global reduction of its targets or by loss/downregulation of a potent competing endogenous RNA, thereby increasing the miRNA:target ratio.
Beyond cell‐cycle regulation, several publications have identified Bcl2 as a direct target of miR‐15 family members in human CLL and other cancer entities 32, 51, raising the question whether reduced apoptosis may contribute to the phenotype we have reported here. However, we have not found any evidence for a regulation of Bcl2 by miR‐15 by qPCR or on protein level, which is in line with other reports that have failed to show any increased expression of Bcl‐2 upon loss or reduction of miR‐15 family members 48, 52. Moreover, we found no evidence for a direct anti‐apoptotic effect upon miR‐15 sequestration, but report enhanced survival only in the context of growth factor withdrawal. This further supports our hypothesis that miR‐15 sequestration mainly affects cell‐cycle regulation and growth factor signaling to interfere with the pre‐B‐to‐immature B cell transition.
Consistent with our findings, mice deficient for miR‐15a/miR‐16‐1 or miR‐15b/miR‐16‐2 cluster expression develop a lymphoproliferative disease that closely recapitulates the phenotype associated with human CLL, including a prominent expansion of CD5+ B cells 25, 53. A detailed analysis of an NK‐specific miR‐15a/miR‐16‐1 deletion revealed a perturbation of terminal NK cell maturation, resulting in an accumulation of immature NK precursors in the periphery 54. Along the same line, the application of a miR‐15 inhibitor in the mouse forebrain resulted in increased neural progenitor cell proliferation and defective neuronal differentiation 55. Thus, the emerging theme is that miR‐15 family members control the proliferation of a diverse set of tissues and, possibly in an indirect manner, also affect cellular differentiation. Clearly, this raises the question why no other obvious alteration beyond the CLL‐like phenotype and NK cell maturation have been reported in mice deficient for miR‐15a/miR‐16‐1 or miR‐15b/miR‐16‐2. It is tempting to speculate that the miR‐15 family can compensate for the loss of one or two members in most tissues under homeostatic conditions, thereby masking a potential phenotype. With respect to early B cells, miRNA transcriptome analyses revealed that at least the miR‐15a/miR‐16‐1 as well as the miR‐15b/miR‐16‐2 clusters are equally well expressed 22, 56. This may explain why, for example, B cell development is not affected by loss of either the miR‐15a/miR‐16‐1 or the miR‐15b/16‐2 clusters alone, but why our sponge‐approach was able to reveal this putative redundancy. Other tissues, however, such as NK cells or the cells involved in CLL pathogenesis, may be exceptionally dependent on a high level of overall miR‐15 family activity, thereby already provoking a phenotype upon deletion of one cluster.
In summary, our findings suggest a bi‐phasic model in which the miR‐15 family is regulated by both pre‐BCR and IL‐7R signaling throughout early B cell development (Fig EV5). Upon entering the pre‐B stage, cells are mainly controlled by the IL‐7R and by proliferative, Syk‐mediated signals from the pre‐BCR 57, 58. These signals keep the miR‐15 family in check, resulting in derepression of its target genes such as cyclin E1 and cyclin D3. In consequence, pre‐B cells are enabled to undergo several rounds of cell divisions, which appears to have a suppressive effect on differentiation on its own. However, this proliferative phase is limited by the signals emanating simultaneously from the pre‐BCR, mediated by tumor suppressors SLP‐65/BLNK, Btk, and PLC‐γ2 3. Signaling through this module initiates a regulatory program that not only induces a set of transcription factors which synergize to promote cell‐cycle arrest, light chain locus accessibility, and transcription, but also attenuates IL‐7R signaling 27, 29, 37, 43, 59, 60. This is mainly accomplished by pre‐BCR‐mediated activation of FOXO1, which directly lowers expression of the IL‐7R and thereby reduces its signaling capacity 61. In consequence, the miR‐15 family becomes active, which feeds back onto and further lowers signaling downstream of the IL‐7R. This promotes full miR‐15 family function, resulting in the efficient repression of cell cycle regulating target genes and thus consolidating the switch from a cycling to a resting stage as a prerequisite for efficient differentiation.
Materials and Methods
Cells, cell culture, and retroviral transduction
The pre‐B cell lines wk3, 1587, and 1677 were derived by extended culture of total bone marrow of SLP‐65−/− and SLP‐65−/−LAT−/− mice, respectively, in IL‐7‐supplemented medium. The pre‐B cell lines 1676 and 74 have already been described 23, 62. Pre‐B cells were cultured in IMDM (Sigma) containing 7.5% FCS (Biochrom Superior), 100 U/ml penicillin, 100 U/ml streptomycin (PAN), and 50 μM 2‐ME. If not stated otherwise, pre‐B cell medium was supplemented with IL‐7, either using the supernatant of IL‐7‐expressing J558L cells in excess or defined amounts of recombinant IL‐7 (PeproTech).
For the generation of retroviruses, Plat‐E packaging cells 63 were transfected with DNA plasmids using PEI (polyethyleneimine, Polysciences) at a 4:1 PEI:DNA ratio. Supernatants were harvested after 36 and 60 h, mixed with polybrene (8 μg/ml final concentration), and used for spin‐infection of pre‐B cells in a tabletop centrifuge (400 g at 30°C for 3 × 30 min).
Puromycin selection at a concentration of 1 μg/ml was done for 3–4 days or until the culture fully recovered. In experimental settings, cells were always cultured in the absence of selection agent. Syk was inhibited using R406 (Selleckchem) at a concentration of 2 μM.
Plasmid constructs
For the miRNA sponge library, DNA fragments containing about 15–30 repeats of miRNA‐binding sites with a central bulged mismatch region at position 9–12 were generated by concatemeric PCR. In short, overlapping forward and reverse PCR primers were designed based on the mature miRNA sequence, for example, CAAGACCTGTgatTCTACCTTCCGT and atcACAGGTCTTGACGGAAGGTAGA (lower cases mark the mismatch region, the linker between the binding sites in underlined) for miR‐1839 (AAGGTAGATAGAACAGGTCTTG). Using this pair of oligonucleotides, a first round of PCR was performed with an annealing temperature of 36°C and 35 cycles. Five μl of this PCR was used as a template for a second round of PCR with the same conditions but only 10–15 cycles. PCR products were separated on an agarose gel, and DNA fragments of the appropriate size (about 500 bp; corresponding to about 20 repeats of the miRNA‐binding sites) were isolated and blunt‐end cloned into pJET1.2. After sequence verification, sponge fragments were cloned by standard ligation 3′ of a dsRedExpress2 or tailless CD8 cDNA into a modified LMP vector in which the miR‐30‐based shRNA cassette had been deleted. A detailed protocol is available upon request. For hsa‐miR‐15a/16‐1 and miR‐15b/16‐2 overexpression, sequences amplified from human genomic DNA were cloned via XhoI and EcoRI digest into LMP‐dsRedExpress2 vector in exchange for the miR‐30‐based shRNA cassette. Expression constructs for human cyclin D3 and cyclin E1 were generated by cloning the respective cDNAs out of a pDONR‐based cDNA library into MSCV‐based vectors such as pMIG. For the 3′‐UTR assay, UTRs of interest were cloned by PCR and ligated into LMP‐destGFP 62 3′ of the destabilized GFP cDNA via XhoI and ClaI. Likewise, miRNA sensors were generated by ligation of annealed primers encoding the respective binding sites into LMP‐destGFP. All oligonucleotide sequences are available in the Appendix Table S4.
Flow cytometry
Single‐cell suspensions were stained using anti‐IgM (eB121‐15F9; eBioscience and II/41, BD Pharmingen), anti‐kappa light chain (RMK‐12; Biolegend), anti‐CD8a (53‐6.7; Biolegend), anti‐IL7Ra/CD127 (A7R34; Biolegend), and streptavidin‐APC (Biolegend). Apoptotic cells were quantified by exclusion of dead cells (positive for 7‐AAD, PI or DAPI) combined with either binding of fluorescence‐labeled Annexin V (eBioscience) or by staining of condensed DNA with 1 μg/ml Hoechst 33342 for 8 min at 37°C 64. For cell proliferation assays, pre‐B cells were pulsed with 10 μM EdU for 45 min before antibody staining, fixation, permeabilization and labeling of incorporated nucleoside analogs using the EdU Flow Cytometry Cell Proliferation Assay (Life Technologies). For intracellular pStat5 analysis, cells were fixed in 1.5% methanol‐free formaldehyde for 20 min at room temperature and permeabilized in cold 90% methanol at −20°C over night. Single‐cell suspensions were stained using anti‐pY694 Stat5 (#D47E7; Cell Signaling Technologies) and a rabbit IgG isotype control (DA1E; Cell Signaling Technologies) followed by Alexa Fluor 647‐labeled anti‐rabbit IgG (Cell Signaling Technologies). Data were acquired on an LSR Fortessa (Becton Dickinson) or CyAn ADP Analyzer (Beckman Coulter) and analyzed with FlowJo software (Tree Star).
Microarray analysis
Wk3 pre‐B cells were selected for expression of the scrambled sponge or the sponge targeting the miR‐15 family (based on the mature miR‐16 sequence), respectively. Total RNA was isolated from cells cultured with IL‐7 and after IL‐7 withdrawal for 36 h using the RNeasy system (Qiagen). RNA processing, microarray hybridization (Agilent G3 Mouse 8 × 60K array) and data quality control were performed by IMGM (Martinsried, Germany). Microarray data have been deposited under GEO accession number GSE72767. Data were analyzed using the Subio software platform. In short, data files for two biological replicates, each comprising four different samples (wk3 scrambled sponge cultured with or without IL‐7 for 36 h, wk3 miR‐15 family sponge cultured with or without IL‐7 for 36 h), were quantile normalized, log2‐transformed, normalized to the control sample of each experiment (wk3 scrambled sponge cells cultured +IL‐7) and averaged. As a quality control, measurements that did not exceed background levels in all eight samples were discarded. Moreover, measurements whose Ch1 raw signal did not exceed a mean value of 100 in at least two out of the four sample groups were filtered out. For the identification of differentially expressed genes (DEGs), pairs of sample groups were filtered for annotated genes whose expression changed by at least twofold (P‐value < 0.05). For the tree, measurement IDs corresponding to annotated DEGs in wk3 scrambled sponge cells cultured with versus without IL‐7 (representing the normal transcriptional changes upon IL‐7 withdrawal) were clustered according to Euclidean distance. For the miRNA target identification, pairs of sample groups (miR‐15 sponge versus scrambled sponge; with or without IL‐7) were compared with each other and annotated genes upregulated by at least 1.25‐fold (P‐value < 0.05) were filtered out. These lists of unique genes were then separately filtered against lists of computationally predicted miR‐15a, miR‐15b, and miR‐16 targets based on the PicTar (401 genes with conserved miRNA‐binding sites in total) and the miRanda (dataset from 2010, “good mirSVR score; conserved miRNA”, 4,480 unique genes in total). For gene ontology analysis, a list of 109 core genes predicted to be targeted by miR‐15a, miR‐15b, and/or miR‐16 was fed into the PANTHER classification pipeline 65.
Quantitative PCR analysis
For coding and miRNA genes, total RNA isolated either using the RNeasy system (Qiagen) or Trizol (Life Technologies) was reverse transcribed with random hexamer or specific primers, respectively, followed by SYBR green‐based quantitative PCR. Analysis was performed based on the ΔΔC t method. Oligonucleotide sequences are available in the Appendix Table S4.
Western blot analysis
For Western blot analysis, about 1–2 × 106 wk3 pre‐B cells were lysed in ice‐cold RIPA buffer (50 mM Tris–HCl, pH 7.4, 1% NP‐40, 0.25% sodium deoxycholate, 150 mM NaCl, 1 mM EDTA (pH 8), protease inhibitor cocktail; Sigma), mixed with reducing sample buffer, boiled for 10 min, and subjected to SDS–PAGE and Western blotting onto PVDF membranes. Proteins were detected using anti‐cyclin E1 (MBS222338, MyBioSource), anti‐cyclin D3 (1/cyclin D3; BD Biosciences), anti‐Bcl2 (7/Bcl‐2; BD Biosciences), anti‐pY (4G10; Millipore), anti‐Akt/PKB (C67E7; Cell Signaling Technologies), and anti‐actin antibodies (13E5; Cell Signaling Technologies). Immunoreactive proteins were visualized with HRP‐labeled secondary antibodies and the ECL system (Advansta) on light‐sensitive film (Amersham, GE).
Statistical analyses
Depending on the experimental setup, paired or unpaired two‐tailed Student's t‐tests or one‐way ANOVA (repeated measures whenever applicable) followed by Bonferroni testing (95% confidence interval) were applied for statistical analyses using the Prism 5 software (GraphPad). P‐values < 0.05 were considered statistically significant and labeled according to the following scheme: ***P < 0.001, **P < 0.01, *P < 0.05, ns = not significant. Values in figures represent mean + standard deviation.
Adoptive transfer experiments
Animal experiments were performed in accordance with Austrian legislation (BMWF: 66‐011/0006‐II/3b/2014). Mice were housed in individualized ventilated cages with a combination of hardwood bedding and nesting material and were maintained on a 12:12 h light:dark cycle. For the adoptive transfer, a mixture of 9 × 106 scrambled sponge‐expressing (dsRed+), miR‐15 sponge‐expressing (GFP+), and non‐transduced wk3 cells were injected into the tail vain of five female C57BL/6 Rag1 −/− mice (6–10 weeks old). Due to this dual fluorescence strategy, no randomization method was used and the investigators were not blinded to the group allocation. Note that the sponge used in this experiment combined binding site repeats corresponding to miR‐16 and miR‐15a to induce maximal inhibition of the miR‐15 family (Appendix Table S2). Mice were sacrificed and analyzed by flow cytometry after 12 days.
Author contributions
SH conceived the concept of this work, designed and performed experiments, and analyzed data. SEL, ML, and BK performed experiments and analyzed data. FS assisted with in vivo experiments. ZK assisted with experiments. SEL, AV, and SH interpreted and discussed results. AV and SH wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 5
Acknowledgements
This work was funded by the Deutsche‐Forschungsgemeinschaft through Grant EXC294 (the Centre for Biological Signalling Studies, BIOSS), the Tiroler Zukunftsstiftung, and the elite postdoctoral program of the Baden‐Württemberg foundation to SH and the Austrian Science Fund (FWF; P23510‐B19; I1298) to AV. We thank Günther Böck for cell sorting and Katharina Rossi for animal care.
EMBO Reports (2017) 18: 1604–1617
References
- 1. Schlissel MS (2003) Regulating antigen‐receptor gene assembly. Nat Rev Immunol 3: 890–899 [DOI] [PubMed] [Google Scholar]
- 2. Nishimoto N, Kubagawa H, Ohno T, Gartland GL, Stankovic AK, Cooper MD (1991) Normal pre‐B cells express a receptor complex of mu heavy chains and surrogate light‐chain proteins. Proc Natl Acad Sci USA 88: 6284–6288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Herzog S, Reth M, Jumaa H (2009) Regulation of B‐cell proliferation and differentiation by pre‐B‐cell receptor signalling. Nat Rev Immunol 9: 195–205 [DOI] [PubMed] [Google Scholar]
- 4. Torres RM, Flaswinkel H, Reth M, Rajewsky K (1996) Aberrant B cell development and immune response in mice with a compromised BCR complex. Science 272: 1804–1808 [DOI] [PubMed] [Google Scholar]
- 5. Clark MR, Mandal M, Ochiai K, Singh H (2013) Orchestrating B cell lymphopoiesis through interplay of IL‐7 receptor and pre‐B cell receptor signalling. Nat Rev Immunol 14: 69–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Küppers R, Dalla‐Favera R (2001) Mechanisms of chromosomal translocations in B cell lymphomas. Oncogene 20: 5580–5594 [DOI] [PubMed] [Google Scholar]
- 7. Zhang L, Reynolds TL, Shan X, Desiderio S (2011) Coupling of V(D)J recombination to the cell cycle suppresses genomic instability and lymphoid tumorigenesis. Immunity 34: 163–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mijušković M, Chou Y‐F, Gigi V, Lindsay CR, Shestova O, Lewis SM, Roth DB (2015) Off‐target V(D)J recombination drives lymphomagenesis and is escalated by loss of the Rag2 C terminus. Cell Rep 12: 1842–1852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Papaemmanuil E, Rapado I, Li Y, Potter NE, Wedge DC, Tubio J, Alexandrov LB, Van Loo P, Cooke SL, Marshall J et al (2014) RAG‐mediated recombination is the predominant driver of oncogenic rearrangement in ETV6‐RUNX1 acute lymphoblastic leukemia. Nat Genet 46: 116–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Friedman RC, Farh KKH, Burge CB, Bartel DP (2008) Most mammalian mRNAs are conserved targets of microRNAs. Genome Res 19: 92–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Xiao C, Rajewsky K (2009) MicroRNA control in the immune system: basic principles. Cell 136: 26–36 [DOI] [PubMed] [Google Scholar]
- 12. Ventura A, Young AG, Winslow MM, Lintault L, Meissner A, Erkeland SJ, Newman J, Bronson RT, Crowley D, Stone JR et al (2008) Targeted deletion reveals essential and overlapping functions of the miR‐17∼92 family of miRNA clusters. Cell 132: 875–886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhou B, Wang S, Mayr C, Bartel DP, Lodish HF (2007) miR‐150, a microRNA expressed in mature B and T cells, blocks early B cell development when expressed prematurely. Proc Natl Acad Sci USA 104: 7080–7085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Xiao C, Calado DP, Galler G, Thai T‐H, Patterson HC, Wang J, Rajewsky N, Bender TP, Rajewsky K (2007) MiR‐150 controls B cell differentiation by targeting the transcription factor c‐Myb. Cell 131: 146–159 [DOI] [PubMed] [Google Scholar]
- 15. Rao DS, O'Connell RM, Chaudhuri AA, Garcia‐Flores Y, Geiger TL, Baltimore D (2010) MicroRNA‐34a perturbs B lymphocyte development by repressing the forkhead box transcription factor foxp1. Immunity 33: 48–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Thai T‐H, Calado DP, Casola S, Ansel KM, Xiao C, Xue Y, Murphy A, Frendewey D, Valenzuela D, Kutok JL et al (2007) Regulation of the germinal center response by microRNA‐155. Science 316: 604–608 [DOI] [PubMed] [Google Scholar]
- 17. Rodriguez A, Vigorito E, Clare S, Warren MV, Couttet P, Soond DR, van Dongen S, Grocock RJ, Das PP, Miska EA et al (2007) Requirement of bic/microRNA‐155 for normal immune function. Science 316: 608–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Vigorito E, Perks KL, Abreu‐Goodger C, Bunting S, Xiang Z, Kohlhaas S, Das PP, Miska EA, Rodriguez A, Bradley A et al (2007) microRNA‐155 regulates the generation of immunoglobulin class‐switched plasma cells. Immunity 27: 847–859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Teng G, Hakimpour P, Landgraf P, Rice A, Tuschl T, Casellas R, Papavasiliou FN (2008) MicroRNA‐155 is a negative regulator of activation‐induced cytidine deaminase. Immunity 28: 621–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. de Yébenes VG, Belver L, Pisano DG, González S, Villasante A, Croce C, He L, Ramiro AR (2008) miR‐181b negatively regulates activation‐induced cytidine deaminase in B cells. J Exp Med 205: 2199–2206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ebert MS, Neilson JR, Sharp PA (2007) MicroRNA sponges: competitive inhibitors of small RNAs in mammalian cells. Nat Methods 4: 721–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Spierings DC, McGoldrick D, Hamilton‐Easton AM, Neale G, Murchison EP, Hannon GJ, Green DR, Withoff S (2011) Ordered progression of stage‐specific miRNA profiles in the mouse B2 B‐cell lineage. Blood 117: 5340–5349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Flemming A, Brummer T, Reth M, Jumaa H (2003) The adaptor protein SLP‐65 acts as a tumor suppressor that limits pre‐B cell expansion. Nat Immunol 4: 38–43 [DOI] [PubMed] [Google Scholar]
- 24. Calin GA (2002) Frequent deletions and down‐regulation of micro‐ RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci USA 99: 15524–15529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Klein U, Lia M, Crespo M, Siegel R, Shen Q, Mo T, Ambesi‐Impiombato A, Califano A, Migliazza A, Bhagat G et al (2010) The DLEU2/miR‐15a/16‐1 cluster controls B cell proliferation and its deletion leads to chronic lymphocytic leukemia. Cancer Cell 17: 28–40 [DOI] [PubMed] [Google Scholar]
- 26. Grimson A, Farh KK‐H, Johnston WK, Garrett‐Engele P, Lim LP, Bartel DP (2007) MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell 27: 91–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lu R, Medina KL, Lancki DW, Singh H (2003) IRF‐4,8 orchestrate the pre‐B‐to‐B transition in lymphocyte development. Genes Dev 17: 1703–1708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ma S, Turetsky A, Trinh L, Lu R (2006) IFN regulatory factor 4 and 8 promote Ig light chain locus activation in pre‐B cell development. J Immunol 177: 7898–7904 [DOI] [PubMed] [Google Scholar]
- 29. Ma S, Pathak S, Trinh L, Lu R (2008) Interferon regulatory factors 4 and 8 induce the expression of Ikaros and Aiolos to down‐regulate pre‐B‐cell receptor and promote cell‐cycle withdrawal in pre‐B‐cell development. Blood 111: 1396–1403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Krek A, Grün D, Poy MN, Wolf R, Rosenberg L, Epstein EJ, MacMenamin P, da Piedade I, Gunsalus KC, Stoffel M et al (2005) Combinatorial microRNA target predictions. Nat Genet 37: 495–500 [DOI] [PubMed] [Google Scholar]
- 31. Betel D, Wilson M, Gabow A, Marks DS, Sander C (2008) The microRNA.org resource: targets and expression. Nucleic Acids Res 36: D149–D153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cimmino A, Calin GA, Fabbri M, Iorio MV, Ferracin M, Shimizu M, Wojcik SE, Aqeilan RI, Zupo S, Dono M et al (2005) miR‐15 and miR‐16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci USA 102: 13944–13949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cooper AB, Sawai CM, Sicinska E, Powers SE, Sicinski P, Clark MR, Aifantis I (2006) A unique function for cyclin D3 in early B cell development. Nat Immunol 7: 489–497 [DOI] [PubMed] [Google Scholar]
- 34. Hwang HC, Clurman BE (2005) Cyclin E in normal and neoplastic cell cycles. Oncogene 24: 2776–2786 [DOI] [PubMed] [Google Scholar]
- 35. Kato J, Matsushime H, Hiebert SW, Ewen ME, Sherr CJ (1993) Direct binding of cyclin D to the retinoblastoma gene product (pRb) and pRb phosphorylation by the cyclin D‐dependent kinase CDK4. Genes Dev 7: 331–342 [DOI] [PubMed] [Google Scholar]
- 36. Toyoshima H, Hunter T (1994) p27, a novel inhibitor of G1 cyclin‐Cdk protein kinase activity, is related to p21. Cell 78: 67–74 [DOI] [PubMed] [Google Scholar]
- 37. Mandal M, Powers SE, Ochiai K, Georgopoulos K, Kee BL, Singh H, Clark MR (2009) Ras orchestrates exit from the cell cycle and light‐chain recombination during early B cell development. Nat Immunol 10: 1110–1117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Malin S, McManus S, Cobaleda C, Novatchkova M, Delogu A, Bouillet P, Strasser A, Busslinger M (2010) Role of STAT5 in controlling cell survival and immunoglobulin gene recombination during pro‐B cell development. Nat Immunol 11: 171–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jiang Q, Li WQ, Hofmeister RR, Young HA, Hodge DR, Keller JR, Khaled AR, Durum SK (2004) Distinct regions of the interleukin‐7 receptor regulate different Bcl2 family members. Mol Cell Biol 24: 6501–6513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bednarski JJ, Nickless A, Bhattacharya D, Amin RH, Schlissel MS, Sleckman BP (2012) RAG‐induced DNA double‐strand breaks signal through Pim2 to promote pre‐B cell survival and limit proliferation. J Exp Med 209: 11–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mandal M, Powers SE, Maienschein‐Cline M, Bartom ET, Hamel KM, Kee BL, Dinner AR, Clark MR (2011) Epigenetic repression of the Igk locus by STAT5‐mediated recruitment of the histone methyltransferase Ezh2. Nat Immunol 12: 1212–1220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Powers SE, Mandal M, Matsuda S, Miletic AV, Cato MH, Tanaka A, Rickert RC, Koyasu S, Clark MR (2012) Subnuclear cyclin D3 compartments and the coordinated regulation of proliferation and immunoglobulin variable gene repression. J Exp Med 209: 2199–2213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lazorchak AS, Schlissel MS, Zhuang Y (2006) E2A and IRF‐4/Pip promote chromatin modification and transcription of the immunoglobulin kappa locus in pre‐B cells. Mol Cell Biol 26: 810–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Stadhouders R, de Bruijn MJW, Rother MB, Yuvaraj S, Ribeiro de Almeida C, Kolovos P, Van Zelm MC, van Ijcken W, Grosveld F, Soler E et al (2014) Pre‐B cell receptor signaling induces immunoglobulin κ locus accessibility by functional redistribution of enhancer‐mediated chromatin interactions. PLoS Biol 12: e1001791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Herzog S, Hug E, Meixlsperger S, Paik J‐H, DePinho RA, Reth M, Jumaa H (2008) SLP‐65 regulates immunoglobulin light chain gene recombination through the PI(3)K‐PKB‐Foxo pathway. Nat Immunol 9: 623–631 [DOI] [PubMed] [Google Scholar]
- 46. Amin RH, Schlissel MS (2008) Foxo1 directly regulates the transcription of recombination‐activating genes during B cell development. Nat Immunol 9: 613–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Liu Q, Fu H, Sun F, Zhang H, Tie Y, Zhu J, Xing R, Sun Z, Zheng X (2008) miR‐16 family induces cell cycle arrest by regulating multiple cell cycle genes. Nucleic Acids Res 36: 5391–5404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Linsley PS, Schelter J, Burchard J, Kibukawa M, Martin MM, Bartz SR, Johnson JM, Cummins JM, Raymond CK, Dai H et al (2007) Transcripts targeted by the microRNA‐16 family cooperatively regulate cell cycle progression. Mol Cell Biol 27: 2240–2252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Li G, Miskimen KL, Wang Z, Xie XY, Brenzovich J, Ryan JJ, Tse W, Moriggl R, Bunting KD (2010) STAT5 requires the N‐domain for suppression of miR15/16, induction of bcl‐2, and survival signaling in myeloproliferative disease. Blood 115: 1416–1424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mukherji S, Ebert MS, Zheng GXY, Tsang JS, Sharp PA, van Oudenaarden A (2011) MicroRNAs can generate thresholds in target gene expression. Nat Genet 43: 854–859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Xia L, Zhang D, Du R, Pan Y, Zhao L, Sun S, Hong L, Liu J, Fan D (2008) miR‐15b and miR‐16 modulate multidrug resistance by targeting BCL2 in human gastric cancer cells. Int J Cancer 123: 372–379 [DOI] [PubMed] [Google Scholar]
- 52. Fulci V, Chiaretti S, Goldoni M, Azzalin G, Carucci N, Tavolaro S, Castellano L, Magrelli A, Citarella F, Messina M et al (2007) Quantitative technologies establish a novel microRNA profile of chronic lymphocytic leukemia. Blood 109: 4944–4951 [DOI] [PubMed] [Google Scholar]
- 53. Lovat F, Fassan M, Gasparini P, Rizzotto L, Cascione L, Pizzi M, Vicentini C, Balatti V, Palmieri D, Costinean S et al (2015) miR‐15b/16‐2 deletion promotes B‐cell malignancies. Proc Natl Acad Sci USA 112: 11636–11641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sullivan RP, Leong JW, Schneider SE, Ireland AR, Berrien‐Elliott MM, Singh A, Schappe T, Jewell BA, Sexl V, Fehniger TA (2015) MicroRNA‐15/16 antagonizes Myb To control NK cell maturation. J Immunol 195: 2806–2817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lv X, Jiang H, Liu Y, Lei X, Jiao J (2014) MicroRNA‐15b promotes neurogenesis and inhibits neural progenitor proliferation by directly repressing TET3 during early neocortical development. EMBO Rep 15: 1305–1314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kuchen S, Resch W, Yamane A, Kuo N, Li Z, Chakraborty T, Wei L, Laurence A, Yasuda T, Peng S et al (2010) Regulation of MicroRNA expression and abundance during lymphopoiesis. Immunity 32: 828–839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Fleming HE, Paige CJ (2002) Cooperation between IL‐7 and the pre‐B cell receptor: a key to B cell selection. Semin Immunol 14: 423–430 [DOI] [PubMed] [Google Scholar]
- 58. Marshall AJ, Fleming HE, Wu GE, Paige CJ (1998) Modulation of the IL‐7 dose‐response threshold during pro‐B cell differentiation is dependent on pre‐B cell receptor expression. J Immunol 161: 6038–6045 [PubMed] [Google Scholar]
- 59. Lin YC, Jhunjhunwala S, Benner C, Heinz S, Welinder E, Mansson R, Sigvardsson M, Hagman J, Espinoza CA, Dutkowski J et al (2010) A global network of transcription factors, involving E2A, EBF1 and Foxo1, that orchestrates B cell fate. Nat Immunol 11: 635–643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Heizmann B, Kastner P, Chan S (2013) Ikaros is absolutely required for pre‐B cell differentiation by attenuating IL‐7 signals. J Exp Med 210: 2823–2832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ochiai K, Maienschein‐Cline M, Mandal M, Triggs JR, Bertolino E, Sciammas R, Dinner AR, Clark MR, Singh H (2012) A self‐reinforcing regulatory network triggered by limiting IL‐7 activates pre‐BCR signaling and differentiation. Nat Immunol 13: 300–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Knackmuss U, Lindner SE, Aneichyk T, Kotkamp B, Knust Z, Villunger A, Herzog S (2015) MAP3K11 is a tumor suppressor targeted by the oncomiR miR‐125b in early B cells. Cell Death Differ 23: 242–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Morita S, Kojima T, Kitamura T (2000) Plat‐E: an efficient and stable system for transient packaging of retroviruses. Gene Ther 7: 1063–1066 [DOI] [PubMed] [Google Scholar]
- 64. Belloc F, Dumain P, Boisseau MR, Jalloustre C, Reiffers J, Bernard P, Lacombe F (1994) A flow cytometric method using Hoechst 33342 and propidium iodide for simultaneous cell cycle analysis and apoptosis determination in unfixed cells. Cytometry 17: 59–65 [DOI] [PubMed] [Google Scholar]
- 65. Mi H, Muruganujan A, Casagrande JT, Thomas PD (2013) Large‐scale gene function analysis with the PANTHER classification system. Nat Protoc 8: 1551–1566 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 5