

Abstract
The RNA-dependent protein kinase (PKR) is an interferon-induced, RNA-activated enzyme that phosphorylates the α-subunit of eukaryotic initiation factor 2 (eIF2α), inhibiting the function of the eIF2 complex and continued initiation of translation. When bound to an activating RNA and ATP, PKR undergoes autophosphorylation reactions at multiple serine and threonine residues. This autophosphorylation reaction stimulates the eIF2α kinase activity of PKR. The binding of certain viral RNAs inhibits the activation of PKR. Wild-type PKR is obtained as a highly phosphorylated protein when overexpressed in Escherichia coli. We report here that treatment of the isolated phosphoprotein with the catalytic subunit of protein phosphatase 1 dephosphorylates the enzyme. The in vitro autophosphorylation and eIF2α kinase activities of the dephosphorylated enzyme are stimulated by addition of RNA. Thus, inactivation by phosphatase treatment of autophosphorylated PKR obtained from overexpression in bacteria generates PKR in a form suitable for in vitro analysis of the RNA-induced activation mechanism. Furthermore, we used gel mobility shift assays, methidiumpropyl-EDTA·Fe footprinting and affinity chromatography to demonstrate differences in the RNA-binding properties of phospho- and dephosphoPKR. We found that dephosphorylation of PKR increases binding affinity of the enzyme for both kinase activating and inhibiting RNAs. These results are consistent with an activation mechanism that includes release of the activating RNA upon autophosphorylation of PKR prior to phosphorylation of eIF2α.
INTRODUCTION
The RNA-dependent protein kinase (PKR) is a component of the interferon signaling system, a collection of pathways that lead to growth inhibition in a number of different cell lines in response to viral infection (1). In vitro PKR is activated by binding to RNA molecules with extensive duplex secondary structure (2). In vivo the enzyme is believed to be activated by viral double-stranded RNA (dsRNA) or viral replicative intermediates comprising dsRNA (3). Activated PKR can phosphorylate several protein substrates, including the α-subunit of the heterotrimeric eukaryotic translation initiation factor 2 (eIF2α) (4–6). Phosphorylation of eIF2α has the effect of inhibiting continued initiation of protein synthesis by the eIF2 complex (7). Viruses that infect eukaryotic cells have evolved mechanisms by which they circumvent the activity of this antiviral kinase (8,9). Some, such as adenovirus and Epstein–Barr virus, synthesize highly structured RNAs that bind PKR and block activation (VA and EBER RNAs, respectively) (10,11). However, once activated by dsRNA, the eIF2α kinase activity of PKR is not inhibited by these viral RNAs (3). Certain eukaryotic mRNAs have also been shown to activate PKR and evidence for roles for PKR in cell growth control and apoptosis has been presented (12–14).
Human PKR is 68 kDa with an ∼20 kDa N-terminal dsRNA-binding domain (dsRBD) and a C-terminal protein kinase domain (Fig. 1; 15). The dsRBD is composed of two copies of the dsRNA-binding motif (dsRBM I and dsRBM II), a sequence motif found in many dsRNA-binding proteins (16). These proteins bind dsRNA in a largely sequence-independent fashion and are involved in a myriad of biological processes, such as RNA editing, RNA trafficking, RNA processing and transcriptional and translational regulation (17–20). Binding of RNA to the PKR dsRBD causes a conformational change in the enzyme that alters the ATP-binding site in the kinase domain and leads to autophosphorylation at multiple serine and threonine residues throughout the PKR sequence (Fig. 1; 4,21–24). Once autophosphorylated, PKR can phosphorylate its protein substrates, such as eIF2α (4). PKR activation by autophosphorylation is a reversible process, as dephosphorylation of activated PKR with a type 1 protein phosphatase has been shown to inactivate the kinase and this inactivation can be reversed by RNA-stimulated autophosphorylation in vitro (25). However, once autophosphorylated, PKR is only minimally stimulated by further addition of dsRNA (4,25). Indeed, autophosphorylated PKR has been shown to maintain its active conformation even when activator RNA is removed (4). Five of the 14 autophosphorylation sites in PKR described to date reside in the 20 kDa dsRBD (Fig. 1; 23,24). Furthermore, one of these residues (S33) has been shown to be in proximity to the RNA in the bound complex (26). These results suggested that autophosphorylation may have an effect on the RNA-binding properties of the enzyme (26). Interestingly, although wild-type PKR and several site-directed mutants have been overexpressed using a variety of different expression systems in various organisms, the extent to which kinase active recombinant enzyme can be stimulated by RNA in vitro has been disputed (6,27). Thus, convenient, efficient sources of recombinant PKR in its dephosphorylated form that responds to RNA activation in vitro are needed. The lack of such sources has impeded detailed biophysical studies of the RNA activation mechanism due to the quantity of enzyme required for these experiments.
Figure 1.

Domain make-up of human PKR (15). The double-stranded RNA-binding motifs (dsRBMs) are shown as dark grey boxes. The 11 kinase sub-domains of PKR are shown as light grey boxes and the eIF2α kinase insert domain (an amino acid stretch of 32 residues N-terminal to sub-domain VI) is shown as a black box. The third basic region is depicted as an open box between dsRBD and the kinase domain. Serine and threonine autophosphorylation sites that have been identified to date are depicted as lines above their respective locations (S33, S83, T88, T89, T90, S242, T255, T258, T446 and T451) (21–24).
Here we show that inactivation by phosphatase treatment of autophosphorylated PKR obtained from overexpression in Escherichia coli generates a dephosphorylated form of the enzyme whose in vitro autophosphorylation and eIF2α kinase activities are stimulated by addition of RNA. Furthermore, we demonstrate differences in the RNA-binding properties of autophosphorylated PKR and dephosphorylated PKR. Autophosphorylated PKR binds RNA weakly relative to the dephosphorylated enzyme. This result was observed with both a kinase activating synthetic RNA and a kinase inhibiting viral RNA using multiple techniques (gel mobility shift assays, MPE·Fe footprinting and affinity chromatography). These results are consistent with an activation mechanism that includes release of the activating RNA upon autophosphorylation of PKR prior to phosphorylation of eIF2α and explains the observation that the eIF2α kinase activity of autophosphorylated PKR is not regulated by RNA.
MATERIALS AND METHODS
General
Distilled, deionized water was used for all aqueous reactions and dilutions. Biochemical reagents were obtained from Sigma/Aldrich unless otherwise noted. [γ-32P]ATP (6000 Ci/mmol) was obtained from DuPont NEN. Imaging plates for storage phosphor autoradiography were purchased from Kodak. All data from phosphorimaging plates were obtained using a Molecular Dynamics STORM 840 PhosphorImager and ImageQuant software. Liquid scintillation counting was carried out with a Beckman LS 6500 Scintillation Counter and Bio-Safe II cocktail from Research Products International.
Generation of phosphorylated and dephosphorylated PKR samples
Escherichia coli BL21 (DE3) pLysS cells containing an expression vector for FLAG epitope-tagged PKR (pET-PKR) were grown in LB containing 100 µg/ml ampicillin and 34 µg/ml chloramphenicol to an OD600 of 0.6 (28). Protein overexpression was induced by addition of 0.5 mM IPTG and the cells were incubated for an additional 2 h at 37°C. The cells were then collected by centrifugation. For every 100 ml of cell culture, 1 ml of cell resuspension buffer (phosphate-buffered saline, pH 7.4, 1 mM PMSF, 0.1% Triton X-100, 100 µg/ml lysozyme) was added. The cell resuspensions were frozen at –80°C for 12 h, following which they were allowed to thaw at room temperature for 0.5 h. DNase I was added to a final concentration of 25 µg/ml. The cell lysates were centrifuged for 0.5 h at 16 000 g. The clarified lysates were then added to a DEAE–Sepharose FF (Pharmacia) column. Unbound proteins were removed with successive washes of phosphate-buffered saline, pH 7.4, 1 mM DTT, 0.1% Triton X-100, 1 mM PMSF, 1 µg/ml aprotinin, 1 µg/ml pepstatin and 1 µg/ml leupeptin and 0.3 µg/ml Bacterial Protease Inhibitor cocktail (Sigma) (buffer A). FLAG–PKR was eluted with a gradient of NaCl (10–500 mM) in buffer A. Fractions were assayed for FLAG–PKR by anti-FLAG western blotting. Fractions containing FLAG–PKR were pooled and concentrated using an AMICON concentrator (MWCO 10 kDa). The protein was then bound to anti-FLAG M2 antibody–agarose (20 µl) (Sigma) by incubating the antibody matrix with the pooled fractions for a period of 2 h at 4°C. The antibody matrix was gently centrifuged and the supernatant discarded. The matrix was washed with 50 mM Tris–HCl, pH 7.0, 0.1 mM EDTA, 5 mM DTT, 0.01% Brij-35 and 1 mM MnCl2 and resuspended in 49 µl of the same buffer. Matrix-bound FLAG–PKR was then dephosphorylated by addition of 1 U recombinant catalytic subunit of protein phosphatase 1α from rabbit (PP1cα) (New England Biolabs) and incubation for 45 min at 30°C. After this reaction, phosphatase was removed with successive washes with buffer A. The dephosphorylated form of FLAG–PKR was then eluted from the matrix with 200 µM FLAG peptide in buffer A. The supernatant containing the protein was stored at 4°C. To obtain the phosphorylated form of FLAG–PKR, PP1cα was omitted from the above reaction. The FLAG-tagged kinase-deficient mutant PKR (K296R) was expressed in E.coli using the expression vector pAR (ΔRI)-PKR (K296R) obtained from Prof. Nahum Sonenberg (McGill University) (29). Protein samples were electrophoresed on a 10% SDS–PAGE gel and samples were visualized either by anti-FLAG western blotting or by SyproOrange (BioRad) staining. A standard curve for protein concentration was generated by resolving known amounts of bovine serum albumin on a 10% SDS–PAGE gel, visualizing the bands by SyproOrange staining and quantifying the protein bands using a Molecular Dynamics STORM 840 PhosphorImager and ImageQuant software. This standard curve was then used to determine the concentration of PKR samples.
Kinase assays
Poly(I)·poly(C) at a concentration of 1 µg/ml was incubated with 100 nM PKR in 20 mM Tris–HCl, pH 7.6, 10 mM KCl, 10 mM MgCl2 and 10% glycerol for 5 min at room temperature. [γ-32P]ATP was added to a final concentration of 2 µM. The autophosphorylation reaction was allowed to proceed for 5 min at 30°C, following which His6–eIF2α, obtained by overexpression in bacteria and purified via published procedures (30) with minor modifications, was added to a final concentration of 588 nM. Aliquots of the reaction mixture were removed at 30, 60, 90 and 120 s and quenched with pre-heated (95°C) SDS–PAGE gel loading buffer. The protein samples were electrophoresed on a 13.5% SDS–PAGE gel and the labeled proteins visualized and band intensities quantified using a STORM PhosphorImager and ImageQuant software. To determine the molar amount of phosphate incorporated into eIF2α per unit time, bands were cut from the SDS–PAGE gels and scintillation counted. A standard curve correlating counts per minute to moles of phosphate was obtained using serial dilutions of the [γ-32P]ATP used in each reaction. Rates were obtained from the slopes of linear fits to the data plotted as nmol phosphate incorporated into eIF2α per min. Each experiment was carried out in triplicate and plotted values are averages of the three experiments.
Gel mobility shift experiments
PKR RNA ligands were obtained by transcription using T7 RNA polymerase as previously described (26,31). The dissociation constants for PP1-treated FLAG–PKR binding to a 92 nt RNA activator and an RNA inhibitor derived from adenovirus (VAI RNA) were obtained by native gel shift assays as previously described (32). In a typical binding experiment, PKR at varying concentrations was incubated with 5′-end-labeled RNA in 25 mM HEPES, pH 7.5, 10 mM NaCl, 5% glycerol, 5 mM DTT, 0.1 mM EDTA (1× binding buffer) and 30 µg/ml yeast tRNAPhe for 7 min at room temperature (20 µl final volume). At the end of the incubation, 4 µl of 50% glycerol, 0.025% bromophenol blue, 0.025% xylene cyanol was added to each binding reaction. PKR–RNA complexes were resolved on a 10% (79:1 acrylamide:bisacrylamide) native gel. Data were obtained by exposing the dried gel to a storage phosphor plate and quantifying the bands using ImageQuant software. All labeled RNA that migrated slower than free RNA in the absence of added protein was considered to be bound by PKR. The fraction of RNA bound (q) as a function of protein concentration was fitted to equation 1, where [PKR] represents protein concentration, Kd is the dissociation constant and n is the Hill coefficient. The reported dissociation constant and Hill coefficient represents the average from three binding experiments ± SD. The fitted Hill coefficients were 1.6 ± 0.5 for the 92 nt activating RNA and 1.6 ± 0.1 for VAI RNA
q = [PKR]n/([PKR]n + Kdn) 1
Methidiumpropyl-EDTA (MPE)·Fe footprinting experiments
A footprinting assay was employed to assess differences in affinity between the phosphorylated and dephosphorylated forms of PKR for viral inhibitor RNA, VAI RNA and the 92 nt RNA activator. A cleavage reagent capable of cleaving duplex nucleic acids, MPE·Fe, was used as the footprinting reagent (33). In a typical protection experiment, PKR at varying concentrations was incubated with 5′-end-labeled RNA in 25 mM Tris–HCl, pH 7.5, 10 mM NaCl and 30 µg/ml yeast tRNAPhe in a final reaction volume of 20 µl for 7 min at room temperature. MPE was added to a final concentration of 0.5 µM and Fe(NH4)2(SO4)2·(H2O)6 was added to a final concentration of 1 µM. Cleavage of the phosphodiester backbone was initiated by addition of sodium ascorbate to a final concentration of 20 mM. The reaction was allowed to proceed for 7 min, followed by addition of water and extraction with phenol:CHCl3. The RNA was recovered by ethanol precipitation. The dried pellets obtained after ethanol precipitation were resuspended in 99% formamide and 0.025% xylene cyanol. The cleavage products were then electrophoresed on a denaturing polyacrylamide sequencing gel and visualized by exposing the gel to a storage phosphor plate.
Affinity chromatography experiments
Phosphorylated and dephosphorylated forms of FLAG–PKR were generated as described above. However, before elution with FLAG peptide, the solid phase immobilized proteins were washed several times with cold 1× binding buffer and transferred to two BioRad-30 microchromatography spin columns. 5′-End-labeled activator RNA was then added to the two protein samples and was allowed to bind at room temperature for 10 min. Unbound RNA was removed from the two samples by successive washes with 1× binding buffer and centrifugation at 3000 r.p.m. Under these conditions, no labeled RNA is retained by the anti-FLAG antibody matrix in the absence of PKR. Bound RNA was eluted from the immobilized PKR samples with increasing concentrations of NaCl. Fractions were collected and counted by scintillation counting. After elution of all bound RNA, the two protein samples were eluted with 200 µM FLAG peptide and electrophoresed on a 10% SDS–PAGE gel. The protein samples were then transferred to a PVDF membrane and visualized by western blotting.
RESULTS
Dephosphorylated PKR can be obtained by overexpression of the wild-type enzyme in bacteria and treatment with PP1cα
Wild-type human PKR is isolated from E.coli or Saccharomyces cerevisiae overexpression systems as a highly phosphorylated protein (6,21,22,27). Since E.coli lacks eukaryotic kinases and a kinase-inactive mutant of PKR (K296R) is not phosphorylated during overexpression in bacteria or yeast, the phosphorylation observed is attributed to PKR autophosphorylation (22,27). This autophosphorylation during overexpression is believed to arise from the binding of endogenous RNAs that serve to activate the enzyme (27). Phosphorylation of PKR leads to a slower mobility on SDS–PAGE gels relative to kinase-inactive mutants and observation of this mobility difference is a convenient method to assess the phosphorylation state of the enzyme (22,27). We found that the catalytic subunit of the α-isoform of type 1 protein phosphatase (PP1cα) efficiently converted the slowly migrating phosphorylated form of bacterially expressed wild-type PKR to a form of the enzyme that co-migrated with the kinase-deficient mutant (PKR K296R) (Fig. 2A). A type 1 protein phosphatase had been previously shown to dephosphorylate and inactivate PKR (25). Other phosphatases, such as calf intestinal phosphatase (CIP), are known to dephosphorylate autophosphorylated PKR in vitro (22,34). We found PP1cα to be the most efficient in comparison to CIP, shrimp alkaline phosphatase and calcineurin (data not shown).
Figure 2.

Dephosphorylation of PKR by PP1cα. (A) Anti-FLAG western blot of a SDS–PAGE gel used to resolve PKR samples. Lane 1, wild-type FLAG epitope-tagged PKR overexpressed in E.coli; lane 2, FLAG epitope-tagged kinase-deficient K296R mutant of PKR overexpressed in E.coli; lane 3, wild-type FLAG epitope-tagged PKR overexpressed in E.coli and treated with PP1cα. (B) SyproOrange stained gel used to resolve phosphorylated and dephosphorylated PKR samples after purification. Lane 1, protein molecular weight markers; lane 2, wild-type FLAG epitope-tagged PKR overexpressed in E.coli; lane 3, wild-type FLAG epitope-tagged PKR overexpressed in E.coli and treated with PP1cα. (C) Time course for the phosphorylation of eIF2α by PKR samples. (Top) Storage phosphor autoradiograms of SDS–PAGE gels used to resolve products from kinase reactions with phosphorylated PKR. Lanes 1–4, no RNA added; lanes 5–8, poly(I)·poly(C) added to a final concentration of 1 µg/ml. Lanes 1 and 5, 30 s time points; lanes 2 and 6, 60 s time points; lanes 3 and 7, 90 s time points; lanes 4 and 8, 120 s time points. (Bottom) Plot of product formation as a function of time in the presence (blue colored boxes) and absence (blue open circles) of 1 µg/ml poly(I)·poly(C). The data were fitted to a line using the least squares method of KaleidaGraph. Data points reported are the average of three independent experiments. (D) (Top) Storage phosphor autoradiograms of SDS–PAGE gels used to resolve products from kinase reactions with PP1cα-treated PKR. Lanes 1–4, no RNA added; lanes 5–8, poly(I)·poly(C) added to a final concentration of 1 µg/ml. Lanes 1 and 5, 30 s time points; lanes 2 and 6, 60 s time points; lanes 3 and 7, 90 s time points; lanes 4 and 8, 120 s time points. (Bottom) Plot of product formation as a function of time in the presence (red filled boxes) and absence (red open circles) of 1 µg/ml poly(I)·poly(C).
To compare the kinase activities of dephosphorylated and autophosphorylated PKR obtained as described above, we purified each of these two forms of the protein (Fig. 2B). The key step in this purification was binding of autophosphorylated, FLAG epitope-tagged PKR to an anti-FLAG affinity purification matrix and dephosphorylation of PKR while solid phase immobilized. The phosphatase was removed with successive buffer washes and PKR was eluted with soluble FLAG peptide. For isolation of autophosphorylated PKR, the phosphatase was simply omitted from the dephosphorylation reaction. To assess the effect PP1cα treatment had on the eIF2α kinase activity of PKR, we measured the rates of eIF2α phosphorylation by the two forms of the protein in the presence and absence of activating RNA [poly(I)·poly(C)] (Fig. 2C and D and Table 1). As expected, autophosphorylated PKR was more active (∼20-fold) than PP1cα-treated PKR in the absence of RNA and was not significantly stimulated by addition of activating RNA. In contrast, the rate of eIF2α phosphorylation by PP1cα-treated PKR was increased 11-fold by prior incubation with activating RNA and ATP. Importantly, the rate reached by this sample was only ∼2-fold less than that observed for PKR without PP1cα treatment. These results are similar to those observed with native PKR isolated from rabbit reticulocytes treated with a type 1 protein phosphatase and indicate that human PKR expressed in bacteria can also undergo reversible activation by autophosphorylation/phosphatase-mediated dephosphorylation (25).
Table 1. Initial rates of eIF2α phosphorylation by PKR samplesa.
| PKR sample |
Rateb (nM min–1) |
Rate enhancement |
| PhosphoPKRc, no RNA | 10.4 ± 4.0 | |
| PhosphoPKR + RNAd | 11.5 ± 4.1 | 1.1-fold |
| DephosphoPKRe, no RNA | 0.48 ± 0.36 | |
| DephosphoPKR + RNAd | 5.3 ± 3.5 | 11-fold |
aThe eIF2α phosphorylations were carried out with 100 nM PKR, 588 nM eIF2α and 2 µM ATP in 20 mM Tris–HCl, pH 7.6, 10 mM KCl, 10 mM MgCl2 and 10% glycerol at 30°C.
bRates are reported with the units nmol phosphorylated eIF2α formed per min as described in Materials and Methods.
cPhosphoPKR was generated by overexpression of the wild-type enzyme in E.coli as previously described (27,28).
dPKR was incubated with ATP and poly(I)·poly(C) for 5 min at 30°C prior to addition of eIF2α.
eDephosphoPKR was generated by treatment of phosphoPKR with the catalytic subunit of protein phosphatase 1α.
The RNA-binding activity of PKR is stimulated by dephosphorylation with PP1cα
To compare the RNA-binding properties of autophosphorylated and dephosphorylated PKR, gel mobility shift assays were performed using a 92 nt RNA ligand identified as a PKR activator from a sequence-randomized RNA library (Fig. 3; 32). This RNA was chosen for these studies because it binds to the dsRBD of PKR with high affinity to form a saturatable complex well-suited for gel mobility shift experiments (32). Furthermore, photocrosslinking experiments involving this RNA and a PKR isolated dsRBD indicated that a previously identified autophosphorylation site (Ser33) was located near the protein/RNA interface in the complex, suggesting that autophosphorylation may play a role in regulating RNA binding (26).
Figure 3.
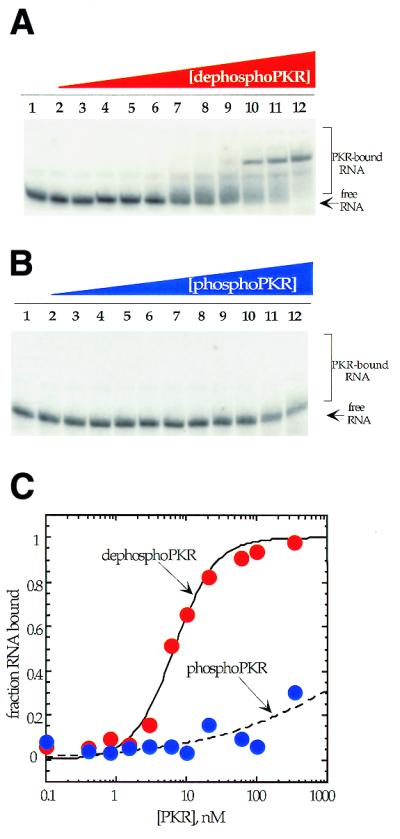
Native gel mobility shifts for phosphorylated and dephosphorylated PKR binding to a 92 nt RNA activator (32). (A) Storage phosphor autoradiogram of a representative gel used to separate PKR-bound from free RNA for PP1cα-treated PKR. Lanes 1–12, 0, 0.1, 0.4, 0.8, 1.5, 3, 6, 10, 20, 60, 100 and 330 nM PKR added, respectively. (B) Storage phosphor autoradiogram of a representative gel used to separate PKR-bound from free RNA for phosphorylated PKR. Lanes 1–12, 0, 0.4, 0.8, 1.5, 3, 6, 10, 20, 60, 100, 330 and 570 nM PKR added, respectively. (C) Plot of fraction RNA bound by dephosphorylated PKR (red filled circles) and phosphorylated PKR (blue filled circles) as a function of protein concentration. The data were fitted to the equation: fraction bound = [PKR]n/([PKR]n + Kdn) using the least squares method of KaleidaGraph.
Dephosphorylated PKR, which has relatively low kinase activity, bound tightly to this RNA with a dissociation constant of 7.1 ± 1.8 nM (Fig. 3). In contrast, although phosphorylated PKR is an active kinase, little binding to this RNA was observed at concentrations up to 300 nM. At 570 nM enzyme a smear is detected in the gel above free RNA, with no more than 30% of the RNA bound. Thus, the Kd for this RNA for the phosphorylated form of PKR must be >570 nM. This corresponds to at least an 80-fold stimulation in binding to this RNA activator by dephosphorylation of PKR.
MPE is a synthetic intercalating agent that cleaves duplex nucleic acids in the presence of ferrous ion and oxygen (33,35). Cleavage by MPE·Fe exhibits little sequence selectivity, a characteristic that makes it well-suited for protection studies. MPE·Fe footprinting has previously been used to determine nucleotides of DNA involved in complex formation with proteins and small molecules (36–39). We surmised that the same technique could be generally applicable to duplex RNA-binding proteins as well. MPE·Fe footprinting was employed to determine the difference in binding between the phospho and dephospho forms of PKR. Results from the gel shift experiments with the 92 nt activator RNA led us to believe that we may observe differences in the extent of protection from MPE·Fe cleavage when this RNA was bound by phosphoPKR versus dephosphoPKR. Figure 4A shows the results of an MPE·Fe footprinting reaction with the activator RNA in the presence of increasing amounts of dephospho- and phosphoPKR (lanes 4–6 and 7–9, respectively). The nucleotides most efficiently cleaved by MPE·Fe correspond to those making up the 3′ base paired stem (regions labeled a and b in Figure 4B). Importantly, dephosphoPKR protects this region of the RNA from MPE·Fe cleavage more effectively than phosphoPKR. As can be seen in Figure 4A (lanes 6 and 9) and C, there is an ∼2-fold difference in the intensity of the cleavage bands in the stem of the activating RNA in region a in the presence of 330 nM dephosphoPKR as compared to the same concentration of the phosphorylated form. A similar effect was observed for region b. The fact that this region of the RNA was protected from MPE·Fe cleavage by PKR was not surprising, as the duplex formed by nucleotides in regions a and b had been shown previously to be a binding site for PKR (31,32).
Figure 4.

MPE·Fe footprinting of a 92 nt activator RNA of PKR using phosphorylated and dephosphorylated forms of the enzyme. (A) Storage phosphor autoradiogram of a polyacrylamide gel showing the effect of dephosphorylated and phosphorylated PKR on MPE·Fe cleavage. OH–, alkaline hydrolysis; T1, T1 RNase; lane 1, RNA with no added MPE·Fe or Na ascorbate; lane 2, RNA in the presence of 500 nM MPE·Fe with no added Na ascorbate; lane 3, RNA in the presence of 500 nM MPE·Fe and 20 mM Na ascorbate; lanes 4–6, RNA in the presence of 500 nM MPE·Fe and 20 mM Na ascorbate and increasing concentrations of dephosphoPKR (lane 4, 10 nM; lane 5, 100 nM; lane 6, 330 nM); lanes 7–9, RNA in the presence of 500 nM MPE·Fe and 20 mM Na ascorbate and increasing concentrations of phosphoPKR (lane 7, 10 nM; lane 8, 100 nM; lane 9, 330 nM). The mapping of the major cleavage sites by MPE·Fe (identified by letters and brackets) is shown on the gel. Protection from cleavage at this site by increasing concentrations of dephosphoPKR was reproducible in five independent determinations with a representative gel shown. (B) Sites of protection from MPE·Fe cleavage are identified on the predicted secondary structure of the 92 nt activator RNA as letters and brackets. (C) Quantification of the cleavage intensities in the presence of no protein (black line), phosphoPKR at 330 nM (blue line), dephosphoPKR at 330 nM (red line). The nucleotide positions are denoted on the x-axis of the graph. The y-axis represents counts on the phosphor plate as arbitrary units (a.u.).
In addition to the two techniques discussed above (gel shift and MPE·Fe footprinting), affinity chromatography experiments were performed to highlight the differences in binding between phospho- and dephosphoPKR. An anti-FLAG antibody matrix was used to immobilize the two forms of the protein, following which 5′-end-labeled activator RNA was bound to the solid phase-immobilized proteins. The bound RNA–protein complex was washed several times to remove unbound RNA. Increasing concentrations of NaCl were then used to elute the bound RNA and fractions were analyzed by scintillation counting. It was observed that at 500 mM NaCl, very little labeled RNA remained bound to the phosphoPKR matrix whereas a large fraction of the RNA remained bound to dephosphoPKR under the same conditions (Fig. 5). Indeed, 1.5 M NaCl was required to elute all of the RNA from the dephosphoPKR matrix. To show that the difference in binding between the two forms of protein in this experiment was a result of the phosphorylation state of PKR and not due to varying amounts of protein on the two matrices, a western blot was performed on the two samples post-elution. The western blot shows no significant difference in the amounts of protein present in the two samples (Fig. 5, inset). These results corroborate the findings of the earlier two experiments: dephosphoPKR binds to the 92 nt activator RNA more tightly than phosphoPKR.
Figure 5.
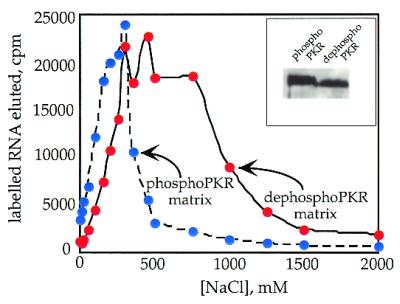
RNA binding to phosphorylated and dephosphorylated PKR demonstrated using affinity chromatography. The graph depicts the effect of increasing NaCl concentration on the ability of phosphoPKR matrix (blue circles) versus dephosphoPKR matrix (red circles) to retain 5′-end-labeled 92 nt activator RNA. Plotted values are the average of two independent experiments. (Inset) A western blot carried out to determine the amounts of phospho- and dephosphoPKR present on each matrix.
An important factor to consider while interpreting the above results is the fact that the 92 nt activator RNA was selected from a randomized RNA library raised against the dsRBD of PKR (32). To state the results that we obtained as a general property of the enzyme, it would be necessary to perform similar experiments on a naturally occurring RNA ligand of PKR. For this purpose we chose to study VAI RNA, an inhibitor of PKR derived from adenovirus (3). Native gel shift experiments were performed using the two forms of PKR and VAI RNA (Fig. 6). DephosphoPKR bound tightly to this RNA with a dissociation constant of 55 ± 10 nM, while little binding to this RNA was observed at concentrations up to 100 nM phosphoPKR. At 330 nM, no more than 25% of RNA was bound. Therefore, the Kd for VAI RNA binding to the phosphorylated form of PKR must be greater than 330 nM, corresponding to at least a 6-fold stimulation in binding by dephosphorylation.
Figure 6.
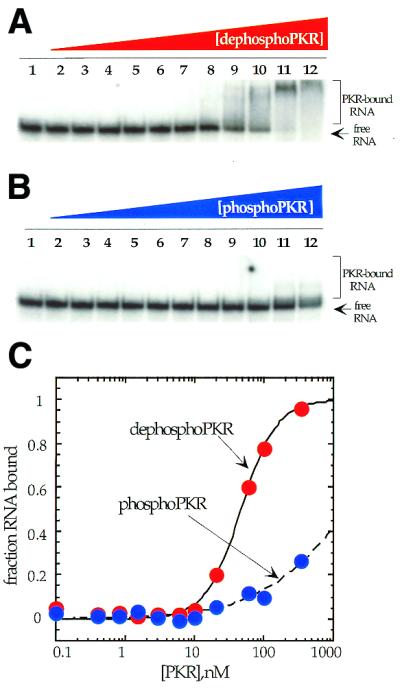
Native gel mobility shifts for phosphorylated and dephosphorylated PKR binding to a viral RNA inhibitor (VAI RNA). (A) Storage phosphor autoradiogram of a representative gel used to separate PKR-bound from free RNA for PP1cα-treated PKR. Lanes 1–12, 0, 0.1, 0.4, 0.8, 1.5, 3, 6, 10, 20, 60, 100 and 330 nM PKR added, respectively. (B) Storage phosphor autoradiogram of a representative gel used to separate PKR-bound from free RNA for phosphorylated PKR. Lanes 1–12, 0, 0.1, 0.4, 0.8, 1.5, 3, 6, 10, 20, 60, 100 and 330 nM PKR added, respectively. (C) Plot of fraction RNA bound by dephosphorylated PKR (red filled circles) and phosphorylated PKR (blue filled circles) as a function of protein concentration. The data were fitted to the equation: fraction bound = [PKR]n/([PKR]n + Kdn) using the least squares method of KaleidaGraph.
MPE·Fe footprinting of VAI RNA confirmed the difference in binding between the two forms of PKR to VAI RNA (Fig. 7). Figure 7 shows the effect of increasing concentrations of the phosphorylated and dephosphorylated forms of PKR on cleavage of VAI RNA by MPE·Fe. As was seen with the 92 nt activating RNA and expected for the intercalator MPE·Fe, duplex regions of VAI RNA are the most efficiently cleaved by this footprinting reagent. At the highest concentration of dephosphoPKR (330 nM), clear protection is observed as compared to the sample without protein added (Fig. 7A, compare lanes 3 and 6, and C). The footprint is observed in the apical stem–loop region of the RNA (designated a and b in Fig. 7). Importantly, this region of the RNA corresponds to the previously identified binding site for PKR (31,40). Furthermore, phosphoPKR at the same concentration has little effect on the MPE·Fe cleavage reaction at this location in VAI RNA (Fig. 7A, lane 9, and C).
Figure 7.

MPE·Fe footprinting of a viral RNA inhibitor (VAI RNA) using phosphorylated and dephosphorylated forms of the enzyme. Storage phosphor autoradiogram of a representative gel showing the effect of dephosphorylated and phosphorylated PKR on MPE·Fe cleavage. OH–, alkaline hydrolysis; T1, T1 RNase; lane 1, RNA with no added MPE·Fe or Na ascorbate; lane 2, RNA in the presence of 500 nM MPE·Fe with no added Na ascorbate; lane 3, RNA in the presence of 500 nM MPE·Fe and 20 mM Na ascorbate; lanes 4–6, RNA in the presence of 500 nM MPE·Fe and 20 mM Na ascorbate and increasing concentrations of dephosphoPKR (lane 4, 10 nM; lane 5, 100 nM; lane 6, 330 nM); lanes 7–9, RNA in the presence of 500 nM MPE·Fe and 20 mM Na ascorbate and increasing concentrations of phosphoPKR (lane 7, 10 nM, lane 8, 100 nM; lane 9, 330 nM). The mapping of the major cleavage sites by MPE·Fe (identified by letters and brackets) is shown on the gel. Protection from cleavage at this site by increasing concentrations of dephosphoPKR was reproducible in six independent determinations with a representative gel shown. (B) Sites of protection from MPE·Fe cleavage are identified on the predicted secondary structure of the VAI RNA as letters and brackets. (C) Quantification of the cleavage intensities in the presence of no protein (black line), phosphoPKR at 330 nM (blue line), dephosphoPKR at 330 nM (red line). The nucleotide positions are denoted on the x-axis of the graph. The y-axis represents counts on the phosphor plate as arbitrary units (a.u.). Underlined nucleotides represent sequence changes made to facilitate cloning. This region of VAI RNA is outside the previously described PKR-binding site (40).
DISCUSSION
It has been reported that PKR expressed in bacteria is fully activated and does not respond to RNA regulators in vitro (6). However, another report indicated that bacterially expressed PKR is only partially activated and does show in vitro stimulation by RNA activators (27). We have observed that the ability of bacterially expressed PKR to respond to RNA in vitro correlates with the extent of phosphorylation of the enzyme (Fig. 2). Previously, Barber et al. had shown that the level of PKR phosphorylation was related to the length of time protein expression was allowed to proceed post-induction in E.coli (27). Indeed, we have observed that the in vitro responsiveness of bacterially expressed PKR to RNA regulators also correlates with expression time, undoubtedly due to the different extents of autophosphorylation at different expression times (data not shown). It is possible that the apparently conflicting reports regarding the responsiveness of bacterially expressed PKR to RNA arise from differences in the times of protein expression post-induction or the presence or absence of phosphatase inhibitors during purification. Our results demonstrate that inactivation by phosphatase treatment of autophosphorylated PKR obtained from overexpression in bacteria generates PKR in a form suitable for in vitro analysis of the RNA-induced activation mechanism. Our technique generates ∼100 µg purified dephosphoPKR per liter of culture whose in vitro eIF2α kinase activity is stimulated 11-fold by the presence of an activating RNA [poly(I)·poly(C)].
The advent of a technique for generating wild-type PKR in a dephosphorylated state allowed us to directly assess the effect of autophosphorylation on RNA binding affinity. The discovery of autophosphorylation sites in the dsRBD of PKR and the observation that one of those sites is near the protein/RNA interface in a complex between the dsRBD and an RNA activator led us to speculate that phosphorylation could influence the RNA-binding properties of the enzyme (23,24,26). This is consistent with previous reports indicating that once autophosphorylated, PKR no longer responds to RNA regulators, including kinase inhibiting RNAs (41). Given the number of autophosphorylation sites found in PKR to date (10 sites mapped, possibly 14 total), we concluded that identification of sites involved in controlling RNA binding by site-directed mutagenesis may be problematical, particularly given the possibility that the phosphorylation effect on RNA affinity may be cumulative, with no particularly important single site (21–24). Therefore, we chose to analyze the effect of global dephosphorylation of the active, wild-type enzyme using protein phosphatase 1. Although our dephosphorylated sample has a mobility during electrophoresis similar to that of a kinase-inactive mutant (K296R), which is not autophosphorylated during bacterial expression (27), we cannot rule out the presence of PP1cα-resistant phosphates remaining on the enzyme. Indeed, mass spectrometry experiments are underway in our laboratory to determine the extent of phosphorylation for both forms of the protein studied here. Nevertheless, our binding studies with two different RNAs using three different experimental techniques support the conclusion that when PKR is dephosphorylated by phosphatase treatment, its affinity for RNA increases.
Gel shift experiments with two different RNAs demonstrated that phospho- and dephosphoPKR have distinct RNA binding affinities. Dephosphorylated PKR exhibited tight binding to a 92 nt activator RNA and for a naturally occurring inhibiting RNA (7.1 ± 1.8 nM for the activator and 55 ± 10 nM for VAI RNA), whereas the phosphorylated form of the protein did not show high affinity binding to either RNA. For VAI RNA binding to PKR, a Kd of ∼2 nM has been reported (42). An earlier study approximated the Kd for VAI RNA to be ∼0.3 nM (10). Both studies used a catalytically inactive (K296R) version of full-length PKR. Our measured Kd for binding of dephosphorylated wild-type PKR to VAI RNA is higher than these values. It is possible that differences in VAI RNA preparation methods contribute to the differences in binding affinities observed. Another possible explanation may be unequal amounts of active enzyme in the different PKR samples. We do observe that dephosphorylated PKR has a specific activity (after re-autophosphorylation in the presence of RNA) that is ∼2-fold lower than phosphoPKR (Fig. 2 and Table 1). This could arise from some denaturation during phosphatase treatment. However, the fact that the RNA binding affinity of PKR increases after phosphatase treatment indicates that this effect is independent of any denaturation that may be occurring.
Interestingly, when the RNA-binding properties of wild-type PKR expressed in bacteria (and presumably highly phosphorylated) were previously compared to that of a kinase-inactive mutant, little difference was observed (27). Furthermore, no significant difference in RNA binding was reported when a mutant of PKR, unable to be autophosphorylated at four sites in the RBD, was compared to wild-type phosphorylated enzyme (24). In both cases, co-immunoprecipitation experiments were used and the amounts of labeled RNA retained on PKR–antibody matrices were compared. We have shown that both autophosphorylated and dephosphorylated PKR, when immobilized on an antibody matrix, can retain a labeled RNA ligand whose affinity for the two forms of the enzyme differs by >80-fold (Fig. 5). However, by varying the ionic strength of the wash buffer, we show that the antibody matrix bound to phosphorylated PKR retains the RNA ligand less effectively (RNA is eluted at lower NaCl concentrations) than that bound to dephosphorylated PKR. It appears that co-immunoprecipitation experiments can underestimate differences in affinity and are highly dependent on the nature of the buffer used to wash the immunocomplexes.
MPE·Fe is an intercalator that has been used extensively to study the interaction of duplex DNA-binding proteins with their respective substrates (36–38). MPE·Fe has also been used as a base pair-specific probe for structured RNAs (35). Use of this intercalator is advantageous because it is not sequence specific and its cleavage efficiency is dependent on the presence of duplex regions in the nucleic acid of interest. We decided to exploit this property of MPE·Fe as a footprinting agent to study the effect of PKR binding to two highly structured RNAs. For both RNAs studied here we found that dephosphoPKR is more effective than phosphoPKR at protecting the RNA from cleavage by MPE·Fe in the known PKR-binding site. In addition, to the best of our knowledge, this constitutes the first example of using MPE·Fe as a footprinting agent for dsRBM-containing proteins. This reagent should be generally applicable to the study of any member of this important class of RNA-binding proteins.
The difference in RNA binding affinities between phospho- and dephosphoPKR may have an effect on the way viruses inhibit the enzyme. Viral RNA inhibitors cannot inhibit PKR once it is autophosphorylated and active (41). A virus that uses an RNA inhibitor of PKR apparently needs to intercept the enzyme in its dephospho state. Therefore, such a virus may benefit from activation of a phosphatase. It has been shown that the γ134.5 protein from herpes simplex virus complexes with protein phosphatase 1α to dephosphorylate eIF2α, thereby precluding PKR-mediated translational inhibition (43). Given the results presented here, it is also possible that viruses have evolved similar mechanisms to recruit a PP1-like phosphatase to dephosphorylate PKR, thereby making PKR susceptible to RNA-mediated viral inhibition. The phosphatase activation could be transient in nature, occurring at early stages of infection before build-up of the viral RNA inhibitor. Further experiments will be necessary to test this hypothesis.
Our experiments show that increasing levels of phosphorylation result in a lowering of the affinity of PKR for an RNA activator. This could result in release of the activator during the autophosphorylation reaction and serve as a switch in kinase specificity from autophosphorylation to eIF2α phosphorylation. A model for the activation and deactivation of PKR including these elements is shown in Figure 8. Binding of the activator to the dsRBD causes a conformational change in the kinase domain, allowing productive binding to ATP (4). Autophosphorylation generates a form of the protein with low affinity for RNA and high eIF2α kinase activity. This form can be dephosphorylated by a PP1-like phosphatase to regenerate the form of PKR with high affinity for RNA and low eIF2α kinase activity. This model for PKR activation is consistent with the finding that autophosphorylation of PKR, but not phosphorylation of eIF2α by autophosphorylated PKR, is inhibited by the adenovirus RNA inhibitor VAI RNA (41).
Figure 8.

Model for the activation and deactivation of PKR involving interconversion of a low eIF2α kinase activity, high RNA affinity form and a high eIF2α kinase activity, low RNA affinity form of the enzyme.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Mihir K. Bhayani for technical assistance and Prof. Ronald Wek (Indiana University Medical School) for the (His)6-eIF2α expression plasmid p240. This work was supported by a grant from the National Institutes of Health to P.A.B. (GM-57214).
References
- 1.Jaramillo M.L., Abraham,N. and Bell,J.C. (1995) The interferon system: a review with emphasis on the role of PKR in growth control (Glossary). Cancer Invest., 13, 327–338. [DOI] [PubMed] [Google Scholar]
- 2.Manche L., Green,S.R., Schmedt,C. and Mathews,M.B. (1992) Interactions between double-stranded RNA regulators and the protein kinase DAI. Mol. Cell. Biol., 12, 5238–5248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.O’Malley R.P., Mariano,T.M., Siekieka,J. and Mathews,M.B. (1986) A mechanism for the control of protein synthesis by adenovirus VA RNAI. Cell, 44, 391–400. [DOI] [PubMed] [Google Scholar]
- 4.Galabru J. and Hovanessian,A. (1987) Autophosphorylation of the protein kinase dependent on double-stranded RNA. J. Biol. Chem., 262, 15538–15544. [PubMed] [Google Scholar]
- 5.Samuel C.E., Knutson,G.S., Berry,M.J., Atwater,J.A. and Lasky,S.R. (1986) Purification of double-stranded RNA-dependent protein kinase from mouse fibroblasts. Methods Enzymol., 119, 499–516. [DOI] [PubMed] [Google Scholar]
- 6.Kumar A., Haque,J., Lacoste,J., Hiscott,J. and Williams,B.R. (1994) Double-stranded RNA-dependent protein kinase activates transcription factor NF-κB by phosphorylating IκB. Proc. Natl Acad. Sci. USA, 91, 6288–6292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wek R.C. (1994) eIF-2 kinases: regulators of general and gene-specific translation initiation. Trends Biochem. Sci., 19, 491–496. [DOI] [PubMed] [Google Scholar]
- 8.Clemens M.J., Laing,K.G., Jeffrey,I.W. and Schofield,A. (1994) Regulation of the interferon-inducible eIF-2 protein kinase by small RNAs. Biochimie, 76, 770–778. [DOI] [PubMed] [Google Scholar]
- 9.Jagus R. and Gray,M.M. (1994) Proteins that interact with PKR. Biochimie, 76, 779–791. [DOI] [PubMed] [Google Scholar]
- 10.Sharp T.V., Schwemmle,M., Jeffrey,I., Laing,K. and Mellor,H. (1993) Comparative analysis of the regulation of the interferon-inducible protein kinase PKR by Epstein–Barr virus RNAs EBER-1 and EBER-2 and adenovirus VAI RNA. Nucleic Acids Res., 21, 4483–4490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kitajewski J., Schneider,R.J., Safer,B., Munemitsu,S.M., Samuel,C.E., Thimmappaya,B. and Shenk,T. (1986) Adenovirus VAI RNA antagonizes the antiviral action of interferon by preventing activation of the interferon-induced eIF-2α kinase. Cell, 45, 195–200. [DOI] [PubMed] [Google Scholar]
- 12.Davis S. and Watson,J.C. (1996) In vitro activation of the interferon-induced, double-stranded RNA-dependent protein kinase PKR by RNA from the 3′ untranslated regions of human α-tropomyosin. Proc. Natl Acad. Sci. USA, 93, 508–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meurs E.F., Galabru,J., Barber,G.N., Katz,M.G. and Hovanessian,A.G. (1993) Tumor suppressor function of the interferon-induced double-stranded RNA-activated protein kinase. Proc. Natl Acad. Sci. USA, 90, 232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koromilas A.E., Roy,S., Barber,G.N., Katze,M.G. and Sonenberg,N. (1992) Malignant transformation by a mutant of the IFN-inducible dsRNA-dependent protein kinase. Science, 257, 1685–1689. [DOI] [PubMed] [Google Scholar]
- 15.Meurs E., Chong,K., Galabru,J., Thomas,N.S.B., Kerr,I.M., Williams,B.R.G. and Hovanessian,A.G. (1990) Molecular cloning and characterization of the human double-stranded RNA-activated protein kinase induced by interferon. Cell, 62, 379–390. [DOI] [PubMed] [Google Scholar]
- 16.Fierro-Monti I. and Mathews,M.B. (2000) Proteins binding to duplexed RNA: one motif, multiple functions. Trends Biochem. Sci., 25, 241–246. [DOI] [PubMed] [Google Scholar]
- 17.Bass B.L., Nishikura,K., Keller,W., Seeburg,P.H., Emeson,R.B., O’Connell,M.A., Samuel,C.E. and Herbert,A. (1997) A standardized nomenclature for adenosine deaminases that act on RNA. RNA, 3, 947–949. [PMC free article] [PubMed] [Google Scholar]
- 18.Bycroft M., Gruenert,S., Murzin,A.G. and Proctor,M. (1995) NMR solution structure of a dsRNA binding domain from Drosophila staufen protein reveals homology to the N-terminal domain of ribosomal protein S5. EMBO J., 14, 3563–3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kharrat A., Macias,M.J., Gibson,T.J. and Nilges,M. (1995) Structure of the dsRNA binding domain of E.coli RNase III. EMBO J., 14, 3572–3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Langland J.O., Kao,P.N. and Jacobs,B.L. (1999) Nuclear factor-90 of activated T-cells: a double-stranded RNA-binding protein and substrate for the double-stranded RNA-dependent protein kinase, PKR. Biochemistry, 38, 6361–6368. [DOI] [PubMed] [Google Scholar]
- 21.Taylor D.R., Lee,S.B., Romano,P.R., Marshak,D.R., Hinnebusch,A.G., Esteban,M. and Mathews,M.B. (1996) Autophosphorylation sites participate in the activation of the double-stranded-RNA-activated protein kinase PKR. Mol. Cell. Biol., 16, 6295–6302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Romano P.R., Garcia-Barrio,M.T., Zhang,X., Wang,Q., Taylor,D.R., Zhang,F., Herring,C., Mathews,M.B., Qin,J. and Hinnebusch,A.G. (1998) Autophosphorylation in the activation loop is required for full kinase activity in vivo of human and yeast eukaryotic initiation factor 2-α kinases PKR and GCN2. Mol. Cell. Biol., 18, 2282–2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang X., Herring,C.J., Romano P.R., Szczepanowska,J., Brzeska,H., Hinnebusch,A.G. and Qin,J. (1998) Identification of phosphorylation sites in proteins separated by polyacrylamide gel electrophoresis. Anal. Chem., 70, 2050–2059. [DOI] [PubMed] [Google Scholar]
- 24.Taylor D.R., Tian,B., Romano,P.R., Hinnebusch,A.G., Lai,M.M.C. and Mathews,M.B. (2001) Hepatitis C virus envelope protein E2 does not inhibit PKR by simple competition with autophosphorylation sites in the RNA-binding domain. J. Virol., 75, 1265–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Szyszka R., Kudlick,W., Kramer,G., Hardesty,B., Galabru,J. and Hovanessian,A. (1989) A type 1 phosphoprotein phosphatase active with phosphorylated Mr = 68,000 initiation factor 2 kinase. J. Biol. Chem., 264, 3827–3831. [PubMed] [Google Scholar]
- 26.Spanggord R.J. and Beal,P.A. (2000) Site-specific modification and RNA crosslinking of the RNA-binding domain of PKR. Nucleic Acids Res., 28, 1899–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barber G.N., Tomita,J., Hovanessian,A.G., Meurs,E. and Katze,M.G. (1991) Functional expression and characterization of the interferon-induced double-stranded RNA activated P68 protein kinase from Escherichia coli. Biochemistry, 30, 10356–10361. [DOI] [PubMed] [Google Scholar]
- 28.Vuyisich M. and Beal,P.A. (2000) Regulation of the RNA-dependent protein kinase by triple helix formation. Nucleic Acids Res., 28, 2369–2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cosentino G.P., Venkatesan,S., Serluca,F.C., Green,S.R., Mathews,M.B. and Sonenberg,N. (1995) Double-stranded-RNA-dependent protein kinase and TAR RNA-binding protein form homo- and heterodimers in vivo. Proc. Natl Acad. Sci. USA, 92, 9445–9449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhu S., Sobolev,A.Y. and Wek,R.C. (1996) Histidyl-tRNA synthetase-related sequences in GCN2 protein kinase regulate in vitro phosphorylation of eIF-2. J. Biol. Chem., 271, 24989–24994. [DOI] [PubMed] [Google Scholar]
- 31.Spanggord R.J. and Beal,P.A. (2001) Selective binding by the RNA binding domain of PKR revealed by affinity cleaving. Biochemistry, 40, 4272–4280. [DOI] [PubMed] [Google Scholar]
- 32.Bevilacqua P.C., George,C.X., Samuel,C.E. and Cech,T.R. (1998) Binding of the protein kinase PKR to RNAs with secondary structure defects: role of the tandem A-G mismatch and noncontiguous helixes. Biochemistry, 37, 6303–6316. [DOI] [PubMed] [Google Scholar]
- 33.Hertzberg R.P. and Dervan,P.B. (1984) Cleavage of DNA with methidiumpropy-EDTA-iron(II): reaction conditions and product analysis. Biochemistry, 23, 3934–3945. [DOI] [PubMed] [Google Scholar]
- 34.Thomis D.C. and Samuel,C.E. (1993) Mechanism of interferon action: evidence for intermolecular autophosphorylation and autoactivation of the interferon-induced, RNA-dependent protein kinase PKR. J. Virol., 67, 7695–7700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vary C.P. and Vournakis,J.N. (1984) RNA structure analysis using methidiumpropyl-EDTA-Fe(II): a base-pair specific RNA structure probe. Proc. Natl Acad. Sci. USA, 81, 6978–6982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lenzmeier B.A., Baird,E.E., Dervan,P.B. and Nyborg,J.K. (1999) The Tax protein-DNA interaction is essential for HTLV-I transactivation in vitro. J. Mol. Biol., 291, 731–744. [DOI] [PubMed] [Google Scholar]
- 37.Chepanoske C.L., Porello,S.L., Fujiwara,T., Sugiyama,H. and David,S.S. (1999) Substrate recognition by Escherichia coli MutY using substrate analogs. Nucleic Acids Res., 27, 3197–3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kahl B.F., Li,H. and Paule,M.R. (2000) DNA melting and promoter clearance by eukaryotic RNA polymerase I. J. Mol. Biol., 299, 75–89. [DOI] [PubMed] [Google Scholar]
- 39.Van Dyke M.W. and Dervan,P.B. (1983) Chromomycin, mithramycin and olivomycin binding sites on heterogeneous deoxyribonucleic acid. Footprinting with (methidiumpropyl-EDTA)iron(II). Biochemistry, 22, 2373–2377. [DOI] [PubMed] [Google Scholar]
- 40.Clarke P.A. and Mathews,M.B. (1995) Interactions between the double-stranded RNA binding motif and RNA: definition of the binding site for the interferon-induced protein kinase DAI (PKR) on adenovirus VA RNA. RNA, 1, 7–20. [PMC free article] [PubMed] [Google Scholar]
- 41.Galabru J., Katz,M.G., Robert,N. and Hovanessian,A. (1989) The binding of double-stranded RNA and adenovirus VAI RNA to the interferon-induced protein kinase. Eur. J. Biochem., 178, 581–589. [DOI] [PubMed] [Google Scholar]
- 42.McCormack S.J. and Samuel,C.E. (1995) Mechanism of interferon action: RNA-binding activity of full-length and R-domain forms of the RNA-dependent protein kinase PKR—determination of Kd values for VAI and TAR RNAs. Virology, 206, 511–519. [DOI] [PubMed] [Google Scholar]
- 43.He B., Gross,M. and Roizman,B. (1997) The γ134.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1α to dephosphorylate the α subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activated protein kinase. Proc. Natl Acad. Sci. USA, 94, 843–848. [DOI] [PMC free article] [PubMed] [Google Scholar]