

Abstract

Pyruvate formate-lyase activating enzyme (PFL-AE) is a radical S-adenosyl-l-methionine (SAM) enzyme that installs a catalytically essential glycyl radical on pyruvate formate-lyase. We show that PFL-AE binds a catalytically essential monovalent cation at its active site, yet another parallel with B12 enzymes, and we characterize this cation site by a combination of structural, biochemical, and spectroscopic approaches. Refinement of the PFL-AE crystal structure reveals Na+ as the most likely ion present in the solved structures, and pulsed electron nuclear double resonance (ENDOR) demonstrates that the same cation site is occupied by 23Na in the solution state of the as-isolated enzyme. A SAM carboxylate-oxygen is an M+ ligand, and EPR and circular dichroism spectroscopies reveal that both the site occupancy and the identity of the cation perturb the electronic properties of the SAM-chelated iron–sulfur cluster. ENDOR studies of the PFL-AE/[13C-methyl]-SAM complex show that the target sulfonium positioning varies with the cation, while the observation of an isotropic hyperfine coupling to the cation by ENDOR measurements establishes its intimate, SAM-mediated interaction with the cluster. This monovalent cation site controls enzyme activity: (i) PFL-AE in the absence of any simple monovalent cations has little–no activity; and (ii) among monocations, going down Group 1 of the periodic table from Li+ to Cs+, PFL-AE activity sharply maximizes at K+, with NH4+ closely matching the efficacy of K+. PFL-AE is thus a type I M+-activated enzyme whose M+ controls reactivity by interactions with the cosubstrate, SAM, which is bound to the catalytic iron–sulfur cluster.
Introduction
Pyruvate formate-lyase activating enzyme (PFL-AE) is a member of the large and diverse radical S-adenosyl-l-methionine (SAM) superfamily, members of which use an iron–sulfur cluster and SAM to initiate difficult radical transformations in all kingdoms of life.1−3 Radical SAM enzymes share a common CX3CX2C motif or variation thereof, and the conserved cysteines coordinate three irons of a [4Fe-4S] cluster, while SAM coordinates the fourth iron through its amino and carboxylate moieties.4,5 Radical SAM catalysis is thought to be initiated by electron transfer from the reduced [4Fe-4S]+ cluster to the sulfonium of SAM, inducing S–C(5′) bond cleavage to give methionine and a 5′-deoxyadenosyl radical (5′-dAdo•) intermediate that abstracts a hydrogen atom from substrate. While the 5′-dAdo• intermediate has never been observed for PFL-AE nor for any other radical SAM enzyme, recent results reveal the involvement of a novel organometallic intermediate in which the SAM-derived dAdo moiety is directly bound via the 5′-C to the unique iron of the [4Fe-4S] cluster.6 This organometallic intermediate is proposed to undergo homolytic Fe–C bond cleavage to yield the 5′-dAdo•.6 In the case of PFL-AE, the 5′-dAdo• radical then abstracts a hydrogen atom from glycine 734 of pyruvate formate-lyase (PFL), activating it for the catalytic conversion of pyruvate and coenzyme A (CoA) to formate and acetyl-CoA under anaerobic conditions (Figure 1).7,8 Careful studies of the PFL-AE/PFL system allowed quantitative determination of the product of H atom abstraction, the stable glycyl radical on PFL, demonstrating the 1:1 stoichiometry between electron loss from the [4Fe-4S]+ cluster and product formation on PFL.9
Figure 1.

Reaction catalyzed by PFL-AE. The PFL-AE substrate PFL, is activated by H atom abstraction from G734, concomitant with conversion of SAM to methionine and 5′-deoxyadenosine.
Two crystal structures of PFL-AE have been reported: one of PFL-AE/SAM and another of PFL-AE/SAM with a bound 7-mer PFL peptide substrate analogue of PFL containing the target glycine residue.10 The structures reveal a partial (β/α)6 triosephosphate isomerase (TIM) barrel with a wide opening able to accommodate the large substrate PFL, a domain of which is proposed to bind in the active site of PFL-AE.10 SAM is bound through the amino and carboxylate moieties to the unique iron of the cluster, a binding mode first revealed by electron nuclear double resonance (ENDOR) spectroscopy.5 Two motifs conserved in the radical SAM superfamily interaction with SAM: the GGE motif (PFL-AE residues G77, G78, and E79) directly binds the methionine portion of SAM, while the GXIXGXXE motif (G167VIVGXXE175 in PFL-AE) stabilizes the adenine moiety binding site. The PFL peptide substrate is bound across the lateral opening of the partial TIM barrel primarily by contacts between the peptide backbone and PFL-AE side chains. One of these interactions involves the highly conserved DGXGXR motif located on PFL-AE loop A (residues 10–20), and upon peptide binding, this loop undergoes a large conformational change by swinging up into the active site to establish these interactions. The movement of this loop is thought to be essential to the activation of PFL, possibly by orienting the glycine loop of PFL in the active site.10
The PFL substrate of PFL-AE is a homodimer wherein each monomer consists of a 10-stranded α/β barrel with the active site located at the center.11,12 Within the active site are two catalytically essential cysteine residues, C418 and C419, as well as the site of the glycyl radical, G734. This active site is buried 8 Å from the surface of the protein; however, G734 must be directly accessed by the active site of PFL-AE in order for the stereospecific H atom abstraction that activates PFL. Taken together, the structures of PFL and PFL-AE thereby suggest that a significant PFL conformational change is required for the glycine-containing loop to be accessible to the active site of PFL-AE. Experimental evidence for such a conformational change has been provided by a study utilizing fluorescence and electron paramagnetic resonance (EPR) spectroscopies and activity assays that together point to an open conformation of PFL in the presence of PFL-AE.13 In the proposed open conformation of PFL, the radical domain containing G734 flips out of the center of PFL and becomes accessible to PFL-AE.13
Our studies of PFL-AE over many years provided a detailed understanding of the EPR spectroscopic properties of the active site [4Fe-4S]+ in the absence and presence of SAM. We were therefore surprised when we began to see significant batch–batch variations in these spectra, and we ultimately drew a connection between these spectroscopic differences and changes in buffer salt composition. Subsequently, close inspection of the structure of PFL-AE in complex with the PFL model 7-mer peptide revealed the presence of a bound monovalent cation in the active site.
To identify this cation, to characterize the cation binding site, to determine the likely identity of the in vivo metal occupying that site, and to discover the functional significance of this cation, we have carried out detailed structural refinement and analysis, EPR, ENDOR, and circular dichroism (CD) spectroscopic studies of cation binding, and studies of the variations in enzymatic function with choice of cation. Instances where a monovalent metal ion interacts with a protein-bound metal center14 and even more importantly, is essential for enzymatic catalytic function,15,16 are rare. However, in yet another parallel between the radical SAM and B12 radical enzymes, assays of PFL-AE activity reveal that its catalytic function is controlled by the presence and identity of the bound monovalent cation, requiring a K+ ion bound in the active site for optimal activity, as is the case for the B12 enzyme dioldehydrase.16,17
Materials and Methods
All chemicals were obtained from commercial sources and were of the highest purity commercially available. The enzymes citrate synthase and malic dehydrogenase were purchased from Sigma.
Refinement of Metal Ions/Water Molecule in the Putative Metal Site
The previously reported PFL-AE crystallization conditions consisted of 100 mM HEPES, 3.5 M sodium formate and 0.2 mM 2,6-dimethyl-4-heptyl-beta-d-maltopyranoside, pH 6.8, and the resulting peptide-PFL-AE model was refined in CNS.10 To further investigate a putative metal ion binding site, ten additional cycles of refinement of the deposited structure (PDB entry 3CB8) were carried out in parallel in REFMAC518 in the CCP4i program suite19 with the putative site modeled as a water molecule, sodium ion, magnesium ion, potassium ion or calcium ion (note that other than water, of these ions, only sodium was known to be present in the crystallization conditions). Additionally, a round of refinement was carried out in CNS20 using the same starting model with water, sodium, magnesium, potassium or calcium modeled in the metal site, this time, refining the occupancy of metal site with the site’s B-factor set to 76.00 (the average B-factor of the surrounding atoms). The B-factors, R-factors, occupancies, distances to nearby atoms (Table S1–S5), and difference density maps (Figures S1 and S2) of the resulting models were examined to judge likelihood of each atom type’s presence in the site in question.
Expression and Purification of PFL and PFL-AE and Generation of Variants
PFL-AE wild-type and variants were expressed and purified as previously described5,21−24 with modifications as described here and in the SI. The pCAL-n-EK plasmid containing the PFL-AE gene was transformed into E. coli BL21(DE3)pLysS (Stratagene) cells for overexpression. Mutagenesis was carried out using the QuikChange site-directed mutagenesis kit from Stratagene and plasmids containing the variant genes were transformed into BL21(DE3)pLysS cells. Mutagenesis primers to introduce the D104A change (CAT TCA TAC CTG TCT GGC CAC CAA CGG TTT TGT TC) and the D129A change (CGT TCA TCT GTT TGA GAG CGA GCA TTA CCA GGT C) were synthesized by Integrated DNA Technologies. Purification of “metal-free” PFL-AE involved the omission of alkali metal cations in all purification buffers, in order to minimize occupancy of the monovalent cation site. The Tris buffers were made by using Tris base, and adjusting the pH with HCl. In addition, no alkali metal salts were added to the PFL-AE purification buffers. Excess monovalent cations were removed from the PFL-AE substrate, namely PFL, by dialysis. All further details are provided in the SI.
PFL-AE Activity Assays
PFL-AE activity was assayed in a Unilab MBraun anaerobic chamber containing ≤1 ppm of O2 using a modified version of the coupled enzyme assays previously published.21,25,26 For assaying the effect of each metal ion on activity, a 100 mM Tris pH 7.6 buffer containing 100 mM of monovalent cation salt (NaCl, KCl, NH4Cl, RbCl, CsCl, or LiCl) or 1 mM of divalent cation salt (CaCl2, MgCl2, or Zn(NO3)2) was used. A control lacking any salt addition was also conducted with each assay. To carry out the assay, 450 μL of activation solution was mixed containing 0.05 μM PFL-AE purified in the absence of M+, 5 μM PFL, 0.1 mM SAM, 10 mM oxamate (or 10 mM pyruvate for assays conducted with CaCl2), 8 mM DTT and 25 μM 5-deazariboflavin (added last in the dark) in a 100 mM Tris, pH 7.6 buffer containing different cations as described above (all concentrations given as final concentrations). This mixture was added to a shortened, acid-washed NMR tube and PFL-AE was photoreduced by illumination with a 300 W halogen lamp and the protein was kept in a 30 °C ± 2 °C water bath for 5 min and then covered with foil to prevent further reduction. To assay the activity, 5 μL of the resulting activation solution was placed on a lid of an anaerobic cuvette containing 895 μL of coupling solution (3 mM NAD+, 55 μM CoA, 0.05 mg/mL BSA, 10 mM pyruvate, 10 mM malate, 2 U/mL citrate synthase, 30 U/mL malic dehydrogenase, and 10 mM DTT in a 100 mM Tris, pH 8.1 buffer) prior to sealing the cuvette and removing it from the chamber. The two solutions were mixed by inverting the cuvette just prior to placing in a thermostated (at 30 °C) Cary 60 UV–vis spectrophotometer. The production of NADH was monitored at 340 nm for 90 s. Rates were calculated from the slope of absorbance vs time from 18 to 90 s, when the slopes stabilized and were the most linear. One unit of PFL activity corresponds to the production of one μmole of pyruvate per minute, and 35 units of PFL is equivalent to 1 nmole of PFL active sites.27 The definition of one unit of PFL-AE activity is the amount that catalyzes the production of 1 nmole of active PFL per minute.21 All assays were conducted in triplicate.
To examine the dependence of PFL-AE activity on the concentration of K+, PFL-AE was assayed with 500, 300, 200, 100, 75, 50, 25, 10, or 0 mM KCl in a 100 mM Tris, pH 7.6 buffer with 1 mM DTT. The ionic strength was maintained at a constant value by adding choline chloride for those with less than 100 mM KCl and activity was monitored.
EPR Sample Preparation and Spectroscopy
In an MBraun anaerobic chamber with ≤1 ppm of O2, 300 μM PFL-AE, 100 μM 5-deazariboflavin (added last in the dark), 5 mM DTT in a 100 mM Tris, pH 7.6 buffer containing either no cation, or 100 mM of monovalent cation salt (NaCl, KCl, NH4Cl, RbCl, CsCl, or LiCl), or 1 mM of divalent cation salt (CaCl2, MgCl2, or Zn(NO3)2) was placed in an EPR tube. Photoreduction was accomplished by illumination with a 300 W halogen lamp in an ice bath for 1 h, and samples were either frozen as-is or SAM was added to a final concentration of 2 mM and incubated for ∼5 min before freezing in liquid nitrogen.
EPR spectra were recorded on a Bruker EMX X-band spectrometer equipped with a liquid helium cryostat and temperature controller from Oxford Instruments. Typical experimental parameters were 12 K and 9.37 GHz, with 1.0 mW microwave power, 100 kHz modulation frequency, and 10 G modulation amplitude. Each spectrum is the average of four scans. The EPR spectra in the presence of SAM were simulated using Easy Spin version 5.1.11.28
Circular Dichroism
CD experiments were conducted under anaerobic conditions using a Jasco-710 spectropolarimeter at ambient temperature (23 °C ± 2 °C). Measurements were collected in a 1 cm path length cuvette from 300 to 800 nm with a sensitivity of 100 millidegrees, 0.1 nm data pitch, continuous scan mode with a speed of 100 nm/min, a response time of 1 s, 1.0 nm bandwidth, and accumulation of 3 scans. PFL-AE was added to an anaerobic cuvette in an MBraun anaerobic chamber to a final concentration of 50 μM containing 1 mM DTT in a 100 mM Tris, pH 7.6 buffer and either no cation or 100 mM of NaCl, KCl, NH4Cl, RbCl, CsCl, or LiCl. After initial scans without SAM were conducted, SAM was added to a final concentration of 500 μM. For determination of the KD for K+, a final concentration of 50 μM PFL-AE and 500 μM SAM was used prior to KCl addition. KCl was titrated to final concentrations of 0, 2, 3, 6, 12, 21, 35, 49, 301, and 500 mM.
ENDOR Measurements
Q-band CW ENDOR spectra were collected as described.29,30 Pulsed ENDOR spectra were collected at 2 K on a previously described spectrometer.30 The Mims pulse sequence (π/2−τ–π/2–T–π/2−τ–echo, RF applied during interval T) was used for small hyperfine couplings.31,32
For a nucleus with hyperfine coupling, A, Mims pulsed ENDOR has a response R which depends on the product, Aτ, according to the equation,
| 1 |
The response function R (eq 1) equals zero, corresponding to minima in the Mims ENDOR response (hyperfine “suppression holes”), when Aτ = n, n = 0, 1, ..., and maxima at Aτ = (2n + 1)/2; n = 0, 1, ...33 The “holes” at n = 1, 2, 3, ... can be adjusted by varying τ, but the “central”, n = 0, hole at ν = νN persists regardless. This can be of significance in distinguishing a tensor that is dominated by anisotropic interactions from one that is dominated by isotropic ones. The latter would never lead to ENDOR intensity near νN; the former does so for certain orientations, but the ν = 0 Mims hole tends to diminish the differences between the two cases. Simulation of the ENDOR patterns employed the program, ENDORSIM.34
Results
Refinement of Metal Ions/Water Molecule in the Putative Metal Site
The X-ray crystal structure of PFL-AE/SAM/7-mer PFL peptide10 reveals a previously unreported putative metal binding site in the vicinity of the active site. Two aspartic acid side chains (D104 and D129), two carbonyl oxygens from residues T105 and M127, and one oxygen from the carboxylate of SAM point toward the unidentified electron density in a distorted trigonal bipyramidal geometry, with each of the potential oxygen ligands within 3 Å of the center of the site (Figures 2 and S1). We thus carried out further refinements directed toward characterizing this putative metal site, using an approach similar to that described previously.35
Figure 2.

Putative metal site with sodium modeled (purple sphere). Protein amino acids are shown in gray, a portion of SAM is shown in green, and [4Fe-4S] cluster is shown in yellow and rust. (a) 2Fo–Fc electron density map contoured at 0.75 σ and displayed as a blue mesh within 2 Å of the sodium ion. Refined distances to sodium are also displayed. (b) Angles between metal ligands. (c) Stereoview of the putative metal site with sodium modeled.
The most likely candidate ion to occupy this site in the crystal structure is Na+, as this cation is present in the crystallization buffer as well as throughout the protein purification. The protein also is exposed to Mg2+ in early purification steps and so this cation was considered as well. In addition, we considered the possibility that a water molecule might account for the electron density observed in this site. For completeness and comparison, we also carried out refinements with the species in this site assigned as K+ and Ca2+.
The locations (Figure S2) and B-factors (Table S4) of the four protein-derived oxygen ligands do not change significantly after refinement in REFMAC, regardless of whether water, sodium, potassium, magnesium, or calcium was modeled into the site (Figure S2), though small shifts do occur to accommodate the different atoms’ radius (for example, compare the distances listed in Tables S1 and S2). To examine whether the presence of water or a metal ion provides better agreement with the crystallographic data, we set the occupancies of these ions/molecule to 100%, and compared the B-factors for each following refinement (Tables S3 and S4). A resulting B-factor lower than the average of the surrounding area indicates that the ions/molecule has too little scattering to account for the density, whereas if the B-factor is higher, that is an indication of too much scattering. The best fit is the ion/molecule with a B-factor that is most similar to the average in that area. We find that water has significantly lower than the average B-factor compared to the surrounding atoms, calcium and potassium are significantly higher, and sodium and magnesium are the closest to average (Table S4). In addition, more extensive positive Fo–Fc electron density is present in this site when modeled with water than when modeled with sodium or magnesium (Figure S2), and negative Fo–Fc electron density appears when the site is modeled as potassium or calcium (Figure S2). These observations are consistent with the presence of sodium or magnesium in this site.
As an additional approach, we set the B-factor of the ions/molecule to the average of the surrounding area, allowed the occupancies to refine, and compared the resulting occupancies (Table S5). An occupancy value of higher than 100% would indicate that the modeled species has too little scattering, while a lower occupancy would reflect that the modeled species has too much scattering power. Whereas water had too high an occupancy, the other ligands had occupancies of less than 100%. As expected, sodium and magnesium had the most reasonable occupancy values. Again, these observations are consistent with presence of sodium or magnesium in this site, with sodium more likely because it was known to be present in our crystallization buffer at high concentrations (3.5 M sodium formate) and exposure of the protein to magnesium was limited to early purification steps.
We also considered coordination geometry and the average distance of the oxygen ligands (Tables S1 and S2) in our assignment of the likely ion/molecule to occupy this site. On the basis of these criteria, water is not a good candidate, as it would not be capable of forming the number (5) and the geometry (distorted trigonal bipyramidal) of interactions observed. In contrast, trigonal bipyramidal is a common geometry for metal ions. The average metal–oxygen distance (2.5 Å) agrees with known sodium ion sites in protein structures as well as the Cambridge Structural Database.36 Our conclusion is therefore that Na+ occupies this metal binding site in the PFL-AE/SAM/peptide crystal structure (Figure 2). However, a different metal ion might occupy this site in vivo.
EPR and the Effect of Cations on the Electronic Structure of PFL-AE
The proximity of the monovalent cation site to the [4Fe-4S] cluster suggested the cation might affect the electronic and/or metrical structure of the active site of PFL-AE. This was investigated using EPR spectroscopy of PFL-AE in the presence of a variety of monovalent and divalent cations, and with and without SAM.
In the absence of SAM, the signal from the [4Fe-4S]+ cluster changes with the presence and identity of the cation (Figure S3). The M+-free signal is slightly heterogeneous, and the heterogeneity varies with the identity of M+.
As has been previously shown,9,37 the addition of SAM to PFL-AE causes a marked change in the cluster signal. The majority rhombic component of the SAM-free signal converts to a majority axial signal, with g|| = 2.00, g⊥ = 1.89, with a minority rhombic component (see Table S6 for all g-values), and an intensification of the [4Fe-4S]+ EPR signal.
The effects of the monovalent cation on the EPR signal are even more pronounced in the presence of SAM, which arranges the spectra in order of increase M+ ionic radius (Figure 3). The EPR signals for M+-free PFL-AE in the absence of SAM and for all the PFL-AE samples in the presence of different monovalent cations and SAM were simulated as a sum of spectra from two different enzyme states (Figure S4). One state has a near-axial g-tensor, g1 = [2.01, 1.89, 1.88], similar to those previously reported for photoreduced PFL-AE9,37 and independent of the added cation; the second state has a rhombic g-tensor, but with precise values that depend slightly on the identity of the cation: g2 ≈ [2.01, 1.92, 1.87] (Table S6). The relative populations of the two states varies strongly with cation. For both the smallest (Li+) and largest (Cs+) cations the near-axial signal is the majority component of the signal. Indeed, those spectra are little different from the spectrum in the absence of M+, although ENDOR spectroscopy shows that both cations do bind (see below). The signal from the sample with K+ has by far the largest contribution from the rhombic component, with g2 = 1.920; activity measurements presented below suggest this represents the active conformation.
Figure 3.

EPR spectra of PFL-AE in the presence of SAM and the indicated monovalent cation. Spectra are stacked in order from smallest (Li+) to largest (Cs+) cation. Solid vertical lines are drawn at g values of the nearly axial signal (2.005 and 1.889), and dashed vertical lines are drawn at typical g-values of the more rhombic signal (2.007, 1.920, 1.868). The origin of the modest variation in signal intensity is unknown. Each sample contained 300 μM PFL-AE in the presence of 100 mM of the cation indicated and 2 mM SAM. EPR parameters: 12 K, 9.37 GHz, 1.0 mW.
EPR spectra of PFL-AE in the presence of Ca2+ or Mg2+ were essentially identical to those for the M+-free PFL-AE both for SAM-free and SAM-bound enzyme (Figure S5), and activity measurements (see below) support the suggestion that unlike the alkali metal cations, divalent cations do not bind.
To further analyze how different cations affect the active site in PFL-AE, CD spectroscopy in the visible region was performed on the M+-free purified protein in the presence and absence of different monovalent cations and SAM. CD spectroscopy in the visible region is used to analyze d–d transitions of metals in chiral environments such as the [4Fe-4S] clusters in PFL-AE. In the absence of SAM, the M+-free spectrum is unaffected by the addition of a cation (Figure S6). On the other hand, when SAM is present, new spectral features appear in the presence of K+, NH4+, Rb+, and Na+ (Figure S7), with the most prominent at approximately 400 and 500 nm. These features grow in with increasing concentrations of K+ (Figure S8), plateauing after 50 mM KCl. A fit of these spectral changes yielded, KD = 7.6 ± 1.2 mM (Figure S8). Above ∼180 mM KCl the intensity drops off drastically, which we attribute to perturbation of the active site due to the high ionic strength.
Altering the Cation Binding Site
To further characterize the cation binding site in PFL-AE, the two conserved aspartate (D104 and D129) residues coordinating the monovalent cation were changed to alanine (D104A and D129A, respectively). D129 of PFL-AE hydrogen bonds to the ribose hydroxyls of SAM in addition to coordinating the monovalent cation;10 the corresponding residue in the radical SAM enzyme biotin synthase is a catalytically essential asparagine (N153) that also hydrogen bonds to a ribose hydroxyl of SAM but does not coordinate a monovalent cation.38,39
PFL-AE activity assays in the presence of 100 mM KCl revealed that the D104A variant had very low activity (5.5 ± 0.2 U/mg, compared to 54.2 ± 1.5 for WT enzyme). EPR analysis of this variant confirms that D104 is important for cation binding. First, none of the monovalent cations had a significant effect on the EPR signals for [4Fe-4S]+-PFL-AE(D104A) or [4Fe-4S]+-PFL-AE(D104A)/SAM, with the EPR spectra in all cases looking very similar to the buffer exchanged M+-free D104A variant (Figure S9). In addition, the EPR signal observed for D104A in the presence of SAM is similar to D104A M+-free in the absence of SAM, suggesting SAM does not bind well in this variant (Figure 4).
Figure 4.

EPR spectra of the PFL-AE wild type (solid line) and variants D104A (dashed line) and D129A (dotted line) in the presence of SAM, and in the absence or presence of 100 mM K+ or Na+. EPR parameters are as for Figure 3.
The PFL-AE(D129A) variant, on the other hand, retains the ability to bind cations. The EPR spectra of PFL-AE(D129A) vary with the presence and identity of cation, particularly in the presence of SAM (Figure 4), giving rise to spectra that are very similar to those of the WT enzyme, indicating that this variant binds M+ and SAM in a manner similar to WT. Further, in the presence of K+ this variant exhibits only moderately lower enzymatic activity than WT enzyme (42.7 ± 3.4 vs 54.2 ± 1.5 U/mg).
ENDOR Studies of Cation Binding Near the Cluster
To identify and further characterize cation binding to the newly identified site, we carried out ENDOR studies characterizing the interactions between the cation nuclear spin and the electron spin of the [4Fe-4S]+ cluster of PFL-AE. To our knowledge, the only previous study showing hyperfine coupling to a monocation located nearby an FeS cluster involved a Cs+ ion bound to a residue near the S = 1/2 [Fe3S4]+ cluster of the hyperthermophilic archaeon Pyrococcus furiosus.14 The present ENDOR studies both directly examine the cation bound in the active site of PFL-AE, and probe how the presence and identity of the cation influences the positioning of SAM.
M+ Binding
The first result of this approach is confirmation that Na+ occupies this metal binding site in the PFL-AE/SAM complex in a solution of enzyme isolated with buffers that contain Na+. A 35 GHz Mims ENDOR spectrum collected from such a sample (Figure 5A) exhibits a doublet centered at the 23Na Larmor frequency, split by a hyperfine coupling, A = 0.14 MHz. As shown, this doublet is barely detectable in a sample purified in the absence of 23Na+ (or any alkali metal cation) added to the buffer. The full 23Na+ intensity is restored by 23Na+ loading, and the weak signal is completely eliminated upon treatment with, and replacement of 23Na+, by a range of other monovalent cations, as illustrated for Tl+. Clearly, studies to date have invariably involved enzyme with 23Na+ bound in the active site, since standard purification procedures involve buffers containing NaCl. Indeed, in hindsight, this signal was initially seen, although not assigned, in our early studies of PFL-AE.23
Figure 5.

35 GHz Mims 23Na ENDOR spectra of PFL-AE/SAM: (A) 23Na ENDOR spectra at g2 (g ∼ 1.89) for PFL-AE/SAM/as-isolated (black), Na+ treated (red), M+-free (brown) and Tl+ treated (green). (B) 2D field-frequency pattern of 23Na ENDOR spectra for PFL-AE/SAM/as-isolated (black). Experimental fields as indicated. ENDOR simulations are shown in blue. Experimental conditions: microwave frequency ∼34.8 GHz; microwave pulse length (π/2) = 50 ns; τ = 1250 ns; and T = 2 K. Simulation parameters: aiso = −0.03 MHz, T = 0.11 MHz, Euler angle β = 30°.
Figure 5B shows the 2D pattern of 23Na+ Mims ENDOR spectra collected across the EPR envelope of the PFL-AE/Na+/SAM complex. The field-dependent features are well described by an axial hyperfine-coupling matrix, A = [0.19, −0.14, −0.14] (MHz), which can be decomposed into an axial anisotropic interaction, T = [2T, −T, −T], T = 0.11 MHz, and, surprisingly an isotropic coupling, aiso = −0.03 MHz. The presence of this latter coupling requires that the spin-density delocalized onto the SAM carboxylate ligand to the unique Fe is further delocalized onto Na+. Whether this delocalization involves weak covalency or merely “Pauli delocalization”40−43 through intimate contact between carboxylate-O and Na+, its presence does require direct interaction/contact between Na+ and this ligand, with spin delocalization as indicated in Scheme 1. Incidentally, the isotropic coupling to a monocation equals the spin density in its “valence” s-orbital, ρs multiplied by the coupling for a single electron in that orbital, As, aiso = Asρs, and use of a tabulated value for As(44) shows that the aiso for 23Na+ corresponds to only ∼3 × 10–5 of a spin in the 23Na 3s orbital.
Scheme 1. M+-SAM-Cluster Interaction.
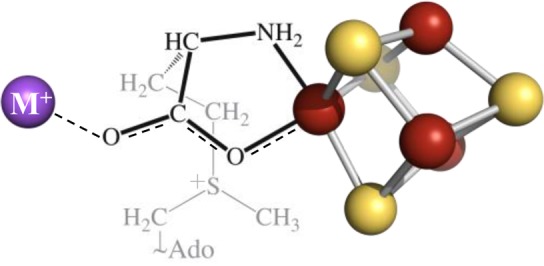
M+ is shown in purple, iron in rust, and sulfur in yellow.
The dipolar constant, T, is somewhat larger (+0.03 MHz) than would be expected for the through-space interaction spin on the unique Fe ion that is 6.5 Å away from Na+ (or all Fe ions on the cluster), which can be explained by additional interaction with the small spin densities on the carboxylate. The dipolar interaction with a center bearing spin-density ρ at distance r from the nucleus scales as T ∼ ρ/r3, and the difference between the measured T and that calculated for ρ = 1 at 6.5 Å is equivalent to an interaction with ρ = 1% of a spin on the carboxylate-O at r = 2.4 Å.
We next examined the samples treated with the other M+ ions. Hyperfine coupling between the reduced cluster and the cation is observed for Li+, Cs+, and Tl+, as well as Na+ (Figure 6), demonstrating that these three also bind to the cation site. Figure S10 shows the 2D field-frequency pattern of Mims ENDOR spectra for Li+, Cs+, and Tl+ ions, and Table S8 contains the hyperfine-tensor parameters and ns-orbital spin densities derived from their simulation. The spin densities for the other three ions bound to PFL-AE are within a factor of ∼3 of that for Na+. Although no signal was observed for K+ or Rb+, their presence abolished the residual 23Na+ signal; that, and the influences of those ions on the 13C ENDOR of 13CH3–SAM (see below), show that they likewise occupy the cation binding site. The major isotope of each of these two nuclei has an extremely small nuclear g-factor; it is the resulting low Larmor frequencies and electron–nuclear hyperfine interactions that render their signals unobservable.
Figure 6.
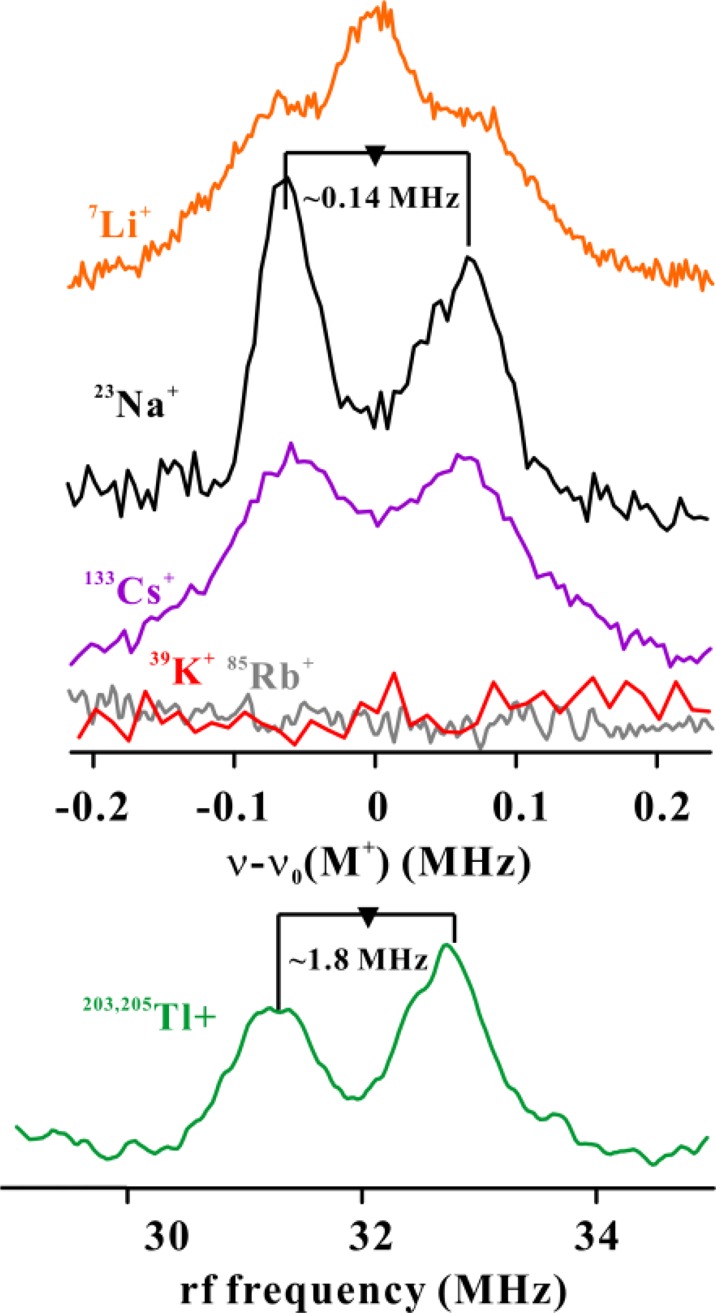
(top) Mims ENDOR spectra at g2 are centered at each nuclei Larmor frequency. (bottom) CW ENDOR spectrum at first g2 (g ∼ 1.94). Larmor frequencies for 203Tl (30% natural abundance) and 205Tl (70% natural abundance) are 31.93 and 32.23 MHz, respectively. Conditions: For Mims ENDOR, same as Figure 5. For CW ENDOR, microwave frequency ∼34.97 GHz, microwave power 1 mW, 100 kHz modulation amplitude 0.3 G, rf sweep rate 2 MHz/s and T = 2 K.
The 2D field-frequency patterns of ENDOR spectra were collected for each of the three ENDOR-observable alternative ions (Li+, Cs+, and Tl+), and analyzed as for 23Na+. The data reveal that all four cations, whether small (Li+), large (Cs+, Tl+), or in between (Na+), are able to bind in the cation site as a tight ion-pair with the carboxylate-O, as indicated by the presence of a small isotropic contribution to the hyperfine interaction, Figure S10. The coupling to Tl+ is ∼50 times larger than those to the other ions, A ∼ 2 MHz, simply because As, the coupling per s-orbital electron for this high atomic-number ion is much larger than those for the other ions.44 For comparison, we note that Tl+ actually incorporated into the [Fe3S4]+ cluster of the hyperthermophilic archaeon Pyrococcus furiosus to form a S = 1/2 [TlFe3S4]+ cluster displays a hyperfine coupling of A ∼ 370 MHz.14 Whereas ENDOR of the radical-pair intermediate trapped during reaction of the B12 enzyme dioldehydratase shows no hyperfine coupling to Tl+ bound in the monocation site of this enzyme.45
Finally, the Li+ and Cs+ spectra show additional intensity at the Larmor frequency associated with “distant” ions, presumably present in the solvent-accessible cavity at the active site.
ENDOR Study of M+ Influence on SAM Binding Geometry
To test for a possible influence of the cation on the positioning of the sulfonium target for SAM cleavage relative to the PFL-AE [4Fe-4S] cluster, we used ENDOR to study the PFL-AE/SAM complex with the sulfonium methyl labeled with 13C. We had previously studied the CH3 positioning for the Na+-bound PFL-AE, although the presence of the cation was not recognized at the time.23 Analysis of 2D field-frequency 13C ENDOR patterns revealed an isotropic hyperfine contribution, which requires that SAM lies in direct contact with the cluster. From the ENDOR-derived anisotropic hyperfine coupling contributions for 2H and 13C we estimated the distance from the closest methyl proton of SAM to the closest iron of the cluster to be ∼3.0–3.8 Å, while the distance from the methyl carbon to the nearest iron to be ∼4–5 Å.23 The subsequent X-ray structure determination confirmed, and made precise this picture.10
Figure 7 shows 13C Mims ENDOR spectra for PFL-AE\methyl-13C-SAM\M+ collected at g2, arranged according to the ionic radius of M+, as suggested by reactivity measurements described immediately below. For each M+, the spectrum is a doublet with hyperfine coupling, A ∼ 0.3 MHz. However, the shapes of the 13C doublets clearly vary with M+, and do so in a fashion that roughly reflects the M+ ionic radius. Thus, for Li+, the smallest M+, the individual peaks of the doublet are smooth and unstructured. The peaks for Na+ clearly instead show structure, and this structure is most evident in the spectrum for K+, which ion also appears to cause a slight increase in the doublet splitting, suggesting a slightly stronger interaction with the cluster spin. The structure persists through Cs+; it not only vanishes with Tl+, but the individual peaks are the narrowest of all for this ion. For completeness, the included spectrum with no M+ is also structured.
Figure 7.

35 GHz 13C Mims ENDOR spectra at g2 for PFL-AE/methyl-13C SAM/M+. Experimental conditions: As for Figure 5 except τ = 600 ns.
This “structure” in the 13C-methyl ENDOR spectra is clearly related to the heterogeneity in the EPR signals (Figure 3). We interpret it to mean that the ENDOR spectra are a superposition of distinct 13C signals from the two major active-site conformations associated with the two major components of the EPR signal, States 1 and 2 (Figure 3). This in turn implies that the position of the sulfonium methyl relative to the cluster Fe, and thus its ENDOR spectrum, is different in these two conformations. K+ stands out among the ions in Group 1 of the periodic table in the high occupancy of the “rhombic” configuration and the corresponding “structure” in the 13C-methyl ENDOR spectrum.
Effect of Cations on PFL-AE Activity
The cation-dependence of the EPR spectrum of PFL-AE/(M+)/SAM, the ion-pairing of the cation with the carboxylate-ligand of the iron–sulfur cluster, and the cation-dependence of the SAM positioning in the PFL-AE active site as shown by ENDOR, all suggested a possible role for this cation in the enzyme-catalyzed reaction. To test this, enzyme activity assays were carried out in the presence of each of six monovalent cations (Na+, K+, NH4+, Rb+, Cs+, and Li+) and three divalent cations (Ca2+, Mg2+, and Zn2+), as well as in the absence of any monatomic or ammonium cations. These experiments were carried out with PFL-AE that had been purified in the absence of such cations, using a Tris-Cl buffer with no added alkali metal salt. The results reveal a strong dependence of PFL-AE activity on the identity of the cation, with K+ providing the highest activity (Figure 8 and Table S7). Among monocations, going down Group 1 of the periodic Table from Li+ to Cs+, Figure 8 shows a sharp maximum at the ionic radius of K+, with NH4+ closely matching the efficacy of K+. The 13C ENDOR measurements correspondingly suggest that K+ likewise has a distinctive effect on the positioning of the sulfonium target for SAM cleavage (Figure 7). The lack of stimulation by Li+ and Cs+ is not due to an absence of cation binding, as ENDOR provides evidence that both of these cations bind in the metal site. In the presence of divalent cations (Ca2+, Mg2+, Zn2+) PFL-AE showed little-to-no activity (Figure 8 and Table S7).
Figure 8.

PFL-AE specific activity (U/mg) is influenced by the ionic radius of the cation present in the activity assay and thus the active site. Monovalent cations stimulate PFL-AE activity while divalent cations (blue box) inhibit PFL-AE activity relative to that in the absence of these cations (green-dashed line). The black dotted line is at zero activity. The red line is provided to guide the eye. For these assays, a concentration of 5 μM PFL, 0.05 μM PFL-AE, and 0.1 mM SAM in the presence of 100 mM of the specified monovalent cation or 1 mM of divalent cation was used. Data are the average of at least three different trials with each trial done in triplicate. “M+-free” is PFL-AE purified in the absence of any alkali metal cation and no cation is added to the assay.
By carrying out the PFL-AE assay at a range of K+ concentrations, we were able to estimate a KD of 44. ± 11. mM for K+ binding to the PFL-AE/SAM complex under assay conditions (Figure S8). Although this value is higher than that estimated via the CD titrations described above (7.6 ± 1.2 mM), the experimental conditions vary (including temperature, presence and concentrations of various buffer components, presence of the PFL-AE substrate PFL in the activity assays) by necessity between these two experiments, and we attribute the differing values to these disparities. We previously used EPR spectroscopy to estimate KD = 6.5 mM for K+ binding to PFL-AE/SAM,46 a value in line with the CD measurement reported herein. Regardless, KD in the range of 7–44 mM puts the binding affinity in a range that would provide near maximal PFL-AE activity at the typical in vivo K+ concentrations in E. coli of ∼200 mM.47
Discussion
Close examination of the X-ray crystal structure revealed the presence of a monovalent cation in the PFL-AE active site coordinated by 5 oxygens: two carboxylate oxygens from conserved aspartate residues (D104 and D129), two backbone carbonyls from T105 and M127, and one carboxyl oxygen of SAM. This cation binding site is located at the back of the active site against the barrel, and given its interactions with both conserved amino acid residues and SAM, we hypothesized that this cation likely played an important role in active site structure, SAM binding, and enzymatic activity.
To probe monovalent cation binding in solution, we used EPR and ENDOR spectroscopies. Clear perturbations in the EPR lineshapes and g-values were observed that depended on the presence and identity of the cation, with the most dramatic effects occurring when SAM is also present. These observations are consistent with the binding of monovalent cations in the PFL-AE active site as identified by X-ray crystallography, with SAM providing one of the ligands to the monovalent cation. The maximum in activity at K+ seen in going down Group 1 of the periodic table (Figure 8) correlates with the maximum contribution of a “rhombic” component to the EPR of the enzyme with bound SAM/K+ (Figure 3). 13C ENDOR spectroscopy correspondingly shows that the location of the target sulfonium of SAM is different in the “rhombic” conformation (Figure 7), which suggests that the size of the cation plays a key role in the optimal positioning of SAM relative to the [4Fe-4S] cluster (Figure 7), and that the positioning in the “rhombic” conformation enhances reactivity.
ENDOR spectroscopy has also been used to probe the electronic coupling of the monovalent cations to the [4Fe-4S] cluster. As shown in Figure 6, an ENDOR signal due to the interaction between the nuclear spin of the cation and the electron spin of the [4Fe-4S]+ cluster is observed for cations that bind in the metal site identified by X-ray crystallography. Such a signal is observed for Li+, Na+, Cs+, and Tl+, indicating that these cations are indeed bound at this metal-binding site in close proximity to the [4Fe-4S]+ cluster. While we do not observe ENDOR signals in the case of K+ and Rb+, this is attributed to the low Larmor frequencies and electron–nuclear hyperfine interactions for these nuclei rather than to lack of binding; in fact, addition of either K+ or Rb+ abolishes the Na+ ENDOR signal for PFL-AE purified in Na+-containing buffers, providing indirect confirmation that these cations bind in the same site. The existence of an isotropic hyperfine coupling of the spin on the [4Fe-4S] cluster to the ions with observed ENDOR response shows the existence of intimate contact between the Fe-bound carboxylate-O of SAM and the M+ ion, as indicated in Scheme 1.
Focusing on the strong dependence of steady-state rates on the presence and identity of the monovalent cation, they show that monovalent alkali metal ions stimulate PFL-AE activity in a pattern that correlates with their position in Group 1 of the periodic table. Figure 8 suggests this correlates with a tight activity optimum at the ionic radius of K+, with gradual falloff to lower radius and a sharp falloff to higher. Potassium is generally the most abundant alkali metal cation in E. coli cells, with concentrations typically ∼200 mM.47 Half-maximal activation was obtained at [K+] = 44 ± 10.5 mM, reflecting a binding constant in the physiologically relevant range.
Divalent cations do not activate PFL-AE, and relative to enzyme isolated in the absence of monatomic or ammonium cations, divalents appear to be slightly inhibitory (Figure 8). However, although our data appears to show that in the absence of monovalent cation PFL-AE retains a small amount (∼10%) of activity, it is instead likely that this residual activity is due to trace amounts of monovalent cations present even in “metal-free” protein. This conclusion is supported by the weak but unambiguous Na+ ENDOR signal from nominally “M+-free” PFL-AE, Figure 5. Thus, we suggest that the divalent cations should be viewed as being nonactivating, rather than inhibitory. Indeed, there is no direct evidence that these divalent cations really bind.
The monovalent cation binding site in PFL-AE involves two conserved aspartic acid residues, D104 and D129. Changing either of these amino acids to alanine decreased activity, although the decrease was modest for D129A and dramatic for D104A. Analysis of the EPR spectra of the two aspartate variants suggest that D104 is required for cation binding while D129 plays a less critical role. First, changing the monovalent cation induces almost no change in the EPR signal for D104A, suggesting cations are not binding effectively to this site. Second, the EPR signal of D104A is not significantly altered in the presence of SAM, indicating that the lack of cation binding negatively impacts SAM binding. In contrast, EPR spectroscopic studies of the D129A variant indicate that the monovalent cations bind, and SAM coordinates in a manner similar to that of the WT.
Observation of an active-site monovalent cation in PFL-AE that directly interacts with SAM and the [4Fe-4S] cluster and affects catalytic activity is an unexpected and significant finding. To evaluate the prevalence of such a cation site in the radical SAM superfamily, we performed an alignment of PFL-AE with other radical SAM enzymes, including other glycyl radical enzyme-activating enzymes (GRE-AEs), with a focus on the DTX21MXD motif that spans the ligands to the monovalent cation in PFL-AE (Figure 9). The key acidic residues that serve as ligands to the monovalent cation in PFL-AE are conserved in only a subset of the activating enzymes, and are absent in other radical SAM sequences examined, although in some cases alternative potential ligands replace the aspartate residues in the DTX21MXD motif (Figure 9). A third ligand to the cation site in PFL-AE is a backbone carbonyl of T105; interestingly, this residue is conserved in many of the radical SAM sequences, including several for which the acidic residue ligands are not conserved. On the basis of the limited conservation of the acidic residue ligands to the monovalent cation, it appears that this monovalent cation is present in many, but not all, of the GRE-AE enzymes in the radical SAM superfamily. ENDOR spectroscopy appears to provide a means of rapidly screening other enzymes for cation binding. Understanding the detailed mechanism by which the monovalent cation serves to enhance catalytic activity of PFL-AE, and presumably several other GRE-AEs, awaits further studies.
Figure 9.

Sequence alignment of PFL-AE with other activating enzymes and selected radical SAM enzymes. This alignment focuses on the PFL-AE DTX21MXD motif, and has been truncated to show only the region between beta2 to alpha4 for clarity. In the sequences that have been characterized structurally, beta strands are highlighted in yellow and alpha helices are highlighted in cyan. The residues of the PFL-AE DTX21MXD motif are highlighted in pink and shown in bold text. Residues of this motif that are conserved in the other sequences are highlighted in pink, and conservative substitutions of residues in the motif are highlighted in maroon with bold white lettering. Note that at the positions corresponding to D104 and D129, acidic residues are highlighted in pink, and any other amino acid side chain that could act as a hard metal ligand is highlighted as a conservative substitution. The sequences aligned are E. coli pyruvate formate-lyase activating enzyme (UniProtKB ID P0A9N4), T. aromatica benzylsuccinate synthase activating enzyme (O87941), P. difficile 4-hydroxyphenylacetate decarboxylase activating enzyme (C9XIS7), C. butyricum glycerol dehydratase activator (Q8GEZ7), E. coli anaerobic ribonucleoside-triphosphate reductase-activating protein (P0A9N8), C. perfringens anaerobic sulfatase-maturating enzyme (Q0TTH1), E. coli oxygen-independent coproporphyrinogen III oxidase (P32131), E. coli biotin synthase (P12996), S. aureus molybdenum cofactor biosynthesis protein A (P65388), C. subterminale lysine 2,3-aminomutase (Q9XBQ8), C. hydrogenoformans HydG protein (Q3ABV3), T. maritima [FeFe]-Hydrogenase matures HydE (Q9X0Z6), B. circulans BtrN (Q8G907), E. coli ribosomal RNA large subunit methyltransferase N (P36979), and G. thermodenitrificans spore photoproduct lyase (A4IQU1).
Enzymes activated by metal cations such as Na+ or K+, including the many enzymes whose catalytic mechanism does not directly incorporate a metal center, can be classified as cofactor-like (type I) or allosteric (type II).48 In type I enzymes, the M+ is absolutely required for enzyme activity and helps to anchor the substrate in the active site, often acting in tandem with a divalent cation. In type II enzymes, no direct interaction between M+ and substrate occurs and the M+ is not absolutely required for activity or substrate binding. Our observation that PFL-AE most likely is inactive in the absence of a monovalent cation and that the cation binds in the active site and is coordinated by SAM, a cosubstrate with PFL in this reaction, plus the evidence from EPR spectroscopy that monovalent cation binding is required for SAM binding, points to PFL-AE being a type I M+ activated enzyme. It is interesting to note, however, that as yet there are few reported cases of metalloenzymes that are activated by a monovalent cation binding in the active site in proximity to the catalytic metal center. Among these are the Mn(II) fosfomycin resistance enzyme, FosA,15 and the B12-dependent dioldehydrase.16 We speculate that this may well change.
The monovalent cation might be playing one or more of a variety of roles. It clearly plays an important role in SAM binding and positioning in the active site of PFL-AE. The bridging coordination of the SAM carboxylate between the unique iron of the [4Fe-4S] cluster and the monovalent cation could affect the SAM-cluster interaction and thus reactivity. This would be consistent with the proposed role for the K+ bound in the B12-dependent dioldehydrase, where the binding energy of the cation is thought to induce a conformational change that stimulates homolytic Co–C bond cleavage.16 We have recently shown that PFL-AE catalysis involves an organometallic intermediate in which the 5′-C of the 5′-deoxyadenosine is directly bound to an iron of the [4Fe-4S] cluster;6 this intermediate is analogous to the B12 cofactor, which has a bond between the 5′-C of 5′-deoxyadenosine and the cobalt of cobalamin. By extension then, one might suggest that the monovalent cation in the active site of PFL-AE promotes Fe–C bond cleavage in this intermediate. Regardless, the observation that both the radical-generating B12 enzyme dioldehydrase and the radical SAM enzyme PFL-AE incorporate a monovalent cation in the active site, near the catalytic metal and in contact with substrate, potentially with the role of influencing conformation and promoting metal–carbon bond cleavage, provides yet one more intriguing parallel between radical SAM and B12 enzymes.
The role of the monocation could also include interacting with the methionine after SAM cleavage, perhaps orchestrating its departure from the active site. The presence of the monovalent cation and its interaction with SAM affects the charge balance in the active site, and how this might affect catalysis is unclear. Finally, and at the most basic level it does appear that the monovalent cation confers some stability to PFL-AE: the M+-free enzyme is more susceptible to stresses such as freeze–thaw events than the enzyme purified in the presence of monovalent cations.
Conclusion
We describe here for the first time the presence of a catalytically important monovalent cation in the active site of a radical SAM enzyme, PFL-AE. This monovalent cation is observed in the crystal structures of PFL-AE and identified as Na+ in these structures. We have probed the solution state of the enzyme using EPR and ENDOR spectroscopies, and have shown that a wide range of monovalent cations, including all but the largest alkali metals, is able to bind in this site, but going down Group 1 of the periodic table from Li+ to Cs+ there is a sharp activity maximum at K+, suggesting a precise requirement on the ionic radius. Indeed, the monovalent cation is coordinated by a carboxylate oxygen of SAM, with the other carboxylate oxygen of SAM coordinated to the unique iron of the [4Fe-4S] cluster, in a direct linkage between the monovalent cation and the catalytic cluster that allows spin transfer from the cluster to the monocation.
The presence of a bound cation appears to be essential for catalytic function, indicating that PFL-AE is a type I M+ activated enzyme with K+ being the physiologically most-active cation. This behavior links this radical SAM enzyme to the B12-dependent dioldehydratase, yet another parallel between the two classes of radical-generating enzymes, while the multiple approaches to the characterization of the PFL-AE cation provide even greater depth to its characterization. The cation’s location, catalytic importance, and modulation of substrate position all indicate its central mechanistic role. Indeed, this role might even be the same in both enzymes, control of an M-C bond cleavage to generate the 5′-dAdo radical.
Acknowledgments
This work was supported by the National Institutes of Health grants GM54608 (to J.B.B.) and GM111097 (to B.M.H.), and by the National Science Foundation grant MCB-0543833 (to C.L.D.). C.L.D. is a Howard Hughes Medical Institute Investigator.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b04883.
Details on protein production and purification; Figures S1–S11; Tables S1–S8 (PDF)
Author Present Address
⊥ Department of Applied Biochemistry and Food Science, Saga University, Saga 840–8502, Japan.
The authors declare no competing financial interest.
Supplementary Material
References
- Sofia H. J.; Chen G.; Hetzler B. G.; Reyes-Spindola J. F.; Miller N. E. Nucleic Acids Res. 2001, 29, 1097–1106. 10.1093/nar/29.5.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey P. A.; Hegeman A. D.; Ruzicka F. J. Crit. Rev. Biochem. Mol. Biol. 2008, 43, 63–88. 10.1080/10409230701829169. [DOI] [PubMed] [Google Scholar]
- Broderick J. B.; Duffus B. R.; Duschene K. S.; Shepard E. M. Chem. Rev. 2014, 114, 4229–4317. 10.1021/cr4004709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs C.; Broderick W. E.; Henshaw T. F.; Broderick J. B.; Huynh B. H. J. Am. Chem. Soc. 2002, 124, 912–913. 10.1021/ja017562i. [DOI] [PubMed] [Google Scholar]
- Walsby C. J.; Ortillo D.; Broderick W. E.; Broderick J. B.; Hoffman B. M. J. Am. Chem. Soc. 2002, 124, 11270–11271. 10.1021/ja027078v. [DOI] [PubMed] [Google Scholar]
- Horitani M.; Shisler K. A.; Broderick W. E.; Hutcheson R. U.; Duschene K. S.; Marts A. R.; Hoffman B. M.; Broderick J. B. Science 2016, 352, 822–825. 10.1126/science.aaf5327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knappe J.; Neugebauer F. A.; Blaschkowski H. P.; Gänzler M. Proc. Natl. Acad. Sci. U. S. A. 1984, 81, 1332–1335. 10.1073/pnas.81.5.1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner A. F. V.; Frey M.; Neugebauer F. A.; Schäfer W.; Knappe J. Proc. Natl. Acad. Sci. U. S. A. 1992, 89, 996–1000. 10.1073/pnas.89.3.996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henshaw T. F.; Cheek J.; Broderick J. B. J. Am. Chem. Soc. 2000, 122, 8331–8332. 10.1021/ja002012q. [DOI] [Google Scholar]
- Vey J. L.; Yang J.; Li M.; Broderick W. E.; Broderick J. B.; Drennan C. L. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 16137–16141. 10.1073/pnas.0806640105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker A.; Fritz-Wolf K.; Kabsch W.; Knappe J.; Schultz S.; Wagner A. F. V. Nat. Struct. Biol. 1999, 6, 969–975. 10.1038/13341. [DOI] [PubMed] [Google Scholar]
- Becker A.; Kabsch W. J. Biol. Chem. 2002, 277, 40036–40042. 10.1074/jbc.M205821200. [DOI] [PubMed] [Google Scholar]
- Peng Y.; Veneziano S. E.; Gillispie G. D.; Broderick J. B. J. Biol. Chem. 2010, 285, 27224–27231. 10.1074/jbc.M109.096875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu W.; Telser J.; Hoffman B. M.; Smith E. T.; Adams M. W. W.; Finnegan M. G.; Conover R. C.; Johnson M. K. J. Am. Chem. Soc. 1994, 116, 5722–5729. 10.1021/ja00092a024. [DOI] [Google Scholar]
- Rife C. L.; Pharris R. E.; Newcomer M. A.; Armstrong R. N. J. Am. Chem. Soc. 2002, 124, 11001–11003. 10.1021/ja026879v. [DOI] [PubMed] [Google Scholar]
- Schwartz P. A.; Frey P. A. Biochemistry 2007, 46, 7293–7301. 10.1021/bi700078z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata N.; Masuda J.; Tobimatsu T.; Toraya T.; Suto K.; Morimoto Y.; Yasuoka N. Structure 1999, 7, 997–1008. 10.1016/S0969-2126(99)80126-9. [DOI] [PubMed] [Google Scholar]
- Murshudov G. N.; Skubák P.; Lebedev A. A.; Pannu N. S.; Steiner R. A.; Nicholls R. A.; Winn M. D.; Long F.; Vagin A. A. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67, 355–67. 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potterton E.; Briggs P.; Turkenburg M.; Dodson E. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2003, 59, 1131–7. 10.1107/S0907444903008126. [DOI] [PubMed] [Google Scholar]
- Brünger A. T.; Adams P. D.; Clore G. M.; DeLano W. L.; Gros P.; Grosse-Kunstleve R. W.; Jiang J. S.; Kuszewski J.; Nilges M.; Pannu N. S.; Read R. J.; Rice L. M.; Simonson T.; Warren G. L. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1998, 54, 905–21. 10.1107/S0907444998003254. [DOI] [PubMed] [Google Scholar]
- Broderick J. B.; Henshaw T. F.; Cheek J.; Wojtuszewski K.; Smith S. R.; Trojan M. R.; McGhan R. M.; Kopf A.; Kibbey M.; Broderick W. E. Biochem. Biophys. Res. Commun. 2000, 269, 451–456. 10.1006/bbrc.2000.2313. [DOI] [PubMed] [Google Scholar]
- Nnyepi M. R.; Peng Y.; Broderick J. B. Arch. Biochem. Biophys. 2007, 459, 1–9. 10.1016/j.abb.2006.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsby C. J.; Hong W.; Broderick W. E.; Cheek J.; Ortillo D.; Broderick J. B.; Hoffman B. M. J. Am. Chem. Soc. 2002, 124, 3143–3151. 10.1021/ja012034s. [DOI] [PubMed] [Google Scholar]
- Krebs C.; Henshaw T. F.; Cheek J.; Huynh B.-H.; Broderick J. B. J. Am. Chem. Soc. 2000, 122, 12497–12506. 10.1021/ja003335p. [DOI] [Google Scholar]
- Conradt H.; Hohmann-Berger M.; Hohmann H.-P.; Blaschkowski H. P.; Knappe J. Arch. Biochem. Biophys. 1984, 228, 133–142. 10.1016/0003-9861(84)90054-7. [DOI] [PubMed] [Google Scholar]
- Brush E. J.; Lipsett K. A.; Kozarich J. W. Biochemistry 1988, 27, 2217–2222. 10.1021/bi00406a061. [DOI] [PubMed] [Google Scholar]
- Külzer R.; Pils T.; Kappl R.; Hüttermann J.; Knappe J. J. Biol. Chem. 1998, 273, 4897–4903. 10.1074/jbc.273.9.4897. [DOI] [PubMed] [Google Scholar]
- Stoll S.; Schweiger A. J. Magn. Reson. 2006, 178, 42–55. 10.1016/j.jmr.2005.08.013. [DOI] [PubMed] [Google Scholar]
- Werst M. M.; Davoust C. E.; Hoffman B. M. J. Am. Chem. Soc. 1991, 113, 1533–1538. 10.1021/ja00005a011. [DOI] [Google Scholar]
- Davoust C. E.; Doan P. E.; Hoffman B. M. J. Magn. Reson., Ser. A 1996, 119, 38–44. 10.1006/jmra.1996.0049. [DOI] [Google Scholar]
- Doan P. E.; Hoffman B. M. Chem. Phys. Lett. 1997, 269, 208–214. 10.1016/S0009-2614(97)00293-5. [DOI] [Google Scholar]
- Schweiger A.; Jeschke G.. Principles of Pulse Electron Paramagnetic Resonance; Oxford University Press: Oxford, U.K., 2001. [Google Scholar]
- Doan P. E.; Lees N. S.; Shanmugam M.; Hoffman B. M. Appl. Magn. Reson. 2010, 37, 763–779. 10.1007/s00723-009-0083-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doan P. E.The past, present, and future of orientation-selected ENDOR analysis: Solving the challenges of dipolar-coupled nuclei. In Paramagnetic Resonance of Metallobiomolecules; Telser J., Ed.; American Chemical Society: Washington, D.C., 2003; pp 55–81. [Google Scholar]
- Wong C.; Fujimori D. G.; Walsh C. T.; Drennan C. L. J. Am. Chem. Soc. 2009, 131, 4872–4879. 10.1021/ja8097355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H.; Chruszcz M.; Lasota P.; Lebioda L.; Minor W. J. Inorg. Biochem. 2008, 102, 1765–1776. 10.1016/j.jinorgbio.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsby C. J.; Ortillo D.; Yang J.; Nnyepi M.; Broderick W. E.; Hoffman B. M.; Broderick J. B. Inorg. Chem. 2005, 44, 727–741. 10.1021/ic0484811. [DOI] [PubMed] [Google Scholar]
- Farrar C. E.; Jarrett J. T. Biochemistry 2009, 48, 2448–2458. 10.1021/bi8022569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkovitch F.; Nicolet Y.; Wan J. T.; Jarrett J. T.; Drennan C. L. Science 2004, 303, 76–79. 10.1126/science.1088493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horitani M.; Byer A. S.; Shisler K. A.; Chandra T.; Broderick J. B.; Hoffman B. M. J. Am. Chem. Soc. 2015, 137, 7111–7121. 10.1021/jacs.5b00498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horitani M.; Offenbacher A. R.; Carr C. A. M.; Yu T.; Hoeke V.; Cutsail G. E. III; Hammes-Schiffer S.; Klinman J. P.; Hoffman B. M. J. Am. Chem. Soc. 2017, 139, 1984–1997. 10.1021/jacs.6b11856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foner S. N.; Cochran E. L.; Bowers V. A.; Jen C. K. J. Chem. Phys. 1960, 32, 963–971. 10.1063/1.1730905. [DOI] [Google Scholar]
- Morton J. R.; Preston K. F.; Strach S. J.; Adrian F. J.; Jette A. N. J. Chem. Phys. 1979, 70, 2889–2893. 10.1063/1.437825. [DOI] [Google Scholar]
- Weil J. A.; Bolton J. R.; Wertz J. E.. Electron Paramagnetic Resonance: Elementary Theory and Practical Applications; John Wiley & Sons: New York, 1994. [Google Scholar]
- Schwartz P. A.; LoBrutto R.; Reed G. H.; Frey P. A. Protein Sci. 2007, 16, 1157–1167. 10.1110/ps.072768007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutcheson R. A. U.Mechanistic and spectroscopic investigations of pyruvate formate-lyase activating enzyme. Dissertation, Montana State University, 2012. [Google Scholar]
- Shabala L.; Bowman J.; Brown J.; Ross T.; McMeekin T.; Shabala S. Environ. Microbiol. 2009, 11, 137–148. 10.1111/j.1462-2920.2008.01748.x. [DOI] [PubMed] [Google Scholar]
- Page M. J.; Di Cera E. Physiol. Rev. 2006, 86, 1049–1092. 10.1152/physrev.00008.2006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.